 Harry Petrushkin
Harry Petrushkin Md. Samiul Hasan
Md. Samiul Hasan Miles R. Stanford2
Miles R. Stanford2 Graham R. Wallace
Graham R. Wallace- 1Clinical and Diagnostic Oral Sciences, Queen Mary University of London, London, UK
- 2Academic Unit of Ophthalmology, St Thomas’s Hospital, London, UK
- 3Centre for Translational Inflammation Research, University of Birmingham, Birmingham, UK
Behçet’s disease (BD) is a complex inflammatory disease, of unknown etiology. While disease pathogenesis remains unclear, a strong relationship between BD and HLA-B*51 has been established over the last 30 years. A number of theories exist regarding the cause of BD; however, few are able to account for the increased rates of HLA-B*51 positive individuals, particularly around the Mediterranean basin and Middle-East where the prevalence is highest. This review outlines current immunogenetic data on BD and the immunoregulatory role natural killer cells may play. It also describes the interaction of the killer immunoglobulin-like receptor – KIR3DL1 with its ligand Bw4, which is found on HLA-B51. Finally, CD94/NKG2D, MICA, and ERAP are outlined with regard to their potential roles in BD.
Behçet’s Disease
Behçet’s disease (BD) is a complex systemic inflammatory disorder, consisting of oral aphthous ulcers, genital ulcers, papulopustular and erythema nodosum-like skin lesions, uveitis, retinal vasculitis, thrombophlebitis, arterial aneurysms, and arthritis. The disease is found primarily along the ancient Silk Route from the Mediterranean Basin across Asia to Japan (1, 2). The prevalence of BD in UK is around 0.64/100,000, whereas in Istanbul 370/100,000 individuals are affected by this disease (3).
Natural Killer Cell Biology
Natural killer (NK) cells are lymphocytes of the innate immune system. They are involved in the early stages of defense against foreign and self-cells undergoing stress. NK cells comprise 10–15% circulating lymphocytes in humans, falling mostly into two distinct populations. About 90% are CD56dimCD16+ve cells, capable of killing and antibody dependent cytotoxicity. These cells produce relatively little IFNγ (4, 5). NK cells identify their targets through a set of activatory or inhibitory receptors, which recognize non-self targets as well as self-proteins that are upregulated as a result of stress. They can also kill cells that down regulate expression of self-MHC molecules during times of infection or transformation (6, 7).
The other 10% are CD56brightCD16−ve and are weakly cytotoxic. These cells do not proliferate vigorously in response to IL-2, but produce substantial amounts of IFNγ, granulocyte–macrophage colony-stimulating factor (GM-CSF), chemokines such as chemokine ligand 3 (CCL3) and CCL4 (8). It has been suggested that CD56brightCD16−ve cells utilize their cytokine production to play a regulatory role in immune responses while CD56dimCD16+ve cells are terminally differentiated cytotoxic effector cells (9).
Natural killer cells express a number of activatory and inhibitory receptors. Killer immunoglobulin-like receptors (KIR) and NKG2D are discussed in detail below with respect to their role in BD. Natural cytotoxicity receptors (NCR) are activatory receptors found on NK cells. There are three known receptors; NKp46, NKp30, and NKp44. NKp46 and NKp30 are constitutively expressed, whereas NKp44 is expressed during NK cell activation by IL-2 (10, 11). NCR have been investigated in the context of tumor biology and cytokine production; however, they do not interact directly with class-I ligands and there is limited evidence for their role in autoimmune/autoinflammatory disease.
IL-15 and IL-15Rα are essential for normal development of naive CD8+ve T cells, intestinal intraepithelial lymphocytes, and NK cells. IL-15 shares structural similarity with IL-2 and both cytokines signal through CD122 to increase proliferation of NK cells (12).
Elevated IL-15 levels have been reported in serum, cerebrospinal fluid, and aqueous humor from patients with BD (13–15). Serum IL-15 levels have been found to correlate with erythrocyte sedimentation rate but not C-reactive protein levels and IL-15Rα expression has been found to be significantly reduced in leukocytes from patients compared to controls (12). While these findings suggest a possible role for IL-15 in BD pathogenesis, the mechanism remains unclear. It would be useful to establish whether there is a link between BD clinical activity and serum/organ specific IL-15/IL-15Rα levels. The effect of altered levels of IL-15 and the IL-15R complex is known to impact NK cell proliferation thus suggesting a possible pathway for NK cell control in the disease.
NK Cell Licensing
Natural killer cells are “licensed” to recognize and kill cells that do not express self-MHC. In 1986, Karre et al. described this phenomenon as the “missing self” hypothesis (16). This states that NK cells are activated to detect and kill cells that are thought to be non-self, i.e., infected cells or those undergoing neoplastic changes. Over the last 30 years, a number of authors have demonstrated that NK cells from MHC-deficient mice and humans do not effectively kill target cells (17, 18).
Natural killer cell receptors such as KIR have loci that segregate independently of MHC loci. Thus, in any one individual, some NK cells do not have a corresponding inhibitory HLA ligand, i.e., KIR3DL1+ve cells in a Bw6 homozygote. It is unclear how such cells could be prevented from killing autologous cells. Yokoyama et al. proposed that KIR on NK cells must recognize their cognate HLA ligand in order to acquire functional competence through licensing (19). An alternate “disarming” hypothesis proposes that NK cells that fail to recognize MHC class-I via inhibitory KIR become anergic (20). Hence, paradoxically, an inhibitory receptor interaction appears to be required for an NK cell to acquire function.
In the context of infection, unlicensed NK cells may become activated in response to cytokines from infected cells. This may permit a broader repertoire of NK cells to contribute to the response to pathogens without the risk of autoimmunity (21). Moreover, the licensing concept embraces gene dosage, i.e., KIR3DL1+ve cells in donors who are homozygous for Bw4 display increased responsiveness to tumor stimulation compared to heterozygotes and those who lack the ligand. In contrast, NK cells lacking KIR3DL1 show no difference in activity (22).
Genetic Studies in Behçet’s Disease
Evidence for a genetic association in BD first came about in 1982, when Ohno et al. published their results suggesting that HLA-B*51 was associated with BD in Japanese patients (23). This association has become recognized as the most frequently observed genetic factor in BD and is present in between 13 and 80% of patients (24). HLA-B51 presents antigen to CD8+ve cytotoxic T cells, but is also known to interact with the KIR3DL1 via its Bw4 epitope (25) (Figure 1). KIR interacts with position 7 and 8 of the HLA-bound peptide and with positions 77–83 of HLA molecules that have the Bw4 epitope (26, 27). Bw4 is present on a third of all HLA-B alleles and is defined by leucine at position 82 and arginine at position 83. It is associated with strong inhibition of NK cell activity via KIR3DL1 interaction. The Bw6 epitope (Ser 77, Asn 80, Leu 81, Arg 82, and Gly 83) does not interact with KIR3DL1 (28). There is considerable variation within the KIR haplotype. KIR genes differ in content and copy number and not all alleles are present in every individual.
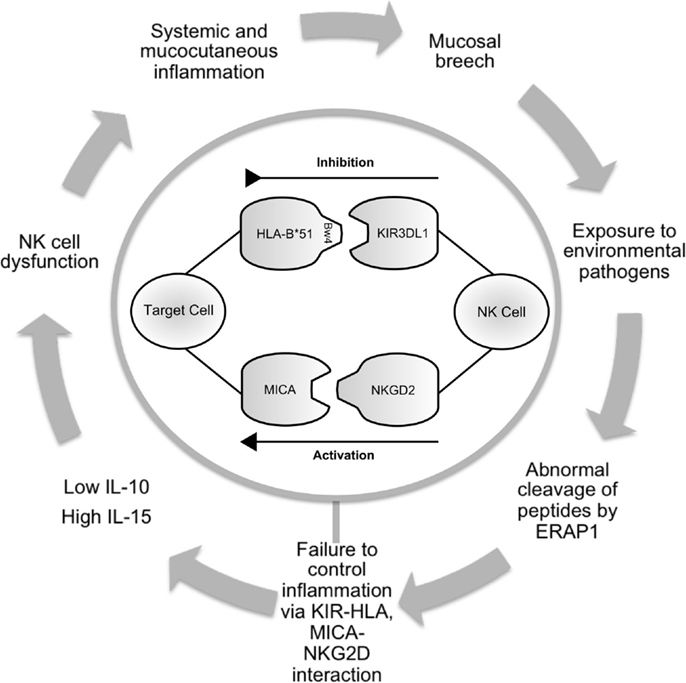
Figure 1. Schematic illustrating the potential role of NK cells in Behçet’s disease. Infection or inflammation at mucocutatneous surfaces may persist due to dysfunction of the innate immune response in predisposed individuals. ERAP1 may have a role in processing peptides that are presented by HLA-class-I molecules. KIR3DL1 inhibits cell-mediated cytotoxicity via interaction with HLA-B*51. This effect is balanced by activatory interactions via NKG2D and MICA. Production of cytokines including IL-10 and IL-15 contribute to abnormal NK cell function leading to prolonged inflammation and further episodes. NK cell, natural killer cell; ERAP, endoplasmic reticulum aminopeptidase 1; HLA, human leukocyte antigen; KIR, killer immunoglobulin-like receptor; MICA, MHC class-I polypeptide-related sequence A; NKG2D, natural killer group 2; receptor D; IL, interleukin.
The Bw4 epitope can be found on other HLA alleles associated with BD, such as HLA-B*2702 (29) as well as those thought to be protective, such as HLA-B*58 or HLA-A*0301 (30). The association with HLA-B*51 may be due to peptide binding with low affinity leading to reduced tolerance and/or inefficient NK cell licensing (31).
Four genome-wide association studies (GWAS) investigating genetic associations in BD have been carried out to date and all have confirmed association with the MHC region (32–35). In the last 3 years, data from these studies have been subjected to imputational analysis (a process using known haploytpes to allow testing of untyped genetic variants for association with a trait of interest) and a number of genes were found to achieve significance including: C–C motif chemokine receptor 1 (CCR1), STAT4, killer cell lectin-like receptor subfamily C (KLRC4), and endoplasmic reticulum amino peptidase 1 (ERAP1) (35). All of these genes are involved in cytotoxicity in some capacity. Of note, KLCRC4 is located on a haplotype block, which also codes for other NK cell receptors, such as NKG2D (36). A functional role for this gene has yet to be established, but there is evidence for reduced cytotoxicity in NK cells from patients carrying this haplotype (37) (Table 1).

Table 1. Genes implicated in the pathogenesis of Behçet’s disease and their potential mechanisms of action.
In 2013, Hughes et al. closely examined the HLA region using deep sequencing, and identified rs116799036, a SNP in the HLA-B*51 promoter region, 24 kb upstream from HLA-B*51 and 18 kb upstream of MICA, which was found to achieve genome-wide significance even when controlled for the effects of HLA-B*51. Furthermore, when controlling for the effects of rs116799036, the association with HLA-B*51 was abrogated, suggesting that the association between HLA-B*51 and BD may not be causal (38). However, this finding was not replicated in a larger cohort, which confirmed HLA-B*51 as the strongest association (46). In addition, this study found that HLA-A*03, HLA-B*15, HLA-B*27, HLA-B*49, HLA-B*57, and HLA-A*26 each contributed independently to the risk of developing BD. This data support HLA-B*51 as the strongest association in BD.
Functional Activity of NK Cells in Behçet’s Disease
In order to investigate the functional activity of NK cells in BD, Yamaguchi et al. assessed 47 patients with BD (10 with active disease). CD69+ve NK cells were significantly increased in active BD, but their cytotoxic function was similar to inactive and control subjects. IL-12Rβ2 mRNA levels were decreased in NK cells from patients with active BD, compared to inactive patients or healthy controls. Furthermore, NK cells from inactive patients suppressed IFNγ production by CD4+ve T cells from patients with active disease (45). These findings suggest that while NK cells proliferate in active BD, they may be rendered hyporesponsive due to an abnormality in IL-12 signaling. Hamzaoui et al. also described hyporesponsive NK cells in bronchoalveolar lavage (BAL) samples from patients with pulmonary BD. The authors did not examine the effects of IL-12, but instead focused on IL-2/IL-15Rβ (CD122), which was found to have reduced surface expression. CD122 is shared by the IL-15 receptor, which is partly responsible for maintenance and cytotoxicity of NK cells (47).
Treusch et al. evaluated the effect of IFNα-2a on the NK cell repertoire in BD. IFNα-2a is often used as a second or third line therapeutic agent in BD, particularly in the presence of ocular disease. The investigators found a significant decrease in circulating NK cells in active BD patients after treatment compared to controls. This reduction may be a direct effect of IFNα-2a or secondary induction of remission of BD. IFNα-2a has been postulated to work directly on NK cells by inducing apoptosis and indirectly by reducing IL-18, IL-12, and/or IL-21 (48).
NK Cell Receptors
Killer Immunoglobulin-Like Receptors
In humans, receptors belong to either the immunoglobulin-like superfamily (IgSF) or the C-type lectin-like receptor (CTLR) superfamily. KIR are part of the IgSF and have specific ligands in the HLA-class-I family (49).
Inhibitory KIR (i.e., KIR2DL and KIR3DL), contain immunoreceptor tyrosine-based inhibition motifs (ITIMs) in their cytoplasmic domains. Activating KIR molecules (i.e., KIR2DS and KIR3DS) lack ITIMs and have a charged residue in their transmembrane domains, which likely pair with the DAP12 signaling adapter. KIR2DL4 is unusual as it consists of a long intracellular region but only one ITIM and a positively charged amino acid in the transmembrane region. Unlike other clonally distributed KIR, KIR2DL4 is transcribed by all NK cells and acts as an activating receptor on recognition of HLA-G (50, 51). Inhibitory KIR consistently binds with a higher affinity than their activating counterparts (52).
Saruhan-Direskeneli et al. found no association of KIR3DL1 expression on NK or T cells in patients with BD, a finding not altered by the presence of Bw4 motif in patients (53). Similar results were found by Middleton et al. after analyzing 14 KIR genes in a cohort of Turkish patients and ethnically matched controls using a sequence-specific oligonucleotide probe (SSOP) method. They reported an increased frequency of KIR3DL1 and its ligand Bw4 (p = 0.0003) and a corresponding decreased frequency of KIR3DL1 without Bw4 (p = 0.00004) in patients compared to controls. However, this difference was abrogated when the presence of HLA-B*51 was controlled for (p = 0.7075) (54).
The frequency of 16 KIR genes (2DL1, 2DL2, 2DL3, 2DL4, 2DL5, 3DL1, 3DL2, 3DL3, 2DS1, 2DS2, 2DS3, 2DS4, 2DS5, 3DS1, 2DP1, and 3DP1) was assessed in Korean patients with either HLA-B27-associated ankylosing spondylitis and uveitis or BD related uveitis. In the patients with ankylosing spondylitis, the frequency of KIR3DL1 was significantly lower than healthy controls (p = 0.043), while there was no difference in patients with BD (55).
These studies support a role for KIR3DL1 and Bw4 in BD; however, there is currently insufficient evidence to suggest that this is via KIR3DL1–HLA-B51 interaction. The KIR cluster is highly polymorphic, second only to the HLA in complexity and is an unsuitable candidate for GWAS analysis as SNP probes targeting the region are unable to bind due to allelic and copy number variation within the gene. Other methods including targeted sequencing and functional analysis of different alleles must be explored further.
CD94/NKG2
The CD94/NKG2 heterodimer is a member of the CTLR family and recognizes HLA-E (56). It is thought that CD94/NKG2 work in conjunction with KIR by responding to changes in HLA expression and can also transduce both activating and inhibitory signals (57). The ligand for CD94/NKG2A is the non-classical HLA-E. Peptides bound to HLA-E function as modulators of NK cell activity. The NKG2D homodimer, is expressed at various levels on NK cells and recognizes MICA and UL16-binding protein (ULBP) leading to NK cell activation through DAP10 signaling.
Saruhan-Direskeneli et al. found CD94 expression was increased on CD56+CD16+ve and CD56+CD3+ve NK cells in patients with BD (53). It has been subsequently demonstrated that both CD94 (c.-134*T) and NKG2A (c.-4258*C, c.338-90*G) polymorphisms are associated with reduced risk of BD, an effect enhanced when combined with expression of HLA-E*0101. Individuals without this genotype had nearly a fivefold increased risk of developing BD. Furthermore, the NKG2C c.305*T polymorphism resulted in an increased rate of both ocular disease and arthritis (p = 0.0001), whereas the CD94 c.-134*A polymorphism was associated with oral, skin, genital, and gastrointestinal manifestations (58). Of note, HLA-E binds peptides derived from HLA-class-1 molecules and are also recognized by NK cells via CD94/NKG2A, resulting in an inhibition of cell lysis, thus providing another mechanism for NK cell regulation (59).
While frequently overlooked in the field of BD, HLA-E, and HLA-G have important roles in regulating inflammation. HLA-E is an activator of NK cells via its interaction with CD94/NKG2A and HLA-G inhibits the cytolytic activities of NK cells via its interaction with KIR2DL4 and LIR-1/LIR-2. A study in Korean patients with BD indicated that the variants; HLA-E*0101 and HLA-G*010101 (p = 0.0002, OR 0.7, p = 0.002, OR 0.7, respectively) were associated with a reduced risk of developing the disease, whereas HLA-E*010302, HLA-G*010102G*0105N alleles, and 3741_3754ins14bp were all associated with an increased risk of BD (p < 0.0001, OR 1.6; p = 0.002, OR 1.8; p = 0.024, OR = 2.0; and p = 0.003, OR 1.4, respectively). This data indicates that target cells expressing HLA-E*0101 and HLA-G*010101 are protected against NK and CD8+ve cell-mediated cytotoxicity. Conversely polymorphisms in HLA-E and HLA-G may lead to an imbalance of lymphocyte functions and to the development of BD (60).
MICA in Behcet’s Disease
MICA is a stress-induced antigen and is recognized by the lymphoid stress surveillance system. It is a ligand for NKG2D, and involved in NK cell activation (Figure 1). While it is generally agreed that the association between MICA and BD is largely due to linkage disequilibrium (LD) with HLA-B*51 (61–63), MICA may have an independent functional role in disease pathogenesis. In Korean patients, MICA*A6 homozygosity was associated with BD in both HLA-B*51+ve and HLA-B*51−ve patients. The authors also assessed the susceptibility risk of MICA*A6 homozygosity for BD in HLA-B*51−ve patients and found an independent association (p < 0.001, RR-22.27) (64).
The affinity of MICA as a ligand for NKG2D is not uniform. Low affinity MICA have a valine at codon 129 and include MICA*019 and MICA*009; these receptors have a 10- to 50-fold weaker affinity for NKG2D compared to those with methionine in position 129 (65). Moreover, an individual’s response to different MICA alleles is “tuned” and maintained over time (44).
Interestingly Munoz-Saa et al. commented that none of their 42 BD patients expressed a high affinity phenotype for NKG2D compared to 14% of controls, whereas other autoimmune diseases such as anterior uveitis, ankylosing spondylitis, and psoriatic arthritis are all associated with high affinity alleles (42, 66–68).
The association of MICA and MHC class-I haplotypes with BD, suggests a major role in the disease for cells regulated by these molecules including NK cells and subsets of T cells (40, 69).
Endoplasmic Reticulum Aminopeptidase
As discussed above, a polymorphism at the ERAP1 locus was associated with BD by imputation of GWAS data (35). The most documented function of ERAP1 and ERAP2 is to trim peptides that bind to MHC class-I molecules (Figure 1). Different ERAP1 variants can produce a different peptide pool, which in turn can affect folding and expression of MHC class-I on the cell surface and CD8 T-cell cytotoxicity. Furthermore, KIR and CD94/NKG2A function are affected by the peptide bound to MHC class-I (70). At present, there is no published data reporting the functional effects of endoplasmic reticulum aminopeptidase (ERAP) polymorphisms in BD. Future work addressing this area is warranted.
Conclusion
Several potential mechanisms explaining the pathogenesis of BD involve NK cells:
1. Defects in the NK cell repertoire may permit persistent viral infections, resulting in a chronic inflammatory response leading to BD.
2. NK cells lacking appropriate inhibitory KIR (such as KIR3DL1) may fail to recognize self-MHC and cause autologous tissue damage (71).
The role of NK cells in the pathogenesis of BD has been greatly advanced by genetic studies and the association of MICA and ERAP support involvement of the innate immune system.
However, major challenges remain, such as the lack of association between inhibitory KIR receptors and HLA-B51. I. In the last decade, over 50 new KIR3DL1 allotypes have been described, only a small proportion of which are present in different world populations (28, 72). It would be useful to establish the geographic spread of KIR3DL1 and correlate it to previously published work on HLA-B*51 (2).
It is possible that future work investigating KIR3DL1 polymorphisms will uncover allotypes previously untested for. Such studies, carried out in well-defined and characterized cohorts with adequate numbers to confirm the basic findings, to date, will not only strengthen the hypotheses outlined in this review but also create a body of evidence that may lead to potential new therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sakane T, Takeno M, Suzuki N, Inaba G. Behcet’s disease. N Engl J Med (1999) 341:1284–91. doi: 10.1056/NEJM199910213411707
2. Verity DH, Marr JE, Ohno S, Wallace GR, Stanford MR. Behcet’s disease, the silk road and HLA-B51: historical and geographical perspectives. Tissue Antigens (1999) 54:213–20. doi:10.1034/j.1399-0039.1999.540301.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Azizlerli G, Kose AA, Sarica R, Gul A, Tutkun IT, Kulac M, et al. Prevalence of Behcet’s disease in Istanbul, Turkey. Int J Dermatol (2003) 42:803–6. doi:10.1046/j.1365-4362.2003.01893.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Caligiuri MA. Human natural killer cells. Blood (2008) 112:461–9. doi:10.1182/blood-2007-09-077438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Inngjerdingen M, Kveberg L, Naper C, Vaage JT. Natural killer cell subsets in man and rodents. Tissue Antigens (2011) 78:81–8. doi:10.1111/j.1399-0039.2011.01714.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Raulet DH. Interplay of natural killer cells and their receptors with the adaptive immune response. Nat Immunol (2004) 5:996–1002. doi:10.1038/ni1114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: “l’union fait la force”. Blood (2005) 106:2252–8. doi:10.1182/blood-2005-03-1154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood (2001) 97:3146–51. doi:10.1182/blood.V97.10.3146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc Natl Acad Sci U S A (2011) 108:728–32. doi:10.1073/pnas.1012356108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Moretta A, Parolini S, Castriconi R, Bottino C, Vitale M, Sivori S, et al. Function and specificity of human natural killer cell receptors. Eur J Immunogenet (1997) 24:455–68. doi:10.1046/j.1365-2370.1997.d01-117.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Sivori S, Vitale M, Morelli L, Sanseverino L, Augugliaro R, Bottino C, et al. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J Exp Med (1997) 186:1129–36. doi:10.1084/jem.186.7.1129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Choi J, Lee ES, Choi B, Sohn S. Therapeutic potency of poly I:C in HSV-induced inflammation through up-regulation of IL-15 receptor alpha. Immunobiology (2013) 218:1119–30. doi:10.1016/j.imbio.2013.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Ahn JK, Yu HG, Chung H, Park YG. Intraocular cytokine environment in active Behcet uveitis. Am J Ophthalmol (2006) 142:429–34. doi:10.1016/j.ajo.2006.04.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Hamzaoui K, Hamzaoui A, Ghorbel I, Khanfir M, Houman H. Levels of IL-15 in serum and cerebrospinal fluid of patients with Behcet’s disease. Scand J Immunol (2006) 64:655–60. doi:10.1111/j.1365-3083.2006.01844.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Curnow SJ, Pryce K, Modi N, Knight B, Graham EM, Stewart JE, et al. Serum cytokine profiles in Behcet’s disease: is there a role for IL-15 in pathogenesis? Immunol Lett (2008) 121:7–12. doi:10.1016/j.imlet.2008.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature (1986) 319:675–8. doi:10.1038/319675a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Bix M, Liao NS, Zijlstra M, Loring J, Jaenisch R, Raulet D. Rejection of class I MHC-deficient haemopoietic cells by irradiated MHC-matched mice. Nature (1991) 349:329–31. doi:10.1038/349329a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Furukawa H, Yabe T, Watanabe K, Miyamoto R, Miki A, Akaza T, et al. Tolerance of NK and LAK activity for HLA class I-deficient targets in a TAP1-deficient patient (bare lymphocyte syndrome type I). Hum Immunol (1999) 60:32–40. doi:10.1016/S0198-8859(98)00097-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Yokoyama WM, Kim S. How do natural killer cells find self to achieve tolerance? Immunity (2006) 24:249–57. doi:10.1016/j.immuni.2006.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Raulet DH. Missing self recognition and self tolerance of natural killer (NK) cells. Semin Immunol (2006) 18:145–50. doi:10.1016/j.smim.2006.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature (2005) 436:709–13. doi:10.1038/nature03847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Kim S, Sunwoo JB, Yang L, Choi T, Song YJ, French AR, et al. HLA alleles determine differences in human natural killer cell responsiveness and potency. Proc Natl Acad Sci U S A (2008) 105:3053–8. doi:10.1073/pnas.0712229105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Ohno S, Ohguchi M, Hirose S, Matsuda H, Wakisaka A, Aizawa M. Close association of HLA-Bw51 with Behcet’s disease. Arch Ophthalmol (1982) 100:1455–8. doi:10.1001/archopht.1982.01030040433013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Gul A. Genome-wide association studies in Behcet’s disease: expectations and promises. Clin Exp Rheumatol (2011) 29:S3–5.
25. Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 public epitope of HLA-B molecules confers reactivity with natural killer cell clones that express NKB1, a putative HLA receptor. J Exp Med (1995) 181:1133–44. doi:10.1084/jem.181.3.1133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Peruzzi M, Parker KC, Long EO, Malnati MS. Peptide sequence requirements for the recognition of HLA-B*2705 by specific natural killer cells. J Immunol (1996) 157:3350–6.
27. Peruzzi M, Wagtmann N, Long EO. A p70 killer cell inhibitory receptor specific for several HLA-B allotypes discriminates among peptides bound to HLA-B*2705. J Exp Med (1996) 184:1585–90. doi:10.1084/jem.184.4.1585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Parham P, Norman PJ, Abi-Rached L, Guethlein LA. Variable NK cell receptors exemplified by human KIR3DL1/S1. J Immunol (2011) 187:11–9. doi:10.4049/jimmunol.0902332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Gul A, Uyar FA, Inanc M, Ocal L, Barrett JH, Aral O, et al. A weak association of HLA-B*2702 with Behcet’s disease. Genes Immun (2002) 3:368–72. doi:10.1038/sj.gene.6363863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Montes-Cano MA, Conde-Jaldon M, Garcia-Lozano JR, Ortiz-Fernandez L, Ortego-Centeno N, Castillo-Palma MJ, et al. HLA and non-HLA genes in Behcet’s disease: a multicentric study in the Spanish population. Arthritis Res Ther (2013) 15:R145. doi:10.1186/ar4328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Gebreselassie D, Spiegel H, Vukmanovic S. Sampling of major histocompatibility complex class I-associated peptidome suggests relatively looser global association of HLA-B*5101 with peptides. Hum Immunol (2006) 67:894–906. doi:10.1016/j.humimm.2006.08.294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Fei Y, Webb R, Cobb BL, Direskeneli H, Saruhan-Direskeneli G, Sawalha AH. Identification of novel genetic susceptibility loci for Behcet’s disease using a genome-wide association study. Arthritis Res Ther (2009) 11:R66. doi:10.1186/ar2695
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Mizuki N, Meguro A, Ota M, Ohno S, Shiota T, Kawagoe T, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behcet’s disease susceptibility loci. Nat Genet (2010) 42:703–6. doi:10.1038/ng.624
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Remmers EF, Cosan F, Kirino Y, Ombrello MJ, Abaci N, Satorius C, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet’s disease. Nat Genet (2010) 42:698–702. doi:10.1038/ng.625
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Genome-wide association analysis identifies new susceptibility loci for Behcet’s disease and epistasis between HLA-B*51 and ERAP1. Nat Genet (2013) 45:202–7. doi:10.1038/ng.2520
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Karasneh J, Gul A, Ollier WE, Silman AJ, Worthington J. Whole-genome screening for susceptibility genes in multicase families with Behcet’s disease. Arthritis Rheum (2005) 52:1836–42. doi:10.1002/art.21060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Hayashi T, Imai K, Morishita Y, Hayashi I, Kusunoki Y, Nakachi K. Identification of the NKG2D haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res (2006) 66:563–70. doi:10.1158/0008-5472.CAN-05-2776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Hughes T, Coit P, Adler A, Yilmaz V, Aksu K, Duzgun N, et al. Identification of multiple independent susceptibility loci in the HLA region in Behcet’s disease. Nat Genet (2013) 45:319–24. doi:10.1038/ng.2551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature (2000) 405:537–43. doi:10.1038/35014520
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Yabuki K, Mizuki N, Ota M, Katsuyama Y, Palimeris G, Stavropoulos C, et al. Association of MICA gene and HLA-B*5101 with Behcet’s disease in Greece. Invest Ophthalmol Vis Sci (1999) 40:1921–6.
41. Hughes EH, Collins RW, Kondeatis E, Wallace GR, Graham EM, Vaughan RW, et al. Associations of major histocompatibility complex class I chain-related molecule polymorphisms with Behcet’s disease in Caucasian patients. Tissue Antigens (2005) 66:195–9. doi:10.1111/j.1399-0039.2005.00465.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Munoz-Saa I, Cambra A, Pallares L, Espinosa G, Juan A, Pujalte F, et al. Allelic diversity and affinity variants of MICA are imbalanced in Spanish patients with Behcet’s disease. Scand J Immunol (2006) 64:77–82. doi:10.1111/j.1365-3083.2006.01780.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Mizuki N, Meguro A, Tohnai I, Gul A, Ohno S, Mizuki N. Association of major histocompatibility complex class I chain-related gene A and HLA-B alleles with Behcet’s disease in Turkey. Jpn J Ophthalmol (2007) 51:431–6. doi:10.1007/s10384-007-0473-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Shafi S, Vantourout P, Wallace G, Antoun A, Vaughan R, Stanford M, et al. An NKG2D-mediated human lymphoid stress surveillance response with high interindividual variation. Sci Transl Med (2011) 3:113ra124. doi:10.1126/scitranslmed.3002922
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Yamaguchi Y, Takahashi H, Satoh T, Okazaki Y, Mizuki N, Takahashi K, et al. Natural killer cells control a T-helper 1 response in patients with Behcet’s disease. Arthritis Res Ther (2010) 12:R80. doi:10.1186/ar3005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Ombrello MJ, Kirino Y, De Bakker PI, Gul A, Kastner DL, Remmers EF. Behcet disease-associated MHC class I residues implicate antigen binding and regulation of cell-mediated cytotoxicity. Proc Natl Acad Sci U S A (2014) 111:8867–72. doi:10.1073/pnas.1406575111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Hamzaoui K, Berraies A, Kaabachi W, Ammar J, Hamzaoui A. Pulmonary manifestations in Behcet disease: impaired natural killer cells activity. Multidiscip Respir Med (2013) 8:29. doi:10.1186/2049-6958-8-29
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Treusch M, Vonthein R, Baur M, Gunaydin I, Koch S, Stubiger N, et al. Influence of human recombinant interferon-alpha2a (rhIFN-alpha2a) on altered lymphocyte subpopulations and monocytes in Behcet’s disease. Rheumatology (Oxford) (2004) 43:1275–82. doi:10.1093/rheumatology/keh311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Lanier LL. NK cell receptors. Annu Rev Immunol (1998) 16:359–93. doi:10.1146/annurev.immunol.16.1.359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Rajagopalan S, Fu J, Long EO. Cutting edge: induction of IFN-gamma production but not cytotoxicity by the killer cell Ig-like receptor KIR2DL4 (CD158d) in resting NK cells. J Immunol (2001) 167:1877–81. doi:10.4049/jimmunol.167.4.1877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Faure M, Long EO. KIR2DL4 (CD158d), an NK cell-activating receptor with inhibitory potential. J Immunol (2002) 168:6208–14. doi:10.4049/jimmunol.168.12.6208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Katz G, Markel G, Mizrahi S, Arnon TI, Mandelboim O. Recognition of HLA-Cw4 but not HLA-Cw6 by the NK cell receptor killer cell Ig-like receptor two-domain short tail number 4. J Immunol (2001) 166:7260–7. doi:10.4049/jimmunol.166.12.7260
53. Saruhan-Direskeneli G, Uyar FA, Cefle A, Onder SC, Eksioglu-Demiralp E, Kamali S, et al. Expression of KIR and C-type lectin receptors in Behcet’s disease. Rheumatology (2004) 43:423–7. doi:10.1093/rheumatology/keh063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Middleton D, Meenagh A, Sleator C, Gourraud PA, Ayna T, Tozkir H, et al. No association of KIR genes with Behcet’s disease. Tissue Antigens (2007) 70:435–8. doi:10.1111/j.1399-0039.2007.00929.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Moon SJ, Oh EJ, Kim Y, Kim KS, Kwok SK, Ju JH, et al. Diversity of killer cell immunoglobulin-like receptor genes in uveitis associated with autoimmune diseases: ankylosing spondylitis and Behcet disease. Ocul Immunol Inflamm (2013) 21:135–43. doi:10.3109/09273948.2012.754905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Lee N, Llano M, Carretero M, Ishitani A, Navarro F, Lopez-Botet M, et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A (1998) 95:5199–204. doi:10.1073/pnas.95.9.5199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Winter CC, Gumperz JE, Parham P, Long EO, Wagtmann N. Direct binding and functional transfer of NK cell inhibitory receptors reveal novel patterns of HLA-C allotype recognition. J Immunol (1998) 161:571–7.
58. Seo J, Park JS, Nam JH, Bang D, Sohn S, Lee ES, et al. Association of CD94/NKG2A, CD94/NKG2C, and its ligand HLA-E polymorphisms with Behcet’s disease. Tissue Antigens (2007) 70:307–13. doi:10.1111/j.1399-0039.2007.00907.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Borrego F, Ulbrecht M, Weiss EH, Coligan JE, Brooks AG. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med (1998) 187:813–8. doi:10.1084/jem.187.5.813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Park KS, Park JS, Nam JH, Bang D, Sohn S, Lee ES. HLA-E*0101 and HLA-G*010101 reduce the risk of Behcet’s disease. Tissue Antigens (2007) 69:139–44. doi:10.1111/j.1399-0039.2006.00742.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Tian W, Boggs DA, Ding WZ, Chen DF, Fraser PA. MICA genetic polymorphism and linkage disequilibrium with HLA-B in 29 African-American families. Immunogenetics (2001) 53:724–8. doi:10.1007/s00251-001-0392-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Gao X, Single RM, Karacki P, Marti D, O’Brien SJ, Carrington M. Diversity of MICA and linkage disequilibrium with HLA-B in two North American populations. Hum Immunol (2006) 67:152–8. doi:10.1016/j.humimm.2006.02.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Cambra A, Munoz-Saa I, Crespi C, Serra A, Etxagibel A, Matamoros N, et al. MICA-HLA-B haplotype diversity and linkage disequilibrium in a population of Jewish descent from Majorca (the Balearic Islands). Hum Immunol (2009) 70:513–7. doi:10.1016/j.humimm.2009.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Park SH, Park KS, Seo YI, Min DJ, Kim WU, Kim TG, et al. Association of MICA polymorphism with HLA-B51 and disease severity in Korean patients with Behcet’s disease. J Korean Med Sci (2002) 17:366–70. doi:10.3346/jkms.2002.17.3.366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, et al. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics (2001) 53:279–87. doi:10.1007/s002510100325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Ricci-Vitiani L, Vacca A, Potolicchio I, Scarpa R, Bitti P, Sebastiani G, et al. MICA gene triplet repeat polymorphism in patients with HLA-B27 positive and negative ankylosing spondylitis from Sardinia. J Rheumatol (2000) 27:2193–7.
67. Gonzalez S, Brautbar C, Martinez-Borra J, Lopez-Vazquez A, Segal R, Blanco-Gelaz MA, et al. Polymorphism in MICA rather than HLA-B/C genes is associated with psoriatic arthritis in the Jewish population. Hum Immunol (2001) 62:632–8. doi:10.1016/S0198-8859(01)00242-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Amroun H, Djoudi H, Busson M, Allat R, El Sherbini SM, Sloma I, et al. Early-onset ankylosing spondylitis is associated with a functional MICA polymorphism. Hum Immunol (2005) 66:1057–61. doi:10.1016/j.humimm.2005.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Mizuki N, Ota M, Kimura M, Ohno S, Ando H, Katsuyama Y, et al. Triplet repeat polymorphism in the transmembrane region of the MICA gene: a strong association of six GCT repetitions with Behcet disease. Proc Natl Acad Sci U S A (1997) 94:1298–303. doi:10.1073/pnas.94.4.1298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Vivian JP, Duncan RC, Berry R, O’Connor GM, Reid HH, Beddoe T, et al. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature (2011) 479:401–5. doi:10.1038/nature10517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Takeno M, Shimoyama Y, Kashiwakura J, Nagafuchi H, Sakane T, Suzuki N. Abnormal killer inhibitory receptor expression on natural killer cells in patients with Behcet’s disease. Rheumatol Int (2004) 24:212–6. doi:10.1007/s00296-003-0352-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Tao SD, He YM, Ying YL, He J, Zhu FM, Lv HJ. KIR3DL1 genetic diversity and phenotypic variation in the Chinese Han population. Genes Immun (2014) 15:8–15. doi:10.1038/gene.2013.55
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Behçet’s syndrome, Behçet’s disease, Behçet’s, NK cells, KIR3DL1, KIR, HLA-B*51, HLA-B antigens
Citation: Petrushkin H, Hasan MS, Stanford MR, Fortune F and Wallace GR (2015) Behçet’s disease: do natural killer cells play a significant role? Front. Immunol. 6:134. doi: 10.3389/fimmu.2015.00134
Received: 08 December 2014; Accepted: 10 March 2015;
Published online: 24 March 2015.
Edited by:
Eleanor Riley, London School of Hygiene and Tropical Medicine, UKReviewed by:
Paul J. Norman, Stanford University School of Medicine, USAJohn Trowsdale, University of Cambridge, UK
Michael J. Ombrello, National Institute of Arthritis and Musculoskeletal and Skin Diseases, USA
Copyright: © 2015 Petrushkin, Hasan, Stanford, Fortune and Wallace. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harry Petrushkin, Clinical and Diagnostic Oral Sciences, Queen Mary University of London, Blizard Building, London E1 2AT, UK e-mail: h.j.d.petrushkin@qmul.ac.uk