 Chad L. Mayer1
Chad L. Mayer1 Caitlin S. L. Parello1Benjamin C. Lee1
Caitlin S. L. Parello1Benjamin C. Lee1 Kiyoshi Itagaki2
Kiyoshi Itagaki2 Shinichiro Kurosawa1*
Shinichiro Kurosawa1* Deborah J. Stearns-Kurosawa1
Deborah J. Stearns-Kurosawa1
- 1Department of Pathology and Laboratory Medicine, Boston University School of Medicine, Boston, MA, USA
- 2Department of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA
Hemolytic uremic syndrome (HUS) from enterohemorrhagic Escherichia coli infection is a leading cause of kidney failure in otherwise healthy U.S. children. The bacterial Shiga toxins (Stx) induce the characteristic coagulopathy of HUS, but the damage to toxin-receptor expressing cells and organ injury due to ischemia likely also releases inflammatory damage-associated molecular patterns (DAMPs), which may exacerbate injury along with the toxins. To examine this, human aortic and renal glomerular cell anti-coagulant and barrier functions were studied after in vitro challenge with Stx1, Stx2, and DAMPs. There was significant loss of surface anti-coagulant protein C pathway molecules, increased expression of pro-thrombotic PAR1 and reduced protein C activation capability by 15–27%. Histones nearly completely prevented the activated protein C protection of endothelial cells from thrombin-induced permeability. In mice, lethal Stx2 challenge elevated plasma HMGB1 (day 2, 321 ± 118%; p < 0.01) and extracellular histones (day 3, 158 ± 62%; p < 0.01). Mice colonized with Stx2-expressing Citrobacter rodentium developed increased HMGB1 (day 5, 155 ± 55%; p < 0.01) and histones (day 3, 378 ± 188%; p < 0.01). Anti-histone antibody reduced both DAMPs to baseline, but was not sufficient to improve survival outcome or kidney function. Together, these data suggest a potential role Stx to produce DAMPs, and DAMPs to produce endothelial injury and a pro-thrombotic environment.
Introduction
Enterohemorrhagic Escherichia coli (EHEC) are toxigenic intestinal bacteria that cause vomiting, diarrhea, edema, and hemorrhagic colitis. In some patients, the disease can progress to a potentially life-threatening syndrome known as diarrhea-associated hemolytic uremic syndrome (D + HUS), characterized by thrombotic microangiopathy, thrombocytopenia, and hemolytic anemia, all of which contribute to acute kidney injury (1). In the U.S., D + HUS is a leading cause of acute kidney failure in otherwise healthy children (2). EHEC produce and secrete Shiga toxin 1 (Stx1), Shiga toxin 2 (Stx2), or both, and serotypes that secrete Stx2 are associated with more clinically severe disease (3). Much of the pathogenesis observed during EHEC infection is attributed to the toxins, which are considered primary virulence factors of EHEC. The toxins bind to globotriaosylceramide (Gb3, CD77) receptors whose distribution is particularly high on renal glomerular endothelial cells in humans and on renal tubular epithelium in mice (4–6). The toxins are then internalized and transported to the endoplasmic reticulum, and the A subunit is activated to generate RNA N-glycosidase activity (7). Within the cytosol, the active A subunit cleaves an adenine residue from 28S ribosomal RNA, which prevents protein synthesis (8–10) and initiates ribotoxic (11, 12) and endoplasmic reticular-stress responses (13), leading to apoptosis and inflammation.
A major clinical feature of D + HUS is thrombotic microangiopathy, in which clots form inappropriately in the microvasculature, resulting in ischemic consequences and organ injury. As endothelial cells are crucial for maintaining blood fluidity and preventing leakage through the vessel walls, it is possible that Stx- and inflammation-induced perturbations in these functions contribute to the observed thrombotic microangiopathy. A critical pathway mediating these functions is the protein C pathway, which provides anti-coagulant, anti-inflammatory, and barrier protection activities (14). The activated form of the protein C zymogen mediates these activities, and activated protein C (APC) is generated by the protease action of thrombin, which is localized to the endothelial cell surface through binding to thrombomodulin (TM). Generation of APC is further facilitated by the endothelial protein C receptor (EPCR), which presents protein C zymogen to the cell surface thrombin/TM complex for more efficient activation (15, 16). During in vitro studies using human renal glomerular endothelial cells (HRGEC), Stx2-induced a small decrease in TM antigen expression (17), but resultant functional changes were not determined.
As an anti-coagulant, APC inhibits coagulation cofactors Va and VIIIa (18), but its barrier-protective activity is mediated by its occupation of EPCR and subsequent activation of protease-activated receptor 1 (PAR1). PAR1 is intimately involved in endothelial barrier function, and signaling by this discriminatory receptor is protease-specific depending on whether it is activated by APC, thrombin, or other proteases (19–22). PAR1 activation by thrombin contributes to thrombosis while also increasing endothelial barrier permeability; however, PAR1 activation by APC in concert with EPCR elicits an opposite barrier-protective effect. Although human endothelial cells express the Gb3 toxin receptor, little is known about how the Shiga toxins impact expression and function of PAR1, EPCR, and TM, and disruption of these molecules can have significant consequences (23, 24).
Enterohemorrhagic Escherichia coli are generally non-invasive, but the intestinal damage observed during EHEC infection can be considerable, with inflammation, hemorrhage, edema, and focal necrosis predominating (25). Often released by damaged cells are molecules termed damage-associated molecular patterns (DAMPs) (26): normal, endogenous molecules that can be extruded from the cell into the blood or tissue. Examples of DAMPs include histones, which can circulate or localize in neutrophil extracellular traps (27), or HMGB1 from monocytes (28). DAMPs also are released from necrotic cells, and circulating DAMPs can activate many of the same receptors as pathogen-associated molecular patterns to propagate inflammation and tissue damage (29, 30). Some DAMPs also can cause endothelial dysfunction manifested by increased permeability (31) or increased platelet adhesion (27). Although it has not been repeatedly demonstrated that DAMPs are released in the context of EHEC infection or Shiga toxin release, DAMPs from damaged tissue increase in several patient and animal models of sepsis and trauma (32–35). Given the extent of intestinal and kidney injury after EHEC toxin challenge (36–38), and the relative paucity of Shiga toxins observed in serum during hemolytic uremic syndrome (HUS) (39), we hypothesized that injury to any cell expressing the Gb3 receptor for Shiga toxins would release DAMPs, and that those DAMPs compromise the antithrombotic and barrier-protective properties of endothelial cells, leading to thrombotic microangiopathy and HUS.
Materials and Methods
Reagents
Plasma levels of HMGB1 and extracellular histones were measured using ELISAs for HMGB1 (IBL-international, Toronto, ON, Canada, and Chondrex Inc., Redmond, WA, USA) and cell-death detection (Roche, Indianapolis, IN, USA), respectively. Human aortic endothelial cells (Cascade Biologics, Grand Island, NY, USA) or HRGEC (Sciencell, Carlsbad, CA, USA) were purchased and grown in endothelial cell medium (Sciencell) supplemented with 5% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, endothelial cell growth supplements according to the manufacturer’s instructions. These cell lines are morphologically similar, but HAEC grow more quickly and form tighter junctions, in addition to not being fenestrated as renal glomerular endothelial cells are. All experiments were performed between passage 2 and 6. Antibodies were purchased against PAR1 (ATAP2, Santa Cruz Biotechnology, Dallas, TX, USA, un/conjugated to Alexa 488), and TM, clone 1029, conjugated to Oregon green, for flow cytometry, and clone 1A4 for on-cell western (Becton Dickinson, Franklin Lakes, NJ, USA). Anti-EPCR (JRK 1494, un/conjugated to Alexa 488) were available from C.T. Esmon (Oklahoma Medical Research Foundation, Oklahoma City, OK, USA). Goat anti-mouse IRDye 800CW for use in on-cell westerns was purchased from Li-COR (Lincoln, NE, USA). Stx1 and Stx2 were purchased from Tufts University and endotoxin was removed by incubation with polymyxin B-agarose (Sigma Aldrich, St. Louis, MO, USA). Residual endotoxin levels were <0.015 ng/mL (LAL Chromogenic Endotoxin Quantitation Kit, Thermo Scientific, Rockford, IL, USA). Hybridomas producing anti-histone IgG clone BWA3 (40) were kindly provided by Dr. Ann Rothstein (Department of Immunology and Virology, University of Massachusetts Medical School, Worcester, MA, USA) and the antibody was purified from conditioned media using HiTrap Protein G columns (GE Healthcare, Piscataway, NJ, USA) and standard methods. Calf thymus histones and TNFα were purchased from Sigma Aldrich. Human plasma-derived protein C was a kind gift from Kaketsuken (Kumamoto, Japan). Spectrozyme PCa was purchased from American Diagnostica (Lexington, MA, USA), human α-thrombin, and anti-thrombin III were from Hematologic Technologies, Inc. (Essex Junction, VT, USA). All other reagents such as bovine serum albumin (BSA), ethlyenediaminetetraacetic acid (EDTA), and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were laboratory or research grade and purchased from Sigma Aldrich and ThermoFisher Scientific.
Mouse Model of Stx2 Toxemia
Mice were purchased from the Jackson Laboratories (Bar Harbor, ME, USA) for all experiments. All animal studies were approved by the Boston University Institutional Animal Care and Use Committee. Mice (6-week-old male C57BL/6J) were injected intraperitoneally with 1 ~ 1.2 ng Stx2/20 g bodyweight at day 0 and day 3. Control mice received equal volumes of normal saline by intra-peritoneal injection. Blood was collected into 5 mM EDTA by facial vein bleed prior to each Stx2 challenge, and periodically until euthanasia for downstream analyses, consistent with the IACUC-approved protocols. Animals were monitored and weighed daily and plasma blood urea nitrogen (BUN) was quantified as a marker of acute kidney injury using a QuantiChrome Urea Assay Kit (BioAssay Systems, Hayward, CA, USA).
Mouse Model of EHEC Infection
Mice (6-week-old female C57BL/6J) were inoculated with Citrobacter rodentium. This is a Gram negative mouse pathogen that either was lysogenized with an Stx2-producing phage (strain DBS770) in order to express the Shiga toxin, or a control strain that does not express the toxin (strain DBS771). C. rodentium strains were kindly provided by Dr. John M. Leong (Department of Molecular Biology and Microbiology, Tufts University Medical Center, Boston, MA, USA) and have been described in detail elsewhere (41). Briefly, bacteria were grown in LB broth containing chloramphenicol (final 10 μg/mL) or both chloramphenicol and kanamycin (25 μg/mL) to an OD600 of 0.75–0.90. Bacteria were washed, re-suspended in sterile saline, and mice were challenged with 1 × 109 CFU by oral gavage on day 0. Weight was monitored daily and plasma collected periodically by facial vein bleed. CFU/g feces was determined by mixing feces with 10 volumes of phosphate buffered saline (PBS) (ThermoScientific), homogenizing with a sterile toothpick, centrifuging 30 s at 4000 rpm, and plating serially diluted supernatants on LB agar with appropriate restrictive antibiotics. Plates were grown overnight at 32°C and colonies were counted the next day.
Flow Cytometry
Human aortic endothelial cells between passage 2 and 6 were seeded on 0.1% gelatin-coated plates at 1.5 × 104 cells/cm2. The next day, media was replaced with fresh media containing Stx1 or Stx2 (100 ng/mL), extracellular histones (50 μg/mL), or combinations. Cells maintained ≥85% viability under these conditions. Cells were incubated for 24 h, washed with PBS and then detached using Versene (Invitrogen) and passed through a 40 μm nylon mesh. All subsequent steps were done on ice. Cells were washed in PBS with 25 mm HEPES, 0.5% BSA, and 5 mM EDTA, pH 7.4 (flow buffer) before staining with primary conjugated antibodies (2 μg/mL) for 30 min. Cells were washed again and re-suspended in flow buffer. Dead cells were excluded by propidium iodide staining. Cell associated fluorescence was determined using a FACSCalibur flow cytometer (Becton Dickinson) and data analyzed using FlowJo X (Treestar Inc., Ashland, OR, USA) software. Based on peak morphology (one peak vs. bimodal), the geometric mean or an M1 region expressed as a percentage was used for data analysis. One-way ANOVA with a Dunnett posttest was used to determine significant differences between geometric means or M1 regions at p < 0.05.
On-Cell Western
Antigen expression on HRGEC was done by on-cell western due to difficulty in producing large numbers through passaging. On-cell western was performed with aortic endothelial cells as well, specifically to compare surface antigen expression with HRGEC at baseline. Optimal toxin and histone concentrations that impacted surface phenotype while maintaining cell viability were determined in preliminary dose–response studies. Cell viability was maintained at 82–96% under experimental conditions (data not shown). Black-walled, clear-bottomed 96-well tissue culture plates (Corning, Tewksbury, MA, USA), of the type used for fluorescent experiments, were coated with 0.1% gelatin and then seeded with HRGEC (Sciencell) grown in endothelial cell media. When cells were near confluent, media were replaced and challenges were added. After 24 h, cells were washed once with PBS then fixed for 20 min at room temperature in PBS with 3.7% formaldehyde. After fixation, cells were washed twice with 50 mM Tris, pH 7.4, 150 mM NaCl, 0.05% Tween-20 (TBST), and blocked with Odyssey blocking buffer (Li-Cor, Lincoln, NE, USA) at room temperature with gentle shaking. Following the blocking step, cells were washed twice with TBST and incubated with primary unconjugated antibodies for EPCR (JRK 1494), PAR1 (ATAP2), or TM (1A4) at 2 μg/mL in Odyssey blocking buffer. Cells were washed twice with TBST and incubated with goat anti-mouse IRDye 800CW (Li-Cor) diluted 1:800 in Odyssey blocking buffer and Draq5 (5 mM stock, Thermo Scientific) diluted 1:10,000. Plates were washed with TBST, and all liquid aspirated before reading fluorescent values on a Li-Cor Odyssey dual-channel infrared imager at 700 and 800 nm. Images were analyzed with Odyssey 2.1 software (LiCor), and fluorescent intensities at 800 nm were normalized to intact cell number based on Draq5 staining at 700 nm. One-way ANOVA with a Dunnett posttest was used to determine significance.
Protein C Activation Assay
Confluent human aortic endothelial cells (HAEC) were incubated with Stx1 (100 ng/mL), Stx2 (100 ng/mL), calf thymus histones (50 μg/mL), or combinations. Confluent HRGEC were challenged with Stx1 (50 ng/mL), Stx2 (50 ng/mL), or histones (50 μg/mL). For experiments with anti-histone BWA3 monoclonal antibody, endothelial cells were challenged with 30 μg/mL histones with or without BWA3 at 20 μg/mL. All challenges were done for 24 h at 37°C in 5% CO2. Cells were washed with Hank’s balanced salt solution (HBSS; Thermo) and protein C was added at a final concentration of 0.4 μM in 0.1 mM HEPES, pH 7.4, 0.6 mM MgCl2, and 1 mM CaCl2. Human α-thrombin (10 nM final) was added and the reaction was stopped after 30 min at 37°C with HBSS, 0.1 mM HEPES, 0.5 mg/mL human ATIII, and 1.4 U/mL heparin. Spectrozyme PCa (American Diagnostica, Lexington, MA) was added (0.2 μM final) and the change in absorbance 405 nm with time was performed on a VersaMax ELISA Microplate Reader and analyzed with SoftMax Pro 5.4 (Molecular Devices, Sunnyvale, CA, USA). APC levels were determined by reference to a standard curve prepared with purified human APC, and normalized to intact cell numbers determined using Draq5 staining of a parallel plate, then normalized to 100% activation by unchallenged cells. One-way ANOVA with a Dunnett posttest was used to determine significant differences.
Endothelial Cell Permeability
Endothelial cell monolayer permeability changes were quantified from changes in electrical resistance across a human aortic endothelial monolayer as monitored by electric cell-substrate impedance sensing using an ECIS® ZO array station (Applied Biophysics, Troy, NY, USA). For each experiment, two 40 electrode/well 8W10E+ electrode chamber slides (Applied Biophysics) were coated with 10 mM l-cysteine/well for 10 min, washed 2 times with HBSS, coated with 5 μM fibronectin in HBSS for 10 min, then washed 2 times more with HBSS and allowed to dry. HAEC between passages 2 and 6 were seeded at a density of 2.5 × 105 cells/chamber and allowed to adhere and form a monolayer as determined by the stabilizing capacitance at 64 kHz. For experiments with pre-treatments, histones (25 μg/mL) or Stx2 (100 ng/mL) were added to the appropriate wells 12 h before thrombin addition. Thrombin was added to 1 U/mL (16.85 nM) and the experiment was followed for at least 12 h after thrombin addition. For experiments using APC, concentrations of thrombin were lowered to 2 nM in order for the protective effect of APC on PAR1 to be observed (19). Pre-incubation with APC (10 nM) was done for 3 h prior to thrombin challenge. The area under the curve for normalized resistance over the selected range of time was determined and one-way ANOVA with a Dunnett posttest was used to determine significance.
Results
Large Vessel Endothelium Expresses More Surface EPCR, PAR1, and TM Antigen than Microvascular Renal Endothelium
Differential endothelial expression of EPCR and TM is well-recognized, and while TM is widely expressed, surface EPCR expression is known to increase with vessel size (42). Relative quantitation of TM and EPCR levels in different vascular beds is not well established, particularly for renal endothelium. In order to better interpret the impact of Stx and DAMPs on receptor expression, quantitative comparisons of baseline surface EPCR, PAR1, and TM on the surface of normal, unchallenged HAEC, representing large vessel endothelium, and HRGEC, representing small vessel endothelium, were performed. These experiments differed from those involving Stx and DAMP challenges in that they were accomplished by performing on-cell westerns with both cell types. We observed that at baseline HAEC expressed significantly higher antigen amounts of EPCR (7.4-fold), PAR1 (1.8-fold), and TM (3.2-fold) when compared to HRGEC (Figure 1).
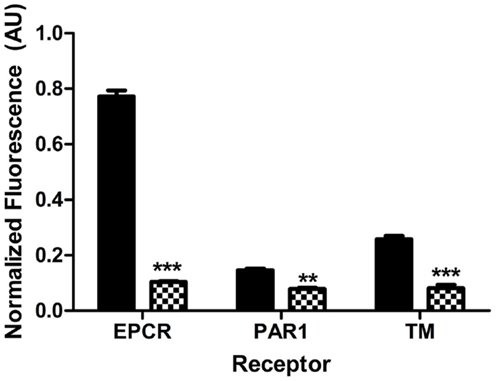
Figure 1. Different endothelium express different amounts of PAR1, EPCR, and TM. Surface expression of EPCR, PAR1, and TM on large vessel human aortic endothelial cells (HAEC, black) and microvascular human renal glomerular endothelial cells (HRGEC, checkered). The significance is determined by comparing the amount of EPCR, PAR1, or TM on each cell type in unchallenged cells at 24 h. Constitutive expression of EPCR, PAR1, and TM on both HAEC and HRGEC for this figure was determined by on-cell western, as described in Section “Materials and Methods.” Comparison was done by two-way ANOVA with a Bonferroni posttest. **p < 0.01, ***p < 0.001.
Stx and DAMPs Shift Endothelial Expression of PAR1 and Protein C Pathway Molecules
The abilities of the Shiga toxins and histone DAMPs to alter surface expression of endothelial cell molecules have important implications for maintaining blood fluidity and were determined by flow cytometry with HAEC (Figure 2). HAEC challenged with histones demonstrated significantly increased expression of thrombin receptor PAR1 relative to control cells (Figure 2A; p < 0.001), whereas challenge with either Stx1 or Stx2 caused a subset of cells (35–40%) to significantly lower their surface expression of PAR-1 (Figure 2B; p < 0.001). EPCR surface antigen expression was reduced significantly relative to media alone in cells incubated with histones or Stx (Figures 2C,D; p < 0.001). TM expression was reduced after histone treatment (Figure 2E; p < 0.001); however, Shiga toxins did not affect TM levels (Figure 2F).
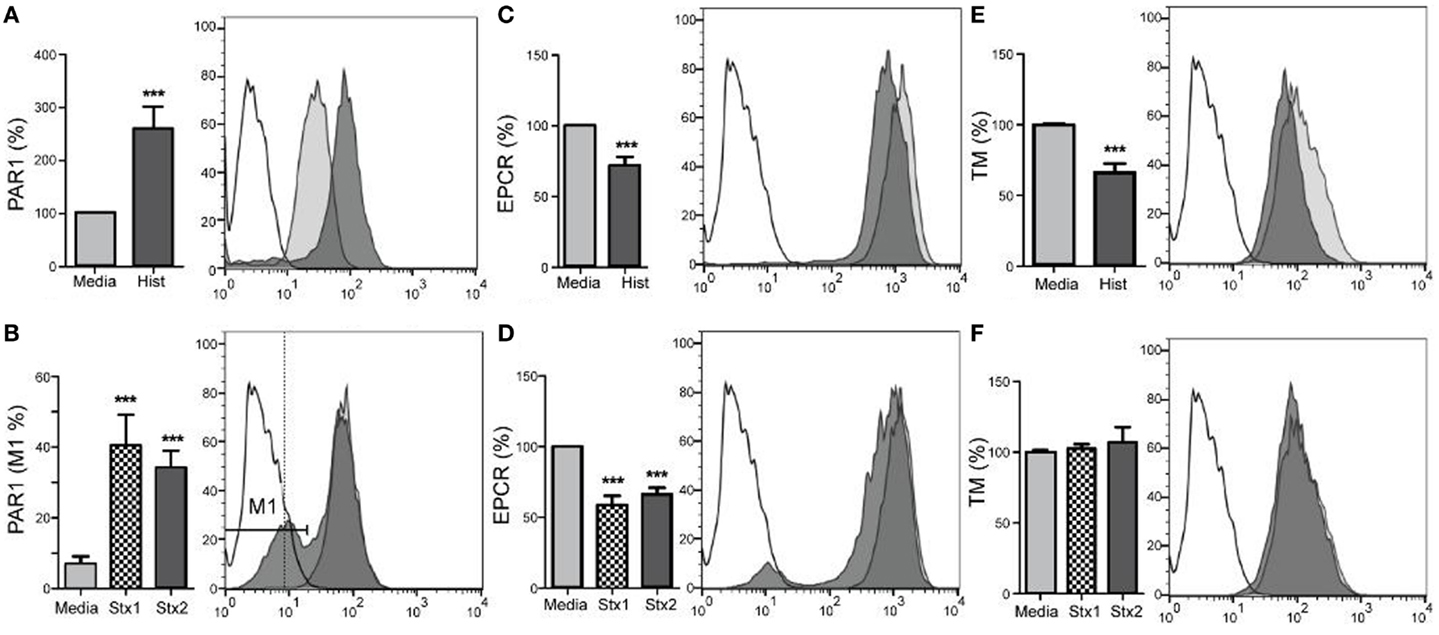
Figure 2. Stx1, Stx2, and histones induce changes in aortic endothelial expression of surface molecules. Human aortic endothelial cells were challenged with 50 μg/mL histones (A,C,E) or 100 ng/mL Stx1 (B,D,F) and surface expression of PAR-1, EPCR, and TM were quantified by flow cytometry using antigen-specific antibodies. Histogram overlays show representative staining of unchallenged cells (gray) and challenged cells (dark) compared to background staining (unfilled), with challenges represented using Stx1, as there were no differences in effect with respect to which Shiga toxin was used. Bar graphs show geometric means ± SD of three to four experiments each, except (B), which compares the M1 region 1, expressed as a percentage. Differences from constitutive expression (media only = 100%) are shown as ***p < 0.001.
As the primary target organ of the toxins in humans is the kidney glomerular endothelium, HRGEC were challenged similarly to aortic endothelial cells (Figure 3). The renal cells are more sensitive to the histones and toxins in vitro (data not shown), so challenge concentrations were decreased to maintain >80% viability and on-cell western was used to minimize manipulation of the cells. Stx1 and Stx2 challenge reduced PAR1 (Figure 3A), EPCR (Figure 3B), and TM surface expression (Figure 3C). Histones did not appreciably alter PAR1 or TM expression, but did decrease EPCR expression on HRGEC.
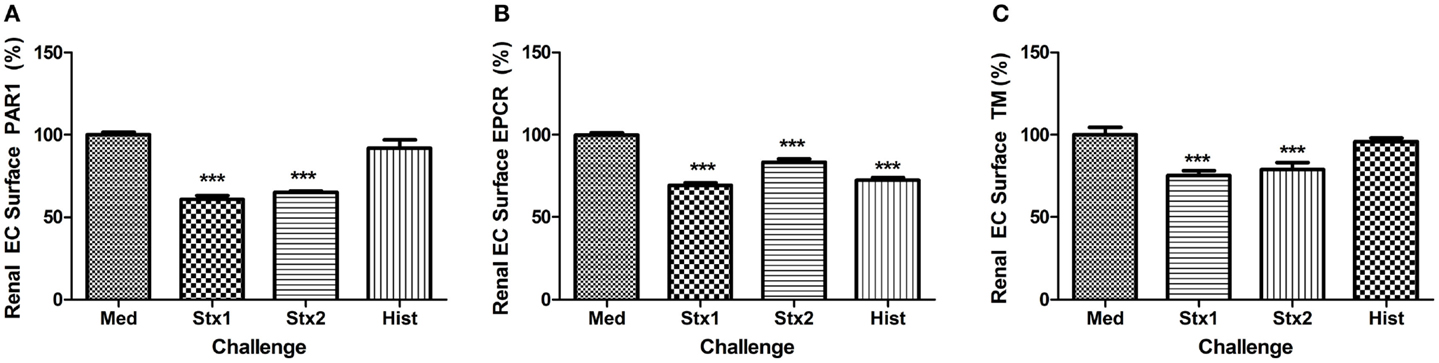
Figure 3. Stx1, Stx2, and histones induce changes in renal glomerular endothelial expression of surface molecules. Surface expression of (A) PAR1, (B) EPCR, and (C) TM were quantified by on-cell western after 24 h incubation with Stx1 (50 ng/mL), Stx2 (100 ng/L), or 50 μg/mL histones. Antigen data were normalized to cell number using Draq5 staining. Differences from media alone are shown as ***p < 0.001.
Histones Reduce Endothelial Activated Protein C Generation
Functional changes resulting from alterations to surface expression of TM or EPCR ought to include changes to the rate of protein C activation. To determine if surface expression level changes of TM or EPCR correlated with functional changes that could be extrapolated to have significance in the development of HUS, the rate of protein C activation was determined on endothelial cells after challenge with Stx or histone DAMPs. Only Stx1 or histones had significant effects on the activation of protein C in aortic endothelial cells (Figure 4A). Histone challenge of HRGEC led to a significant decrease in the rate of protein C activation at 24 h post-challenge (Figure 4B; p < 0.001); however, neither Shiga toxin alone caused a significant difference in the amount of protein C activated. The suppression of protein C pathway function by histones was successfully prevented in the presence of anti-histone monoclonal antibody BWA3, which inhibits H4 and H2a (40), and protein C activation was restored on both aortic and glomerular endothelial cells in the presence of the blocking antibody during challenge with histones (Figures 4C,D).
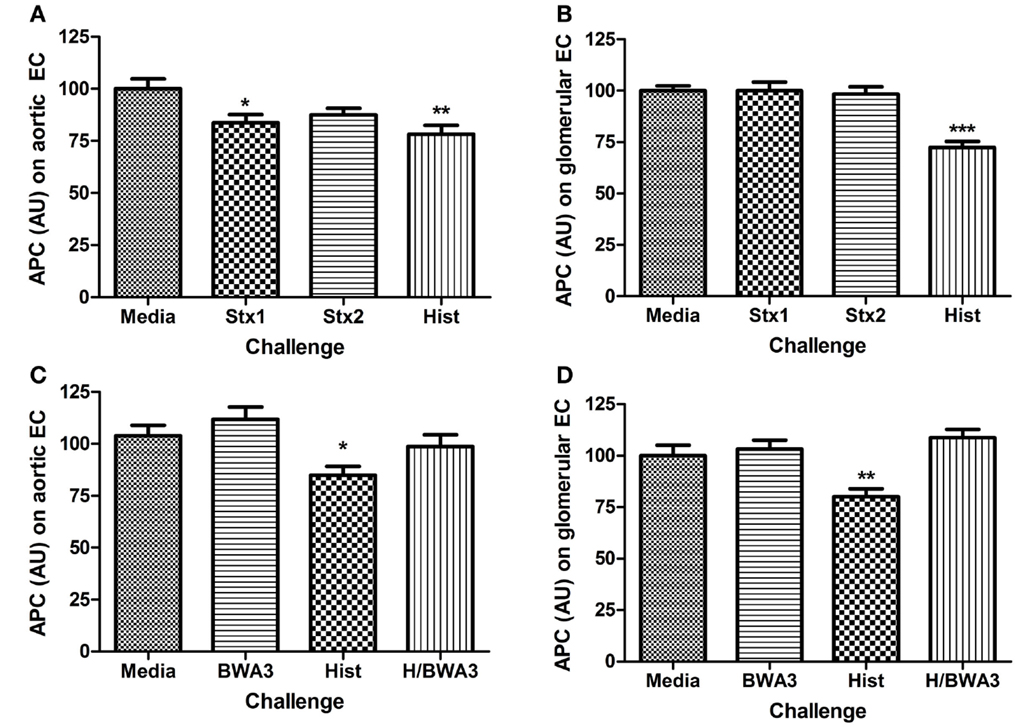
Figure 4. Challenge with Stx or histones decreases activation of protein C. The ability of human aortic endothelial cells (A,C) or renal endothelial cells (B,D) to activate protein C after 24 h challenge with Stx1, Stx2, or histones was determined on confluent monolayers with 10 nM human alpha-thrombin and 0.2 μM human protein C at 37°C. Activated protein C was quantified in cell supernatants with spectrozyme PCa substrate and converted to nM APC using a standard curve, then adjusted for cell number with Draq5 staining. Anti-histone antibody BWA3 (20 μg/mL) was added before challenge in some wells. Differences relative to media alone are shown as *p < 0.05, **p < 0.01, ***p < 0.001.
Histones but not Stx Enhance Thrombin-Induced Loss of Endothelial Barrier Function
Along with their roles in anticoagulation, both endothelium and APC have important roles in maintaining a selective barrier in the vasculature. Endothelial cell permeability changes in response to Stx and histones were examined by monitoring the resistance of a human aortic endothelial monolayer in a continuous fashion using an electrical cell impedance system. As expected, thrombin (1 U/mL final) stimulation resulted in a rapid decrease in resistance (Figure 5A), which correlates with increased permeability. However, in cells pre-treated for 12 h with histones (25 μg/mL) prior to thrombin stimulation, the thrombin-induced barrier function decrease was prolonged (Figure 5B). Recovery to pre-treatment levels following thrombin stimulation occurred after ~2 h; recovery from thrombin on histone-treated endothelium had not yet occurred by 18 h post-challenge (data not shown for presentation purposes). Pre-treatment with APC abrogated thrombin’s effect on monolayer permeability (Figure 5C), consistent with the known barrier protection properties of APC. APC was not able to rescue the loss of cell permeability due to thrombin when the cells were pre-treated with histones (Figure 5C), and treatment with histones alone did not increase permeability (data not shown). Stx2 did not change the duration of increased permeability of HAEC to thrombin, and APC was able to restore cells from thrombin-induced leak regardless of whether or not Stx2 was present (Figure 5D). Challenge with Stx1 did not alter these results. The area under the curves was calculated to quantify the relative effects (Figure 5E). Thus, under these conditions, histone DAMPs greatly exacerbate thrombin’s impact and prevent APC from being protective, though the toxins do not appreciably change endothelial barrier function.
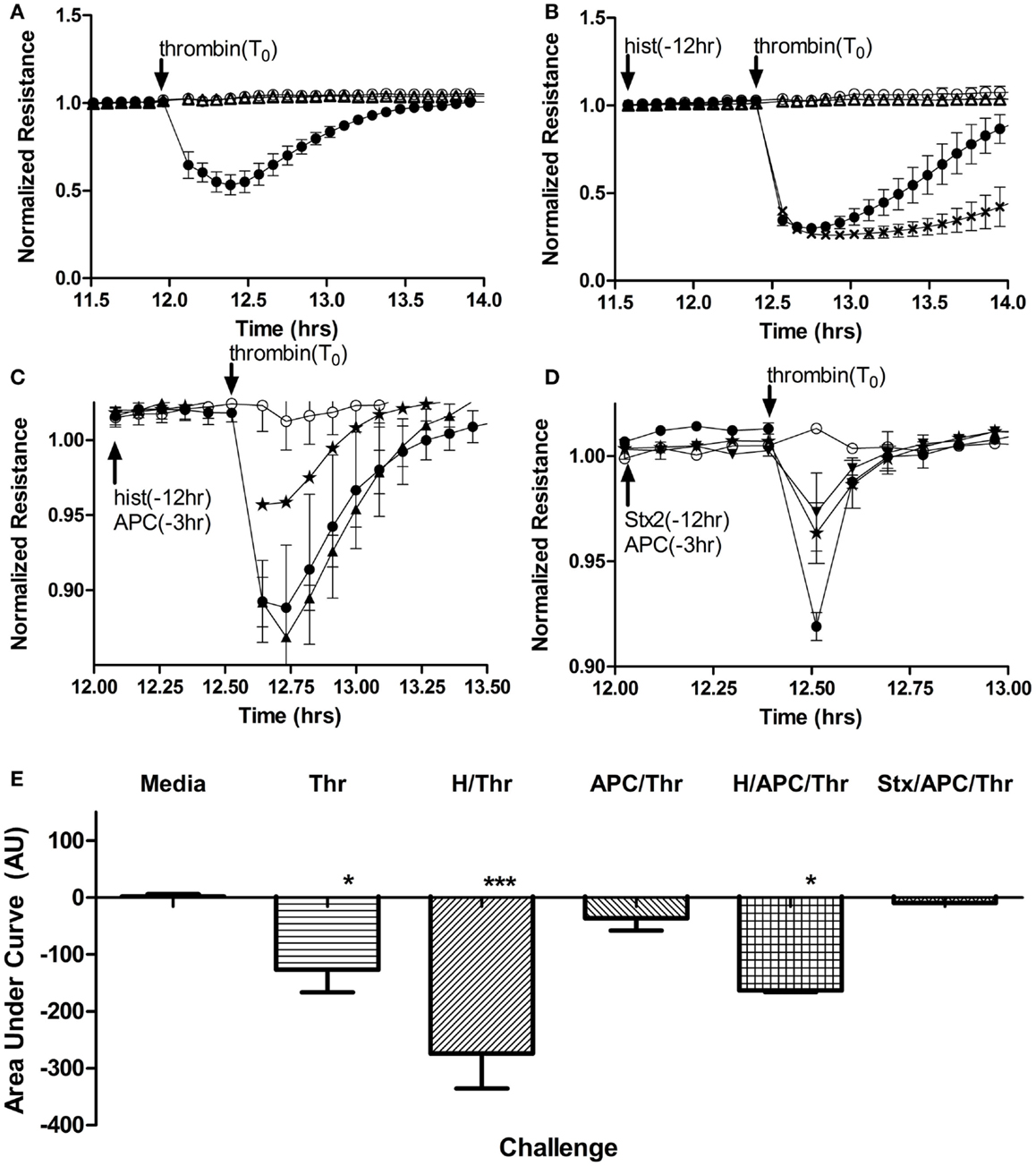
Figure 5. Histones, but not Stx, contribute to increased endothelial cell permeability and block APC rescue. Permeability of human aortic endothelial cells was determined by monitoring changes in electrical resistance across a monolayer as described in Section “Materials and Methods.” (A) Steady state resistance observed with media (○) decreases after addition of 1 U/mL (16.85 nM) human thrombin (●), reflecting increased permeability. Challenge with Stx2 (∆) lead to no change in permeability. (B) Increased permeability after thrombin (●) challenge is significantly prolonged if cells are pre-exposed to 50 μg/mL histones (X) for 12 h, but again, Stx2 causes no appreciable change in permeability (∆). (C) Compared to thrombin (2 nM) alone (●), APC pre-exposure (⋆) attenuates thrombin effects on permeability, but cannot rescue permeability of the cells if they have also been exposed to histones (▲). (D) Thrombin increased permeability (●) and APC rescued cells (⋆), but adding 100 ng/mL Stx2 (▼) did not change the protective effect of APC. (E) Permeability data were quantified as area under the curves; mean ± SD of three to four experiments each. Neither Stx1 nor Stx2 alone altered electrical resistance of the monolayers and so Stx2 is shown on graphs as representative of either Shiga toxin. *p < 0.05, ***p < 0.001.
DAMPs are Present in the Plasma of Stx2-Challenged Mice
In order to determine whether Stx2 challenge produces DAMPs in mice, we used two different mouse models: a Stx2-toxemia model, and a Stx2-producing bacterial model. Mice challenged with Stx2 (1 ng/20 g body weight; i.p.) on day 0 and day 3 exhibited decreased survival (Figure 6A, dashed line) and kidney injury evidenced by increasing BUN relative to saline controls (Figure 6B; p < 0.001 on day 4). Plasma histone levels in Stx2 challenged mice were significantly elevated on day 3 post-challenge (258% ± 62% n = 24) when compared to day 0 (Figure 6C). Plasma levels of HMGB1, another DAMP associated with disease severity in sepsis models, were significantly elevated by 2 days after Stx2 challenge (421% ± 117%; n = 13) when compared to day 0 (Figure 6E). Pre-treatment with anti-histone BWA3 antibody prevented the significant rise in plasma histones (Figure 6D). Surprisingly, pre-treatment with anti-histone BWA3 antibody also abrogated the rise in HMGB1 (Figure 6F). Despite the reduction in circulating DAMPs by antibody, there was no alteration in survival outcome after lethal Stx2 challenge (Figure 6A, dotted). This result was not wholly unexpected, as the renal damage in mice is centered more in the tubular epithelium and collecting ducts, as opposed to the renal glomerular endothelium in humans, and the mice do not display thrombotic microangiopathy.
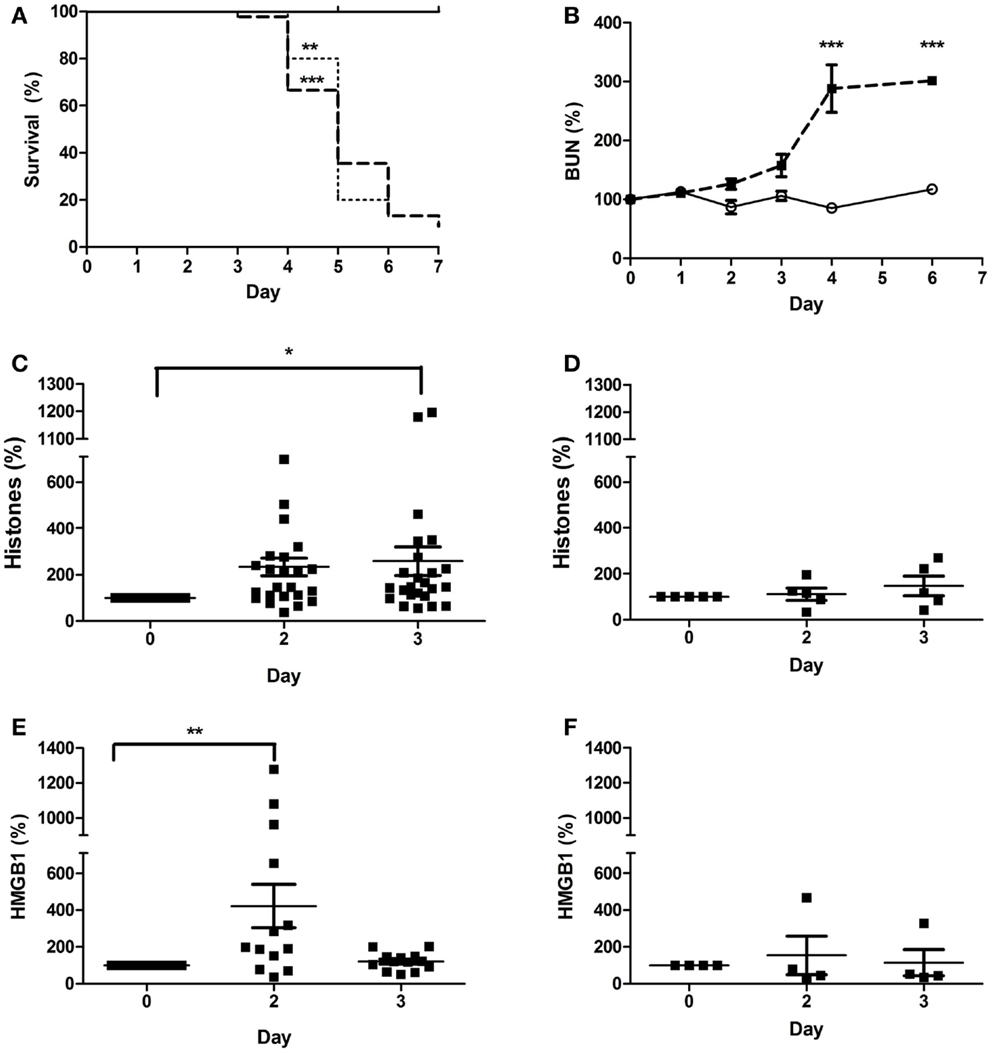
Figure 6. Plasma DAMPs increase after Stx2 challenge in mice. Mice were challenged i.p. with 1 ng/20 g body weight Stx2 [bold dashed line in (A), black squares in (B–F), n = 45] or saline [solid line in (A), open circle in (B), n = 15] on Day 0 and Day 3 and monitored for (A) survival and (B) plasma BUN. Plasma histones (C,D) and HMGB1 (E,F) were measured by ELISA. One group received a bolus dose of anti-histone BWA3 (400 μg i.p.) before challenge [(D,F); n = 5]. Survival was not influenced by BWA3 [(A), dotted; n = 5]. *p < 0.05, **p < 0.01, ***p < 0.001.
During EHEC infection, Stx exposure occurs over days as the gut is colonized by EHEC that produce and release the Shiga toxins, so we also utilized a murine model in which mice are infected by oral gavage with C. rodentium strains that either do or do not express Stx2. The intestinal attaching/effacing lesions and contributions of Stx2 toward organ injury have been described (41), and this model has been shown to produce similar A/E lesions and thus be more relevant than a Stx2-alone model. Inoculating the mice by oral gavage with ~1 × 109 CFU of C. rodentium-Stx2 resulted in decreased survival (Figure 7A; dashed line) compared to mice that received control C. rodentium, which does not express the toxin. As expected, kidney injury was toxin-dependent as evidenced by elevated plasma BUN by days 7–9 with the toxin-producing strain (Figure 7B; p < 0.001 on day 9). Mice challenged with non-Stx2 producing C. rodentium (open circles) did not develop changes in plasma levels of extracellular histones for the duration of the experiment when compared to day 0 (Figure 7C). In contrast, mice challenged with C. rodentium-Stx2 (black squares) developed significantly increased plasma levels of histones on day 3. This effect was significant when compared their own day 0 values and was also significant compared to plasma histone levels in non-Stx2 producing C. rodentium challenged mice (Figure 7D). Plasma HMGB1 did not increase significantly in mice challenged with non-Stx2 producing C. rodentium (Figure 7F), in contrast to elevated levels of HMGB1 by day 5 in C. rodentium-Stx2 mice (Figure 7G). BWA3 anti-histone antibody pre-treatment (dotted line) did not alter survival. Survival was still significantly decreased (p < 0.001) compared to animals challenged with C. rodentium not expressing Stx2, and there was no significant difference when compared with mice given Stx2-expressing C. rodentium and no antibody pre-treatment. The observed increase in plasma histone and HMGB1 levels were abolished by treatment with the anti-histone BWA3 antibody (Figures 7E,H). In conclusion, both murine models demonstrated that DAMPs were consistently elevated when Stx2 was present, and in both cases the rise occurred prior to rise in BUN, confirming that in the presence of Stx2, DAMPs are produced.
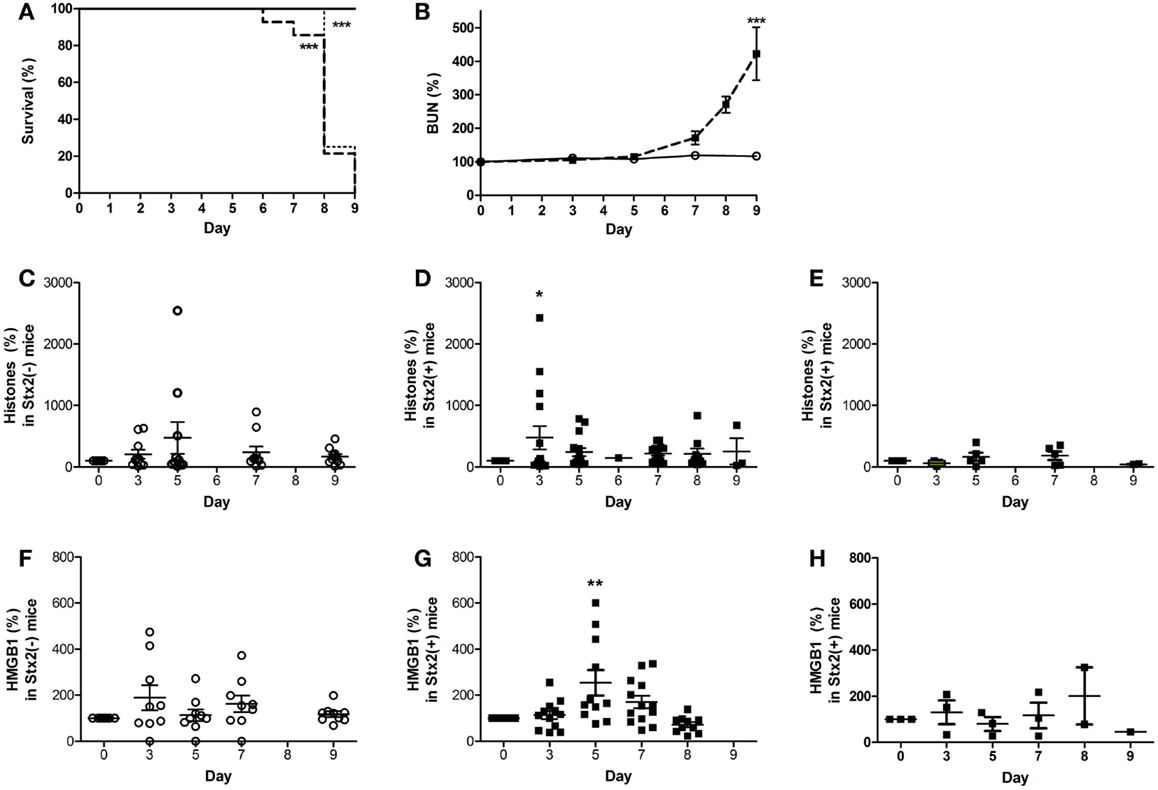
Figure 7. Plasma DAMPs increase during intestinal infection with Stx2+ Citrobacter rodentium. Mice were gavaged with 1 × 109 CFU Stx2+ C. rodentium [bold dashed line in (A), black square in (B,D,E,G,H), n = 15] or Stx2− strain C. rodentium [solid line in (A), open circle in (B,C,F), n = 10] on Day 0 and monitored for (A) survival and (B) plasma BUN. Plasma DAMPs were measured as histones or HMGB1 in Stx2− mice (C,F) and Stx2+ mice (D,G). Baseline levels of both DAMPs were observed when anti-histone antibody BWA3 (400 μg; i.p.) was injected 10 min prior to challenge with Stx2+ bacteria [(E,H); n = 5/group]. Pre-treatment with BWA3 did not affect survival after challenge with the Stx2+ strain [(A), dotted line; n = 5]. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Low Relative Expression of Protective EPCR and TM on Renal Glomerular Endothelial Cells
Differential expression of EPCR and TM between large vessel endothelium and the microvasculature is known from immunohistochemistry studies, particularly for EPCR, which is expressed relatively more on large vessel endothelium (42). Here, we provide quantitative assessment of EPCR, TM, and PAR1 expression on cultured primary endothelial cells using on-cell western. Our results show that there are 3.2-fold lower and 7.4-fold lower expressions of TM and EPCR, respectively, on renal glomerular endothelial cells when compared with aortic endothelial cells in the same experiment. APC is protective in acute kidney injury murine models (43), and the organ system most vulnerable to the sequelae of low EPCR expression is the kidney, which demonstrates enhanced albuminuria and profound renal hemorrhage after LPS challenge (44). The kidneys are already a toxin target due to glomerular Gb3 expression, and the current data suggest that further reduction due to toxin and DAMP exposure of already low TM and EPCR expression may provide an environment highly susceptible to thrombus production.
In vitro Loss of EPCR and TM Surface Expression on Endothelial Cells Due to Stx and DAMPs
D + HUS is characterized by thrombotic microangiopathy, or a forming of microthombi in the small vasculature of the glomerulus, suggesting loss of endothelial anti-coagulant capacity. The receptors and enzymes that make up the protein C pathway of endothelial cells are critical contributors to intact anti-coagulant function of the endothelium and loss of these anti-coagulant and cytoprotective molecules could have significant pro-thrombotic consequences. Surface expression levels of protein C pathway molecules are changed in Shiga toxin- or DAMPs-challenged human endothelial cells, as demonstrated here in vitro. Shiga toxin and/or DAMP challenge of human endothelial cells resulted in a shift toward a pro-coagulant environment, and this has functional consequences with respect to the generation of APC, an enzyme critical in controlling thrombus formation. We made several significant observations, including reduced APC generation, enhancement of endothelial barrier permeability, and reduction of the ability of APC to protect barrier function in the context of Stx or histone DAMP challenge. These data provide insights into the mechanism of Stx-induced endothelial dysfunction in D + HUS and strongly suggest a potential role for tissue damage induced DAMPs in this process.
Thrombin bound to TM preferentially cleaves and activates protein C instead of fibrinogen (45), creating APC and ultimately reducing fibrin clot formation. EPCR makes the thrombin/TM cleavage of protein C even more favorable. EPCR also plays a crucial role in endothelial function because APC/EPCR complex activation of PAR1 leads to a cytoprotective signaling cascade (19, 46, 47). We previously demonstrated in a non-human primate model of HUS that Shiga toxins alone can cause HUS and acute renal failure with fibrin deposition and swelling and detachment of glomerular endothelial cells (48, 49). Cellular mechanisms are difficult to study in non-human primates and mouse models do not develop the coagulopathy of HUS, so the current study with human cells extends the in vivo observations by demonstrating in vitro loss of EPCR and TM surface expression on endothelial cells due to Stx and DAMPs, particularly histones, and reduced capacity to generate APC. A pro-thrombotic environment is further created by DAMP-induced increased expression of PAR-1, the pro-thrombotic thrombin receptor (50). Both histones and Shiga toxins reduced EPCR expression, and Shiga toxins also lowered TM on HRGEC, with the end result that both Stx1 and histones affected the amount of APC generated by HRGEC in HAEC, with histones alone affecting APC production in HRGEC.
Loss of In vitro Endothelial Barrier Function Due to Histones but not Stx
D + HUS patients can develop edema, and in our system we observed loss of in vitro endothelial barrier function primarily due to histones, rather than through any effect of Stx1 or Stx2 on endothelial cells. Histones increased PAR-1 and lowered EPCR expression and pre-treatment of human aortic endothelial monolayers with histones significantly delayed the ability of the endothelial barrier to recover to baseline after thrombin challenge. Histones also significantly inhibited the barrier-protective effects of APC. It is possible that histones might compete as a substrate for APC (32), but we did not find any effect of histones on APC chromogenic activity even at histone concentrations up to fourfold higher than those used here (data not shown). PAR1 can function in both barrier protective and disruptive capacities, depending on which protease cleaves it and whether EPCR is occupied by a ligand. Shiga toxins and DAMPs both decrease the surface ratio of EPCR:PAR1 by lowering EPCR; however, only histones also raise PAR1, which could account for the differential effects of histones vs. Shiga toxins with respect to APC inhibition. We hypothesize that the combination of decreased EPCR along with increased PAR1 creates more opportunity for thrombin signaling while dampening the potential protective effects of EPCR occupied with APC, helping to explain the prolongation of the permeability effects of thrombin despite its use.
DAMPs are Produced by the Action of Shiga Toxins in Mice
The purpose of our mouse models was to demonstrate that DAMPs are produced by the action of Shiga toxins. The finding of significantly elevated DAMPs is to the best of our knowledge a novel finding in mouse models of Shiga toxin challenge and is consistent with prior studies in our non-human primate Stx-HUS models (51). The finding that levels of plasma HMGB1 in both mouse models decreased after an initial significant rise instead of continuing to rise with disease severity could be due to the limitations of current mouse models for this disease, because the distribution of Gb3 receptors in the mouse differs from that in humans or non-human primates (4, 5, 49). Unlike in human kidneys, Shiga toxins target renal tubule epithelial cells in mice (6) giving rise to the possibility that HMGB1 was excreted as urination becomes increasingly excessive in the mouse models due to tubular injury. In contrast, humans and baboons express Gb3 on endothelial cells, particularly in the glomeruli, and disease severity is accompanied by progressive anuria so that the baboon Stx challenge models have and increase in certain plasma DAMPs such as mitochondrial DNA over time (35). However, in contrast, we have previously shown that HMGB1 in non-human primate models also initially rises before dropping (51), so the waxing or waning pattern of different DAMPs in the serum with Shiga toxin challenge likely relies on mechanisms that remain to be understood and fall outside the scope of this discussion.
In our experiments, we introduced the anti-histone antibody BWA3, which targets H4 and H2a, and is derived from an autoimmune mouse model (40). A bolus dose of 400 μg BWA3 prior to Stx2 or C. rodentium-Stx2 significantly decreased plasma levels of histones and HMGB1, although in neither case was the outcome altered for the model. These results suggest that these DAMPs may function more as markers of cell injury in these murine models rather than driving pathology as they might in other models; however, it was not our intention to investigate endothelial injury in our murine models, for reasons, which have been stated already. For this purpose, we turned to in vitro models of endothelial cell injury. Our human endothelial cell experiments show a significant impact by DAMPs with respect to APC generation and endothelial dysfunction, but this is not possible to replicate in the mice, as these models exhibit Stx-induced tubular epithelial injury (5), with no evidence of glomerular endothelial damage, thrombotic microangiopathy, or thrombocytopenia of HUS as stated before. These are limitations of all mouse models using endotoxin-free Stx challenge. Other investigators have shown that in mice challenged with LPS (a model of endothelial cell injury) BWA3 reduced circulating histones and improved survival (32). The current experiments with human endothelial cells raise the possibility that BWA3 might have more of an effect on outcomes in models of kidney injury driven by endothelial rather than tubular damage, such as a non-human primate model that more closely mimics human pathology (36, 37, 49); however, there are clear limitations to doing the experiments described in this paper in non-human primates, and so we chose to progress as described.
Elevated HMGB1 and histones have been shown experimentally to be involved in SIRS development by the fact that inhibition of either is beneficial in animal models of LPS-induced inflammation (32, 33). Currently, development of HUS in patients during EHEC infection is largely attributed to the bacterially produced Shiga toxin effects on endothelium or platelets (52), but the current data suggest that the host response to injury contributes as well. During active infection, Shiga toxins rapidly become undetectable in the plasma days before development of HUS (39). During this time, cells internalize the toxins, the intestinal bacteria damage the intestinal epithelium, and both processes may lead to release of DAMPs. The observations made here cannot link DAMP activities directly to susceptibility of patients to develop HUS, but they do provide insight into a possible molecular basis for development of increased vascular permeability and a pro-thrombotic environment in the context of Stx2-induced tissue damage. We showed this by demonstrating that DAMPs are produced by the Shiga toxins, and that histone DAMPs cause demonstrable endothelial dysfunction in two types of human endothelial cells.
Limitations of the Current Study and Future Directions
While we have made several important observation in the experiments described here promoting a possible role for Stx2-induced DAMPs in the development of endothelial dysfunction underlying HUS, there are still important questions that remain unanswered. We did not identify cellular mechanisms linking Shiga toxins and extracellular histones to the regulation of EPCR, TM, and PAR1 endothelial expression, as we were more interested in the end consequence of reduced protein C activation; however, this mechanism could also be a source of potential therapeutic targets and warrants investigation. Matrix metalloproteinases and MAPK pathways may contribute to EPCR shedding (53, 54), though how these may be impacted by Stx or DAMPs in this context was not addressed. Histones act through TLR4 (55), which might induce PAR1 through a similar mechanism, though we do not address this here. Finally, Stx1 and Stx2 interact with human neutrophils via TLR4 (56), but how that might impact histone or other DAMP activities is not known. Pilot experiments using TAK-1, JNK, and IRAK1/4 inhibitors demonstrated no significant prevention of Shiga toxin effects on EPCR, PAR1, or TM (data not shown). The largest limitation of our studies was the lack of a mouse model demonstrating full-spectrum HUS, which to date has only been reproducibly done by adding LPS to the Stx challenge (6, 57, 58). Ideally, we would test our hypothesis in vivo using animal models that demonstrate kidney injury driven by endothelial dysfunction, and then we could show whether DAMPs are playing a significant role by using therapies such as BWA3 and quantifiable outcomes such as biomarkers and survival. Additionally, quantifying the presence of DAMPs in the blood of patients with HUS would be a good next step prior to experiments with full-spectrum HUS animal models. Previous work has already amply demonstrated the pathology and tissue damage (25, 59), and quantifying the DAMPs that are almost certainly present would provide the evidence needed to investigate whether they function as markers of disease impact or drivers of disease. The data here lay groundwork, giving evidence that this could be a fruitful path for future investigations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Carl J. Hauser (Beth Israel Deaconess Medical Center, Boston, MA, USA) for the generous use of his ECIS Z⊖ system. The authors gratefully acknowledge Priya Jani for technical assistance. CM was supported by NHLBI T32 training grant HL07969 (K. Ravid, PI), U01AI075386 (SK, PI) and R01Ai102931 (SK, PI). CP was supported by NIH/NIAID T32AI089673 (C. Genco, PI).
References
1. Paton JC, Paton AW. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin Microbiol Rev (1998) 11:450–79.
2. Williams DM, Sreedhar SS, Mickell JJ, Chan JC. Acute kidney failure: a pediatric experience over 20 years. Arch Pediatr Adolesc Med (2002) 156:893–900. doi: 10.1001/archpedi.156.9.893
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Boerlin P, McEwen SA, Boerlin-Petzold F, Wilson JB, Johnson RP, Gyles CL. Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J Clin Microbiol (1999) 37:497–503.
4. Boyd B, Lingwood C. Verotoxin receptor glycolipid in human renal tissue. Nephron (1989) 51:207–10. doi:10.1159/000185286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Rutjes NW, Binnington BA, Smith CR, Maloney MD, Lingwood CA. Differential tissue targeting and pathogenesis of verotoxins 1 and 2 in the mouse animal model. Kidney Int (2002) 62:832–45. doi:10.1046/j.1523-1755.2002.00502.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Psotka MA, Obata F, Kolling GL, Gross LK, Saleem MA, Satchell SC, et al. Shiga toxin 2 targets the murine renal collecting duct epithelium. Infect Immun (2009) 77:959–69. doi:10.1128/IAI.00679-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Garred O, van Deurs B, Sandvig K. Furin-induced cleavage and activation of Shiga toxin. J Biol Chem (1995) 270:10817–21. doi:10.1074/jbc.270.18.10817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara T, Igarashi K. Site of action of a vero toxin (vt2) from Escherichia coli o157:H7 and of Shiga toxin on eukaryotic ribosomes. Rna n-glycosidase activity of the toxins. Eur J Biochem (1988) 171:45–50.
9. Obrig TG, Moran TP, Brown JE. The mode of action of Shiga toxin on peptide elongation of eukaryotic protein synthesis. Biochem J (1987) 244:287–94.
10. Fraser ME, Fujinaga M, Cherney MM, Melton-Celsa AR, Twiddy EM, O’Brien AD, et al. Structure of Shiga toxin type 2 (stx2) from Escherichia coli o157:H7. J Biol Chem (2004) 279:27511–7. doi:10.1074/jbc.M401939200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Smith WE, Kane AV, Campbell ST, Acheson DW, Cochran BH, Thorpe CM. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and jnk activation and induction of apoptosis in intestinal epithelial cells. Infect Immun (2003) 71:1497–504. doi:10.1128/IAI.71.3.1497-1504.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Jandhyala DM, Ahluwalia A, Obrig T, Thorpe CM. Zak: a map3kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell Microbiol (2008) 10:1468–77. doi:10.1111/j.1462-5822.2008.01139.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Lee S-Y, Lee M-S, Cherla RP, Tesh VL. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol (2008) 10:770–80. doi:10.1111/j.1462-5822.2007.01083.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein c pathway. Blood (2007) 109:3161–72. doi:10.1182/blood-2006-09-003004
15. Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein c receptor augments protein c activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci U S A (1996) 93:10212–6. doi:10.1073/pnas.93.19.10212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Fukudome K, Ye X, Tsuneyoshi N, Tokunaga O, Sugawara K, Mizokami H, et al. Activation mechanism of anticoagulant protein c in large blood vessels involving the endothelial cell protein c receptor. J Exp Med (1998) 187:1029–35. doi:10.1084/jem.187.7.1029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Fernández G, Te Loo MM, Velden TA, Heuvel LW, Palermo M, Monnens LA. Decrease of thrombomodulin contributes to the procoagulant state of endothelium in hemolytic uremic syndrome. Pediatr Nephrol (2003) 18:1066–8. doi:10.1007/s00467-003-1236-8
18. Marlar RA, Kleiss AJ, Griffin JH. Mechanism of action of human activated protein C, a thrombin-dependent anticoagulant enzyme. Blood (1982) 59:1067–72.
19. Bae JS, Yang L, Manithody C, Rezaie AR. The ligand occupancy of endothelial protein c receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood (2007) 110:3909–16. doi:10.1182/blood-2007-06-096651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Bae JS, Rezaie AR. Thrombin inhibits nuclear factor kappab and rhoa pathways in cytokine-stimulated vascular endothelial cells when epcr is occupied by protein c. Thromb Haemost (2009) 101:513–20. doi:10.1160/TH08-09-0568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Camerer E, Huang W, Coughlin SR. Tissue factor- and factor x-dependent activation of protease-activated receptor 2 by factor viia. Proc Natl Acad Sci U S A (2000) 97:5255–60. doi:10.1073/pnas.97.10.5255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Sen P, Gopalakrishnan R, Kothari H, Keshava S, Clark CA, Esmon CT, et al. Factor viia bound to endothelial cell protein c receptor activates protease activated receptor-1 and mediates cell signaling and barrier protection. Blood (2011) 117:3199–208. doi:10.1182/blood-2010-09-310706
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Faust SN, Levin M, Harrison OB, Goldin RD, Lockhart MS, Kondaveeti S, et al. Dysfunction of endothelial protein c activation in severe meningococcal sepsis. N Engl J Med (2001) 345:408–16. doi:10.1056/NEJM200108093450603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Boldt J, Wollbruck T, Sonneborn S, Welters A, Hempelmann G. Thrombomodulin in intensive care patients. Intensive Care Med (1995) 21:645–50. doi:10.1007/BF01711542
25. Griffin PM, Olmstead LC, Petras RE. Escherichia coli o157:H7-associated colitis. A clinical and histological study of 11 cases. Gastroenterology (1990) 99:142–9.
26. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi:10.1146/annurev.immunol.12.1.991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A (2010) 107:15880–5. doi:10.1073/pnas.1005743107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Lotze MT, Tracey KJ. High-mobility group box 1 protein (hmgb1): nuclear weapon in the immune arsenal. Nat Rev Immunol (2005) 5:331–42. doi:10.1038/nri1594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Rosin DL, Okusa MD. Dangers within: damp responses to damage and cell death in kidney disease. J Am Soc Nephrol (2011) 22:416–25. doi:10.1681/ASN.2010040430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Bianchi ME. Damps, pamps and alarmins: all we need to know about danger. J Leukoc Biol (2007) 81:1–5. doi:10.1189/jlb.0306164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Wolfson RK, Chiang ET, Garcia JGN. Hmgb1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc Res (2011) 81:189–97. doi:10.1016/j.mvr.2010.11.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med (2009) 15:1318–21. doi:10.1038/nm.2053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. Hmg-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285:248–51. doi:10.1126/science.285.5425.248
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial damps cause inflammatory responses to injury. Nature (2010) 464:104–7. doi:10.1038/nature08780
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Sursal T, Stearns-Kurosawa DJ, Itagaki K, Oh SY, Sun S, Kurosawa S, et al. Plasma bacterial and mitochondrial DNA distinguish bacterial sepsis from sterile systemic inflammatory response syndrome and quantify inflammatory tissue injury in nonhuman primates. Shock (2013) 39:55–62. doi:10.1097/SHK.0b013e318276f4ca
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Stearns-Kurosawa DJ, Collins V, Freeman S, Tesh VL, Kurosawa S. Distinct physiologic and inflammatory responses elicited in baboons after challenge with Shiga toxin type 1 or 2 from enterohemorrhagic Escherichia coli. Infect Immun (2010) 78:2497–504. doi:10.1128/IAI.01435-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Stearns-Kurosawa DJ, Oh SY, Cherla RP, Lee MS, Tesh VL, Papin J, et al. Distinct renal pathology and a chemotactic phenotype after enterohemorrhagic Escherichia coli Shiga toxins in non-human primate models of hemolytic uremic syndrome. Am J Pathol (2013) 182:1227–38. doi:10.1016/j.ajpath.2012.12.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Stone SM, Thorpe CM, Ahluwalia A, Rogers AB, Obata F, Vozenilek A, et al. Shiga toxin 2-induced intestinal pathology in infant rabbits is a-subunit dependent and responsive to the tyrosine kinase and potential zak inhibitor imatinib. Front Cell Infect Microbiol (2012) 2:135. doi:10.3389/fcimb.2012.00135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Lopez EL, Contrini MM, Glatstein E, Ayala SG, Santoro R, Ezcurra G, et al. An epidemiologic surveillance of Shiga-like toxin-producing Escherichia coli infection in argentinean children: risk factors and serum Shiga-like toxin 2 values. Pediatr Infect Dis J (2012) 31:20–4. doi:10.1097/INF.0b013e31822ea6cf
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Monestier M, Fasy TM, Losman MJ, Novick KE, Muller S. Structure and binding properties of monoclonal antibodies to core histones from autoimmune mice. Mol Immunol (1993) 30:1069–75. doi:10.1016/0161-5890(93)90153-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Mallick EM, McBee ME, Vanguri VK, Melton-Celsa AR, Schlieper K, Karalius BJ, et al. A novel murine infection model for Shiga toxin-producing Escherichia coli. J Clin Invest (2012) 122:4012–24. doi:10.1172/JCI62746
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Laszik Z, Mitro A, Taylor FB Jr, Ferrell G, Esmon CT. Human protein c receptor is present primarily on endothelium of large blood vessels: implications for the control of the protein c pathway. Circulation (1997) 96:3633–40. doi:10.1161/01.CIR.96.10.3633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Park SW, Chen SW, Kim M, D’Agati VD, Lee HT. Human activated protein C attenuates both hepatic and renal injury caused by hepatic ischemia and reperfusion injury in mice. Kidney Int (2009) 76:739–50. doi:10.1038/ki.2009.255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. von Drygalski A, Furlan-Freguia C, Ruf W, Griffin JH, Mosnier LO. Organ-specific protection against lipopolysaccharide-induced vascular leak is dependent on the endothelial protein c receptor. Arterioscler Thromb Vasc Biol (2013) 33:769–76. doi:10.1161/ATVBAHA.112.301082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Esmon CT, Esmon NL, Harris KW. Complex formation between thrombin and thrombomodulin inhibits both thrombin-catalyzed fibrin formation and factor v activation. J Biol Chem (1982) 257:7944–7.
46. Schuepbach RA, Madon J, Ender M, Galli P, Riewald M. Protease-activated receptor-1 cleaved at r46 mediates cytoprotective effects. J Thromb Haemost (2012) 10:1675–84. doi:10.1111/j.1538-7836.2012.04825.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at arg46. Blood (2012) 120:5237–46. doi:10.1182/blood-2012-08-452169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Siegler RL, Pysher TJ, Tesh VL, Taylor FB Jr. Response to single and divided doses of Shiga toxin-1 in a primate model of hemolytic uremic syndrome. J Am Soc Nephrol (2001) 12:1458–67.
49. Taylor FB Jr, Tesh VL, DeBault L, Li A, Chang AC, Kosanke SD, et al. Characterization of the baboon responses to Shiga-like toxin: descriptive study of a new primate model of toxic responses to stx-1. Am J Pathol (1999) 154:1285–99. doi:10.1016/S0002-9440(10)65380-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell (1991) 64:1057–68. doi:10.1016/0092-8674(91)90261-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lee BC, Mayer CL, Leibowitz CS, Stearns-Kurosawa DJ, Kurosawa S. Quiescent complement in nonhuman primates during E. coli Shiga toxin-induced hemolytic uremic syndrome and thrombotic microangiopathy. Blood (2013) 122(5):803–6. doi:10.1182/blood-2013-03-490060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Melton-Celsa A, Mohawk K, Teel L, O’Brien A. Pathogenesis of Shiga-toxin producing Escherichia coli. Curr Top Microbiol Immunol (2012) 357:67–103. doi:10.1007/82_2011_176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Qu D, Wang Y, Esmon NL, Esmon CT. Regulated endothelial protein c receptor shedding is mediated by tumor necrosis factor-alpha converting enzyme/adam17. J Thromb Haemost (2007) 5:395–402. doi:10.1111/j.1538-7836.2007.02347.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Menschikowski M, Hagelgans A, Eisenhofer G, Siegert G. Regulation of endothelial protein c receptor shedding by cytokines is mediated through differential activation of map kinase signaling pathways. Exp Cell Res (2009) 315:2673–82. doi:10.1016/j.yexcr.2009.05.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through tlr2 and tlr4 in mouse fatal liver injury. J Immunol (2011) 187(5):2626–31. doi:10.4049/jimmunol.1003930
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Brigotti M, Carnicelli D, Arfilli V, Tamassia N, Borsetti F, Fabbri E, et al. Identification of tlr4 as the receptor that recognizes Shiga toxins in human neutrophils. J Immunol (2013) 191:4748–58. doi:10.4049/jimmunol.1300122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Keepers TR, Psotka MA, Gross LK, Obrig TG. A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J Am Soc Nephrol (2006) 17:3404–14. doi:10.1681/ASN.2006050419
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Mayer CL, Leibowitz CS, Kurosawa S, Stearns-Kurosawa DJ. Shiga toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals. Toxins (Basel) (2012) 4:1261–87. doi:10.3390/toxins4111261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Richardson SE, Karmali MA, Becker LE, Smith CR. The histopathology of the hemolytic uremic syndrome associated with verocytotoxin-producing Escherichia coli infections. Hum Pathol (1988) 19:1102–8. doi:10.1016/S0046-8177(88)80093-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: hemolytic uremic syndrome, thrombotic microangiopathy, Shiga toxin 1, Shiga toxin 2, protein C, damage-associated molecular patterns, endothelial cells
Citation: Mayer CL, Parello CSL, Lee BC, Itagaki K, Kurosawa S and Stearns-Kurosawa DJ (2015) Pro-coagulant endothelial dysfunction results from EHEC Shiga toxins and host damage-associated molecular patterns. Front. Immunol. 6:155. doi: 10.3389/fimmu.2015.00155
Received: 19 December 2014; Accepted: 23 March 2015;
Published online: 07 April 2015.
Edited by:
Katharina F. Kubatzky, University Hospital Heidelberg, GermanyReviewed by:
Vernon L. Tesh, Texas A&M University Health Science Center, USAFumiko Obata, University of Maryland School of Medicine, USA
Sowmya Balasubramanian, Tufts University, USA
Copyright: © 2015 Mayer, Parello, Lee, Itagaki, Kurosawa and Stearns-Kurosawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shinichiro Kurosawa, Department of Pathology and Laboratory Medicine, Boston University School of Medicine, 670 Albany Street, 4th floor, Boston, MA 02118, USA e-mail: kurosawa@bu.edu