 Prathibha Reddy Gajjala
Prathibha Reddy Gajjala Maryam Sanati
Maryam Sanati Joachim Jankowski
Joachim Jankowski- Institute for Molecular Cardiovascular Research, Universitätsklinikum RWTH Aachen, Aachen, Germany
Chronic kidney disease (CKD), diabetes mellitus (DM), and cardiovascular diseases (CVD) are complex disorders of partly unknown genesis and mostly known progression factors. CVD and DM are the risk factors of CKD and are strongly intertwined since DM can lead to both CKD and/or CVD, and CVD can lead to kidney disease. In recent years, our knowledge of CKD, DM, and CVD has been expanded and several important experimental, clinical, and epidemiological associations have been reported. The tight cellular and molecular interactions between the renal, diabetic, and cardiovascular systems in acute or chronic disease settings are becoming increasingly evident. However, the (patho-) physiological basis of the interactions of CKD, DM, and CVD with involvement of multiple endogenous and environmental factors is highly complex and our knowledge is still at its infancy. Not only single pathways and mediators of progression of these diseases have to be considered in these processes but also the mutual interactions of these factors are essential. The recent advances in proteomics and integrative analysis technologies have allowed rapid progress in analyzing complex disorders and clearly show the opportunity for new efficient and specific therapies. More than a dozen pathways have been identified so far, including hyperactivity of the renin–angiotensin (RAS)–aldosterone system, osmotic sodium retention, endothelial dysfunction, dyslipidemia, RAS/RAF/extracellular-signal-regulated kinase pathway, modification of the purinergic system, phosphatidylinositol 3-kinase (PI 3-kinase)-dependent signaling pathways, and inflammation, all leading to histomorphological alterations of the kidney and vessels of diabetic and non-diabetic patients. Since a better understanding of the common cellular and molecular mechanisms of these diseases may be a key to successful identification of new therapeutic targets, we review in this paper the current literature about cellular and molecular mechanisms of CKD.
Introduction
A healthy person could be defined as “a man with highly/tightly regulated and coordinated complex biological networks.” The crosstalk between the organs and the systems via molecular, cellular, paracrine, endocrine, and neuronal factors are essential in regulating these networks (1). However, this tight–knit relationship is deregulated at some point when your body goes through a disease or disorder, which in turn influences the function of other organs and the challenge is to find the right target for that specific pathway (2). One example is the crosstalk between the pancreas, heart, and kidney organs, leading to vascular and renal diseases as well as diabetic mellitus (DM), which are strongly entwined with each other. Although, many mediators and pathways have been reported in the literature in regard to these diseases, their genesis is still a puzzle (3, 4). In this paper, we reviewed the current incidence, pathways, and the mediators involved in these intricate diseases and made an attempt to compel the common risk factors from the current literature.
Current Status on Incidence and Prevalence of CKD, DM, and CVD
Since two decades, the shift in the mortality and morbidity is being increased from infectious diseases to non-communicable disease worldwide (5). This rise in the number of patients mostly reflects to vascular and renal disease as well as DM, which are the major public health problem both in developed and developing countries, imposing burden on economy (6).
Chronic Kidney Disease
It is defined as a change in the kidney function or structure for more than 3 months that affects the health of an individual irrespective of the cause (7). Based on the glomerular filtration rate (GFR) and albuminuria content it is set into five stages. According to KDIGO guidelines people with GFR ≥90 ml/min/1.73 m2 are categorized into first stage where the kidney functions normally, 60–89 ml/min/1.73 m2 falls in the second stage with mildly decreased function. The third stage is sub divided into two; people with 45–59 ml/min/1.73 m2 and 30–44 ml/min/1.73 m2, where the GFR is mildly to severely decreased. In stage 4, the GFR decreases to 15–29 ml/min/1.73 m2 and in the fifth stage (<15 ml/min/1.73 m2), kidney fails to function. The early stages are asymptomatic and the end-stage is treated by dialysis or transplantation (8). Diabetes, hypertension, cardiovascular diseases (CVD), and cigarette smoking are the common risk factors of chronic kidney disease (CKD) (9, 10). CKD arises due to many pathological insults that affects the renal function and destroys some of the nephrons. As a result, the other nephrons compensate the function of injured nephrons by hyper filtration. Over a period of time, it develops glomerular hypertension, proteinuria, and eventually loss of renal function (11). An increase in the glomerular capillary pressure leads to the destruction of glomerular capillary wall leading to the dysfunction of podocytes that covers the capillaries and allow the permeability of macromolecules (12). These series of insults result in the release of inflammatory mediators and stimulate the proliferation of cells involved in fibrosis. Protenuria impairs the reparative mechanisms resulting in the scar formation due to the accumulation of extracellular matrix (ECM) molecules finally leading to renal failure.
Incidence and Prevalence
The incidence of CKD is not clearly known since a large number of patients die due to cardiovascular disease before they progress to end-stage renal disease. In Europe and North America, the prevalence was found to be ~10% and in the US, the prevalence was found to be 13.1% (13). Zhang et al. (14) reviewed 26 population-based studies conducted on the prevalence of CKD and reported that in the elderly, the prevalence was between 23.4 and 35.8%. A meta-analysis by Nitsch et al. studied the association of GFR, albuminuria with mortality, and renal failure by sex, and observed that the increased risk of CKD is equal between both sexes (15).
Diabetes Mellitus
Diabetes mellitus is a group of metabolic disorders resulting from hyperglycemia caused by genetic, molecular, or biochemical factors and activation of renin–angiotensin system (RAS), which eventually leads to the damage of end-organs like kidney (16). Diabetic nephropathy (DN), caused by DM, is one of the progressive kidney diseases characterized by damaged vessels (angiopathy) due to type 1 or type 2 diabetes, hypertension, or dyslipidemia affecting kidney filtering system that might progress to end-stage chronic kidney disease (CKD) (17). DN is also associated with CVD increasing mortality of DM patients (18). The hallmarks of DN are the abnormalities in the glomerulus that alter the structure of podocytes, the decrease in nephrin expression, and the thickening of basement, tubular, and glomerular membranes by the extracellular deposition causing tubulointerstitial and glomerular fibrosis (18).
Incidence and Prevalence
According to 2014 statistics, 387 million people are suffering from DM and the prevalence is around 8.3% worldwide, North America being on top (11.4%). 4.9 million deaths were reported in the year 2014 and 46.3% people are undiagnosed (19). DM accounts for 4% of global deaths below the age of 70 years (20). About 25–40% of diabetic patients suffer from DN and/or some degree of CKD (17, 21).
Cardiovascular Disease
Cardiovascular diseases is a group of disorders that is associated with the circulatory system including coronary heart disease (CHD), cerebrovascular disease, peripheral arterial disease, rheumatic heart disease, congenital heart disease, cardiomyopathies, cardiac arrhythmias, and deep vein thrombosis and pulmonary embolism (22). The major risk factors associated with CVD are hypertension, hypercholesterolemia, dyslipidemia, diabetes, smoking, physical inactivity, and obesity (23). Although, few studies have shown that the decrease in the kidney function and higher albuminuria are independent risk factors and not related to diabetes and hypertension (24, 25).
Incidence and Prevalence
It is estimated that more than 17 million deaths were reported in the year 2012 globally, which accounts for 31% of all deaths and might increase to 23.3 million by the year 2030. Among these deaths, 7.4 million deaths were due to CHD and the rest were due to stroke, mostly observed in low-income and middle-income countries (22).
Key Cellular and Molecular Events
RAAS System
One of the key players that have been implicated in the pathogenesis of cardiorenal disease is renin–angiotensin–aldosterone system (RAAS). In response to the decrease in the renal perfusion pressure, renin (protease) is produced by the juxtaglomerular cells of the kidney. It acts on an inactive peptide called angiotensinogen produced by the liver, converting it to angiotensin-I, which is a rate limiting and initial step of RAAS (26). Apart from juxtaglomerular cells, the highest expression of renin (Prorenin) is observed in the connecting tubules and collecting ducts in diabetes patients (27). Angiotensin-I is catalyzed into octapeptides by angiotensin converting enzyme (ACE-dipeptidyl carboxypeptidase), produced in the lungs and lymphocytes (28), to form angiotensin II, which is an active peptide (29). Angiotensin II is also produced by chymase and cathepsin G, which are independent of ACE activity (30). Furthermore, angiotensin II is cleaved to different peptides by the endopeptidases like ACE2. Recently, many new components have been identified in the RAAS system like angioprotectin, Ang III, IV, V, Ang-(1–7), Angiotensin-A, Alamandine, and their co-factors like vasodilation inducing factor (VIF) (31–34), They are mainly formed by the action of several endopeptidase that act via Mas receptors or Mas-related G-protein-coupled receptor membrane D (MrgD) receptors and they have shown to have vasodilatory effects (33, 35). The effects of Angiotensin II is mediated by the G-protein coupled receptors, i.e., angiotensin type 1 (AT1), angiotensin type 2 (AT2) receptors (30), or the MAS receptor (28). The former being expressed in several tissues like cardiovascular system, kidney, and the sympathetic nervous system (35), and the latter is expressed during fetal life and in adults restricted to the adrenals, ovary, brain, heart, and uterus (36). Angiotensin receptors share overall 30% homology and as a result they have different functions and are adapted to different signal transduction pathways (37). Ang II-AT1 axis has a role in vasoconstriction, cell proliferation, and oxidative stress in the kidney and is counteracted by AT2 receptor, which possesses vasodilatory, antiproliferative, and apoptotic properties (38, 39). Activation of AT1 receptors by angiotensin II in the kidney constricts the efferent arteriole, which results in a decrease of the blood flow that affects the glomerular filtration by raising the glomerular capillary pressure. This in turn results in glomerular injury and an increase in the production of nephrotoxic reactive oxygen species (ROS), profibrotic cytokines, and growth factors. The production of these components stimulates mitogenesis of fibroblast cells that deposit the ECM (renal fibrosis) and inhibits the turnover, leading to CKD (40, 41). Angioprotectin antagonized the contractile actions of Ang II, mediated by the Mas receptor, and Ang A has the same affinity to the AT1 receptor as Ang II, but has higher affinity toward AT2 receptor thus may modulate the harmful effects of Ang II (31, 32). Alamandine acts through the Mrg receptor and shows its vasodilatory effects (33). Furthermore, the recently identified factor vasodilation inducing factor (VIF), a chromogranin peptide, reduces the direct vasoconstrictive effects of Ang II, independent of NO in human plasma (34).
In adrenal cortex, Ang II stimulates the release of aldosterone from zona glomerulosa via AT1, which sequentially regulates the blood pressure, fluid, and electrolyte balance through mineralocorticoid receptors of the distal tubule and collecting duct (42). Aldosterone binds to the mineralocorticoid receptor that regulates the sodium–potassium pumps (43) and since it has cell proliferative and profibrotic properties, it directly increases the expression and production of the profibrotic cytokine “transforming growth factor β” (TGF-β). In kidney disease models, aldosterone synthesis is increased and implicated in the proliferation of fibroblast cells, renal fibrosis, and induction of hypertension due to the sodium overload (44). All these effects synergistically provoke renal damage.
The notion of RAAS acting systemically has been changed by the discovery of local RAAS with paracrine or autocrine action during pathogenesis (32, 45), e.g., during hyperglycemia and proteinuria, each component of RAAS was observed in the proximal tubular cells that synthesize Ang II from angiotensinogen into interstitial and luminal side, leading to the activation of sodium pumps besides aldosterone (46, 47). In β cells of pancreas, the deleterious axis of RAAS, i.e., Ang II-ACE-AT1R-aldosterone increases the oxidative stress, promotes apoptosis, decreases the uptake of glucose (by suppressing the GLUT2 through AT1R), and increases the production of ROS through NADPH oxidase (NOX), thereby decreasing the production of insulin leading to hyperglycemia (4). Figure 1 gives the overview of the mediators in RAAS leading to CKD, DM, and CVD.
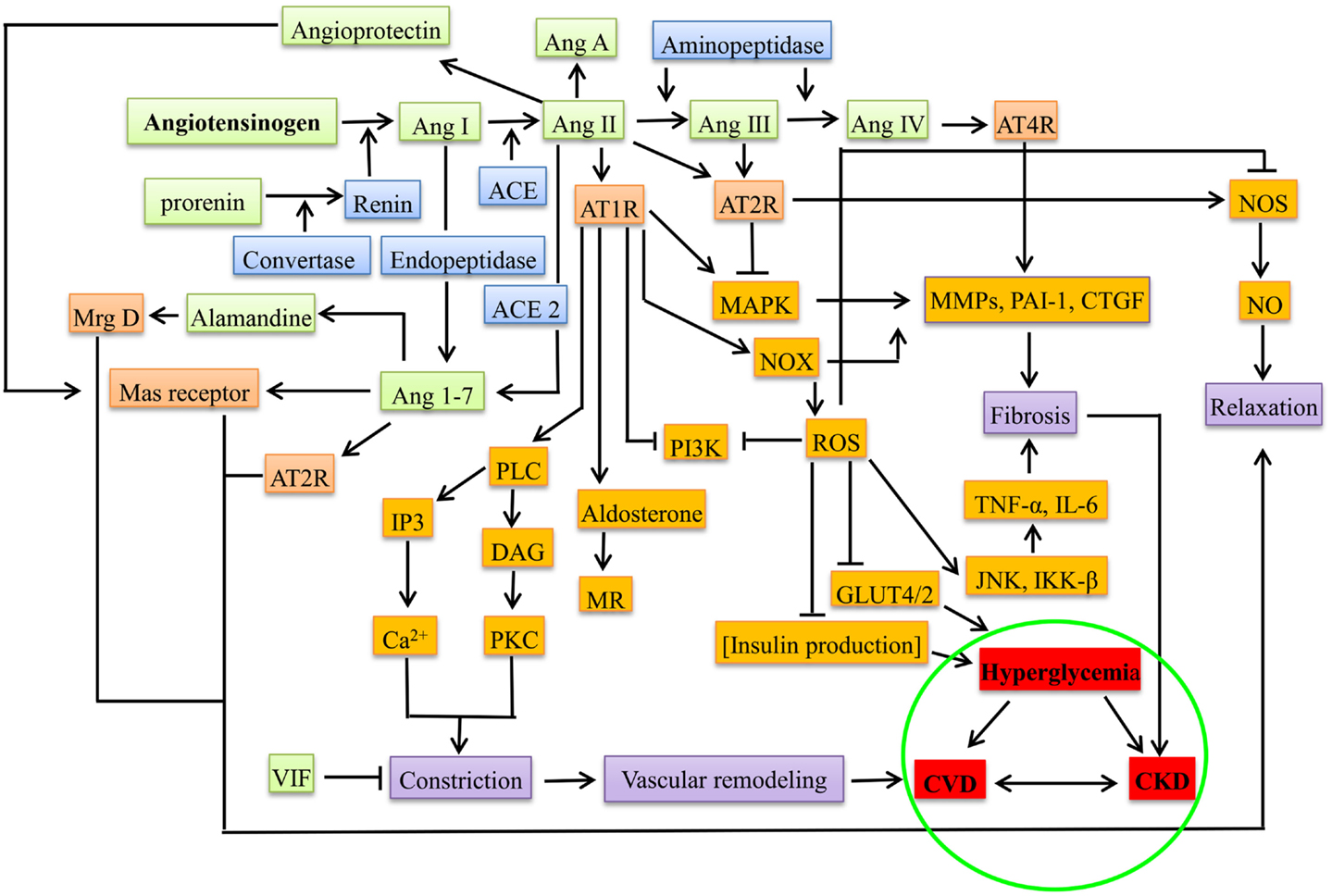
Figure 1. Overview of RAAS system under pathological conditions (CKD, DM, and CVD). The green boxes represent the peptide components of RAAS system, blue represents the enzymes involved, orange represents the receptors, brown represents the downstream molecules/effectors, violet boxes represent the presentation before the disease, and red represents diseased state. Ang, angiotensin; ACE, angiotensin converting enzyme; AT1R, angiotensin type 1 receptor; AT2R, angiotensin type 2 receptor; AT4R, angiotensin type 4 receptor; Mrg D, Mas-related G-protein coupled receptor membrane D; VIF, vasodilation inducing factor; PLC, phospho lipase C; DAG, diacyl glycerol; IP3, inositol triphosphate; PKC, protein kinase C; MR, mineralocorticoid receptor; PI3K, phosphoinositide 3-kinase; MAPK, mitogen-activated protein kinase; NOX, NADPH oxidase; ROS, reactive oxygen species; MMPs, matrix metalloproteinases; PAI-1, plasminogen activator inhibitor-1; CTGF, connective tissue growth factor; TNF-α, tumor necrosis factor alpha; JNK, c-Jun N-terminal kinases; IKK-β, inhibitor of nuclear factor-kappa-B kinase; NOS, nitric oxide synthase; CVD, cardiovascular disease; CKD, chronic kidney disease.
Wnt/β-catenin signaling pathway plays an essential role in the organogenesis and tissue homeostasis, which upon activation translocate β-catenin into nucleus, that binds to the T-cell factor (TCF) or lymphoid enhancing factor (LEF) along with other co-factors like CREB-binding protein (CBP), which eventually transcribes the target genes. In healthy subjects, this pathway is silent in kidneys, but in CKD, it is reactivated (48). By bioinformatics approach, Zhou et al. (49) demonstrated that all RAS genes have putative TCF/LEF binding sites at their promoter regions. This is a likely link between the activation of RAS by Wnt/β-catenin during CKD.
Inflammation
Chronic inflammation is observed in patients with CKD and it contributes to the CVD morbidity and mortality by accelerating the vascular inflammation (50). Release of cytokines at the site of injury recruits the activated immune cells, which further enhances the inflammatory state by producing additional mediators (51). CXCL12 is a chemokine that binds to the CXCR4/7 and has a role in homing of stem and progenitor cells in the bone marrow to the site of injury or into the circulation. CXCL12/CXCR4 axis shows atheroprotective properties by mobilizing the endothelial progenitor cells to the site of injury (52). Local production of Ang II has been implicated in the pathophysiology of inflammation (53). Ang II stimulates the production of proinflammatory molecules like NF-κB by Toll-like receptor 4 in addition to Ang III and Ang IV via AT1 and AT2 receptors (54, 55). Ang II also upregulates the “vascular cellular adhesion molecule-1” (VCAM), “intracellular adhesion molecule-1” (ICAM), and NF-κB, and thereby mediates the production of chemokines like monocyte chemoattractant protein-1 (MCP-1), which recruit the immune cells (56). In mesangial cells, the expression of ETS-1 (E26 transformation-specific sequence), a key regulator of vascular inflammation, is upregulated in the presence of Ang II via ROS production by NADPH oxidase and it participates in inflammation by recruiting T-cells and monocytes/macrophages into the vessel walls (57). In CKD patients, the cytokines; IL-6, IL-1β, and TNF-α are elevated causing the cardiovascular outcomes (58–60). The TAM (Tyro 3, Axl, Mer) ligand-receptor pathway is involved in the regulation of inflammatory processes. This pathway is deregulated in CKD patients and plays a role in atherosclerosis and thrombosis (50). TAM ligands (Gas-6 and Protein S) and receptors are expressed by the innate immune cells that limit the production of proinflammatory cytokines, which are stimulated through toll-like receptors (TLR) by the activation of suppressor of cytokine signaling (SOCS) (61, 62). TAM ligand–receptor signaling is linked to the elevation of cytokines in CKD patients, endotoxemia-mediated TLR activation, chronic monocyte activation, and the involvement of macrophages in atherosclerosis in CKD (50). ROS formation and activation of TLR increases the expression of Gas-6 and shedding of TAM (sTAM) receptors from monocytes binding to Gas-6, which results in the deregulation of TAM receptor signaling, the cytokine, and TLR cascades getting activated in CKD patients and finally chronic inflammation (50). Due to over nutrition and low physical activity, the cells are overloaded with glucose and free fatty acids (FFA), the β cells are unable to produce more insulin, and this causes insulin resistance leading to impaired glucose tolerance (63). The cell avoids citric acid cycle when the caloric intake is more than the required energetics. Acetyl-CoA formed by oxidation of glucose or FFA, combines with oxaloacetate to form citrate, which enters the citric acid cycle, generating an excess amount of NADH that impairs the electron transport in mitochondria resulting in the formation of ROS. The cell avoids it by inhibiting the entry of fatty acids into mitochondria as a result FFA build up in the cytosol (64). Furthermore, the increase in intracellular FFA leads to a decrease in the transportation of GLUT4 to the plasma membrane and therefore the buildup of glucose concentration in the blood (65). Constant exposure to glucose leads to dysfunctioning of β cells and endothelial cells (66). In hyperglycemic conditions, the endothelial cell metabolism is modified and implicated in endothelial dysfunction, e.g., presence of high glucose is activating the NADPH oxidase resulting in the increased production of ROS and an increase in oxidative stress by reducing the entry of glucose-6-phosphate into the pentose phosphate pathway thereby reducing the NADPH formation (67, 68). Excess of glucose activates the arginase uncoupling the endothelial nitric oxide synthase (eNOS) resulting in the increase of superoxide anion production. Increased levels of glucose also diverts to polyol pathway where it is converted to sorbitol by utilizing the NADPH, increasing the ROS production, and activating NF-κB signaling pathway (69–71). Due to changes in the endothelial metabolism, advanced glycation products (AGEs) are formed, which crosslink the extracellular molecules, causing increased vessel stiffness, resulting in vascular complications (72). The AGE products bind to their receptors (RAGE) that are expressed on the monocytes, endothelial cells, and smooth muscle cells. They are involved in the increased expression of scavenger receptor class A in macrophages, which results in the uptake of the oxidized LDL leading to the formation of foam cells, a characteristic feature of atherosclerosis (69). Cholesteryl fatty esters are the main component of foam cells that induce cytotoxicity and are involved in lesion development in atherosclerosis (73). The late stages of macrophages release the lipid content and tissue factors leading to the formation of pro-thrombotic core, resulting in rupture of plaque, leading to blood clot and contributing to myocardial infarction and stroke (73). AGE–RAGE interaction with vascular smooth muscle cells (VSMCs) is involved in cell proliferation calcification process and also activates the NF-κB signaling pathway where it controls the expression of cytokines (71). RAGE also acts as an adhesive receptor in the endothelial cells, attracting the immune cells at the site of injury (74). Nitric oxide (NO) is formed from the arginine by eNOS in endothelium and is involved in vasodilation. Asymmetrical dimethyl arginine (ADMA), an endogenous analog of arginine, which is elevated in atherosclerotic patients competes to bind at the active site of eNOS thereby decreasing the NO (75). ROS produced due to eNOS uncoupling and NADPH oxidase activity triggers the expression of adhesion molecules, transmigration of immune cells, VSMCs proliferation and migration, endothelial apoptosis, and oxidation of lipids (69). Oxidized LDL results in the mitochondrial DNA damage and dysfunction in the endothelial cells, leading to high production of ROS (76). Xu et al. (77) analyzed cohorts of elderly adults and reported that a proinflammatory diet is associated with the systemic inflammation and reduced kidney function.
Fibrosis
At the site of cell injury or tissue damage, the cells are replaced by the same cell type or with fibrous tissue after the clearance of the inflammatory response. The kidneys have an intrinsic capacity to repair cell death by the de-differentiation and proliferation of tubular epithelial cells. Failure of these processes results in fibrosis during infarction/ischemia or toxic insult (78, 79). Renal fibrosis is a prominent feature of every stage of CKD where an excessive accumulation and deposition of ECM are observed. At the beginning of inflammatory response in interstitium, infiltrates of macrophage population can be observed, which links inversely with the kidney function (80), that could be either deleterious (M1 macrophages) or advantageous (M2 macrophages). This is followed by transdifferentiation of interstitial cell population to myofibroblasts. These myofibroblasts have characteristics of both smooth muscle cells and fibroblast cells, which produce ECM proteins, like collagen or fibronectin, that eventually results in scar formation (81, 82). Myofibroblasts are the primary effector cells involved in both tissue remodeling and fibrosis (83). They are mainly derived from the de-differentiation of resident pericytes and fibroblast cells in the presence of fibrogenic factors that promote cell to cell interaction. These fibrogenic factors are secreted by endothelial cells, epithelial cells, and myeloid leukocytes especially the monocyte-derived cells and they are also dependent on the environmental stimuli like hyperglycemia and hypoxia (84, 85). The M1 and M2 macrophages are derived from monocytes based on the local stimuli. For example, interferon gamma (IFN γ), TLR, tumor necrosis factor, and granulocyte-macrophage colony-stimulating factor stimulate M1 macrophages. M1 macrophages are involved in tissue injury that secretes proinflammatory cytokines such as IL-12, IL-23, IL-1, IL-6, type-I interferon, and also produce reactive oxygen intermediates and nitric oxide. M2 macrophages, on the other hand, play a role in tissue repair and are stimulated by IL-4, IL-10, IL-13, corticosteroids, vitamin D, macrophage colony-stimulating factor, and TGF β (82, 86). Tubular epithelium is involved in the production of ROS and inflammatory mediators that evade the interstitium via basolateral secretion or paracrine pathways (82). They secrete mediators like monocyte chemoattractant protein-1 (MCP-1), IL-8, fractalkine, TGF β, endothelin by triggering megalin receptor-mediated protein endocytosis during protein overload. They also activate specific pathways with other co-receptors, cubilin and amnionless, leading to interstitial inflammation, fibrosis, and loss of nephron (87).
TGF-β1
The cytokine TGF-β1 is a prominent and powerful factor mediating myofibroblast activation, which integrates the effects of other fibrogenic factors (88). TGF-β1 is synthesized by all types of cells in the kidney and released in association with the latency-associated peptide (LAP) binding to latent TGF-β-binding protein (LTBP). Upon stress stimuli like hypoxia (89), RAAS (90), oxidative stress (91), and TGF-β1 are activated, binding to the Type-II TGF-β receptor. Type II TGF-β receptor is a kinase that recruits Type-I TGF-β and phosphorylates the downstream molecules like Smad2/3, which in turn forms a complex with Smad4 and then translocate to the nucleus to transcribe the target genes along with other factors (88, 92). TGF-β1 shows profibrotic effects on kidney through different mechanisms where it induces the production of ECM through Smad3 by binding to the promoter region of collagen or Smad-independent pathways. This leads to an inhibition of its degradation by inducing tissue inhibitor of metalloproteinase (TIMPs) and inhibiting matrix metalloproteinases (MMPs). TGF-β1 is also involved in transdifferentiation of different types of kidney cells to myofibroblast cells (88, 93, 94). It has a role in podocytopenia where the podocytes undergo apoptosis and detach from the glomerular basement membrane resulting in the loss of integrity of microvasculature (95). In hyperglycemic conditions, the AGEs and Ang II induce transdifferentiation of epithelial to mesenchymal transition (EMT) mediated through Smad3 phosphorylation (96, 97). TGF-β1 stimulates the differentiation of epithelial, endothelial, and macrophage cells to mesenchymal transition synthesizing ECM (81, 98). Stimulation of myofibroblast with TGF-β1 increases the expression of cannabinoid receptor 1 that increases collagen expression (99). The latent form of TGF-β1 is protective against inflammation and fibrosis by phosphorylating Smad7 (inhibitory Smad) (100). TGF-β1 shows effects via Smad-independent pathways like p38, JNK, extracellular-signal-regulated kinase (ERK), mitogen-activated protein kinase (MAPK), integrin-like kinase (ILK) PI3K/Akt (101–103). TGF-β activates PI3K that phosphorylates Akt and is involved in fibroblast proliferation and ECM deposition (92). Inhibition of PI3K activity results in a decrease in TGF-β-Smad2 phosphorylation and has an effect on EMT and cell migration (104). In mesangial cells, TGF-β1 activates phosphatidylinositol 3-kinase PI3K/Akt signaling, resulting in the mesangial cell hypertrophy and fibrosis in diabetes (105, 106). High glucose levels, protein glycation end-products, Ang II, endothelin-1, and growth factors like PDGF and EGF, induce cell proliferation through PI3K/Akt signal transduction (92).
Upregulation of renin and angiotensinogen was observed in glomeruli and involved in the renal scarring or repair by the action of Ang II (107). Transfection of glomeruli with renin and angiotensinogen stimulates the proliferation of fibroblast cells, increases the production of ECM (by inducing the mRNA of proteins like type I procollagen and fibronectin in cultured mesangial cells) and also transcribes collagen type 1 (IV) and 3 (IV) but not type I in cultured proximal tubular cells by Ang II which in turn depends on TGF-β expression (107). Renin alone is able to stimulate the expression of TGF-β in mesangial cells and this could contribute to renal fibrosis despite of Ang II blockade (108). Expression of “connective tissue growth factor” (CTGF), a fibrotic mediator is increased in the kidney induced by TGF-β and Ang II (109). Ang II induces the “plasminogen activator inhibitor-1” (PAI-1) and “tissue inhibitor of matrix metalloproteinases-1” (TIMP-1) via AT1R, which inhibits metalloproteinase affecting the matrix turnover, resulting in accumulation of ECM. PAI-1 is also stimulated by Ang IV via AT4R in proximal tubules thereby playing a role in renal fibrosis (110). In DN, Ang IV is formed by the degradation of Ang II when in high concentrations by different enzymes and this induces PAI-I (111). Most of the fibroblast cells in the interstitium originate from tubular epithelial cells through a process called EMT. This process is mediated by TGF-β and the fibroblast cells are involved in interstitial fibrosis and tubular atrophy (112). Figure 2 shows the cells and the mediators involved in the fibrosis.
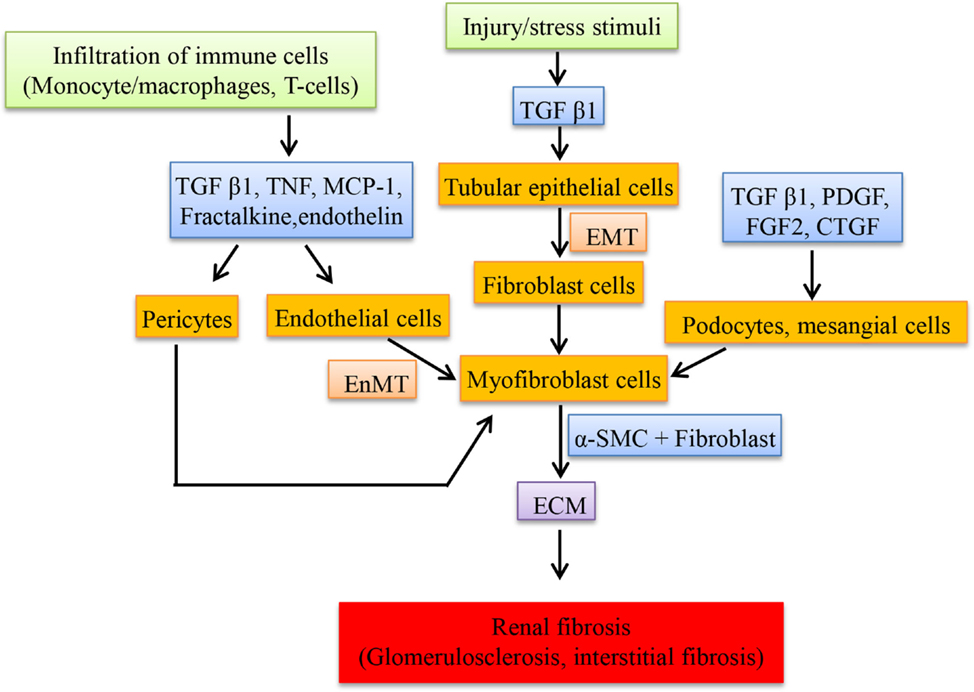
Figure 2. Schematic representation of cellular mediators involved in renal fibrosis. The infiltration of macrophages secretes a set of mediators at the injured sites of kidney resulting in the transition of resident cells to myofibroblast cells, which proliferate and secrete the extracellular matrix compounds.
All these factors line up, and as a result the kidney loses its function and faces oxidative stress, subsequently leading to the accumulation of toxic substrates. Some of these substrates are involved in the post-translational modification of proteins like carbamylation and oxidation of LDL and HDL, which aggravate the progression of atherosclerosis thus leading to CVD (113, 114). RAAS blockade has been shown to be effective for the treatment of CKD, CVD, DM, hypertension with proteinuria. With an increase in the plasma aldosterone concentrations, the left ventricular mass has shown to be larger in early CKD stages in hypertensive patients (115).
Uremic Toxins
Uremic toxins are in general the waste products that accumulate in the body fluid due to dysfunction of the kidney. They are divided into small water soluble compounds, middle molecules, and protein-bound uremic compounds (116). Among protein-bound uremic toxins, para-cresol sulfate (PCS) and indoxyl sulfate (IS) are linked to the cardiovascular comorbidities (117). p-cresol, a metabolite of p-cresol sulfate showed reduced contraction rates of cardiomyocytes, resulting in the irregular beating mediated by protein kinase C (PKC) by increasing the intracellular calcium levels (118). Furthermore, p-cresol is involved in the increased endothelial micro-particle shedding mediated by Rho-kinase in hemodialysis patients, leading to endothelial dysfunction, which is also observed in acute coronary syndromes, acute ischemic stroke, and venous thromboembolism (119–122).
Indoxyl sulfate also contributes to atherosclerosis and peripheral artery diseases. Indoxy sulfate stimulates NADPH oxidase in endothelial cells, increasing the ROS formation and decreasing the levels of glutathione, thus elevating oxidative stress (123). The uptake of IS through organic anion transporter 3 (OAT 3), further stimulates the expression of “prorenin receptors” (PRR) in the aorta of a CKD mouse model, thereby activating aryl hydrocarbon receptors (AhR) and NF-κ B in VSMCs. Activation of PRR by IS promotes cell proliferation and expression of tissue factors like platelet-derived growth factor and receptor (PDGF/R) in VSMCs. It also activates MAPK pathways, thus linking the uremia-induced cardiovascular events (124, 125). Moreover, IS stimulates the production of IL-1, IL-6, and TNF-α in THP-1 cells. Lekawanvijit et al. (126) demonstrated that IS plays a critical role in cardiac remodeling by acting as profibrotic, prohypertrophic, proinflammatory factor, mediated by the activation of MAPK (p38, p42/44) and NF-κ B pathway. IS also upregulates the expression of ICAM and MCP-1 through ROS production, activating NF-κ B pathway in endothelial cells. PCS and IS stabilize the active form of epidermal growth factor (EGFR) and help in dimerization and phosphorylation, leading to an increase in the MMP2/9 expression either directly or by its downstream molecules. Therefore, they are both involved in renal tissue remodeling (127). Under oxidative stress, the protein-bound uremic solutes have an impact on the cardiac tissue architecture as well as on the kidney tissue. For example, uridine adenosine tetraphosphate (Up4A), which is synthesized by the endothelial cells acts via purinergic receptors P2Y that activates MEK, ERK1/2 by phosphorylation, and regulates the expression of vascular calcification (VC) mediators like Cbfa, Msx2, Osx, OCN, osteopontin (OPN) leading to the transdifferentiation of vascular smooth vessel cells to osteochondrogenic cells (128, 129). In VSMCs, Up4A stimulates the production of ROS by Nox1 dependent way and increases the expression of MCP-1 through phosphorylation of MAPK via P2Y (2) receptor (130). The uremic toxin phenyl acetic acid is associated with arterial vascular properties in patients with CKD that are undergoing hemodialysis (131). It also inhibits the inducible nitric oxide synthase (iNOS) and impairs the macrophage function, thus increasing the incidence of infection in uremia (132, 133).
Vascular Calcification: A Link between CKD, DM, and CVD
The mechanism of VC is multifactorial and is poorly understood. The unusual deposition of calcium is observed in media and intima of the vessel, which changes the elasticity and the hemodynamics of the vessel wall that result in stroke and ischemic heart disease (134). This ectopic calcification is noticed in atherosclerosis, diabetes, hypertension, dyslipidemia, and CKD (134). The main presentations of calcification are atherosclerotic lesions, medial calcification, aortic stenosis, and calciphylaxis, which are the most dramatic and often fatal entity. Atherosclerosis, CKD, hyperphosphatemia, and vitamin K-targeting oral anticoagulants have emerged as key contributors to VC.
Vascular calcification is an active and highly regulated process, closely resembling osteogenesis. The factors contributing to the VC are the high levels of calcium and phosphorous due to abnormal bone metabolism, transition of VSMCs to chondrocyte or osteoblast-like cells, imbalance in the levels of inhibitors of calcification, low levels of klotho expression, high levels of parathyroid hormone (PTH), and fibroblast growth factor-23 (FGF-23) expression, Vitamin D, and ECM remodeling (135, 136). VC is characterized by transdifferentiation of VSMCs to osteoblast-like cells where they express a number of bone matrix proteins (137). High plasma calcium phosphate product (Ca × P) contributes to this active process and hyperphosphatemia is considered a key driver of VC in CKD. VC is observed due to imbalance between inhibitory and inducing mediators. The most relevant are OPN, osteoprotegerin (OPG), fibroblast growth factor-23 (FGF-23), bone morphogenetic proteins (BMP), matrix-GLA protein (MGP), CD73, aldosterone, fetuin, pyrophosphate, magnesium, and uremic toxins like Up4A. Increased expression of OPN is an indicator of VC and transformation of VSMC to osteoblast-like cells (138). Unphosphorylated OPN enhances whereas phosphorylated OPN decreases VC (138). The amount of OPN can be pharmacologically modified by Mg2+ (139, 140). OPG is a decoy receptor for receptor activator of nuclear factor-kappaB ligand (RANKL) that inhibits RANKL-induced VC and bone loss (141). OPG−/− mice display osteoporosis and VC. The phosphaturic hormone FGF-23 activates the FGFR1 receptor in the presence of Klotho (142). Klotho has FGF-23-independent phosphaturic actions. However, it is still unclear whether FGF-23 or Klotho exert direct effects on VSMC phenotype (143). Klotho-deficient mice display accelerated aging, hyperphosphatemia, osteoporosis, and VC. A human klotho mutation was associated with VC in a teenager (144). Both, systemic inflammation and CKD decrease Klotho expression. In CKD, FGF-23 levels increase more than a 100-fold in response to hyperphosphatemia, and elevated FGF-23 levels are predictive of cardiovascular events (145).
Bone morphogenetic proteins are crucial mediators of vascular remodeling and neovascularization (146). BMPs regulate calcification in mesenchymal progenitor cells for cartilage and bone (147) and modify expression of osteoblast markers like alkaline phosphatase in VSMC. The expression of BMPs is increased at VC sites. In addition, BMP and osteoblast homeoprotein Msx2 signaling pathway promotes the differentiation of myofibroblasts into the osteogenic pathway and enhances VC. BMP7 is a calcification inhibitor whereas BMP-2 promotes VC (148). Protein C and S deficit and therapy with vitamin K-targeting anticoagulants are risk factors for calciphylaxis (calcific uremic arteriolopathy) (149). Vitamin K-dependent factors, including proteins C and S, require glutamic acid carboxylation to yield gamma carboxyglutamic residues (Gla) for activation. MGP is a vitamin K2-dependent calcification inhibitor. MGP−/− mice die a few weeks after birth from aortic rupture due to massive calcification. Vitamin K deficiency has been linked to VC in in vitro and in vivo mouse studies (150). Warfarin, an anticoagulant decreases the active form of MGP and increases VC in CKD rats while vitamin K increased active MGP and prevented VC in warfarin-treated rats (151).
Aldosterone contributes to VC by activating PIT1-dependent osteo-inductive signaling (152). α2-HS-glycoprotein/fetuin levels are decreased in CKD and associate with VC. Uremic toxins, such as IS, induce expression of osteoblast-specific proteins in human VSMC and associate with aortic calcification in CKD, whereas in hypertensive rats, it promotes aortic calcification (153). Leukocyte activation evoked by CKD directly contributes to VC and pyrophosphate acts as an inhibitor of VC (154). A low extracellular pyrophosphate level induces VC in murine model of progeria (155).
Recently, protective effects of VC by magnesium through multiple mechanisms by decreasing the intimal media thickness and aortic pulse wave velocity were described (139). In CKD rats, treatment with a magnesium-based phosphate binder significantly reduced aortic calcification compared to sevelamer-treated rats (156). However, randomized controlled studies should address the impact of magnesium administration in CKD patients.
Metabolic Derangements in CKD Modulating VC
In CKD, hyperphosphatemia, iatrogenic hypercalcemia, abnormal levels of PTH and FGF/23, diabetes, and inflammation may contribute to VC.
Hyperphosphatemia. High serum phosphate is associated to progression of VC and increased cardiovascular risks in patients with CKD and in dialysis. In the general population, serum phosphorus in the high normal range is associated with increased mortality (157). Exposure of VSMCs to high phosphorus concentrations results in loss of smooth muscle proteins and differentiation into osteogenic cells (158). Osteogenic transdifferentiation may end up in cell apoptosis with production of apoptotic bodies acting as nucleation sites for mineral deposition (159). Hyperphosphatemia-induced nanocrystals upregulate the expression of BMP-2 and OPN VSMC (160). Phosphorus binders are used to reduce phosphorus levels as a key factor in the management of CKD-MBD (mineral and bone disorder). The use of calcium-containing phosphate binders that induce positive calcium balance is associated with increased arterial calcification in the majority of studies (161). The amount of calcium binders required to control phosphate results in positive calcium balance (162) and most studies reveal that the progression of VC is slower with non-calcium-based binders as compared with calcium-containing binders. Excessive load of calcium may favor the development of VC directly or through an inhibition in PTH secretion with the consequent reduction of bone turnover.
Hypercalcemia. In CKD patients, hypercalcemia is usually a consequence of excess calcium administration or vitamin D overdosing and is associated to VC. High-calcium medium increases VSMC calcification (159) and potentiates the effect of phosphate. Calcium induces the release of matrix vesicles by VSMC that become calcified in the absence of MGP (163).
Hypermagnesemia. In dialysis patients, high serum magnesium concentration was associated with less VC. Low serum magnesium was associated with increased mortality of hemodialysis and non-dialysis CKD patients in observational studies (164). Magnesium-containing phosphate binder-induced positive calcium balance is associated with increased arterial calcification in the majority of studies (165). Many interventional studies showed that magnesium-based compounds are more effective in reducing the phosphate level that improve the survival and progression of VC (166).
Vitamin D. Vitamin D may have dose-dependent opposing effects on VC. Vitamin D3 increases calcium transport into the cells and upregulates calcification-enhancing genes, osteocalcin, osterix, and Runx2 (167). Protective effects of Vitamin D3 on VC may be expected by its re-differentiating and anti-inflammatory properties (inhibition of TGFbeta and IL-6) (168), as well as by increasing the expression of VC inhibitors (MGP and Osteopontin).
Experimental studies in vitro and in vivo have shown that high doses of calcitriol may induce VC (169, 170). Knockout mouse models of FGF-23 and Klotho, which exhibit increased arterial calcification, have elevated calcitriol levels (171). In mice, deficiency in both FGF23 and renal 1-alpha-hydroxylase appear to blunt arterial calcification (171). The effect of calcitriol on VC is mainly due to the increase in phosphate absorption that in renal failure results in hyperphosphatemia.
FGF-23-klotho. The hormone fibroblast growth factor-23 (FGF-23) induces phosphaturia and inhibits calcitriol synthesis and PTH production. Serum levels of FGF-23 increase early in the development of CKD. Several clinical studies demonstrated associations between higher levels of FGF23 and VC (172). However, neutralization of FGF-23 in uremic rats results in increased VC (173). A more recent human study including a large population showed that FGF-23 is not associated with VC (174). Some authors have not been able to identify klotho, co-receptor for FGF-23 in human arteries (174). Whether FGF23 has a direct effect on VC is still under debate.
Parathyroid hormone. Hyperparathyroidism is a major risk factor for medial calcification in renal disease (175). In uremic animals, PTH infusion produced osteogenic transdifferentiation of VSMCs with increased calcium deposition regardless of phosphate intake. The severity of medial calcium deposition in the aorta correlates with the levels of serum PTH in uremic rats with ovariectomy and with the loss of cortical bone. High and low PTH is associated with high turnover and adynamic bone disease, respectively. They both lead to decreased bone formation, a situations in which the bone is not efficient in incorporating excess calcium and phosphorus (176). This may in turn increase the risk for soft-tissue calcifications.
Diabetes mellitus. In diabetic patients, VC is commonly seen in coronary and vascular arteries of lower limbs (177, 178). Monckeberg already described medial artery calcification in an autopsy series that was identified in older age, renal failure, and diabetes as the factors associated with calcifications (179). In patient with diabetes, increased serum creatinine concentration, older age, and poor glucose control are associated with VC and mortality. High cholesterol levels, smoking, and body mass index were not associated with increased VCs. In the Ldlr−/− diabetic mice, a high fat diet produces aortic calcification with concomitant upregulation of osteogenic gene expression (180). Incubation of bovine VSMCs with high glucose led to osteogenic transdifferentiation and calcification (181). In diabetic arteriosclerosis, Msx1 and Msx2 promote vascular mineralization (182). Furthermore, elevation of glucose levels increased the BMP-2/Msx2-Wnt pathway, which promoted osteogenic differentiation (181). Thus, it is likely that high glucose may have direct effects on the osteogenic differentiation of VSMCs and per se VC formation.
Dyslipidemia. From clinical studies, lipids do not appear to play a major role on VC formation. In vitro, HDL inhibits the osteogenic differentiation; among the fatty acids, stearate, promoted mineralization, whereas inhibition of acetyl-CoA carboxylase reduced mineralization (183). As mentioned above, in diabetes, cells are loaded with FFAs. One of the abundantly found long-chain saturated fatty acid is palmitic acid, induces the expression of BMP-2, Msx2, and OPN in human aortic smooth cells through the expression of long chain acyl-coA synthetase 3 and NF-κB that resulted in osteoblastic differentiation (184). Oxidized lipids are involved in VC. Oxidized LDL promotes the calcification process in human coronary artery smooth muscle cells by upregulating the expression of mineral matrix proteins like osterix (Osx) and Runx2 mediated through nuclear factor of activated T cells (185, 186).
Inflammation-oxidative stress. Inflammatory cytokines and oxidative stress promote VSMC expression of osteogenic transcription factors. In a murine model of uremia, phenotypic changes in VSMCs occur prior to calcium deposition, accompanied by elastin degradation, possibly due to upregulation of proteases, including matrix metalloproteinases (187).
Oxidative stress and oxidized lipids induce osteochondrogenic differentiation of vascular cells in vitro. Oxidative stress induces Cbfa1/Runx2 expression through modulation of PI3-kinase/Akt signaling (188). In vitro studies show that TNF-α induces calcification in VSMC. TNF-α promotes phosphate-induced calcification by reducing efflux of a calcification inhibitor, pyrophosphate, and its transport protein, ankylosis protein homolog (189). The important role of oxidant stress on VC is demonstrated in experiments showing that antioxidants block vascular cell calcification in vitro.
Clinical Link of the Diseases
Epidemiological studies have so far confirmed and reported that there is an association between CKD and CVD (190). Go et al. (191) conducted a study on the relationship between GFR and cardiovascular events in a large group who had not undergone dialysis or transplantation and demonstrated an inverse relationship. The key mechanisms accounting for cardiovascular events apart from the traditional risk factors could be disturbances in mineral metabolism, anemia, ADMA (asymmetric omega NG, NG-dimethylarginine), inflammation, and oxidative stress, which are observed even in CKD. Whether CKD causes CVD or acts as a marker is still controversial (192). Due to disturbances in the metabolism in diabetes, the most effected cells are endothelial and β cells. Since the micro and macro vessels are lined with endothelial cells, the whole vascular system is damaged leading to CKD and CVD. Therefore, disturbance in one of these diseases will affect the other. Figure 3 shows how these diseases are interlinked with each other in a clinical perspective.
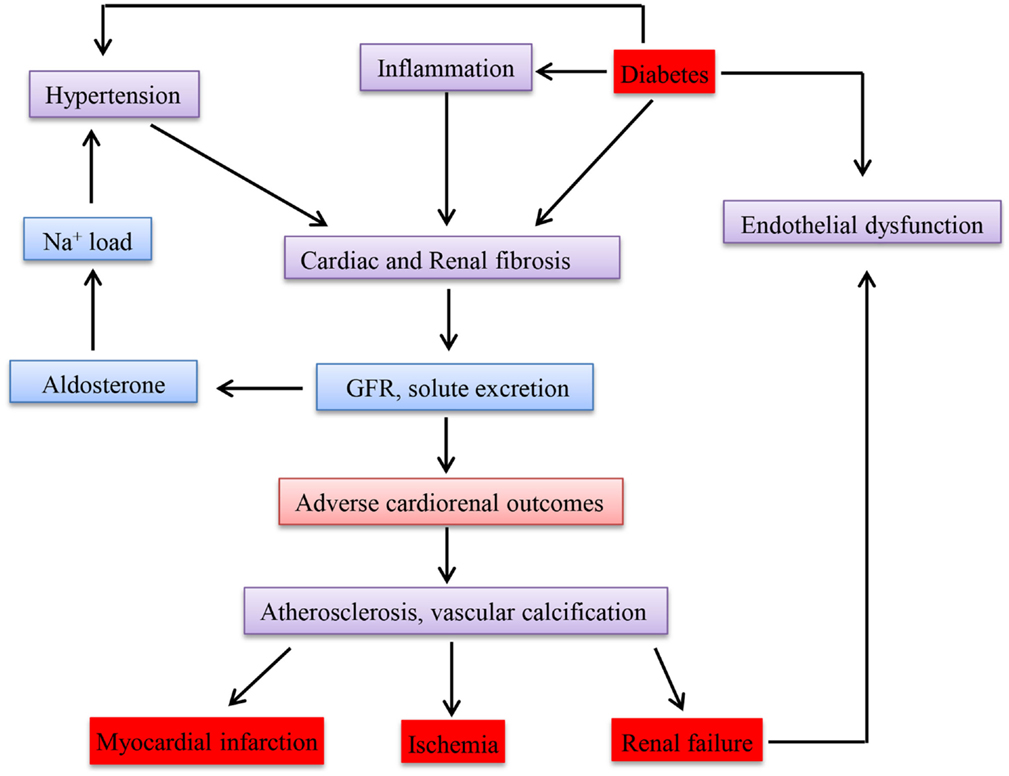
Figure 3. Schematic representation of clinical link between chronic kidney disease, diabetes mellitus, and cardiovascular disease.
Conclusion
The heart, kidney, and the vascular system are strongly related and help in maintaining hemostasis and cardiorenal equilibrium. Unfavorable outcomes are most common in CKD and for that reason it may deserve a specific approach to evaluate its progression. Due to the advancements in the basic research, many mechanistic pathways have been elucidated in the context of these diseases recently but none of the pathway could interpret the genesis of CKD. Therefore, a key factor in intervention for CKD patients is having an insight on the mechanisms of risk factors associated with it. Our knowledge about the underlying mechanism is increasing, but obviously still at its infancy. However, knowing the key components involved in the pathology of the diseases, their pathophysiological significance and their trend during acute phases, is essential as the basis for new prevention and treatment strategies to combat these diseases in the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by a grant from European Union’s seventh frame work FP7 to Marie Curie Initial Training Network “iMODE-CKD” (FP7-PEOPLE-2013-ITN-608332).
References
1. Virzí GM, Clementi A, Brocca A, de Cal M, Vescovo G, Granata A, et al. The hemodynamic and nonhemodynamic crosstalk in cardiorenal syndrome type 1. Cardiorenal Med (2014) 4(2):103–12. doi:10.1159/000362650
2. Viswanathan G, Gilbert S. The cardiorenal syndrome: making the connection. Int J Nephrol (2010) 2011:283137. doi:10.4061/2011/283137
3. Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation (2007) 116(1):85–97. doi:10.1161/CIRCULATIONAHA.106.678342
4. Favre GA, Esnault VL, Van Obberghen E. Modulation of glucose metabolism by the renin-angiotensin-aldosterone system. Am J Physiol Endocrinol Metab (2015) 308(6):E435–49. doi:10.1152/ajpendo.00391.2014
5. Beaglehole R, Yach D. Globalisation and the prevention and control of non-communicable disease: the neglected chronic diseases of adults. Lancet (2003) 362(9387):903–8. doi:10.1016/S0140-6736(03)14335-8
6. Atkins RC. The epidemiology of chronic kidney disease. Kidney Int Suppl (2005) 94:S14–8. doi:10.1111/j.1523-1755.2005.09403.x
7. Stevens PE, Levin A. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann Intern Med (2013) 158(11):825–30. doi:10.7326/0003-4819-158-11-201306040-00007
8. KDIGO consortium. Chapter 1: Definition and classification of CKD. Kidney Int Suppl (2013) 3(1):19–62. doi:10.1038/kisup.2012.64
9. Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, Jafar TH, Heerspink HJ, Mann JF, et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet (2013) 382(9889):339–52. doi:10.1016/S0140-6736(13)60595-4
10. García-Esquinas E, Loeffler LF, Weaver VM, Fadrowski JJ, Navas-Acien A. Kidney function and tobacco smoke exposure in US adolescents. Pediatrics (2013) 131(5):e1415–23. doi:10.1542/peds.2012-3201
11. Metcalfe W. How does early chronic kidney disease progress? A background paper prepared for the UK consensus conference on early chronic kidney disease. Nephrol Dial Transplant (2007) 22(Suppl 9):ix26–30. doi:10.1093/ndt/gfm446
12. Zoja C, Abbate M, Remuzzi G. Progression of renal injury toward interstitial inflammation and glomerular sclerosis is dependent on abnormal protein filtration. Nephrol Dial Transplant (2015) 30(5):706–12. doi:10.1093/ndt/gfu261
13. Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, et al. Prevalence of chronic kidney disease in the United States. JAMA (2007) 298(17):2038–47. doi:10.1001/jama.298.17.2038
14. Zhang QL, Rothenbacher D. Prevalence of chronic kidney disease in population-based studies: systematic review. BMC Public Health (2008) 8:117. doi:10.1186/1471-2458-8-117
15. Nitsch D, Grams M, Sang Y, Black C, Cirillo M, Djurdjev O, et al. Associations of estimated glomerular filtration rate and albuminuria with mortality and renal failure by sex: a meta-analysis. BMJ (2013) 346:f324. doi:10.1136/bmj.f324
16. Zain M, Awan FR. Renin angiotensin aldosterone system (RAAS): its biology and drug targets for treating diabetic nephropathy. Pak J Pharm Sci (2014) 27(5):1379–91.
17. Lizicarova D, Krahulec B, Hirnerova E, Gaspar L, Celecova Z. Risk factors in diabetic nephropathy progression at present. Bratisl Lek Listy (2014) 115(8):517–21. doi:10.4149/BLL_2014_101
18. Duran-Salgado MB, Rubio-Guerra AF. Diabetic nephropathy and inflammation. World J Diabetes (2014) 5(3):393–8. doi:10.4239/wjd.v5.i3.393
19. International Diabetes Federation. IDF Diabetes Atlas. 6th ed. Brussels: International Diabetes Federation (2013). Available from: http://www.idf.org/diabetesatlas
20. Global Status Report on Noncommunicable Diseases (2014). Available from: www.who.int/mediacentre/factsheets/fs355/en/
21. Koro CE, Lee BH, Bowlin SJ. Antidiabetic medication use and prevalence of chronic kidney disease among patients with type 2 diabetes mellitus in the United States. Clin Ther (2009) 31(11):2608–17. doi:10.1016/j.clinthera.2009.10.020
22. Fact Sheets on Cardiovascular Disease WHO (2015. Available from: www.who.int/mediacentre/factsheets/fs317/en/
23. Wong ND. Epidemiological studies of CHD and the evolution of preventive cardiology. Nat Rev Cardiol (2014) 11(5):276–89. doi:10.1038/nrcardio.2014.26
24. Fox CS, Matsushita K, Woodward M, Bilo HJ, Chalmers J, Heerspink HJ, et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without diabetes: a meta-analysis. Lancet (2012) 380(9854):1662–73. doi:10.1016/S0140-6736(12)61350-6
25. Mahmoodi BK, Matsushita K, Woodward M, Blankestijn PJ, Cirillo M, Ohkubo T, et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without hypertension: a meta-analysis. Lancet (2012) 380(9854):1649–61. doi:10.1016/S0140-6736(12)61272-0
26. Braun-Menendez E, Page IH. Suggested revision of nomenclature – angiotensin. Science (1958) 127(3292):242. doi:10.1126/science.127.3292.242-a
27. Kang JJ, Toma I, Sipos A, Meer EJ, Vargas SL, Peti-Peterdi J. The collecting duct is the major source of prorenin in diabetes. Hypertension (2008) 51(6):1597–604. doi:10.1161/HYPERTENSIONAHA.107.107268
28. Jankowski V, Vanholder R, van der Giet M, Henning L, Tölle M, Schönfelder G, et al. Detection of angiotensin II in supernatants of stimulated mononuclear leukocytes by matrix-assisted laser desorption ionization time-of-flight/time-of-flight mass analysis. Hypertension (2005) 46(3):591–7. doi:10.1161/01.HYP.0000177436.09733.d4
29. Skeggs LT Jr, Kahn JR, Lentz K, Shumway NP. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. J Exp Med (1957) 106(3):439–53. doi:10.1084/jem.106.3.439
30. Lavoie JL, Sigmund CD. Minireview: overview of the renin-angiotensin system – an endocrine and paracrine system. Endocrinology (2003) 144(6):2179–83. doi:10.1210/en.2003-0150
31. Jankowski V, Tölle M, Santos RA, Günthner T, Krause E, Beyermann M, et al. Angioprotectin: an angiotensin II-like peptide causing vasodilatory effects. FASEB J (2011) 25(9):2987–95. doi:10.1096/fj.11-185470
32. Jankowski V, Vanholder R, van der Giet M, Tölle M, Karadogan S, Gobom J, et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler Thromb Vasc Biol (2007) 27(2):297–302. doi:10.1161/01.ATV.0000253889.09765.5f
33. Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, et al. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res (2013) 112(8):1104–11. doi:10.1161/CIRCRESAHA.113.301077
34. Salem S, Jankowski V, Asare Y, Liehn E, Welker P, Raya-Bermudez A, et al. Identification of the vasoconstriction-inhibiting factor (VIF), a potent endogenous cofactor of angiotensin II acting on the angiotensin II type 2 receptor. Circulation (2015) 131(16):1426–34. doi:10.1161/CIRCULATIONAHA.114.013168
35. Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev (2003) 24(3):261–71. doi:10.1210/er.2003-0001
36. Calò LA, Schiavo S, Davis PA, Pagnin E, Mormino P, D’Angelo A, et al. Angiotensin II signaling via type 2 receptors in a human model of vascular hyporeactivity: implications for hypertension. J Hypertens (2010) 28(1):111–8. doi:10.1097/HJH.0b013e328332b738
37. Duncan JA, Scholey JW, Miller JA. Angiotensin II type 1 receptor gene polymorphisms in humans: physiology and pathophysiology of the genotypes. Curr Opin Nephrol Hypertens (2001) 10(1):111–6. doi:10.1097/00041552-200101000-00017
38. Pernomian L, Baraldi Araujo Restini C. Counter-regulatory effects played by the ACE – Ang II – AT1 and ACE2 – Ang-(1-7) – Mas axes on the reactive oxygen species-mediated control of vascular function: perspectives to pharmacological approaches in controlling vascular complications. Vasa (2014) 43(6):404–14. doi:10.1024/0301-1526/a000387
39. Arima S, Ito S. Angiotensin II type 2 receptors in the kidney: evidence for endothelial-cell-mediated renal vasodilatation. Nephrol Dial Transplant (2000) 15(4):448–51. doi:10.1093/ndt/15.4.448
40. Dzau VJ, Re R. Tissue angiotensin system in cardiovascular medicine. A paradigm shift? Circulation (1994) 89(1):493–8. doi:10.1161/01.CIR.89.1.493
41. Wolf G. Angiotensin II as a mediator of tubulointerstitial injury. Nephrol Dial Transplant (2000) 15(Suppl 6):61–3. doi:10.1093/ndt/15.suppl_6.61
42. Quinn SJ, Williams GH. Regulation of aldosterone secretion. Annu Rev Physiol (1988) 50:409–26. doi:10.1146/annurev.ph.50.030188.002205
43. Schweda F. Salt feedback on the renin-angiotensin-aldosterone system. Pflugers Arch (2015) 467(3):565–76. doi:10.1007/s00424-014-1668-y
44. Wenzel U. Aldosterone and progression of renal disease. Curr Opin Nephrol Hypertens (2008) 17(1):44–50. doi:10.1097/MNH.0b013e3282f29028
45. Ruster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol (2006) 17(11):2985–91. doi:10.1681/ASN.2006040356
46. Beutler KT, Masilamani S, Turban S, Nielsen J, Brooks HL, Ageloff S, et al. Long-term regulation of ENaC expression in kidney by angiotensin II. Hypertension (2003) 41(5):1143–50. doi:10.1161/01.HYP.0000066129.12106.E2
47. Zhuo JL, Imig JD, Hammond TG, Orengo S, Benes E, Navar LG. Ang II accumulation in rat renal endosomes during Ang II-induced hypertension: role of AT(1) receptor. Hypertension (2002) 39(1):116–21. doi:10.1161/hy0102.100780
48. He W, Kang YS, Dai C, Liu Y. Blockade of Wnt/beta-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J Am Soc Nephrol (2011) 22(1):90–103. doi:10.1681/ASN.2009121236
49. Zhou L, Li Y, Hao S, Zhou D, Tan RJ, Nie J, et al. Multiple genes of the renin-angiotensin system are novel targets of Wnt/beta-catenin signaling. J Am Soc Nephrol (2015) 26(1):107–20. doi:10.1681/ASN.2014010085
50. Lee IJ, Hilliard BA, Ulas M, Yu D, Vangala C, Rao S, et al. Monocyte and plasma expression of TAM ligand and receptor in renal failure: links to unregulated immunity and chronic inflammation. Clin Immunol (2015) 158(2):231–41. doi:10.1016/j.clim.2015.01.012
51. Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med (1997) 336(15):1066–71. doi:10.1056/NEJM199704103361506
52. Döring Y, Pawig L, Weber C, Noels H. The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front Physiol (2014) 5:212. doi:10.3389/fphys.2014.00212
53. Wennmann DO, Hsu HH, Pavenstadt H. The renin-angiotensin-aldosterone system in podocytes. Semin Nephrol (2012) 32(4):377–84. doi:10.1016/j.semnephrol.2012.06.009
54. Wolf G, Wenzel U, Burns KD, Harris RC, Stahl RA, Thaiss F. Angiotensin II activates nuclear transcription factor-kappaB through AT1 and AT2 receptors. Kidney Int (2002) 61(6):1986–95. doi:10.1046/j.1523-1755.2002.00365.x
55. Bondeva T, Roger T, Wolf G. Differential regulation of toll-like receptor 4 gene expression in renal cells by angiotensin II: dependency on AP1 and PU.1 transcriptional sites. Am J Nephrol (2007) 27(3):308–14. doi:10.1159/000102551
56. Androulakis ES, Tousoulis D, Papageorgiou N, Tsioufis C, Kallikazaros I, Stefanadis C. Essential hypertension: is there a role for inflammatory mechanisms? Cardiol Rev (2009) 17(5):216–21. doi:10.1097/CRD.0b013e3181b18e03
57. Pearse DD, Tian RX, Nigro J, Iorgulescu JB, Puzis L, Jaimes EA. Angiotensin II increases the expression of the transcription factor ETS-1 in mesangial cells. Am J Physiol Renal Physiol (2008) 294(5):F1094–100. doi:10.1152/ajprenal.00458.2007
58. Knight EL, Rimm EB, Pai JK, Rexrode KM, Cannuscio CC, Manson JE, et al. Kidney dysfunction, inflammation, and coronary events: a prospective study. J Am Soc Nephrol (2004) 15(7):1897–903. doi:10.1097/01.ASN.0000128966.55133.69
59. Descamps-Latscha B, Herbelin A, Nguyen AT, Roux-Lombard P, Zingraff J, Moynot A, et al. Balance between IL-1 beta, TNF-alpha, and their specific inhibitors in chronic renal failure and maintenance dialysis. Relationships with activation markers of T cells, B cells, and monocytes. J Immunol (1995) 154(2):882–92.
60. Pecoits-Filho R, Lindholm B, Axelsson J, Stenvinkel P. Update on interleukin-6 and its role in chronic renal failure. Nephrol Dial Transplant (2003) 18(6):1042–5. doi:10.1093/ndt/gfg111
61. Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol (2013) 5(11):a009076. doi:10.1101/cshperspect.a009076
62. Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol (2008) 8(5):327–36. doi:10.1038/nri2303
63. Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia (2003) 46(1):3–19. doi:10.4239/wjd.v6.i4.598
64. Ceriello A, Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol (2004) 24(5):816–23. doi:10.1161/01.ATV.0000122852.22604.78
65. Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, Bashan N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes (1998) 47(10):1562–9. doi:10.2337/diabetes.47.10.1562
66. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes (2003) 52(1):1–8. doi:10.2337/diabetes.52.1.1
67. Zhang Z, Apse K, Pang J, Stanton RC. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J Biol Chem (2000) 275(51):40042–7. doi:10.1074/jbc.M007505200
68. Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol Metab (2014) 25(9):452–63. doi:10.1016/j.tem.2014.06.012
69. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res (2015) 116(7):1231–44. doi:10.1161/CIRCRESAHA.116.302855
70. Lorenzi M. The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp Diabetes Res (2007) 2007:61038. doi:10.1155/2007/61038
71. Chuah YK, Basir R, Talib H, Tie TH, Nordin N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int J Inflam (2013) 2013:403460. doi:10.1155/2013/403460
72. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation (2006) 114(6):597–605. doi:10.1161/CIRCULATIONAHA.106.621854
73. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol (2013) 13(10):709–21. doi:10.1038/nri3520
74. Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med (2003) 198(10):1507–15. doi:10.1084/jem.20030800
75. Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol (2000) 20(9):2032–7. doi:10.1161/01.ATV.20.9.2032
76. Tang X, Luo YX, Chen HZ, Liu DP. Mitochondria, endothelial cell function, and vascular diseases. Front Physiol (2014) 5:175. doi:10.3389/fphys.2014.00175
77. Xu H, Sjögren P, Ärnlöv J, Banerjee T, Cederholm T, Risérus U, et al. A proinflammatory diet is associated with systemic inflammation and reduced kidney function in elderly adults. J Nutr (2015) 145(4):729–35. doi:10.3945/jn.114.205187
78. Bonventre JV. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J Am Soc Nephrol (2003) 14(Suppl 1):S55–61. doi:10.1097/01.ASN.0000067652.51441.21
80. Yu F, Wu LH, Tan Y, Li LH, Wang CL, Wang WK, et al. Tubulointerstitial lesions of patients with lupus nephritis classified by the 2003 international society of nephrology and renal pathology society system. Kidney Int (2010) 77(9):820–9. doi:10.1038/ki.2010.13
81. LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med (2013) 19(8):1047–53. doi:10.1038/nm.3218
82. Eddy AA. Overview of the cellular and molecular basis of kidney fibrosis. Kidney Int Suppl (2011) (2014) 4(1):2–8. doi:10.1038/kisup.2014.2
83. Mezni I, Galichon P, Bacha MM, Sfar I, Hertig A, Goucha R, et al. [The epithelial-mesenchymal transition and fibrosis of the renal transplant]. Med Sci (Paris) (2015) 31(1):68–74. doi:10.1051/medsci/20153101015
84. Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Annu Rev Pathol (2013) 8:241–76. doi:10.1146/annurev-pathol-020712-163930
85. Qi W, Chen X, Poronnik P, Pollock CA. The renal cortical fibroblast in renal tubulointerstitial fibrosis. Int J Biochem Cell Biol (2006) 38(1):1–5. doi:10.1016/j.biocel.2005.09.005
86. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol (2014) 5:614. doi:10.3389/fimmu.2014.00614
87. Christensen EI, Birn H, Storm T, Weyer K, Nielsen R. Endocytic receptors in the renal proximal tubule. Physiology (Bethesda) (2012) 27(4):223–36. doi:10.1152/physiol.00022.2012
88. Meng XM, Tang PM, Li J, Lan HY. TGF-beta/Smad signaling in renal fibrosis. Front Physiol (2015) 6:82. doi:10.3389/fphys.2015.00082
89. Orphanides C, Fine LG, Norman JT. Hypoxia stimulates proximal tubular cell matrix production via a TGF-beta1-independent mechanism. Kidney Int (1997) 52(3):637–47. doi:10.1038/ki.1997.377
90. Wolf G. Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-beta pathway. Kidney Int (2006) 70(11):1914–9. doi:10.1038/sj.ki.5001846
91. Shin DM, Jeon JH, Kim CW, Cho SY, Lee HJ, Jang GY, et al. TGFbeta mediates activation of transglutaminase 2 in response to oxidative stress that leads to protein aggregation. FASEB J (2008) 22(7):2498–507. doi:10.1096/fj.07-095455
92. Lan A, Du J. Potential role of Akt signaling in chronic kidney disease. Nephrol Dial Transplant (2015) 30(3):385–94. doi:10.1093/ndt/gfu196
93. Vindevoghel L, Lechleider RJ, Kon A, de Caestecker MP, Uitto J, Roberts AB, et al. SMAD3/4-dependent transcriptional activation of the human type VII collagen gene (COL7A1) promoter by transforming growth factor beta. Proc Natl Acad Sci U S A (1998) 95(25):14769–74. doi:10.1073/pnas.95.25.14769
94. Wu CF, Chiang WC, Lai CF, Chang FC, Chen YT, Chou YH, et al. Transforming growth factor beta-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am J Pathol (2013) 182(1):118–31. doi:10.1016/j.ajpath.2012.09.009
95. Lopez-Hernandez FJ, Lopez-Novoa JM. Role of TGF-beta in chronic kidney disease: an integration of tubular, glomerular and vascular effects. Cell Tissue Res (2012) 347(1):141–54. doi:10.1007/s00441-011-1275-6
96. Chung AC, Zhang H, Kong YZ, Tan JJ, Huang XR, Kopp JB, et al. Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J Am Soc Nephrol (2010) 21(2):249–60. doi:10.1681/ASN.2009010018
97. Yang F, Huang XR, Chung AC, Hou CC, Lai KN, Lan HY. Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J Pathol (2010) 221(4):390–401. doi:10.1002/path.2721
98. Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol (2010) 21(2):212–22. doi:10.1681/ASN.2008121226
99. Lecru L, Desterke C, Grassin-Delyle S, Chatziantoniou C, Vandermeersch S, Devocelle A, et al. Cannabinoid receptor 1 is a major mediator of renal fibrosis. Kidney Int (2015). doi:10.1038/ki.2015.63
100. Huang XR, Chung AC, Wang XJ, Lai KN, Lan HY. Mice overexpressing latent TGF-beta1 are protected against renal fibrosis in obstructive kidney disease. Am J Physiol Renal Physiol (2008) 295(1):F118–27. doi:10.1152/ajprenal.00021.2008
101. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature (2003) 425(6958):577–84. doi:10.1038/nature02006
102. Li Y, Tan X, Dai C, Stolz DB, Wang D, Liu Y. Inhibition of integrin-linked kinase attenuates renal interstitial fibrosis. J Am Soc Nephrol (2009) 20(9):1907–18. doi:10.1681/ASN.2008090930
103. Loeffler I, Wolf G. Transforming growth factor-beta and the progression of renal disease. Nephrol Dial Transplant (2014) 29(Suppl 1):i37–45. doi:10.1093/ndt/gft267
104. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem (2000) 275(47):36803–10. doi:10.1074/jbc.M005912200
105. Ghosh Choudhury G, Abboud HE. Tyrosine phosphorylation-dependent PI 3 kinase/Akt signal transduction regulates TGFbeta-induced fibronectin expression in mesangial cells. Cell Signal (2004) 16(1):31–41. doi:10.1016/S0898-6568(03)00094-9
106. Mahimainathan L, Das F, Venkatesan B, Choudhury GG. Mesangial cell hypertrophy by high glucose is mediated by downregulation of the tumor suppressor PTEN. Diabetes (2006) 55(7):2115–25. doi:10.2337/db05-1326
107. Wolf G. Link between angiotensin II and TGF-beta in the kidney. Miner Electrolyte Metab (1998) 24(2–3):174–80. doi:10.1159/000057367
108. Huang Y, Wongamorntham S, Kasting J, McQuillan D, Owens RT, Yu L, et al. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int (2006) 69(1):105–13. doi:10.1038/sj.ki.5000011
109. Rodríguez-Vita J, Sánchez-López E, Esteban V, Rupérez M, Egido J, Ruiz-Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation (2005) 111(19):2509–17. doi:10.1161/01.CIR.0000165133.84978.E2
110. Abrahamsen CT, Pullen MA, Schnackenberg CG, Grygielko ET, Edwards RM, Laping NJ, et al. Effects of angiotensins II and IV on blood pressure, renal function, and PAI-1 expression in the heart and kidney of the rat. Pharmacology (2002) 66(1):26–30. doi:10.1159/000063252
111. Wolf G, Mentzel S, Assmann KJ. Aminopeptidase A: a key enzyme in the intrarenal degradation of angiotensin II. Exp Nephrol (1997) 5(5):364–9.
112. Galichon P, Finianos S, Hertig A. EMT-MET in renal disease: should we curb our enthusiasm? Cancer Lett (2013) 341(1):24–9. doi:10.1016/j.canlet.2013.04.018
113. Gajjala PR, Fliser D, Speer T, Jankowski V, Jankowski J. Emerging role of post-translational modifications in chronic kidney disease and cardiovascular disease. Nephrol Dial Transplant (2015). doi:10.1093/ndt/gfv048
114. Speer T, Owala FO, Holy EW, Zewinger S, Frenzel FL, Stähli BE, et al. Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur Heart J (2014) 35(43):3021–32. doi:10.1093/eurheartj/ehu111
115. Mulè G, Nardi E, Guarino L, Cacciatore V, Geraci G, Calcaterra I, et al. Plasma aldosterone and its relationship with left ventricular mass in hypertensive patients with early-stage chronic kidney disease. Hypertens Res (2015) 38(4):276–83. doi:10.1038/hr.2014.171
116. Chao CT, Chiang CK. Uremic toxins, oxidative stress, and renal fibrosis: an interwined complex. J Ren Nutr (2015) 25(2):155–9. doi:10.1053/j.jrn.2014.10.010
117. Lin CJ, Pan CF, Liu HL, Chuang CK, Jayakumar T, Wang TJ, et al. The role of protein-bound uremic toxins on peripheral artery disease and vascular access failure in patients on hemodialysis. Atherosclerosis (2012) 225(1):173–9. doi:10.1016/j.atherosclerosis.2012.07.012
118. Peng YS, Ding HC, Lin YT, Syu JP, Chen Y, Wang SM. Uremic toxin p-cresol induces disassembly of gap junctions of cardiomyocytes. Toxicology (2012) 302(1):11–7. doi:10.1016/j.tox.2012.07.004
119. Bernal-Mizrachi L, Jy W, Jimenez JJ, Pastor J, Mauro LM, Horstman LL, et al. High levels of circulating endothelial microparticles in patients with acute coronary syndromes. Am Heart J (2003) 145(6):962–70. doi:10.1016/S0002-8703(03)00103-0
120. Simak J, Gelderman MP, Yu H, Wright V, Baird AE. Circulating endothelial microparticles in acute ischemic stroke: a link to severity, lesion volume and outcome. J Thromb Haemost (2006) 4(6):1296–302. doi:10.1111/j.1538-7836.2006.01911.x
121. Chirinos JA, Heresi GA, Velasquez H, Jy W, Jimenez JJ, Ahn E, et al. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J Am Coll Cardiol (2005) 45(9):1467–71. doi:10.1016/j.jacc.2004.12.075
122. Meijers BK, Van Kerckhoven S, Verbeke K, Dehaen W, Vanrenterghem Y, Hoylaerts MF, et al. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am J Kidney Dis (2009) 54(5):891–901. doi:10.1053/j.ajkd.2009.04.022
123. Dou L, Jourde-Chiche N, Faure V, Cerini C, Berland Y, Dignat-George F, et al. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J Thromb Haemost (2007) 5(6):1302–8. doi:10.1111/j.1538-7836.2007.02540.x
124. Yisireyili M, Saito S, Abudureyimu S, Adelibieke Y, Ng HY, Nishijima F, et al. Indoxyl sulfate-induced activation of (pro)renin receptor promotes cell proliferation and tissue factor expression in vascular smooth muscle cells. PLoS One (2014) 9(10):e109268. doi:10.1371/journal.pone.0109268
125. Yamamoto H, Tsuruoka S, Ioka T, Ando H, Ito C, Akimoto T, et al. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int (2006) 69(10):1780–5. doi:10.1038/sj.ki.5000340
126. Lekawanvijit S, Adrahtas A, Kelly DJ, Kompa AR, Wang BH, Krum H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur Heart J (2010) 31(14):1771–9. doi:10.1093/eurheartj/ehp574
127. Sun CY, Young GH, Hsieh YT, Chen YH, Wu MS, Wu VC, et al. Protein-bound uremic toxins induce tissue remodeling by targeting the EGF receptor. J Am Soc Nephrol (2015) 26(2):281–90. doi:10.1681/ASN.2014010021
128. Schuchardt M, Tölle M, Prüfer J, Prüfer N, Huang T, Jankowski V, et al. Uridine adenosine tetraphosphate activation of the purinergic receptor P2Y enhances in vitro vascular calcification. Kidney Int (2012) 81(3):256–65. doi:10.1038/ki.2011.326
129. Jankowski V, Tölle M, Vanholder R, Schönfelder G, van der Giet M, Henning L, et al. Uridine adenosine tetraphosphate: a novel endothelium-derived vasoconstrictive factor. Nat Med (2005) 11(2):223–7. doi:10.1038/nm1188
130. Schuchardt M, Prüfer J, Prüfer N, Wiedon A, Huang T, Chebli M, et al. The endothelium-derived contracting factor uridine adenosine tetraphosphate induces P2Y(2)-mediated pro-inflammatory signaling by monocyte chemoattractant protein-1 formation. J Mol Med (Berl) (2011) 89(8):799–810. doi:10.1007/s00109-011-0750-6
131. Scholze A, Jankowski V, Henning L, Haass W, Wittstock A, Suvd-Erdene S, et al. Phenylacetic acid and arterial vascular properties in patients with chronic kidney disease stage 5 on hemodialysis therapy. Nephron Clin Pract (2007) 107(1):c1–6. doi:10.1159/000105137
132. Schmidt S, Westhoff TH, Krauser P, Ignatius R, Jankowski J, Jankowski V, et al. The uraemic toxin phenylacetic acid impairs macrophage function. Nephrol Dial Transplant (2008) 23(11):3485–93. doi:10.1093/ndt/gfn266
133. Jankowski J, van der Giet M, Jankowski V, Schmidt S, Hemeier M, Mahn B, et al. Increased plasma phenylacetic acid in patients with end-stage renal failure inhibits iNOS expression. J Clin Invest (2003) 112(2):256–64. doi:10.1172/JCI200315524
134. Belmadani S, Matrougui K. Can the NK family of osteoblast homeodomain transcription factors signaling be a magic bullet to reverse calcification-induced vasculopathy in diabetes? Diabetes (2014) 63(12):4011–2. doi:10.2337/db14-1366
135. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res (2011) 109(6):697–711. doi:10.1161/CIRCRESAHA.110.234914
136. Moe SM, Chen NX. Mechanisms of vascular calcification in chronic kidney disease. J Am Soc Nephrol (2008) 19(2):213–6. doi:10.1681/ASN.2007080854
137. Mazière C, Savitsky V, Galmiche A, Gomila C, Massy Z, Mazière JC. Oxidized low density lipoprotein inhibits phosphate signaling and phosphate-induced mineralization in osteoblasts. Involvement of oxidative stress. Biochim Biophys Acta (2010) 1802(11):1013–9. doi:10.1016/j.bbadis.2010.07.010
138. Poggio P, Grau JB, Field BC, Sainger R, Seefried WF, Rizzolio F, et al. Osteopontin controls endothelial cell migration in vitro and in excised human valvular tissue from patients with calcific aortic stenosis and controls. J Cell Physiol (2011) 226(8):2139–49. doi:10.1002/jcp.22549
139. Salem S, Bruck H, Bahlmann FH, Peter M, Passlick-Deetjen J, Kretschmer A, et al. Relationship between magnesium and clinical biomarkers on inhibition of vascular calcification. Am J Nephrol (2012) 35(1):31–9. doi:10.1159/000334742
140. Menendez-Castro C, Hilgers KF, Amann K, Daniel C, Cordasic N, Wachtveitl R, et al. Intrauterine growth restriction leads to a dysregulation of Wilms’ tumour supressor gene 1 (WT1) and to early podocyte alterations. Nephrol Dial Transplant (2013) 28(6):1407–17. doi:10.1093/ndt/gfs517
141. Rennenberg RJ, Schurgers LJ, Kroon AA, Stehouwer CD. Arterial calcifications. J Cell Mol Med (2010) 14(9):2203–10. doi:10.1111/j.1582-4934.2010.01139.x
142. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, et al. Suppression of aging in mice by the hormone Klotho. Science (2005) 309(5742):1829–33. doi:10.1126/science.1112766
143. Rattazzi M, Bertacco E, Del Vecchio A, Puato M, Faggin E, Pauletto P. Aortic valve calcification in chronic kidney disease. Nephrol Dial Transplant (2013) 28(12):2968–76. doi:10.1093/ndt/gft310
144. Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest (2007) 117(9):2684–91. doi:10.1172/JCI31330
145. Seiler S, Reichart B, Roth D, Seibert E, Fliser D, Heine GH. FGF-23 and future cardiovascular events in patients with chronic kidney disease before initiation of dialysis treatment. Nephrol Dial Transplant (2010) 25(12):3983–9. doi:10.1093/ndt/gfq309
146. Larrivée B, Prahst C, Gordon E, del Toro R, Mathivet T, Duarte A, et al. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell (2012) 22(3):489–500. doi:10.1016/j.devcel.2012.02.005
147. Takei S, Ichikawa H, Johkura K, Mogi A, No H, Yoshie S, et al. Bone morphogenetic protein-4 promotes induction of cardiomyocytes from human embryonic stem cells in serum-based embryoid body development. Am J Physiol Heart Circ Physiol (2009) 296(6):H1793–803. doi:10.1152/ajpheart.01288.2008
148. Liberman M, Johnson RC, Handy DE, Loscalzo J, Leopold JA. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem Biophys Res Commun (2011) 413(3):436–41. doi:10.1016/j.bbrc.2011.08.114
149. Brandenburg VM, Sinha S, Specht P, Ketteler M. Calcific uraemic arteriolopathy: a rare disease with a potentially high impact on chronic kidney disease-mineral and bone disorder. Pediatr Nephrol (2014) 29(12):2289–98. doi:10.1007/s00467-013-2746-7
150. Krüger T, Oelenberg S, Kaesler N, Schurgers LJ, van de Sandt AM, Boor P, et al. Warfarin induces cardiovascular damage in mice. Arterioscler Thromb Vasc Biol (2013) 33(11):2618–24. doi:10.1161/ATVBAHA.113.302244
151. McCabe KM, Booth SL, Fu X, Shobeiri N, Pang JJ, Adams MA, et al. Dietary vitamin K and therapeutic warfarin alter the susceptibility to vascular calcification in experimental chronic kidney disease. Kidney Int (2013) 83(5):835–44. doi:10.1038/ki.2012.477
152. Voelkl J, Alesutan I, Leibrock CB, Quintanilla-Martinez L, Kuhn V, Feger M, et al. Spironolactone ameliorates PIT1-dependent vascular osteoinduction in klotho-hypomorphic mice. J Clin Invest (2013) 123(2):812–22. doi:10.1172/JCI64093
153. Barreto FC, Barreto DV, Liabeuf S, Meert N, Glorieux G, Temmar M, et al. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol (2009) 4(10):1551–8. doi:10.2215/CJN.03980609
154. Villa-Bellosta R, Wang X, Millán JL, Dubyak GR, O’Neill WC. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am J Physiol Heart Circ Physiol (2011) 301(1):H61–8. doi:10.1152/ajpheart.01020.2010
155. Villa-Bellosta R, Rivera-Torres J, Osorio FG, Acín-Pérez R, Enriquez JA, López-Otín C, et al. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation (2013) 127(24):2442–51. doi:10.1161/CIRCULATIONAHA.112.000571
156. De Schutter TM, Behets GJ, Geryl H, Peter ME, Steppan S, Gundlach K, et al. Effect of a magnesium-based phosphate binder on medial calcification in a rat model of uremia. Kidney Int (2013) 83(6):1109–17. doi:10.1038/ki.2013.34
157. Tonelli M, Curhan G, Pfeffer M, Sacks F, Thadhani R, Melamed ML, et al. Relation between alkaline phosphatase, serum phosphate, and all-cause or cardiovascular mortality. Circulation (2009) 120(18):1784–92. doi:10.1161/CIRCULATIONAHA.109.851873
158. Shroff R, Long DA, Shanahan C. Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol (2013) 24(2):179–89. doi:10.1681/ASN.2011121191
159. Shroff RC, McNair R, Skepper JN, Figg N, Schurgers LJ, Deanfield J, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol (2010) 21(1):103–12. doi:10.1681/ASN.2009060640
160. Sage AP, Lu J, Tintut Y, Demer LL. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int (2011) 79(4):414–22. doi:10.1038/ki.2010.390
161. Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Suppl (2009) 113:S1–130. doi:10.1038/ki.2009.188
162. Spiegel DM, Brady K. Calcium balance in normal individuals and in patients with chronic kidney disease on low- and high-calcium diets. Kidney Int (2012) 81(11):1116–22. doi:10.1038/ki.2011.490
163. Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res (2011) 109(1):e1–12. doi:10.1161/CIRCRESAHA.110.238808
164. Van Laecke S, Nagler EV, Verbeke F, Van Biesen W, Vanholder R. Hypomagnesemia and the risk of death and GFR decline in chronic kidney disease. Am J Med (2013) 126(9):825–31. doi:10.1016/j.amjmed.2013.02.036
165. Moe SM, Chertow GM. The case against calcium-based phosphate binders. Clin J Am Soc Nephrol (2006) 1(4):697–703. doi:10.2215/CJN.00560206
166. Spiegel DM. Magnesium in chronic kidney disease: unanswered questions. Blood Purif (2011) 31(1–3):172–6. doi:10.1159/000321837
167. Wu-Wong JR, Noonan W, Ma J, Dixon D, Nakane M, Bolin AL, et al. Role of phosphorus and vitamin D analogs in the pathogenesis of vascular calcification. J Pharmacol Exp Ther (2006) 318(1):90–8. doi:10.1124/jpet.106.101261
168. Becker LE, Koleganova N, Piecha G, Noronha IL, Zeier M, Geldyyev A, et al. Effect of paricalcitol and calcitriol on aortic wall remodeling in uninephrectomized ApoE knockout mice. Am J Physiol Renal Physiol (2011) 300(3):F772–82. doi:10.1152/ajprenal.00042.2010
169. Jono S, Nishizawa Y, Shioi A, Morii H. 1,25-Dihydroxyvitamin D3 increases in vitro vascular calcification by modulating secretion of endogenous parathyroid hormone-related peptide. Circulation (1998) 98(13):1302–6. doi:10.1161/01.CIR.98.13.1302
170. Mizobuchi M, Finch JL, Martin DR, Slatopolsky E. Differential effects of vitamin D receptor activators on vascular calcification in uremic rats. Kidney Int (2007) 72(6):709–15. doi:10.1038/sj.ki.5002406
171. Ohnishi M, Nakatani T, Lanske B, Razzaque MS. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1alpha-hydroxylase. Kidney Int (2009) 75(11):1166–72. doi:10.1038/ki.2009.24
172. Desjardins L, Liabeuf S, Renard C, Lenglet A, Lemke HD, Choukroun G, et al. FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos Int (2012) 23(7):2017–25. doi:10.1007/s00198-011-1838-0
173. Shalhoub V, Shatzen EM, Ward SC, Davis J, Stevens J, Bi V, et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest (2012) 122(7):2543–53. doi:10.1172/JCI61405
174. Scialla JJ, Lau WL, Reilly MP, Isakova T, Yang HY, Crouthamel MH, et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int (2013) 83(6):1159–68. doi:10.1038/ki.2013.3
175. Jean G, Bresson E, Lorriaux C, Mayor B, Hurot JM, Deleaval P, et al. Increased levels of serum parathyroid hormone and fibroblast growth factor-23 are the main factors associated with the progression of vascular calcification in long-hour hemodialysis patients. Nephron Clin Pract (2012) 120(3):c132–8. doi:10.1159/000334424
176. Davies MR, Lund RJ, Mathew S, Hruska KA. Low turnover osteodystrophy and vascular calcification are amenable to skeletal anabolism in an animal model of chronic kidney disease and the metabolic syndrome. J Am Soc Nephrol (2005) 16(4):917–28. doi:10.1681/ASN.2004100835
177. Lilly SM, Qasim AN, Mulvey CK, Churchill TW, Reilly MP, Eraso LH. Non-compressible arterial disease and the risk of coronary calcification in type-2 diabetes. Atherosclerosis (2013) 230(1):17–22. doi:10.1016/j.atherosclerosis.2013.06.004
178. Patsch JM, Zulliger MA, Vilayphou N, Samelson EJ, Cejka D, Diarra D, et al. Quantification of lower leg arterial calcifications by high-resolution peripheral quantitative computed tomography. Bone (2014) 58:42–7. doi:10.1016/j.bone.2013.08.006
179. Yoshida T, Yamashita M, Horimai C, Hayashi M. High glucose concentration does not modulate the formation of arterial medial calcification in experimental uremic rats. J Vasc Res (2013) 50(6):512–20. doi:10.1159/000355263
180. Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, et al. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr−/− mice. Arterioscler Thromb Vasc Biol (2007) 27(12):2589–96. doi:10.1161/ATVBAHA.107.153668
181. Chen NX, Duan D, O’Neill KD, Moe SM. High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol Dial Transplant (2006) 21(12):3435–42. doi:10.1093/ndt/gfl429
182. Cheng SL, Behrmann A, Shao JS, Ramachandran B, Krchma K, Bello Arredondo Y, et al. Targeted reduction of vascular Msx1 and Msx2 mitigates arteriosclerotic calcification and aortic stiffness in LDLR-deficient mice fed diabetogenic diets. Diabetes (2014) 63(12):4326–37. doi:10.2337/db14-0326
183. Ting TC, Miyazaki-Anzai S, Masuda M, Levi M, Demer LL, Tintut Y, et al. Increased lipogenesis and stearate accelerate vascular calcification in calcifying vascular cells. J Biol Chem (2011) 286(27):23938–49. doi:10.1074/jbc.M111.237065
184. Kageyama A, Matsui H, Ohta M, Sambuichi K, Kawano H, Notsu T, et al. Palmitic acid induces osteoblastic differentiation in vascular smooth muscle cells through ACSL3 and NF-kappaB, novel targets of eicosapentaenoic acid. PLoS One (2013) 8(6):e68197. doi:10.1371/journal.pone.0068197
185. Goettsch C, Rauner M, Hamann C, Sinningen K, Hempel U, Bornstein SR, et al. Nuclear factor of activated T cells mediates oxidised LDL-induced calcification of vascular smooth muscle cells. Diabetologia (2011) 54(10):2690–701. doi:10.1007/s00125-011-2219-0
186. Taylor J, Butcher M, Zeadin M, Politano A, Shaughnessy SG. Oxidized low-density lipoprotein promotes osteoblast differentiation in primary cultures of vascular smooth muscle cells by up-regulating Osterix expression in an Msx2-dependent manner. J Cell Biochem (2011) 112(2):581–8. doi:10.1002/jcb.22948
187. Pai A, Leaf EM, El-Abbadi M, Giachelli CM. Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am J Pathol (2011) 178(2):764–73. doi:10.1016/j.ajpath.2010.10.006
188. Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem (2008) 283(22):15319–27. doi:10.1074/jbc.M800021200
189. Zhao G, Xu MJ, Zhao MM, Dai XY, Kong W, Wilson GM, et al. Activation of nuclear factor-kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int (2012) 82(1):34–44. doi:10.1038/ki.2012.40
190. van der Velde M, Matsushita K, Coresh J, Astor BC, Woodward M, Levey A, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with all-cause and cardiovascular mortality. A collaborative meta-analysis of high-risk population cohorts. Kidney Int (2011) 79(12):1341–52. doi:10.1038/ki.2010.536
191. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med (2004) 351(13):1296–305. doi:10.1056/NEJMoa041031
Keywords: cardiovascular diseases, diabetes mellitus, inflammation, fibrosis, chronic kidney diseases
Citation: Gajjala PR, Sanati M and Jankowski J (2015) Cellular and molecular mechanisms of chronic kidney disease with diabetes mellitus and cardiovascular diseases as its comorbidities. Front. Immunol. 6:340. doi: 10.3389/fimmu.2015.00340
Received: 06 May 2015; Accepted: 17 June 2015;
Published: 08 July 2015
Edited by:
Mario Mellado, Consejo Superior de Investigaciones Científicas, SpainCopyright: © 2015 Gajjala, Sanati and Jankowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joachim Jankowski, Institute of Molecular Cardiovascular Research, RWTH University of Aachen, Pauwelsstrasse 30, Aachen D-52074, Germany, jjankowski@ukaachen.de