 Guillermo Valencia Pacheco1*
Guillermo Valencia Pacheco1* Irene B. Novelo Noh2
Irene B. Novelo Noh2 Rubí M.-H. Velasco Cárdenas2Angélica V. Angulo Ramírez3
Rubí M.-H. Velasco Cárdenas2Angélica V. Angulo Ramírez3 Ricardo F. López Villanueva4
Ricardo F. López Villanueva4 Irma G. Quintal Ortiz1Ligia G. Alonso Salomón1
Irma G. Quintal Ortiz1Ligia G. Alonso Salomón1 Norma Pavía Ruz1
Norma Pavía Ruz1 Nubia A. Rivero Cárdenas1
Nubia A. Rivero Cárdenas1
- 1Laboratorio de Hematología, Centro de Investigaciones Regionales Dr. Hideyo Noguchi, Universidad Autónoma de Yucatán, Mérida, Mexico
- 2Facultad de Química, Universidad Autónoma de Yucatán, Mérida, Mexico
- 3Hospital General Dr. Agustín O’Horán, Mérida, Mexico
- 4Hospital General Regional ISSSTE, Servicios de Salud de Yucatán (SSY), Mérida, México
Background: Systemic lupus erythematosus (SLE) is a chronic inflammatory autoimmune disease involving multiple organs. It is currently accepted that several genetic, environmental, and hormonal factors are contributing to its development. Innate immunity may have a great influence in autoimmunity through Toll-like receptors. TLR-7 recognizing single-strand RNA has been involved in SLE. Its activation induces intracellular signal with attraction of MyD88 and NF-kBp65, leading to IFN-α synthesis which correlate with disease activity.
Objective: To assess the expression of TLR-7, MyD88, and NF-kBp65 in B lymphocytes of Mayan women with SLE.
Methods: One hundred patients with SLE and 100 healthy controls, all of them Mayan women, were included. TLR-7 was analyzed on B and T lymphocytes, and MyD88 and NF-kB only in B lymphocytes. Serum INF-α level was evaluated by ELISA.
Results: Significant expression (p < 0.0001) of TLR-7 in B and T lymphocytes and serum IFN-α increased (p = 0.034) was observed in patients. MyD88 and NF-kBp65 were also increased in B lymphocytes of patients. TLR-7 and NF-kBp65 expression correlated, but no correlation with INF-α and disease activity was detected.
Conclusion: Data support the role of TLR-7 and signal proteins in the pathogenesis of SLE in the Mayan population of Yucatán.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease of connective tissue characterized by B lymphocytes hyperactivity and autoantibodies against nuclear self-antigens. The disease has a worldwide distribution and predominantly affects women. The SLE pathogenesis is yet unknown but several genetic, hormonal, and environmental factors are contributing to its development (1–4). The incidence of SLE patients varies according to the population studied (5, 6). Several studies have been conducted in patients from different populations (Asian, European, and American), but few in Mexican population. Mexico has an admixed Mestizo population with a genetic pool from the Amerindian and the Spanish (7). Mexican individuals with SLE appear to have a more severe disease and a lower age of onset than European women and a higher frequency of disease activity flares. Moreover, it has been reported that the prevalence of SLE in Yucatán (0.7%) is slightly higher than the national prevalence (0.6%) (8, 9), but immune studies have not been conducted in the Mayan population.
Studies have shown that abnormal stimulation of innate immunity may have a great influence on immunopathogenesis of SLE through Toll-like receptors (TLRs) (10, 11). Those are pattern-recognition receptors (PRR) that identify a broad range of pathogen-associated molecular patterns (PAMPs) (12, 13). So far, 11 human TLRs have been identified, and TLR-7 has been associated with SLE in both human and mouse models (14–19). This receptor is found on endosomes of several immune cells, mainly antigen-presenting cells, such as dendritic and B cells (20). The recognition and internalization, through the B cell receptor, of nuclear self-antigens released as a consequence of apoptosis in SLE patients, can activate TLR-7 in endosomes of B lymphocytes supporting its role in the production of autoantibodies (21–24). RNA-containing complexes must access the interior of the plasmacytoid dendritic cells (pDCs), through the Fc receptors, thus providing a route of entry for RNA to reach TLR-7, with the resulting INF-α production (25, 26). INF-α influences the development, progression, and pathogenesis of SLE (27–30). As a result of TLR-7 ligation, INF-α enhances TLR-7 signaling in pDCs forming a positive feedback loop (31, 32).
The TLR-7 ligation induce signal transduction via the myeloid differentiation primary-response protein 88 (MyD88), a common adaptor protein, which interacts with IRAK1/4 (Interleukin-1 receptor-associated kinase 1/4) and TRAF6 (TNF receptor-associated factor 6) to form the MyD88/IRAK1/IRAK4/TRAF6 complex. Subsequently, IRAK1 and TRAF6 dissociate from the receptor complex and interact with kinases IKKB (IκB kinases) resulting in the activation of NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells), permitting the expression of genes of proinflammatory cytokine and chemokines (33, 34). On the other hand, the transcription factor IRF-7 (Interferon regulatory factor 7) can bind to the MyD88/IRAK1/IRAK4 complex, and its activation is dependent upon TLR-7 requiring the TRAF3 (TNF receptor-associated factor 3) protein, which joins IRAK1 and IKKα kinases to produce IFN-α (34).
Previously, the copy number variation (CNV) of TLR-7 gene in 80 Mayan women with SLE was analyzed in our laboratory. We found that 10% of patients had more than two copies of the TLR-7 gene. These data suggest that increased CNV of the TLR7 gene may be a risk factor in this population (35). However, the expression of TLR-7 and signaling proteins has not been analyzed in B lymphocytes of our patients. Our aim was to assess the TLR-7, MyD88, and NF-kBp65 expression in B cells of Mayan women with SLE and to compare them to healthy controls. Protein expressions were correlated with serum INF-α and disease activity.
Materials and Methods
SLE Patients
One hundred SLE women of Mayan origin were recruited at the Rheumatology outpatient of the Agustin O’Horán and ISSSTE Regional Hospital, Yucatán. Diagnosis was established according to the American College of Rheumatology (ACR) criteria (36), and disease activity was evaluated by SLEDAI score (37). One hundred healthy women of the same origin were studied as controls. All selected subjects included in the study gave their informed consent, according to the Declaration of Helsinki. The study was approved by the Research Ethics Committee of the Agustin O’Horán Hospital of Yucatán (CIE-008-1-11). All women gave 15 ml of venous peripheral blood in one collection.
Cell Isolation
Ten milliliters of venous peripheral blood were collected in heparinized tubes. Peripheral blood mononuclear cells (PBMC) were isolated from each subject, either patient and control, by gradient centrifugation on Ficoll-Hypaque (NycoPrep 1.077, Axis-Shield PoC AS, Oslo, Norway), and the cell viability and concentration was determined by staining with trypan blue and counted in a Neubauer chamber. Cells were adjusted to a concentration of 1 × 106 cells/ml in complete medium [RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2.0 mM l-glutamine].
TLR-7, MyD88, and NF-kBp65 Expression
Isolated PBMC (1 × 106 cells/tube) were first incubated with surface monoclonal antibodies against CD19 (Clone 1D3) and CD4 (Clone RPA-T4) conjugated with fluorescein isothiocyanate (FITC) and allophycocyanin (APC), respectively (IMGENEX, San Diego, CA, USA), in darkness at 4°C for 30 min. The cells were then fixed and permeabilized using the IC-Flow kit (IMGENEX, 10083K Cat, San Diego, CA, USA), and incubated with monoclonal antibodies against TLR-7 (Clone 4G6), MyD88 (Clone 4D6), and NF-kBp65 (Clone 2J10D7) conjugated with phycoerythrin (PE), in darkness at 4°C for 30 min. Mouse IgG conjugated with FITC, PE, and APC were included as isotype controls (all from IMGENEX, San Diego, CA, USA). The cells were finally washed and assessed by flow cytometry. A total of 10,000 cells were analyzed in the flow cytometer (FACScalibur, BD Biosciences Corp., San Jose, CA, USA) using the Cell Quest software. The lymphocytes population was identified using the forward scatter (FSC) versus side scatter (SSC) distribution. The percentages of CD19+ B and CD4+ cells expressing TLR-7 and CD19+ B cells expressing MyD88 and NF-kBp65 were assessed. The relative fluorescence intensity (rFI) of TLR-7, MyD88, and NF-kBp65 was calculated based on the mean fluorescence intensity of the sample (MFIs) compared with isotype control (MFIc), using the formula: rFI = MFIs − MFIc/MFIc.
Interferon-Alpha
Five milliliters of venous peripheral blood (without anticoagulant) were selected from each patient and control subject to obtain serum. Serum levels of IFNα in were determined by VeriKine Human IFN-alpha Serum Sample ELISA kit, following the directions of the supplier (PBL Assay Science Piscataway, NJ, USA). The kit quantitates human IFNα using a sandwich immunoassay, with an anti-secondary antibody conjugated to horseradish peroxidase (HRP) and tetramethylbenzidine (TMB) as substrate. The detection range of 12.5–1000 pg/ml was calculated using a standard curve. Each standard, blank, and sample test was run in duplicate. The absorbance was determined at 450 nm, using a microplate reader (BIOTEK Instrument, Inc., VT, USA).
Statistical Analysis
Wilcoxon matched-pairs signed rank test was used to assess the significance of any difference in values of TLR-7, MyD88, NF-kBp65 expression and IFNα serum, among SLE patients and control subjects (p < 0.05). Correlation analysis was done using the Pearson correlation coefficient. In all comparisons, the level of significance was p < 0.05, using the Graph Pad Prism 5 software.
Results
Characteristics of SLE Patients
All patients were under treatment, 54% of them had active disease determined by SLEDAI (>4), and most residents of the Merida city (54%), the rest from the surrounding Yucatán state. The average age of patients was 39.73 years with different times of evolution (Table 1).
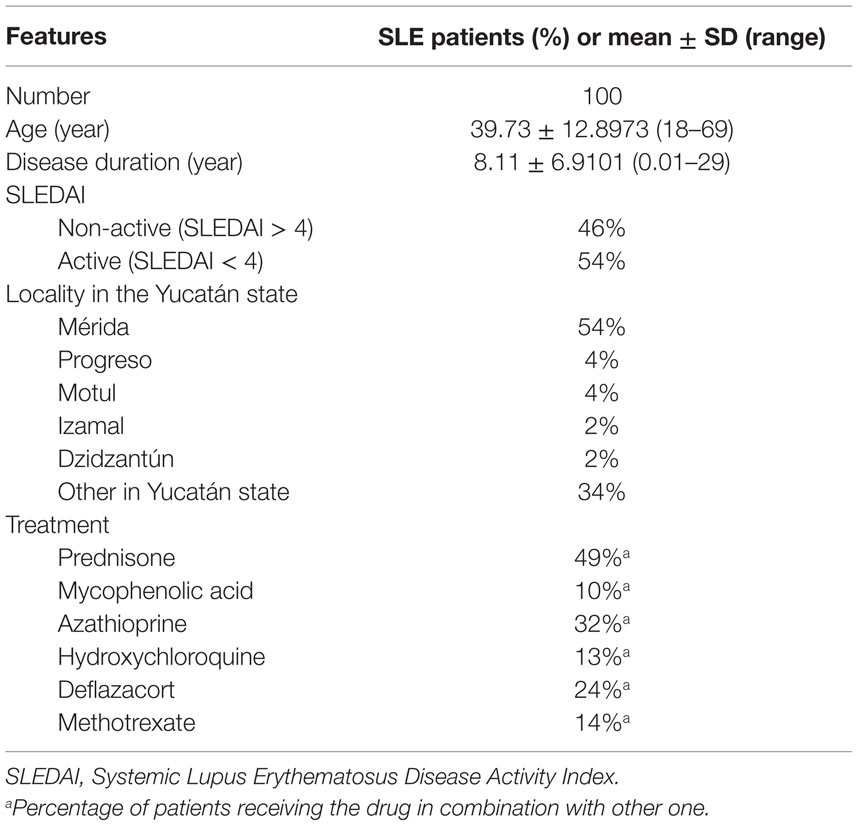
Table 1. Characteristics of SLE patients.
Expression of TLR-7, MyD88, and NF-kBp65
A representative figure of the analysis by flow cytometry, as described in Section “Materials and Methods,” is shown in Figure S1 in Supplementary Material. A higher percentage of CD19+ B lymphocytes expressing TLR-7 were found in patients compared to controls (p < 0.0001) (Figure 1A). The rFI of TLR-7 was significantly higher in B lymphocytes of patients, but no difference was found between patients and controls (p = 0.1882) (Figure 1B). Furthermore, significant expression of TLR-7 was found in CD4+ T lymphocytes of patients with respect to controls (p < 0.0001), and the rFI of TLR-7 was higher in patients. Additionally, no correlation between TLR-7 expression in CD19+ B lymphocytes and active disease (SLEDAI > 4) was observed (Figure 2).
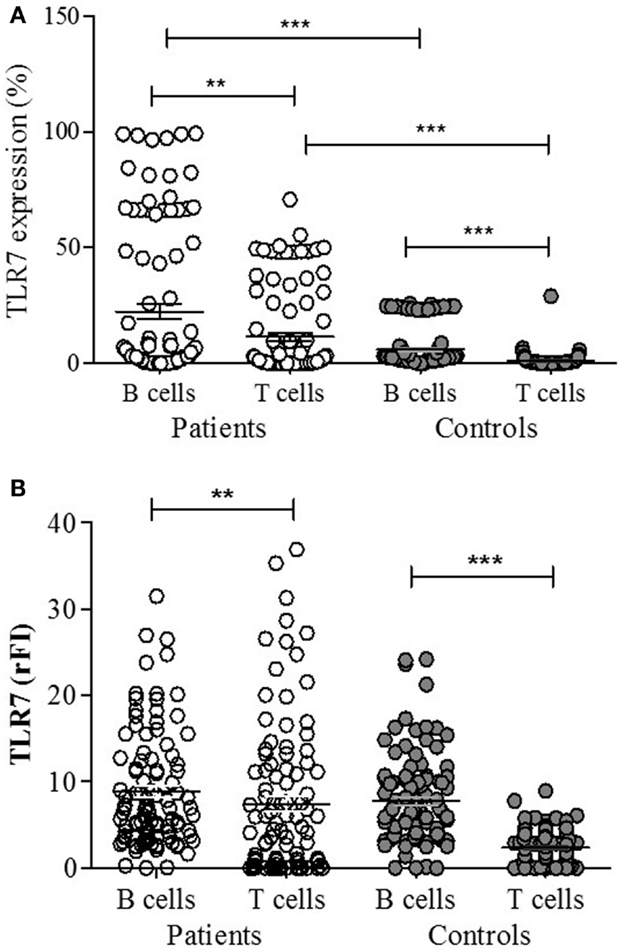
Figure 1. TLR-7 expression in B (CD19+) and T (CD4+) lymphocytes of SLE patients (n = 100) and control subjects (n = 100), analyzed by flow cytometry. Results expressed as the percentage (A) and relative fluorescence intensity (rFI) (B) of TLR-7 are presented in scatter plots and mean with SEM. Wilcoxon matched-pairs signed rank test was used to assess the difference of expression among SLE patients and control subject. **p = 0.0004, ***p < 0.0001.
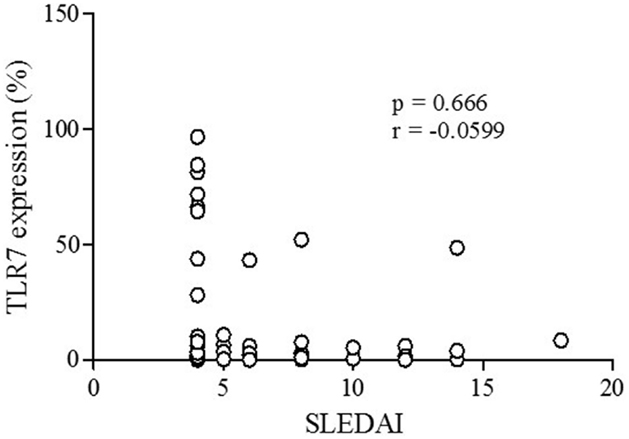
Figure 2. Correlation analysis of TLR-7 expression (%) with active disease (SLEDAI > 4) in SLE patients. Results are presented in scatter plots. Pearson correlation test was used to assess correlation, r = Pearson correlation coefficient; p < 0.05.
Regarding MyD88 and NF-kBp65, both were expressed more in CD19+ B lymphocytes of patients (n = 50) than of controls (n = 50) (p < 0.0001) (Figure 3A). Only NF-kBp65 correlated with TLR-7 expression in B lymphocytes of patients (Figure 4). The rFI of both proteins was significantly increased in CD19+ B lymphocytes from patients (p < 0.0001) (Figure 3B), but no correlation with TLR-7 expression was observed (Figure S2 in Supplementary Material). No correlation with active disease (SLEDAI > 4) was observed with both proteins.
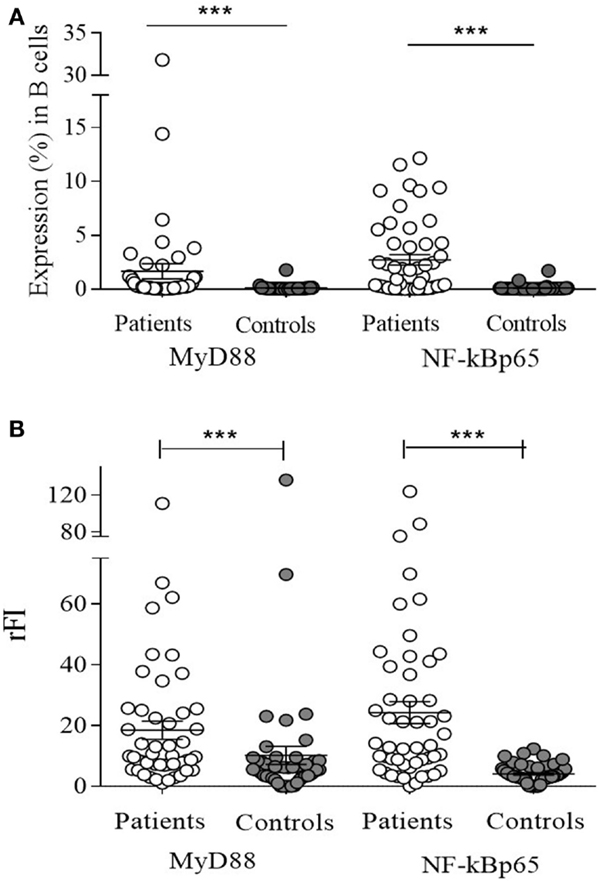
Figure 3. MyD88 and NF-kBp65 expression in B lymphocytes of SLE patients (n = 50) and control subjects (n = 50), analyzed by flow cytometry. Results expressed as the percentage (A) and relative fluorescence intensity (rFI) (B) of MyD88 and XF-kBp65 is presented in scatter plots and mean with SEM. Wilcoxon matched-pairs signed rank test was used to assess the difference of expression among SLE patients and control subject. ***p < 0.0001.
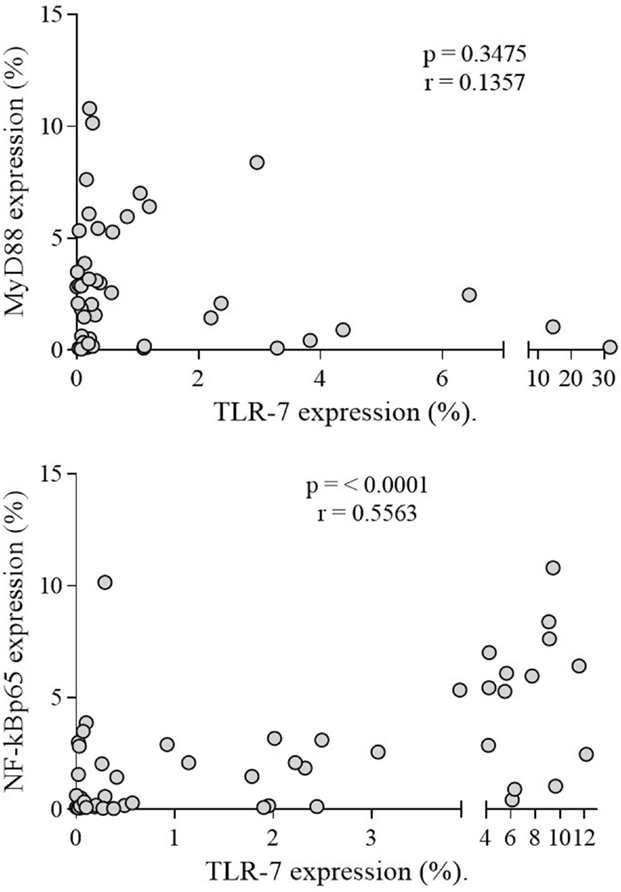
Figure 4. Correlation analysis of TLR-7 with MyDSS and NF-kBp65 expression in SLE patients. Results are presented in scatter plots. Pearson correlation test was used to assess correlation, r = Pearson correlation coefficient; p < 0.05.
Serum IFN-α
IFN-α was variable in patients (7.6 ± 3.54 ng/μl) but significantly higher (p = 0.0001) compared to undetectable levels in controls (Figure 5). However, no correlation with TLR-7 expression and disease activity (SLEDAI > 4) was observed (Figure 6).
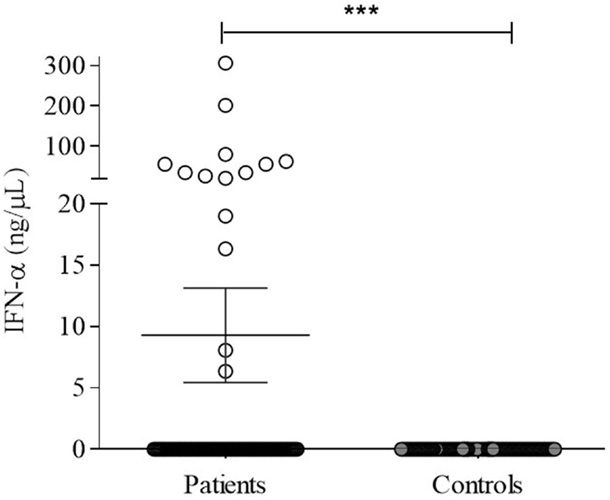
Figure 5. Serum IFN-α in SLE patients (n = 100) and controls subjects (n = 100) were detected by human IFN-α ELISA, as described in Section “Materials and Methods.” Results expressed as nanograms per picoliter of IFN-α are presented in scatter plots and mean with SEM. Wilcoxon matched-pairs signed rank test was used to assess the difference among SLE patients and control subject. ***p = 0.0001.
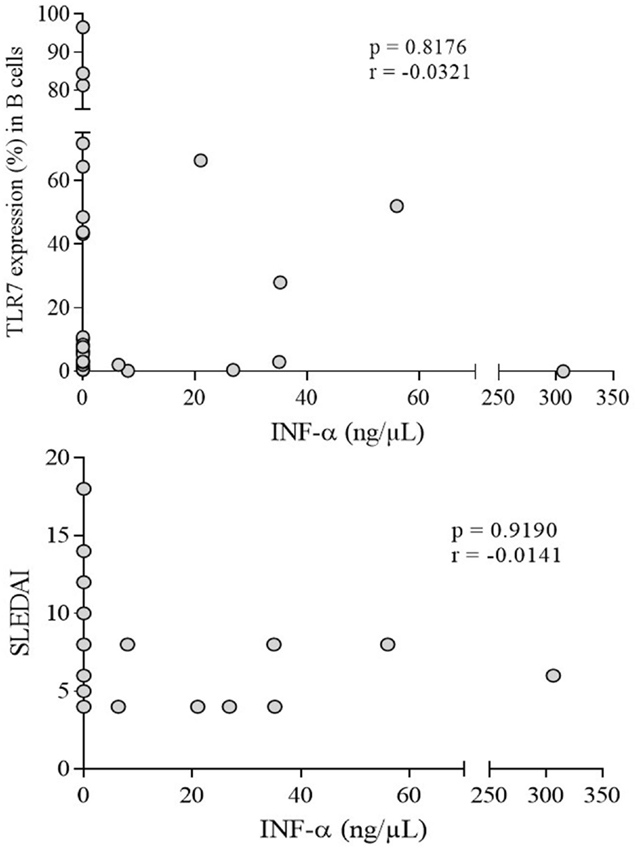
Figure 6. Correlation analysis of serum IFN-α with TLR-7 expression (%) and active disease (SLEDAI > 4) in SLE patients. Results are presented in scatter plots. Pearson correlation test was used to assess correlation, r = Pearson correlation coefficient; p < 0.05.
Discussion
This research represented the first report on the expression of TLR-7 and signal proteins MyD88 and NF-kBp65 in B lymphocytes of Mayan women with SLE. We observed significant increase of TLR-7 expression in B lymphocytes of SLE patients compared to control subjects, which was consistent with previous studies, even with different analysis procedures (38–41). The rFI of TLR7 also was increased in B cells from patients. This suggested that overexpression of TLR-7 in B lymphocytes may be a common characteristic of SLE patients since TLR-7 are the main source of pathological antibodies for the disease. Moreover, correlation between the TLR-7 expression and disease severity has been reported in SLE patients; however, we did not find correlation probably due to therapy they already received. Further study is needed to clarify this discrepancy.
Although a high percentage of B lymphocytes expressing TLR-7 were observed in patients, different subtypes of CD4+ T cells also express TLRs (42). The TLR-7 has been reported on CD4+CD25+ T regulatory (Treg) cells, and TLR-7 activation increase their suppressor function by suppressing autoreactive lymphocytes, but defects in their number and function may contribute to pathogenesis of SLE (43–45). It has been reported IL-17 secretion by human CD4 T cells stimulated with TLR-7 agonist, suggesting that TLR-7 ligation generates proinflammatory cytokines that induces Th17 differentiation and establishes a link between TLR-7 interaction and Th17 cell differentiation (46). An imbalanced Th17/Treg ratio favoring Th17 cells has been reported in SLE patients (47). We observed higher expression of TLR-7 in CD4+ T cells in SLE patients, but subtypes of Treg and Th17 cells were not identified. Further researches are needed to strengthen the role of TLR-7 on these cells as a mechanism of action in autoimmunity.
Few studies have evaluated the expression of MyD88 in cells of SLE patients. Nakano et al. assessed the mRNA of MyD88 in Chinese patients B lymphocytes, and found no significant difference with controls (48). Chen et al. studied the role of the TLR-7 signaling pathway in the pathogenesis of adult-onset Still’s disease (AOSD) and SLE, finding increased levels of mRNA of MyD88, TRAF6, IRAK-4, and IFN-α in mononuclear cell of SLE patients, which correlated with disease activity (49). Data suggested that overexpression of MyD88-dependent signaling molecules may be a pathogenesis mechanism in SLE. We found significant expression levels of MyD88 in B lymphocytes of patients by flow cytometry, but no correlation with TLR-7 expression and disease activity was found, suggesting that activation of TLR-7 signaling pathway in our SLE patients appeared to be unaffected by the disease activity. Further studies are needed to establish whether MyD88 expression levels correlate with its mRNA or are influenced by the activation of other intracellular receptors that share the molecule, and if the treatment received by patients has any impact on MyD88.
The NF-kBp65 is an inducible transcription factor that controls genes involved in inflammatory responses and play an important role in B lymphocytes maintenance (50–52). Genetic associations have been found between genes involved in NF-kBp65 signaling pathway in Chinese SLE patients, highlighting the role of NF-kBp65 in autoimmunity (53). We found significantly higher expression levels of NF-kBp65 in B lymphocytes from SLE patients consistent with those reported (53), suggesting its constitutive activation in B cells of patients; however, no correlation with TLR-7 expression and disease activity was observed. Data support its role in the mechanisms of autoimmunity, but further studies are needed to identify which receptor induces NF-kBp65 activation, promoting the survival of autoreactive B lymphocytes in SLE patients, despite treatment received.
A hallmark of SLE is the elevated levels of INF-α in serum. Approximately 50% of patients have been shown to have dysregulated expression of genes involved in the INF pathway, which correlates with disease activity (54, 55). High levels of INF-α were detected in our patients, but no correlation with disease activity was found. It is important to note that although 54% of patients had active disease, and all of them showed variable levels of serum INF-α, inactive patients showed low and undetectable levels. This variability is likely due to the effect of the drugs they received. Studies in murine models have reported the inhibitory effect of chloroquine and corticosteroids on the immune response. Acidification of lysosomes and function of TLR-7 and TLR-9 are inhibited by hydroxychloroquine, and its activation and release of cytokines is suppressed by prednisone (56, 57). In this regard, 54% of our patients were receiving prednisone in combination with other drugs, suggesting that the combined effect of drugs may modify the inflammatory response and inhibit the synthesis of cytokines, including INF-α. Longitudinal studies are needed to determine the effect of therapy on the synthesis of INF-α and disease activity.
Conclusion
Our results show increased expression of TLR-7, MyD88, and NF-kBp65 in B lymphocytes from Mayan women, which supports its role in the pathogenesis of SLE in this ethnic population of southeast of Mexico.
Author Contributions
Conceived and designed the experiments: GP. Analyzed the data: CO, IN, and RC. Contributed to the writing of the manuscript: AR and RV. All authors reviewed and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by CONACYT (National Council of Science and Technology), grant FONSEC SALUD 2010-1-139788. We are grateful to Nicole R. Van Wynsberghe for editing.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org//article/10.3389/fimmu.2016.00022
References
1. Mills JA. Systemic lupus erythematosus. N Engl J Med (1994) 330:1871–9. doi: 10.1056/NEJM199406303302608
2. Vyse TJ, Kotzin BL. Genetic susceptibility to systemic lupus erythematosus. Annu Rev Immunol (1998) 16:261–92. doi:10.1146/annurev.immunol.16.1.261
3. Lispky PE. Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nat Immunol (2001) 2:764–6. doi:10.1038/ni0901-764
4. Crispín JC, Nick S, Liossis C, Kis-Toth K, Lieberman LA, Kyttaris Y, et al. Pathogenesis of human systemic lupus erythematosus: recent advances. Trends Mol Med (2010) 16(2):47–57. doi:10.1016/j.molmed.2009.12.005
5. McCarty DJ, Manzi S, Medsger TA Jr, Ramsey-Goldman R, LaPorte RE, Kwoh CK. Incidence of systemic lupus erythematosus. Race and gender differences. Arthritis Rheum (1995) 38(9):1260–70. doi:10.1002/art.1780380914
6. Danchencho N, Satia JA, Anthony SM. Lupus around the world. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus (2006) 15:308–18. doi:10.1191/0961203306lu2305xx
7. Yang N, Li H, Criswell LA, Gregersen PK, Alarcon-Riquelme ME, Kittles R, et al. Examination of ancestry and ethnic affiliation using highly informative diallelic DNA markers: application to diverse and admixed populations and implications for clinical epidemiology and forensic medicine. Hum Genet (2005) 118:382–92. doi:10.1007/s00439-005-0012-1
8. Peláez-Ballestas I, Sanin L, Moreno-Montoya J, Alvarez-Nemegyei J, Burgos-Vargas R, Garza-Elizondo M, et al. Epidemiology of the rheumatic diseases in Mexico. A study of 5 regions based on the COPCORD methodology. J Rheumatol (2011) 38(86):3–8. doi:10.3899/jrheum.100951
9. Álvarez-Nemegyei J, Peláez-Ballestas I, Sanin L, Cardel M, Ramirez-Angulo A, Goycochea-Robles M. Prevalence of musculoskeletal pain and rheumatic diseases in the southeastern region of Mexico. A COPCORD-based community survey. J Rheumatol Suppl (2011) 37(86):21–5. doi:10.3899/jrheum.100954
10. Akira T, Takeda K, Kaisho T. Toll-like receptors: proteins linking innate and acquired immunity. Nat Immunol (2001) 2(8):675–80. doi:10.1038/90609
11. Theofilopouluos AN, Gonzalez-Quintal R, Lawson BR, Koh YT, Stern ME, Kono DH, et al. Sensors of innate immune system; their link to rheumatic diseases. Nat Rev Rheumatol (2010) 6:146–56. doi:10.1038/nrrheum.2009.278
12. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol (2003) 9:335–76. doi:10.1146/annurev.immunol.21.120601.141126
13. Guggino G, Giardina A, Ciccia F, Triolo G, Dieli F, Sireci G. Are toll-like receptors and decoy receptors involved in the immunopathogenesis of systemic lupus erythematosus and lupus-like syndromes? Clin Dev Immunol (2012) 2012:1–5. doi:10.1155/2012/135932
14. Richez C, Blanco P, Rifkin I, Moreau JK, Schaeverbeke T. Role for toll-like receptors in autoimmune disease: the example of systemic lupus erythematosus. Joint Bone Spine (2011) 78(2):124–30. doi:10.1016/j.jbspin.2010.09.005
15. Hurst J, von Landenberg P. Toll-like receptors and autoimmunity. Autoimmun Rev (2008) 7(3):204–8. doi:10.1016/j.autrev.2007.11.006
16. Santiago-Raber ML, Dunand-Sauthier I, Wu T, Li QZ, Uematsu S, Akira S, et al. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun (2010) 34:339–48. doi:10.1016/j.jaut.2009.11.001
17. Pisitikun P, Deane JA, Difilippantonio MJ, Taransenko T, Satterthwaite AB, Bolland S. Autoreactive B cells responses to RNA-related antigens due to TLR7 gene duplication. Science (2006) 312:1669–72. doi:10.1126/science.1124978
18. Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhoua J, et al. A TLR7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A (2006) 103:9970–5. doi:10.1073/pnas.0603912103
19. Hwang SH, Lee H, Yamamoto M, Jones LA, Dayalan J, Hopkins R, et al. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J Immunol (2012) 189:5786–96. doi:10.4049/jimmunol.1202195
20. Iwasaki A, Medzhitov R. Regulation of adaptative immunity by the innate immune system. Science (2010) 327:291–5. doi:10.1126/science.1183021
21. Bijil M, Limburg PC, Kallenberg CGM. New insights into the pathogenesis of systemic lupus erythematosus (SLE); the role of apoptosis. Neth J Med (2001) 59(2):66–75. doi:10.1016/S0300-2977(01)00131-0
22. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll like receptors. Nature (2002) 416:603–7. doi:10.1038/416603a
23. Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol (2010) 184:1840–8. doi:10.4049/jimmunol.0902592
24. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity (2006) 25:417–28. doi:10.1016/j.immuni.2006.07.013
25. Lövgren T, Eloranta ML, Kastner B, Wahren-Herlenius M, Alm GV, Rönnblom L. Induction of interferon alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjogren’s syndrome autoantigen-associated RNA. Arthritis Rheum (2006) 54(6):1917–27. doi:10.1002/art.21893
26. Batteux F, Palmer P, Däeron M, Weill B, Lebon P. FCγRII (CD32)-dependent induction of interferon-alpha by serum from patients with lupus erythematosus. Eur Cytokine Netw (1999) 10(4):509–14.
27. Kirou KA, Lee C, George S, Louca K, Peterson MGE, Crow MK. Activation of the interferon-α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum (2005) 52(5):1491–503. doi:10.1002/art.21031
28. Dall’Era MC, Cardarelli PM, Preston BT, Witte A, Davis JC Jr. Type I interferon correlates with serological and clinical manifestations of SLE. Ann Rheum Dis (2005) 64(12):1692–7. doi:10.1136/ard.2004.033753
29. Feng X, Wu H, Grossman JM, Hanvivadhanakul P, FitzGerald JD, Park GS, et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum (2006) 54(9):2951–62. doi:10.1002/art.22044
30. Zhuang H, Narain S, Sobel E, Lee PY, Nacionales DC, Kelly KM, et al. Association of antinucleoprotein autoantibodies with upregulation of type I interferon-inducible gene transcripts and dendritic cell maturation in systemic lupus erythematosus. Clin Immunol (2005) 117(3):238–50. doi:10.1016/j.clim.2005.07.009
31. Rajagopal D, Paturel C, Morel Y, Uematsu S, Akira S, Diebold SS. Plasmacytoid dendritic cell-derived type I interferon is crucial for the adjuvant activity of Toll-like receptor 7 agonists. Blood (2010) 115(10):1949–57. doi:10.1182/blood-2009-08-238543
32. Asselin-Paturel C, Brizard G, Chemin K, Boonstra A, O’Garra A, Vicari A, et al. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med (2005) 201(7):1157–67. doi:10.1084/jem.20041930
33. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi:10.1016/j.cell.2006.02.015
34. West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol (2006) 22:409–37. doi:10.1146/annurev.cellbio.21.122303.115827
35. Guillermo VP, Darig CC, Lizbeth GH, Gerardo PM, Guadalupe AA, Yumi NU, et al. Copy number variation of TLR-7 gene and its association with the development of systemic lupus erythematosus in female patients from Yucatan Mexico. Genet Epigenet (2014) 6:31–6. doi:10.4137/GEG.S16707
36. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum (1997) 40:1725. doi:10.1002/art.1780400928
37. Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI: a disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum (1992) 35:630–40. doi:10.1002/art.1780350606
38. Klonowska-Szymczyk A, Wolska A, Robak T, Cebula-Obrzut B, Smolewski P, Robak E. Expression of toll-like receptors 3, 7, and 9 in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Mediators Inflamm (2014) 2014:381418. doi:10.1155/2014/381418
39. Komatsuda A, Wakui H, Iwamoto K, Ozawa M, Togashi M, Masai R, et al. Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cell from patients with systemic lupus erythematosus. Clin Exp Immunol (2008) 152:482–7. doi:10.1111/j.1365-2249.2008.03646.x
40. Wong CK, Wong PT, Tam LS, Li EK, Chen DP, Lam CW. Activation profile of Toll-like receptors of peripheral blood lymphocytes in patients with systemic lupus erythematosus. Clin Exp Immunol (2010) 159(1):11–22. doi:10.1111/j.1365-2249.2009.04036.x
41. Lyn-Cook BD, Xie C, Oates J, Treadwell E, Word B, Hammons G, et al. Increased expression of Toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol Immunol (2014) 61:38–43. doi:10.1016/j.molimm.2014.05.001
42. Liu G, Zhang L, Zhao Y. Modulation of immune responses through direct activation of toll-like receptors to T cells. Clin Exp Immunol (2010) 160:168–75. doi:10.1111/j.1365-2249.2010.04091.x
43. Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengol J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med (2003) 197:403–11. doi:10.1084/jem.20021633
44. Forward NA, Furlong SJ, Yang Y, Lin TJ, Hoskin DW. Signaling through TLR7 enhances the immunosuppressive activity of murine CD4+CD25+ T regulatory cells. J Leukoc Biol (2010) 87(1):117–25. doi:10.1189/jlb.0908559
45. Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol (2015) 45:344–55. doi:10.1002/eji.201344280
46. Kattah MG, Wong MT, Yocum MD, Utz PJ. Cytokines secreted in response to toll-like receptor ligand stimulation modulate differentiation of human Th17 cells. Arthritis Rheum (2008) 58:1619–20. doi:10.1002/art.23497
47. Alunno A, Bartoloni E, Bistoni O, Nocentini G, Ronchetti S, Caterbi S, et al. Balance between regulatory T and Th17 cells in systemic lupus erythematosus: the old and the new. Clin Dev Immunol (2012) 2012:823085. doi:10.1155/2012/823085
48. Nakano S, Morimoto S, Suzuki S, Watanabe T, Amano H, Takasaki Y. Up-regulation of the endoplasmic reticulum transmembrane protein UNC93B in the B cells of patients with active systemic lupus erythematosus. Rheumatology (Oxford) (2010) 49(5):876–81. doi:10.1093/rheumatology/keq001
49. Chen DY, Lin CC, Chen YM, Lan JL, Hung WT, Chen HH, et al. Involvement of TLR7 MyD88-dependent signaling pathway in the pathogenesis of adult-onset Still’s disease. Arthritis Res Ther (2013) 15(2):R39. doi:10.1186/ar4193
51. Gerondakis S, Strasser A. The role of ReI/NF-kappaB transcription factors in B lymphocytes survival. Sem Immunol (2003) 15(3):159–66. doi:10.1016/S1044-5323(03)00036-8
52. Siebenlist U, Brown K, Claudio E. Control of lymphocyte development by nuclear factor-kappaB. Nat Rev Immunol (2005) 5(6):435–45. doi:10.1038/nri1629
53. Zhang W, Shi Q, Xu X, Chen H, Lin W, Zhang F, et al. Aberrant CD40-induced NF-kB activation in human lupus B lymphocytes. PLoS One (2012) 7(8):e41644. doi:10.1371/journal.pone.0041644
54. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A (2003) 100(5):2610–5. doi:10.1073/pnas.0337679100
55. Niewold TB, Clark DN, Salloum R, Poole BD. Interferon alpha in Systemic Lupus Erythematosus. J Biomed Biotechnol (2010) 2010:8. doi:10.1155/2010/948364
56. Rönnblom L. The type I interferon system in the etiopathogenesis of autoimmune diseases. Ups J Med Sci (2011) 116:227–37. doi:10.3109/03009734.2011.624649
Keywords: innate immunity, Toll-like receptor 7, Interferon-α, systemic lupus erythematosus, pathogenesis
Citation: Pacheco GV, Novelo Noh IB, Velasco Cárdenas RM-H, Angulo Ramírez AV, López Villanueva RF, Quintal Ortiz IG, Alonso Salomón LG, Ruz NP and Rivero Cárdenas NA (2016) Expression of TLR-7, MyD88, NF-kB, and INF-α in B Lymphocytes of Mayan Women with Systemic Lupus Erythematosus in Mexico. Front. Immunol. 7:22. doi: 10.3389/fimmu.2016.00022
Received: 09 November 2015; Accepted: 15 January 2016;
Published: 02 February 2016
Edited by:
Timothy B. Niewold, Mayo Clinic, USAReviewed by:
Geanncarlo Lugo-Villarino, Centre National de la Recherche Scientifique (CNRS), FrancePhilippe Georgel, Strasbourg University, France
Copyright: © 2016 Pacheco, Novelo Noh, Velasco Cárdenas, Angulo Ramírez, López Villanueva, Quintal Ortiz, Alonso Salomón, Ruz and Rivero Cárdenas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guillermo Valencia Pacheco, vpacheco@correo.uady.mx