 Michelle J. Hickey
Michelle J. Hickey Nicole M. Valenzuela
Nicole M. Valenzuela Elaine F. Reed
Elaine F. Reed- Department of Pathology and Laboratory Medicine, UCLA Immunogenetics Center, University of California Los Angeles, Los Angeles, CA, USA
Allorecognition is the activation of the adaptive immune system to foreign human leukocyte antigen (HLA) resulting in the generation of alloantibodies. Due to a high polymorphism, foreign HLA is recognized by the immune system following transplant, transfusion, or pregnancy resulting in the formation of the germinal center and the generation of long-lived alloantibody-producing memory B cells. Alloantibodies recognize antigenic epitopes displayed by the HLA molecule on the transplanted allograft and contribute to graft damage through multiple mechanisms, including (1) activation of the complement cascade resulting in the formation of the MAC complex and inflammatory anaphylatoxins, (2) transduction of intracellular signals leading to cytoskeletal rearrangement, growth, and proliferation of graft vasculature, and (3) immune cell infiltration into the allograft via FcγR interactions with the FC portion of the antibody. This review focuses on the generation of HLA alloantibody, routes of sensitization, alloantibody specificity, and mechanisms of antibody-mediated graft damage.
Introduction
The immune response is designed to recognize antigens that are distinct from self – termed “non-self” or “altered self” – be they protein, lipid, or carbohydrate. Allorecognition is the activation of the transplant recipient’s adaptive immune response to foreign histocompatibility antigens following transplant (1, 2). This review focuses on the recognition of allogeneic human leukocyte antigen (HLA) and non-HLA molecules by the humoral immune response in the context of transplantation. We discuss the generation of alloantibodies, and how they mediate graft injury and rejection.
Human Leukocyte Antigen: Genomic Organization, Structure, Polymorphism, and Function
The human major histocompatibility complex (MHC), located on chromosome 6, is composed of highly polymorphic HLA class I genes (HLA-A, -B, and -C), HLA class II genes (HLA-DR, -DQ, and -DP), non-classical class I genes (HLA-E, -F, and -G), and class I-like genes (MICA and MICB) (3). The HLA class I molecules function to present peptide derived from intracellular antigens to CD8+ T lymphocytes and serve as ligands for receptors on natural killer (NK) cells. The HLA class II molecules present antigens from the extracellular space to CD4+ T cells.
Human leukocyte antigen molecules are heterodimers formed by polypeptides encoded by two distinct genetic loci (Figure 1) (3). The HLA class I molecule consists of one heavy β-chain that is non-covalently bound to a β2-microglobulin (β2m) light chain at the cell surface for stability. β2m is highly conserved and does not exhibit polymorphism. The HLA class II molecule is composed of two transmembrane glycoprotein chains – an α-chain and β-chain. The α-chain shared by all HLA-DR molecules (DRA1) has limited polymorphism (seven alleles identified to date, with only two different proteins/amino acid sequences; the amino acid polymorphism is V217L in the cytoplasmic domain) and is not a known target of humoral alloresponses. By contrast, both the α- and β-chains of HLA-DP and HLA-DQ are polymorphic (3, 4).
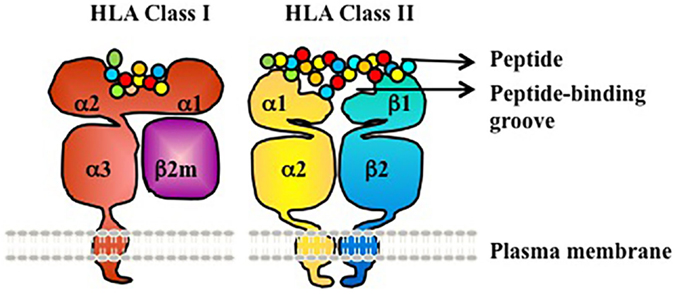
Figure 1. HLA class I and II are heterodimeric transmembrane proteins. HLA Class I is made up of a heavy chain with three globular domains (a1, a2, and a3) non-covalently bound to β2m. HLA Class II is made up of two heavy chains (a-chain and b-chain) each with two globular domains (a1 and a2 or b1 and b2). The a1and a2 domains of HLA class I, and the a1 and b1 domains of HLA class II, make up the peptide-binding groove.
Globular domains of the HLA Class I and II molecules form the peptide-binding cleft that accommodates peptide antigens and interacts with the T cell receptor (TCR). The remarkable polymorphism of HLA Class I and II molecules allows for the presentation of a vast array of antigenic peptides within the human population. Each HLA molecule binds distinct peptides. At the protein level, HLA molecules are defined as antigens by either low-resolution (two digit, serologic level) or high resolution (four digit, allele level) nomenclature (Table 1). At the serologic level, there are about 20 HLA-A, 50 HLA-B, 10 HLA-Cw, 18 HLA-DR, and 7 HLA-DQ antigens. However, at the allele level of resolution, the number of HLA antigens in each serogroup is tremendously expanded due to genetic polymorphism within each serogroup – ~2000–3000 distinct proteins for each of HLA-A, B, and C, ~500–2000 for each of DRB1, DQB1, DPB1, and ~10–50 for each of DRB3 (DR52), DRB4 (DR53), DRB5 (DR51), and DQA1(4). Amino acid differences between HLA alleles enable presentation of a diverse array of peptides, and represent the basis for alloimmune recognition of non-self HLA by both T cells and antibodies (3).

Table 1. Typing of HLA molecules can be at low or high resolution.
Mechanisms of Allorecognition and Generation of Allospecific Antibodies
Three distinct pathways of allorecognition have been defined (Figure 2). The direct, indirect, and semidirect pathways can occur independently or simultaneously. Activation of the recipient’s CD4+ T lymphocytes is a pivotal step in the initiation of the immune response to alloantigen following transplantation leading to downstream activation of cytotoxic CD8+ T lymphocytes and antibody-producing B cells.
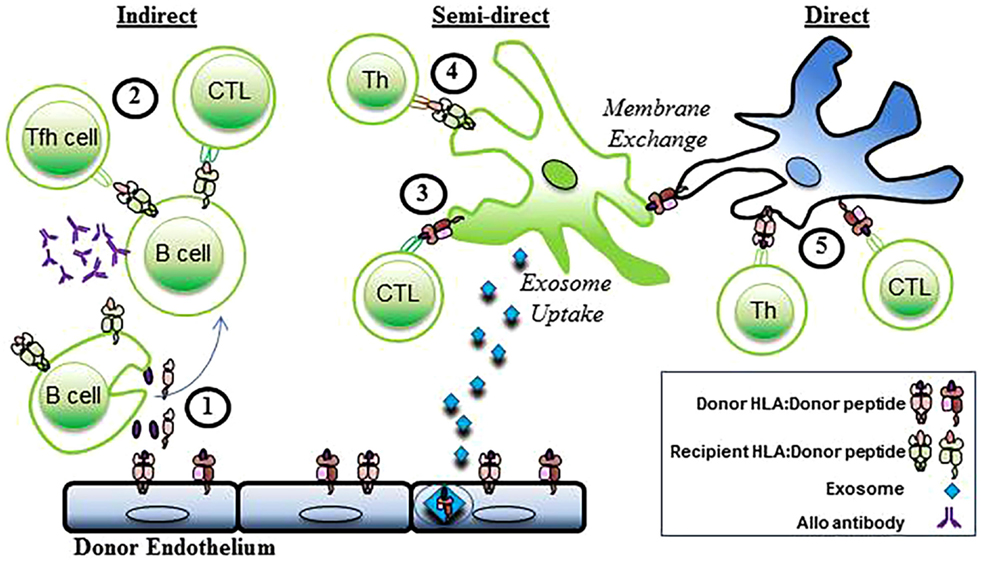
Figure 2. Indirect, semidirect, and direct allorecognition. In the indirect pathway, (1) donor alloantigens are processed by recipient B cells and (2) presented to recipient T follicular helper (TFH) cells and CTL. Alloantibodies are generated when alloreactive B cells interact with CD4+ T cells. The semidirect pathway involves (3) intact donor HLA class I:peptide complexes that are presented on the DC of the recipient (through either membrane exchange or exosome uptake) to recipient CD8+ T cells (CTL). Simultaneously, (4) processed donor peptide is presented in the context of the recipient’s HLA class II to the recipient’s helper CD4+ T cells (Th). In the direct pathway, (5) allogeneic MHC class I and II antigens are presented to recipient CD4+ and cytotoxic CD8+ T cells (CTL) by donor APCs. Recipient cells, green. Donor Cells, blue.
Indirect Allorecognition
Indirect allorecognition is the activation of the transplant recipient’s CD4+ T cells by alloantigen that is processed and presented in the context of the recipients HLA as occurs with the normal immune response to foreign pathogen (2). Donor antigens, shed by the grafted organ, are processed and presented in the context of self-restricted HLA class II by the recipient’s B cells. The recipient’s follicular helper CD4+ T cells are then activated to provide help leading to the generation of alloreactive CD8+ effector T cells and antibody-producing B cells (1, 5, 6). The immune response engendered by this pathway is credited with driving chronic rejection and due to lower frequency of T cells with indirect allospecificity, and requirement for antigen processing, is physio-dynamically slower than the response to presentation through the direct pathway (7–10).
Direct Allorecognition
Direct allorecognition is the activation of the transplant recipient’s CD4+ T cells by donor HLA:peptide complexes (2). Antigen presentation is mediated by the donor’s dendritic cells that are transplanted as passengers with the organ. In the context of inflammatory signals subsequent to the transplantation surgery, the donor’s DC, presenting intact donor allo-histocompatibility antigens, migrate to the secondary lymph nodes of the recipient and present antigen to the recipients CD4+ T cells (11, 12). The strength of the immune response elicited by the direct allorecognition pathway correlates to the high frequency of recipient allogeneic T cells that become activated during the first few weeks following transplant (13, 14) mediating acute rejection. The immune response weakens as the passenger DC leave the graft (15, 16). CD4+ T cells activated through the direct pathway are capable of providing help to effector CD8+ T cells, therefore, promoting rejection of the transplanted organ (5). However, activation of B cells and production of alloantibody does not occur in the context of direct allorecognition as there is no cognate interaction between the T helper cell and B cell (5).
Semi-Direct Allorecognition
The semi-direct pathway of allorecognition is presented as a hypothesis to describe events of apparent overlap between the direct and indirect pathways. Evidence from animal models of transplant rejection indicate that indirect allospecific CD4+ T cells can provide help to direct allospecific CD8+ T cells (17, 18). In principle, this would require a “four cell” model in which CD4+ T cells activated via the indirect pathway by processed alloantigen in the context of self-restricted HLA class II provide help to effector CD8+ T cells activated via the direct pathway by donor passenger APC bearing intact HLA:peptide. The “four cell model” challenges the dogma of the “three cell” or “linked” model whereby the primary mechanism by which activated helper T cells provide help to effector CD8+ T cells is by providing signals to the APC that result in the upregulation of presented antigens (19–21). Helper CD4+ T cells, therefore, “license” APC to more effectively present peptide in the context of HLA class I. The “three cell” model requires that both antigenic determinants recognized by CD4+ and CD8+ T cells be presented on the same APC.
However, the mechanism underling the phenomena of semi-direct allorecognition more likely lies in the exchange of membrane proteins between immune cells (22). After transplantation, the recipients DC acquire intact donor HLA class I:peptide complexes from donor passenger DC or endothelial cells through either cell–cell interactions or by uptake of exosomes containing the antigen that are shed from donor tissue (23, 24). In following, the recipients DC now bears intact donor HLA class I molecules as well as recipient HLA class II molecules, and is capable of stimulating the recipients CD4+ and CD8+ T cells via the indirect and direct pathways in a “three cell” model. Soluble MHC class I can be taken up by DC in vitro, and then presented leading to the production of alloantibody (25). The work by Curry et al. implies that soluble alloantigen can be taken up and presented intact to direct B cells, and can simultaneously be processed and presented to indirect CD4+ T cells.
Generation of HLA Alloantibody
Conlon et al. (6) definitively showed that production of alloantibody occurs exclusively through the indirect pathway. In a murine heart allograft model, C57B/6 mice (H-2b) lacking intact TCRs were transplanted with a BALB/c allograft (H-2Kd). Subsequent reconstitution with TCR transgenic CD4+ T cells engineered to specifically recognize an immunodominant BALB/c peptide (H-2Kd54–68) processed and presented by MHC Class II resulted in a strong anti-H-2Kd IgG alloantibody response to the allograft. Furthermore, the adoptively transferred CD4+ T cells were found in germinal centers (GC), having acquired the phenotype of T follicular helper (TFH) cells (CXCR5+CCR7−), and anti-H-2Kd plasma cells were found in the bone marrow. By contrast, direct-pathway CD4+ T cells were unable to provide help to allospecific B cells and alloantibody was not produced.
Formation of the Germinal Center and Generation of Long-Lived Memory
B cells residing in the secondary lymphoid organs can be exposed to small antigens directly through diffusion from the lymphatic system, or to large immune complexed antigens presented by follicular dendritic cells or by macrophages. Regulation of B cell immunity and generation of antibody-secreting plasma cells is primarily dependent on interactions with TFH cells in the GC of the secondary lymphoid organs (26). Antigen-specific TFH cells and antigen-primed B cells migrate to follicular regions of the secondary lymph nodes and form stable contacts through the signal lymphocyte activation molecule (SLAM)-associated protein (SAP) (27). Integrins and the SLAM protein CD84 are also involved in the interaction between TFH cells and pre-GC B cells (28). These interactions ultimately lead to significant proliferation of antigen-specific B cells and the formation of the GC. CD4+ T cells recognize their cognate peptide antigen presented in HLA class II by the B cell, and provide help through costimulation and cytokines to drive activation and clonal expansion of B cells.
In the GC, B cells make contact with TFH cells that are both transient and stable resulting in selection of B cells that will that enter the long-lived memory component of the immune system (29, 30). Here, through somatic hypermutation, GC B cells that have high antigen affinity differentiate into memory B cells or antibody-producing long-lived plasma cells (31). Activated B cells differentiate into low-affinity antibody-producing plasmablasts, or undergo class-switch recombination and somatic hypermutation to form affinity matured, class-switched memory B cells or plasma cells. Long-lived plasma cells residing in the bone marrow contribute to much of the circulating antigen-specific immunoglobulin and can persist for decades. There is also evidence that memory B cells can be maintained in the circulation without a requirement for continuous antigen exposure (32), ready for rapid recall upon repeated stimulation with antigen.
Recent work has aimed at detection of circulating allospecific memory B cells to predict durable sensitization and anamnestic responses in patients awaiting transplantation. One recent report (33) found that circulating HLA-specific B cells were found only in patients with a history of sensitization, and were detectable in nearly half of such patients. Interestingly, patients with circulating HLA antibodies but no known sensitization event had no detectable circulating B cells. Transfusion also resulted in little to no detectable circulating anti-HLA memory B cells, consistent with the theory that transfusion is a less vigorous sensitizing event compared with pregnancy or transplantation (see below) (34). Snanoudj et al. were able to detect circulating B cells targeting prior donor antigens many decades after transplantation and even after graft removal (33), supporting the paradigm that memory B cells do not require persistent antigen for survival. Finally, and most notably, several patients had detectable HLA antibody secreting B cells in circulation but no detectable circulating antibodies in their sera.
Kinetics of Allorecognition
Direct pathway-activated donor-specific T cells are associated with acute T cell-mediated rejection in renal transplant patients (35). CD4+ T cells isolated from the recipient’s pre-transplant blood that were responsive to direct allostimulation with donor cells were also found to be predictive of early post-transplant outcomes (35, 36). However, T cells activated via the direct pathway were found to be predominantly hyporesponsive in patients with transplant coronary artery disease (TCAD), chronic allograft nephropathy (CAN), or chronic rejection following liver transplant indicating that these cells are not contributing to chronic rejection (7, 10, 37). In comparison, T cells primed by the indirect pathway are thought to mediate chronic rejection and are found in high frequency in patients with CAN, and in heart transplant patients with chronic rejection (7, 9, 10, 38).
Notably, T cells stimulated by the indirect allorecognition pathways are also capable of contributing to acute rejection during the early post-transplant period. Circulating allopeptide-reactive T cells were predictive of rejection in heart transplant patients studied during the first 10 weeks post-transplant (39). Furthermore, T cells responsive to allopeptide were found in significant quantities above that found in circulation when isolated from biopsies of graft tissue, suggesting that indirect pathway T cells can contribute directly to acute graft rejection (39).
Alloantibody Antigen Specificity
Antibody Structure and Function
Antibodies are heterodimers composed of a light chain and heavy chain encoded by distinct loci on different chromosomes. Each chain contains a constant region that is invariant, and a variable region that undergoes both recombination and somatic hypermutation to yield clonally unique sequences. The variable regions of both heavy and light chain form the antigen binding region (“complementarity determining region”), or paratope, which binds its cognate epitope on the antigen. Human immunoglobulins are divided into five isotypes (IgM, IgD, IgA, IgE, and IgG). Several of these isotypes are further divided into subclasses (IgG1, IgG2, IgG3, and IgG4; IgA1 and IgA2). Antibody isotype and subclass are determined by the constant region.
The subclasses were identified and numbered according to their predominance in circulation rather than order on the genome. Early in the GC reaction, IgM+ B cells class switch first to IgG3 or IgG1, then IgG2, and rarely IgG4 [immunoglobulin sequential class switching is described in Ref. (40, 41)].
Functionally, the subclasses of IgG are distinct. IgG1 has the highest concentration in circulation, and fixes complement well. IgG2 is the next most abundant in circulation and is not an efficient complement fixer. IgG3 is unique with its long hinge region that confers the highest affinity for C1q compared with other subclasses, making it a potent effector [extensively reviewed in Ref. (42)]. However, IgG3 has the shortest half-life in circulation and, being first in order of class switching, has typically the lowest affinity for antigen but is the most potent activator of complement (43). IgG1, IgG3, and IgG4 mostly recognize protein antigens, while IgG2 is canonically efficient at recognizing carbohydrate antigens (in the absence of T cell help) and allergens. It is thought that IgG2 and IgG4 appear later after class switching and affinity maturation, as they have higher affinity for antigen but generally less effective activation of Fc-mediated effector functions, to temporally limit the immune response (41).
Antibodies Are Specific for Antigenic Epitopes
Alloantibodies can be generated against any of the polymorphic loci, i.e., HLA-A, -B, -Cw, DRB1, DRB3 (DR52), DRB4 (DR53), DRB5 (DR51), DQB1, DQA1, DPB1, and DPA1. Antibodies recognize three-dimensional arrangements of amino acids on antigens, called epitopes. Fifteen to 25 amino acid residues form epitopes that are not necessarily adjacent in linear sequence, but are generally within 4 Å (44) (Figure 3). Many of the amino acid polymorphisms within HLA molecules lie within and around the peptide-binding groove at exposed residues on the alpha helices of the α1 and α2 chains of HLA class I, and on the α1 and β1 chains of HLA class II, enabling presentation of diverse peptides. The host–pathogen arms race is believed to have driven this polymorphism to prevent pathogen immune escape and protect populations from epidemics (45). Interestingly, antibody reactivity may also be influenced by the bound peptide (46), which can alter the overall three-dimensional conformation of HLA.
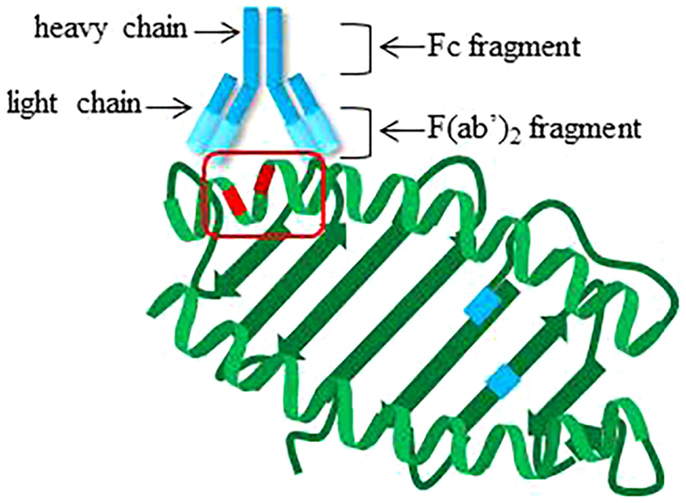
Figure 3. Cartoon structure of antibody engaging epitope on HLA class I. Theoretical locations of polymorphisms impacting peptide binding are indicated in blue on the beta sheet of HLA class I. Locations of polymorphic amino acid residues available for recognition by antibodies on the alpha helices are highlighted in red. Red box, epitope.
Immunogenicity of HLA
Alloantibodies recognize three-dimensional amino acid epitopes on non-self HLA molecules. Because of the evolution of the HLA system, these epitopes can be shared by many different antigens, leading to broad antibody sensitization after exposure to a limited repertoire of non-self HLA. Antibodies are also sensitive enough to recognize single amino acid differences, resulting in intra-allele antibody production (47). Furthermore, molecular differences between HLA antigens can affect expression levels at the surface of the cell. Below, we describe in greater detail the mechanisms governing these various aspects of alloantibody recognition of epitopes.
Cross-Reactive Antibody Groups
The diversity of HLA has been driven by several genetic processes during positive selection. One major mechanism is gene conversion via homologous recombination. Gene conversion results in large segments of genetic material being shared between alleles, giving rise to multiple proteins with the same or similar amino acid epitopes that can be recognized by the alloantibody response (48). This epitope sharing also results in cross-reactive antibody groups (CREGs), and indeed phylogenetic grouping of HLA based on nucleotide sequences generally mirrors serological cross-reactivity (49). Broad sensitization against many HLA antigens can, thus, occur even when the immune system is only exposed to a single non-self HLA antigen. For example, exposure to HLA-A11 may result in the generation of an antibody that is specific to an epitope carried by multiple HLA antigens belonging to the A1 CREG including HLA-A1, A3, A23, A24, A36, and A80 as well as A11 (50, 51). In another example demonstrating inter-locus reactivity, sensitization to HLA-Cw can lead to antibody production to antigens of HLA-B (52) as HLA-B and HLA-Cw are more closely related to one another than to HLA-A. Similarly, DP antigens share epitopes with DR (53).
An extreme example of broad sensitization is in response to the mutually exclusive public epitopes Bw4 and Bw6, which are present on many different HLA-B (as well as some HLA-A, for Bw4) antigens. An individual exposed to a Bw6 positive antigen, such as B7, may produce antibodies against the Bw6 epitope that react with more than 20 different HLA-B antigens, carried by more than 50% of the population. These determinants, as well as C1 and C2 determinants on HLA-Cw molecules, are critical for NK cell receptor (KIR) binding, and so have likely been conserved through co-evolution of HLA and KIR receptors to prevent loss of self recognition (54).
In seminal work, Parham and McLean (55) described serological reactivity in relationship to known amino acid sequence data, first raising the idea of molecular matching. Differences in epitopes or “structural compatibility” between self and potential donor HLA antigens, also known as “eplets,” could portend the likelihood of an antibody response. HLA typing for solid organ transplantation is generally reported at the serologic (two digit) level. However, epitope matching is best accomplished with higher resolution HLA typing such that amino acid sequences that may be different within serologically equivalent groups are defined. Several groups have advocated for the use of structural epitope or eplet matching strategies in organ allocation, over serologic level matching (56, 57). For example, Wiebe et al. reported a lower incidence of de novo DSA production in patients who were HLA class II epitope matched (58), and immunogenicity of HLA-DP (59) also appears to be strongly based on epitope recognition.
Allele-Specific Antibodies
Antibodies can be produced against epitopes within antigens that differ from self by as little as one amino acid. Therefore, in addition to antibodies against serologic level HLA molecules, individuals can produce antibodies to other alleles of “self” antigens, if amino acid sequences in key positions are sufficiently disparate. For example, a patient who displays HLA-DQ6 at the serologic level may also be defined through higher resolution typing methods as DQB1*06:01 at the allele level. The patient may become sensitized to other alleles of DQ6 and display allele-specific antibodies to alleles, such as DQB1*06:04, that are distinct from self (60).
Epitopes Formed by Specific DQA1/DQB1 Pairings
It is also possible for individuals to make antibodies against an epitope that is formed by the pairing of specific DQα1 and DQβ1 chains (61). The majority of HLA-DQ reactive antibodies recognize the DQβ chain, while a minority (<20%) bind DQα chain or a combination epitope formed by specific DQα/β pairings (61). Importantly, such antibodies do not produce positive crossmatches against donors who carry only one of the DQα or DQβ alleles in a different pairing (62), emphasizing the specificity of such antibodies.
Molecular Contributions to Immunogenicity
Differences in antigen availability may necessarily influence immunogenicity. Cell surface expression levels are known to vary among different loci, and even different alleles, of HLA. Certainly, expression of HLA-Cw (63, 64) and HLA-DP (65) is less abundant than proteins of other loci on endothelial cells that make up the vascular walls of the transplanted organ. In addition, HLA-A was found to be more highly expressed than HLA-B in HEK293T cells as it is hypothesized to form a more stable interaction with β2m throughout the terminal region of the alpha 2 domain and the entire alpha 3 domain (66) of the molecules.
Furthermore, Ramsuran et al. recently reported wide variation in mRNA levels between different antigens of HLA-A; for example, individuals homozygous for HLA-A24 had higher expression of HLA-A than those homozygous for HLA-A3, which was attributed to polymorphic CpG sites and increased DNA methylation in the lower expressing alleles (67). Finally, lower expression of HLA-Cw may be the result of reduced affinity for β2 microglobulin, resulting in less stable protein at the cell surface (68, 69), increased degradation of mRNA (63), or differential regulation by miRNA (70). Accordingly, sensitization to HLA-Cw is reportedly less frequent compared with other HLA class I molecules (71).
Sensitizing Events Leading to HLA Immunization: Routes and Rates of HLA Sensitization
Antibody responses to allogeneic HLA molecules can occur after any exposure to non-self tissues, such as transfusion, pregnancy, or transplantation. However, the durability and nature of the sensitization may vary depending on the alloimmunizing event.
Transfusion
Interestingly, the incidence of alloimmunization in the general population with a history of prior transfusion is less than 2% (72, 73), while in comparison Hyun et al. (74) reported that one-third of transplant candidates with a history of transfusion were sensitized. The discrepancy indicates that transplant patients may have a more robust response to sensitization via transfusion, or may have more transfusions compared with non-transplant candidates.
Transfusion alone is considered poorly immunogenic. Sensitization to HLA antigens via transfusion requires very large blood volumes or multiple events to induce persistent HLA allosensitization in otherwise non-sensitized individuals (34). Paradoxically, a protective “transfusion effect” was reported in the early transplantation literature (75, 76), initially suggesting that donor-specific transfusion is immunomodulatory and improved graft outcomes. Animal models have suggested that graft passenger leukocytes are important in this process, thus, providing tolerance prior to transplant (77). However, transplant recipients sensitized by third party transfusion have poorer 1-year survival compared with non-sensitized recipients (78). A modern meta-analysis of that era concluded that higher rates of HLA sensitization are found in patients with a history of transfusion compared with those without, and that there is a neutral to negative effect on allograft outcome after sensitization by transfusion (79).
Pregnancy
Both full-term pregnancy and spontaneous miscarriage induce alloantibodies (80). Anti-paternal alloantibodies appear around or after the 28th week of gestation during pregnancy (80). Sensitized women have higher rates of parity (pregnancy) compared with non-sensitized patients (81). One-third to half of women develop HLA immunization after delivery during their first pregnancy (73, 74, 82), and immunization frequency increases with parity (82). Antibodies to HLA class I were slightly more frequent than those to class II, although both were produced.
Female patients receiving kidney allografts from their male partners or their offspring experienced higher rejection rates (83), pointing to increased immunological risk in women upon re-exposure to paternal antigens on the allograft. Generally, antibodies induced by pregnancy declined in the circulation over time. Even so, post-transplant antibody increases occurred in the pregnancy cohort even decades after the last pregnancy (84).
Transplantation
Transplantation itself is a significant alloimmunizing event (81), and previously non-donor sensitized solid organ transplant recipients develop de novo donor-specific HLA antibodies at a rate of about 8–10% in the first year for liver and renal transplants (85, 86), and 15–25% of renal and cardiac transplant by 10 years post-transplant (85, 87, 88). Removal (transplantectomy) of failed renal-allografts appears to stimulate a large increase in circulating DSA (89), whether from increased immune activation in response to surgical trauma, removal of the antigen “sink” provided by the allograft and/or immunosuppression, is unclear.
When evaluating a patient for re-transplantation, it is important to consider the presence of donor-specific alloantibodies that were formed via sensitization to the first allograft in relationship to the donor antigens carried by the second potential donor – the so-called “repeat mismatches.” Repeat mismatched donor HLA antigens against which a recipient has preformed alloantibody, particularly to HLA-DR, were found to have a detrimental effect on renal-allograft survival (90–92). While Farney et al. did not uncover a deleterious effect on graft survival of retransplantation with donors who shared mismatches in the presence of alloantibodies with prior donors (93), a more recent study found that re-exposure to mismatched HLA class I antigens increased the risk of early graft loss in renal transplant recipients (94). Typically, repeat HLA mismatches in donors against which a recipient has made antibodies are avoided by transplant programs (95).
Allografts are also used for vascular reconstruction in many forms of congenital heart disease and have been demonstrated to cause persistent sensitization to HLA antigens (96). These findings have implications for those in whom heart transplant is considered late in the clinical course.
Ventricular Assist Devices
Ventricular Assist Devices (VADs) are associated with increased production of HLA antibodies. The current paradigm is that the VAD provides a continual antigenic or inflammatory stimulus that promotes generation of de novo HLA antibodies when patients are exposed to blood and/or platelet transfusions or heightens existing HLA antibody levels. In a recent study, we observed that patients implanted with the older pulsatile VAD (BiVad) showed increased HLA sensitization vs. patients implanted with the Heart MateII Axial VAD suggesting the older, pulsatile devices had greater sensitization potential (97).
Natural Antibodies
It has been suggested that HLA antibodies may be formed by means other than the typical routes of sensitization discussed above. Antibodies to HLA are found in non-transfused males at a rate of nearly 50% in one study (98) and often react with a restricted subset of HLA antigens that are uncommon in the general population. There is some evidence pointing to cross-reactivity of pathogens (especially viral) with HLA by T cells (99–102). The abundant viral-specific memory T cell repertoire may, therefore, contribute to alloantibody production. Viral cross-reactivity with HLA may also occur at the protein level. For example, antibodies to HIV-1 may recognize HLA (103), and immunization with the HepB vaccine caused HLA antibody positivity in approximately half of previously negative, healthy adults 1 month after vaccination (104).
It has been proposed that “natural antibodies” against the non-classical HLA-E molecule can cross-react with HLA class I molecules (105). Alternatively, it is possible that antibodies detected are false-positive reactions with denatured antigen, a known limitation of the single-antigen bead assay commonly used to identify HLA antibodies in the sera (106–108). Additional evidence shows these antibodies do not often react with native antigen on cells (108, 109), and the clinical significance and durability of such natural antibody responses remain unclear (110, 111).
Non-HLA Antibodies
Non-HLA antibodies can be directed toward either alloantigens, such as the major histocompatibility complex class I chain-related gene A (MICA) or B (MICB), or tissue-specific autoantigens, such as vimentin, cardiac myosin (CM), collagen V (Col V), agrin and angiotensin II receptor type I (AT1R). Additional non-HLA targets recently identified by Jackson et al. include anti-endothelial cell targets, including endoglin, EGF-like repeats, Fms-like tyrosine kinase-3 ligand, and ICAM-4. The principle antigenic targets of non-HLA antibodies are expressed on cells of the allograft, including endothelium and epithelium. Therefore, donor cells are in direct contact with the recipients circulating peripheral blood lymphocytes, and have been shown to be the major immunological targets for the pathogenesis of allograft rejection. Prevalence of anti-endothelial cell antibodies (AECA) among renal recipients was nearly one quarter in pre-transplant sera (112). AECAs correlated with post-transplant HLA DSA and AMR. Sun et al. observed that anti-endothelial cell antibodies were found in patients pre-transplant, but that they did not correlate with outcome or rejection; by contrast, de novo development of AECAs was significantly associated with early and severe acute rejection, but not C4d (113). AECA were implicated as the cause of acute antibody-mediated rejection (AMR) in 30% of heart transplant recipients without DSA to HLA (114).
Mechanisms of Graft Damage by HLA Antibodies
High-titered pre-transplant DSA directed against HLA class I antigens can cause catastrophic hyperacute rejection and immediate graft loss (115), whereas high titer class II DSA mediate graft rejection 2–4 days after transplant, upon re-expression of HLA class II antigens on the endothelium of the allograft (116–118). By contrast, pre-transplant DSA of low titer are often associated with development of acute AMR during the first 3 months after transplantation and/or lower long-term graft survival (119). If left untreated, patients with AMR are at risk of graft loss and/or markedly shortened overall graft survival time. Patients producing de novo anti-HLA antibodies against their donor following transplantation are also at increased risk of graft failure unless their response can be controlled or abrogated (120).
There are three major effector functions carried out by antibodies that can impact the graft. First, bivalent IgG can dimerize or crosslink its target upon binding. Collective studies indicate that IgG binding to HLA agonistically crosslinks HLA molecules and triggers downstream activation of the target cells. Second, antibodies can activate the classical complement cascade through binding to the Fc fragment to trigger production of potent anaphylatoxins, chemoattractants, opsonins, and cell-damaging factors. Thirdly, HLA IgG bound to target cells can engage Fc receptors on myeloid and lymphoid cells, to employ a host of Fc receptor-mediated effector functions, including antibody-dependent cell cytotoxicity, antibody-dependent phagocytosis, and augment recruitment. These effector functions work in concert, and there is substantial interplay between them, as we will discuss below.
HLA Antibody-Induced Signaling in Graft Vascular Cells
Antibodies are capable of agonistically crosslinking their protein targets at the cell surface [recently reviewed in Ref. (121)]. In vascular cells, crosslinking of HLA induces intracellular signaling cascades that lead to functional changes, such as increased cell migration, cytoskeletal rearrangement, growth and proliferation, endothelial activation and exocytosis, and increased recruitment of leukocytes. These functional changes parallel the histological findings in clinical AMR, including microvascular inflammation, endothelial dysfunction, expansion of the neointima, and infiltration of mononuclear cells (Table 2).
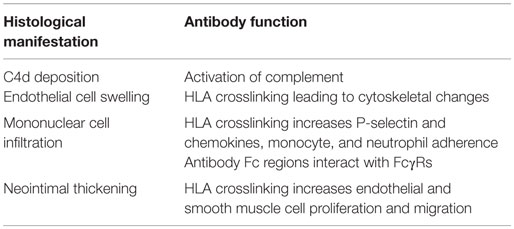
Table 2. Function of HLA antibodies leading to histological manifestations of AMR.
HLA class I and II do not have intrinsic kinase activity and, therefore, partner with other proteins to transduce intracellular signals. Ligation of HLA class I with antibodies increases its association with integrin β4 which, in turn, activates intracellular signaling cascades (122). Integrin β4 is an important cell adhesion protein regulating cell adhesion, proliferation, migration, and survival. Blockade of integrin β4 impairs HLA antibody-stimulated signal transduction. Protein(s) that partner with HLA class II to transduce signaling are not yet reported. However, ligation of either HLA class I or class II with antibodies activates mammalian target of rapamycin (mTOR) signaling through the SRC/FAK–PI3K–AKT pathway and increases Akt-dependent cell survival signaling, through upregulation of Bcl-2 and HO-1 (123–125). Furthermore, activation of key signaling proteins in endothelium, including S6K, S6RP, and ERK, demonstrated in human cardiac allografts with AMR and in murine models of MHC antibody-mediated injury (126–128) suggests pro-survival signals that may increase endothelial persistence, stress fiber formation (129, 130), and resistance to complement-induced cell death (125), contributing to neointimal formation during chronic rejection. Additional work by Galvani et al. points to a direct effect of MHC antibodies on smooth muscle cells. Crosslinking of HLA I with antibodies provokes mitogenic signaling through matrix metalloproteinases in vitro, and contributes to neointimal thickening of human arterial grafts in vivo in murine recipients (131, 132). An additional feature of alloantibody crosslinking of HLA is increased intracellular calcium levels, leading to exocytosis of endothelial Weibel–Palade body vesicles and increased cell surface P-selectin (133–135). P-selectin captures neutrophils and monocytes (133, 134), facilitating recruitment of immune cells into the allograft.
Complement
The complement system is an ancient form of innate immunity that relies on proteolytic cleavage of active components. Complement proteins are always present in the circulation, but become rapidly activated upon exposure to target molecules. There are three main pathways of complement, which differ by the activating stimulus. The lectin pathway becomes activated upon recognition by mannose binding lectin (MBL) of pathogen-specific glycan residues on the surface of bacteria, fungi, and viruses. The alternative pathway of complement is initiated at the surface of non-host cells due to the presence of such factors as lipopolysaccharides on Gram-negative bacteria, zymosans on fungi and yeast, and other pathogen-associated molecules. Complexed human immunoglobulin has also been shown to activate the alternative pathway. The classical complement pathway is initiated exclusively by antigen-bound antibody through binding of the Fc portion of certain isotypes and subclasses to C1q. All of these pathways rely on sequential enzymatic reactions that produce active split products involved in inflammation, and all of these pathways converge on the terminal component C5.
Activation of complement by antibodies was one of the earliest methods used to detect donor-specific HLA antibodies, and positive cytotoxic crossmatch is still often considered to be a contraindication to transplant, as antibodies detected by this method can mediate hyperacute rejection of solid organ transplants (115). Although the end result of complement activation, namely deposition of MAC and cell cytotoxicity, has been a focus, it is now thought to be a rare event (136). Endothelial cells express complement regulatory proteins (CD55/DAF, CD59, Crry) that antagonize complement activation by inactivating split products. C3d and C4d are generated by such inhibitory receptors and mark early complement activation. Attention has turned to the activity and predictive value of other complement proteins. Products of complement activation, in particular C4d, have proven histological utility in detecting donor-specific antibody bound to the graft (137, 138). Other split products, including C4a, C3a, and C3b, are potent inflammatory signals that promote immune cell recruitment and opsonization.
FcγR-Bearing Immune Cells
Many cells express surface receptors that can interact with the constant region heavy chain (Fc) of antibodies. The human Fc receptor system consists of several classes that can bind to IgG (FcγR, CD64, CD32, CD16), IgA (FcαR, CD89), and IgE (FcϵR, CD23). The human receptor for IgM (FcμR) had been elusive until relatively recently (139). Fc receptors serve to bridge the humoral and cellular arms of the immune system, and provide innate immune cells with a target, and are critical for a variety of functions, including antibody-dependent cell-mediated phagocytosis (ADCP), antibody-dependent cell-mediated cytotoxicity (ADCC), cell–cell tethering and degranulation.
Given that IgG is thought to be the most clinically relevant isotype of HLA antibodies, we will focus on Fc-gamma receptors (FcγR) that bind to this isotype of immunoglobulins. FcγRs are expressed broadly in both the myeloid and lymphoid compartments. There are three major classes of FcγRs, FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16). FcγRII and FcγRIII are further composed of several functionally disparate isoforms, most of which are dimorphic in the human population (140, 141). Polymorphisms in human FcγRs influence susceptibility to autoimmune disease and response to anti-tumor therapeutics (142–146), and may also influence susceptibility of transplant recipients to rejection (147, 148), although a thorough evaluation of the role of different FcγR alleles in antibody-mediated transplant rejection has not been reported.
Due to their lower affinity, the majority of FcγRs do not bind monomeric IgG very efficiently. Only the high-affinity FcγRI (CD64) is the exception, and cells with this receptor have been shown to carry monomeric IgG in circulation. FcγRs do bind to antigen-associated IgG, however, such as in immune complexes or immobilized on a (cell) surface. Once bound, FcγRs become crosslinked as they physically colocalize at high antibody-antigen density. This promotes intracellular signaling in the FcγR-bearing cell leading to activation and maturation, and mediates effector functions such as phagocytosis or cytotoxicity.
The relevance of FcγR-bearing innate immune cells to antibody-mediated graft injury is reflected in the diagnostic criteria and histological manifestations of AMR. For example, infiltration of CD68+ macrophages is included in the AMR diagnostic criteria in cardiac transplantation (149), where macrophage staining is found intravascularly (150, 151). Indeed, increased macrophage burden is correlative with worse prognosis (152). Although not currently included in the AMR diagnostic criteria for renal transplantation, macrophage infiltration during rejection is also predictive of worse outcome in kidney allografts (153, 154). Our recent studies are consistent with these clinical findings and show that monocyte recruitment to HLA-Ab-activated endothelium is mediated by HLA-induced Weibel–Palade exocytosis and P-selectin expression (134). Blockade of P-selectin potently inhibited leukocyte recruitment to the allograft during AMR underscoring its therapeutic potential (134). Furthermore, HLA-Ab augmented monocyte recruitment by the interaction of monocyte FcγRs with the Fc portion of the HLA-Abs (135). This interaction was IgG subclass dependent and influenced by monocyte FcγRIIa allelic variants. Monocytes from donors carrying the high-affinity FcγRIIa-H131 allele had greater FcγR-dependent adhesion to ECs activated with HLA-Abs of both IgG1 and IgG2 subclasses compared with monocytes expressing only FcγRIIa-R131. These results are clinically relevant and suggest that recipients producing DSA and carrying high-affinity FcγR alleles may be pre-disposed to acute AMR accompanied by increased monocyte infiltration.
Summary
Taken together, antibodies to donor proteins, including HLA, can cause graft damage through three major mechanisms, including direct activation of endothelial, smooth muscle, and epithelial cells to promote proliferation and inflammation; activation of the complement system to generate inflammatory split products; and engagement of FcγRs on NK cells, monocytes, and neutrophils.
Conclusion
Allorecognition by the humoral immune system results in formation of antibodies to HLA and a variety of non-HLA proteins, and occurs after exposure to non-self tissues through pregnancy, transfusion, or transplantation. Alloantibody formation is dependent upon T cell interactions and is primarily driven by indirect allorecognition by T cells. In addition, “natural” antibodies or anti-viral antibodies may cross-react with HLA, although the clinical significance of such antibodies is not clear. Antibodies to donor HLA mediate allograft injury through Fc-dependent as well as Fc-independent mechanisms, which closely reflect the diagnostic criteria for AMR. Non-HLA antibodies can be against polymorphic proteins, such as MICA, or against autoantibodies, and also associate with worse graft outcome, although their etiology is less clear than for HLA DSA.
Author Contributions
MH was responsible for outlining, research, and writing of the manuscript. NV was responsible for outlining, research, and writing of the manuscript. ER reviewed and critically examined the manuscript and shaped its final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
NV is supported by a Histocompatibility Laboratory Director Trainee Fellowship from the UCLA Department of Pathology. ER is supported by NIH RO1 AI042819.
References
1. Steele DJ, Laufer TM, Smiley ST, Ando Y, Grusby MJ, Glimcher LH, et al. Two levels of help for B cell alloantibody production. J Exp Med (1996) 183(2):699–703. doi: 10.1084/jem.183.2.699
2. Afzali B, Lombardi G, Lechler RI. Pathways of major histocompatibility complex allorecognition. Curr Opin Organ Transplant (2008) 13(4):438–44. doi:10.1097/MOT.0b013e328309ee31
3. Zhang JQ HM, Valenzuela NM, Zhang X, Lan JH, Cecka M, Reed EF. Histocompatibility and Immunogenetics for Solid Organ Transplantation. London: Springer Science+Business Media (2015).
4. IMGT/HLA. Available from: http://www.ebi.ac.uk/ipd/imgt/hla/stats.html.
5. Taylor AL, Negus SL, Negus M, Bolton EM, Bradley JA, Pettigrew GJ. Pathways of helper CD4 T cell allorecognition in generating alloantibody and CD8 T cell alloimmunity. Transplantation (2007) 83(7):931–7. doi:10.1097/01.tp.0000257960.07783.e3
6. Conlon TM, Saeb-Parsy K, Cole JL, Motallebzadeh R, Qureshi MS, Rehakova S, et al. Germinal center alloantibody responses are mediated exclusively by indirect-pathway CD4 T follicular helper cells. J Immunol (2012) 188(6):2643–52. doi:10.4049/jimmunol.1102830
7. Hornick PI, Mason PD, Yacoub MH, Rose ML, Batchelor R, Lechler RI. Assessment of the contribution that direct allorecognition makes to the progression of chronic cardiac transplant rejection in humans. Circulation (1998) 97(13):1257–63. doi:10.1161/01.CIR.97.13.1257
8. Benichou G, Valujskikh A, Heeger PS. Contributions of direct and indirect T cell alloreactivity during allograft rejection in mice. J Immunol (1999) 162(1):352–8.
9. Hornick PI, Mason PD, Baker RJ, Hernandez-Fuentes M, Frasca L, Lombardi G, et al. Significant frequencies of T cells with indirect anti-donor specificity in heart graft recipients with chronic rejection. Circulation (2000) 101(20):2405–10. doi:10.1161/01.CIR.101.20.2405
10. Baker RJ, Hernandez-Fuentes MP, Brookes PA, Chaudhry AN, Cook HT, Lechler RI. Loss of direct and maintenance of indirect alloresponses in renal allograft recipients: implications for the pathogenesis of chronic allograft nephropathy. J Immunol (2001) 167(12):7199–206. doi:10.4049/jimmunol.167.12.7199
11. Richters CD, van Gelderop E, du Pont JS, Hoekstra MJ, Kreis RW, Kamperdijk EW. Migration of dendritic cells to the draining lymph node after allogeneic or congeneic rat skin transplantation. Transplantation (1999) 67(6):828–32. doi:10.1097/00007890-199903270-00008
12. Sandner SE, Salama AD, Houser SL, Palmer E, Turka LA, Sayegh MH. New TCR transgenic model for tracking allospecific CD4 T-cell activation and tolerance in vivo. Am J Transplant (2003) 3(10):1242–50. doi:10.1046/j.1600-6143.2003.00220.X
13. Kaminski E, Hows J, Man S, Brookes P, Mackinnon S, Hughes T, et al. Prediction of graft versus host disease by frequency analysis of cytotoxic T cells after unrelated donor bone marrow transplantation. Transplantation (1989) 48(4):608–13.
14. Schwarer AP, Jiang YZ, Deacock S, Brookes PA, Barrett AJ, Goldman JM, et al. Comparison of helper and cytotoxic antirecipient T cell frequencies in unrelated bone marrow transplantation. Transplantation (1994) 58(11):1198–203. doi:10.1097/00007890-199412270-00011
15. Lechler RI, Batchelor JR. Immunogenicity of retransplanted rat kidney allografts. Effect of inducing chimerism in the first recipient and quantitative studies on immunosuppression of the second recipient. J Exp Med (1982) 156(6):1835–41. doi:10.1084/jem.156.6.1835
16. Braun MY, McCormack A, Webb G, Batchelor JR. Mediation of acute but not chronic rejection of MHC-incompatible rat kidney grafts by alloreactive CD4 T cells activated by the direct pathway of sensitization. Transplantation (1993) 55(1):177–82. doi:10.1097/00007890-199301000-00033
17. Lee RS, Grusby MJ, Glimcher LH, Winn HJ, Auchincloss H Jr. Indirect recognition by helper cells can induce donor-specific cytotoxic T lymphocytes in vivo. J Exp Med (1994) 179(3):865–72. doi:10.1084/jem.179.3.865
18. Wise MP, Bemelman F, Cobbold SP, Waldmann H. Linked suppression of skin graft rejection can operate through indirect recognition. J Immunol (1998) 161(11):5813–6.
19. Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med (1997) 186(1):65–70. doi:10.1084/jem.186.1.65
20. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature (1998) 393(6684):474–8. doi:10.1038/30989
21. Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature (1998) 393(6684):480–3. doi:10.1038/31002
22. Smyth LA, Afzali B, Tsang J, Lombardi G, Lechler RI. Intercellular transfer of MHC and immunological molecules: molecular mechanisms and biological significance. Am J Transplant (2007) 7(6):1442–9. doi:10.1111/j.1600-6143.2007.01816.x
23. Herrera OB, Golshayan D, Tibbott R, Salcido Ochoa F, James MJ, Marelli-Berg FM, et al. A novel pathway of alloantigen presentation by dendritic cells. J Immunol (2004) 173(8):4828–37. doi:10.4049/jimmunol.173.8.4828
24. Smyth LA, Herrera OB, Golshayan D, Lombardi G, Lechler RI. A novel pathway of antigen presentation by dendritic and endothelial cells: implications for allorecognition and infectious diseases. Transplantation (2006) 82(1 Suppl):S15–8. doi:10.1097/01.tp.0000231347.06149.ca
25. Curry AJ, Pettigrew GJ, Negus MC, Easterfield AJ, Young JL, Bolton EM, et al. Dendritic cells internalise and re-present conformationally intact soluble MHC class I alloantigen for generation of alloantibody. Eur J Immunol (2007) 37(3):696–705. doi:10.1002/eji.200636543
26. McHeyzer-Williams LJ, Pelletier N, Mark L, Fazilleau N, McHeyzer-Williams MG. Follicular helper T cells as cognate regulators of B cell immunity. Curr Opin Immunol (2009) 21(3):266–73. doi:10.1016/j.coi.2009.05.010
27. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature (2008) 455(7214):764–9. doi:10.1038/nature07186
28. Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, et al. Optimal germinal center responses require a multistage T cell: B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity (2010) 32(2):253–65. doi:10.1016/j.immuni.2010.01.010
29. Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity (2007) 27(2):190–202. doi:10.1016/j.immuni.2007.07.009
30. Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science (2007) 315(5811):528–31. doi:10.1126/science.1136736
31. Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, et al. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med (2006) 203(11):2419–24. doi:10.1084/jem.20061254
32. Maruyama M, Lam KP, Rajewsky K. Memory B-cell persistence is independent of persisting immunizing antigen. Nature (2000) 407(6804):636–42. doi:10.1038/35036600
33. Snanoudj R, Claas FH, Heidt S, Legendre C, Chatenoud L, Candon S. Restricted specificity of peripheral alloreactive memory B cells in HLA-sensitized patients awaiting a kidney transplant. Kidney Int (2015) 87(6):1230–40. doi:10.1038/ki.2014.390
34. Scornik JC, Meier-Kriesche HU. Blood transfusions in organ transplant patients: mechanisms of sensitization and implications for prevention. Am J Transplant (2011) 11(9):1785–91. doi:10.1111/j.1600-6143.2011.03705.x
35. Bestard O, Crespo E, Stein M, Lucia M, Roelen DL, de Vaal YJ, et al. Cross-validation of IFN-gamma Elispot assay for measuring alloreactive memory/effector T cell responses in renal transplant recipients. Am J Transplant (2013) 13(7):1880–90. doi:10.1111/ajt.12285
36. Hricik DE, Rodriguez V, Riley J, Bryan K, Tary-Lehmann M, Greenspan N, et al. Enzyme linked immunosorbent spot (ELISPOT) assay for interferon-gamma independently predicts renal function in kidney transplant recipients. Am J Transplant (2003) 3(7):878–84. doi:10.1034/j.1600-6143.2003.00132.x
37. de Haan A, van den Berg AP, Hepkema BG, van Dijk E, Haagsma EB, The TH, et al. Donor-specific hyporeactivity after liver transplantation: prominent decreases in donor-specific cytotoxic T lymphocyte precursor frequencies independent of changes in helper T lymphocyte precursor frequencies or suppressor cell activity. Transplantation (1998) 66(4):516–22. doi:10.1097/00007890-199808270-00017
38. Vella JP, Spadafora-Ferreira M, Murphy B, Alexander SI, Harmon W, Carpenter CB, et al. Indirect allorecognition of major histocompatibility complex allopeptides in human renal transplant recipients with chronic graft dysfunction. Transplantation (1997) 64(6):795–800. doi:10.1097/00007890-199712270-00033
39. Liu Z, Colovai AI, Tugulea S, Reed EF, Fisher PE, Mancini D, et al. Indirect recognition of donor HLA-DR peptides in organ allograft rejection. J Clin Invest (1996) 98(5):1150–7. doi:10.1172/JCI118898
40. Jackson KJ, Wang Y, Collins AM. Human immunoglobulin classes and subclasses show variability in VDJ gene mutation levels. Immunol Cell Biol (2014) 92(8):729–33. doi:10.1038/icb.2014.44
41. van Zelm MC. B cells take their time: sequential IgG class switching over the course of an immune response? Immunol Cell Biol (2014) 92(8):645–6. doi:10.1038/icb.2014.48
42. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol (2014) 5:520. doi:10.3389/fimmu.2014.00520
43. Devey ME, Bleasdale-Barr KM, Bird P, Amlot PL. Antibodies of different human IgG subclasses show distinct patterns of affinity maturation after immunization with keyhole limpet haemocyanin. Immunology (1990) 70(2):168–74.
44. Duquesnoy RJ. A structurally based approach to determine HLA compatibility at the humoral immune level. Hum Immunol (2006) 67(11):847–62. doi:10.1016/j.humimm.2006.08.073
45. Hertz T, Nolan D, James I, John M, Gaudieri S, Phillips E, et al. Mapping the landscape of host-pathogen coevolution: HLA class I binding and its relationship with evolutionary conservation in human and viral proteins. J Virol (2011) 85(3):1310–21. doi:10.1128/JVI.01966-10
46. Mulder A, Eijsink C, Kester MG, Franke ME, Kardol MJ, Heemskerk MH, et al. Impact of peptides on the recognition of HLA class I molecules by human HLA antibodies. J Immunol (2005) 175(9):5950–7. doi:10.4049/jimmunol.175.9.5950
47. Nakayama S, Kawaguchi G, Karaki S, Nagao T, Uchida H, Kashiwase K, et al. Effect of single amino acid substitution at residue 167 of HLA-B51 on binding of antibodies and recognition of T cells. Hum Immunol (1994) 39(3):211–9. doi:10.1016/0198-8859(94)90262-3
48. Rodey GE, Neylan JF, Whelchel JD, Revels KW, Bray RA. Epitope specificity of HLA class I alloantibodies. I. Frequency analysis of antibodies to private versus public specificities in potential transplant recipients. Hum Immunol (1994) 39(4):272–80. doi:10.1016/0198-8859(94)90270-4
49. McKenzie LM, Pecon-Slattery J, Carrington M, O’Brien SJ. Taxonomic hierarchy of HLA class I allele sequences. Genes Immun (1999) 1(2):120–9. doi:10.1038/sj.gene.6363648
50. Wade JA, Hurley CK, Takemoto SK, Thompson J, Davies SM, Fuller TC, et al. HLA mismatching within or outside of cross-reactive groups (CREGs) is associated with similar outcomes after unrelated hematopoietic stem cell transplantation. Blood (2007) 109(9):4064–70. doi:10.1182/blood-2006-06-032193
51. Marrari M, Mostecki J, Mulder A, Claas F, Balazs I, Duquesnoy RJ. Human monoclonal antibody reactivity with human leukocyte antigen class I epitopes defined by pairs of mismatched eplets and self-eplets. Transplantation (2010) 90(12):1468–72. doi:10.1097/TP.0b013e3182007b74
52. Lomago J, Jelenik L, Zern D, Howe J, Martell J, Zeevi A, et al. How did a patient who types for HLA-B*4403 develop antibodies that react with HLA-B*4402? Hum Immunol (2010) 71(2):176–8. doi:10.1016/j.humimm.2009.11.013
53. Billen EV, Christiaans MH, Doxiadis II, Voorter CE, van den Berg-Loonen EM. HLA-DP antibodies before and after renal transplantation. Tissue Antigens (2010) 75(3):278–85. doi:10.1111/j.1399-0039.2009.01428.x
54. Norman PJ, Hollenbach JA, Nemat-Gorgani N, Guethlein LA, Hilton HG, Pando MJ, et al. Co-evolution of human leukocyte antigen (HLA) class I ligands with killer-cell immunoglobulin-like receptors (KIR) in a genetically diverse population of sub-Saharan Africans. PLoS Genet (2013) 9(10):e1003938. doi:10.1371/journal.pgen.1003938
55. Parham P, McLean J. Characterization, evolution, and molecular basis of a polymorphic antigenic determinant shared by HLA-A and B products. Hum Immunol (1980) 1(2):131–9. doi:10.1016/0198-8859(80)90100-7
56. Tambur AR, Buckingham M, McDonald L, Luo X. Development of donor-specific and non-donor-specific HLA-DP antibodies post-transplant: the role of epitope sharing and epitope matching. Clin Transpl (2006):399–404.
57. Claas FH, Rahmel A, Doxiadis II. Enhanced kidney allocation to highly sensitized patients by the acceptable mismatch program. Transplantation (2009) 88(4):447–52. doi:10.1097/TP.0b013e3181b04a5f
58. Wiebe C, Pochinco D, Blydt-Hansen TD, Ho J, Birk PE, Karpinski M, et al. Class II HLA epitope matching-A strategy to minimize de novo donor-specific antibody development and improve outcomes. Am J Transplant (2013) 13(12):3114–22. doi:10.1111/ajt.12478
59. Laux G, Mansmann U, Deufel A, Opelz G, Mytilineos J. A new epitope-based HLA-DPB matching approach for cadaver kidney retransplants. Transplantation (2003) 75(9):1527–32. doi:10.1097/01.TP.0000061759.57702.8A
60. Muro M, Gonzalez-Soriano MJ, Salgado G, Lopez R, Boix F, Lopez M, et al. Specific “intra-allele” and “intra-broad antigen” human leukocyte antigen alloantibodies in kidney graft transplantation. Hum Immunol (2010) 71(9):857–60. doi:10.1016/j.humimm.2010.05.018
61. Tambur AR, Leventhal JR, Friedewald JJ, Ramon DS. The complexity of human leukocyte antigen (HLA)-DQ antibodies and its effect on virtual crossmatching. Transplantation (2010) 90(10):1117–24. doi:10.1097/00007890-201007272-00536
62. Tambur AR, Leventhal JR, Zitzner JR, Walsh RC, Friedewald JJ. The DQ barrier: improving organ allocation equity using HLA-DQ information. Transplantation (2013) 95(4):635–40. doi:10.1097/TP.0b013e318277b30b
63. McCutcheon JA, Gumperz J, Smith KD, Lutz CT, Parham P. Low HLA-C expression at cell surfaces correlates with increased turnover of heavy chain mRNA. J Exp Med (1995) 181(6):2085–95. doi:10.1084/jem.181.6.2085
64. Apps R, Meng Z, Del Prete GQ, Lifson JD, Zhou M, Carrington M. Relative expression levels of the HLA class-I proteins in normal and HIV-infected cells. J Immunol (2015) 194(8):3594–600. doi:10.4049/jimmunol.1403234
65. Muczynski KA, Ekle DM, Coder DM, Anderson SK. Normal human kidney HLA-DR-expressing renal microvascular endothelial cells: characterization, isolation, and regulation of MHC class II expression. J Am Soc Nephrol (2003) 14(5):1336–48. doi:10.1097/01.ASN.0000061778.08085.9F
66. Dellgren C, Nehlin JO, Barington T. Cell surface expression level variation between two common Human Leukocyte Antigen alleles, HLA-A2 and HLA-B8, is dependent on the structure of the C terminal part of the alpha 2 and the alpha 3 domains. PLoS One (2015) 10(8):e0135385. doi:10.1371/journal.pone.0135385
67. Ramsuran V, Kulkarni S, O’Huigin C, Yuki Y, Augusto DG, Gao X, et al. Epigenetic regulation of differential HLA-A allelic expression levels. Hum Mol Genet (2015) 24(15):4268–75. doi:10.1093/hmg/ddv158
68. Giacomini P, Beretta A, Nicotra MR, Ciccarelli G, Martayan A, Cerboni C, et al. HLA-C heavy chains free of beta2-microglobulin: distribution in normal tissues and neoplastic lesions of non-lymphoid origin and interferon-gamma responsiveness. Tissue Antigens (1997) 50(6):555–66. doi:10.1111/j.1399-0039.1997.tb02913.x
69. Sibilio L, Martayan A, Setini A, Lo Monaco E, Tremante E, Butler RH, et al. A single bottleneck in HLA-C assembly. J Biol Chem (2008) 283(3):1267–74. doi:10.1074/jbc.M708068200
70. Kulkarni S, Savan R, Qi Y, Gao X, Yuki Y, Bass SE, et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature (2011) 472(7344):495–8. doi:10.1038/nature09914
71. Bryan CF, Luger AM, Smith JL, Warady BA, Wakefield M, Schadde E, et al. Sharing kidneys across donor-service area boundaries with sensitized candidates can be influenced by HLA C. Clin Transplant (2010) 24(1):56–61. doi:10.1111/j.1399-0012.2009.01167.x
72. Triulzi DJ, Kleinman S, Kakaiya RM, Busch MP, Norris PJ, Steele WR, et al. The effect of previous pregnancy and transfusion on HLA alloimmunization in blood donors: implications for a transfusion-related acute lung injury risk reduction strategy. Transfusion (2009) 49(9):1825–35. doi:10.1111/j.1537-2995.2009.02206.x
73. De Clippel D, Baeten M, Torfs A, Emonds MP, Feys HB, Compernolle V, et al. Screening for HLA antibodies in plateletpheresis donors with a history of transfusion or pregnancy. Transfusion (2014) 54(12):3036–42. doi:10.1111/trf.12727
74. Hyun J, Park KD, Yoo Y, Lee B, Han BY, Song EY, et al. Effects of different sensitization events on HLA alloimmunization in solid organ transplantation patients. Transplant Proc (2012) 44(1):222–5. doi:10.1016/j.transproceed.2011.12.049
75. Salvatierra O Jr, Vincenti F, Amend W, Potter D, Iwaki Y, Opelz G, et al. Deliberate donor-specific blood transfusions prior to living related renal transplantation. A new approach. Ann Surg (1980) 192(4):543–52. doi:10.1097/00000658-198010000-00012
76. Pfaff WW, Fennell RS, Howard RJ, Ireland JF, Scornik JC. Planned random donor blood transfusion in preparation for transplantation. Sensitization and graft survival. Transplantation (1984) 38(6):701–3. doi:10.1097/00007890-198412000-00030
77. Josien R, Heslan M, Brouard S, Soulillou JP, Cuturi MC. Critical requirement for graft passenger leukocytes in allograft tolerance induced by donor blood transfusion. Blood (1998) 92(12):4539–44.
78. Fuller TC, Delmonico FL, Cosimi B, Huggins CE, King M, Russell PS. Impact of blood transfusion on renal transplantation. Ann Surg (1978) 187(2):211–8. doi:10.1097/00000658-197802000-00020
79. Scornik JC, Bromberg JS, Norman DJ, Bhanderi M, Gitlin M, Petersen J. An update on the impact of pre-transplant transfusions and allosensitization on time to renal transplant and on allograft survival. BMC Nephrol (2013) 14:217. doi:10.1186/1471-2369-14-217
80. Regan L, Braude PR, Hill DP. A prospective study of the incidence, time of appearance and significance of anti-paternal lymphocytotoxic antibodies in human pregnancy. Hum Reprod (1991) 6(2):294–8.
81. Dunn TB, Noreen H, Gillingham K, Maurer D, Ozturk OG, Pruett TL, et al. Revisiting traditional risk factors for rejection and graft loss after kidney transplantation. Am J Transplant (2011) 11(10):2132–43. doi:10.1111/j.1600-6143.2011.03640.x
82. Masson E, Vidal C, Deschamps M, Bongain S, Thevenin C, Dupont I, et al. Incidence and risk factors of anti-HLA immunization after pregnancy. Hum Immunol (2013) 74(8):946–51. doi:10.1016/j.humimm.2013.04.025
83. Higgins R, Lowe D, Hathaway M, Williams C, Lam FT, Kashi H, et al. Human leukocyte antigen antibody-incompatible renal transplantation: excellent medium-term outcomes with negative cytotoxic crossmatch. Transplantation (2011) 92(8):900–6. doi:10.1097/TP.0b013e31822dc38d
84. Higgins R, Lowe D, Daga S, Hathaway M, Williams C, Lam FT, et al. Pregnancy-induced HLA antibodies respond more vigorously after renal transplantation than antibodies induced by prior transplantation. Hum Immunol (2015) 76(8):546–52. doi:10.1016/j.humimm.2015.01.012
85. Everly MJ, Rebellato LM, Haisch CE, Ozawa M, Parker K, Briley KP, et al. Incidence and impact of de novo donor-specific alloantibody in primary renal allografts. Transplantation (2013) 95(3):410–7. doi:10.1097/TP.0b013e31827d62e3
86. Kaneku H, O’Leary JG, Banuelos N, Jennings LW, Susskind BM, Klintmalm GB, et al. De novo donor-specific HLA antibodies decrease patient and graft survival in liver transplant recipients. Am J Transplant (2013) 13(6):1541–8. doi:10.1111/ajt.12212
87. Smith JD, Banner NR, Hamour M, Ozawa M, Goh A, Robinson D, et al. De novo donor HLA-specific antibodies after heart transplantation are an independent predictor of poor patient survival. Am J Transplant (2011) 11(2):312–9. doi:10.1111/j.1600-6143.2010.03383.x
88. Wiebe C, Gibson W, Blydt-Hansen TD, Karpinski M, Ho J, Storsley LJ, et al. Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant (2012) 12(5):1157–67. doi:10.1111/j.1600-6143.2012.04013.x
89. Billen EV, Christiaans MH, Lee J, van den Berg-Loonen EM. Donor-directed HLA antibodies before and after transplantectomy detected by the luminex single antigen assay. Transplantation (2009) 87(4):563–9. doi:10.1097/TP.0b013e3181949e37
90. Cecka JM, Terasaki PI. Repeating HLA antigen mismatches in renal retransplants – a second class mistake? Transplantation (1994) 57(4):515–9. doi:10.1097/00007890-199402000-00007
91. Doxiadis II, de Lange P, D’Amaro J, de Meester J, Schreuder GM, Claas FH. Repeated HLA mismatches in cadaveric renal transplantation: is it safe to transplant? Transplant Proc (1997) 29(1–2):1408–9. doi:10.1016/S0041-1345(96)00612-4
92. Fuller A, Profaizer T, Roberts L, Fuller TC. Repeat donor HLA-DR mismatches in renal transplantation: is the increased failure rate caused by noncytotoxic HLA-DR alloantibodies? Transplantation (1999) 68(4):589–91. doi:10.1097/00007890-199907270-00002
93. Farney AC, Matas AJ, Noreen HJ, Reinsmoen N, Segall M, Schmidt WJ, et al. Does re-exposure to mismatched HLA antigens decrease renal re-transplant allograft survival? Clin Transplant (1996) 10(2):147–56.
94. House AA, Chang PC, Luke PP, Leckie SH, Howson WT, Ball EJ, et al. Re-exposure to mismatched HLA class I is a significant risk factor for graft loss: multivariable analysis of 259 kidney retransplants. Transplantation (2007) 84(6):722–8. doi:10.1097/01.tp.0000281398.41670.1f
95. Coupel S, Giral-Classe M, Karam G, Morcet JF, Dantal J, Cantarovich D, et al. Ten-year survival of second kidney transplants: impact of immunologic factors and renal function at 12 months. Kidney Int (2003) 64(2):674–80. doi:10.1046/j.1523-1755.2003.00104.x
96. O’Connor MJ, Lind C, Tang X, Gossett J, Weber J, Monos D, et al. Persistence of anti-human leukocyte antibodies in congenital heart disease late after surgery using allografts and whole blood. J Heart Lung Transplant (2013) 32(4):390–7. doi:10.1016/j.healun.2012.12.009
97. Kwon MH, Zhang JQ, Schaenman JM, Cadeiras M, Gjertson DW, Krystal CA, et al. Characterization of ventricular assist device-mediated sensitization in the bridge-to-heart-transplantation patient. J Thorac Cardiovasc Surg (2015) 149(4):1161–6. doi:10.1016/j.jtcvs.2015.01.003
98. Morales-Buenrostro LE, Terasaki PI, Marino-Vazquez LA, Lee JH, El-Awar N, Alberu J. “Natural” human leukocyte antigen antibodies found in nonalloimmunized healthy males. Transplantation (2008) 86(8):1111–5. doi:10.1097/01.tp.0000331682.91699.7b
99. Burrows SR, Silins SL, Khanna R, Burrows JM, Rischmueller M, McCluskey J, et al. Cross-reactive memory T cells for Epstein-Barr virus augment the alloresponse to common human leukocyte antigens: degenerate recognition of major histocompatibility complex-bound peptide by T cells and its role in alloreactivity. Eur J Immunol (1997) 27(7):1726–36. doi:10.1002/eji.1830270126
100. Amir AL, D’Orsogna LJ, Roelen DL, van Loenen MM, Hagedoorn RS, de Boer R, et al. Allo-HLA reactivity of virus-specific memory T cells is common. Blood (2010) 115(15):3146–57. doi:10.1182/blood-2009-07-234906
101. Morice A, Charreau B, Neveu B, Brouard S, Soulillou JP, Bonneville M, et al. Cross-reactivity of herpesvirus-specific CD8 T cell lines toward allogeneic class I MHC molecules. PLoS One (2010) 5(8):e12120. doi:10.1371/journal.pone.0012120
102. D’Orsogna LJ, van der Meer-Prins EM, Zoet YM, Roelen DL, Doxiadis II, Claas FH. Detection of allo-HLA cross-reactivity by virus-specific memory T-cell clones using single HLA-transfected K562 cells. Methods Mol Biol (2012) 882:339–49. doi:10.1007/978-1-61779-842-9_19
103. de Santis C, Lopalco L, Robbioni P, Longhi R, Rappocciolo G, Siccardi AG, et al. Human antibodies to immunodominant C5 region of HIV-1 gp120 cross-react with HLA class I on activated cells. AIDS Res Hum Retroviruses (1994) 10(2):157–62. doi:10.1089/aid.1994.10.157
104. Alberu J, Morales-Buenrostro LE, de Leo C, Vargas-Rojas MI, Marino-Vazquez LA, Crispin JC. A non-allogeneic stimulus triggers the production of de novo HLA antibodies in healthy adults. Transpl Immunol (2007) 18(2):166–71. doi:10.1016/j.trim.2007.06.001
105. Ravindranath MH, Kaneku H, El-Awar N, Morales-Buenrostro LE, Terasaki PI. Antibodies to HLA-E in nonalloimmunized males: pattern of HLA-Ia reactivity of anti-HLA-E-positive sera. J Immunol (2010) 185(3):1935–48. doi:10.4049/jimmunol.1000424
106. El-Awar N, Terasaki PI, Nguyen A, Sasaki N, Morales-Buenrostro LE, Saji H, et al. Epitopes of HLA antibodies found in sera of normal healthy males and cord blood. Clin Transpl (2008):199–214. doi:10.1016/j.humimm.2009.06.020
107. Roberts T, Tumer G, Gebel HM, Bray RA. Solid-phase assays for the detection of alloantibody against human leukocyte antigens: panacea or Pandora? Int J Immunogenet (2014) 41(5):362–9. doi:10.1111/iji.12138
108. Visentin J, Guidicelli G, Bachelet T, Jacquelinet C, Audry B, Nong T, et al. Denatured class I human leukocyte antigen antibodies in sensitized kidney recipients: prevalence, relevance, and impact on organ allocation. Transplantation (2014) 98(7):738–44. doi:10.1097/TP.0000000000000315
109. In JW, Rho EY, Shin S, Park KU, Song EY. False-positive reactions against HLA class II molecules detected in Luminex single-antigen bead assays. Ann Lab Med (2014) 34(5):408–10. doi:10.3343/alm.2014.34.5.408
110. Cai J, Terasaki PI, Anderson N, Lachmann N, Schonemann C. Intact HLA not beta2m-free heavy chain-specific HLA class I antibodies are predictive of graft failure. Transplantation (2009) 88(2):226–30. doi:10.1097/TP.0b013e3181ac6198
111. Otten HG, Verhaar MC, Borst HP, van Eck M, van Ginkel WG, Hene RJ, et al. The significance of pretransplant donor-specific antibodies reactive with intact or denatured human leucocyte antigen in kidney transplantation. Clin Exp Immunol (2013) 173(3):536–43. doi:10.1111/cei.12127
112. Jackson AM, Sigdel TK, Delville M, Hsieh SC, Dai H, Bagnasco S, et al. Endothelial cell antibodies associated with novel targets and increased rejection. J Am Soc Nephrol (2015) 26(5):1161–71. doi:10.1681/ASN.2013121277
113. Sun Q, Cheng Z, Cheng D, Chen J, Ji S, Wen J, et al. De novo development of circulating anti-endothelial cell antibodies rather than pre-existing antibodies is associated with post-transplant allograft rejection. Kidney Int (2011) 79(6):655–62. doi:10.1038/ki.2010.437
114. Zhang Q, Cecka JM, Gjertson DW, Ge P, Rose ML, Patel JK, et al. HLA and MICA: targets of antibody-mediated rejection in heart transplantation. Transplantation (2011) 91(10):1153–8. doi:10.1097/TP.0b013e3182157d60
115. Patel R, Terasaki PI. Significance of the positive crossmatch test in kidney transplantation. N Engl J Med (1969) 280(14):735–9. doi:10.1056/NEJM196904032801401
116. Suitters A, Rose M, Higgins A, Yacoub MH. MHC antigen expression in sequential biopsies from cardiac transplant patients – correlation with rejection. Clin Exp Immunol (1987) 69(3):575–83.
117. Hubscher SG, Adams DH, Elias E. Changes in the expression of major histocompatibility complex class II antigens in liver allograft rejection. J Pathol (1990) 162(2):165–71. doi:10.1002/path.1711620115
118. Facoetti A, Nano R, Zelini P, Morbini P, Benericetti E, Ceroni M, et al. Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin Cancer Res (2005) 11(23):8304–11. doi:10.1158/1078-0432.CCR-04-2588
119. Lefaucheur C, Loupy A, Hill GS, Andrade J, Nochy D, Antoine C, et al. Preexisting donor-specific HLA antibodies predict outcome in kidney transplantation. J Am Soc Nephrol (2010) 21(8):1398–406. doi:10.1681/ASN.2009101065
120. Loupy A, Jordan SC. Transplantation: donor-specific HLA antibodies and renal allograft failure. Nat Rev Nephrol (2013) 9(3):130–1. doi:10.1038/nrneph.2013.18
121. Valenzuela NM, Reed EF. Antibodies to HLA molecules mimic agonistic stimulation to trigger vascular cell changes and induce allograft injury. Curr Transplant Rep (2015) 2(3):222–32. doi:10.1007/s40472-015-0065-6
122. Zhang X, Rozengurt E, Reed EF. HLA class I molecules partner with integrin beta4 to stimulate endothelial cell proliferation and migration. Sci Signal (2010) 3(149):ra85. doi:10.1126/scisignal.2001158
123. Jin YP, Fishbein MC, Said JW, Jindra PT, Rajalingam R, Rozengurt E, et al. Anti-HLA class I antibody-mediated activation of the PI3K/Akt signaling pathway and induction of Bcl-2 and Bcl-xL expression in endothelial cells. Hum Immunol (2004) 65(4):291–302. doi:10.1016/j.humimm.2004.07.014
124. Le Bas-Bernardet S, Coupel S, Chauveau A, Soulillou JP, Charreau B. Vascular endothelial cells evade apoptosis triggered by human leukocyte antigen-DR ligation mediated by allospecific antibodies. Transplantation (2004) 78(12):1729–39. doi:10.1097/01.TP.0000147339.31581.99
125. Narayanan K, Jaramillo A, Phelan DL, Mohanakumar T. Pre-exposure to sub-saturating concentrations of HLA class I antibodies confers resistance to endothelial cells against antibody complement-mediated lysis by regulating Bad through the phosphatidylinositol 3-kinase/Akt pathway. Eur J Immunol (2004) 34(8):2303–12. doi:10.1002/eji.200324843
126. Lepin EJ, Zhang Q, Zhang X, Jindra PT, Hong LS, Ayele P, et al. Phosphorylated S6 ribosomal protein: a novel biomarker of antibody-mediated rejection in heart allografts. Am J Transplant (2006) 6(7):1560–71. doi:10.1111/j.1600-6143.2006.01355.x
127. Jindra PT, Hsueh A, Hong L, Gjertson D, Shen XD, Gao F, et al. Anti-MHC class I antibody activation of proliferation and survival signaling in murine cardiac allografts. J Immunol (2008) 180(4):2214–24. doi:10.4049/jimmunol.180.4.2357
128. Li F, Wei J, Valenzuela NM, Lai C, Zhang Q, Gjertson D, et al. Phosphorylated S6 kinase and S6 ribosomal protein are diagnostic markers of antibody-mediated rejection in heart allografts. J Heart Lung Transplant (2015) 34(4):580–7. doi:10.1016/j.healun.2014.09.047
129. Ziegler ME, Jin YP, Young SH, Rozengurt E, Reed EF. HLA class I-mediated stress fiber formation requires ERK1/2 activation in the absence of an increase in intracellular Ca2+ in human aortic endothelial cells. Am J Physiol Cell Physiol (2012) 303(8):C872–82. doi:10.1152/ajpcell.00199.2012
130. Ziegler ME, Souda P, Jin YP, Whitelegge JP, Reed EF. Characterization of the endothelial cell cytoskeleton following HLA class I ligation. PLoS One (2012) 7(1):e29472. doi:10.1371/journal.pone.0029472
131. Galvani S, Auge N, Calise D, Thiers JC, Canivet C, Kamar N, et al. HLA class I antibodies provoke graft arteriosclerosis in human arteries transplanted into SCID/beige mice. Am J Transplant (2009) 9(11):2607–14. doi:10.1111/j.1600-6143.2009.02804.x
132. Galvani S, Trayssac M, Auge N, Thiers JC, Calise D, Krell HW, et al. A key role for matrix metalloproteinases and neutral sphingomyelinase-2 in transplant vasculopathy triggered by anti-HLA antibody. Circulation (2011) 124(24):2725–34. doi:10.1161/CIRCULATIONAHA.111.021790
133. Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, et al. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc Natl Acad Sci U S A (2007) 104(4):1301–6. doi:10.1073/pnas.0602035104
134. Valenzuela NM, Hong L, Shen XD, Gao F, Young SH, Rozengurt E, et al. Blockade of p-selectin is sufficient to reduce MHC I antibody-elicited monocyte recruitment in vitro and in vivo. Am J Transplant (2013) 13(2):299–311. doi:10.1111/ajt.12016
135. Valenzuela NM, Mulder A, Reed EF. HLA class I antibodies trigger increased adherence of monocytes to endothelial cells by eliciting an increase in endothelial P-selectin and, depending on subclass, by engaging FcgammaRs. J Immunol (2013) 190(12):6635–50. doi:10.4049/jimmunol.1201434
136. Wehner J, Morrell CN, Reynolds T, Rodriguez ER, Baldwin WM III. Antibody and complement in transplant vasculopathy. Circ Res (2007) 100(2):191–203. doi:10.1161/01.RES.0000255032.33661.88
137. Nickeleit V, Mihatsch MJ. Kidney transplants, antibodies and rejection: is C4d a magic marker? Nephrol Dial Transplant (2003) 18(11):2232–9. doi:10.1093/ndt/gfg304
138. Rodriguez ER, Skojec DV, Tan CD, Zachary AA, Kasper EK, Conte JV, et al. Antibody-mediated rejection in human cardiac allografts: evaluation of immunoglobulins and complement activation products C4d and C3d as markers. Am J Transplant (2005) 5(11):2778–85. doi:10.1111/j.1600-6143.2005.01074.x
139. Kubagawa H, Oka S, Kubagawa Y, Torii I, Takayama E, Kang DW, et al. Identity of the elusive IgM Fc receptor (FcmuR) in humans. J Exp Med (2009) 206(12):2779–93. doi:10.1084/jem.20091107
140. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood (2009) 113(16):3716–25. doi:10.1182/blood-2008-09-179754
141. Nimmerjahn F, Ravetch JV. FcgammaRs in health and disease. Curr Top Microbiol Immunol (2011) 350:105–25. doi:10.1007/82_2010_86
142. Manger K, Repp R, Spriewald BM, Rascu A, Geiger A, Wassmuth R, et al. Fcgamma receptor IIa polymorphism in Caucasian patients with systemic lupus erythematosus: association with clinical symptoms. Arthritis Rheum (1998) 41(7):1181–9. doi:10.1002/1529-0131(199807)41:7<1181::AID-ART6>3.0.CO;2-C
143. Karassa FB, Trikalinos TA, Ioannidis JP. The role of FcgammaRIIA and IIIA polymorphisms in autoimmune diseases. Biomed Pharmacother (2004) 58(5):286–91. doi:10.1016/j.biopha.2004.04.004
144. Diamantopoulos PT, Kalotychou V, Polonyfi K, Sofotasiou M, Anastasopoulou A, Galanopoulos A, et al. Correlation of Fc-gamma RIIA polymorphisms with latent Epstein-Barr virus infection and latent membrane protein 1 expression in patients with low grade B-cell lymphomas. Leuk Lymphoma (2013) 54(9):2030–4. doi:10.3109/10428194.2012.762512
145. Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol (2013) 6:1. doi:10.1186/1756-8722-6-1
146. Maiga B, Dolo A, Toure O, Dara V, Tapily A, Campino S, et al. Fc gamma receptor IIa-H131R polymorphism and malaria susceptibility in sympatric ethnic groups, Fulani and Dogon of Mali. Scand J Immunol (2014) 79(1):43–50. doi:10.1111/sji.12122
147. Pawlik A, Florczak M, Bak L, Domanski L, Rozanski J, Dabrowska-Zamojcin E, et al. The Fc gamma RIIa polymorphism in patients with acute kidney graft rejection. Ann Transplant (2003) 8(4):24–6.
148. Yuan FF, Watson N, Sullivan JS, Biffin S, Moses J, Geczy AF, et al. Association of Fc gamma receptor IIA polymorphisms with acute renal-allograft rejection. Transplantation (2004) 78(5):766–9. doi:10.1097/01.TP.0000132560.77496.CB
149. Berry GJ, Burke MM, Andersen C, Bruneval P, Fedrigo M, Fishbein MC, et al. The 2013 International Society for Heart and Lung Transplantation Working Formulation for the standardization of nomenclature in the pathologic diagnosis of antibody-mediated rejection in heart transplantation. J Heart Lung Transplant (2013) 32(12):1147–62. doi:10.1016/j.healun.2012.11.005
150. Fishbein GA, Fishbein MC. Morphologic and immunohistochemical findings in antibody-mediated rejection of the cardiac allograft. Hum Immunol (2012) 73(12):1213–7. doi:10.1016/j.humimm.2012.07.011
151. Fedrigo M, Feltrin G, Poli F, Frigo AC, Benazzi E, Gambino A, et al. Intravascular macrophages in cardiac allograft biopsies for diagnosis of early and late antibody-mediated rejection. J Heart Lung Transplant (2013) 32(4):404–9. doi:10.1016/j.healun.2012.12.017
152. Xu L, Collins J, Drachenberg C, Kukuruga D, Burke A. Increased macrophage density of cardiac allograft biopsies is associated with antibody-mediated rejection and alloantibodies to HLA antigens. Clin Transplant (2014) 28(5):554–60. doi:10.1111/ctr.12348
153. Magil AB, Tinckam K. Monocytes and peritubular capillary C4d deposition in acute renal allograft rejection. Kidney Int (2003) 63(5):1888–93. doi:10.1046/j.1523-1755.2003.00921.x
Keywords: human leukocyte antigen, allorecognition, HLA antibody, non-HLA antibody, transplant, Fc receptor, complement, endothelium
Citation: Hickey MJ, Valenzuela NM and Reed EF (2016) Alloantibody Generation and Effector Function Following Sensitization to Human Leukocyte Antigen. Front. Immunol. 7:30. doi: 10.3389/fimmu.2016.00030
Received: 18 November 2015; Accepted: 20 January 2016;
Published: 04 February 2016
Edited by:
Gilles Benichou, Massachusetts General Hospital, USAReviewed by:
Nuala Mooney, Centre National de la Recherche Scientifique, FranceEmmanuel Zorn, Columbia University Medical Center, USA
Copyright: © 2016 Hickey, Valenzuela and Reed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elaine F. Reed, ereed@mednet.ucla.edu