 Tamara Zietek
Tamara Zietek Eva Rath
Eva Rath- 1Department of Nutritional Physiology, Technische Universität München, Freising, Germany
- 2Chair of Nutrition and Immunology, Technische Universität München, Freising, Germany
Chronic diseases, such as obesity and diabetes, cardiovascular, and inflammatory bowel diseases (IBD) share common features in their pathology. Metabolic disorders exhibit strong inflammatory underpinnings and vice versa, inflammation is associated with metabolic alterations. Next to cytokines and cellular stress pathways, such as the unfolded protein response (UPR), alterations in the enteroendocrine system are intersections of various pathologies. Enteroendocrine cells (EEC) have been studied extensively for their ability to regulate gastrointestinal motility, secretion, and insulin release by release of peptide hormones. In particular, the L-cell-derived incretin hormone glucagon-like peptide 1 (GLP-1) has gained enormous attention due to its insulinotropic action and relevance in the treatment of type 2 diabetes (T2D). Yet, accumulating data indicate a critical role for EEC and in particular for GLP-1 in metabolic adaptation and in orchestrating immune responses beyond blood glucose control. EEC sense the lamina propria and luminal environment, including the microbiota via receptors and transporters. Subsequently, mediating signals by secreting hormones and cytokines, EEC can be considered as integrators of metabolic and inflammatory signaling. This review focuses on L cell and GLP-1 functions in the context of metabolic and inflammatory diseases. The effects of incretin-based therapies on metabolism and immune system are discussed and the interrelation and common features of metabolic and immune-mediated disorders are highlighted. Moreover, it presents data on the impact of inflammation, in particular of IBD on EEC and discusses the potential role of the microbiota as link between nutrients, metabolism, immunity, and disease.
Introduction
Metabolically driven pathologies, such as obesity, insulin resistance, and type 2 diabetes, but also immunologically mediated disorders, such as inflammatory bowel diseases (IBD), are considered chronic diseases. Concomitant with the spread of the western lifestyle, the prevalence of these diseases has rapidly increased (1–3) now constituting a global health problem (4). Even though phenotypically different, these diseases share common features in their pathology. Metabolic disorders exhibit strong inflammatory underpinnings and vice versa, inflammation is associated with metabolic alterations. For example, obesity evokes a broad array of inflammatory and metabolic responses leading to low-grade local inflammation and in turn to defective insulin receptor signaling and disruption of metabolic homeostasis (5). Conversely, patients suffering from sepsis and animal models of endotoxin-induced inflammation show a loss of glycemic control (6, 7). On a molecular level, metabolically driven and immunologically mediated disorders converge on cellular stress responses, such as endoplasmic reticulum unfolded protein response (ER UPR) (8). Furthermore, many chronic diseases are associated with increased levels of the pro-inflammatory cytokines tumor necrosis factor (TNF) and interleukin 6 (IL-6) (9, 10) as well as alterations in the intestinal microbiome (11, 12).
During the past years, the intestine and in particular, intestinal epithelial cells (IEC) are more and more recognized as key players in maintaining metabolic and immune homeostasis. Constituting a surface area up to 40 m2 (13) and being the bodies’ most important interface with the external environment, the intestinal epithelium needs to allow efficient nutrient absorption while maintaining barrier function and modulating immunity (12). Different epithelial cell subtypes fulfill distinct functions, with absorptive enterocytes and secretory (mucin-producing) goblet and (antimicrobial peptides-producing) Paneth cells accounting for the largest proportion of epithelial cells. Enteroendocrine cells (EEC) found scattered throughout the intestine comprise approximately only 1% of the epithelium but secrete more than 20 different peptide hormones, making them collectively one of the largest endocrine systems (14). Among the gut hormones, glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypetide (GIP), referred to as incretins, have gained enormous attention due to their insulinotropic action and relevance in the treatment of type 2 diabetes (T2D) (15). Especially GLP-1 and GLP-1-based antidiabetic therapies are in the spotlight of biomedical research. GLP-1 secreted by L cells not only potentiates the glucose-induced insulin response, promotes β-cell survival, slows gastric emptying (GE), and regulates energy expenditure and body weight, but also exerts neuroprotective, cardioprotective, and anti-inflammatory effects. Experimental approaches and treatment of patients with GLP-1 analogs as well as dipeptidyl peptidase-IV (DPP-4) inhibitors, which inhibit the endopeptidase that rapidly degrades GIP and GLP-1, underline the multiple beneficial effects mediated by GLP-1, beyond blood glucose control. While the mechanisms underlying hormone secretion from EEC and their functions in the context of digestion and metabolism are well studied, their role in intestinal inflammation and their interrelation with immune cells is virtually unknown. Recent data indicate that EEC, next to nutrients, also actively sense the microbiota and bacterial products, enlightening a new aspect of the cross-talk between immune and endocrine system.
This review illustrates L cell and GLP-1 functions and discusses the effects of incretin-based therapies on metabolic and inflammatory signaling. It focuses on the interrelation and common features of metabolic and immune-mediated disorders converging on GLP-1. Moreover, it presents data on the impact of intestinal inflammation, in particular of IBD, on EEC and the potential contribution of EEC to these pathologies. In addition, the possible roles of cytokines, UPR, and the microbiota as link between nutrients, metabolism, immunity, and disease are discussed.
Enteroendocrine Cells: Luminal Sensing and L Cell Function
Typically, EEC have been classified based on the primary hormone they contain. EEC secreting GLP-1, GLP-2, and peptide YY (PYY) are referred to as L cells, whereas GIP-producing cells are considered K cells. However, it was demonstrated that expression patterns of L cells differ considerably depending on their location in the intestine (16–18). In the proximal small intestine, L cells also express considerable amounts of GIP, cholecystokinin (CCK), and secretin.
EEC have been studied extensively for their ability to adjust gastrointestinal motility, secretion, and insulin release thereby enabling efficient postprandial assimilation of nutrients (19). To achieve this prime function of creating the optimal absorptive and digestive conditions following nutrient intake, EEC are equipped with specific apical sensor proteins for the detection of distinct luminal nutrients. Depending on the location along the gut axis, luminal stimuli of EEC comprise monosaccharides, free fatty acids (FA), monoacylglycerols, amino acids, di/tripeptides bile acids, short-chain fatty acids (SCFAs), and indole. The following sections briefly summarize receptors and transporters involved in environmental sensing by L cells (Figure 1). For more detailed information, see Ref. (15, 19).
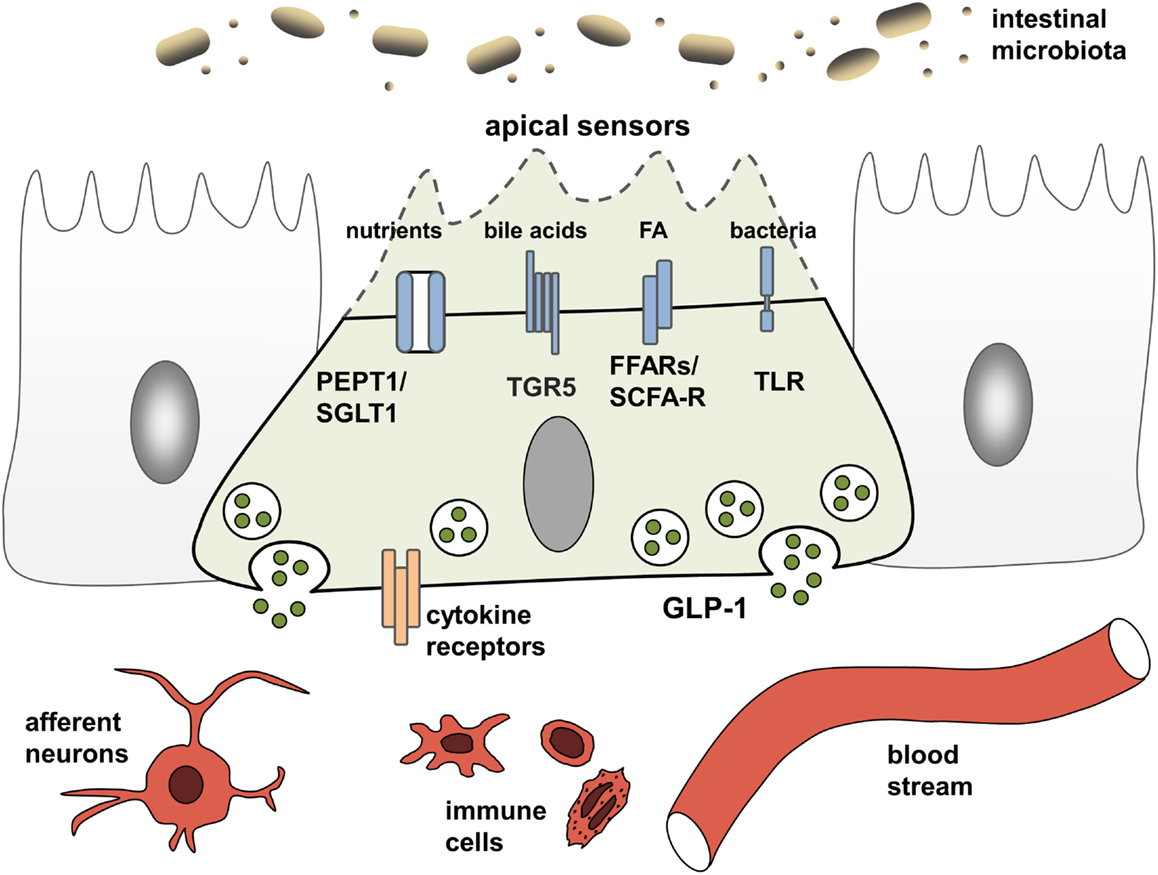
Figure 1. L cells as interface between luminal-derived signals and host metabolism and immune system. L cells sense the luminal and lamina propria environment via receptors and transporters and mediate signals through hormone and cytokine secretion. Fatty acids (FA), free fatty acid receptor (FFAR), short-chain fatty acid (SCFA), sodium-dependent glucose transporter (SGLT1), proton-coupled peptide-transporter (PEPT1), glucagon-like peptide 1 (GLP-1), G-protein-coupled bile acid receptor 1 (TGR5).
Nutrient Sensing
Carbohydrates, lipids, and proteins are detected via their digestion end products. Luminal glucose represents the most potent secretagogue for incretin hormone release in vivo, an effect mainly mediated by the sodium-dependent glucose transporter, SGLT1 (SLC5A1) (15). As indicated by experiments demonstrating concomitant ingestion of proteins and glucose to enhanced GLP-1 responses (20), peptides and amino acids are sensed by EEC via the proton-coupled peptide-transporter PEPT1 (SLC15A1) and the calcium-sensing receptor (CaSR) (15). In the case of SGLT1 and PEPT1, nutrient-sensing is linked to hormone secretion by the electrogenic character of the transporters. Substrate transport is coupled to cation influx causing membrane depolarization, subsequent opening of voltage-gated calcium channels and an increase in intracellular calcium levels, finally resulting in exocytosis and hormone release from vesicles (15). This sensing property of nutrient transporters in EEC has coined the term “transceptors” and has extended the group of sensor proteins. Before SGLT1 was identified as the first transceptor, only G-protein-coupled receptors (GPCR) were known to fulfill such sensory functions in the intestine. GPCR comprise the amino acid- and oligopeptide-sensing receptors CaSR (15), metabotropic glutamate receptors (20), LPAR5/GPR92/93 (21), GPRC6A (22), and taste receptors (TRs) (23). Furthermore, GPCR are responsible for detection of lipid-derived long-chain/medium-chain FA and dietary-fiber derived SCFAs. Predominantly, lipid ingestion causes GIP release. Yet, FA also elicit GLP-1 responses via the free fatty acid receptors (FFAR) FFAR1/GPR40, FFAR4/GPR120, and GPR119. Dietary fibers, after fermentation to SCFAs by the microbiota, are sensed by FFAR2/GPR43 and FFAR3/GPR41 and also trigger GLP-1 secretion (24).
Along the lines of GLP-1 analogs and DPP-4 inhibitors, various agonists of GPR40 and GPR120 have been developed and proven effective in ameliorating glycemic control in animal trials (21, 22), now being in phase I or II clinical trials for the treatment of T2D (23). Interestingly, high fat consumption is associated with enhanced L cell density in obese subjects and mice (25). This increase in the number GLP-1-positive cells observed in mice was attributed to improved L cell differentiation and, furthermore, SCFAs were sufficient to increase L cell numbers in intestinal organoids of human and murine origin (25, 26). It has been suggested that this effect, favoring insulin secretion, might constitute an adaptive response of the intestine to counterbalance diet-induced insulin resistance.
Several other transporters and receptors, such as the facilitative glucose transporter (GLUT2), are thought to play a role in incretin secretion, however, their contribution to luminal compound sensing remains unclear.
Taste Receptors
Taste receptors detecting complex tastes, such as sweet, umami, and bitter taste, are present in the intestinal epithelium including EEC. While taste 1 receptors function as dimers to sense sweet or umami, taste 2 receptors detect a large variety of bitter tastants (26). Consequently, tastants might induce or contribute to the release of peptide hormones from EEC (24, 27). Studies indicate a physiological role for umami (TAS1R1/R3) and bitter TRs, yet, the relevance of the sweet TR T1R2/T1R3 is highly controversial (15, 19, 27). T1R2/T1R3 is activated not only by sugar but also by non-metabolizable sweeteners, such as saccharin (28). Although studies using EEC lines and tissue explants have demonstrated GLP-1 release in response to T1R2/T1R3 ligands, the in vivo relevance of sweet TR activation on incretin secretion still remains unclear, since others have been unable to demonstrate functional activity of T1R2/T1R3 in primary cultured L cells or in perfused intestinal preparations (15, 19). In line, in vivo studies in animals and humans consistently failed to show effects of artificial sweeteners on plasma incretins (28–30). However, sweet TR activation results in increased apical SGLT1 levels and, via this effect, might contribute to incretin secretion (15). Interestingly, a selective upregulation of the bitter TR TR2R138 was shown in the colon of mice fed a high fat diet (31), and T2R38, a human receptor activated by the same ligand, phenylthiocarbaminde, has been demonstrated not only to be expressed in EEC of the colonic mucosa but also to be induced in overweight/obese subjects (32). T2R38 is known to respond to Gram-negative bacterial quorum-sensing molecules in human upper airway cilia thereby regulating innate immune responses (33). It is attractive to speculate that these receptors function as sensors for subpopulation of the intestinal microbiota and might respond to the alterations of gut microbial communities associated with long-term high-fat diet and obesity.
Pattern Recognition and Sensing of Bacterial Products
While a role for T2Rs in microbial sensing of EEC still needs to be addressed, there is clear evidence that EEC respond to bacteria and bacterial products. In particular, EEC possess functional toll-like receptors (TLR) and upon lipopolysaccharide (LPS) stimulation, GLP-1 release is triggered in mice (34). Also, bacterial metabolites, such as SCFA and indole, a product of bacterial tryptophan metabolism involved in interbacterial communication, exert direct signaling actions on colonic L cells (19). Further evidence for the importance of the microbiota for incretin regulation comes from germ-free (GF) and antibiotic-treated mice, which have severely reduced SCFA levels, and concomitantly increased basal GLP-1 plasma levels as well as increased proglucagon expression, specifically in the colon (35). Increasing energy supply suppressed proglucagon expression in GF mice, suggesting that colonic L cells sense energy availability and regulate basal GLP-1 secretion accordingly.
Next to indole, which acts on voltage-gated K+ channels to enhance Ca2+ entry thereby stimulating GLP-1 secretion (36) and microbiota-derived SCFA that are sensed by FFAR2/GPR43 and FFAR3/GPR41 (see above), the importance of bile acid-induced incretin secretion via the bile acid receptor GPBAR/TGR5 has been proven in vitro and in vivo. TGR5 ligands improve insulin sensitivity and glucose homeostasis through the secretion of incretins (27) and the rapid improvement of hyperglycemia after bariatric surgery has been attributed partly to alterations in bile acid metabolism and sensing (29, 30). A central role of bile acid signaling as a novel pharmacological target in the metabolic syndrome and related diseases, such as obesity, T2D, atherosclerosis, liver disease, and cancer, is underscored by the presence of TGR5 in various tissues and cell types. Activation of TGR5 in brown adipose tissue and skeletal muscle increases energy expenditure, while activation in macrophages inhibits production of pro-inflammatory cytokines (37).
Through affecting bile acid metabolism and providing SCFA, the intestinal microbiota might indirectly impact TGR5-signaling and nutrient sensing leading to incretin hormone secrection, yet, directly triggering activation of EEC via TLR also evokes an inflammatory response (38). Upon exposure to flagellin or LPS (39, 40) EEC express pro-inflammatory cytokines. TLR activation evokes a specific gene response indicating EEC to participate in innate immune responses to commensals and pathogens (39). Together with observations that immune cells are in close physical contact with EEC (41), this led to the suggestion of an immunoendocrine axis and a critical role for EEC in orchestrating intestinal immune responses (42).
In the context of bacterial pattern recognition, it is noteworthy that PEPT1, known to mediate di/tripeptide-induced GLP-1 secretion, was also shown to transport small bacterial-derived peptides, such as muramyl dipeptide (MDP). Intracellularly, these peptides can be sensed by nucleotide-binding oligomerization domain (NOD)-like receptors (NLR) that belong to the innate immune system and recognize pathogen-associated molecular patterns (43). Yet, there is neither data on the transport and possible consequences of bacterial-derived proteins in EEC nor on the expression of NLR in EEC. However, Pept1−/− mice do not show any abnormalities in weight or any other anthropometric or clinical chemistry measurement when animals are fed a standard high-carbohydrate diet (44). Furthermore, the role of PEPT1 in intestinal inflammation remains controversial, since there is conflicting data on expression levels under inflammatory conditions in mice and humans (43, 45, 46).
GLP-1 Actions
GLP-1 Effect on Blood Glucose Control
Upon stimulation, L cells secrete different peptide hormones, including the incretin GLP-1. GLP-1 is derived from a transcription product of the proglucagon gene gcg, which also encodes GLP-2 and further factors. Mainly expressed in the ileum and colon, GLP-1 is known as the most powerful incretin in humans and lowers postprandial blood glucose via augmentation of glucose-dependent insulin release from pancreatic β-cells, inhibition of glucagon secretion from pancreatic α-cells, and delay of GE. Furthermore, GLP-1 increases pancreatic β-cell growth by promoting proliferation and reducing apoptosis, an effect that might be predominantly mediated locally via α-cell-derived GLP-1 (47). Conversely, L cells are responsive to insulin and insulin resistance is associated with impaired GLP-1 secretion in vitro and in vivo (48). These properties constitute the basis for GLP-1-based antidiabetic therapies, yet GLP-1 also exerts anorexigenic effects by promoting satiety and reducing food intake.
Glucagon-like peptide 1 and GIP act via G-protein-coupled receptors. The GLP-1R is expressed in many tissues, including pancreatic islets, the central nervous system, lung, kidney, heart, intestine, and also on immune cells (49, 50), underlining the numerous roles for GLP-1-signaling beyond blood glucose control.
When secreted by L cells, GLP-1 either functions in an endocrine manner, being released into the blood stream where it is rapidly inactivated by DPP-4 with a half-life of about 2 min, or exerts paracrine effects like stimulating neurons. Triggering vagal afferents, GLP-1 mediates signaling from gut to brain with anorexigenic effects and via nerve terminals in the hepatoportal region, it can affect metabolic functions in the liver (15). In line, peptide hormones secreted by EEC can modulate immune responses via the nervous system. For instance, is has been shown that nutritional stimulation of CCK receptors attenuated inflammation through inhibition of pro-inflammatory cytokine secretion from macrophages via the vagus nerve (51).
GLP-1 and Immune System
There is a strong cross-talk between immune and endocrine system (Figure 1). Data indicate that immune cells and cytokine mediated-signaling impacts EEC numbers during infection and chronic inflammation of the gut (52, 53). On the other hand, there is a widespread expression of GLP-1R on multiple immune cell populations, particularly on intestinal intraepithelial lymphocytes (IEL) (50, 54). Moreover, in addition to its enzymatic functions, DPP-4 is known as the lymphocyte cell surface protein CD26, which plays a key role in T-cell development, activation, and immune regulation (55, 56). Last but not least, insulin itself was shown to inhibit IL-10-mediated regulatory T-cell functions (57) and also GLP-1 exerts direct anti-inflammatory effects. Supporting a function of GLP-1 in modulating immune responses, circulating GLP-1 was reported to be increased in states of chronic inflammatory disease, including the metabolic syndrome, coronary artery disease, or heart failure (58–60) but also in critically ill patients and patients with sepsis (61). Furthermore, treatment with GLP-1 as well as GLP-1 analogs and DPP-4-inhibitors exhibit beneficial effect in a vast array of diseases.
Incretin-Based Therapies
GLP-1-based therapies and DPP-4-inhibition, which also targets GIP by increasing its half-life, have been proven successful in the treatment of T2D.
Common adverse effects of incretin mimetics and DPP-4 inhibitors include gastrointestinal symptoms, such as nausea and vomiting as well as hypersensitivity reactions. Safety issues have also been raised on a possible association between incretin-based drugs and pancreatitis or pancreatic cancer based on animal studies and data on patients (62). However, animal as well as patient data are inconsistent and suffer from several confounders (63, 64). Since the relationship remains unclear, both the Endocrinologic and Metabolic Drugs Advisory Committee and the FDA’s Division of Metabolism and Endocrinology Products concluded that there is not sufficient evidence available to conclude that incretin-based therapies cause acute pancreatitis or pancreatic cancer (65). Nonetheless, pancreatitis is continued to be considered a risk associated with these drugs until more data are available (65).
In addition, several effects of incretin-based diabetes therapies beyond their glycemic-lowering properties via the endocrine pancreas have been described, including reduced immune cell infiltration and altered cytokine expression (66). Again emphasizing the interrelation of metabolic and inflammatory disorders, beneficial outcomes have also been observed in cardiovascular disease (67), stroke (68), hepatic diseases (69, 70), nephritis (71), neuro-inflammation (72, 73), rheumatoid arthritis (74), lung inflammation (75), and sepsis (7) (Figure 2).
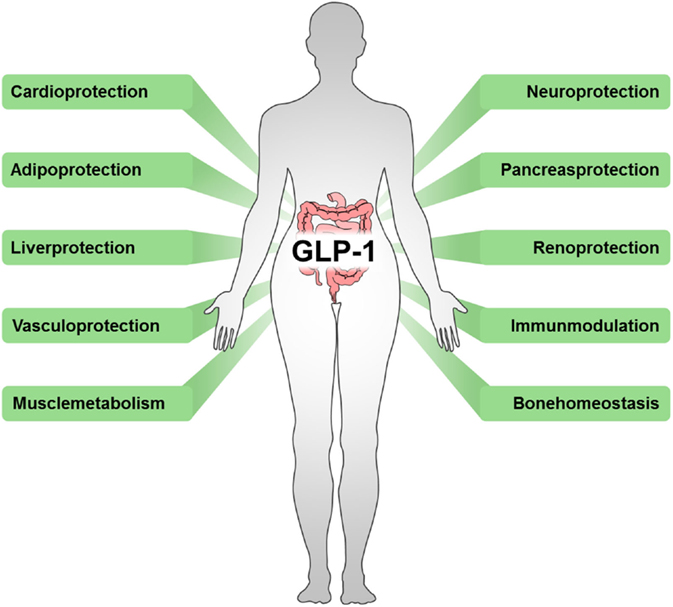
Figure 2. Beneficial outcomes of drugs targeting incretins. Incretin-based therapeutics, such as GLP-1 analogs, GLP-1R agonists, or DPP-4 inhibitors, exert multiple effects in various tissues.
GLP-1 and GLP-1 Analogs
In patients with T2D, GLP-1 and GLP-1 analogs were shown to improve cardiovascular-risk profiles, by reducing body fat content, blood pressure, circulating lipids, and inflammatory markers (76). Furthermore, low-grade inflammation of the endothelium is an early event in the pathogenesis of atherosclerotic cardiovascular disease and the GLP-1 analog liraglutide was shown to exhibited anti-oxidative and anti-inflammatory effects, including upregulation of anti-oxidative enzymes and inhibition of nuclear factor kappa B (NF-κB)-signaling on endothelial cells (67, 77). Through inactivation of NF-κB, a master regulator of inflammatory responses, GLP-1 treatment of mice also significantly alleviated lung inflammation and pulmonary fibrosis (75). In mouse models of Alzheimer’s disease synthetic incretin hormones exerted neuroprotective effects, including reduced amyloid plaque load, reducing oxidative stress and the chronic inflammatory response in the brain, and enhancing neurogenesis (72, 73). In line, the GLP-1 receptor agonist Exendin-4 was suggested to down-regulate pro-inflammatory responses and reduce oxidative stress by suppressing MAPK signaling pathways in peripheral lymphocytes of patients with T2D (78). In addition, Exendin-4 was demonstrated to selectively reduce the production of cytokines from activated IEL, indicating a local enteroendocrine–immune axis (50). In an obese mouse model of diabetes, GLP-1 was shown to reduce macrophage infiltration and directly inhibit inflammatory pathways in adipocytes and adipose tissue macrophages (79). Notably, studies have shown beneficial effects of GLP-1 treatment on glycemia in critically ill patients and patients suffering from septic shock, a phenomenon also seen in patients with T2D. Concomitantly, the incretin effect was reduced in critically ill patients resembling previous findings in patients with T2D (6). In line, experimental evidence indicates beneficial effects of DPP-4-inhibition and GLP-1 analog treatment during sepsis. The improved survival of animals with LPS-induced endotoxemia by treatment with GLP-1 analogs (7) or endotoxin-challenged DPP-4 knockout rats (61) is associated with reduced vascular inflammation/dysfunction. In this context, the diminished levels of oxidative stress and tissue protection were attributed to direct anti-inflammatory capacities of GLP-1. In non-alcoholic fatty liver disease and hepatocyte steatosis, GLP-1 and incretin mimetics have been shown to ameliorate pathology by promoting macroautophagy and SIRT1-mediated signals (69, 70, 80).
DPP-4 Inhibitors
DPP-4 activity is markedly increased in obese subjects and in animal models of obesity (81, 82), suggesting the endopeptidase as target for therapeutic interventions. DPP-4 inhibitors increase the amount of circulating GLP-1 and GIP by delaying their inactivation. On immune cells, inhibition of DPP-4/CD26 results in anti-inflammatory effects via regulation of chemokines and anti-inflammatory cytokines like IL-10 and transforming growth factor (TGF)-β (83, 84) and DPP-4 might also impact macrophage polarization. CD26 is furthermore involved in a variety of human autoimmune diseases, and as a cell surface protease, DPP-4/CD26 plays an important role in tumor progression (85). However, the majority of actions ascribed to DPP-4 in immune cells are attributable to non-enzymatic actions of the enzyme; hence, DPP-4 signaling in immune cells seems independent of its catalytic enzyme activity (86).
Nonetheless, DPP-4 inhibitors improve cardiovascular outcomes by modulating innate and adaptive immunity and suppression of the NLRP3 inflammasome, a multiprotein complex involved in caspase-1 activation and downstream maturation of pro-inflammatory cytokines, TLR4 and IL-1β in human macrophages (87, 88). For ischemic stroke and retinal damage, two major complications of diabetes mellitus, it was shown that DPP-4 inhibition prevented inflammation and mediated neuroprotective effects (68, 89). These properties were attributed to antioxidant, anti-inflammatory, and anti-apoptotic mechanisms, including suppression of NF-κB and downstream of the inflammatory cytokines TNF and IL-6. Concomitantly, the anti-inflammatory cytokine IL-10 was found to be elevated (68). In addition, data indicate that DPP-4 inhibitors also impact immune cell recruitment and reduced macrophage infiltration seemed to be mediated directly via GLP-1-dependent signaling in a rat nephritis model (71).
In summary, these observations underline the tight interrelation of endocrine signaling and immune responses and, furthermore, indicate common disease mechanism for metabolic and immune-mediated disorders.
Common Features of Metabolic and Immune-Mediated Disorders Impacting GLP-1
Immune Mediators/Cytokines
Yet another strong link between metabolic and immune-mediated disorders are elevated levels of the cytokines TNF and IL-6, underlying several pathologies and both being reported to impact on GLP-1 signaling (Figure 3).
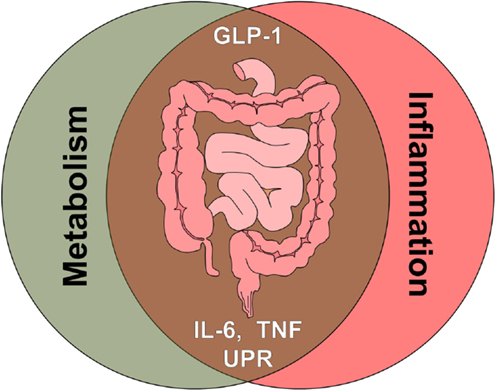
Figure 3. The intestine as crossroad of metabolism and inflammation. Multiple signals associated with metabolic signaling and inflammatory signaling, such as gut hormones, cytokines, and cellular stress pathways, converge at the level of the intestine. Glucagon-like peptide 1 (GLP-1), tumor necrosis factor (TNF), interleukin 6 (IL-6), unfolded protein response (UPR).
Tumor Necrosis Factor
Both TNF and IL-6 are correlated with the body mass index (BMI), and TNF is known to play a role in obesity and in particular in the insulin resistance and diabetes that often accompany obesity (10, 90). Typically, TNF is involved in systemic inflammation and the acute phase reaction. It is able to induce fever, apoptotic cell death, cachexia, inflammation, and to inhibit tumorgenesis. Besides obesity, dysregulation of TNF production has been implicated in a variety of human diseases, including Alzheimer’s disease (91), cancer (92), rheumatoid arthritis (74), and IBD (93). Critically ill patients demonstrate elevated levels of TNF and IL-6 and as mentioned above, these patients and T2D share phenotypical similarities such as hyperglycemia, insulin resistance, and systemic inflammation (6). During obesity, pro-inflammatory macrophages accumulate in adipose tissue representing a chronic low-grade inflammation; these cells are the dominant sources of TNF promoting insulin resistance (9). Neutralization of TNF improves glucose uptake in murine obesity and mice lacking TNF are protected from high-fat-diet-induced insulin resistance (10, 94). L cells express TNF receptor 1 (TNFR1) and chronic exposure to TNF was shown to impair GLP-1 (95) as well as GLP-2 secretion (96). In mice with high-fat diet-induced hyperglycemia, hyperinsulinemia, and associated induction of TNF, a reduction of GLP-1-secretion was observed, which could be reversed by treatment with the TNF-neutralizing antibody etanercept (95). In a human study, infusion of TNF-induced systemic inflammation and concomitantly reduced plasma levels of GLP-1 (97).
Tumor necrosis factor is a therapeutic target in several diseases and anti-TNF treatment is successfully applied in rheumatoid arthritis, psoriasis, ankylosing spondylitis, and IBD. Considering the effects of TNF on L cells and data from animal models, indicating that impaired glucose tolerance following high-fat diet-induced obesity can be ameliorated by anti-TNF therapy (95), one might expect a significant impact of anti-TNF treatment on glucose metabolism. However, inhibition of TNF does not alter the state of insulin resistance in IBD patients (98, 99), and no differences in plasma insulin, glucose, and insulin resistance were noted in pediatric Crohn’s disease (CD) patients when comparing pre- and post infliximab (a chimeric anti-TNF antibody) treatment measurements (100). Furthermore, long-term therapy with etanercept did not alter fasting GIP-1 levels in patients with active rheumatoid arthritis. No changes were observed in these patients before and after anti-TNF treatment in the plasma GLP-1-response to an oral glucose challenge (101). In summary, these data underline that there is no simple correlation between TNF, GLP-1, and glycemic control under physiological conditions, but an interrelated disease network, including numerous factors.
Interleukin 6
Next to TNF, the cytokine IL-6 possesses the best-documented impact on incretin hormone secretion. IL-6 exerts pro-inflammatory as well as anti-inflammatory effects depending on its source and the context of secretion. T cells, macrophages, adipocytes, and myocytes are among the cells secreting IL-6. As a pro-inflammatory cytokine, IL-6 participates in the acute phase reaction and stimulates immune responses, especially during infection and tissue damage (102). In this context, inhibition of the IL-6 pathway was successfully applied in the treatment of rheumatoid arthritis (103). The anti-inflammatory properties comprise inhibition of TNF and IL-1 as well as activation of IL-10 (102). Within the intestine, IL-6 has been shown to prevent epithelial apoptosis and promote epithelial proliferation during injury and inflammation (104). IL-6 is also produced in muscle, with increased levels following muscle contraction. During exercise, it is thought to act in a hormone-like manner to stimulate energy mobilization, leading to increased body temperature and highlighting its metabolic functions (102). Obesity and T2D are associated with elevated plasma concentrations of IL-6, with adipose tissue being the major source under these conditions (105, 106). Several studies demonstrated IL-6 to regulate glucose tolerance and insulin action (107, 108) and furthermore, IL-6 was shown to enhance insulin secretion by increasing GLP-1 production in L cells and alpha cells, leading to improved glucose tolerance (47). Hence, IL-6 was suggested to mediate crosstalk between insulin-sensitive tissues, intestinal L cells, and pancreatic islets through GLP-1. In the CNS, IL-1 and IL-6 were demonstrated to mediate GLP-1 receptor-induced suppression of food intake, and body weight (109). Additionally, significant associations of GLP-1 were found with markers of inflammation, including IL-6 and C-reactive protein (CRP) in critically ill patients, and endotoxin, IL-1, and IL-6 were sufficient to induce GLP-1 secretion in mice (61). Conversely, endotoxin-dependent hyperinsulinemia was markedly blunted by the use of GLP-1 receptor antagonists or in IL-6 knockout mice (61).
Several other cytokines and mechanisms have been implicated at the interface of metabolism and inflammation. For example, IL-10 has been shown to prevent diet-induced insulin resistance (110), and IL-6 and TNF are known to decrease levels of the protective adipokine adiponectin (111). RANTES, a pro-inflammatory chemokine, was demonstrated to reduce glucose-dependent secretion of GLP-1 and GLP-2, thereby impairing glucose-induced insulin secretion in mice (112).
ER Unfolded Protein Response
A cellular condition on which metabolically mediated and inflammation-driven pathologies converge is ER stress and the associated UPR. Underlying various diseases, such as neurodegenerative disorders, diabetes, cancer, atherosclerosis, and IBD, the ER UPR has gained enormous attention during the past years (8). In mammalian cells, the ER is essential for cholesterol production, for calcium homeostasis, and for the transit of correctly folded proteins to the extracellular space. Hence, a functional ER UPR is essential to all secretory cells. Among the conditions that challenge ER functions and elicit ER stress responses are changes in calcium homeostasis or redox status, elevated protein synthesis, accumulation of unfolded or misfolded proteins, energy deficiency, and microbial infections (113). The purpose of the ER UPR is to restore ER homeostasis by enhancing protein degradation, reducing protein synthesis and expanding the protein folding capacity by upregulation of chaperones that help proteins in the ER lumen to fold (114). However, if the ER stress is prolonged or excessive, ER UPR can ultimately lead to cell death via apoptotic pathways (115, 116).
Inflammation and ER stress are linked at many levels; inflammation is characterized by the production of large amounts of proteins, such as cytokines or chemokines and, furthermore, studies using mice deficient in ER UPR-mediators link ER stress in the highly secretory subtypes of IEC, antimicrobial peptides-producing Paneth cells and mucin-producing goblet cells, with antimicrobial defense and intestinal inflammation (117–119). In addition, ER UPR-signaling can directly intersect with inflammatory pathways, including NF-κB, TLR-mediated signaling, and production of reactive oxygen species (ROS) (120–123). Yet, ER UPR is involved not only in inflammatory responses but also in metabolic processes and adipocyte differentiation (124, 125).
It has been suggested that the ER is essential in the coordination of metabolic responses through its ability to control the synthetic and catabolic pathways of various nutrients (126). These features are reflected by the responsiveness of the ER UPR to the nutritional state of mammalian cells (127). ER UPR impacts glucose as well as lipid metabolism (8). Consequently, there is strong evidence that ER UPR plays a critical role in pancreatic β cell survival and is relevant in the pathology of diabetes, obesity, and insulin resistance. Pancreatic β cells need to dramatically increase their insulin production to fit the demand under chronic insulin resistance and, hence, pancreatic islets from mice and humans with T2D show signs of ER UPR (128). Several factors associated with obesity and T2D, such as inflammatory cytokines and free FA, can induce ER UPR (129, 130). In particular, ER UPR-mediated activation of JNK has been linked to the development of insulin resistance and diabetes by inhibition of insulin receptor signaling (131); and various animal models bearing modifications in ER UPR-associated functions demonstrate defects in pancreatic β cells and impaired glucose metabolism (131–134).
Notably, several chemicals used to treat T2D like PPAR agonists or salicylates have been shown to affect ER UPR-associated pathways (135, 136). In murine models of obesity and diabetes, administration of the chemical chaperones phenyl butyric acid (PBA) and tauro-ursodeoxycholic acid (TUDCA), increased systemic insulin sensitivity, established normoglycemia, reduced fatty liver disease, and suppressed inflammatory signaling (137). PBA and TUDCA were furthermore shown to prevent ER stress-induced inhibition of apoB100 secretion, a feature contributing to hepatic steatosis (138) and to ameliorate atherosclerosis in mouse models (139). In the context of IBD, oral administration of either PBA or TUDCA reduced the severity of dextran sulfate sodium (DSS)-induced acute colitis as well as chronic colitis in IL-10-deficient mice (140). During intestinal inflammation, the loss of secretory Paneth and goblet cell is a well-described phenomenon. This effect has been linked to ER UPR-signaling (118, 119, 141), and ER stress has also been demonstrated to counteract epithelial stemness (142). Interestingly, intestinal L cells have been reported to be prone to gluco- and lipotoxicity, with lipotoxicity being associated with activated ER stress response (143). Yet, there are virtually no reports on ER stress, ER UPR, and the functional consequences in L cells under disease conditions. However, it has been shown that GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing UPR and promoting macroautophagy (69), and that postoperative increases in circulating cholic acid concentration contribute to improvements in glucose homeostasis after IT surgery by ameliorating ER stress (144). In conclusion, ER stress and the associated signaling might be a promising target for further research and future therapeutic interventions on the level of EEC.
Intestinal Inflammation and Enteroendocrine Cells
Intestinal epithelial cells are crucial for maintaining intestinal homeostasis, constituting an interface between the two major factors influencing intestinal inflammation, the gut microbiota and the immune system (12). IEC directly sense enteric luminal bacteria and interact with immune cells of the lamina propria as well as IELs and are, therefore, considered to be a constitutive component of the mucosal immune system (12, 145). As mentioned above, alterations in IEC subpopulations have been described previously in the context of intestinal inflammation, in particular for mucin-producing goblet cell and antimicrobial-peptide-producing Paneth cells. Yet, alterations in EEC numbers and secretion of gut hormones have also been observed in intestinal inflammation and were implicated in the changes in feeding patterns often accompanying these conditions (146, 147) (Figure 4).
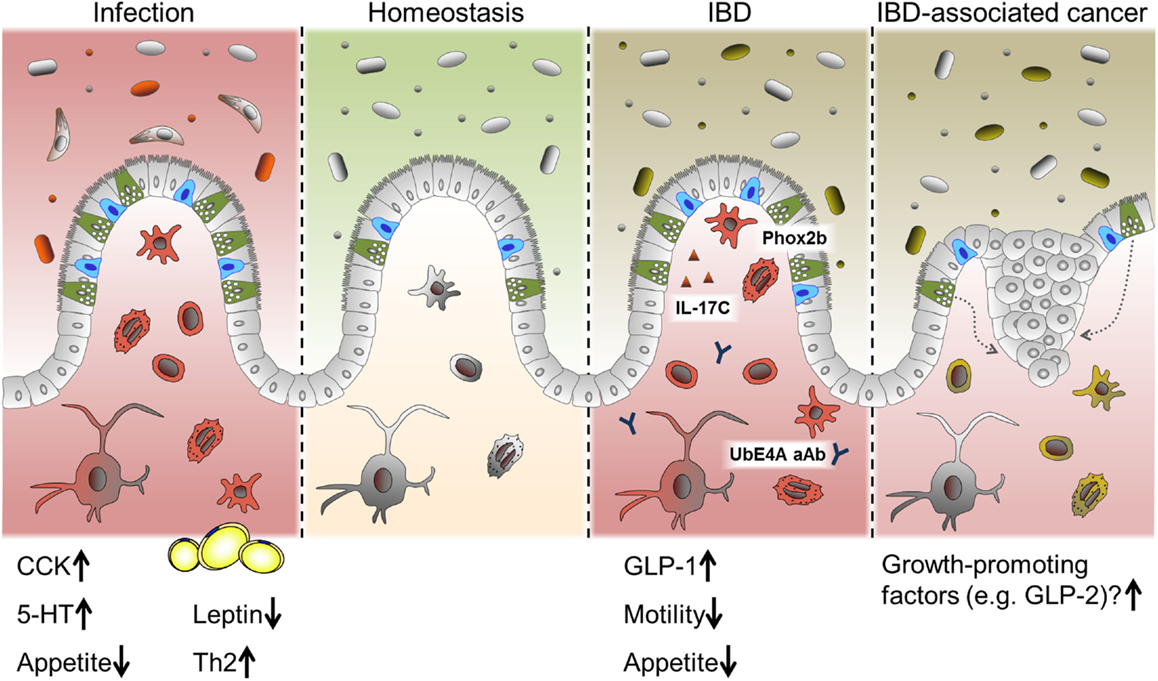
Figure 4. EEC in intestinal pathologies. Alterations in EEC and EEC-derived hormones have been reported under conditions of acute intestinal inflammation such as infections and chronic intestinal inflammation like IBD. While CCK and 5-HT are predominantly induced during infection, several roles for EEC are proposed in IBD. Suggested mechanism include a SNIP in the EEC-associated transcription factor Phox2b, auto-antibodies (aAB) against the EEC protein UbE4A, IL-17C secretion and increased levels of GLP-1. Changes in gut motility and appetite are also reported during intestinal inflammation. Long-standing inflammation like in persistent IBD is associated with an increased risk of developing carcinomas. Growth-promoting factors produced by EEC might contribute to this progression.
Enteroendocrine Cells during Infection
In intestinal infection, responses on the level of EEC seem to be a conserved mechanism throughout different species and infectious agents. In this context, CCK-positive cells appear to be a main target of regulation, and increases in this cell type have been reported in fish, lamb, pigs, mice, and humans during infection (42, 53, 148). Interestingly, hyperplasia of selected EEC subtypes in response to intestinal infection is probably dependent on immune cells, since serotonin (5-HT)-producing cell hyperplasia associated with Citrobacter rodentium infection was absent in mice lacking adaptive immunity (53) and CD4+ T cells were necessary to increase numbers of 5-HT producing enterochromaffin cells during helminth infection (149). Of note, increased levels of CCK are correlated with the period of hypophagia seen during enteritis. In an elegant study, Worthington et al. demonstrated that the reduction of fat mass and the resulting decrease in the adipokine leptin due to hyperphagia, was necessary for an effective Th2 immune response and, subsequently, efficient parasite expulsion (146). Intriguingly, in human lymphocytic colitis, a condition with unclear etiology and characterized by accumulation of lymphocytes in the colonic epithelium and lamina propria, both chromogranin A (CgA) and PYY-positive cell densities were reported to be increased (150, 151). These results illustrate a different layer of the interaction of EEC/gut hormones, metabolism, and immunmodulation.
Enteroendocrine Cells in IBD
Inflammatory bowel diseases and its two main idiopathic pathologies ulcerative colitis (UC) and CD are chronic, immunologically mediated disorders of the gastrointestinal tract. They are characterized by relapsing inflammations of the colon (UC) and the whole gastrointestinal tract (CD), respectively. UC as well as CD are multifactorial diseases and are associated with alterations of the innate and adaptive immune system, luminal and mucosa-associated microbiota as well as epithelial function (3). A failure to control inflammatory processes at the IEC level may critically contribute to IBD pathogenesis. So far, EEC have not attracted much interest in the research on IBD pathology. Yet, a gene wide association study (GWAS) has identified a single nuclear polymorphism (SNIP) in the enteroendocrine-associated transcription factor Phox2b as risk factor for Crohn’s disease (152) and auto-antibodies to the enteroendocrine protein UbE4A are associated with disease behavior (153). Utilizing double immunofluorescence techniques, a co-localization of Phox2b with CgA and GLP-1 was demonstrated (154). In this context, it is noteworthy that EEC have been observed to be a specific target of immune responses resulting in EEC depletion during acute small intestinal allograft rejection (155). Moreover, EEC have recently been identified as producers of IL-17C in IBD, a novel member of the IL-17 cytokine family, and most likely involved in the pathogenesis of active IBD (156).
Patients suffering from IBD often display metabolic changes concomitantly to altered adipokine levels and increased inflammatory parameters (157–160). While leptin level was shown to be inversely related to disease activity, resistin level was increased in patients with active disease and was identified as independent predictor of disease activity in CD (157, 160, 161). Insulin resistance and changes in lipid metabolism are a common phenomenon in IBD. Of note, the relative insulin resistance observed is in most cases due to increased insulin levels, whereas serum glucose remains normal (158–160, 162). Interestingly, Valentini et al. reported hyperinsulinemia in IBD patients to be associated with a decrease in adiponectin and proved hyperinsulinemia to be an independent protective factor for 6-month maintenance of remission (160). Intestinal inflammation is associated with elevated levels of circulating free FA in IBD. It has been suggested that these alterations reflect an “energy appeal reaction” of the organism providing free energy in the circulation, which is needed by inflammatory cells (158). Besides, reduced appetite, anorexia and altered intestinal motility often accompany intestinal inflammation, and might be linked to EEC, since PYY, GLP-1, and CCK signal satiety and modify motility (14, 163, 164).
Increased serum levels of human pancreatic polypeptide (HPP), gastrin, motilin, CCK, PYY, ghrelin, and also GIP, GLP-1, and GLP-2 have been reported either at baseline or postprandially in UC and/or CD (154, 163, 165–168). Several of these studies show conflicting results, which might be partly due to technical advancements, since some of the results have been published more than 30 years ago. However, some alterations in serum levels were reflected by changes in EEC-subtype numbers found by immunohistochemistry, substantiating these observations (154, 166, 169). Overall, the total number of studies and patients is limited, especially when regarding the heterogeneity of IBD pathologies. Yet, an increase in total EEC numbers defined as CgA-positive cells has been reported for ileal CD (154, 169). Furthermore underlining the link between EEC and chronic intestinal inflammation, T-cell receptor α knockout mice displaying reduced cytokine levels and decreased EEC numbers, develop an UC-like phenotype (52). For GLP-1, elevated levels were found in CD (165) and UC (167), whereas no differences were found in CD specifically affecting the small bowel (163). Noteworthy, Moran et al. showed increased numbers of GLP-1 and CgA-positive cells in terminal ileal CD, which was confined to the site of active inflammation. Neither in the presence of active colitis (without ileal involvement) nor in quiescent ileitis, EEC numbers were changed in the terminal ileum (154). This might indicate total changes in hormone secretion to be too minor to be recovered in serum due to the limited tissue affected. Yet, the changes in EEC numbers and activity observed seemed to be cell-type specific, since PYY expression remained unaltered, suggesting a selective induction of GLP-1 rather than a simple global upregulation of all EEC lineages and their products (154). On the other hand, DPP-4, the incretin degrading endopeptidase, has been found to be reduced in tissue and plasma in active CD (170) and further inhibition of DPP-4 accelerated mucosal healing in a murine model of colitis (171). Locally increased GLP-1 levels might exert paracrine functions on IEL (50) and afferent neurons, impacting mucosal immune responses and feeding patterns, respectively. Appetite is reduced in active small bowel CD, and in a subset of patients with IBD, GE is delayed, and prolonged GE is associated with higher disease activity and increased secretion of GLP-1. Conversely, GE is accelerated and GLP-1 release decreases significantly, following effective therapy (164).
The upregulation of GLP-1 positive cells might be linked to an increase in GLP-2, an L-cell-derived peptide and a further splice product of the gcg gene. GLP-2 is an epithelial growth factor implicated in epithelial homeostasis, barrier function, and repair following injury. GLP-2 exerts anti-inflammatory functions, such as sustaining proper Paneth cell function (172), and has been shown to ameliorate experimental colitis in several animal models (173–176). More important, a GLP-2 analog was proven effective in the treatment of active moderate to severe CD (177) and improved intestinal functions in patients with short bowel syndrome (178, 179).
In the context of epithelial homeostasis and wound healing, it is remarkable that incretin gene expression is regulated through the Wnt signaling pathway (180, 181). Wnt signals determine the intestinal stem cell niche and regulate intestinal stemness and proliferation (182). Chronic and acute intestinal inflammation is associated with alterations in cell proliferation most likely due to tissue insults and IEC loss. GLP-1 itself is capable of stimulating the Wnt pathway in an autocrine manner (183), suggesting a further potential mechanism linking incretins to intestinal tissue homeostasis.
On the other hand, clinical data enlighten further aspects of EEC function and metabolism. Surgical resection of affected parts of the intestine and colectomy are part of IBD treatment. Ileostomists show a significant reduction in circulating GLP-1 associated with elevated glucagon concentrations, indicating the colonic endocrine tissue to have an important role in postprandial metabolism (184). Enlightening a different aspect of the role of EEC under pathological conditions, long-standing intestinal inflammation is associated with an increased risk of developing adenocarcinomas. It has been suggested that an increased EEC mass in response to chronic inflammatory injury drives neoplasia by producing trophic hormones (185).
Enteroendocrine cells alterations rather seem to be a general feature of intestinal inflammation than a specific mechanism of IBD. To clarify if these alterations represent only a consequence or causatively contribute to intestinal inflammation, more research is needed. Yet, EEC and their products might modulate IBD pathology through orchestrating a metabolic-inflammatory response (Figures 1 and 5).
Microbiota and Enteroendocrine Cells
In recent years, the intestinal microbiota and its correlation with numerous human pathologies has gained increasing attention. There is a complex interaction between host genetic and metabolic makeup, diet, and microbiota (Figure 5). Changes in terms of diversity and richness have been associated with conditions, such as obesity, T2D, Parkinson’s disease, cardiovascular disease, and IBD. Manipulating the microbial composition is, therefore, an attractive therapeutic approach; probiotics as well as prebiotics have been demonstrated to exert multiple beneficial effects in IBD (186) and T2D (187), including increased GLP-1 release (188). Concurrently, evidence from studies with GF mice indicates a huge impact of the microbiota on EEC numbers (189). More specifically, Glp1r knockout mice display an altered composition of their intestinal microbiota (50). As described above, EEC can directly sense their microbial environment via TLR and through metabolites generated the microbiota. Regarding GLP-1 expression, SCFA and bile acids are the best-studied links between microbiota and gut hormone secretion.
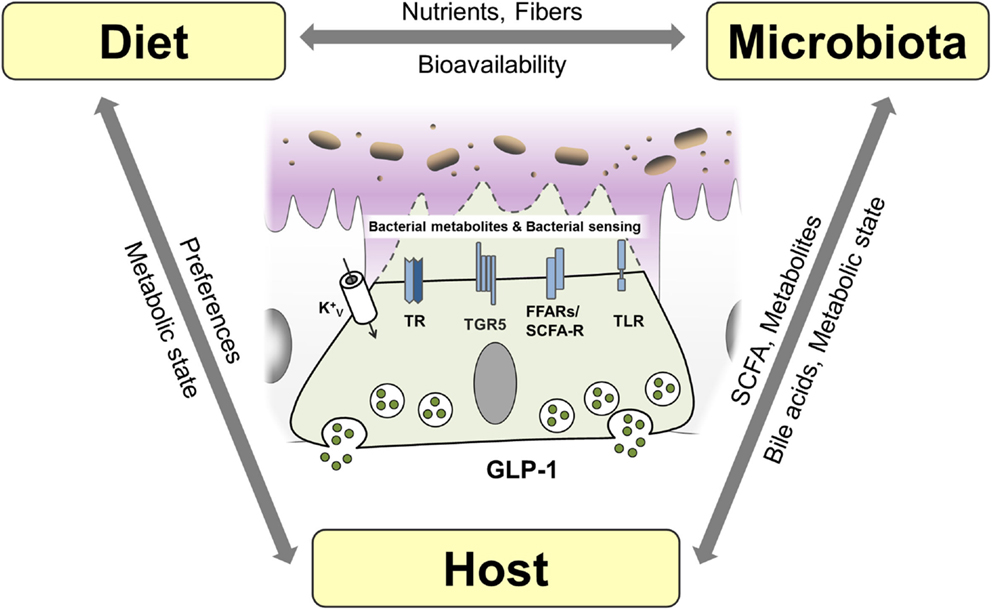
Figure 5. Interaction of host, diet, and microbiota and proposed mechanisms of bacterial sensing in EEC. EEC are able sense bacterial products such as short-chain fatty acids (SCFA) and the bacterial metabolite indole as well as bacteria through taste receptors (TR), free fatty acid receptors (FFAR)/SCFA receptors and K+ channels. Bile acids are sensed by EEC via TGR5 and can be metabolized and conjugated by the microbiota impacting host metabolism. Diet, microbiota and host genetic and metabolic makeup are interrelated factors influencing EEC function.
Short-chain fatty acids are produced by bacteria in the distal gut by fermentation of fibers and exert various beneficial effects on human health (190). In particular, butyrate has been shown to ameliorate mucosal inflammation and oxidative status, improving intestinal barrier, preventing colorectal cancer and beyond these intestinal effects, to reduce food intake, improve hypercholesterolemia, insulin resistance, and ischemic stroke (191). SCFA are able to modulate cytokine and chemokine expression of immune cells and adipocytes (190). They are sensed by L cells via FFAR2/GPR43 and FFAR3/GPR41, and activation of these receptors triggers GLP-1 secretion (15). The importance of SCFA-mediated GLP-1 secretion is highlighted by the observation that FFAR2 knockout mice exhibit reduced insulin levels and impaired glucose tolerance due to reduced GLP-1 levels (24). Furthermore, butyrate-induced GLP-1 secretion is attenuated in FFAR3 knockout mice (51). Interestingly, it was demonstrated that different compositions of the intestinal microbiota via their distinct abilities to produce SCFA might in turn affect expression of FFAR by epigenetic gene regulation (192). Conversely, modulating the composition of the microbiota using the probiotic VLS#3, stimulated production of butyrate, promoting GLP-1 secretion, and improving the metabolic state in mice (193). Of note, VSL#3 has also been shown to be effective in the treatment of IBD (186).
There is only limited data on the role of specific bacterial species on incretins; however, in an elegant study, Everard et al. showed that the abundance of Akkermansia muciniphila was associated with L cell number and secretory capacity (194). A. muciniphila is a mucin-degrading, acetate and propionate-producing bacterium residing in the mucus layer, thus being in relative close proximity to the intestinal epithelial layer (195), hence having the potential capacity to activate GPR43. Interestingly, A. muciniphila has been shown to be reduced in obese and T2D mice and that administration of A. muciniphila for 4 weeks was sufficient to reverse high-fat diet-induced obesity and T2D (196).
In contrast to SCFA, bile acids are produced by the host. Synthetized from cholesterol in the liver, bile acids facilitate formation of micelles in the intestine, promoting absorption of dietary fat (197), but additionally, they are increasingly recognized to act as signaling molecules and receptor ligands (198). The hepatic bile acid synthesis is tightly regulated by negative feedback mechanisms (199) and by transforming primary into secondary bile acid species, the intestinal microbiota strongly affects bile acid metabolism (200). Thus, modulating the bioavailability and resorption of bile acids, the microbiota impacts on enterohepatic bile acid-signaling and greatly impacts the whole-body metabolic homeostasis (199). Next to the bile acid receptor TGR5, which is directly linked to GLP-1 secretion from L cells, the farnesoid X receptor (FXR) was implicated in bile acid-induced metabolic alterations. Activation of FXR by the gut microbiota was shown to reduce the expression levels of most bile acid synthesis enzymes and inhibit the expression of gluconeogenic genes (201, 202), thereby linking the intestinal microbiota to the regulation of bile acids and metabolic homeostasis. Conversely, bile acids impact the bacterial composition of the microbiota by exhibiting strong antimicrobial functions (203).
Bile-acid sequestrants are used to sequester bile acids in the intestine, leading to increased bile acid synthesis and consequently to a reduction in low-density lipoprotein cholesterol. In patients with T2D, bile-acid sequestrants have furthermore been shown to improve glucose control. This effect was suggested to be due to multiple mechanisms, including alteration of the bile acid pool, changes in microbiota composition, improvement of hepatic glucose metabolism, and increased release of incretin hormones (204, 205). Also, some of the remarkable metabolic effects of bariatric surgery have been attributed to altered bile acid signaling (199). In line, TGR5 and FXR agonists have shown promising results in the treatment of metabolic and inflammatory diseases, such as IBD and T2D (37). Modulating the microbiota/SCFA/bile acid-signaling to impact metabolism and inflammatory processes via GLP-1 and other mechanisms, therefore, represents a new strategy for the treatment of several chronic diseases.
Conclusion and Perspective
Exploring the role of EEC and incretin hormones beyond blood glucose control is still in its infancy. In the context of inflammation, incretins might be a double-edged sword, since peptide hormones secreted by EEC not only exert direct immunomodulatory effects on diverse immune cell subsets (14) but GIP signaling has also been associated with pro-inflammatory processes and insulin resistance in mice (206). The complexity of the immune–endocrine axis is further highlighted by the unexpected finding that mice overexpressing TNF are protected from high-fat diet-induced insulin resistance (207). Along this line, IL-6 knockout mice only display an altered metabolic phenotype when fed a high-fat diet (208). To tackle the question on the role of EEC under inflammatory conditions, in particular if and how they are involved in the onset of diseases, more research is needed.
Multiple drugs targeting GLP-1 functions, ER UPR, bile acid signaling, and the microbiota have already proven to be effective in the treatment of T2D and IBD (Figure 6). Yet, there seem to be much more possibilities and fields of applications for these therapeutics and more still to be developed. Promising new experimental tools and models, such as intestinal organoids (209), will allow elucidating remaining questions on EEC functions and may permit identification of new therapeutic and nutriceutical targets.
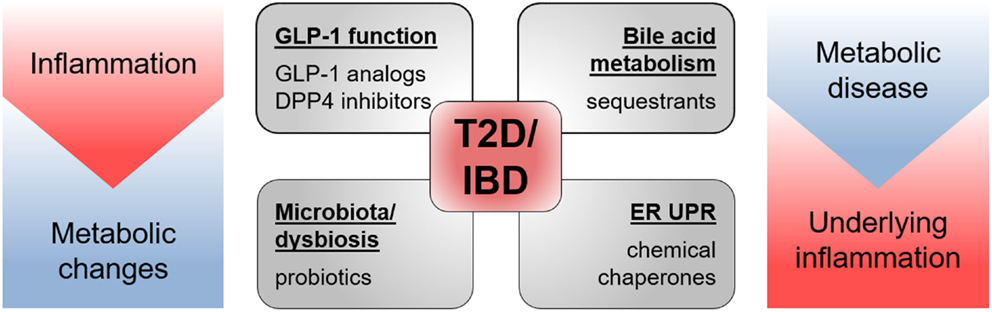
Figure 6. Drugs targeting T2D and IBD pathology. Drugs shown to be effective in animal models of and/or in patients with T2D as well as IBD target GLP-1 function, bile acid metabolism, microbiota, and endoplasmic reticulum unfolded protein response (ER UPR). Both pathologies share metabolic changes and inflammatory processes in their etiology.
Author Contributions
All authors listed, have made substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Isabel Gosch and Johannes Zang for their helpful suggestions on the figures and support.
Funding
This work was supported by the German Research Foundation (DFG) and the Technical University of Munich (TUM) in the framework of the Open Access Publishing Program.
References
1. Ogden CL, Lamb MM, Carroll MD, Flegal KM. Obesity and socioeconomic status in children and adolescents: United States, 2005-2008. NCHS Data Brief (2010) (51):1–8.
2. Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world – a growing challenge. N Engl J Med (2007) 356(3):213–5. doi:10.1056/NEJMp068177
3. Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol (2006) 3(7):390–407. doi:10.1038/ncpgasthep0528
4. Rodgers A, Ezzati M, Vander Hoorn S, Lopez AD, Lin RB, Murray CJ, et al. Distribution of major health risks: findings from the Global Burden of Disease study. PLoS Med (2004) 1(1):e27. doi:10.1371/journal.pmed.0010027
5. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444(7121):860–7. doi:10.1038/nature05485
6. Nielsen ST, Janum S, Krogh-Madsen R, Solomon TP, Møller K. The incretin effect in critically ill patients: a case-control study. Crit Care (2015) 19(1):402. doi:10.1186/s13054-015-1118-z
7. Steven S, Hausding M, Kröller-Schön S, Mader M, Mikhed Y, Stamm P, et al. Gliptin and GLP-1 analog treatment improves survival and vascular inflammation/dysfunction in animals with lipopolysaccharide-induced endotoxemia. Basic Res Cardiol (2015) 110(2):6. doi:10.1007/s00395-015-0465-x
8. Rath E, Haller D. Inflammation and cellular stress: a mechanistic link between immune-mediated and metabolically driven pathologies. Eur J Nutr (2011) 50(4):219–33. doi:10.1007/s00394-011-0197-0
9. Brestoff JR, Artis D. Immune regulation of metabolic homeostasis in health and disease. Cell (2015) 161(1):146–60. doi:10.1016/j.cell.2015.02.022
10. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (1993) 259(5091):87–91. doi:10.1126/science.7678183
11. Clavel T, Desmarchelier C, Haller D, Gérard P, Rohn S, Lepage P, et al. Intestinal microbiota in metabolic diseases: from bacterial community structure and functions to species of pathophysiological relevance. Gut Microbes (2014) 5(4):544–51. doi:10.4161/gmic.29331
12. Clavel T, Haller D. Bacteria- and host-derived mechanisms to control intestinal epithelial cell homeostasis: implications for chronic inflammation. Inflamm Bowel Dis (2007) 13(9):1153–64. doi:10.1002/ibd.20174
13. Helander HF, Fandriks L. Surface area of the digestive tract – revisited. Scand J Gastroenterol (2014) 49(6):681–9. doi:10.3109/00365521.2014.898326
14. Worthington JJ. The intestinal immunoendocrine axis: novel cross-talk between enteroendocrine cells and the immune system during infection and inflammatory disease. Biochem Soc Trans (2015) 43(4):727–33. doi:10.1042/BST20150090
15. Zietek T, Daniel H. Intestinal nutrient sensing and blood glucose control. Curr Opin Clin Nutr Metab Care (2015) 18(4):381–8. doi:10.1097/MCO.0000000000000187
16. Habib AM, Richards P, Cairns LS, Rogers GJ, Bannon CA, Parker HE, et al. Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology (2012) 153(7):3054–65. doi:10.1210/en.2011-2170
17. Egerod KL, Engelstoft MS, Grunddal KV, Nøhr MK, Secher A, Sakata I, et al. A major lineage of enteroendocrine cells coexpress CCK, secretin, GIP, GLP-1, PYY, and neurotensin but not somatostatin. Endocrinology (2012) 153(12):5782–95. doi:10.1210/en.2012-1595
18. Sykaras AG, Demenis C, Cheng L, Pisitkun T, Mclaughlin JT, Fenton RA, et al. Duodenal CCK cells from male mice express multiple hormones including ghrelin. Endocrinology (2014) 155(9):3339–51. doi:10.1210/en.2013-2165
19. Gribble FM, Reimann F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu Rev Physiol (2016) 78:277–99. doi:10.1146/annurev-physiol-021115-105439
20. Nakamura E, Hasumura M, Uneyama H, Torii K. Luminal amino acid-sensing cells in gastric mucosa. Digestion (2011) 83(Suppl 1):13–8. doi:10.1159/000323399
21. Choi S, Lee M, Shiu AL, Yo SJ, Halldén G, Aponte GW. GPR93 activation by protein hydrolysate induces CCK transcription and secretion in STC-1 cells. Am J Physiol Gastrointest Liver Physiol (2007) 292(5):G1366–75. doi:10.1152/ajpgi.00516.2006
22. Oya M, Kitaguchi T, Pais R, Reimann F, Gribble F, Tsuboi T. The G protein-coupled receptor family C group 6 subtype A (GPRC6A) receptor is involved in amino acid-induced glucagon-like peptide-1 secretion from GLUTag cells. J Biol Chem (2013) 288(7):4513–21. doi:10.1074/jbc.M112.402677
23. Wang JH, Inoue T, Higashiyama M, Guth PH, Engel E, Kaunitz JD, et al. Umami receptor activation increases duodenal bicarbonate secretion via glucagon-like peptide-2 release in rats. J Pharmacol Exp Ther (2011) 339(2):464–73. doi:10.1124/jpet.111.184788
24. Margolskee RF, Dyer J, Kokrashvili Z, Salmon KS, Ilegems E, Daly K, et al. T1R3 and gustducin in gut sense sugars to regulate expression of Na+-glucose cotransporter 1. Proc Natl Acad Sci U S A (2007) 104(38):15075–80. doi:10.1073/pnas.0706678104
25. Aranias T, Grosfeld A, Poitou C, Omar AA, Le Gall M, Miquel S, et al. Lipid-rich diet enhances L-cell density in obese subjects and in mice through improved L-cell differentiation. J Nutr Sci (2015) 4:e22. doi:10.1017/jns.2015.11
26. Behrens M, Meyerhof W. Gustatory and extragustatory functions of mammalian taste receptors. Physiol Behav (2011) 105(1):4–13. doi:10.1016/j.physbeh.2011.02.010
27. Jeon TI, Zhu B, Larson JL, Osborne TF. SREBP-2 regulates gut peptide secretion through intestinal bitter taste receptor signaling in mice. J Clin Invest (2008) 118(11):3693–700. doi:10.1172/JCI36461
28. Wu T, Zhao BR, Bound MJ, Checklin HL, Bellon M, Little TJ, et al. Effects of different sweet preloads on incretin hormone secretion, gastric emptying, and postprandial glycemia in healthy humans. Am J Clin Nutr (2012) 95(1):78–83. doi:10.3945/ajcn.111.021543
29. Ford HE, Peters V, Martin NM, Sleeth ML, Ghatei MA, Frost GS, et al. Effects of oral ingestion of sucralose on gut hormone response and appetite in healthy normal-weight subjects. Eur J Clin Nutr (2011) 65(4):508–13. doi:10.1038/ejcn.2010.291
30. Steinert RE, Frey F, Töpfer A, Drewe J, Beglinger C. Effects of carbohydrate sugars and artificial sweeteners on appetite and the secretion of gastrointestinal satiety peptides. Br J Nutr (2011) 105(9):1320–8. doi:10.1017/S000711451000512X
31. Vegezzi G, Anselmi L, Huynh J, Barocelli E, Rozengurt E, Raybould H, et al. Diet-induced regulation of bitter taste receptor subtypes in the mouse gastrointestinal tract. PLoS One (2014) 9(9):e107732. doi:10.1371/journal.pone.0107732
32. Latorre R, Huynh J, Mazzoni M, Gupta A, Bonora E, Clavenzani P, et al. Expression of the bitter taste receptor, T2R38, in enteroendocrine cells of the colonic mucosa of overweight/obese vs. lean subjects. PLoS One (2016) 11(2):e0147468. doi:10.1371/journal.pone.0147468
33. Lee RJ, Cohen NA. Taste receptors in innate immunity. Cell Mol Life Sci (2015) 72(2):217–36. doi:10.1007/s00018-014-1736-7
34. Nguyen AT, Mandard S, Dray C, Deckert V, Valet P, Besnard P, et al. Lipopolysaccharides-mediated increase in glucose-stimulated insulin secretion: involvement of the GLP-1 pathway. Diabetes (2014) 63(2):471–82. doi:10.2337/db13-0903
35. Wichmann A, Allahyar A, Greiner TU, Plovier H, Lundén GÖ, Larsson T, et al. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe (2013) 14(5):582–90. doi:10.1016/j.chom.2013.09.012
36. Chimerel C, Emery E, Summers DK, Keyser U, Gribble FM, Reimann F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Rep (2014) 9(4):1202–8. doi:10.1016/j.celrep.2014.10.032
37. Stepanov V, Stankov K, Mikov M. The bile acid membrane receptor TGR5: a novel pharmacological target in metabolic, inflammatory and neoplastic disorders. J Recept Signal Transduct Res (2013) 33(4):213–23. doi:10.3109/10799893.2013.802805
38. Palazzo M, Balsari A, Rossini A, Selleri S, Calcaterra C, Gariboldi S, et al. Activation of enteroendocrine cells via TLRs induces hormone, chemokine, and defensin secretion. J Immunol (2007) 178(7):4296–303. doi:10.4049/jimmunol.178.7.4296
39. Selleri S, Palazzo M, Deola S, Wang E, Balsari A, Marincola FM, et al. Induction of pro-inflammatory programs in enteroendocrine cells by the Toll-like receptor agonists flagellin and bacterial LPS. Int Immunol (2008) 20(8):961–70. doi:10.1093/intimm/dxn055
40. Bogunovic M, Davé SH, Tilstra JS, Chang DT, Harpaz N, Xiong H, et al. Enteroendocrine cells express functional Toll-like receptors. Am J Physiol Gastrointest Liver Physiol (2007) 292(6):G1770–83. doi:10.1152/ajpgi.00249.2006
41. Zhang WJ, Duan JZ, Lei N, Xing H, Shao Y, Yang GB. Cellular bases for interactions between immunocytes and enteroendocrine cells in the intestinal mucosal barrier of rhesus macaques. Cell Tissue Res (2012) 350(1):135–41. doi:10.1007/s00441-012-1464-y
42. Moran GW, Leslie FC, Levison SE, Worthington J, McLaughlin JT. Enteroendocrine cells: neglected players in gastrointestinal disorders? Ther Adv Gastroenterol (2008) 1(1):51–60. doi:10.1177/1756283X08093943
43. Ingersoll SA, Ayyadurai S, Charania MA, Laroui H, Yan Y, Merlin D. The role and pathophysiological relevance of membrane transporter PepT1 in intestinal inflammation and inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol (2012) 302(5):G484–92. doi:10.1152/ajpgi.00477.2011
44. Hu Y, Smith DE, Ma K, Jappar D, Thomas W, Hillgren KM. Targeted disruption of peptide transporter Pept1 gene in mice significantly reduces dipeptide absorption in intestine. Mol Pharm (2008) 5(6):1122–30. doi:10.1021/mp8001655
45. Wuensch T, Ullrich S, Schulz S, Chamaillard M, Schaltenberg N, Rath E, et al. Colonic expression of the peptide transporter PEPT1 is downregulated during intestinal inflammation and is not required for NOD2-dependent immune activation. Inflamm Bowel Dis (2014) 20(4):671–84. doi:10.1097/01.MIB.0000443336.71488.08
46. Dalmasso G, Nguyen HT, Ingersoll SA, Ayyadurai S, Laroui H, Charania MA, et al. The PepT1-NOD2 signaling pathway aggravates induced colitis in mice. Gastroenterology (2011) 141(4):1334–45. doi:10.1053/j.gastro.2011.06.080
47. Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med (2011) 17(11):1481–9. doi:10.1038/nm.2513
48. Lim GE, Huang GJ, Flora N, LeRoith D, Rhodes CJ, Brubaker PL. Insulin regulates glucagon-like peptide-1 secretion from the enteroendocrine L cell. Endocrinology (2009) 150(2):580–91. doi:10.1210/en.2008-0726
49. Bullock BP, Heller RS, Habener JF. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology (1996) 137(7):2968–78. doi:10.1210/endo.137.7.8770921
50. Yusta B, Baggio LL, Koehler J, Holland D, Cao X, Pinnell LJ, et al. GLP-1R agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte GLP-1R. Diabetes (2015) 64(7):2537–49. doi:10.2337/db14-1577
51. Luyer MD, Greve JW, Hadfoune M, Jacobs JA, Dejong CH, Buurman WA. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J Exp Med (2005) 202(8):1023–9. doi:10.1084/jem.20042397
52. Rubin DC, Zhang H, Qian P, Lorenz RG, Hutton K, Peters MG. Altered enteroendocrine cell expression in T cell receptor alpha chain knock-out mice. Microsc Res Tech (2000) 51(2):112–20. doi:10.1002/1097-0029(20001015)51:2<112::AID-JEMT2>3.0.CO;2-D
53. O’Hara JR, Skinn AC, MacNaughton WK, Sherman PM, Sharkey KA. Consequences of Citrobacter rodentium infection on enteroendocrine cells and the enteric nervous system in the mouse colon. Cell Microbiol (2006) 8(4):646–60. doi:10.1111/j.1462-5822.2005.00657.x
54. Hadjiyanni I, Siminovitch KA, Danska JS, Drucker DJ. Glucagon-like peptide-1 receptor signalling selectively regulates murine lymphocyte proliferation and maintenance of peripheral regulatory T cells. Diabetologia (2010) 53(4):730–40. doi:10.1007/s00125-009-1643-x
55. Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science (1993) 261(5120):466–9. doi:10.1126/science.8101391
56. Morimoto C, Schlossman SF. The structure and function of CD26 in the T-cell immune response. Immunol Rev (1998) 161:55–70. doi:10.1111/j.1600-065X.1998.tb01571.x
57. Han JM, Patterson SJ, Speck M, Ehses JA, Levings MK. Insulin inhibits IL-10-mediated regulatory T cell function: implications for obesity. J Immunol (2014) 192(2):623–9. doi:10.4049/jimmunol.1302181
58. Yamaoka-Tojo M, Tojo T, Takahira N, Matsunaga A, Aoyama N, Masuda T, et al. Elevated circulating levels of an incretin hormone, glucagon-like peptide-1, are associated with metabolic components in high-risk patients with cardiovascular disease. Cardiovasc Diabetol (2010) 9:17. doi:10.1186/1475-2840-9-17
59. Piotrowski K, Becker M, Zugwurst J, Biller-Friedmann I, Spoettl G, Greif M, et al. Circulating concentrations of GLP-1 are associated with coronary atherosclerosis in humans. Cardiovasc Diabetol (2013) 12:117. doi:10.1186/1475-2840-12-117
60. Hattori A, Kawamura I, Yamada Y, Kanamori H, Aoyama T, Ushikoshi H, et al. Elevated plasma GLP-1 levels and enhanced expression of cardiac GLP-1 receptors as markers of left ventricular systolic dysfunction: a cross-sectional study. BMJ Open (2013) 3(9):e003201. doi:10.1136/bmjopen-2013-003201
61. Kahles F, Meyer C, Möllmann J, Diebold S, Findeisen HM, Lebherz C, et al. GLP-1 secretion is increased by inflammatory stimuli in an IL-6-dependent manner, leading to hyperinsulinemia and blood glucose lowering. Diabetes (2014) 63(10):3221–9. doi:10.2337/db14-0100
62. Nauck MA, Vilsbøll T, Gallwitz B, Garber A, Madsbad S. Incretin-based therapies: viewpoints on the way to consensus. Diabetes Care (2009) 32(Suppl 2):S223–31. doi:10.2337/dc09-S315
63. Azoulay L. Incretin-based drugs and adverse pancreatic events: almost a decade later and uncertainty remains. Diabetes Care (2015) 38(6):951–3. doi:10.2337/dc15-0347
64. Drucker DJ, Sherman SI, Gorelick FS, Bergenstal RM, Sherwin RS, Buse JB. Incretin-based therapies for the treatment of type 2 diabetes: evaluation of the risks and benefits. Diabetes Care (2010) 33(2):428–33. doi:10.2337/dc09-1499
65. Egan AG, Blind E, Dunder K, de Graeff PA, Hummer BT, Bourcier T, et al. Pancreatic safety of incretin-based drugs – FDA and EMA assessment. N Engl J Med (2014) 370(9):794–7. doi:10.1056/NEJMp1314078
66. Lovshin JA, Zinman B. Blood pressure-lowering effects of incretin-based diabetes therapies. Can J Diabetes (2014) 38(5):364–71. doi:10.1016/j.jcjd.2014.05.001
67. Krasner NM, Ido Y, Ruderman NB, Cacicedo JM. Glucagon-like peptide-1 (GLP-1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS One (2014) 9(5):e97554. doi:10.1371/journal.pone.0097554
68. El-Sahar AE, Safar MM, Zaki HF, Attia AS, Ain-Shoka AA. Sitagliptin attenuates transient cerebral ischemia/reperfusion injury in diabetic rats: implication of the oxidative-inflammatory-apoptotic pathway. Life Sci (2015) 126:81–6. doi:10.1016/j.lfs.2015.01.030
69. Sharma S, Mells JE, Fu PP, Saxena NK, Anania FA. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS One (2011) 6(9):e25269. doi:10.1371/journal.pone.0025269
70. Wang XC, Gusdon AM, Liu H, Qu S. Effects of glucagon-like peptide-1 receptor agonists on non-alcoholic fatty liver disease and inflammation. World J Gastroenterol (2014) 20(40):14821–30. doi:10.3748/wjg.v20.i40.14821
71. Higashijima Y, Tanaka T, Yamaguchi J, Tanaka S, Nangaku M. Anti-inflammatory role of DPP-4 inhibitors in a nondiabetic model of glomerular injury. Am J Physiol Renal Physiol (2015) 308(8):F878–87. doi:10.1152/ajprenal.00590.2014
72. Holscher C. The incretin hormones glucagonlike peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement (2014) 10(1 Suppl):S47–54. doi:10.1016/j.jalz.2013.12.009
73. Iwai T, Sawabe T, Tanimitsu K, Suzuki M, Sasaki-Hamada S, Oka J. Glucagon-like peptide-1 protects synaptic and learning functions from neuroinflammation in rodents. J Neurosci Res (2014) 92(4):446–54. doi:10.1002/jnr.23335
74. Chen CY, Tsai CY. From endocrine to rheumatism: do gut hormones play roles in rheumatoid arthritis? Rheumatology (Oxford) (2014) 53(2):205–12. doi:10.1093/rheumatology/ket255
75. Gou S, Zhu T, Wang W, Xiao M, Wang XC, Chen ZH. Glucagon like peptide-1 attenuates bleomycin-induced pulmonary fibrosis, involving the inactivation of NF-kappaB in mice. Int Immunopharmacol (2014) 22(2):498–504. doi:10.1016/j.intimp.2014.07.010
76. van Genugten RE, Möller-Goede DL, van Raalte DH, Diamant M. Extra-pancreatic effects of incretin-based therapies: potential benefit for cardiovascular-risk management in type 2 diabetes. Diabetes Obes Metab (2013) 15(7):593–606. doi:10.1111/dom.12050
77. Shiraki A, Oyama J, Komoda H, Asaka M, Komatsu A, Sakuma M, et al. The glucagon-like peptide 1 analog liraglutide reduces TNF-alpha-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis (2012) 221(2):375–82. doi:10.1016/j.atherosclerosis.2011.12.039
78. He L, Wong CK, Cheung KK, Yau HC, Fu A, Zhao HL, et al. Anti-inflammatory effects of exendin-4, a glucagon-like peptide-1 analog, on human peripheral lymphocytes in patients with type 2 diabetes. J Diabetes Investig (2013) 4(4):382–92. doi:10.1111/jdi.12063
79. Lee YS, Park MS, Choung JS, Kim SS, Oh HH, Choi CS, et al. Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia (2012) 55(9):2456–68. doi:10.1007/s00125-012-2592-3
80. Xu F, Li Z, Zheng X, Liu H, Liang H, Xu H, et al. SIRT1 mediates the effect of GLP-1 receptor agonist exenatide on ameliorating hepatic steatosis. Diabetes (2014) 63(11):3637–46. doi:10.2337/db14-0263
81. Yang J, Campitelli J, Hu G, Lin Y, Luo J, Xue C. Increase in DPP-IV in the intestine, liver and kidney of the rat treated with high fat diet and streptozotocin. Life Sci (2007) 81(4):272–9. doi:10.1016/j.lfs.2007.04.040
82. Lee SA, Kim YR, Yang EJ, Kwon EJ, Kim SH, Kang SH, et al. CD26/DPP4 levels in peripheral blood and T cells in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab (2013) 98(6):2553–61. doi:10.1210/jc.2012-4288
83. Yazbeck R, Howarth GS, Abbott CA. Dipeptidyl peptidase inhibitors, an emerging drug class for inflammatory disease? Trends Pharmacol Sci (2009) 30(11):600–7. doi:10.1016/j.tips.2009.08.003
84. Reinhold D, Bank U, Bühling F, Lendeckel U, Faust J, Neubert K, et al. Inhibitors of dipeptidyl peptidase IV induce secretion of transforming growth factor-beta 1 in PWM-stimulated PBMC and T cells. Immunology (1997) 91(3):354–60. doi:10.1046/j.1365-2567.1997.d01-2258.x
85. Zhao Y, Yang L, Zhou Z. Dipeptidyl peptidase-4 inhibitors: multitarget drugs, not only antidiabetes drugs. J Diabetes (2014) 6(1):21–9. doi:10.1111/1753-0407.12063
86. Vora KA, Porter G, Peng R, Cui Y, Pryor K, Eiermann G, et al. Genetic ablation or pharmacological blockade of dipeptidyl peptidase IV does not impact T cell-dependent immune responses. BMC Immunol (2009) 10:19. doi:10.1186/1471-2172-10-19
87. Aroor AR, McKarns S, Demarco VG, Jia G, Sowers JR. Maladaptive immune and inflammatory pathways lead to cardiovascular insulin resistance. Metabolism (2013) 62(11):1543–52. doi:10.1016/j.metabol.2013.07.001
88. Dai Y, Dai D, Wang X, Ding Z, Mehta JL. DPP-4 inhibitors repress NLRP3 inflammasome and interleukin-1beta via GLP-1 receptor in macrophages through protein kinase C pathway. Cardiovasc Drugs Ther (2014) 28(5):425–32. doi:10.1007/s10557-014-6539-4
89. Gonçalves A, Marques C, Leal E, Ribeiro CF, Reis F, Ambrósio AF, et al. Dipeptidyl peptidase-IV inhibition prevents blood-retinal barrier breakdown, inflammation and neuronal cell death in the retina of type 1 diabetic rats. Biochim Biophys Acta (2014) 1842(9):1454–63. doi:10.1016/j.bbadis.2014.04.013
90. Fenkci S, Rota S, Sabir N, Sermez Y, Guclu A, Akdag B. Relationship of serum interleukin-6 and tumor necrosis factor alpha levels with abdominal fat distribution evaluated by ultrasonography in overweight or obese postmenopausal women. J Investig Med (2006) 54(8):455–60. doi:10.2310/6650.2006.06010
91. Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta-analysis of cytokines in Alzheimer’s disease. Biol Psychiatry (2010) 68(10):930–41. doi:10.1016/j.biopsych.2010.06.012
92. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell (2001) 104(4):487–501. doi:10.1016/S0092-8674(01)00237-9
93. Brynskov J, Foegh P, Pedersen G, Ellervik C, Kirkegaard T, Bingham A, et al. Tumour necrosis factor alpha converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease. Gut (2002) 51(1):37–43. doi:10.1136/gut.51.1.37
94. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature (1997) 389(6651):610–4. doi:10.1038/39335
95. Gagnon J, Sauvé M, Zhao W, Stacey HM, Wiber SC, Bolz SS, et al. Chronic exposure to TNFalpha impairs secretion of glucagon-like peptide-1. Endocrinology (2015) 156(11):3950–60. doi:10.1210/en.2015-1361
96. Tsukahara T, Watanabe K, Watanabe T, Yamagami H, Sogawa M, Tanigawa T, et al. Tumor necrosis factor alpha decreases glucagon-like peptide-2 expression by up-regulating G-protein-coupled receptor 120 in Crohn disease. Am J Pathol (2015) 185(1):185–96. doi:10.1016/j.ajpath.2014.09.010
97. Lehrskov-Schmidt L, Lehrskov-Schmidt L, Nielsen ST, Holst JJ, Møller K, Solomon TP. The effects of TNF-alpha on GLP-1-stimulated plasma glucose kinetics. J Clin Endocrinol Metab (2015) 100(4):E616–22. doi:10.1210/jc.2014-4244
98. Koutroubakis IE, Oustamanolakis P, Malliaraki N, Karmiris K, Chalkiadakis I, Ganotakis E, et al. Effects of tumor necrosis factor alpha inhibition with infliximab on lipid levels and insulin resistance in patients with inflammatory bowel disease. Eur J Gastroenterol Hepatol (2009) 21(3):283–8. doi:10.1097/MEG.0b013e328325d42b
99. Tigas S, Tsatsoulis A. Endocrine and metabolic manifestations in inflammatory bowel disease. Ann Gastroenterol (2012) 25(1):37–44.
100. Steiner SJ, Pfefferkorn MD, Fitzgerald JF, Denne SC. Carbohydrate and lipid metabolism following infliximab therapy in pediatric Crohn’s disease. Pediatr Res (2008) 64(6):673–6. doi:10.1203/PDR.0b013e318186dde2
101. Chen CY, Tsai CY, Lee PC, Lee SD. Long-term etanercept therapy favors weight gain and ameliorates cachexia in rheumatoid arthritis patients: roles of gut hormones and leptin. Curr Pharm Des (2013) 19(10):1956–64. doi:10.2174/1381612811319100014
102. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol (2015) 16(5):448–57. doi:10.1038/ni.3153
103. Kremer JM, Blanco R, Brzosko M, Burgos-Vargas R, Halland AM, Vernon E, et al. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate: results from the double-blind treatment phase of a randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum (2011) 63(3):609–21. doi:10.1002/art.30158
104. Kuhn KA, Manieri NA, Liu TC, Stappenbeck TS. IL-6 stimulates intestinal epithelial proliferation and repair after injury. PLoS One (2014) 9(12):e114195. doi:10.1371/journal.pone.0114195
105. Herder C, Haastert B, Müller-Scholze S, Koenig W, Thorand B, Holle R, et al. Association of systemic chemokine concentrations with impaired glucose tolerance and type 2 diabetes: results from the Cooperative Health Research in the Region of Augsburg Survey S4 (KORA S4). Diabetes (2005) 54(Suppl 2):S11–7. doi:10.2337/diabetes.54.suppl_2.S11
106. Mohamed-Ali V, Goodrick S, Rawesh A, Katz DR, Miles JM, Yudkin JS, et al. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab (1997) 82(12):4196–200. doi:10.1210/jcem.82.12.4450
107. Holmes AG, Mesa JL, Neill BA, Chung J, Carey AL, Steinberg GR, et al. Prolonged interleukin-6 administration enhances glucose tolerance and increases skeletal muscle PPARalpha and UCP2 expression in rats. J Endocrinol (2008) 198(2):367–74. doi:10.1677/JOE-08-0113
108. Weigert C, Hennige AM, Lehmann R, Brodbeck K, Baumgartner F, Schaüble M, et al. Direct cross-talk of interleukin-6 and insulin signal transduction via insulin receptor substrate-1 in skeletal muscle cells. J Biol Chem (2006) 281(11):7060–7. doi:10.1074/jbc.M509782200
109. Shirazi R, Palsdottir V, Collander J, Anesten F, Vogel H, Langlet F, et al. Glucagon-like peptide 1 receptor induced suppression of food intake, and body weight is mediated by central IL-1 and IL-6. Proc Natl Acad Sci U S A (2013) 110(40):16199–204. doi:10.1073/pnas.1306799110
110. Hong EG, Ko HJ, Cho YR, Kim HJ, Ma Z, Yu TY, et al. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes (2009) 58(11):2525–35. doi:10.2337/db08-1261
111. Chakraborti CK. Role of adiponectin and some other factors linking type 2 diabetes mellitus and obesity. World J Diabetes (2015) 6(15):1296–308. doi:10.4239/wjd.v6.i15.1296
112. Pais R, Zietek T, Hauner H, Daniel H, Skurk T. RANTES (CCL5) reduces glucose-dependent secretion of glucagon-like peptides 1 and 2 and impairs glucose-induced insulin secretion in mice. Am J Physiol Gastrointest Liver Physiol (2014) 307(3):G330–7. doi:10.1152/ajpgi.00329.2013
113. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol (2007) 8(7):519–29. doi:10.1038/nrm2199
114. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem (2005) 74:739–89. doi:10.1146/annurev.biochem.73.011303.074134
115. Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol (2006) 4(11):e374. doi:10.1371/journal.pbio.0040374
116. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep (2006) 7(9):880–5. doi:10.1038/sj.embor.7400779
117. Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, et al. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest (2001) 107(5):585–93. doi:10.1172/JCI11476
118. Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med (2008) 5(3):e54. doi:10.1371/journal.pmed.0050054
119. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell (2008) 134(5):743–56. doi:10.1016/j.cell.2008.07.021
120. Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol (2010) 11(5):411–8. doi:10.1038/ni.1857
121. Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, et al. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol (2004) 24(23):10161–8. doi:10.1128/MCB.24.23.10161-10168.2004
122. Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ (2004) 11(4):372–80. doi:10.1038/sj.cdd.4401378
123. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol (2006) 26(8):3071–84. doi:10.1128/MCB.26.8.3071-3084.2006
124. Burkart A, Shi X, Chouinard M, Corvera S. Adenylate kinase 2 links mitochondrial energy metabolism to the induction of the unfolded protein response. J Biol Chem (2011) 286(6):4081–9. doi:10.1074/jbc.M110.134106
125. Pouyssegur J, Shiu RP, Pastan I. Induction of two transformation-sensitive membrane polypeptides in normal fibroblasts by a block in glycoprotein synthesis or glucose deprivation. Cell (1977) 11(4):941–7. doi:10.1016/0092-8674(77)90305-1
126. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell (2010) 140(6):900–17. doi:10.1016/j.cell.2010.02.034
127. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell (2001) 7(6):1165–76. doi:10.1016/S1097-2765(01)00265-9
128. Huang CJ, Haataja L, Gurlo T, Butler AE, Wu X, Soeller WC, et al. Induction of endoplasmic reticulum stress-induced beta-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am J Physiol Endocrinol Metab (2007) 293(6):E1656–62. doi:10.1152/ajpendo.00318.2007
129. Kharroubi I, Ladrière L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology (2004) 145(11):5087–96. doi:10.1210/en.2004-0478
130. Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res (2006) 47(12):2726–37. doi:10.1194/jlr.M600299-JLR200
131. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science (2004) 306(5695):457–61. doi:10.1126/science.1103160
132. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell (2001) 7(6):1153–63. doi:10.1016/S1097-2765(01)00264-7
133. Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem (2005) 280(1):847–51. doi:10.1074/jbc.M411860200
134. Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab (2009) 10(1):13–26. doi:10.1016/j.cmet.2009.06.002
135. Silva AM, Wang D, Komar AA, Castilho BA, Williams BR. Salicylates trigger protein synthesis inhibition in a protein kinase R-like endoplasmic reticulum kinase-dependent manner. J Biol Chem (2007) 282(14):10164–71. doi:10.1074/jbc.M609996200
136. Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res (2001) 61(16):6213–8.
137. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science (2006) 313(5790):1137–40. doi:10.1126/science.1128294
138. Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest (2008) 118(1):316–32. doi:10.1172/JCI32752
139. Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med (2009) 15(12):1383–91. doi:10.1038/nm.2067
140. Cao SS, Zimmermann EM, Chuang BM, Song B, Nwokoye A, Wilkinson JE, et al. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology (2013) 144(5):989.e–1000.e. doi:10.1053/j.gastro.2013.01.023
141. Zhao F, Edwards R, Dizon D, Afrasiabi K, Mastroianni JR, Geyfman M, et al. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2-/- mice. Dev Biol (2010) 338(2):270–9. doi:10.1016/j.ydbio.2009.12.008
142. Heijmans J, van Lidth de Jeude JF, Koo BK, Rosekrans SL, Wielenga MC, van de Wetering M, et al. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep (2013) 3(4):1128–39. doi:10.1016/j.celrep.2013.02.031
143. Vasu S, Moffett RC, McClenaghan NH, Flatt PR. Differential molecular and cellular responses of GLP-1 secreting L-cells and pancreatic alpha cells to glucotoxicity and lipotoxicity. Exp Cell Res (2015) 336(1):100–8. doi:10.1016/j.yexcr.2015.05.022
144. Cummings BP, Bettaieb A, Graham JL, Kim J, Ma F, Shibata N, et al. Bile-acid-mediated decrease in endoplasmic reticulum stress: a potential contributor to the metabolic benefits of ileal interposition surgery in UCD-T2DM rats. Dis Model Mech (2013) 6(2):443–56. doi:10.1242/dmm.010421
145. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell (2004) 118(2):229–41. doi:10.1016/j.cell.2004.07.002
146. Worthington JJ, Samuelson LC, Grencis RK, McLaughlin JT. Adaptive immunity alters distinct host feeding pathways during nematode induced inflammation, a novel mechanism in parasite expulsion. PLoS Pathog (2013) 9(1):e1003122. doi:10.1371/journal.ppat.1003122
147. Harrison E, Lal S, McLaughlin JT. Enteroendocrine cells in gastrointestinal pathophysiology. Curr Opin Pharmacol (2013) 13(6):941–5. doi:10.1016/j.coph.2013.09.012
148. Bermudez R, Vigliano F, Quiroga MI, Nieto JM, Bosi G, Domeneghini C. Immunohistochemical study on the neuroendocrine system of the digestive tract of turbot, Scophthalmus maximus (L.), infected by Enteromyxum scophthalmi (Myxozoa). Fish Shellfish Immunol (2007) 22(3):252–63. doi:10.1016/j.fsi.2006.05.006
149. Wang H, Steeds J, Motomura Y, Deng Y, Verma-Gandhu M, El-Sharkawy RT, et al. CD4+ T cell-mediated immunological control of enterochromaffin cell hyperplasia and 5-hydroxytryptamine production in enteric infection. Gut (2007) 56(7):949–57. doi:10.1136/gut.2006.103226
150. El-Salhy M, Gundersen D, Hatlebakk JG, Hausken T. High densities of serotonin and peptide YY cells in the colon of patients with lymphocytic colitis. World J Gastroenterol (2012) 18(42):6070–5. doi:10.3748/wjg.v18.i42.6070
151. El-Salhy M, Gundersen D, Hatlebakk JG, Hausken T. Chromogranin A cell density as a diagnostic marker for lymphocytic colitis. Dig Dis Sci (2012) 57(12):3154–9. doi:10.1007/s10620-012-2249-6
152. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39(5):596–604. doi:10.1038/ng2032
153. Sakiyama T, Fujita H, Tsubouchi H. Autoantibodies against ubiquitination factor E4A (UBE4A) are associated with severity of Crohn’s disease. Inflamm Bowel Dis (2008) 14(3):310–7. doi:10.1002/ibd.20328
154. Moran GW, Pennock J, McLaughlin JT. Enteroendocrine cells in terminal ileal Crohn’s disease. J Crohns Colitis (2012) 6(9):871–80. doi:10.1016/j.crohns.2012.01.013
155. Fishbein TM, Novitskiy G, Lough DM, Matsumoto C, Kaufman SS, Shetty K, et al. Rejection reversibly alters enteroendocrine cell renewal in the transplanted small intestine. Am J Transplant (2009) 9(7):1620–8. doi:10.1111/j.1600-6143.2009.02681.x
156. Friedrich M, Diegelmann J, Schauber J, Auernhammer CJ, Brand S. Intestinal neuroendocrine cells and goblet cells are mediators of IL-17A-amplified epithelial IL-17C production in human inflammatory bowel disease. Mucosal Immunol (2015) 8(4):943–58. doi:10.1038/mi.2014.124
157. Chouliaras G, Panayotou I, Margoni D, Mantzou E, Pervanidou P, Manios Y, et al. Circulating leptin and adiponectin and their relation to glucose metabolism in children with Crohn’s disease and ulcerative colitis. Pediatr Res (2013) 74(4):420–6. doi:10.1038/pr.2013.114
158. Karrasch T, Obermeier F, Straub RH. Systemic metabolic signaling in acute and chronic gastrointestinal inflammation of inflammatory bowel diseases. Horm Metab Res (2014) 46(6):445–51. doi:10.1055/s-0034-1374587
159. Korkmaz H, Sahin F, Ipekci SH, Temel T, Kebapcilar L. Increased pulse wave velocity and relationship with inflammation, insulin, and insulin resistance in inflammatory bowel disease. Eur J Gastroenterol Hepatol (2014) 26(7):725–32. doi:10.1097/MEG.0000000000000104
160. Valentini L, Wirth EK, Schweizer U, Hengstermann S, Schaper L, Koernicke T, et al. Circulating adipokines and the protective effects of hyperinsulinemia in inflammatory bowel disease. Nutrition (2009) 25(2):172–81. doi:10.1016/j.nut.2008.07.020
161. Konrad A, Lehrke M, Schachinger V, Seibold F, Stark R, Ochsenkühn T, et al. Resistin is an inflammatory marker of inflammatory bowel disease in humans. Eur J Gastroenterol Hepatol (2007) 19(12):1070–4. doi:10.1097/MEG.0b013e3282f16251
162. Bregenzer N, Hartmann A, Strauch U, Schölmerich J, Andus T, Bollheimer LC. Increased insulin resistance and beta cell activity in patients with Crohn’s disease. Inflamm Bowel Dis (2006) 12(1):53–6. doi:10.1097/01.MIB.0000195975.97673.f5
163. Moran GW, Leslie FC, McLaughlin JT. Crohn’s disease affecting the small bowel is associated with reduced appetite and elevated levels of circulating gut peptides. Clin Nutr (2013) 32(3):404–11. doi:10.1016/j.clnu.2012.08.024
164. Keller J, Binnewies U, Rösch M, Juul Holst J, Beglinger C, Andresen V, et al. Gastric emptying and disease activity in inflammatory bowel disease. Eur J Clin Invest (2015) 45:1234–42. doi:10.1111/eci.12542
165. Besterman HS, Mallinson CN, Modigliani R, Christofides ND, Pera A, Ponti V, et al. Gut hormones in inflammatory bowel disease. Scand J Gastroenterol (1983) 18(7):845–52. doi:10.3109/00365528309182104
166. El-Salhy M, Danielsson A, Stenling R, Grimelius L. Colonic endocrine cells in inflammatory bowel disease. J Intern Med (1997) 242(5):413–9. doi:10.1046/j.1365-2796.1997.00237.x
167. Keller J, Beglinger C, Holst JJ, Andresen V, Layer P. Mechanisms of gastric emptying disturbances in chronic and acute inflammation of the distal gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol (2009) 297(5):G861–8. doi:10.1152/ajpgi.00145.2009
168. Xiao Q, Boushey RP, Cino M, Drucker DJ, Brubaker PL. Circulating levels of glucagon-like peptide-2 in human subjects with inflammatory bowel disease. Am J Physiol Regul Integr Comp Physiol (2000) 278(4):R1057–63.
169. Bishop AE, Pietroletti R, Taat CW, Brummelkamp WH, Polak JM. Increased populations of endocrine cells in Crohn’s ileitis. Virchows Arch A Pathol Anat Histopathol (1987) 410(5):391–6. doi:10.1007/BF00712758
170. Moran GW, O’Neill C, Padfield P, McLaughlin JT. Dipeptidyl peptidase-4 expression is reduced in Crohn’s disease. Regul Pept (2012) 177(1–3):40–5. doi:10.1016/j.regpep.2012.04.006
171. Mimura S, Ando T, Ishiguro K, Maeda O, Watanabe O, Ujihara M, et al. Dipeptidyl peptidase-4 inhibitor anagliptin facilitates restoration of dextran sulfate sodium-induced colitis. Scand J Gastroenterol (2013) 48(10):1152–9. doi:10.3109/00365521.2013.832366
172. Lee SJ, Lee J, Li KK, Holland D, Maughan H, Guttman DS, et al. Disruption of the murine Glp2r impairs Paneth cell function and increases susceptibility to small bowel enteritis. Endocrinology (2012) 153(3):1141–51. doi:10.1210/en.2011-1954
173. Qi KK, Wu J, Wan J, Men XM, Xu ZW. Purified PEGylated porcine glucagon-like peptide-2 reduces the severity of colonic injury in a murine model of experimental colitis. Peptides (2014) 52:11–8. doi:10.1016/j.peptides.2013.11.012
174. Boushey RP, Yusta B, Drucker DJ. Glucagon-like peptide 2 decreases mortality and reduces the severity of indomethacin-induced murine enteritis. Am J Physiol (1999) 277(5 Pt 1):E937–47.
175. Alavi K, Schwartz MZ, Palazzo JP, Prasad R. Treatment of inflammatory bowel disease in a rodent model with the intestinal growth factor glucagon-like peptide-2. J Pediatr Surg (2000) 35(6):847–51. doi:10.1053/jpsu.2000.6861
176. Drucker DJ, Yusta B, Boushey RP, DeForest L, Brubaker PL. Human [Gly2]GLP-2 reduces the severity of colonic injury in a murine model of experimental colitis. Am J Physiol (1999) 276(1 Pt 1):G79–91.
177. Buchman AL, Katz S, Fang JC, Bernstein CN, Abou-Assi SG, Teduglutide Study Group. Teduglutide, a novel mucosally active analog of glucagon-like peptide-2 (GLP-2) for the treatment of moderate to severe Crohn’s disease. Inflamm Bowel Dis (2010) 16(6):962–73. doi:10.1002/ibd.21117
178. Jeppesen PB, Gilroy R, Pertkiewicz M, Allard JP, Messing B, O’Keefe SJ. Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome. Gut (2011) 60(7):902–14. doi:10.1136/gut.2010.218271
179. Jeppesen PB, Pertkiewicz M, Messing B, Iyer K, Seidner DL, O’keefe SJ, et al. Teduglutide reduces need for parenteral support among patients with short bowel syndrome with intestinal failure. Gastroenterology (2012) 143(6):1473.e–81.e. doi:10.1053/j.gastro.2012.09.007
180. García-Martínez JM, Chocarro-Calvo A, Moya CM, García-Jiménez C. WNT/beta-catenin increases the production of incretins by entero-endocrine cells. Diabetologia (2009) 52(9):1913–24. doi:10.1007/s00125-009-1429-1
181. Gustafson B, Smith U. WNT signalling is both an inducer and effector of glucagon-like peptide-1. Diabetologia (2008) 51(10):1768–70. doi:10.1007/s00125-008-1109-6
182. Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev (2005) 19(8):877–90. doi:10.1101/gad.1295405
183. Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev (2007) 87(4):1409–39. doi:10.1152/physrev.00034.2006
184. Robertson MD, Livesey G, Morgan LM, Hampton SM, Mathers JC. The influence of the colon on postprandial glucagon-like peptide 1 (7-36) amide concentration in man. J Endocrinol (1999) 161(1):25–31. doi:10.1677/joe.0.1610025
185. Gledhill A, Hall PA, Cruse JP, Pollock DJ. Enteroendocrine cell hyperplasia, carcinoid tumours and adenocarcinoma in long-standing ulcerative colitis. Histopathology (1986) 10(5):501–8. doi:10.1111/j.1365-2559.1986.tb02501.x
186. Hormannsperger G, von Schillde MA, Haller D. Lactocepin as a protective microbial structure in the context of IBD. Gut Microbes (2013) 4(2):152–7. doi:10.4161/gmic.23444
187. Gomes AC, Bueno AA, de Souza RG, Mota JF. Gut microbiota, probiotics and diabetes. Nutr J (2014) 13:60. doi:10.1186/1475-2891-13-60
188. Delzenne NM, Cani PD, Neyrinck AM. Modulation of glucagon-like peptide 1 and energy metabolism by inulin and oligofructose: experimental data. J Nutr (2007) 137(11 Suppl):2547S–51S.
189. Nogueira AM, Barbosa AJ. Immunocytochemical study of intestinal endocrine cells in germ-free mice. Eur J Histochem (1994) 38(3):213–8.
190. Puddu A, Sanguineti R, Montecucco F, Viviani GL. Evidence for the gut microbiota short-chain fatty acids as key pathophysiological molecules improving diabetes. Mediators Inflamm (2014) 2014:162021. doi:10.1155/2014/162021
191. Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol (2011) 17(12):1519–28. doi:10.3748/wjg.v17.i12
192. Remely M, Aumueller E, Merold C, Dworzak S, Hippe B, Zanner J, et al. Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity. Gene (2014) 537(1):85–92. doi:10.1016/j.gene.2013.11.081
193. Yadav H, Lee JH, Lloyd J, Walter P, Rane SG. Beneficial metabolic effects of a probiotic via butyrate-induced GLP-1 hormone secretion. J Biol Chem (2013) 288(35):25088–97. doi:10.1074/jbc.M113.452516
194. Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Neyrinck AM, et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes (2011) 60(11):2775–86. doi:10.2337/db11-0227
195. Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol (2004) 54(Pt 5):1469–76. doi:10.1099/ijs.0.02873-0
196. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A (2013) 110(22):9066–71. doi:10.1073/pnas.1219451110
197. Hofmann AF, Borgstroem B. The intraluminal phase of fat digestion in man: the lipid content of the micellar and oil phases of intestinal content obtained during fat digestion and absorption. J Clin Invest (1964) 43:247–57. doi:10.1172/JCI104909
198. Fiorucci S, Mencarelli A, Palladino G, Cipriani S. Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol Sci (2009) 30(11):570–80. doi:10.1016/j.tips.2009.08.001
199. Li T, Chiang JY. Bile acids as metabolic regulators. Curr Opin Gastroenterol (2015) 31(2):159–65. doi:10.1097/MOG.0000000000000156
200. Han JL, Lin HL. Intestinal microbiota and type 2 diabetes: from mechanism insights to therapeutic perspective. World J Gastroenterol (2014) 20(47):17737–45. doi:10.3748/wjg.v20.i47.17737
201. Yamagata K, Daitoku H, Shimamoto Y, Matsuzaki H, Hirota K, Ishida J, et al. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J Biol Chem (2004) 279(22):23158–65. doi:10.1074/jbc.M314322200
202. Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab (2013) 17(2):225–35. doi:10.1016/j.cmet.2013.01.003
203. Kurdi P, Kawanishi K, Mizutani K, Yokota A. Mechanism of growth inhibition by free bile acids in lactobacilli and bifidobacteria. J Bacteriol (2006) 188(5):1979–86. doi:10.1128/JB.188.5.1979-1986.2006
204. Sonne DP, Hansen M, Knop FK. Bile acid sequestrants in type 2 diabetes: potential effects on GLP1 secretion. Eur J Endocrinol (2014) 171(2):R47–65. doi:10.1530/EJE-14-0154
205. Hansen M, Sonne DP, Knop FK. Bile acid sequestrants: glucose-lowering mechanisms and efficacy in type 2 diabetes. Curr Diab Rep (2014) 14(5):482. doi:10.1007/s11892-014-0482-4
206. Chen S, Okahara F, Osaki N, Shimotoyodome A. Increased GIP signaling induces adipose inflammation via a HIF-1alpha-dependent pathway and impairs insulin sensitivity in mice. Am J Physiol Endocrinol Metab (2015) 308(5):E414–25. doi:10.1152/ajpendo.00418.2014
207. Gruber L, Kisling S, Lichti P, Martin FP, May S, Klingenspor M, et al. High fat diet accelerates pathogenesis of murine Crohn’s disease-like ileitis independently of obesity. PLoS One (2013) 8(8):e71661. doi:10.1371/journal.pone.0071661
208. Di Gregorio GB, Hensley L, Lu T, Ranganathan G, Kern PA. Lipid and carbohydrate metabolism in mice with a targeted mutation in the IL-6 gene: absence of development of age-related obesity. Am J Physiol Endocrinol Metab (2004) 287(1):E182–7. doi:10.1152/ajpendo.00189.2003
Keywords: incretins, ER stress, diabetes mellitus, microbiota, inflammation, enteroendocrine cells, glucagon-like peptide 1, inflammatory bowel disease
Citation: Zietek T and Rath E (2016) Inflammation Meets Metabolic Disease: Gut Feeling Mediated by GLP-1. Front. Immunol. 7:154. doi: 10.3389/fimmu.2016.00154
Received: 11 January 2016; Accepted: 08 April 2016;
Published: 22 April 2016
Edited by:
Claudio Mauro, Queen Mary University of London, UKCopyright: © 2016 Zietek and Rath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eva Rath, eva.rath@tum.de
†Tamara Zietek and Eva Rath contributed equally to this work.