 Surasak Jittavisutthikul1,2
Surasak Jittavisutthikul1,2 Watee Seesuay2
Watee Seesuay2 Jeeraphong Thanongsaksrikul2,3
Jeeraphong Thanongsaksrikul2,3 Kanyarat Thueng-in2,4Potjanee Srimanote3Rolf G. Werner5
Kanyarat Thueng-in2,4Potjanee Srimanote3Rolf G. Werner5 Wanpen Chaicumpa2,3*
Wanpen Chaicumpa2,3*
- 1Graduate Program in Immunology, Department of Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
- 2Department of Parasitology, Faculty of Medicine Siriraj Hospital, Center of Research Excellence on Therapeutic Proteins and Antibody Engineering, Mahidol University, Bangkok, Thailand
- 3Graduate Program in Biomedical Science, Faculty of Allied Health Sciences, Thammasat University, Pathum-thani, Thailand
- 4School of Pathology, Institute of Medicine, Suranaree University of Technology, Nakhon Ratchasima Province, Thailand
- 5Industrial Technology, Faculty of Science, University of Tuebingen, Tuebingen, Germany
A safe and effective direct acting anti-hepatitis C virus (HCV) agent is still needed. In this study, human single chain variable fragments of antibody (scFvs) that bound to HCV NS3/4A protein were produced by phage display technology. The engineered scFvs were linked to nonaarginines (R9) for making them cell penetrable. HCV-RNA-transfected Huh7 cells treated with the transbodies produced from four different transformed E. coli clones had reduced HCV-RNA inside the cells and in the cell spent media, as well as fewer HCV foci in the cell monolayer compared to the transfected cells in culture medium alone. The transbodies-treated transfected cells also had up-expression of the genes coding for the host innate immune response, including TRIF, TRAF3, IRF3, IL-28B, and IFN-β. Computerized homology modeling and intermolecular docking predicted that the effective transbodies interacted with several critical residues of the NS3/4A protease, including those that form catalytic triads, oxyanion loop, and S1 and S6 pockets, as well as a zinc-binding site. Although insight into molecular mechanisms of the transbodies need further laboratory investigation, it can be deduced from the current data that the transbodies blocked the HCV NS3/4A protease activities, leading to the HCV replication inhibition and restoration of the virally suppressed host innate immunity. The engineered antibodies should be tested further for treatment of HCV infection either alone, in combination with current therapeutics, or in a mixture with their cognates specific to other HCV proteins.
Introduction
Before 2011, patients with hepatitis C virus (HCV) infection were weekly injected with pegylated-interferon-alpha combined with daily oral ribavirin (a nucleoside analog), which is a standard-of-care (SOC). The treatment conferred high rate of sustained virologic response (SVR) against HCV of most genotypes by restoring the virally suppressed host innate immunity and restraining the viral load (1). However, HCV strains of genotype 1 were relatively refractory to the SOC. Besides, the regimen is prolonged, stringent, and costly, as well as induces severe adverse effects, which are poorly tolerated by a significant fraction of the recipients. During the past decade, intense research has focused on development of direct acting anti-HCV agents (DAAs) that interfere with the functions of several HCV proteins. These are, for examples, inhibitors of NS3/4A protease (e.g., telaprevir, boceprevir, simeprevir, asunaprevir, danoprevir, faldaprevir, paritaprevir); NS5B polymerase (e.g., sofosbuvir, dasabuvir); a protein important for membrane web formation, NS4B [e.g., substituted imidazo(1,2-a) pyrimidines, clemizole, piperazinones]; a regulatory phosphoprotein of the viral replication complex, NS5A (e.g., ombitasvir, daclatasvir, ledipasvir, velpatasvir); or capsid protein (2–10). Also, drugs that inhibit HCV–host protein interaction have been tested (11, 12). Presently, several DAAs have received regulatory approval for clinical use, such as protease inhibitors (boceprevir, telaprevir, and semiprevir), polymerase inhibitor (sofosbuvir), and NS5A inhibitor (ledipasvir). Additional DAAs are in various stages of clinical development (9, 13). Many new drug regimens, when administered either singly, together with SOC, or in various combinations among themselves, conferred improved SVR rate compared with SOC (9, 13–16). However, the challenges that the new drugs are facing include complicated treatment protocols, additional severe side effects, drug–drug interaction, and emergence of drug- and cross-drug-resistant HCV mutants. The new regimens are contraindicated for some groups of patients/subjects (16–21). Thus, safe and effective DAAs are needed.
Passive immunization (administration of ready-made antibody) has been practiced for prevention/intervention and treatment of diseases since the late eighteenth century (called serum therapy/serotherapy at the time) (22). Nevertheless, the physicochemical properties of the plasma membrane impede accessibility of antibodies to their intracellular targets (such as proteins/enzymes of replicating virus). To circumvent this obstacle, several peptides called cell-penetrating peptides (CPPs) have been developed for delivering antibodies/antibody fragments and a variety of other cargoes (proteins, drugs, nucleic acids, plasmids, and siRNAs) across the plasma membrane into cytosol and also to different subcellular compartments (23–27). Examples of the CPPs are protein transduction domains (such as penetratin, HIV-1 Tat peptide) (28), amphipathic peptides such as protein-rich motif (SAP) (29), and other CPP type, including nonaarginine (R9) and polylysine (30). In our laboratory, cell-penetrable antibodies (transbodies) specific to viral proteins have been prepared by linking the antibody molecules to either penetratin or R9. These fusion proteins readily entered mammalian cells and bound to their respective intracellular targets (31–35).
Usually, an antibody molecule uses several amino acids in the complementarity determining regions (CDRs) to interact with multiple residues/sites of the target, which makes it difficult for the pathogens to escape the antibody binding. On the contrary, pathogens may resist small molecular inhibitors through a single amino acid mutation (21). In this study, cell-penetrable human single chain antibody variable fragments (transbodies) to HCV protease that inhibit the viral replication and restore the host innate immune response were produced. It is envisaged that a right combination of cell-penetrable human/humanized antibodies that target pivotal enzymes of the HCV, i.e., helicase (31), polymerase (32, 33), and protease (34), a strategy similar to the precedent anti-HIV regimen, should be a novel anti-HCV remedy that is safe, genotype independent, and more tolerable to the viral mutation.
Materials and Methods
Recombinant HCV NS3/4A Fusion Protein Production
The recombinant NS3/4A protein (rNS3/4A) with the HCV-inherent serine protease activity was produced from BL21 (DE3) E. coli carrying pET23b+-NS3/4A, as described previously (34). The fusion protein contained 180 amino acids of the N-terminal one-third of the NS3 of the genotype 3a HCV linked with residues 21–32 of the NS4A of the same genotype. The recombinant NS3/4A protein cleaved a fluorescent peptide substrate that contained nine amino acids at the protease cleavage site as determined by a continuous fluorescence resonance energy transfer (FRET) assay (34).
Mouse Anti-NS3/4A Polyclonal Antibody
The experiments received approval from the Siriraj Bio-safety Risk Management Taskforce (SI 2014-004). Three ICR mice (National Laboratory Animal Center, Mahidol University, Nakhonpathom, Thailand) were housed in a shoe-box typed cage under 12/12 light/dark cycle at 23–25°C and 55–60% humidity. Feed and water were supplied ad libitum. The animals were immunized intraperitoneally with three doses at 2-week intervals of 10 μg purified recombinant NS3/4A protein mixed with alum (Pierce, USA) at ratio 1:3. After the last booster, the mice were bled and their serum titers were assessed against the homologous antigen by indirect ELISA.
Immunological Methods
Indirect ELISA was performed as described previously (33) using 1 μg of purified rNS3/4A protein in 100 μl of carbonate–bicarbonate buffer, pH 9.6 to coat each well of an ELISA plate. Five percent skim milk in PBS (300 μl/well) was used for blocking the well surface before adding antibody preparations (e.g., serial dilutions of the mouse immune sera, HuscFv preparations) into appropriate wells. Antibody control was either normal mouse serum or lysate of original E. coli. Bovine serum albumin (BSA) served as control antigen and PBS as blank. After incubation, all wells were washed with the PBS containing 0.05% Tween-20 (PBST). Horseradish peroxidase (HRP)-conjugated anti-immunoglobulin isotype (Southern Biotech, Birmingham, AL, USA) and chromogenic substrate (KPL, Gaitherburg, MD, USA) were used for color development. The enzymatic reaction was stopped by adding 1% SDS. OD405nm of contents in all wells were determined against blank.
For SDS-PAGE and Western blot analysis (WB), polyacrylamide gel at a desired gel percentage cast in a Mini-PROTEAN-3® Cell System (Bio-Rad, Hercules, CA, USA) was used. Samples prepared in 6 × SDS gel reducing buffer were boiled for 10 min before loading into the gel slots made in the 4% stacking gel. PageRuler® pre-stained protein ladders (Fermentas, Thermo Fisher Scientific, MA, USA) were included in one or two gel slots as molecular weight marker. Electrophoresis was carried out in Tris–glycine running buffer, pH 8.3, initially at 10 mA per gel until the bromphenol blue dye font reached the resolving gel; then the electricity was increased to 20 mA and continued until the dye font reached the lower gel edge. Separated proteins in the gel were either revealed by staining with Coomassie Brilliant Blue G-250 dye or they were electroblotted onto nitrocellulose membrane (NC) for WB. For WB, the blotted NC was blocked either with 3% BSA or 5% skim milk in PBS. The blocked NC was then probed with antibody preparation; then the antigen–antibody reactive bands were revealed using alkaline phosphatase (AP)-labeled anti-isotype antibody and AP chromogenic substrate.
Phage Clones That Bound to the rNS3/4A Protein and the Phage Transformed E. coli Clones
Phage clones bound to the rNS3/4A fusion protein were fished-out from a human scFv phage display library as described previously (33) using purified rNS3/4A (1 μg) as bait. The rNS3/4A-bound phages were put into HB2151 E. coli bacteria, and the bacteria were spread on a 2 × YT-AG plate. Direct colony PCR (36) was used to screen E. coli colonies that carried HuscFv coding genes (huscfvs). Lysates of huscfv-positive E. coli colonies grown in 0.5 mM IPTG-conditioned broth were determined by Western blotting for the presence of the E-tagged-HuscFvs.
Characterization of the Bacterially Derived-HuscFvs
Spectrometrically standardized soluble HuscFvs in the lysates of transformed HB2151 E. coli clones were tested for binding to the rNS3/4A by indirect ELISA and Western blotting. Original HB2151 E. coli (HB) was used as background binding control in both assays. BSA served as control antigen in the indirect ELISA.
Diversity of the huscfvs sequences of the HB2151 E. coli clones were determined by subjecting the PCR amplified huscfvs to MvaI digestion followed by resolving the cut DNA products in 14% polyacrylamide gel containing 0.5% glycerol. Electrophoresis was carried out in TBE buffer at 20 mA per slab gel. The separated DNA bands were stained by ethidium bromide and the restriction fragment length polymorphism (RFLP) was determined using a Bio Doc-ITTM Imaging System, UVP, USA. CDRs and their respective canonical immunoglobulin framework regions (FRs) of the sequenced huscfvs were predicted using an online Internatioanl ImMunoGeneTics (IMGT®) Information System.
Generation of Cell Penetrating HuscFvs (Transbodies)
Because the antibodies must bind to the target inside the HCV-infected cells, they were linked to nonaarginine (R9), which is a cell-penetrating peptide, as follow: the huscfvs were amplified from the pCANTAB5E phagemids using a Q5 High Fidelity DNA polymerase (Thermo Fisher Scientific). The specific primers were forward-R9-huscfv-LIC: 5′-GGTTGGGAATTGCAACGTCGCCGTCGCTCGCCGTCGCCGTCGCCGTGCGGCCCAGCCCGGCC-3′ and reverse-E-tag-LIC: 5′-GGAGATGGGAAGTCATTAACGCGGTTCCAGCGGATCC-3′. The reaction mixture contained 15.75 μl sterile distilled water, 5.0 μl Q5 reaction buffer (5×), 0.5 μl dNTP mix (10 mM each), forward and reverse primers (10 μM in 1.25 μl each), 1.0 μl of huscfv-phagemid template (3 ng DNA template), and 0.25 unit of Q5 Hotstart DNA polymerase. The thermal cycles were first denaturation at 98°C, 30 s; 34 cycles of denaturation at 98°C for 30 s, annealing/extension at 69°C for 10s, and final extension at 72°C, 5 min; and hold at 4°C, several min. The PCR product was stained with ethidium bromide, verified, and purified from agarose gel using DNA extraction kit (Jena Bioscience, Jena, Germany). The 5′- and 3′- overhangs of the R9-huscfv amplicons were generated by setting up the following reaction mixtures: 9 μl of sterile distilled water, 2 μl of 5× LIC buffer, 0.1 pmol of the purified PCR product, and 1 μl T4 DNA polymerase and dGTP. The reaction mixtures were kept at 25°C for 5 min and stopped by adding 0.6 μl of 0.5 M EDTA. Annealing of the DNA products with 15 base pairs (bp) 5′-overhang to the pLATE52 vector containing complementary overhang was performed by mixing 1 μl of the vector and 60 ng (0.02 pmol) LIC ready vector (Thermo Fisher Scientific). The mixtures were kept at 25°C for 5 min before putting into JM109 E. coli by heat-shock method. The transformed E. coli clones carrying the recombinant pLATE52-R9-huscfv plasmids were screened by PCR using the pLATE52 specific primers, i.e., LIC forward sequence: 5′-TAATACGACTCACTATAGGG-3′ and LIC reverse sequence: 5′-GAGCGGATAACAATTTCACACAGG-3′. The PCR reaction mixture was: 12.7 μl sterile distilled water, 2 μl PCR buffer + KCl (10×), 1.2 μl 25 mM MgCl2, 2 μl dNTP mix (10 μM each), 1 μl (10 μM) each of LIC primers, and 0.5 unit of Taq polymerase.
The pLATE52-R9-huscfv were extracted from the PCR positive clones, purified, put in Rosetta™2 (DE3)-competent cells (Novagen, Schwalbach, Germany), and spread onto selective LB agar plates supplemented with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol (Calbiotech, Spring Valley, CA, USA) (LB-AC agar). The sibling E. coli colonies grown on the agar plates were randomly picked and screened for the presence of the pLATE52-R9-huscfv plasmids by PCR using the pLATE52 primers. The Rosetta™2 (DE3) bacteria with the pLATE52-R9-huscfv plasmids were cultured in 0.5 mM IPTG-conditioned broth. The induced bacterial cells were collected and R9-HuscFvs in their homogenates were determined by SDS-PAGE and Western blotting using anti-6× His as the R9-HuscFv-detection reagent. The clones that expressed the R9-HuscFvs (with 6× His and T7 tags at the N-terminal and E-tag at the C-terminal) were kept as stocks in 20% glycerol at −80°C until use.
Large-scale Production, Purification, and Refolding of the Cell Penetrating HuscFvs
The Rosetta™2 (DE3) E. coli clones carrying the pLATE52-R9-huscfv were grown in 2× YT-AC broth at 37°C with shaking at 250 rpm for 16 h. Ten milliliters of the overnight culture were inoculated into 250 ml of 2× YT-AC broth in a 2-liter flask and incubated with shaking aeration at 37°C until OD600nm was approximately 0.8–0.9 (~3 h). The culture was added with IPTG (final concentration of 1 mM), incubated at 30°C for 6 h, and centrifuged at 5,000 × g at 4°C for 20 min. To prepare the bacterial inclusion bodies (IBs), each 2 g of the E. coli wet pellets were lysed with 10 ml of BugBuster™ protein extraction reagent (Novagen, Schwalbach, Germany) and 20 μl of Lysonase™ bioprocessing reagent (Novagen). The preparation was kept at 25°C on a rotator for 20 min and centrifuged at 8,000 × g at 4°C for 30 min. IB in the pellet was washed twice with Wash-100 reagent and once with Wash-114 reagent with shaking at high speed for 40 min and centrifuged. The IB was then washed with Wash-Solvent reagent and Milli Q water on ice also with vigorous shaking and centrifuged.
For HuscFv refolding, 5 ml of buffer [50 mM CAPS, pH 11.0; 0.3% (w/v) N-Lauryl sarcosine; and 1 mM DTT] were added to reconstitute 5 mg of purified IB and kept at 4°C for 16 h. After dissolving completely, the protein was loaded into the Slide-A-Lyzer® 2K Dialysis Cassettes G2 (Thermo Fisher Scientific), and the cassette was subjected to dialysis against 750 ml of a refolding buffer (20 mM imidazole, pH 8.5, supplemented with 0.1 mM DTT) at 4°C with slow stirring. After 3 h, the buffer was changed to fresh refolding buffer, and the dialysis was continued for 16 h. The refolded protein was subsequently dialyzed against a dialysis buffer (20 mM imidazole without DTT) with slow stirring at 4°C for 16 h. The preparation was filtered through a 0.2-μm low protein binding Acrodisc® Syringe Filter (Pall, NY, USA) and kept in 30°C water-bath for additional 2 h before adding with 60 mM trehalose. Protein concentration of the refolded HuscFvs was measured (Pierce® BCA Protein Assay), while quality and purity of the proteins were determined by SDS-PAGE and protein staining. The antibodies were tested for binding to the homologous antigen (rNS3/4A) by indirect ELISA.
Cell Penetrating Ability of the R9-HuscFvs, Their Intracellular Target Binding, and Toxicity
The experiments involving HCV of this study received approval from the Siriraj Bio-safety Risk Management Taskforce. The pJFH-1 (GenBank AB047639), which carried a full-length HCV genotype 2a cDNA, was used for production of HCV RNA, as described previously (32, 33). Briefly, the plasmid was cut with XbaI endonuclease and transcribed using a T7-transcription kit (MEGAscript®, Ambion, Foster City, CA, USA). Ten micrograms of JFH-1 RNA were put in Huh7 cells (4 × 106 cells) suspended in 0.4 ml of reduced serum medium (Opti-MEM, Invitrogen, USA) by means of electroporation; complete DMEM containing 10% heat-inactivated fetal bovine serum (Hyclone, USA) and antibiotics was immediately added to the electroporated cells, which were then distributed onto individual cover slips (2 × 104 cells/slip) placed in wells of a 24-well tissue culture plate After incubating at 37°C in 5% CO2 incubator overnight and rinsed with sterile PBS, the cells were replenished with 500 μl complete DMEM containing 10 μg of individual R9-HuscFvs. After 1 h, culture supernatants were removed; the cells were washed with PBS, fixed with 1% Triton X-100, 30 min; blocked with 3% BSA; and washed. Rabbit anti-6× His polyclonal antibody (Abcam, Cambridge, UK) (1:500) was added to the cells and incubated at 37°C for 1 h, washed, and 1:1000 mouse anti-NS3/4A polyclonal antibody was added. After 1 h, the cells were washed and added with goat anti-rabbit Alexa Flour 488 (1:500) and donkey anti-mouse Alexa Flour®647 (Abcam). DAPI (1:5000) (Invitrogen, Carlsbad, CA, USA) was used to stain nuclei. The preparations were kept at 37°C for 1 h, and the stained cells were observed under a confocal microscope.
CytoTox96® non-radioactive cytotoxicity (lactate dehydrogenase, LDH) assay kit (Promega, Madison, WI, USA) and Cell Titer-Glo® Luminescent Cell Viability Assay kit (Promega) were used for determining cytotoxicity of the R9-HuscFvs to Huh7 cells. The LDH release assay was performed as described previously (35). Huh7 monolayer in individual wells of a 96-well tissue culture plate (2 × 104 cells/well) were washed with sterile PBS, added with 25 μg of R9-HuscFvs in complete DMEM, and incubated for 24 h at 37°C in 5% CO2 atmosphere. Controls were cells in the medium alone (minimal/spontaneous release) and cells added with 10% SDS (maximal release) were included. After 24 h, culture supernatants (triplicate wells of each treatment) were collected separately. Fifty microliters of culture fluid were mixed with the assay buffer provided with the kit and kept at 25°C for 30 min under light protection. The reactions were stopped by adding 1 M acetic acid, and OD490nm was determined. Percent cytotoxicity was (LDH release of test − spontaneous LDH release)/(Maximal LDH release − spontaneous LDH release) × 100 (35). Percent cell viability was calculated by subtracting 100 with percent cytotoxicity.
For the Luminescent Cell Viability Assay that quantifies the number of viable cells based on the amounts of ATP released by metabolically active cells, Huh7 cells cultured in complete DMEM were seeded into wells of a 96-well opaque tissue culture plates (Corning) (2 × 104 cells/well). After incubating at 37°C in 5% CO2 atmosphere for 24 h, the spent media were removed, and the tested cells were added with 25 μg of purified R9-HuscFvs in 100 μl complete DMEM. Medium only was added to the control cells (negative control). After keeping at 37°C in 5% CO2 incubator for 48 h, the plate was brought to room temperature (25°C) for 30 min; the medium in each well was replaced with 100 μl of the Glo reagent, kept on an orbital shaker for 2 min, then at room temperature for 10 more min for luminescence stabilization. The luminescence signal of each well was recorded using Synergy™ H1 Multi-mode Reader (BioTek®). Luminescence of the well containing medium only served as background luminescence. The percentage of cell viability was (Luminescence of test − Background luminescence)/(Luminescence of negative control − Background luminescence) × 100.
For in vivo toxicity testing of the R9-HuscFv, a protocol of Thai Pharmacopia was followed. Two groups of male BALB/c mice (6–8 weeks old) were used. Each mouse of group 1 (n = 5) was injected intraperitoneally with 25 μg of R9-HuscFv/g body weight in 200 μl buffer. This antibody dose is higher than those used for passive immunization of human (37). Three more booster doses were given on every alternate day. Control mice (group 2; n = 4) were injected with buffer alone. The animals were observed daily for any sign of morbidity, including ruffled hair, eye and nasal discharge, diarrhea, loss of appetite, and body temperature. Their body weights were determined also. Seven days post-last booster, all mice were bled and their sera were monitored for some parameters of blood chemistry including liver enzymes (AP, aspartate aminotransferase, and alanine aminotransferase) and total bilirubin.
Transbody-mediated Inhibition of the HCV Replication
Huh7 cells transfected with JFH-1 RNA was prepared as above. Aliquots of the transfected cells (2 × 105 cells/well) were added to individual wells of a 12-well tissue culture plates and kept for 3 h at 37°C in 5% CO2 incubator. After washing with sterile PBS, the cells were added with 25 μg of purified R9-HuscFvs dissolved in complete DMEM. Negative and background inhibition controls were the infected cells added with medium alone and medium containing 25 μg of control R9-HuscFv, respectively. Positive inhibition controls were 100 units of pegylated (PEG) interferon + 250 μM ribavirin, 0.175 μM telaprevir (VX-950) (Selleckem, Houston, TX, USA), and PEG-interferon + 250 μM ribavirin + 0.175 μM telaprevir. After 5 more days of incubation, total RNAs were extracted from the cells and their spent media (Trizol™ reagent, Invitrogen). Amounts of HCV 5′ UTR in all samples were quantified by qRT-PCR (33). Alternatively, culture supernatants of the infected cells in all wells were removed, and the viral foci formed in the respective cell monolayer were enumerated after staining for the HCV core protein (33).
Quantitative Real-time RT-PCR
The qRT-PCR reaction mixture (12.5 μl) was prepared: 6.25 μl 2× Brilliant II SYBR® Green qRT-PCR master mix, 0.5 μl each of the JFH-1 5′ UTR primers, 0.5 μl RT/RNS block enzyme, RNA template up to 4.75 μl (200–900 ng of standard RNA), and sterile DEPC-treated distilled water to 12.5 μl. The amplification was carried out using Mx3000P QPCR System (Agilent Technologies) at 42°C for 30 min, 55°C for 30 min, initial denaturation at 95°C for 10 min, and 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s. To analyze the dissociation curve, a thermal profile of 95°C for 1 min, ramped down to 55°C for 45 s at a speed of 0.5°C/s and ramped up to 95°C was carried out. Ct of 10-fold serially diluted pJFH-1 with full-length HCV genotype 2a cDNA insert, calculated 2.77 × 107 to 0.02 copy numbers, were used for constructing a standard curve. Log10 RNA copies/ml of sample was extrapolated from the standard curve. Data of one of the three independent and reproducible experiments are reported herein.
HCV Foci Assay
Hepatitis C virus foci formed in the JFH-RNA-transfected Huh7 cell monolayer after receiving different treatments were enumerated (34). Briefly, the spent culture fluids of the transfected cells of all treatments were discarded. After washing, the cells were fixed with absolute methanol at 4°C, 15 min; blocked for 1 h with 10% BSA at 37°C; incubated with mouse anti-HCV core polyclonal antibody; washed; added with AP-conjugated-goat anti-mouse isotype; washed again; and added with AP chromogenic substrate (KPL). Untreated transfected cells and non-transfected (normal) cells were included in the experiments as negative inhibition (maximal number of HCV foci) and negative foci control, respectively. Numbers of HCV foci in each well were counted under 40× magnification using an inverted fluorescence microscope (NIS-Element D version 4.10.0.8310 W/camera, Ti-S Intensilight Ril NIS-D, Nikon, Japan).
Response of the HCV-RNA-transfected Cells after Treatment with the Transbodies and Controls
After treating the JFH-1 RNA-infected Huh7 cells with various R9-HuscFvs and controls (control R9-HuscFv, PEG-IFN + ribavirin, telaprevir, PEG-IFN + ribavirin + telaprevir, and medium alone), expressions of innate immune response genes (amounts of mRNAs), including TRIF, TRAF3, IRF3, IL-28B, and IFNβ, were determined by qRT-PCR as described previously (34) using GAPDH mRNA for normalization.
Computerized Simulation for Predicting Residues of the HCV NS3/4A Protein Bound by the HuscFvs
Three-dimensional structure of NS3/4A (PDB ID-1A1R, RCSB database) was retrieved. Tertiary structures of HuscFvs were modeled by subjecting their amino acid sequences to the I-TASSER server (38). The I-TASSER-predicted models were subsequently refined and improved to their nearest native state (39, 40). Molecular docking between the NS3/4A 3D and the HuscFv models was performed by ClusPro2.0 server (41). Pymol software (The PyMOL Molecular Graphics System, version 1.3r1 edu, Schrodinger, LLC) was used to build and visualize the protein complexes that formed the largest cluster size and the minimal local energy.
Statistical Analysis
Means and SDs of tests and controls were compared using unpaired t-test. p < 0.05 was considered significantly different.
Results
Phage-transformed E. coli Clones That Produced HCV NS3/4A-bound HuscFvs and the HuscFv Characteristics
Fifty-two HB2151 E. coli bacteria infected with the rNS3/4A-bound phages that were grown on the selective LB-A agar plate were checked for the presence of huscfvs, and 18 clones (34.6%) revealed the genes at about 1,000 bp (Figure 1A). After growing these E. coli clones under 0.5 mM IPTG-conditioned broth, lysates of 17 clones contained HuscFvs seen as bands at ~27–32 kDa (Figure 1B). The HuscFvs of nine phage-transformed E. coli clones (nos. 6, 10, 21, 25, 27, 30, 34, 41, and 43) gave significant ELISA signal above controls (Figure 1C) and bound to SDS-PAGE separated-rNS3/4A by WB (Figure 1D). The nine clones revealed eight different huscfv patterns (eight RFLP types) after cutting with MvaI; clones no. 6, 10, 21, and 25 had patterns 1–4, respectively; clones no. 27 and 30 had the same pattern 5, while clones no. 34, 41, and 43 showed patterns 6–8, respectively (Figure 1E). DNA sequences of four clones, i.e., 6, 10, 25, and 34, had complete coding regions, i.e., sequences of four FRs and three CDRs of both VH and VL domains with linker nucleotides coding for (G4S)3 in between the domains. Therefore, these clones were selected for further experiments.
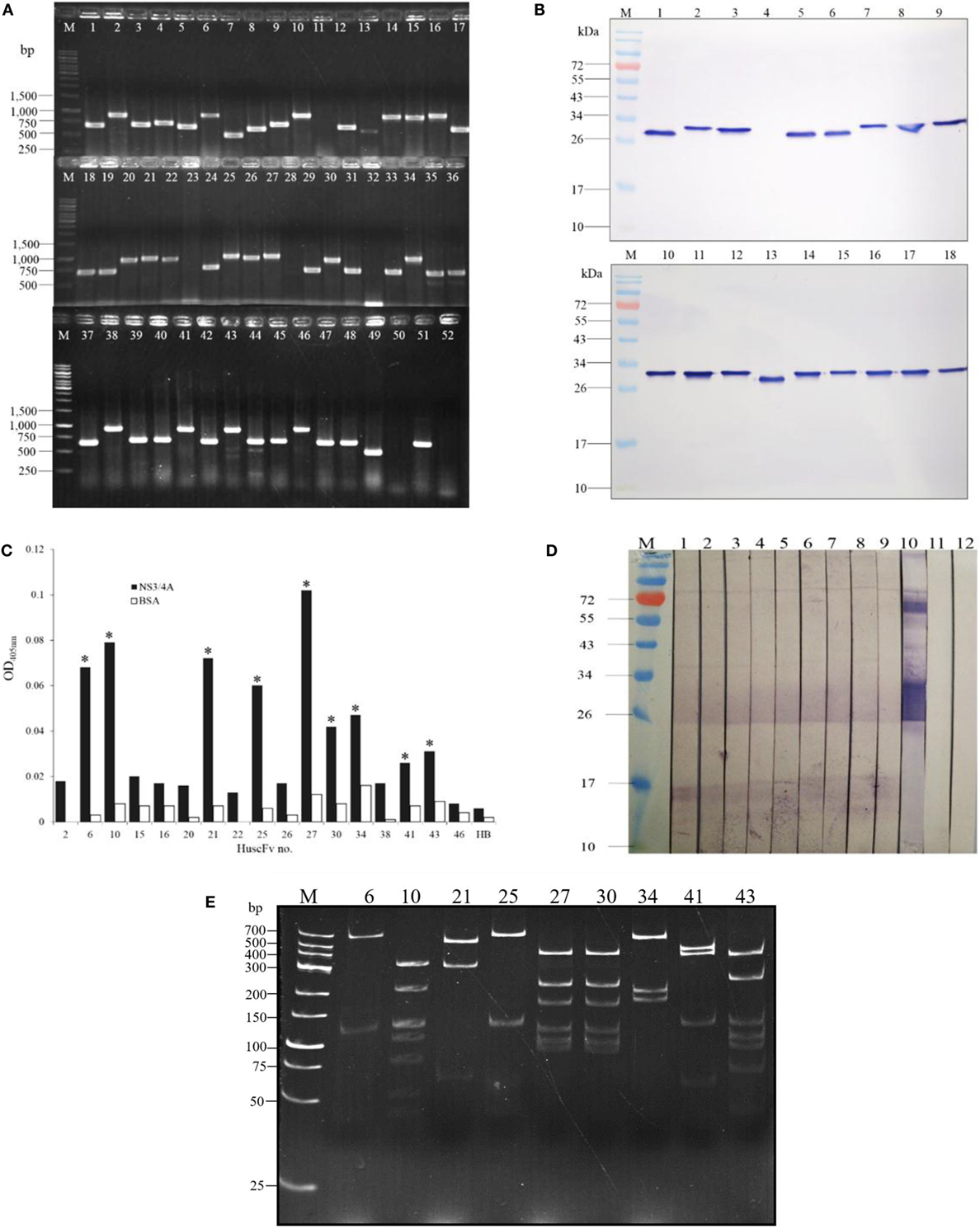
Figure 1. Preparation of E. coli clones that expressed NS3/4A-bound HuscFvs. Results of PCR for amplification of huscfvs using 52 E. coli bacteria infected with individual phages that bound to recombinant NS3/4A as templates; 18 clones (34.6%) revealed the huscfvs at about 1,000 base pairs (bp) (A). After growing the huscfv-positive clones under 0.5 mM IPTG induction, the bacterial lysates were checked for HuscFvs by Western blot analysis. Seventeen of the 18 clones produced the HuscFvs seen as bands at ~27–32 kDa (B). After standardizing spectrophotometrically, the HuscFvs were tested for their ability to bind to the NS3/4A by indirect ELISA. The antibodies produced by nine phage-transformed E. coli clones (nos. 6, 10, 21, 25, 27, 30, 34, 41, and 43) gave significant ELISA signal to the rNS3/4A above control antigen (BSA) and negative HuscFv contol (HB or lysate of original HB2151 E. coli) (C). Binding of the HuscFvs from the nine clones was verified by Western blotting. The HuscFvs were used to probe the SDS-PAGE-separated rNS3/4A, and all of them bound to the antigen seen as diffuse bands at ~26–32 kDa (lanes 1–9) (D); lane 10, positive binding control which the SDS-PAGE-separated rNS3/4A was probed with mouse anti-NS3/4A polyclonal antibodies. Lanes M of (B) and (D), protein marker; numbers at the left are protein molecular masses in kDa. Restriction fragment length polymorphism (RFLP) of the huscfvs of the clones (E). Lanes M of (A) and (E), DNA marker; numbers at the left are DNA sizes in bp.
Cell Penetrable HuscFvs (transbodies) and Their Cell Penetrating Ability and Toxicity
The huscfvs of the clones 6, 10, 25, and 34 in the pCANTAB5E-huscfv recombinant phagemids were subcloned into the pLATE52 plasmids downstream of the R9 gene, and the recombinant plasmids were put into the Rosetta™2 (DE3) E. coli. Figure 2A shows schematic diagram of the R9-HuscFv DNA construct. Examples of the PCR products amplified from the pLATE52-R9-huscfv-transformed Rosetta™2 (DE3) E. coli sibling clones are shown in Figure 2B. The R9-HuscFvs with 6× His and T7 tags at the N-terminal and E-tag at the C-terminal were produced by growing the respective transformed E. coli clones under the 1 mM IPTG induction condition. All clones expressed the R9-HuscFv fusion antibodies at ~32–34 kDa (Figure 2C). Figure 2D shows SDS-PAGE-separated R9-HuscFvs after being purified and refolded. The purified transbodies retained the rNS3/4A binding activity in the indirect ELISA (data not shown).
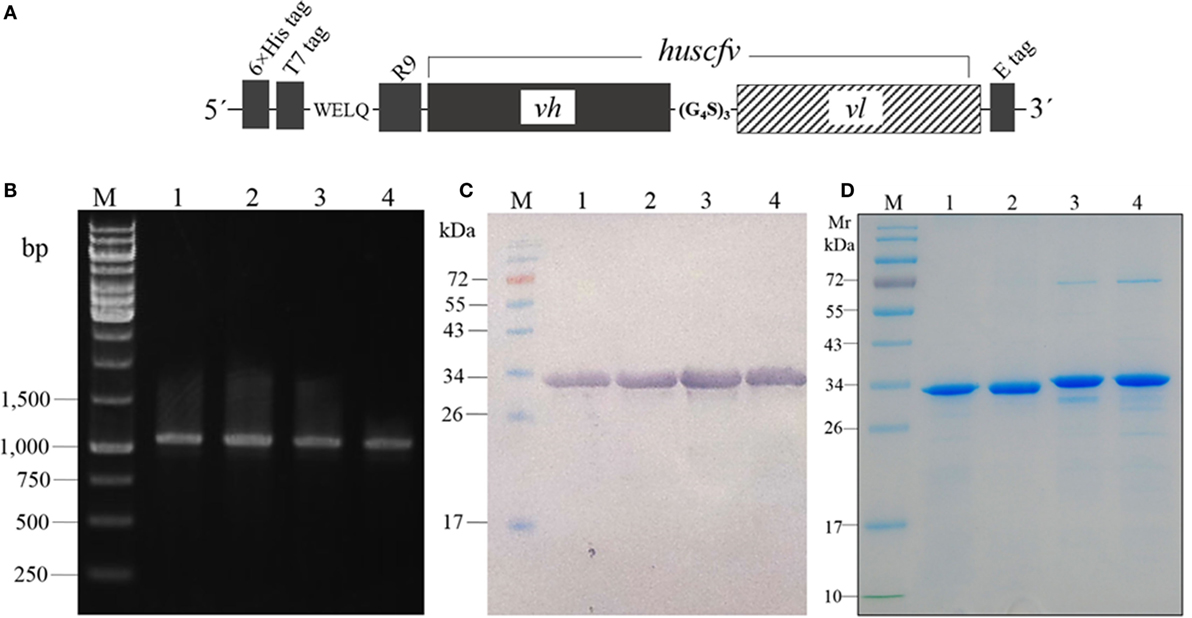
Figure 2. Schematic diagram of the R9-huscfv DNA construct (A) which the DNA sequence coding for R9-HuScFv was flanked with 6× His and T7 tags at the 5′ end and E-tag at the 3′ end; the vh and vl sequences of the huscfv were linked by nucleotides coding for (G4S)3. PCR amplicons of R9-huscfvs contained in the pLATE52-R9-huscfv plasmid-transformed Rosetta™2 (DE3) E. coli clones (B); the PCR product was ~1,140 bp; lane M, GeneRuler™ 1 kb DNA ladder; lanes 1–4, PCR amplicons of R9-huscfv-6, -10, -25, and -34, respectively; numbers at the left are DNA sizes in base pairs. Western blot patterns of homogenates of the four pLATE52-R9-huscfv plasmid-transformed Rosetta™2 (DE3) E. coli clones after growing under IPTG induction (C); the homogenates were subjected to SDS-PAGE and probed with anti-6× His antibody; the R9-HuscFvs were ~34 kDa. Purified R9-HuscFv-6, -10, -25, and -34 from the respective E. coli inclusion bodies (D).
Ability of the R9-HuscFvs to enter the HCV JFH-1 RNA-transfected Huh7 cells and colocalized with the intracellular NS3/4A protein was determined by immunofluorescence assay and confocal microscopy. Representative result is shown in Figure 3. The intracellular R9-HuscFvs probed with rabbit anti-6× His tag followed by goat anti-rabbit immunoglobulin-Alexa Flour®488 gave green fluorescence, while the intracellular NS3/4A probed with mouse anti-NS3/4A followed by donkey anti-mouse immunoglobulin-Alexa Flour®647 appeared in red. The DAPI-stained nuclei are seen in blue. After merging, the R9-HuscFv and the NS3/4A were found to colocalize (orange).
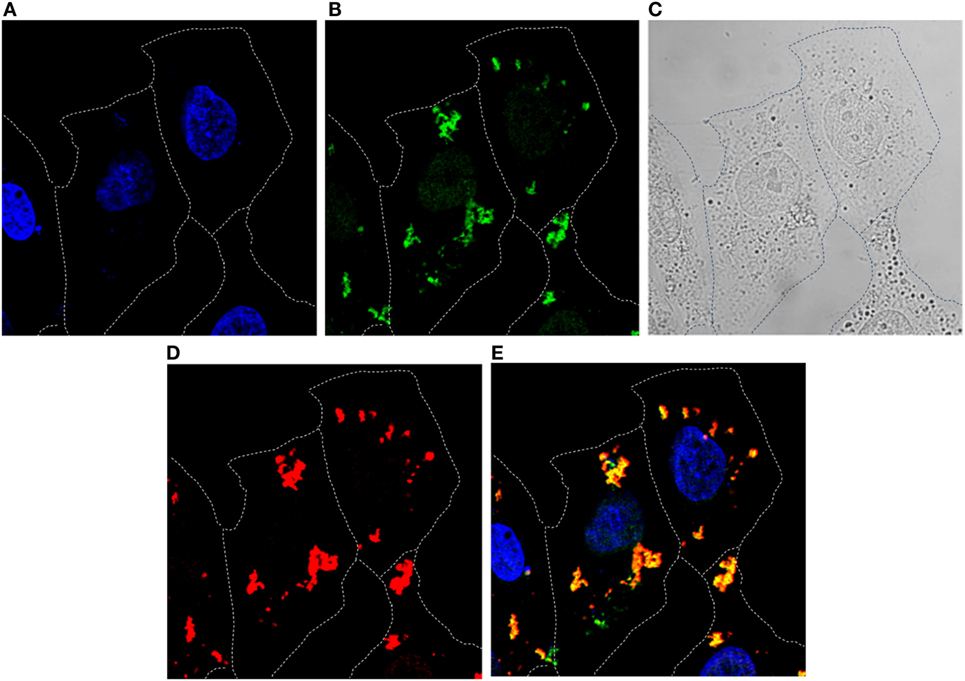
Figure 3. Immunofluorescence staining and confocal microscopy for determining entry of R9-HuscFv10 (as representative) into HCV JFH-1 RNA-transfected Huh7 cells and the R9-HuscFv binding to the intracellular NS3/4A. HCV JFH-1 RNA-transfected Huh7 cells were incubated with the R9-HuscFv then stained (see Materials and Methods). Cells stained with DAPI for localization of nuclei (blue) (A). Intracellular R9-HuscFv probed with rabbit anti-6× His tag followed by goat anti-rabbit immunoglobulin-Alexa Flour®488 (Green) (B). Bright field (C). Intracellular NS3/4A probed with mouse anti-NS3/4A followed by donkey anti-mouse immunoglobulin-Alexa Flour®647 (red) (D). Merged A, B, and D (E). The R9-HuscFv readily entered the HCV-infected cells and colocalized with the intracellular NS3/4A (orange).
Huh7 cells exposed to 25 μg of the respective R9-HuscFvs for 24 h did not release different LDH amounts from cells in medium alone (spontaneous release). The percent calculated viability of the transbody-treated cells was not different from the non-antibody-treated cells (Figure 4A). Likewise, percent cell viability after incubating with the R9-HuscFvs assessed using CellTiter-Glo luminescence assay were not different from the cells in culture medium alone (Figure 4B). Results indicate biocompatibility of the R9-HuscFvs with the mammalian cells.
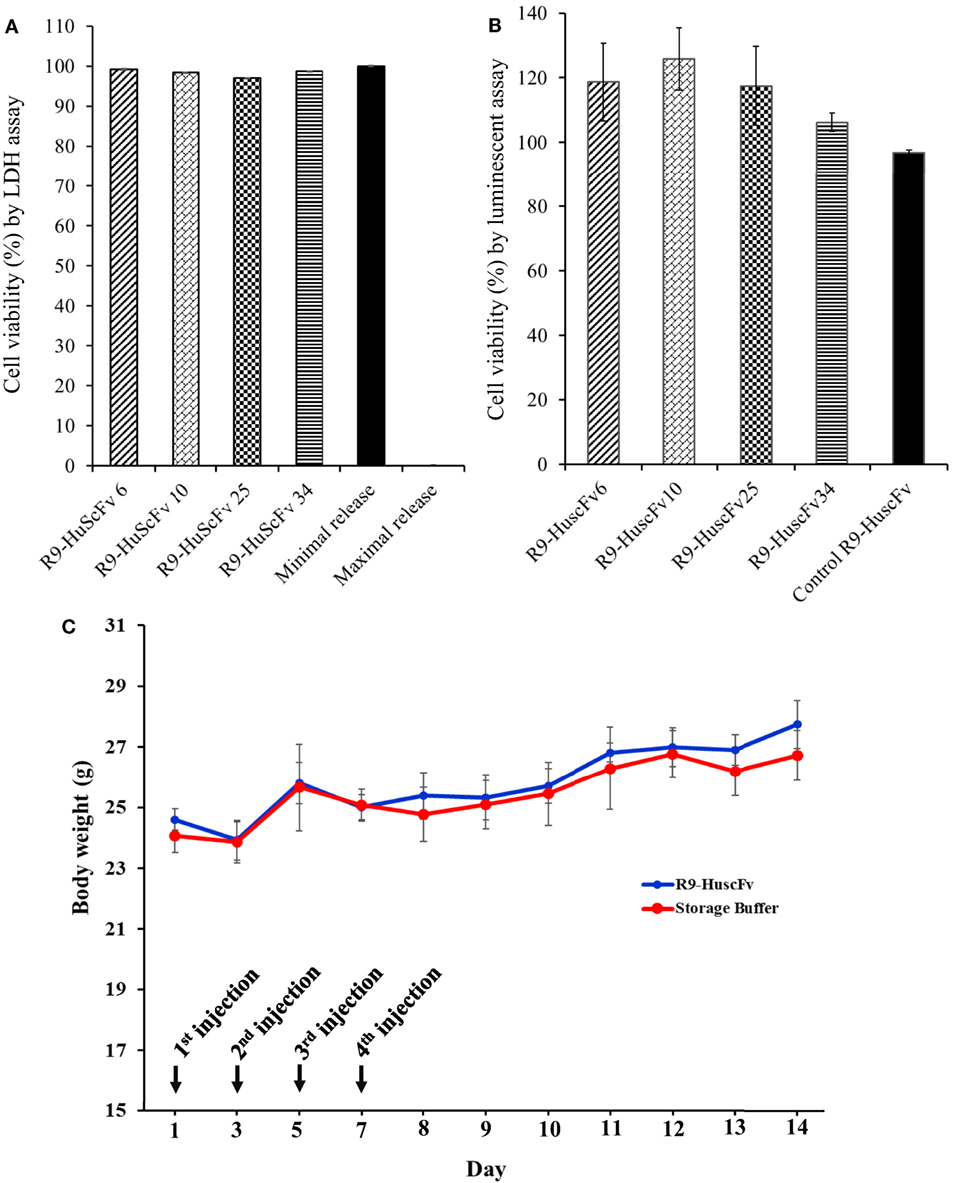
Figure 4. Viability of the Huh7 cells after treating with R9-HuscFvs and controls as determined by LDH assay (A) and CellTiter-Glo® Luminescent assay (B). For (A), Huh7 cells were incubated with 25 μg of R9-HuscFv-6, -10, -25, or -34. LDH in the cell culture medium was detected 24 h after adding the antibodies or controls, i.e., cells in medium alone (minimal/spontaneous LDH release) and cells added with 10% SDS (maximal LDH release). The calculated percent viability of the transbodies-treated cells was not different from the spontaneous (minimal) release. For (B), Huh7 cells were incubated with 25 μg of R9-HuscFvs or medium for 48 h and percent cell viability were determined using the Cell Titer-Glo® Luminescent Cell Viability Assay kit. The R9-HuscFvs did not cause significant reduction of the cell viability compared with the cells cultured in the medium alone. Daily body weights of mice injected intraperitoneally (C) with R9-HuscFv10 (as representative; blue line) compared with control mice that were injected with buffer alone (red line); all mice gained some weights during 14 days of experiment and did not show any sign of morbidity. Some biochemical parameters of the mouse blood samples are shown in Table S1 in Supplementary Material.
None of the mice that received, individually, four doses of 25 μg/g body weight of R9-HuscFv10 (representative) and buffer intraperitoneally on every alternate day showed any sign of morbidity (fever, hair ruffle, or diarrhea). They had normal activity and appetite and gained some weight (Figure 4C). Values of some parameters of the mouse blood chemistry at day 7 post-last booster are shown in Table S1 in Supplementary Material.
Transbody-mediated Inhibition of the HCV Replication
The HCV 5′ UTR copy numbers recovered from the HCV-RNA-transfected Huh7 cells that had been exposed to the NS3/4A-bound-R9-HuscFv-6, -10, -25, and -34 are shown in Figure 5 in comparison with various controls (positive and negative inhibition controls). Among the R9-HuscFvs, the R9-HuscFv10 and R9-HuscFv6 were the most effective transbodies in reducing the amount of the intracellular HCV RNA. Efficacies of all inhibitors and controls in reducing the intracellular HCV RNA are statistically compared as shown in Table 1.
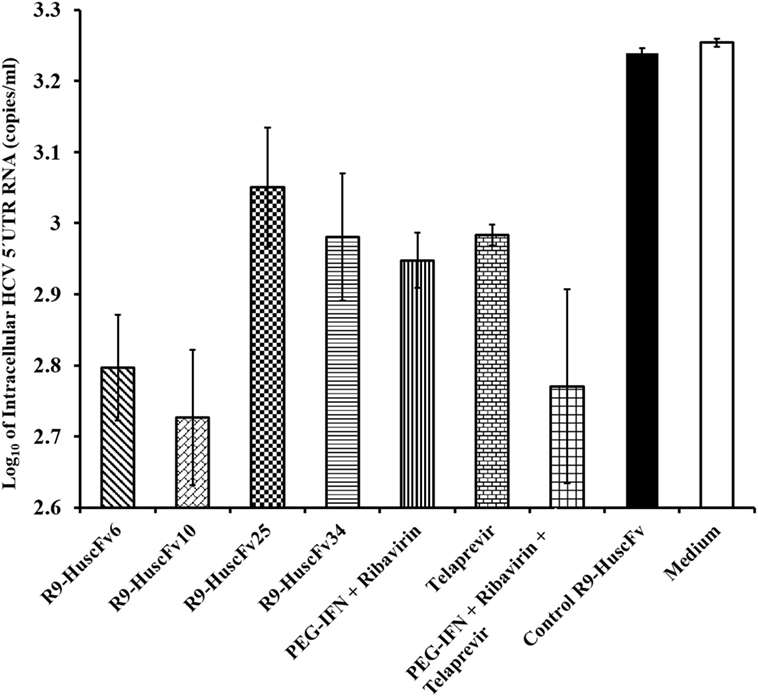
Figure 5. Amounts of HCV 5′ UTR RNA in the JFH-1 RNA-transfected Huh7 cells after treating with the R9-HuscFv-6, -10, -25, and -34 in comparison with controls. Table 1 shows p values for statistical comparison among different treatments.
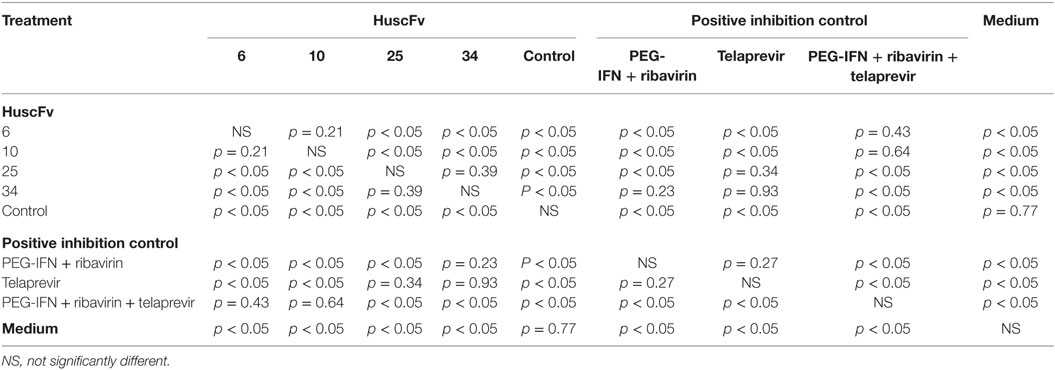
Table 1. Statistical comparison of the results of the HCV 5′ UTR mRNA copy numbers recovered from the HCV-RNA-transfected Huh7 cells that had been exposed to the NS3/4A-bound R9-HuscFvs and various controls.
Amounts of HCV 5′ UTR RNA in culture supernatants of all groups of the treated cells are shown in Figure 6. The most effective R9-HuscFvs in reducing the HCV RNA in the transfected cell culture supernatants were R9-HuscFv10 and R9-HuscFv6. Statistical differences of the effectiveness among groups are shown in Table 2.
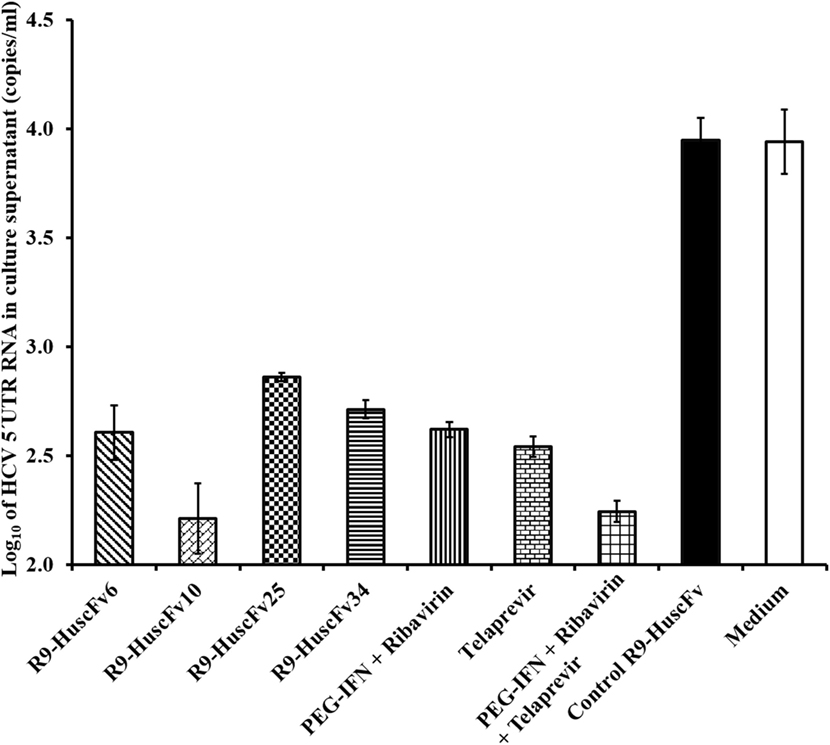
Figure 6. Amounts of HCV 5′ UTR RNA in culture fluids after treating with the R9-HuscFv-6, -10, -25 and -34 in comparison with controls. Table 2 shows p values for statistical comparison among different treatments.
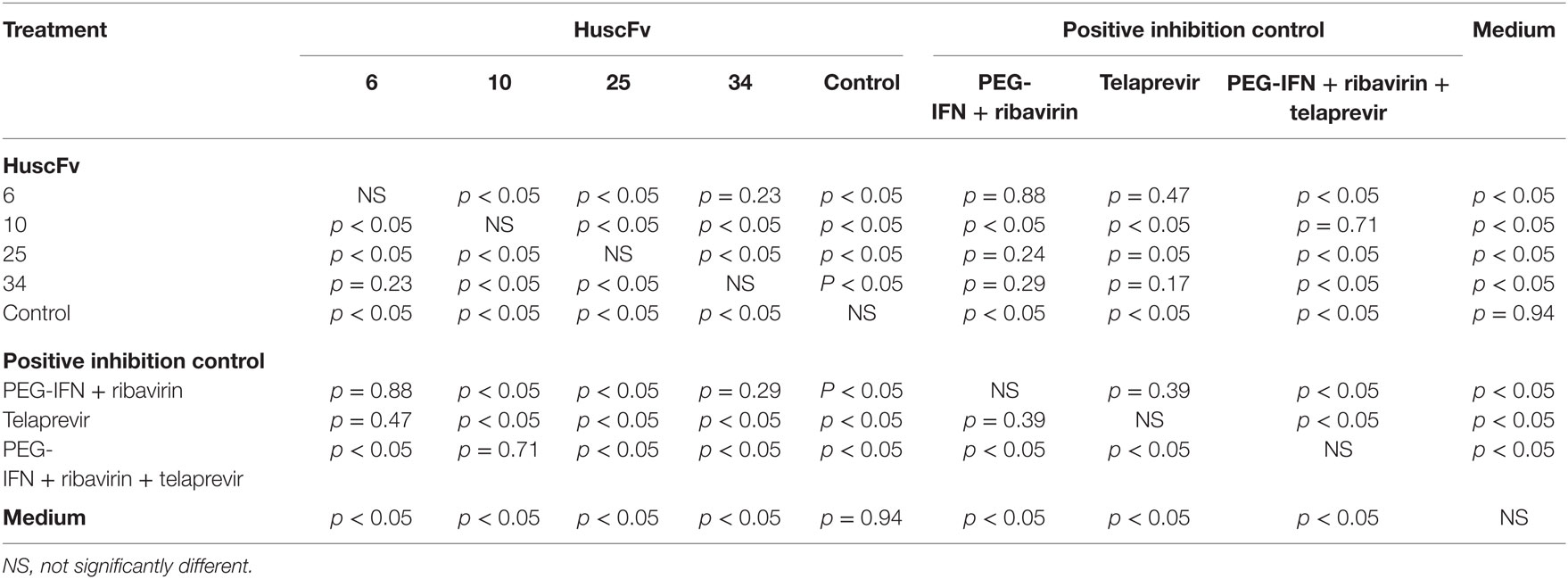
Table 2. Statistical comparison of the results of the HCV 5′ UTR copy numbers recovered from culture supernatants of the HCV-RNA-transfected Huh7 cells that had been treated with R9-HuscFv-6, -10, -25, and -34 and various controls.
Results of the assay for determining the HCV foci in transfected cells treated with R9-HuscFvs and controls are shown in Figure 7. Statistical comparison among all treatment groups are shown in Table 3. Figure S1 in Supplementary Material shows appearance of the HCV foci in cells of individual treatment groups after staining with anti-HCV core protein as the primary antibody. The effectiveness of the transbodies and the controls in reducing the numbers of HCV foci in the transfected cells were conformed to the results of the 5′ UTR RNA expression assay determined by the qRT-PCR. The R9-HuscFv10 was highly effective.
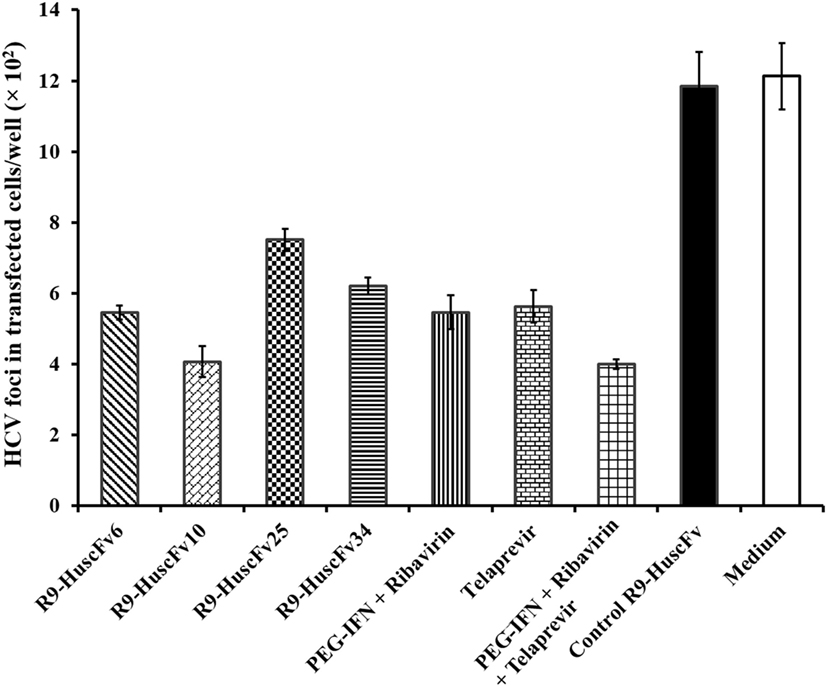
Figure 7. Foci of HCV in the HCV-RNA-transfected Huh7 cells after exposure to the R9-HuscFv-6, -10, -25, and -34 in comparison with controls. Table 3 shows p values for statistical comparison among different treatments.
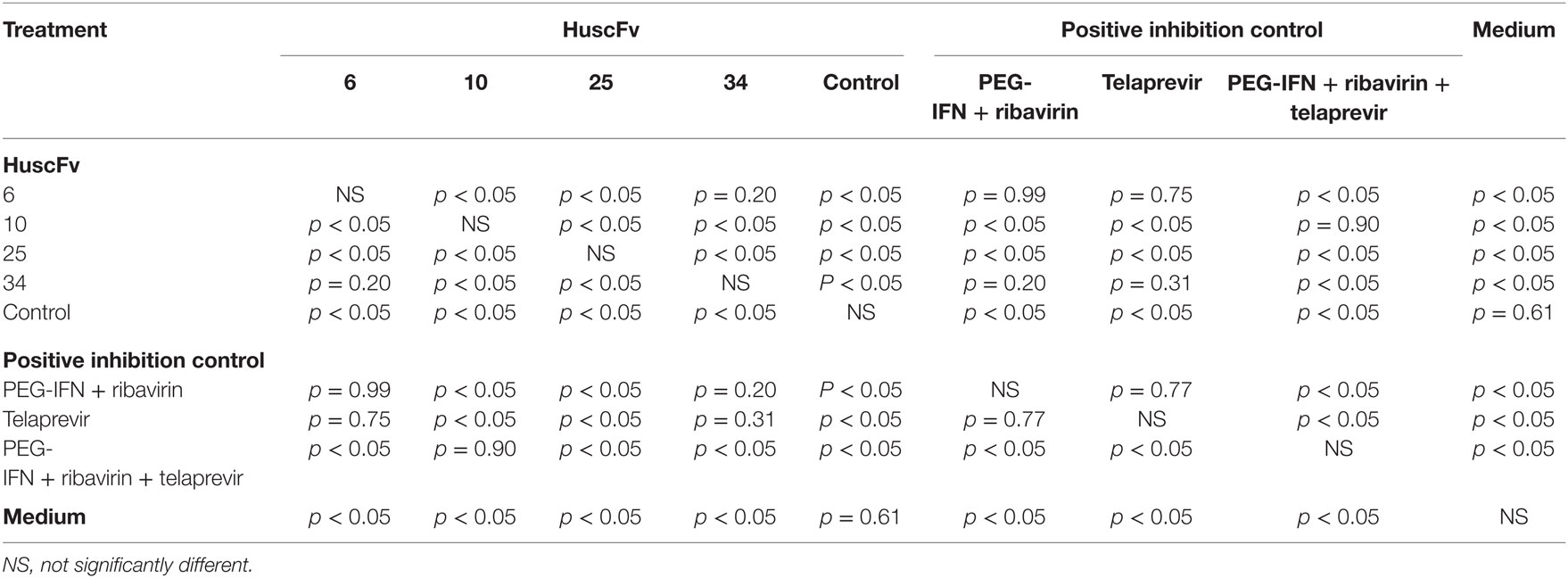
Table 3. Statistical comparison of the results of the HCV foci assay among HCV-RNA-transfected Huh7 cells treated with R9-HuscFvs and various controls.
Response of the HCV-RNA-transfected Cells after Treatment with the Transbodies
Details of the fold change of mRNA expressions of the innate immune response genes, including TRIF, TRAF3, IRF3, IL-28B, and IFN-β, after various treatments in comparison with the non-transfected (normal) Huh7 cells cultured in the medium are shown in Table 4. The R9-HuscFv6 and R9-HuscFv10 restored expressions of all studied host innate immune response genes. The control transbody-exposed transfected cells and the transfected cells maintained in medium alone had lower expressions of the genes than the non-transfected (normal) cells. At the condition and time of the experiments, TRAF3 and IL-28β mRNAs could not be rescued by the R9-HuscFv25 and R9-HuscFv34, respectively. All positive inhibitors, particularly the combined PEG-IFN-α + ribavirin + telaprevir effectively restored expressions of all of the studied genes.
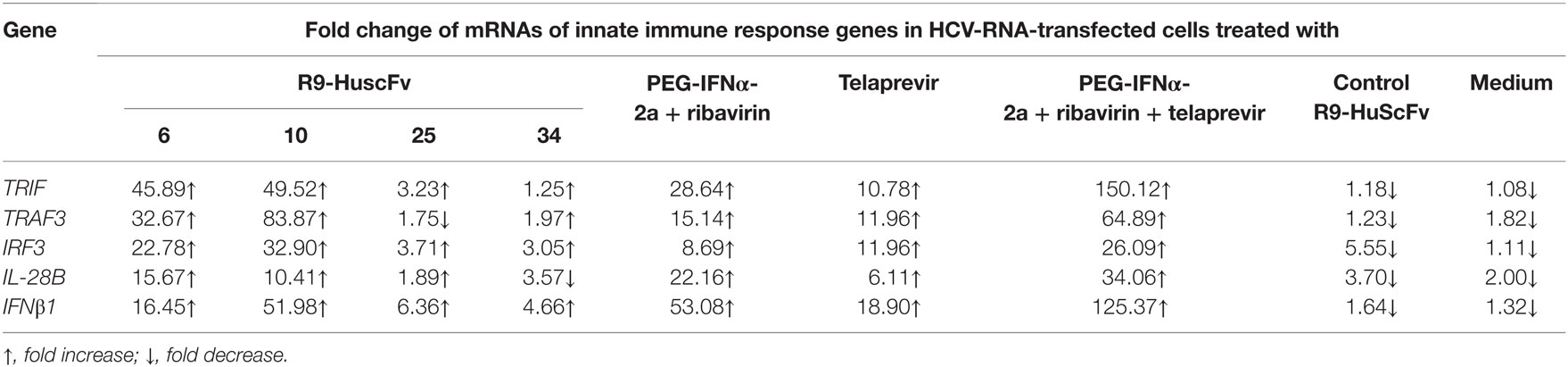
Table 4. Fold change of innate immune response genes in HCV-RNA-transfected human hepatic cells compared to non-transfected cells after treating with NS3/4A specific-R9-HuscFvs and various controls.
Homology Modeling and Intermolecular Docking for Predicting the Presumptive Residues of the HCV NS3/4A Interacted with the HuscFvs
The top 10 treading templates used to model the HuscFv6, HuscFv10, HuscFv25, and HuscFv34 by I-TASSER and the top 10 identified structural analogs of the HuscFvs in the PDB are shown in Tables S2A,B–S5A,B in Supplementary Material, respectively. Table S6 in Supplementary Material gives details of the qualities of the I-TASSER-predicted HuscFv models used in this study. The ClusPro lowest local energies of interactions between the NS3/4A 3D structure and the modeled HuscFv6, HuscFv10, HuscFv25, and HuscFv34 were −356, −428, −290, and −293 kcal/mol, respectively. Details of intermolecular docking, including the interactive residues of the NS3/4A/HCV polyprotein and motives and amino acids and domains of the HuscFvs as well as the interactive bonds between the two parties, are shown in Table 5 and Figures S2–S5 in Supplementary Material. Besides the molecular bonds in Table 5, the amino acid residues of the NS3/4A within 5 Å thresholds of van der Waals radii of HuscFvs are shown separately in Table 6.
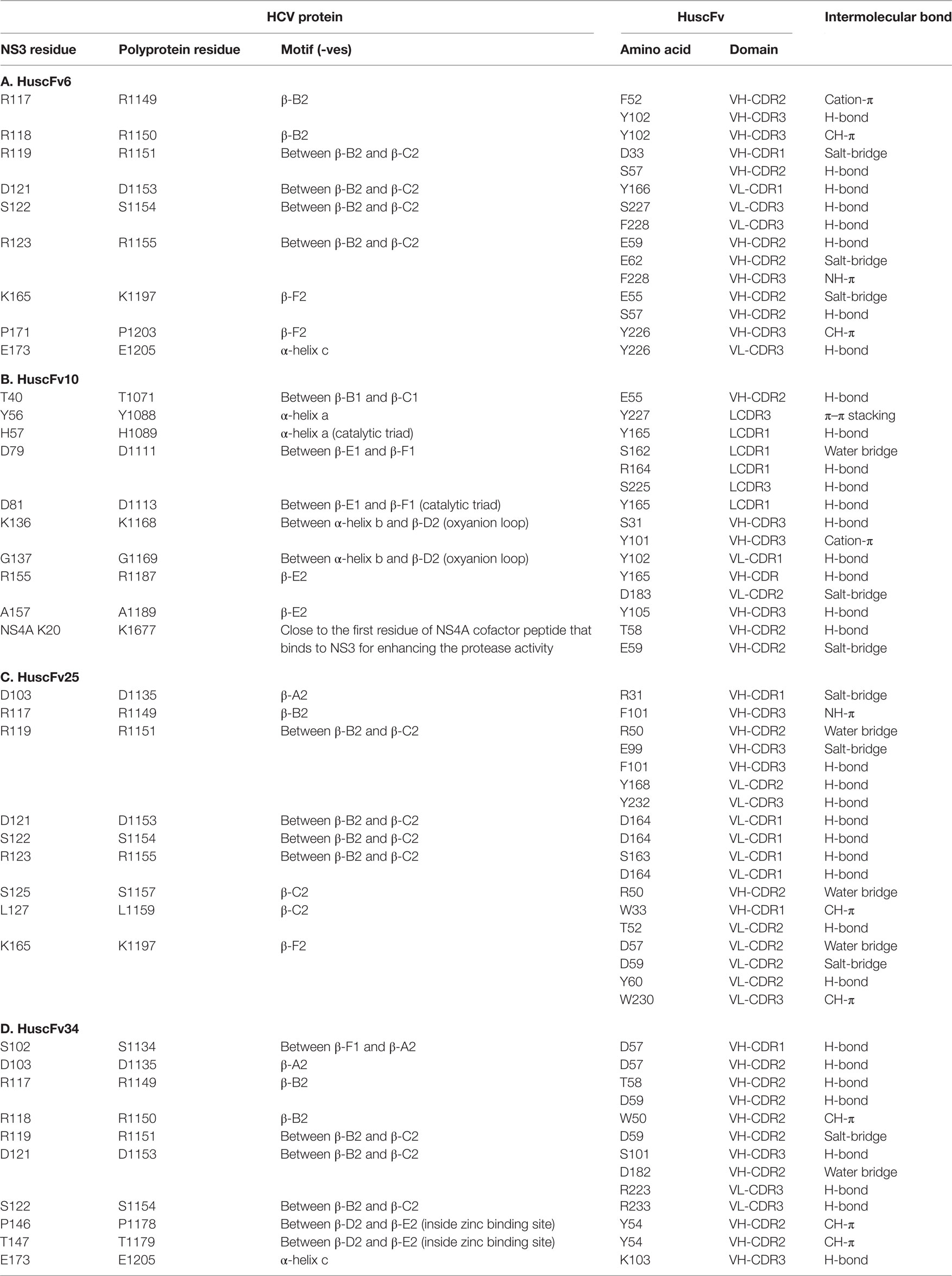
Table 5. Presumptive residues and regions of the NS3/4A and polyprotein of HCV bound by specific HuscFvs.

Table 6. Amino acid residues of NS3 within 5 Å threshold of van der Waals radii of HuscFvs in Table 2.
The most effective HuscFv10 in inhibiting the HCV replication was found to form interface contact with many critical residues of the NS3/4A important for protease activity including H57 and D81 of the catalytic triad, Y56 and D79 near to the catalytic site, K136 and G137 of the oxyanion loop, and K20 of NS4A cofactor which is adjacent to the NS4A residues 21–34 that usually interacts with the NS3 for enhancing the protease activity (Table 5B and Figure S2 in Supplementary Material). Table 6B shows the van de Waals forces that may contribute also to the HuscFv10-target association.
The HuscFv6, which was also highly effective in HCV replication inhibition, did not interact with any residues of the catalytic triad (H57, D81, and S139), oxyanion loop (135LKGSS139), or residues C97, C99, C145, and H149 of the zinc binding site. Instead, center of target binding of this HuscFv6 was located at the basic triad, i.e., 117RRR119, and R123 of the NS3/4A β–B2 and β-C2 motives (Tables 5A and 6A and Figure S3 in Supplementary Material).
The HuscFv25, similar to the HuscFv6, interacted with the basic residues: R117, R119, and R123, although at the higher local energy than the HuscFv6 (Tables 5C and 6C and Figure S4 in Supplementary Material).
The HuscFv34 not only interacted with the 117RRR119 but also with the P146 and T/S147 in the zinc binding site of the protease (Tables 5D and 6D and Figure S5 in Supplementary Material).
Discussion
There is a need of an improved regimen/remedy for treatment of HCV infection. A novel DAA that we would like to offer for future development is the engineered cell-penetrating human/humanized small antibody fragments that interfere with enzymes of the virus. The NS3/4A serine protease is pivotal for releasing mature NS4A, NS4B, NS5A, and NS5B from polyprotein (42) for the HCV replication, assembly, and egress. It also suppresses the host innate antiviral response.
During the early phase of infection, molecular signatures of intracellular viruses usually alarm the host pathogen recognition receptors (PRRs), including endosomal Toll-like receptors (TLRs), and cytosolic retinoic acid-inducible gene 1/helicard (RIG-1/Mda5) to activate various adaptor proteins, i.e., TRIF (Toll-IL-1 receptor domain-containing adaptor inducing IFN-β), Cardif/MAVS, and TRAFs (tumor necrosis factor-associated factors). These factors consequently activate kinases (TBK1 and IKKs) leading to phosphorylation and dimerization of interferon regulatory factor 3/7 (IRF3/7) transcription factor for production of innate interferons, including type-I IFN-β and type-III IFN-λ (IL-28/29) (43–45). The IFNs mediate autocrine and paracrine stimulation of the host cells through JAK/STAT signaling pathways leading to production of various antiviral factors, i.e., 2′, 5′ OAS, PKR, MxA, and ISGs, as well as MHC class I molecules for cytotoxic lymphocyte activation (45). NS3/4A protein of the infecting HCV cleaves TRIF and Cardif/MAVS, binds to TBK1, and inhibits activation of IRF3 (46–48). Moreover, the protein has been implicated in malignant transformation (49–51). The NS3 N-terminal is known to interact with p53 (a tumor suppressor) and inhibited apoptosis of NIH3T3 cells (51, 52). As such, interference with the HCV NS3/4A functions should lead to inhibition of the viral replication as well as restoration of the virally suppressed host innate immune response and might as well reduce the hepatocellular carcinoma incidence among the chronically infected subjects.
In this study, bacterially derived engineered human single chain antibodies that bound to active HCV NS3/4A protein were generated. Because the antibody target is located intracellularly, the antibodies were made into cell penetrable molecules by linking their coding sequences to nonaarginine (R9) nucleotides using a ligase-independent cloning (LIC) method. The R9-HuscFvs were readily purified from the IBs of four E. coli clones (nos. 6, 10, 25, and 34) carrying the R9-huscfv plasmids and successfully refolded using the protocol adapted from (53) and instruction manual of the Protein Refolding Kit of Novagen. The R9-HuscFvs retained their binding to the rNS3/4A as the parental HuscFvs (data not shown) indicating the native refolding of the antibodies. They readily entered the human hepatic cells (Huh7) and bound to the intracellular target. They did not cause significant LDH leakage from the cocultured human hepatic cells compared with the cells in the medium alone indicating compatibility of the antibody preparations with the mammalian cells at the dose that was tested. Safety of the transbodies was also demonstrated in mice after injecting them intraperitoneally (followed the Thai Pharmacopia) at the dose that was relatively high (25 μg/g body weight) for passive immunization in humans (37).
There were significant reductions of the HCV 5′UTR RNA in the Huh7 cells transfected with JFH-RNA and in the cell culture supernatants after treating with the R9-HuscFvs (albeit different degrees of effectiveness among the transbodies from different E. coli clones). The results of foci assay were conformed to the RNA quantification. The most effective transbodies were R9-HuscFv10 and R9-HuscFv6. These transbodies could readily restore expressions of all of the studied innate immune response genes of the HCV-RNA-transfected cells. Taken all together, the results indicate that the transbodies-mediated viral replication inhibition and host innate immune response restoration via the NS3/4A protease interference.
Presumptive residues of the NS3/4A interacted with the HuscFvs were determined by means of computerized homology modeling and intermolecular docking. The lowest local energy of interaction between the HuscFv10 (which was most effective in inhibiting the HCV replication and in restoration of the host innate gene expressions) and the NS3/4A was −428 kcal/mol, indicating relatively higher affinity on the target binding of this antibody than the other three antibodies. The predictive residues of the NS3/4A protease interacted by the HuscFv10 included several residues critical for the protease activity, i.e., H57 and D81 of the catalytic triads and K136 and G137 of the oxyanion loop (54, 55). Binding of the antibody to the H57 and D81 should inhibit deprotonation of the S139 and, thus, failure of the S139 to mediate a nucleophilic attack of the carbonyl carbon of the scissile bond to form a tetrahedral intermediate containing an oxyanion; therefore, the proteolytic cleavage should not be complete, and, hence, no release of the N- and C-peptide products from the polyprotein (56). The HuscFv10 might as well disturb the function of the oxyanion loop by docking on the K136 and G137. Moreover, binding of the antibody to the NS4A K20 might as well interfere with the non-covalent interaction between the cofactor (residues 21–34 of NS4A) to the NS3; thus, impaired the cofactor enhancement of the protease activity. Besides, van de Waals force formed by HuscFv10 and several residues of NS3/4A molecule (Table 6) might have additional negative impact on the protease activity. Possibly by causing steric hindrance of the oxyanion hole via the S138, the S1 pocket through interactive atoms at the L135, F154 and nearby residues, i.e., A156, V158, and C159 (54, 55), and the S6 pocket by means of R123 which is one of the positively charged residues of the S6 that usually interact with the P6 and P5 residues (56, 57).
The HuscFv6 was as effective as the HuscFv10 in inhibiting the HCV replication. The antibody readily restored the host innate immune response genes from the HCV suppression as did the R9-HuscFv10. From computerized simulation, the lowest local binding energy of this antibody to the NS3/4A was as low as −356 kcal/mol. However, computerized simulation indicated that the HuscFv6 did not dock on any residues of the catalytic triad, oxyanion hole, or zinc biding site. Instead, the center of interaction of this antibody with the target was at the basic patch of the NS3 protein formed by R117, R118, and R119. Although the role of this cationic patch has not been elucidated, it is likely that residues in this region might form electrostatic interaction with certain negatively charged surface, such as viral RNA. This interaction might mediate pivotal, yet unraveled, activity of the HCV NS3. Binding of the R9-HuscFv6 to these residues might interfere with the activity. Besides, the positively charged residues R123 and K165 of the NS3/4A S6 pocket are believed to interact with acidic residues at the P6, and sometimes the P5 also (57, 58); blocking of the R123 and K165 by CDR1 and CDR2 of the R9-HuscFv6 VL domain (Table 2) might interfere with the substrate binding of the protease. The HuscFv6 was predicted to form interactive atoms of van de Waals radii with multiple residues of the target (Table 3) that might also interfere with the NS3/4A activity by steric hindrance.
The HuscFv25 was the least effective transbody among the four in inhibiting the HCV replication. Computerized intermolecular docking revealed that the antibody also interacted with the protease basic patch (R117, R118, and R119) and S6 pocket residues (R123 and K165) similar to the HuscFv6. However, the local energy that required for binding of this antibody to the target (−290 kcal/mol) was higher than that of the HuscFv6, which might explain the lower effectiveness of the HuscFv25.
The HuscFv34 interacted with residues of the zinc binding site (C97, C99, C145, P146, and T147), which might cause improper folding of the NS3 and, therefore, deficient in the protease activity (59, 60). The antibody also docked on the NS3 basic patch and, thus, might interfere with the important, but still unknown, activity of the NS3 protein.
In conclusion, bacterially derived cell-penetrating human single chain antibodies specific to NS3/4A protein of HCV produced in this study inhibited replication of the heteologous HCV (genotype 2a of the pJFH-1) in human hepatic cells and restored expressions of the virally suppressed host innate immune response genes. They were not toxic to human cells and mice. The transbodies have potential for developing and testing further for anti-HCV activity, either alone, in combination with the approved HCV therapeutics, or in a mixture with their cognate specific to other HCV proteins.
Ethics Statement
Animal experiments received approval from the Siriraj Bio-safety Risk Management Taskforce (SI 2014-004), Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand. The consent procedure used for human participants is not applicable.
Author Contributions
SJ, WS, JT, and KT-i did experiments and prepared figures; RW, supervised SJ on protein purification; WC conceived the project, designed experiments, analyzed data, and wrote the manuscript; PS helped WC on experimental design.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Takaji Wakita of the Department of Microbiology, Tokyo Metropolitan Institute for Neuroscience, Tokyo, Japan, and Professor Dr. Ralf Batenschlager, Department of Molecular Virology, University of Heidelberg, Germany, for providing the pJFH-1. Huh7 cells were provided by Associate Professor Dr. Thawornchai Limjindaporn, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok.
Funding
The work was cosupported by the NSTDA Chair Professor grant (P-1450624) funded by the Crown Property Bureau; DPG5180001 grant of the Thailand Research Fund (TRF); and the National Research University project of the Office of Commission on Higher Education, Thailand. SJ was a scholar of the TRF Royal Golden Jubilee Ph. D. Program.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2016.00318
References
1. Suresh S. Hepatitis C virus: molecular biology & current therapeutic options. Indian J Med Res (2010) 131:17–34.
2. Kota S, Coito C, Mousseau G, Lavergne JP, Strosberg AD. Peptide inhibitors of hepatitis C virus core oligomerization and virus production. J Gen Virol (2009) 90:1319–28. doi:10.1099/vir.0.008565-0
3. Kota S, Takahashi V, Ni F, Snyder JK, Strosberg AD. Direct binding of a hepatitis C virus inhibitor to the viral capsid protein. PLoS One (2012) 7:e32207. doi:10.1371/journal.pone.0032207
4. Strosberg AD, Kota S, Takahasjhi V, Snyder JK, Mousseau G. Core as a novel viral target for hepatitis C drug. Viruses (2010) 2:1734–51. doi:10.3390/v2081734
5. Gao M, Nettles RE, Belema M, Snyder LB, Hguyen VN, Fridel RA, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature (2010) 65:96–100. doi:10.1038/nature08960
6. Gelman MA, Glenn JS. Mixing the right hepatitis C inhibitor cocktail. Trends Mol Med (2011) 17:34–46. doi:10.1016/j.molmed.2010.10.005
7. Liu X, Huang Y, Cheng M, Pan L, Si Y, Li G, et al. Screening and rational design of hepatitis C virus entry inhibitory peptides derived from GB virus A NS5A. J Virol (2013) 87:1649–57. doi:10.1128/JVI.02201-12
8. Miller MH, Agarwal K, Austin A, Brown A, Barclay ST, Dundas P, et al. Review article: 2014 UK consensus guidelines – hepatitis C management and direct-acting anti-viral therapy. Aliment Pharmacol Ther (2014) 39:1363–75. doi:10.1111/apt.12764
9. Lin MV, Chung R. Recent FDA approval of sofosbuvir and simeprevir. Implications for current HCV treatment. Clin Liver Dis (2014) 3:65–8. doi:10.1002/cld.332
10. Arico-Muendela C, Zhua Z, Dickson H, Parks D, Keicher J, Deng J, et al. Encoded library technology screening of hepatitis C virus NS4B yields a small molecule compound series with in vitro replicon activity. Antimicrob Agents Chemother (2015) 59(6):3450–9. doi:10.1128/AAC.00070-15
11. Lawitz E, Rouzier R, Nguyen T, Nguyen T, Ke J, Praestgaard J, et al. Safety and antiviral efficacy of 14 days of the cyclophilin inhibitor Nim811 in combination with pegylated interferon alpha2a in relapsed genotype 1 HCV infected patients. J Hepatol (2009) 50:S379. doi:10.1016/S0168-8278(09)61047-3
12. Tang H. Cyclophilin inhibitors as a novel HCV therapy. Viruses (2010) 2:1621–34. doi:10.3390/v2081621
13. Kiser JJ, Flexner C. Direct acting antiviral agents for hepatitis C virus infection. Annu Rev Pharmacol Toxicol (2013) 53:427–49. doi:10.1146/annurev-pharmtox-011112-140254
14. Cooper C, Lester R, Thorlund K, Druyts E, El Khoury AC, Yaya S, et al. Direct-acting antiviral therapies for hepatitis C genotype 1 infection: a multiple treatment comparison meta-analysis. Q J Med (2013) 106:153–63. doi:10.1093/qjmed/hcs214
15. Izquierdo L, Helle F, François C, Castelain S, Duverlie G, Brochot E. Simeprevir for the treatment of hepatitis C virus infection. Pharmgenomics Pers Med (2014) 7:241–9. doi:10.2147/PGPM.S52715
16. Feld JJ, Jacobson IM, Hezode C, Asselah T, Ruane PJ, Gruener N, et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med (2015) 373:2599–607. doi:10.1056/NEJMoa1512610
17. Lin C, Gates CA, Rao BG, Brennan DL, Fulghum JR, Luong YP, et al. In vitro studies of cross-resistance mutations against two hepatitis C virus serine protease inhibitors, VX-950 and BILN2061. J Biol Chem (2005) 280:36784–91. doi:10.1074/jbc.M506462200
18. Manns MP, Wedemeyer H, Cornberg M. Treating viral hepatitis C: efficacy, side effects, and complications. Gut (2006) 55:1350–9. doi:10.1136/gut.2005.076646
19. Pawlotsky JM. Therapeutic implications of hepatitis C virus resistance to antiviral drugs. Therap Adv Gastroenterol (2009) 2:205–19. doi:10.1177/1756283X09336045
20. Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med (2011) 364:1207–17. doi:10.1056/NEJMoa1009482
21. Wu S, Kanda T, Nagamoto S, Imazeki F, Yokosuka O. Hepatitis C virus protease inhibitor-resistant mutations: our experience and review. World J Gastroenterol (2013) 19:8940–8. doi:10.3748/wjg.v19.i47.8940
22. Dolman CE. Landmarks and pioneers in the control of diphtheria. Can J Public Health (1973) 64:317–36.
23. Deshayes S, Morris MC, Divita G, Heintz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell Mol Life Sci (2005) 62:1839–49. doi:10.1007/s00018-005-5109-0
24. Morris MC, Deshayes S, Simeoni F, Aldrien-Herrada G, Heitz F, Divita G. A nonocovalent peptide-based carrier for peptide and short interfering RNA delivery. In: Langel Ü, editor. Cell Penetrating Peptides. Boca Raton, FL: CRC Press (2008). p. 387–408.
25. Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol (2009) 157:195–206. doi:10.1111/j.1476-5381.2009.00057.x
26. Morris MC, Deshayes S, Heitz F, Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell (2008) 100:201–17. doi:10.1042/BC20070116
27. Noguchi H, Matsumoto S. Protein transduction technology: a novel therapeutic perspective. Acta Med Okayama (2006) 60:1–11.
28. Snyder EL, Dowdy SF. Recent advances in the use of protein transduction domains for the delivery of peptide, proteins and nucleic acids in vivo. Expert Opin Drug Deliv (2005) 2:43–51. doi:10.1517/17425247.2.1.43
29. Pujals S, Fernandez-Carneado J, Lopez-Iglesias C, Kogan MJ, Giralt E. Mechanistic aspects of CPP-mediated intracellular drug delivery: relevance of CPP-self assembly. Biochim Biophys Acta (2006) 1758:264–79.
30. Noguchi H, Bonner-Weir S, Wei FY, Matsushita M, Matsumoto S. BETA2/NeuroD protein can be transduced into cells due to an arginine- and lysine-rich sequence. Diabetes (2005) 54:2859–66. doi:10.2337/diabetes.54.10.2859
31. Phalaphol A, Thueng-in K, Thanongsaksrikul J, Poungpair O, Bangphoomi K, Sookrung N, et al. Humanized-VH/VHH that inhibit HCV replication by interfering with the virus helicase activity. J Virol Methods (2013) 194:289–99. doi:10.1016/j.jviromet.2013.08.032
32. Thueng-in K, Thanongsaksrikul J, Srimanote P, Bangphoomi K, Poungpair O, Maneewat S, et al. Cell penetrable humanized-VH/VHH that inhibit RNA dependent RNA polymerase (NS5B) of HCV. PLoS One (2012) 7:e49254. doi:10.1371/journal.pone.0049254
33. Thueng-in K, Thanongsaksrikul J, Jittavisutthikul S, Seesuay W, Chulanetra M, Sakolvaree Y, et al. Interference of HCV replication by cell penetrable human monoclonal scFv specific to NS5B polymerase. mAbs (2014) 6:1327–39. doi:10.4161/mabs.29978
34. Jittavisutthikul S, Thanongsaksrikul J, Thueng-in K, Chulanetra M, Srimanote P, Seesuay W, et al. Humanized-VHH transbodies that inhibit HCV protease and replication. Viruses (2015) 7:2030–56. doi:10.3390/v7042030
35. Poungpair O, Pootong A, Maneewatch S, Srimanote P, Tongtawe P, Songserm T, et al. A human single chain transbody specific to matrix protein (M1) interferes with the replication of influenza A virus. Bioconjug Chem (2010) 21:1134–41. doi:10.1021/bc900251u
36. Kulkeaw K, Sakolvaree Y, Srimanote P, Tongtawe P, Maneewatch S, Sookrung N, et al. Human monoclonal ScFv neutralize lethal Thai cobra, Naja kaouthia, neurotoxin. J Proteomics (2009) 72:270–82. doi:10.1016/j.jprot.2008.12.007
37. Tao T, Ma X, Yang J, Zou JY, Ji SM, Tan YS, et al. Humanized anti-CD25 monoclonal antibody treatment of steroid-refractory acute graft-versus-host disease: a Chinese single-center experience in a group of 64 patients. Blood Cancer J (2015) 5:e308. doi:10.1038/bcj.2015.33
38. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc (2010) 5:725–38. doi:10.1038/nprot.2010.5
39. Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J (2011) 101:2525–34. doi:10.1016/j.bpj.2011.10.024
40. Zhang J, Liang Y, Zhang Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure (2011) 19:1784–95. doi:10.1016/j.str.2011.09.022
41. Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, et al. How good is automated protein docking? Proteins (2013) 81:2159–66.
42. Bartenschlager R, Ahlborn-Laake L, Mous J, Jaconson H. Kinetics and structure analysis of hepatitis C virus polyprotein processing. J Virol (1994) 68:5045–55.
43. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature (2001) 413:732–8. doi:10.1038/35099560
44. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol (2004) 5:730–7. doi:10.1038/ni1087
45. Wang Y, Li J, Wang X, Ye L, Zhou Y, Thomas RM, et al. Hepatitis C virus impairs TLR3 signaling and inhibits IFN-λI expression in human hepatoma cell line. Innate Immun (2014) 20:3–11. doi:10.1177/1753425913478991
46. Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AN, Ikeda M, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the toll-like receptor 3 adapter protein TRIF. Proc Natl Acad Sci U S A (2005) 22:2992–7.
47. Li XD, Sun L, Seth RB, Pineda G, Chen Z. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein of the mitochondria to evade innate immunity. Proc Nat Acad Sci U S A (2005) 102:17717–22.
48. Otsuka M, Kato N, Moriyama M, Taniguchi H, Wang Y, Dharel N, et al. Interaction between the HCV NS3 protein and the host TBK1 protein leads to inhibition of cellular antiviral response. Hepatology (2005) 41:1004–12. doi:10.1002/hep.20666
49. Sakamuro D, Furukawa T, Takegami T. Hepatitis C virus nonstructural protein NS3 transforms NIH 3T3 cells. J Virol (1995) 69:3893–6.
50. Zemel R, Gerechet S, Greif H, Bachmatove L, Birk Y, Golan-Goldhirsh A, et al. Cell transformation induced by hepatitis C virus NS3 serine protease. J Viral Hepat (2001) 8:96–102. doi:10.1046/j.1365-2893.2001.00283.x
51. Tanaka M, Nagano-Fujii M, Deng L, Ishido S, Sada K, Hotta H. Single-point mutations of hepatitis C virus NS3 that impair p53 interaction and anti-apoptotic activity of NS3. Biochem Biophys Res Commun (2006) 340:792–9. doi:10.1016/j.bbrc.2005.12.076
52. Fujita T, Ishido S, Muramatsu S, Itoh M, Hotta H. Suppression of actinomycin D-induced apoptosis by the NS3 protein of hepatitis C virus. Biochem Biophys Res Commun (1996) 229:825–8. doi:10.1006/bbrc.1996.1887
53. Rao Dasari VK, Are D, Rao Joginapally V, Mangamoori LN, Rao Adibhatla KSB. Optimization of the downstream process for high recovery of rhG-CSF from inclusion bodies expressed in Escherichia coli. Process Biochem (2008) 43:566–75. doi:10.1016/j.procbio.2008.01.024
54. Love RA, Parge HE, Wickersham JA, Hostomsky Z, Habuka N, Moomaw EW, et al. The crystal structure of the hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell (1996) 87:331–42. doi:10.1016/S0092-8674(00)81350-1
55. Kim JL, Morgenstern KA, Lin C, Fox T, Dwyer MD, Landro JA, et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell (1996) 87:343–55. doi:10.1016/S0092-8674(00)81351-3
56. Raney KD, Sharma SD, Moustafa I, Cameron CE. Hepatitis C virus non-structural protein 3 (HCV NS3): a multifunctional antiviral target. J Biol Chem (2010) 285:22725–31. doi:10.1074/jbc.R110.125294
57. Koch U, Biasiol G, Brunetti M, Fattori D, Pallaoro M, Steinkuhler C. Role of charged residues in the catalytic mechanism of hepatitis C virus NS3 protease: electrostatic precollision guidance and transition-state stabilization. Biochemistry (2001) 40:631–40. doi:10.1021/bi002160t
58. Steinkuhler C, Koch U, Narjes F, Matassa VG. Hepatitis C virus protease inhibitors: current progress and future challenge. Curr Med Chem (2001) 8:919–32.
59. De Franscesco R, Urbani A, Nardi MC, Tomei L, Steinkuhler C, Tramontano A. A zinc binding site in viral serine proteinase. Biochemistry (1996) 35:13282–7.
Keywords: cell-penetrating antibody (transbody), hepatitis C virus, innate immunity, phage display, NS3/4A protease, qRT-PCR, single chain antibody
Citation: Jittavisutthikul S, Seesuay W, Thanongsaksrikul J, Thueng-in K, Srimanote P, Werner RG and Chaicumpa W (2016) Human Transbodies to HCV NS3/4A Protease Inhibit Viral Replication and Restore Host Innate Immunity. Front. Immunol. 7:318. doi: 10.3389/fimmu.2016.00318
Received: 04 March 2016; Accepted: 08 August 2016;
Published: 26 August 2016
Edited by:
Amy Rasley, Lawrence Livermore National Laboratory (DOE), USAReviewed by:
Simone Brogi, University of Siena, ItalyKenichi Morikawa, Hokkaido University Hospital, Japan
Keke Celeste Fairfax, Purdue University, USA
Copyright: © 2016 Jittavisutthikul, Seesuay, Thanongsaksrikul, Thueng-in, Srimanote, Werner and Chaicumpa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wanpen Chaicumpa, wanpen.cha@mahidol.ac.th