Hydrogels with micellar hydrophobic (nano)domains
 Miloslav Pekař
Miloslav Pekař- Faculty of Chemistry, Brno University of Technology, Brno, Czech Republic
Hydrogels containing hydrophobic domains or nanodomains, especially of the micellar type, are reviewed. Examples of the reasons for introducing hydrophobic domains into hydrophilic gels are given; typology of these materials is introduced. Synthesis routes are exemplified and properties of a variety of such hydrogels in relation with their intended applications are described. Future research needs are identified briefly.
General Introduction
Gels are well-known materials of colloidal type in which the dispersion medium (phase) is in liquid state and the dispersed phase (colloidal “particles”) is in solid state. The dispersed phase forms a network throughout the whole gel sample and the liquid is, in fact, entrapped in this mesh. In other words, the liquid swells the solid matter. The entrapment of a liquid within a solid network is the base of specific and unique properties of gels. Gels combine the behavior of liquids and solids in a soft matter, which usually retains its shape, but is relatively easily deformable. Specific behavior of a particular gel sample is determined by the density of its network or the density of crosslinks connecting the network. The crosslinks provide the structure and physical integrity of the gel network. Typically, gels can be characterized as viscoelastic solids (and liquids in a broader sense). If the liquid part is water, the gel is called hydrogel.
Gels in general are soft materials of significant interest for applications in various areas of industry. Hydrogels, composed usually of crosslinked chains of hydrophilic polymers, exhibit distinct property of good swelling capacity in aqueous media. If there are no problems of biocompatibility, they can have widespread applications in different fields related to human health, appearance, or nutrition including pharmaceutical, bioengineering, food industry, medicine, cosmetics, agriculture and horticulture, and so on. Hydrogels, prepared both from synthetic polymers and biopolymers, which are similar to biological tissues and compatible with them have been important materials for drug delivery and tissue engineering. Hydrogels are applied as soft contact lenses, artificial implants, actuators, wound healing dressings, etc. (Shukla et al., 2009). Specifically designed hydrogels capable of a sensitive response to external stimuli such as pH, temperature, ionic strength, magnetic field, electric field, ultraviolet light, and internal stimuli-like chemical architecture, initiators conditions enable them to deserve as potential candidates for biomaterial applications (Shukla et al., 2009).
Hydrogels can be thus be viewed as elastic polymer networks that absorb large amounts of water. Typical hydrogels are prepared by chemical or physical crosslinking of hydrophilic polymers, such as polyacrylamide, poly(ethylene oxide), or poly(2-hydroxyethyl methyl acrylate) (Hao and Weiss, 2011), or biopolymers such as native or modified polysaccharides. Chemically, crosslinked hydrogels are formed from covalent bonds, and thus, they have a permanent shape, which resists to changes in temperature within the range of hydrogel thermal stability. The shape can be deformed by the application of stress and recovered when the stress is removed or if the stress is low enough. Because of the irreversible feature of a covalent bond, such hydrogels cannot be manufactured into controlled shapes by melt processing methods such as injection molding or compression molding, except for cases where the covalent network is formed during that process.
Physical hydrogels have transient crosslinks formed by associations among polymeric chains bearing complementary functional groups that exhibit, for example, electrostatic interactions, chain entanglements, hydrophobic interactions, and hydrogen bonding or produce crystallizing segments (Hao and Weiss, 2011). Because of the reversible nature of non-covalent intermolecular interactions upon temperature changes, the shape of physical hydrogels can be reversibly changed (destroyed and renewed) by changes in temperature and these gels can be melt processed into a desired shape. Although physical hydrogels do not possess a permanent (covalent) network structure, they do exhibit elasticity if the relaxation time of the network is much longer than the application time of stress. That is, physical elastic networks can be achieved when the stresses are either small, the temperature is sufficiently low, or the stress application is of short duration (i.e., short time or high frequency). Under such conditions, a physical gel may be mechanically indistinguishable from a covalent gel, and the network appears to be permanent. The reversible aspect of the physical network structure provides unique properties, such as viscous flow above a critical stress or time that allows the gel to be injectable and improved mechanical toughness compared with a covalent gel.
In general, three-dimensional network structure of (hydro)gels is responsible for their mechanical properties and porous microstructure formed in this network provide permeability and filtration effect.
With the advances of nanotechnology besides the traditional macroscopic gels, macrogels, also micro-, and recently nanogels (Yallapu et al., 2007) have been designed, synthetized, and studied. Microgel is a dispersion of discrete polymeric gel particles, which are in the size range typically from 1 mm to 1 μm. Similarly, nanogels can be described as gel macromolecules in the size range of tens to hundreds nanometers though various definitions of nanogels have been given.
After this Section “General Introduction,” let us move to the main topic of this tractate.
Why Hydrophobic Domains in Hydrogels
This review is focused on hydrogels containing hydrophobic domains or nanodomains, especially of the micellar type. Why to combine hydrogels with something of opposite character and properties? The answer can be simple – to modify, upgrade, or tailor properties of hydrogels. The improved properties are usually dictated by hydrogel applications, many of them coming from the fields of medicine and health-care. Let us review the main application requirements in general and with selected reference examples. The purpose is not to give an exhaustive overview but to point to leading approaches and “hot topics” in this area including their variability and to describe them rather thoroughly. Hydrogels with micellar domains are hybrid colloidal materials of specific properties, which deserve more attention and study, are seen. For example, the research topic phrase “hydrogels incorporating micelles” returned in SciFinder database only about 20 hits (in July 2014); similarly, the phrases “hydrogels and micelles” or “hydrogels and micelles.” It is hoped that this review will encourage more intensive research in this interesting material field.
Nature is a great inspiration and in the same time puts the principal requirements. Biocompatible hydrogels are in dramatically increasing demand that exhibit complex behavior, but are nonetheless conceptually easy to prepare and to process, while having accessible handles with which their mechanical and physiological properties can be tailored (Guo et al., 2014). In general, gels that are highly swollen by a liquid are very weak because they do not have many intrinsic mechanisms to dissipate energy during their deformation. While the frictional properties of hydrogels may be desirable, many hydrogels suffer from low shear strength. Many hydrogels lack the required mechanical properties to be useful as articulating and weight bearing materials. To obtain a hydrogel with a high degree of mechanical toughness, additional dissipative mechanisms at the molecular level is needed. Proper molecular design can impart hydrogels with a suite of attractive properties: improvements in tensile toughness, in strength and resilience while saving the high water uptake, introduction of dynamic physical (i.e., usually non-covalent) crosslinks to promote processability and self-healing, stimuli responsiveness, or shape memory behavior.
Manipulating and controlling hydrogel structures at nanometer level are very effective ways in the design of precisely defined three-dimensional structures, which enable tailoring their mechanical properties. The nanometric scaled structures with different morphologies incorporated into the hydrogel contribute to the control over the hydrogel mechanical and physical properties. The development of hydrogel nanostructuring methods is still challenging because the high water content excludes the use of lithographic techniques. On the other hand, hydrogels nanostructuring can be approached generally and relatively easily by the self-assembly capability of hydrophobic moieties in aqueous environment. Domains formed by the self-assembly impart hydrogels’ new properties not only from the mechanical but also from the functional point of view as will be demonstrated in this review.
The behavior of hydrogels with incorporated hydrophobic (nano)domains resemble the behavior of semi-crystalline polymers, which contain both crystalline regions and amorphous regions (Thomas et al., 2009). Crystallinity is known to have desired effect on the mechanical properties and solubility of polymers. It can be proposed that the hydrophilic segments (resembling amorphous regions) in such a hydrogel could provide the water absorption, fluid flow, and lubricious properties needed while the hydrophobic segments (playing the role of crystalline regions) provide the strength, tear, and shear resistance. Properties may be varied based on the ratio of hydrophobic to hydrophilic composition. The polymers must overcome the heat and entropy factors associated with the crystalline or hydrophobic regions during dissolution, the hydrophobized hydrogels are thus expected to have excellent dissolution resistance in aqueous environment (e.g., in the body). On the other hand, it must be considered that the creation of hydrophobic domains can normally result in a change in the maximum hydration (swelling) as well as in the organization of water molecules within hydrogel structure.
Micelles formed by surfactants at and above the critical micellar concentration are well-known examples of the self-assembly due to hydrophobic interactions in aqueous environment. Moreover, not only low molecular surfactants but also properly structured polymers (or modified biopolymers) may form micelles, called polymeric micelles in this case. Nowadays generally, surfactant and polymer systems play a more and more important role in modern drug delivery, where they may be used to control drug release rate, enhance effective drug solubility, minimize drug degradation, reduce drug toxicity, or facilitate control of drug uptake (Liu and Li, 2005). Moreover, the hydrophobic domains of surfactant micelles allow the solubilization of lipophilic substances. The amphiphilic surfactant structure also provides a potential in increasing the permeability of the drug through biological membranes, resulting in an augmented intracellular drug concentration. In fact, micellar solutions have been proposed as efficient strategies for drug delivery a couple of decades ago. They are able to modulate both the pharmacokinetics and the bioavailability of the drug to result in an overall increase in the drug therapeutic index. Similarly, the incorporation of micelles into hydrogel is expected to result in changes not only of material properties but also in molecular transport (diffusion) within a hydrogel.
With the reference to the medical applications, stimuli-sensitive (sometimes called “intelligent”) and injectable (in situ gelling) hydrogels should be mentioned. The latters can be administered into the body via a minimally invasive route, which is one of their principal advantages. The injectable hydrogels are mostly based on self-assembled nanosized polymeric micelles. When injected into the body using a syringe, sudden changes of surrounding environments such as temperature, pH, and other biochemical signals alter their conformation and/or their non-covalent interactions, resulting in a sol-to-gel phase transition at critical conditions.
These general reasons for the incorporation of hydrophobic (nano)domains into hydrogels will be now illustrated by a selection of published specific examples. Although the main focus is on the micellar domains, related inspiring systems could also be included.
Liu and Li (2005) used micelles in hydrogel for the purpose of drug solubilization, immobilization, and protection. The drug was camptothecin, which exhibits high anti-cancer activity against a wide spectrum of human malignancies. Because it is practically water-insoluble, its full therapeutic potential had not been achieved. Moreover, in vivo and in aqueous solutions buffered at pH above seven it rapidly converts to the respective water-soluble, pharmacologically less active carboxylate form. Camptothecin was solubilized by means of an ionic surfactant, sodium dodecyl sulfate (SDS), and the loaded micelles were mixed into agarose hydrogel. This method not only helped to increase the solubility of the drug but also promoted a good and stable distribution of the drug molecules in the hydrogel, which participated in the controlled released function as well.
Ju et al. (2013) solubilized paclitaxel, a well-known anti-cancer agent with a poor water-solubility, in hydrophobically modified chitosan capable of forming (polymeric) micelles. The loaded micelles were dispersed in an interpenetrated hydrogel formed by a hydrophilic linear triblock polymer and carboxymethyl chitosan, specifically designed for the in situ cancer treatment. The triblock polymer provided gelation at elevated temperature when applied intratumorally. The carboxymethylated chitosan was chemically crosslinked to generate a network preventing the fast erosion of the triblock polymer gel. The gelation and structure are schematically shown in Figure 1. The micellar encapsulation reduced the side-toxicity of the drug and the whole system demonstrated prolonging the in vitro drug release to a large extent.
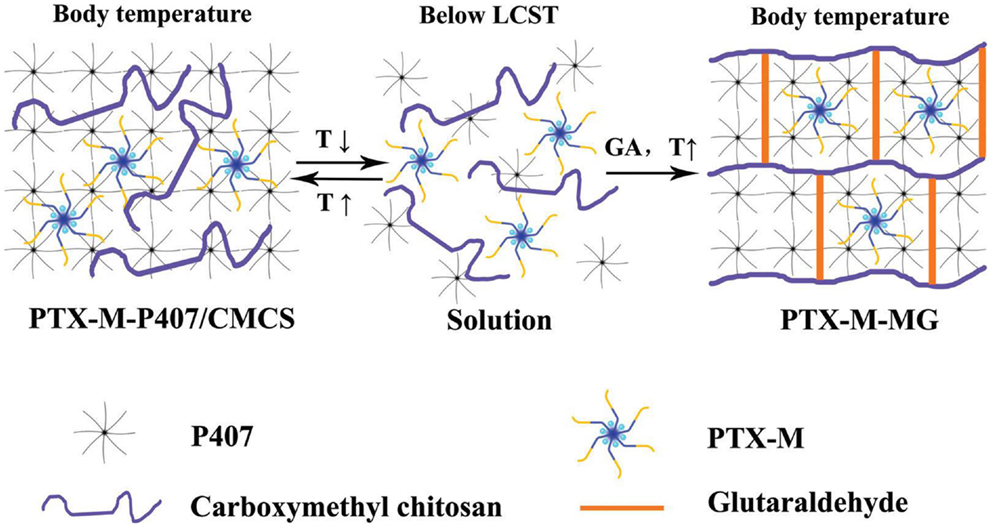
Figure 1. Loaded polymeric micelles (PTX-M) are dispersed in a physical hydrogel formed by triblock copolymer (P407) at elevated temperature. The hydrogel is then reinforced by chemical crosslinks of carboxymethyl chitosan with glutaraldehyde. Reprinted from Ju et al. (2013) with permission.
Wang et al. (2014) tried to develop a system for oral delivery of docetaxel, another anti-cancer agent. This system should provide solution to two main tasks – to overcome the low water-solubility of docetaxel and to target the drug to the intestinal track. Docetaxel was encapsulated in polymeric micelles based on amphiphilic block copolymer, which improved its solubility and permeability. The loaded micelles were incorporated into pH-sensitive hydrogel. The ideal oral delivery hydrogel should shrink with drug entrapped in at low pH environment of stomach, and swell to release drug at high pH environment of intestine. Docetaxel micellar encapsulation should also prevent the identification of docetaxel by proteins, which would cause its efflux and change the transport channel of this drug in the gastrointestinal walls.
Yom-Tov et al. (2014) utilized the self-assembly capability of block and graft copolymers driven by hydrophobic interactions between the blocks for nanostructuring of hydrogels. One of the main observed problems was the limited stability of micelles formed in the hydrogels, mostly attributed to their diffusion out of the hydrogels. A novel method for nanostructuring was therefore developed based on embedding micelles in a hydrogel while anchoring some of their molecules to the surrounding network through their endgroups. The anchored molecules should provide a means to further crosslink the hydrogel and stabilize the network.
Shukla et al. (2009) grafted hydrophobic moieties onto hydrophilic polymer, which forms physical hydrogels. Grafted nanohydrogel was produced, which structurally contained hydrophobic nanodomains. The nanodomains enabled to control water sorption properties and alter physical properties of the hydrogel. Similarly, Inoue et al. (1997) grafted a hydrophobic oligomer on a lightly crosslinked polyelectrolytic hydrogel. The resulting hydrophobic domain structure was intended to be applied as a bioadhesive hydrogel for controlled release of either cationic drugs or hydrophobic drugs. The hydrophobic domains were also supposed to strengthen and control the pore structure within the hydrogel. Also Kim et al. (2014) grafted hydrophobic chains on the hydrophilic backbone of physically crosslinked hydrogel. Micelles were formed from the hydrophobic grafts in the resulting material, which were capable to solubilize a water-insoluble macrophage recruitment agent. The whole system was designed for biomaterial scaffolds, which could provide a suitable environment for cell-based bone regeneration.
Tedeschi et al. (2006) incorporated cationic surfactant micelles (decyltrimethylammonium bromide) into physical poly(vinyl alcohol) hydrogels prepared by the well-known freeze/thaw method. These hydrogels are known for their ability to include inside their porous network variety of water-soluble molecules. This study was aimed at the analysis of the behavior of surfactant molecules in the hydrogel pore matrix and at the study of diffusion of micelles within the hydrogel. The latter is important to understand the dynamics of guest molecules within hydrogels containing micelles especially in relation to their potential use as molecular carriers.
Bromberg (2005) gives an overview of intelligent hydrogels for the oral delivery of (hydrophobic) anti-cancer drugs. Micellar domains within such hydrogels serve to solubilize the hydrophobic agents and participate in their protection during transport within the gastrointestinal tract and in their controlled release. The micelles are of polymeric types and formed by Pluronics, which are a part of gelling copolymer.
Guo et al. (2014) aimed at the improvement of mechanical properties of hydrogels for biomaterial applications, particularly their weakness and brittleness, while retaining their processability. This was achieved by including strong, yet reversible interactions within segregated nanoscopic domains. A multiblock copolymer based on hydrophilic polymer was complemented by ureidopyrimidinone (UPy) moieties contained within the polymeric backbone. These self-complementary units were encapsulated within hydrophobic domains and assembled into strong dimers by fourfold hydrogen bonding, acting to reinforce networks, while the non-covalent nature promoted processability, in contrast to chemical crosslinks in conventional thermosets.
The work by Thomas et al. (2009) was motivated by the design of synthetic replacement for cartilage lost due to osteoarthritis. While hydrogels had shown promise for this application, many of them lacked the required mechanical properties. In this work, hydrogel-forming polymer was blended with a hydrophobic polymeric partner by a mechanical mixing. The hydrophobic regions formed by the hydrophobic component were believed to act as crystalline domains in semi-crystalline polymers with a positive effect on the mechanical properties but without negative effects on the coefficient of friction.
Injectable hydrogels are mostly based on self-assembled micelles composed of amphiphilic block copolymers. Upon injection of the initial sol, sudden changes of surrounding environment induce its gelation. Pluronic copolymers are example of such materials. Their sol–gel transition is induced by the changes in temperature leading to the self-association of these copolymers into spherical micelles. Above the transition temperature, which is around the body temperature, the spherical micelles are closely packed together, resulting in the physically crosslinked hydrogels. Several shortcomings including low mechanical strength, fast dissolution, and rapid drug release under physiological conditions limited their biomedical applications. Chemical crosslinking is one approach to overcome them. To avoid side-effects due to the use of toxic crosslinking agents, Lee et al. (2011) used a bio-inspired, enzyme-mediated crosslinking of functionalized Pluronic micelles for fabricating high-strength injectable hydrogels.
Missirlis et al. (2005) primarily used Pluronic micelles as a solubilization medium for hydrophobic drugs with a prolonged circulation time. They tried to overcome the drawbacks of using these micelles directly (e.g., premature renal excretion or low stability upon dilution in the blood stream) by bridging between micelles to form macromolecular nanogels. This goal was achieved by inverse emulsion photochemical copolymerization with functionalized poly(ethylene glycol) (PEG). Hydrophobic nanodomains within the particles remained after copolymerization and were capable of absorbing large amounts of hydrophobic drugs.
In the work by Hao and Weiss (2011), strong hydrophobic interactions were used to physically crosslink acryl amide-based hydrogels in the swelled state. An acrylic monomer was copolymerized with a fluoroacrylate as the hydrophobic component. The hydrophobic associations of fluorocarbon hydrophobes in water medium created nanodomains that served as multifunctional physical crosslinks (see Figure 2). High stress or slow deformation could be used to achieve viscous flow, but gelation occurred when the stress was removed. These properties, resulting from the dynamic equilibrium nature of the hydrophobic associations, are attractive for injectable hydrogel materials. Moreover, fluorocarbon groups possess excellent chemical and biological inertness, good thermal stability, high fluidity, and low surface energy, which are useful for biomedical applications.
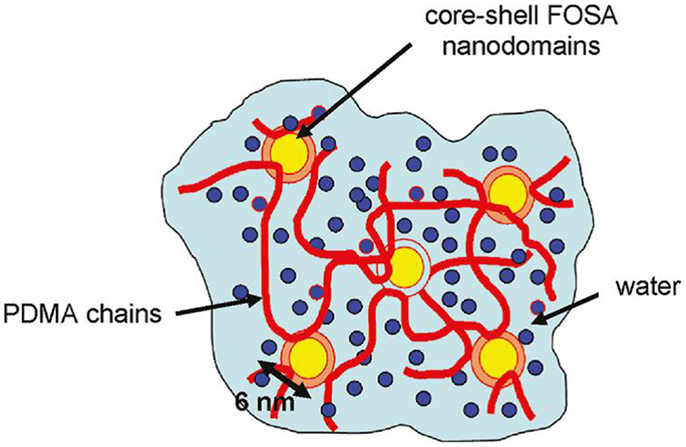
Figure 2. Hydrogel made from a copolymer of dimethyl acrylamide (PDMA) and fluorinated acrylate (FOSA), which forms micelles by strong hydrophobic associations. The micelles also serve as physical crosslinks of the hydrogel. Reprinted from Hao and Weiss (2011) with permission.
In the area of tissue engineering, an ideal scaffold should be able to provide a well-defined microenvironment to promote cell adhesion, proliferation, and differentiation with biodegradability and good biocompatibility. Fabrication of three-dimensional scaffold that can physically support cell infiltration and biologically direct cell behavior remains a challenge. Physical incorporation of polymeric micelles into hydrogels was used to tune the storage modulus, thereby influencing cell behavior in the hydrogel. Li et al. (2012) improved this approach by covalently incorporating nanosized polymeric micelles self-assembled from an amphiphilic block copolymer with crosslinkable functional groups into hydrogel networks.
Frisman et al. (2012) introduced micellar nanostructuring into hydrogels intended as scaffolds for the use in tissue engineering. The nanostructuring should promote cell–matrix interactions and thus provide the ability to control cellular fate and tissue morphogenesis. Scaffold nanostructures guiding cell–matrix interactions mimic environment that is familiar to cells from their native extracellular matrix.
Autonomous damage repair and resulting self-healing in hydrogels require reversible breakable bonds, which prevent the fracture of the molecular backbone. Consequently, self-healing of permanently crosslinked hydrogels is a challenging task because of the irreversible nature of chemical crosslinks. Tuncaboylu et al. (2013) developed self-healing hydrogels by the creation of strong hydrophobic interactions between hydrophilic polymers. Hydrophobe monomer was copolymerized with hydrophilic monomer via micellar polymerization technique in aqueous surfactant solution. Incorporation of hydrophobic sequences within the hydrophilic chains generated strong hydrophobic interactions, which prevented dissolution of the physical gels in water, while the dynamic nature of the junction zones provided homogeneity and self-healing properties.
At the end of this section, let us summarize the reasons for incorporating the hydrophobic (micellar) domains into hydrogels. These domains can
• physically crosslink hydrogels and, when supplied with suitable functional groups, can further serve as structured chemical crosslinks;
• be used for the structuring of hydrogels, which is closely related to;
• controlling (improving, tailoring) of mechanical properties of hydrogels;
• solubilize hydrophobic, usually biologically active, substances, participate on their stabilization, protection, and controlled release;
• impart the self-healing and stimuli-responsive properties to hydrogels.
Moreover, when traditional surfactants are used in hydrogel preparation technology, the general benefits of applying surfactants can be explored like reducing the surface tension, increasing the conductivity, stabilizing the bubble formation, or improving the hydrogel or fiber uniformity.
Classification of Hydrogels with Micellar Hydrophobic (Nano)Domains
As follows from the published examples reviewed in the preceding section, three basic types of hydrogels containing micelles, which form hydrophobic domains or nanodomains can be identified. Of course, materials represented by a combination of the basic types cannot be excluded.
1. Hydrogels with dispersed micelles. They are formed by a simple mixture of micelles and hydrogel matrix. As examples, the systems reported by Liu and Li (2005), Tedeschi et al. (2006), Mangiapia et al. (2007), Stoppel et al. (2011), Frisman et al. (2012), Ju et al. (2013), or Wang et al. (2014) can be given.
2. Hydrogels with integrated micelles. These may be further divided in two subtypes:
a) at least some of dispersed micelles are anchored to the hydrogel network as, for example, in the work by Yom-Tov et al. (2014);
b) micelles (domains) are formed by hydrophobic segments (usually blocks or grafts) located directly on the gel-forming precursor(s), for example, see in references Shukla et al. (2009), Inoue et al. (1997), Kim et al. (2014), Bromberg (2005), Guo et al. (2014), or Thomas et al. (2009).
3. Hydrogels with micellar crosslinks. Micelles (hydrophobic domains in general) form directly either physical or chemical crosslinks of the hydrogel network. Such materials are described, e.g., by Lee et al. (2011), Missirlis et al. (2005), Hao and Weiss (2011), Li et al. (2012), or Tuncaboylu et al. (2013). Here belong also physical hydrogels formed by mixing polyelectrolytes with oppositely charged surfactants under proper ratio and conditions. Surprisingly, these materials almost have not been studied and explored; currently, they are subject of intensive investigation in our laboratory.
Preparation
Various procedures enabling to prepare hydrogels with hydrophobic domains are illustrated in this section by selected examples. In order to give a reader a good idea on their practical realizations, they will be reviewed with adequate details. The order follows the typology introduced in the preceding section.
The preparation used by Liu and Li (2005) was motivated by the solubilization and protection of camptothecin or its derivatives, anti-cancer agents against a wide spectrum of human malignancies. These substances are practically insoluble in water and in vivo or in aqueous solutions buffered at pH above 7, they rapidly convert to the respective water-soluble, pharmacologically less active carboxylate form. First, a homogeneous aqueous solution of camptothecin in micellar surfactant solution of an ionic surfactant, SDS, was prepared. Then, a weighed amount of agarose was added to the solution of camptothecin and surfactant and the mixture was slowly heated to around 90°C until an optically clear solution was obtained. The resultant solution was poured into a flat mold of about 1 mm in thickness, which was kept in an oven at 90°C before use. The warm polymer-surfactant-drug solution in the mold was allowed to cool down naturally at room temperature to form a sheet hydrogel. The experimental procedure and the expected structures are illustrated in Figure 3. For comparison, saturated camptothecin solutions in surfactant solutions below the critical micellar concentration were prepared and incorporated into hydrogels.
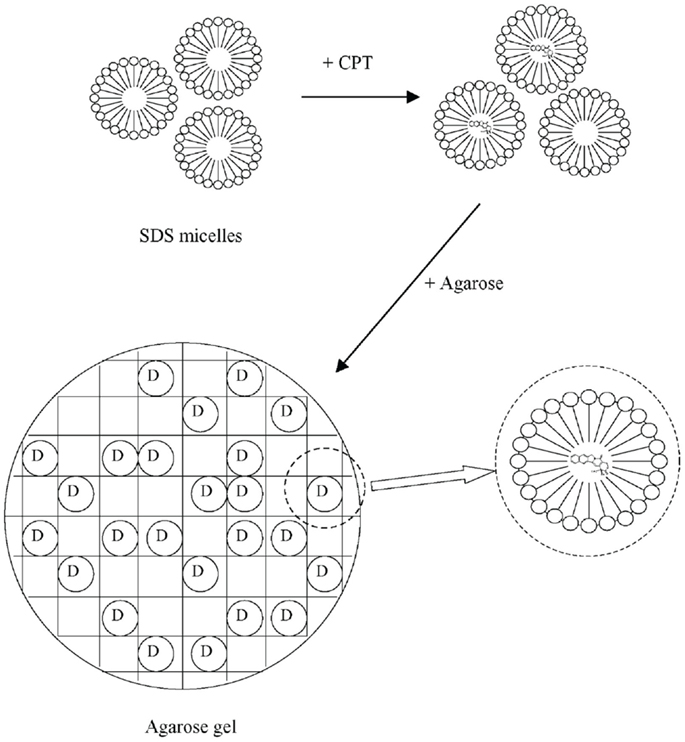
Figure 3. Loading surfactant (SDS) micelles with camptothecin (CPT), the drug (D), and their dispergation in agarose hydrogel. Reprinted from Liu and Li (2005) with permission.
Poly(vinyl alcohol) hydrogel was prepared by Tedeschi et al. (2006) through the standard freeze/thaw procedure from an aqueous polymer solution (averaged Mw of about 115,000, degree of hydrolysis of 98–99%, and concentration of 11% w/w). The polymer was dissolved at elevated temperature, slowly cooled to room temperature, and left overnight at this temperature to enable the escape of air bubbles. The solution was then poured between glass slides with 1 mm spacers and subjected to three repeated freeze/thaw cycles −22°C/25°C, respectively. Dried gels were rehydrated for 24 h at 25°C by a large excess of surfactant aqueous solution at concentrations ranging between 0.006 and 0.276 mol kg−1 (the critical micellar concentration in water of the used surfactant was 0.060 mol kg−1). Very similar system was described also in the study by Mangiapia et al. (2007).
Stoppel et al. (2011) incorporated Pluronic F68 micelles in an alginate hydrogel. Sodium alginate solutions were prepared at 1% (w/v) with the addition of 0.1% glucose and HEPES buffer and stirred for 24 h at room temperature. Pluronic F68 was added to the alginate solution and stirred for an additional 24 h at room temperature. Two common methods were used to crosslink alginate: (1) an inhomogeneous or external method, using CaCl2 or BaCl2, (2) a homogeneous or internal method, which utilizes in situ release of calcium ions from pH-sensitive CaEDTA decomposition. In both methods, the Pluronic-containing alginate solutions were used. The external method was realized by placing the alginate solution into 10 mm dialysis tubing with a molecular weight cut-off of 3–5 kDa (the viscosity-averaged molecular weight of used alginate was declared as about 240 kmol/g) and submerged in either calcium (1 mol/l) or barium (0.5 mol/l) chloride with a specified Pluronic F68 concentration for 48 h. The obtained cylindrical hydrogels were freeze-dried. In the internal gelation technique, the alginate solution was mixed with CaEDTA (50 mmol/l) and glucono-δ-lactone (50 mmol/l) solution and after 12 h of initial crosslinking, hydrogels were immersed in calcium chloride (100 mmol/l) solution containing the same concentration of surfactant (to prevent surfactant leaching) in a humidified chamber (humidity 90% at least) at 25°C for an additional 48 h.
Frisman et al. (2012) prepared a biosynthetic hydrogel scaffold from crosslinked PEG–fibrinogen conjugates modified with the block copolymer Pluronic F127. Pluronic self-assembly into micelles created the desired nanostructures within the hydrogel. PEG-modified fibrinogen with acrylate end groups was used as hydrogel precursor and its solution was thoroughly mixed with Pluronic F127 at 4°C. After equilibration (to ensure micelle formation), the mixture was combined with a photoinitiator solution, equilibrated in a water bath at 37°C, and finally irradiated by UV light (365 nm) to induce crosslinking of the acrylate end groups on the pendant PEG chains.
Ju et al. (2013) upgraded the technique for the solubilization of hydrophobic drugs (paclitaxel in their case) in polymeric micelles formed by Poloxamer 407, which itself also forms hydrogel. Paclitaxel was dissolved in micelles of N-octyl-O-sulfate chitosan, the loaded micelles were dispersed in Poloxamer 407 gels, interpenetrated by a network formed by crosslinking carboxymethyl chitosan with glutaraldehyde. Paclitaxel was solubilized in the micelles by dropping its solution in dehydrated ethanol into the aqueous solution of N-octyl-O-sulfate chitosan (no characteristics given in the original reference) under constant stirring. The resulting mixture was dialyzed against deionized water overnight, filtered through a 450 nm membrane, and freeze-dried. To prepare micelles-containing hydrogel, first solutions of carboxymethyl chitosan (concentration 1.5%, w/v; no characteristics given in the original reference except the carboxylation degree of 45%) and paclitaxel-loaded micelles (4 mg/ml) were mixed and the mixture used to dissolve Poloxamer 407 (Mw = 12600 g/mol, concentration probably 19%, w/v). Finally, glutaraldehyde was added (0.05% in final concentration, w/v) under intense agitation. Gelation was achieved after an incubation at 37°C for 30 min.
Wang et al. (2014) used polymeric micelles formed by triblock copolymer poly(3-caprolactone)–PEG-poly(ε-caprolactone) synthesized by ring-opening polymerization of ε-caprolactone initiated by PEG of molecular weight (Mn) about 2000 g/mol. The final copolymer molar weight was around 3700 g/mol. This amphiphilic, micelles-forming copolymer was used to solubilize docetaxel by thin-film hydration method. Docetaxel and copolymer were co-dissolved in dehydrated ethanol, the solvent was evaporated under reduced pressure and elevated temperature (60°C). Resulting thin layer of homogenous film was hydrated with water under mild stirring. During the process, copolymer could self-assemble into micelles in which docetaxel was encapsulated. Finally, the suspension was filtered through a syringe filter with pore size of 220 nm. As a hydrogel, methoxyl PEG-poly(ε-caprolactone)-acryloyl chloride copolymer was used because it possesses the desired and tunable pH-responsive properties. The hydrogel copolymer was prepared by ring-opening polymerization initiated by PEG methyl ether (Mn = 2000 g/mol) and substitution reaction of acryloyl chloride. The molecular weight of resulting copolymer was about 3000 g/mol. The docetaxel-loaded micelles were incorporated in the hydrogel by water absorption. In fact, the hydrogel matrix was swelled with the micellar solution. A certain volume of docetaxel-loaded micelles solution was completely absorbed in hydrogel owing to the swelling capability of hydrogel. Then the mixture was freeze-dried to obtain the stable docetaxel-micelle-hydrogel system.
Yom-Tov et al. (2014) combined simple embedding of micelles with anchoring some of them to the surrounding hydrogel network. To achieve this goal, part of surfactant monomer molecules were functionalized to create endgroups, which would react with the network. Pluronic F127 was used as a surfactant in pure and diacrylated form. Acrylation was carried by reacting the surfactant with excess (relative to the hydroxyl groups) acryloyl chloride and triethylamine. Mixture of pure and acrylated surfactants at desired ratio was mixed with hydrogel precursor solution at low temperature (4°C) until complete dissolution and then a photoinitiator was added. The resulting solution was heated to induce the formation of micelles (37°C) and then irradiated with UV light to achieve chemically crosslinked material. The total concentration of Pluronics was kept constant at 10% (w/v). The hydrogel precursor solution used in this work was a diacrylated polyethylene glycol-protein (fibrinogen) mixture.
Shukla et al. (2009) prepared hydrogel containing nanosized domains (20–35 nm) of hydrophobic moieties of poly(methyl methacrylate) by grafting crosslinked poly(acrylic acid-co-methyl methacrylate) chains onto polyvinyl alcohol backbone using an efficient redox system. Typically, polyvinyl alcohol, acrylic acid, methyl methacrylate, and water were mixed in a round-bottom flask and homogenized by mechanical mixing. After nitrogen purge at elevated temperature (35°C), N,N′-methylenebis(acrylamide), potassium persulfate, and potassium metabisulfite were added and the mixture was poured into a Petri dish, covered, and kept at 35°C for 72 h to complete gelation.
Inoue et al. (1997) described preparation of hydrophobically modified pH-sensitive hydrogels based on hydrophobic oligomers grafted to a polyelectrolyte hydrogel matrix. Poly(acrylic acid) was used as the bioadhesive polyelectrolyte, oligo(methyl methacrylate) as the hydrophobic component. The grafted hydrogel was prepared by coupling the amino-terminated oligo(methyl methacrylate) onto the poly(acrylic acid) hydrogel backbone through the reaction of the amino group with an activated carboxyl group in the poly(acrylic acid) hydrogel using dicyclohexyl carbodiimide as an activation reagent. The amino-terminated methyl methacrylate oligomer was prepared by free-radical polymerization of methyl methacrylate using 2,2′-butyronitrile as an initiator and 2-amino ethanethiol hydrochloride as a chain transfer agent, respectively. The lightly crosslinked poly(acrylic acid) hydrogel was pre-synthesized by the copolymerization (in aqueous solution) of acrylic acid with added ethylene glycol dimethacrylate as a crosslinking agent (0.5 w/w% to the monomer) and ammonium persulfate as an initiator. The dried poly(acrylic acid) hydrogel was added with the DMF solution of the amino-terminated oligo(methyl methacrylate) and shaken for 24 h. Then, excess solution was removed and a DMF solution of dicyclohexyl carbodiimide was added. The reaction was completed after 24 h at room temperature giving the final graft level from 10 to 50% (w/w).
In the work by Kim et al. (2014), the non-water-soluble macrophage recruiting agent was solubilized in water through micelles formation with l-lactic acid-oligomer grafted gelatin, and the resulting micelles with platelet-rich plasma were incorporated into gelatin hydrogels. First, the l-lactic acid oligomer with a number–average molecular weight of 1000 g/mol was synthesized by ring-opening reaction. The oligomer was grafted on gelatin (of isoelectric point equal to 5) through its amino groups in the average final ratio of about 3 moles/1 mole of gelatin. The macrophage recruiting agent was solubilized in the micelles formed by the grafted gelatin by simple mixing corresponding solutions in DMSO followed by 3 h of stirring; the solubilization was completed by dialysis, centrifugation of undissolved agent, and freeze-drying of final product. The micelles with the solubilized agent were incorporated in gelatin hydrogels by simply mixing a gelatin solution (5% by weight) with the pre-prepared micelles. The mixture was cast in molds, freeze-dried, and the hydrogel was then crosslinked by dehydrothermal treatment at 140°C for 48 h in a vacuum oven. The gelatin hydrogel incorporating the micelles was then impregnated with a solution of the platelet-rich plasma.
Bromberg et al. (2002) prepared Pluronic-poly(acrylic acid) copolymers and crosslinked them by ethylene glycol dimethacrylate to the form of microgel particles. Acrylic acid was partially neutralized in sodium hydroxide solution, Pluronic was dissolved in the resulting solution under nitrogen, and a desired amount of ethylene glycol dimethacrylate was added. Lauroyl peroxide and 4,4′-azobis(4-cyanovaleric acid) dissolved in acrylic acid were then added. The resulting solution was deaerated by nitrogen bubbling and mixed with dodecane containing a dispersion stabilizer. The reaction was run under vigorous stirring at 70°C under nitrogen purge for several hours.
The multisegmented amphiphilic copolymers were prepared by Guo et al. (2014) in a step-growth manner by reacting amino-telechelic PEG with diisocyanate functionalized UPy unit. The aminated PEG was synthesized from imidazole-telechelic precursor, which had been prepared by reaction of telechelic hydroxy-terminated PEG (Mn = 10 kg/mol) with 1,1-carbonyldiimidazole. The amine-terminated PEG contained C12H24 spacer and was prepared by reacting the imidazole precursor with 1,12-dodecyldiamine. The Upy-containing diisocyanate was made from the hydroxyl-functionalized uracil and 1,6-diisocyanatohexane in the presence of pyridine. The basic unit of the final chain-extended copolymers was of the structure [-PEGn-C12H24-urea-C6H12-UPy-C6H12-urea-C12H24-], i.e., had a total of 36 methylene groups per macromolecular repeating unit).
Thomas et al. (2009) produced miscible blends of polymers, which contained hydrophilic and hydrophobic groups. Poly(vinyl alcohol) with an average molecular weight of 124–186 kDa (99% hydrolyzed) or poly(vinyl pyrrolidone) (no other specification except the trade name Plasdone K-90) were used as hydrophilic components, while ethylene units in poly(ethylene-co-vinyl alcohol) based its use as the hydrophobic component. A melt processable miscible blend was created of the polymers utilizing DMSO/water mixtures (80/20 or 90/10) between 80 and 160°C. Mechanical mixing was required at concentrations >30 wt% polymer. Mixing was performed utilizing a twin-screw mixing apparatus followed by extrusion through a small die to form a lyogel (a lyogel is a polymeric gel in which the continuous phase is a solvent other than water.) Poly(vinyl alcohol) (non-blended) was processed using the same method and tested as a control material. After processing, all materials were immersed in reagent grade alcohol for a minimum of 20 min followed by immersion in water. During this solvent-exchange process, the hydrophobic/hydrophilic blended materials turned from a transparent to an opaque, flexible hydrogel as the hydrophobic domains formed. All pure poly(vinyl alcohol) samples remained transparent after this treatment. Samples remained in deionized water for a minimum of 72 h to equilibrate prior to testing.
Lee et al. (2011) induced the enzyme-mediated crosslinking of Pluronic micelles for fabricating high-strength injectable hydrogels. To this end, Pluronic copolymers were end-terminated with tyramine. The tyramine units should be exposed on the surface of Pluronic micelles and can be enzymatically oxidized by tyrosinase giving a crosslinked material. First, Pluronic F127 was transformed to amine-reactive form by a reaction with p-nitrophenyl chloroformate with resulting substitution degree (of end hydroxyl groups) approaching 100%. The modified Pluronic was then reacted with tyramine giving the final substitution also close to 100%. The Pluronic–tyramine conjugate was crosslinked at room temperature by the action of tyrosinase for 48 h.
Pluronic-based chemically cross-linked nanoparticles were developed in Missirlis et al. (2005), which should provide stability toward dilution and drying. They employed photoinitiated polymerization of acrylates as a mean to cross-link aqueous solutions of Pluronic derivatives. Pluronic F127 diacrylate was synthesized by a reaction with acryloyl chloride as described by Cellesi et al. (2002) with 100% conversion of alcohols to acrylates. Hydrogels were prepared either in the form of nanoparticles or as disks. The nanoparticles were synthesized by inverse emulsion photopolymerization. The oil phase for the emulsion preparation was formed by n-hexane with Span 65 surfactant (2% by weight). The aqueous phase contained diacrylated Pluronic F127, PEG diacrylate (average Mn = 575), both in concentration of 6.75% by weight, triethanolamine (2% by weight), and eosin Y (0.02% by weight). Oil-to-water weight ratio was 65/35 and the emulsion was prepared by short sonication. Polymerization occurred under the illumination by laser light of 480–520 nm wavelengths for 1 h. Similar procedure, but without the oil phase, was used to prepare hydrogel disks – the aqueous solution of precursors was placed between two glass slides, illuminated for 30 min, and the disks were finally exposed to water until equilibrium swelling was reached (24 h, at least).
Materials described by Hao and Weiss (2011) are physically crosslinked copolymer hydrogels synthesized from N,N-dimethylacrylamide and 2-(N-ethylperfluorooctane sulfonamido) ethyl acrylate (FOSA). Aggregation of the fluorocarbon hydrophobe moieties produces a physically crosslinked microstructure of core-shell nanodomains, which are composed of an FOSA core surrounded by a water-depleted shell of dimethylacrylamide. The water-poor shell of the dimethylacrylamide presumably comprises the chain segments attached to the FOSA repeat units that have restricted mobility due to the covalent attachment to the FOSA aggregates. These core-shell nanodomains constitute the crosslink junctions in the physical hydrogel that is essentially water-swollen poly(dimethylacrylamide). The copolymer of dimethylacrylamide and FOSA was synthesized by free-radical polymerization of the monomers in dioxane. The resulting molecular weight (Mw) ranged between 50 and 80 kmol/g (with polydispersity between 1.5 and 1.9). The hydrogel samples were prepared by compression-molding dry copolymer films at 150–180°C under vacuum and then immersing the films in deionized water for 7 days to ensure equilibrium hydration.
Li et al. (2012) reported on biodegradable micelles-containing PEG hydrogels synthesized via Michael addition chemistry. For schematic representation of hydrogel synthesis and structure, see Figure 4. The basic polymeric unit of the hydrogel was formed by the four-arm acrylated pol(yethylene glycol) (tetraacrylated PEG), whereas micelles were formed from PEG-polycarbonate diblock copolymer end-capped with vinyl sulfone group (on the PEG block). Due to the scaffold application, also the RGD peptide was chemically built into the hydrogel network as a cell adhesion enhancer. The tetra acrylate-terminated PEG was dissolved in buffer and to this solution, the pre-prepared micellar solution of the vinyl sulfone terminated PEG-polycarbonate in the same buffer was added followed by the addition of peptide solution. The mixture was incubated at 37°C and then added with the buffered solution of tetra-thiolated PEG crosslinker (four-arm tetra sulfhydryl PEG). The reaction mixture was kept at this temperature and the hydrogel formed quickly. Cells in the growth medium (human mesenchymal stem cells) could be added to the gel precursors before the addition of the thiolated crosslinker and thus encapsulated in the hydrogel. Alternatively, a bolaamphiphile/DNA complex as a gene transfection vector could be mixed with the cell suspension and then added to the gel precursors before the addition of the crosslinker. In this way, the effects on gene expression efficiency could be studied directly in the hydrogel matrix.
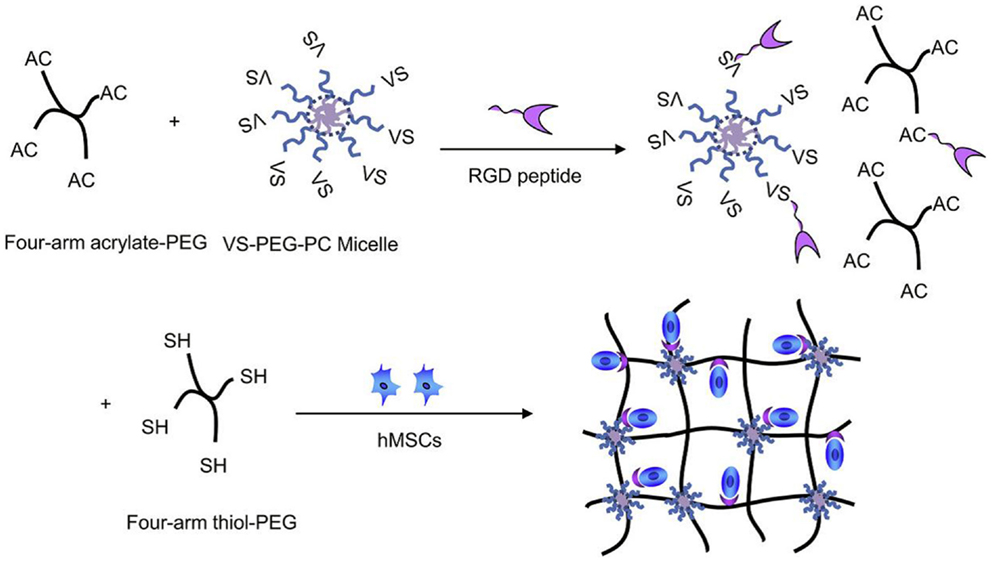
Figure 4. Synthesis of hydrogel with micellar crosslinks and a peptidic structure to bind human mesenchymal stem cells (hMSCs); VS stands for vinyl sulfone. Reprinted from Li et al. (2012) with permission.
Tuncaboylu et al. (2013) copolymerized large hydrophobes such as stearyl methacrylate with the hydrophilic monomer acrylamide in aqueous SDS surfactant solutions. The reaction medium was prepared as a solution of NaCl at the concentration of 0.5 mol/l. The salt led to micellar growth, and hence, solubilization of large hydrophobes within the grown worm-like SDS micelles. A series of hybrid hydrogels was prepared using stearyl methacrylate (2% molar) as a physical crosslinker and various amounts of N,N′-methylenebis(acrylamide) as a chemical crosslinker. The reaction mixture contained SDS at the concentration of 0.24 mol/l, i.e., more than 30 times higher than its critical micellar concentration in water (in the used salt solution, the critical micellar concentration would be about one order of magnitude yet lower). The total concentration of stearyl methacrylate and acrylamide monomers was fixed at 10% (w/v). Specifically, surfactant was dissolved in NaCl solution at elevated temperature (35°C) and then stearyl methacrylate was solubilized in this solution. Acrylamide and bis(acrylamide) were added and after their dissolution, the redox initiator system was added. The micellar copolymerization ran at 25°C for 24 h.
Properties
In this section, concrete examples of the benefits coming from the incorporation of hydrophobic domains into hydrogels are given. The examples correspond to works referred in preceding sections and thus complete this review.
Liu and Li (2005) confirmed that solubilization of camptothecin in surfactant micelles dispersed in agarose hydrogel greatly affected the release behavior of drug molecules. The release of camptothecin was slowed down significantly with increasing surfactant concentration as a result of lowered drug diffusion coefficient. The drug diffusion coefficient could be approximately expressed as an exponential function of surfactant concentration. The authors separated the effects of drug in the aqueous and micellar phases on its diffusion coefficient. At lower surfactant concentrations (up to about twice its critical micellar concentration in pure water), the free drug molecules showed a great influence on diffusion coefficient. With the concentration of surfactant increasing, this effect decreased sharply due to the solubilization of drug molecules into micelles. The influence of the micellarly solubilized drug on its diffusion became prominent particularly at surfactant concentration about three times higher than its critical micellar concentration in pure water. The diffusion coefficient of micelles might be lowered as the amounts of micelles in the system increase, due to the increased friction in crowded environment or due to other possible interaction between micelles. The notable influence of temperature on the release was also observed and was even stronger than that in the case when no surfactant was used. The release was significantly accelerated increasing the temperature from 23 to 37°C; at higher temperature, its effect became negligible.
Results published by Tedeschi et al. (2006) on physical poly(vinyl alcohol) hydrogels with incorporated micelles showed that surfactant micellization was almost unperturbed regardless the confinement of its molecules within the hydrogel network. No direct surfactant–polymer interactions were detected. Thus, the hydrogel maximum swelling was not affected by addition of surfactant and was also independent of surfactant concentration. Surfactant concentrations measured in the hydrogel samples were equal to that in the surfactant aqueous solution used to rehydrate the gels – no surfactant preference for the hydrogel matrix was found. The critical micellar concentration was found to slightly decrease in the polymer solution and it decreased even more in hydrogel. This finding was attributed to the increased surfactant effective concentration in the limited space created by the excluded volume of the polymer. EPR spin probe and small-angle neutron scattering techniques showed that the micelles formed within the pores of the hydrogel matrix possess the same properties as micelles formed in bulk water. NMR diffusion study revealed that diffusion of micelles in hydrogels is much slower in comparison to bulk water. Because no surfactant–hydrogel interactions were found, this was explained by the effect of the intrinsic structural features of hydrogels in which the pores available for the diffusion are filled by the polymer-rich phase.
From the point of view of possible applications, this work confirmed that micelle-containing hydrogels have a complex multidomain structure, which is able to host both hydrophilic and hydrophobic molecules. Diffusion of the guest molecules from such carrier matrix is quite slow. Such systems can be thus promising candidates for the design of new systems for controlled release and/or adsorption of various guest molecules. Very similar results on the surfactant role in poly(vinyl alcohol) hydrogels were reported in the study by Mangiapia et al. (2007). The main aim of this work was detailed structural study of pure hydrogels by small (SANS) and ultrasmall (USANS) angle neutron scattering techniques. Some hydrogel samples were rehydrated using surfactant solution as in the work by Tedeschi et al. (2006) but in this case anionic sodium decylsulfate was used. The authors reported on the lack of any detectable interaction between the polymer chains and surfactant micelles. Also, the characteristics of micelles like the structural parameters, size, aggregation number, and actual charge were not affected by the presence of the poly(vinyl alcohol). Thus, the behavior of surfactant micelles within the poly(vinyl alcohol) hydrogels prepared by the freeze/thaw technique seems to be general and independent on the surfactant nature.
The study by Stoppel et al. (2011) was devoted to the effect of non-ionic surfactants on protein and small molecule transport within hydrogel matrices. Understanding this effect is crucial for the use of these systems in the design and formulation of drug delivery systems, biomaterials, and tissue engineering scaffolds. In their study, Pluronic F68 micelles were incorporated in an alginate hydrogel. The experimental results showed that small molecule transport within hydrogels improved slightly by the addition of Pluronic F68 (evidenced by the increase in their effective diffusion coefficient). The authors expressed expectation that use of a higher surfactant concentration or larger surfactant molecular weight, e.g., Pluronic F108 or F127, would continue to enhance diffusion coefficient and improve transport characteristics, as long as the material properties of the hydrogel would not be strongly affected by the change in formulation. They also note that in the case of a more hydrophobic small molecule, changes in the diffusion behavior would be highly influenced by the size of the complex micelle-solubilized hydrophobic small molecule. From the point of view of mechanical properties, it was found that addition of Pluronic F68 above 2% (w/v) may lead to undesirable material properties. Thus, the incorporation of 5% Pluronic F68 (w/v) reduced the storage modulus by 57% and increased the loss angle tangent by 24% (at a frequency of 1 Hz). Interestingly, while significant changes in solution viscosity were observed for Pluronic F68 addition below 2% (w/v), these interactions were not dominant in the hydrogel form. However, the solution interactions did translate to the gel state through the increased loss angle tangent and increased shrinkage ratio, which ultimately had an effect on the loading capacity through changes in water retention properties. Pluronic addition had minimal effects on riboflavin transport and loading. Conversely, significant effects were observed for bovine serum albumin where loading capacity decreased and release either increased or decreased depending on the gelation method.
Frisman et al. (2012) nanostructured hydrogels based on fibrinogen with Pluronic F127 micelles. Resulting materials were comprehensively analyzed using small-angle X-ray scattering (SAXS) and transmission electron cryo-microscopy and leaching test of the Pluronic micelles from the crosslinked hydrogel network under tissue culture conditions. Cryo-microscopy and SAXS experiments revealed formation of partially ordered micellar structures, surrounded by a PEG–fibrinogen network. Micelles diameter was determined to be approximately 14 nm, which was close to the dimension of micelles (10 nm) formed in the solution of modified fibrinogen (hydrogel precursor). The structure and the ordering of the micelles in the hydrogels were dependent on the concentration of Pluronic F127. At a relatively low concentration of Pluronic, micelles were distributed homogenously and randomly throughout the hydrogel matrix. Organized arrangement of the micelles was observed in samples containing higher concentrations of Pluronic. The arrangement was proposed in the form of clusters of micelles surrounded by areas containing mainly the modified fibrinogen backbone. The shape and the size of the micelles and the distances between them were found not to be affected by the hydrogel crosslinking density. Pluronic F127 also decreased the size distribution of the fibrinogen precursor in the hydrogel. One of the radii determined for the fibrinogen structure was reduced from 3.8 nm in the neat hydrogel to 2.3 nm at the highest Pluronic F127 concentration. The other (smaller) radii of 1.5 nm remained unchanged.
Swelling increased upon the addition of Pluronic and with its increasing concentration – probably due to the hydrophilic nature of this additive. Leaching tests showed that more than 50% of the added Pluronic was released from the hydrogel during the first few hours. The remaining fraction was leached over the next 2–3 days and after 4 days only a very small amount of Pluronic remained entrapped inside the hydrogel matrix.
A fibroblast cell culture was inoculated in the prepared hydrogels. A non-linear dependence of the cell shape on the amount of the added Pluronic was found; hydrogels containing 7% (by weight) of Pluronic provided an optimal environment for cell growth. Increased amount of Pluronic resulted in the decrease of the cell shape factor as a consequence of the above mentioned effect of the surfactant on the size of the fibrinogen precursor. Smaller radius of the fibrinogen proteins backbone reflects a lesser tendency of the hydrogel precursor toward aggregation. This means that more bioactive fibrinogen polypeptidic structures can be exposed to resident cells.
Ju et al. (2013) presented an upgraded system based on Poloxamer 407 for localized delivery of paclitaxel. Although paclitaxel can be solubilized in Poloxamer 407 solution, the solubility is too low to achieve the desired treatment threshold. Therefore, paclitaxel was solubilized in N-octyl-O-sulfate chitosan micelles and these were dispersed in Poloxamer hydrogels interpenetrated by crosslinked network of carboxymethyl chitosan. The introduction of paclitaxel-loaded micelles had no influence on the lower critical solution temperature and gelation time of Poloxame hydrogel, whereas the loading capability for paclitaxel was greatly enhanced. The incorporation of chitosan-based network resulted in higher swelling ratio, stronger mechanical properties, and longer term drug release both in vitro and in vivo. Prolonged retention was revealed at tumor sites, lasting for 20 days, as well as superior tumor inhibition rate with reduced toxicity compared to Taxol (a commercial paclitaxel preparation) or Poloxamer hydrogels not reinforced by chitosan-based networks and containing the same paclitaxel-loaded micelles.
Wang et al. (2014) designed a system for oral delivery of anti-cancer drugs, docetaxel in their case. The pH-responsive hydrogel carrying micelles with solubilized drug was selected as an ideal candidate with following hypothesis on the delivery:
1. Primarily, the loaded micelles pass through the stomach under the protection of pH-shrinked hydrogel and diffuse from the hydrogel in intestinal tract.
2. The hydrogel swells at high pH environment of intestine. The micelles are released and absorbed by Peyer’s patches in small intestine owing to absorption enhancer and subsequently they circulate longer in blood.
3. The micelles are passively targeted to tumor tissue by enhanced permeation and retention effect.
4. At last, docetaxel is released from micelles and suppresses the growth of tumor.
Hydrogels containing loaded micelles showed similar mesh size compared to the blank hydrogels and their pH-responsibility was not impacted. With the pH rising from 1 to 8, more micelles were exposed on the surface of the network, which was primarily ascribed to the expansion of the hydrogel. The diffusion rate of micelles in pH equal to 6.8 was much faster than that in acidic pH (1.2) and also the diffusion in simulated intestinal fluid was much faster than that in simulated gastric fluid. The docetaxel-loaded micelles were confirmed to be absorbed in the intestine, which was attributed to their small size (about 20 nm). The oral bioavailability of docetaxel reached up to 75.6% after oral administration of docetaxel-micelles-hydrogel, about 10 times higher than that of free docetaxel-loaded micelles. The pharmacokinetic area under curve of docetaxel improved accordingly as the administration does increased from 10 to 30 mg/kg in mice. In the subcutaneous breast cancer model, oral docetaxel-micelles-hydrogel inhibited tumor growth dose-dependently; in high dose group, the oral hydrogel system was comparable with intravenous injection of Taxotere (a marketed formulation of docetaxel).
Yom-Tov et al. (2014) covalently linked part of the Pluronic micelles into the hydrogel matrix by replacing part of the Pluronic F127 molecules with a diacryl derivative. They found that the influence of the diacryl derivative on the hydrogel nanostructure is pronounced only after covalently integrating its micelles in the network. By fine tuning the ratio of Pluronic-diacrylate to unmodified Pluronic molecules, different degrees of crosslinking were achieved, which influenced the hydrogels weight gain ability and Young’s modulus. Larger diacryl derivative percentages led to smaller swelling degree, which is in line with the integration of acrylated micelles within the hydrogel network, thus enhancing its crosslinking density and diminishing its weight gain ability. It was also observed that after about 2 h of swelling, the unbound (unmodified) Pluronic molecules leached out of the hydrogel creating cavities and network imperfections. An increase in Pluronic diacryl derivative percentage gave higher values of Young’s modulus as expected due to the increased network density. The authors concluded that the higher porosity and mesh size resulting from the release of unbound unmodified Pluronic molecules from the fully hydrated hydrogels had the most pronounced impact on the hydrogels’ characteristics.
An innovative hydrogel synthesized by Shukla et al. (2009) contained nanosized domains of the size 20–35 nm of hydrophobic moieties of poly(methyl methacrylate) grafted onto hydrophilic poly(vinyl alcohol) backbone and investigated its water sorption properties. Increasing concentration of hydrophobic monomer gave materials with decreasing swelling degree while the diffusion coefficient of water increased. The effects of various parameters on water sorption were then studied on hydrogels prepared with a fixed (“medium”) amount of hydrophobic monomer. The water imbibition capacity increased when pH of the swelling media varied in the range 2–11, but a significant rise in swelling was seen only beyond pH equal to 9. The swelling was favored by increasing temperature or by the presence of chloride or carbonate ions; on contrary, a fall in water sorption was observed during the presence of trivalent phosphate ions.
Inoue et al. (1997) grafted oligo(methyl methacrylate) as a hydrophobic domains forming unit onto hydrophilic poly(acrylic acid) as a hydrogel-forming polymer and tested swelling and drug release properties of final materials. The graft level and molecular weight of this oligomer influenced swelling of resulted hydrogels. Higher graft levels and lower molecular weights of the grafted chains yielded lower swelling ratios. This was explained by the higher concentration of hydrophobic grafts and their aggregation into domains within the hydrogel. Drugs of various polarities and a model protein (positively charged), lysozyme, were solubilized in the hydrogels. Hydrophobic drugs and lysozyme were more slowly released from the hydrogels when compared to the ungrafted poly(acrylic acid) hydrogel. This was probably a consequence of favored absorption of the hydrophobes into the oligomer-formed domains and the lack of interconnections between these domains. Positively charged lysozyme might associate both with the negative charges on acrylic acid moieties and the hydrophobic domains. Consequently, also its release was decelerated. On the other hand, uncharged hydrophilic drugs released faster because of their lower hydrophobic or ionic interactions within the hydrogel. The formation of hydrophobic domains might also enlarge the aqueous pore sizes of the hydrogel, thus permitting the hydrophilic drug to diffuse out more rapidly.
Kim et al. (2014) solubilized a non-water-soluble macrophage recruitment agent in micelles formed within hydrogels of gelatin grafted with hydrophobic chains. Platelet-rich plasma was also incorporated into these hydrogels, which were designed for the bone tissue engineering. Dual release of the agent and plasma from hydrogel was expected to stimulate macrophage recruitment and modulate inflammation, resulting in enhanced bone regeneration. The results of that study experimentally confirmed this hypothesis and dual release in a controlled fashion. The higher number of macrophages recruited was observed around gelatin hydrogels incorporating mixture of micelles-loaded agent and plasma compared with those incorporating either the loaded micelles or the plasma. The hydrogels also decreased the production of pro-inflammatory cytokine, but increased that of anti-inflammatory cytokines. The controlled dual release thus recruited macrophages into the bone defect and modulated inflammatory responses thereat, resulting in promoted bone regeneration.
The development of novel systems for the oral chemotherapy was overviewed by Bromberg (2005). Solubilization in micelles was combined with large macromolecular structures preventing such carriers from being transported into the systemic circulation. The systems were based on the copolymers of Pluronics and acrylic acid prepared in the form of spherical microgel particles with controllable diameter from tens to hundreds of microns. Dangling chains of Pluronics created the hydrophobic domains in microgels. At elevated temperatures, the dangling Pluronic chains rearranged creating tighter micellar aggregates that acted as physical crosslinks, lowering the equilibrium swelling of the microgels. The microgels were then crosslinked by both chemical and physical crosslinks. The size of micellar crosslinks filled the gap between macromolecules (Stokes radii below 10 nm) and microparticles (size 100 nm and higher). These micelles were shown to enhance solubility of hydrophobic drugs such as steroid hormones or camptothecins. The solubilizing micelles also provided an effective barrier against drug decomposition. The gelled material enabled a prolonged contact with mucous tissue due to enhanced bioadhesion and the decelerated diffusion enhanced the bioavailability of the drug.
Formation of hydrophobic domains within a multiblock PEG-based hydrogel was combined with strong hydrogen-bonding associations of UPy units contained in the same copolymer (Guo et al., 2014). The strength of hydrogels significantly improved and could additionally be adjusted by implementing different lengths and composition of PEG precursor. The hydrophobic segments as observed by transmission electron microscopy appeared to be dispersed as small spherical compartments with diameter about 2–5 nm. Although the weight fraction of PEG in these materials was around 0.9, they did not dissolve (at ambient temperature) in water in contrast to pure PEG; only swelling was observed with equilibrium water weight fraction somewhat above 0.8. A short-chain hydrophobic oligo-methylene spacers were used to shield the UPy units and to facilitate their aggregation such that hydrogen-bonding disruption caused by the hydrated matrix could be prevented. The integrity of crosslinking was thus maintained through this molecular shielding. When combined with long PEG segments, this feature translated to low crosslink densities, allowing relatively large elongation-recovery to be achieved with minimal diminishment of modulus compared with pristine PEG. Tensile tests revealed a remarkably strong hydrogel that exhibited nearly perfect strength recovery even at large deformation (exceeding 300%). The cumulative mechanical properties were driven in part by the incompatibility between hydrophilic and hydrophobic chain segments in concert with the strong tendency for hydrogen bonding of the UPy motifs. For example, these hydrogels were substantially stronger and tougher than similar hydrogels formed by chemically crosslinked copolymer micelles with very similar hydrophobic versus hydrophilic content. The developed materials also showed a shape memory behavior. The shape recovery from temporary deformed states was demonstrated to be stimulated either by heating above the melting temperature of PEG or by immersing in water.
Thomas et al. (2009) prepared hydrogels combining both hydrophilic and hydrophobic structures by injection molding with the intention of their application as cartilage replacement materials. Unfortunately, many of their investigated hydrogels lacked the required shear, tear, and creep properties. However, the introduction of hydrophobic groups in the materials was found to have a positive effect on mechanical properties with a minimal effect on coefficient of friction and contact angle. Particularly, the strength and creep resistance were greatly improved. Thus, hydrophobic domains-containing hydrogels were proved to exhibit signs of hydrodynamic lubrication, which are stronger than the reference poly(vinyl alcohol) hydrogels used in this study.
Lee et al. (2011) reinforced poor mechanical properties of Pluronic hydrogels by using Pluronic copolymers terminally conjugated with tyramine, which can be enzymatically crosslinked between neighboring micelles containing the modified copolymers (Figure 5). The enzyme crosslinked hydrogels showed far lower critical gelation concentration, concomitantly showing enhanced gel strength compared to unmodified Pluronic copolymer hydrogels. Rheological studies demonstrated that the enzyme crosslinked hydrogels exhibited a fast and reversible sol–gel transition in response to temperature while maintaining sufficient mechanical strength at the gel state. These materials were thus suitable for injectable, in situ gelling applications. In situ formed hydrogels were eroded gradually, releasing fluorescently labeled dextran in an erosion-controlled manner. Moreover, they showed tissue-adhesive properties due to the presence of unreacted catechol groups in the gel structure.
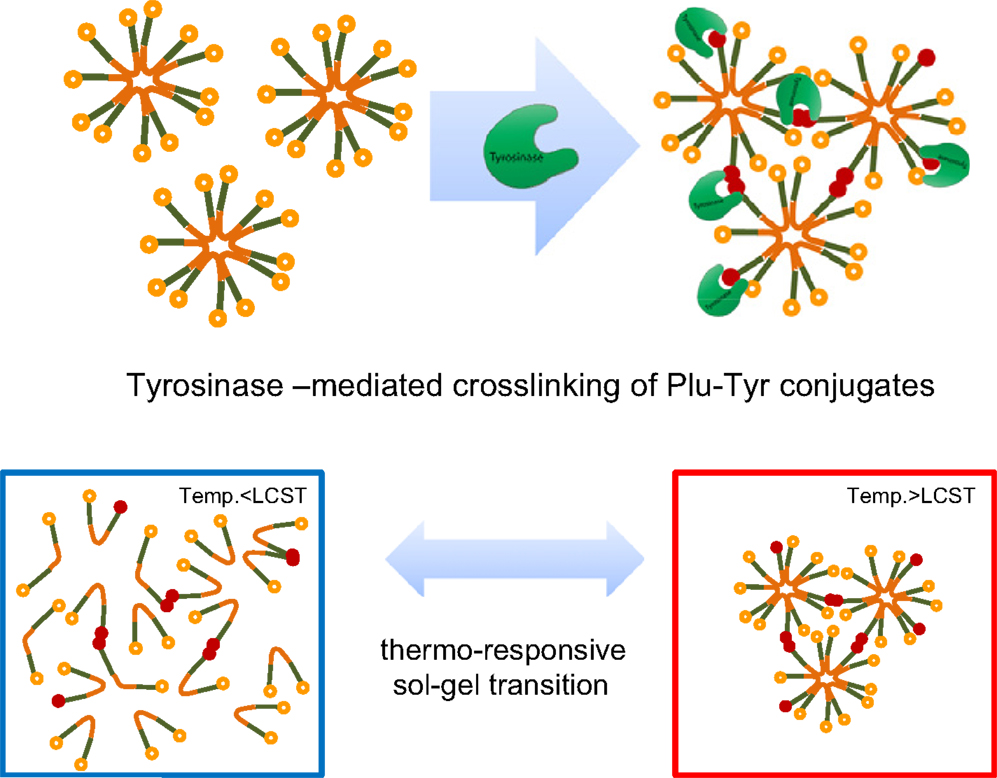
Figure 5. Thermal and enzymatic gelation of tyramine-terminated Pluronic copolymers (Plu-Tyr). Reprinted from Lee et al. (2011) with permission.
Pluronic-based nanoparticles were obtained in Missirlis et al. (2005) by inverse emulsion photocopolymerization with a PEG diacrylate. The size of nanoparticles could be varied between 50 and 500 nm. The hydrophobic domains within these nanoparticles solubilized doxorubicin, an anti-cancer drug, up to almost 10% (w/w). The nanoparticles also appeared stable to freeze-drying and re-suspending in water.
Hao and Weiss (2011) used a fluoroacrylate, capable of strong hydrophobic interactions, as the hydrophobic modifier in poly(alkylacrylamide) hydrogels. Microphase separation of a hydrophobic nanophase due to incompatibility of the fluoroacrylate groups with the hydrogel network served as multifunctional physical crosslinks. This resulted in excellent mechanical properties of final materials, providing the water concentration in the gel was not too low (in this case the hydrogel became more brittle). The strength of these physical hydrogels increased with increasing concentration of the fluoroacrylate moieties. The unusually high stiffness and toughness was attributed also to the ability of these gels to dissipate energy when deformed because of a reversible disengagement of the hydrophobic groups from the nanodomains. That provided a viscous mechanism for stress relaxation and effective energy dissipation. The physical nature of the hydrogels also allowed for their viscous flow if the applied stress was sufficiently high or the deformation time was sufficiently long (i.e., low deformation frequency). The physical gels were more viscous than comparable chemical gels and were much more efficient at dissipating stress, which resulted in higher tensile toughness. At 25°C, the hydrogels were highly elastic (the storage modulus was higher than the loss modulus), but as the temperature increased, viscous behavior increased and a moduli crossover occurred at 55°C and the rheological characteristics of the material changed from a viscoelastic solid to a viscoelastic liquid.
The temperature behavior reflected the glass transition of these hydrogels temperature of which was determined at 45°C. At room temperature, the hydrophobic nanodomains were glassy and the relaxation time of interactions holding them together could be expected to be relatively long. As temperature increased above the glass transition, the hydrophobic domains started to be more flexible and their relaxation times decreased. Observed mechanical behavior is useful for designing an injectable hydrogel where a high stress or slow deformation can be used to achieve viscous flow, but gelation occurs when the stress is removed or sufficiently decreased.
Li et al. (2012) covalently incorporated nanosized polymeric micelles into PEG hydrogel network to tune its properties relevant for cell-related applications in tissue engineering. Increasing the content of the micelles from 0 to 80% led to increased porosity and tunable mechanical property of the hydrogels. It was observed that the size of pores in hydrogel increased with increasing micelle content due to decreased crosslinking degree in this system. At the content of the micelles about 80%, the pores were highly interconnected. The hydrogel with 20% micelles provided the best balance among hydrogel stiffness and porosity for cell survival, leading to the highest viability of human mesenchymal stem cells. When the micelle content was increased up to 80%, storage modulus and gel yield decreased significantly, whereas swelling ratio increased dramatically. The cationic bolaamphiphile/DNA complexes induced higher gene expression efficiency in the hydrogels than the traditional polyethylenimine/DNA complexes, yet showed no cytotoxicity. The gene expression level in the cells in the hydrogel with 20% micelles was the highest as compared to that in the other hydrogels. Overall, the hydrogel with 20% micelles offered an optimal scaffold with ideal physical properties for cell growth and transfection. Nanoparticles incorporated into the hydrogels thus represent a useful strategy to control cellular behavior in a three-dimensional environment. Developed biodegradable nanostructured hydrogels can be an excellent platform for the delivery of human mesenchymal stem cells.
A series of hybrid hydrogels was prepared by the micellar copolymerization of acrylamide stearyl methacrylate as a physical crosslinker forming micelle-like domains and a bis(acrylamide)-based chemical crosslinker, in the presence of a surfactant (Tuncaboylu et al., 2013). Rheological measurements showed that the dynamic reversible physical crosslinks consisting of hydrophobic associations surrounded by surfactant micelles are also effective within the covalent network of the hybrid hydrogels. A significant enhancement in the compressive mechanical properties of the hybrid gels was observed with increasing chemical crosslinker content as expected. Cyclic tensile and compression tests showed that the fraction of hydrophobic associations reversibly broken under an external force decreased with increasing the ratio of chemical crosslinker to the monomer. The results showed that self-healing in hybrid gels can be observed at sufficiently low chemical crosslink densities where the network chains are sufficiently flexible to allow the re-formation of broken hydrophobic associations on the cut surfaces. The hydrophobic associations surrounded by surfactant micelles acted as reversible breakable crosslinks responsible for rapid self-healing of the hydrogels at room temperature without the need for any stimulus or healing agent. No self-healing observed in hybrids formed at the chemical crosslinker ratio equal and greater than 0.01 was explained by the decreased mobility of hydrophobic blocks in the inhomogeneous network structure. The authors also proposed that the use of hydrophobic monomers with longer alkyl side chains would produce enhanced self-healing properties due to the increased mobility of the hydrophobes.
Conclusion
This review focused on rather unexplored colloidal materials. Hydrogels with hydrophobic, particularly micellar, (nano)domains are hybrid materials from the point of view of polarity of their constituents. This hybrid character is the base of their specific material, structural, and colloidal properties. Manipulating the two basic constituents – their contents, chemistry, size, incorporation into hydrogel matrix – enables tailoring of characteristics of these hybrid gels. They can be then favorably employed in variety of applications, especially in the fields of (bio)medicine, biomaterials, drug delivery, cosmetics, but even in agriculture or environment protection. The number of published works is still low and most of them are focused on synthetic and compositional aspects. More information on their physico-chemical or colloidal properties, like (long-term) stability or transport of both hydrophobic and hydrophilic molecules within and from (into) these systems, is needed badly. Further, there is a lack of detailed biology- and cell-related studies in the case of materials intended for biomedical applications.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The support from the project no. LO1211 (National Programme for Sustainability I, Ministry of Education, Youth and Sports) is acknowledged.
References
Bromberg, L. (2005). Intelligent hydrogels for the oral delivery of chemotherapeutics. Expert. Opin. Drug. Deliv. 2, 1003–1013. doi: 10.1517/17425247.2.6.1003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bromberg, L., Temchenko, M., and Hatton, T. A. (2002). Dually responsive microgels from polyether-modified poly(acrylic acid): swelling and drug loading. Langmuir 18, 4944–4952. doi:10.1021/la011868l
Cellesi, F., Tirelli, N., and Hubbell, J. A. (2002). Materials for cell encapsulation via a new tandem approach combining reverse thermal gelation and covalent crosslinking. Macromol. Chem. Phys. 203, 1466–1472. doi:10.1002/1521-3935(200207)203:10/11<1466::AID-MACP1466>3.0.CO;2-P
Frisman, I., Seliktar, D., and Havazelet, B.-P. (2012). Nanostructuring biosynthetic hydrogels for tissue engineering: a cellular and structural analysis. Acta Biomater. 8, 51–60. doi:10.1016/j.actbio.2011.07.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, M., Pitet, L. M., Wyss, H. M., Vos, M., Dankers, P. Y. W., and Meijer, E. W. (2014). Tough stimuli-responsive supramolecular hydrogels with hydrogen-bonding network junctions. J. Am. Chem. Soc. 136, 6969–6977. doi:10.1021/ja500205v
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hao, J., and Weiss, R. A. (2011). Viscoelastic and mechanical behavior of hydrophobically modified hydrogels. Macromolecules 44, 9390–9398. doi:10.1021/ma202130u
Inoue, T., Chen, G., Nakamae, K., and Hoffman, A. S. (1997). A hydrophobically-modified bioadhesive polyelectrolyte hydrogel for drug delivery. J. Control. Release 49, 167–176. doi:10.1016/S0168-3659(97)00072-2
Ju, C., Sun, J., Zi, P., Jin, X., and Zhang, C. (2013). Thermosensitive micelles–hydrogel hybrid system based on poloxamer 407 for localized delivery of paclitaxel. J. Pharm. Sci. 102, 2707–2717. doi:10.1002/jps.23649
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, Y.-H., Furuya, H., and Tabata, Y. (2014). Enhancement of bone regeneration by dual release of a macrophage recruitment agent and platelet-rich plasma from gelatin hydrogels. Biomaterials 35, 214–224. doi:10.1016/j.biomaterials.2013.09.103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S. H., Lee, Y., Lee, S.-W., Ji, H.-Y., Lee, D. S., and Park, T. G. (2011). Enzyme-mediated cross-linking of pluronic copolymer micelles for injectable and in situ forming hydrogels. Acta Biomater. 7, 1468–1476. doi:10.1016/j.actbio.2010.11.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Y., Yang, C., Khan, M., Liu, S., Hedrick, J. L., Yang, Y.-Y., et al. (2012). Nanostructured PEG-based hydrogels with tunable physical properties for gene delivery to human mesenchymal stem cells. Biomaterials 33, 6533–6541. doi:10.1016/j.biomaterials.2012.05.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, J., and Li, L. (2005). SDS-aided immobilization and controlled release of camptothecin from agarose hydrogel. Eur. J. Pharm. Sci. 25, 237–244. doi:10.1016/j.ejps.2005.02.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mangiapia, G., Ricciardi, R., Auriemma, F., De Rosa, C., Lo Celso, F., Triolo, R., et al. (2007). Mesoscopic and microscopic investigation on poly(vinyl alcohol) hydrogels in the presence of sodium decylsulfate. J. Phys. Chem. B 111, doi:10.1021/jp0663107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Missirlis, D., Tirelli, N., and Hubbell, J. A. (2005). Amphiphilic hydrogel nanoparticles. preparation, characterization, and preliminary assessment as new colloidal drug carriers. Langmuir 21, 2605–2613. doi:10.1021/la047367s
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shukla, S. K., Shaikh, W., Gunari, N., Bajpai, A. K., and Kulkarni, R. A. (2009). Self assembled hydrophobic nanoclusters of poly(methylmethacrylate) embedded into polyvinyl alcohol based hydrophilic matrix: preparation and water sorption study. J. Appl. Polym. Sci. 111, 1300–1310. doi:10.1002/app.29155
Stoppel, W. L., White, J. C., Horava, S. D., Bhatia, S. R., and Roberts, S. A. (2011). Transport of biological molecules in surfactant–alginate composite hydrogels. Acta Biomater. 7, 3988–3998. doi:10.1016/j.actbio.2011.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tedeschi, A., Auriemma, F., Ricciardi, R., Mangiapia, G., Trifuoggi, M., Franco, L., et al. (2006). A Study of the microstructural and diffusion properties of poly(vinyl alcohol) cryogels containing surfactant supramolecular aggregates. J. Phys. Chem. B 110, 23031–23040. doi:10.1021/jp061941m
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomas, B. H., Fryman, J. C., Liu, K., and Mason, J. (2009). Hydrophilic–hydrophobic hydrogels for cartilage replacement. J. Mech. Behav. Biomed. Mater. 2, 588–595. doi:10.1016/j.jmbbm.2008.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tuncaboylu, D. C., Argun, A., Algi, M. P., and Okay, O. (2013). Autonomic self-healing in covalently crosslinked hydrogels containing hydrophobic domains. Polymer 54, 6381–6388. doi:10.1016/j.polymer.2013.09.051
Wang, Y. J., Chen, L. J., Tan, L. W., Zhao, Q., Luo, F., Wei, Y. Q., et al. (2014). PEG-PCL based micelle hydrogels as oral docetaxel delivery systems for breast cancer therapy. Biomaterials 35, 6972–6985. doi:10.1016/j.biomaterials.2014.04.099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yallapu, M. M., Reddy, M. K., and Labhasetwar, V. (2007). “Nanogels: chemistry to drug delivery,” in Biomedical Applications of Nanotechnology, eds V. Labhasetwar, and D. L. Leslie-Pelecky (Hoboken: Wiley), 131–171.
Keywords: amphiphilic materials, hydrogel, hydrophobic domains, micelle, nanostructuring, surfactant
Citation: Pekař M (2015) Hydrogels with micellar hydrophobic (nano)domains. Front. Mater. 1:35. doi: 10.3389/fmats.2014.00035
Received: 30 August 2014; Accepted: 17 December 2014;
Published online: 28 January 2015.
Edited by:
P. Davide Cozzoli, University of Salento, ItalyReviewed by:
Gerardino D’Errico, Università degli Studi di Napoli Federico II, ItalyYatender Kumar Bhardwaj, Government of India, India
Copyright: © 2015 Pekař. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miloslav Pekař, Faculty of Chemistry, Brno University of Technology, Purkyňova 118, Brno 612 00, Czech Republic e-mail: pekar@fch.vutbr.cz