Controlled Parallel Crystallization of Lithium Disilicate and Diopside Using a Combination of Internal and Surface Nucleation
 Markus Rampf
Markus Rampf Marc Dittmer
Marc Dittmer Christian Ritzberger
Christian Ritzberger Wolfram Höland
Wolfram Höland- Ivoclar Vivadent AG, Schaan, Liechtenstein
In the mid-twentieth century, Dr. Donald Stookey identified the importance and usability of nucleating agents and mechanisms for the development of glass-ceramic materials. Today, a number of various internal and surface mechanisms as well as combinations thereof have been established in the production of glass-ceramic materials. In order to create new innovative material properties, the present study focuses on the precipitation of CaMgSi2O6 as a minor phase in Li2Si2O5-based glass-ceramics. In the base glass of the SiO2–Li2O–P2O5–Al2O3–K2O–MgO–CaO system, P2O5 serves as nucleating agent for the internal precipitation of Li2Si2O5 crystals, while a mechanical activation of the glass surface by means of ball milling is necessary to nucleate the minor CaMgSi2O6 crystal phase. For a successful precipitation of CaMgSi2O6, a minimum concentration of MgO and CaO in the range between 1.4 and 2.9 mol% in the base glasses was determined. The nucleation and crystallization of both crystal phases takes place during sintering a powder compact. Dependent on the quality of the sintering process, the dense Li2Si2O5–CaMgSi2O6 glass-ceramics show a mean biaxial strength of up to 392 ± 98 MPa. The microstructure of the glass-ceramics is formed by large (5–10 μm) bar-like CaMgSi2O6 crystals randomly embedded in a matrix of small (≤1 μm) plate-like Li2Si2O5 crystals arranged in an interlocking manner. While there is no significant influence of the minor CaMgSi2O6 phase on the strength of the material, the translucency of the material decreases upon precipitation of the minor phase.
Introduction
In the 1950s, Dr. Donald Stookey succeeded in the production of the first glass-ceramic of the lithium disilicate type worldwide (Stookey, 1959; Höland and Beall, 2012). His discovery and following research on this new class of materials paved the way for a number of innovative technical solutions affecting daily life, high-tech industry, and fundamental research in various fields. Stove tops and kitchenware belong to the most famous applications of glass-ceramics just as telescope mirrors, the nosecones of spaceships, and dental prostheses (Höland and Beall, 2012). In light of this wide field of applications for Donald Stookey’s invention, the impact of his work on not only the technological progress of humankind becomes obvious but also has his research motivated and driven the mind and work of a huge number of scientists in various fields of research ever since. In this spirit, the authors of the present publication want to recognize the outstanding activities of Dr. Donald Stookey.
To identify and use the nucleating effect of Ag clusters in the development of glass-ceramics in the SiO2–Li2O system, as well as transferring the general principles of nucleation and crystallization to other glass systems, was a crucial part of his great achievement (Stookey, 1959). Besides enhancing the mechanical strength and machinability of glasses, the precipitation of certain crystal phases helped to control a number of different material properties including optical and electrical properties of glass matrix materials (Beall, 1971, 2014; Mauro et al., 2013).
Initiated after the development of the first lithium disilicate glass-ceramic by Donald Stookey, this system was the subject matter in a multitude of fundamental research topics. A molar ratio SiO2/Li2O of 2, similar to the stoichiometric composition of lithium disilicate, was the basis for most of the compositions investigated (Höland and Beall, 2012). Moreover, glass-ceramics of the lithium disilicate type from multicomponent base glass systems were developed as well. There, the molar ratio of SiO2/Li2O considerably deviated from the stoichiometric value of 2. Technical glass-ceramics as well as consumer applications were developed this way (Beall, 1992, 2014; Echeverría, 1992). Likewise, Höland et al. (1994, 2005, 2007, 2008, 2009) succeeded in the development of lithium disilicate glass-ceramics for dental applications using such multicomponent base glass systems.
Based on this research and development of lithium disilicate glass-ceramics for dental applications and driven by the quest for innovative combinations of material properties, the authors of the present publication pursued the objective of the parallel controlled nucleation and crystallization of lithium disilicate and diopside CaMgSi2O6 from a multicomponent base glass. The fundamental extension of the chemical composition of the lithium disilicate base glass system by adding MgO and CaO was mandatory just as the enhancement of nucleation and crystallization mechanisms. From a glass-ceramic engineering point of view, the authors assumed that controlling two separate nucleation mechanisms is mandatory to precipitate both a layer silicate and a chain silicate from a base glass. Only the development of a powder compact processing facilitated the crystallization of diopside according to the nucleation and crystallization mechanisms reported by Kokubo et al. (1989), Müller et al. (2000), and Reinsch et al. (2008) for different glass systems. The influence of the parallel precipitation of lithium disilicate and diopside on nucleation and crystallization principles as well as on properties crucial for dental applications, e.g., biaxial strength and translucency, were investigated.
Materials and Methods
Glass Formation and Powder Production
Four glasses with different compositions in the SiO2–Li2O–P2O5–Al2O3–K2O–MgO–CaO system were melted from the raw materials quartz, lithium carbonate, aluminum metaphosphate, aluminum oxyhydroxyhydrate, potassium carbonate, alkaline magnesium carbonate, and calcium carbonate in batches of 100 g in an uncovered Pt–Rh crucible using an electric furnace. The melts were held for 2 h at 1500°C and subsequently casted into water. The glass frit was dried in an oven preheated at 150°C for about 2 h. A chemical analysis of the glass frit was conducted by means of X-ray fluorescence (XRF S8 Tiger; Bruker, Karlsruhe, Germany). The dried glass granules were comminuted using a porcelain ball mill loaded with a mixture of steatite, Al2O3, and porcelain milling beads measuring 10–30 mm in diameter. Milling was conducted until a mean grain size of 20 ± 2 μm was reached. The grain size distribution was determined using a particle size analyzer (CILAS 1064, Quantachrome GmbH & Co. KG, Odelzhausen, Germany). Thermal analysis of the glass powder was conducted by means of differential scanning calorimetry (DSC) (STA 449 F3, Netzsch, Selb, Germany) using a heating rate of 10 K/min. Heating microscope analysis (HMA) was used to investigate the sintering characteristics of the glass powder. Therefore, cylinders of 3 mm height and a diameter of 2 mm were pressed and heated with a rate of 10 K/min simultaneously recording its silhouettes (EMI V 2.3, Hesse Instruments, Osterode, Germany). The crystallization behavior of the glass powders was investigated by means of high temperature X-ray diffraction (HT-XRD). The glass powder was heated to 400°C with a rate of 30 K/min. Subsequently, the powder was further heated to 1200°C in 20 K steps. Diffraction patterns were recorded from 10° to 60° 2θ and a step size of 0.014° with a D8-Advance diffractometer (Bruker, Karlsruhe, Germany) at each step. Therefore, the temperature was held for approximately 30 min at each step during recording the diffraction pattern. Qualitative analysis was carried out using the software Bruker Diffrac.Suite EVA. The PDF entries 70-4856 (Li2Si2O5), 29-0828 (Li2SiO3), 15-0760 (Li3PO4), and 89-0830 (CaMgSi2O6) were used for phase identification.
To investigate the crystallization behavior of the bulk glass A and D, glass was casted into a graphite mold measuring 12 mm × 12 mm × 40 mm and immediately put in a muffle furnace preheated at 480°C for 20 min before slowly cooling the glass in the closed furnace. A disk of approximately 3 mm thickness was sawn from the glass, heated with a rate of 10 K/min to 860°C in a furnace of the Programat® type (Ivoclar Vivadent AG, Schaan, Liechtenstein). The temperature was held for 5 min before slowly opening the oven. One side of the crystallized glass-ceramic disk was ground using a 125 μm diamond grit grinding disk. The ground and the “as fired” surface were analyzed using room temperature XRD applying the parameters described above. Subsequently, the glass-ceramic disk was broken, and the fracture surface was investigated by means of scanning electron microscopy (SEM) after etching for 30 s with vapor of 40% hydrofluoric acid and coating with a 1 to 2 nm Au–Pd layer.
Production and Sintering of Powder Compacts
Very small amounts of water, acting as binder, were added to the glass powders before uniaxial pressing of specimen. In this way, powder compacts with different geometries were produced via uniaxial pressing using different press molds. Cubes with an edge length of 18 mm, bars measuring 36 mm × 4.8 mm × 6 mm, and disks with a diameter of 24 and 4 mm thickness were made. The powder compacts were sintered in vacuum to dense glass-ceramics using a furnace of the Programat® type (Ivoclar Vivadent AG, Schaan, Liechtenstein). The powder compacts were heated to 480°C with a rate of 30 K/min and a subsequent dwell time of 20 min. Subsequently, the powder compact was further heated with 10 K/min to 860°C. This temperature was held for 5 min before slowly opening the furnace and thereby cooling the glass-ceramic to room temperature.
Characterization of the Glass-Ceramics
Sintered glass-ceramics were pulverized using a mortar grinder (Mortar Grinder RM 200, Retsch, Haan, Germany) and subsequently sieved <45 μm. In order to qualitatively investigate the crystal phases, diffraction patterns were recorded from 10° to 60° 2θ applying a step size of 0.014°. Furthermore, a quantitative study of the crystal phase formation was conducted applying Rietveld refinement. Therefore, the pulverized glass-ceramic material was elutriated with approximately 20% of its mass of α-Al2O3 (Alfa Aesar, 99.9%, 20–50 μm//ICSD number: 31548) as internal standard in acetone. The solvent was evaporated in an oven preheated at 80°C. After recording the powder XRD patterns from 10° to 100° 2θ in 0.014° steps, the quantification of the crystal phases was done with the TOPAS software from Bruker.
The biaxial fracture strength was determined according to ISO 6872, including a surface finishing with a 15 μm diamond grit grinding disk. The specimen were milled from the sintered glass-ceramic cubes using a CEREC® InLab milling machine (Sirona, Bensheim, Germany). The biaxial strengths are given as mean values of 10 data sets ±SD.
The thermal expansion of the glass ceramics was determined according to ISO 6872 by applying dilatometric analysis using a sapphire reference. The sintered glass-ceramic bars were ground to the required dimensions using a 40 μm diamond grit grinding disk. The error of the measurements is in the range of ±0.5 × 10−6 K−1.
The contrast ratio (CR) of the glass-ceramic disks was determined using a spectrometer of the type CM-3700d (Konica-Minolta, Tokyo, Japan). The sintered disks were prepared according to BS5612. The translucency of a material decreases with increasing CR value.
Micrographs of the glass-ceramics were taken by means of SEM. The surface of the glass-ceramic samples was therefore polished using a 0.5 μm diamond grit grinding disk and subsequently etched with the vapor of 40% hydrofluoric acid for 30 s.
The porosity of the sintered glass-ceramics was estimated using the line intercept method. Therefore, three fragments of the samples broken within testing the biaxial strength of each glass-ceramic were polished using a 0.5 μm diamond grit grinding disk and examined by SEM. The linear ratio of pores is given as means of three data sets ±SD.
Results
Properties and Crystallization Behavior of the Base Glasses
The chemical composition of the base glasses calculated in mol% from the results obtained from XRF analysis is given in Table 1. The concentration of MgO and CaO in the base glasses increases from glasses A to D. The results of the thermal analysis of the base glass powders are presented in Figure 1. The glass transition temperatures Tg as well as the exothermic and endothermic peak temperatures are summarized in Table 2. Base glass A shows a Tg, which is increased by approximately 10 K compared to the base glasses B–D. The first exothermic peak temperatures Texo1 increase from base glasses A to D, while the opposite trend can be observed for the second exothermic peak temperatures Texo2. The third exothermal process can only be detected for the base glasses B–D. Compared to the rather sharp first and second exothermal signals, the third exothermal reaction of the base glasses is characterized by a very wide peak in the temperature range between 780 and 840°C. The first endothermic heat flow could be detected at approximately 947°C for base glass A. Significantly lower endothermic peak temperatures Tendo1 between 887 and 899°C were determined for base glasses B–D. The second endothermic peak Tendo2 could be detected for base glasses B–D.
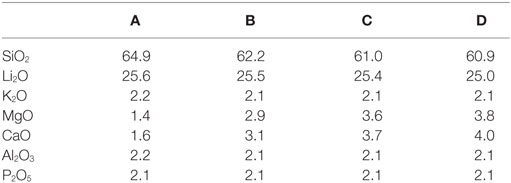
Table 1. Chemical compositions of the base glasses in mol% calculated from the composition determined by XRF analysis.
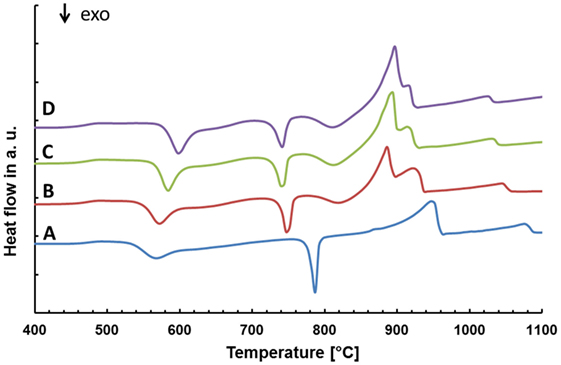
Figure 1. Thermal analysis of the different base glasses.
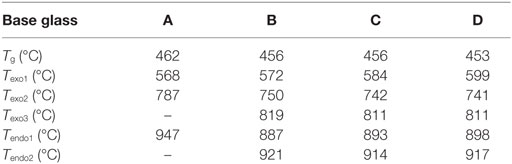
Table 2. Thermal analysis of the base glasses by means of DSC.
Figure 2 shows the data obtained by HMA. The area of the silhouette is plotted as a function of the temperature. The first reduction of the silhouette area occurs for all base glasses in approximately the same temperature range between 500 and 550°C. After running through a plateau between 550 and 700°C, the area again decreases for all glasses within the same temperature range between 700 and 800°C. However, while glasses B–D again show a plateau between 800 and 850°C, no such second plateau can be found for glass A. A continuous reduction of the shadow area can be observed for all glasses at temperatures exceeding 860°C.
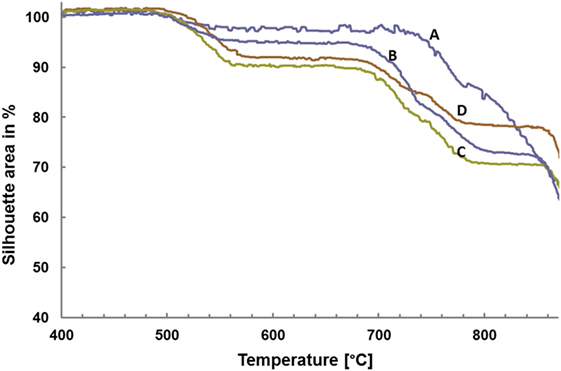
Figure 2. Sintering behavior of the glass powders determined by high temperature microscopy.
Diffractograms of glass D obtained by HT-XRD are presented in Figure 3. The diffraction patterns obtained at 29 and 500°C reveal no crystalline phases. First indications for the crystallization of Li2SiO3 could be observed at 540°C. Characteristic Li2Si2O5 peaks could be analyzed in the diffractogram recorded at 680°C. At 700°C, no Li2SiO3 peaks were detected. However, the intensity of the Li2Si2O5 peaks is strongly increased and Li3PO4 can be detected. Reflections characteristic for diopside occur at 760°C. The intensity of the diopside peaks increases until 860°C. A similar evaluation of the HT-XRD results obtained for glass-ceramics A–D is shown in Table 3, where those temperatures are summarized at which a first evidence for the crystallization of a certain crystal phase was found. No diopside formation was detected for glass A. Compared to B–D, glass A shows significantly increased crystallization temperatures for Li2SiO3 and Li2Si2O5.
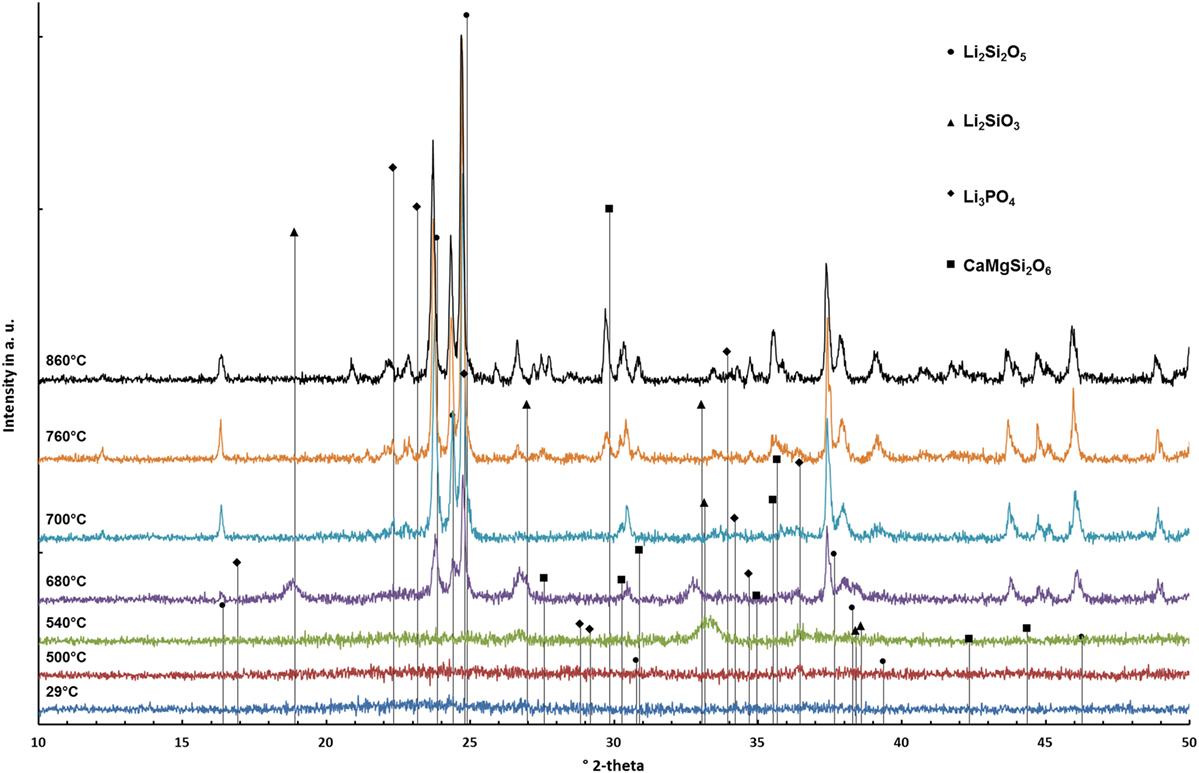
Figure 3. Diffractograms of glass D surveyed at different temperatures.
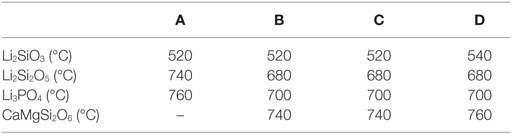
Table 3. Sequence of crystal phase formation as a function of temperature determined by HT-XRD.
Investigating the crystallization behavior of the bulk base glass D by means of room temperature, XRD revealed Li2Si2O5 and CaMgSi2O6 for the “as fired” sample surface. No diopside crystals were detected in the diffraction pattern recorded of the ground sample.
The additional investigation of the micrograph of the fracture surface in Figure 4 shows small (≤1 μm) plate-like crystals in the bulk of the material and large bar-like crystals growing from the surface into the bulk of the material. The investigation of bulk base glass A revealed solely the formation of Li2Si2O5, and no surface crystallization of diopside could be observed.
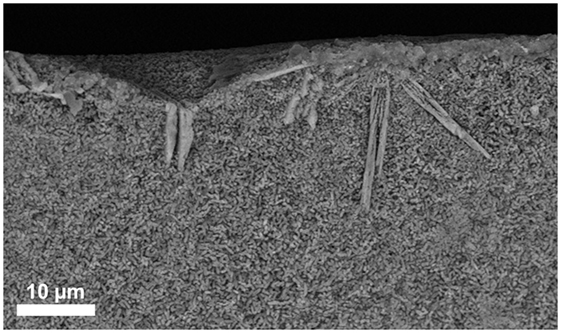
Figure 4. SEM micrograph taken from the fracture surface of a bulk base glass sample D crystallized at 860°C for 5 min.
Characterization of the Glass-Ceramics
Diffraction patterns of the sintered glass-ceramics recorded at room temperature are shown in Figure 5. Li2Si2O5 and Li3PO4 could be detected in all glass-ceramics. However, glass-ceramics B–D reveal the presence of diopside.
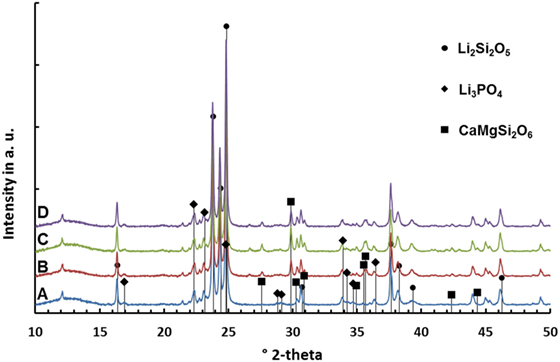
Figure 5. Diffractograms of glass-ceramics after sintering at 860°C for 5 min.
The results of the quantitative crystal phase analysis by means of Rietveld refinement are shown in Table 4. Li2Si2O5 is the main crystal phase in the glass-ceramics. The content of Li2Si2O5 decreases from A to D, while the content of diopside increases from B to C. With respect to the accuracy of measurement and refinement, there is no difference in the ratio of diopside in glass-ceramics C and D. Glass-ceramic A comprises a considerably higher ratio of amorphous residual glass phase than B–D.
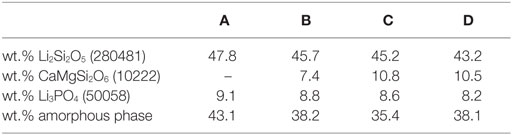
Table 4. Quantitative composition of glass-ceramics after sintering and crystallization at 860°C for 5 min (ICSD number).
Micrographs of the glass-ceramics A–D displayed in Figure 6 reveal an interlocking microstructure of small (≤1 μm) plate-like crystals. The microstructure of glass-ceramics B–D additionally comprises bar-like crystals with a length between 3 and 10 μm. The large bar-like crystals are randomly distributed within the microstructure. The analysis of the porosity revealed thoroughly dense glass-ceramics A and D, while B and C showed a linear ratio of pores of 4.5 ± 3.4 or 0.9 ± 0.4%, respectively.

Figure 6. Micrographs of the glass-ceramics (A–D) after sintering at 860°C for 5 min.
The biaxial strength of the glass-ceramics is presented in Table 5. Glass-ceramic D shows the highest mean biaxial strength. With respect to the SD of the measurement results, the biaxial strength of glass-ceramic D is significantly higher than that of glass-ceramics B and C. The strength of glass-ceramics A and D is in a similar range.
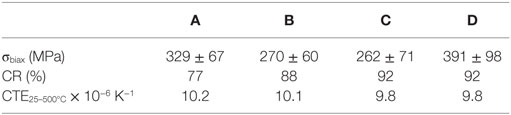
Table 5. Properties of the glass-ceramics.
No significant difference in the thermal expansion properties of the different glass-ceramics could be analyzed. The results are presented in Table 5. CTE25–500°C values in the range between 9.8 × 10−6 K−1 and 10.2 × 10−6 K−1 were determined.
All glass-ceramics show a low degree of translucency. Glass-ceramic A revealed a considerably lower opacity than glass-ceramics B–D, which are rather similar (Table 5).
Discussion
The extension of the chemical composition of the base glass by certain amounts of MgO and CaO is the prerequisite for the precipitation of CaMgSi2O6 as a minor crystal phase in Li2Si2O5 glass-ceramics. Increasing the content of MgO and CaO from approximately 1.4 to 2.9 mol% yielded the precipitation of diopside in the present study. However, no internal nucleation and crystallization of diopside was found in the present base glasses, but surface crystallization of diopside could be verified in the preliminary study of the crystallization behavior of the bulk base glass D (Figure 4). Hence, an activation of the glass surface was mandatory to nucleate the crystallization of diopside. In the present study, the activation of the glass surface was achieved by means of ball milling. It is important to note that the magnitude of activation of a glass surface is strongly dependent on the method and processing parameters used. There exist a lot of different milling techniques as well as there are various alternative ways to initiate surface nucleation and crystallization for different material systems, e.g., by means of tribochemistry (Henicke, 1984). Hence, the concentration of MgO and CaO that is mandatory for the crystallization of diopside can deviate dependent on the method used for the activation of the glass surface.
The analysis of the base glasses by means of DSC (Figure 1; Table 2) reveals two rather sharp exothermal peaks for all the base glasses. According to the data obtained by HT-XRD (Table 3), these peaks can be correlated to the crystallization of Li2SiO3 and Li2Si2O5. The rather sharp characteristic of these peaks suggests that an internal nucleation and crystallization mechanism is dominant. The effect of P2O5 as internal nucleating agent for lithium silicates at a glassy amorphous interphase was previously reported in literature (Höland and Beall, 2012). HT-XRD studies in the present glass-ceramics reveal that crystalline Li3PO4 can only be detected after heat treatments above 700°C, whereas the crystallization of Li2SiO3 already starts at ≥520°C (Table 3). Hence, a mechanism reported by Höland and Beall (2012) describing the nucleation of lithium silicate at the interphase of a glassy amorphous or disordered nanocrystalline early phase of Li3PO4 seems most likely.
A rather wide exothermal signal in the DSC curves of glass B–D between 780 and 860°C are an indication for a surface nucleation and crystallization mechanism. The data obtained by HT-XRD show that diopside forms in exactly this temperature range. Hence, the authors conclude that diopside crystallizes on the activated surface of the glass grains. Furthermore, surface nucleation and crystallization display well-known mechanisms found in the investigations on diopside glass-ceramics (Kokubo et al., 1989; Zanotto, 1991; Müller et al., 2000).
In summary, the precipitation of two different silica-based crystal phases from the present glass system requires the control of two essentially different nucleation mechanisms. On the one hand, the layer silicate Li2Si2O5 is nucleated by an internal nucleation mechanism, and on the other hand, surface activation nucleates the crystallization of the chain silicate CaMgSi2O6.
To produce bulk glass-ceramics from internal and surface crystallizing base glasses, a powder compact process was developed. Crystallization of the glass powder and sintering of the powder compacts to achieve dense bulk glass-ceramics has to be controlled in parallel. Analyzing the sintering behavior of the glass powders by means of HMA (Figure 2), together with the results of the HT-XRD studies (Table 3), was the basis to develop an adequate firing cycle. According to the HMA curves, densification starts at approximately 500°C for all base glasses. Since the Tg of the base glass powders is between 450 and 460°C, the starting densification is clearly a result of the softening and beginning viscous flow of the glass. Crystallization of Li2SiO3 and Li2Si2O5 between 540 and 700°C clearly increase the viscosity of the glasses and hence stop the densification as can be seen in the first plateau of the HMA curve. Base glasses B–D show a second plateau ending at approximately 855°C, which can be explained by the crystallization of diopside hindering the sintering process. Dissolution of crystal phases and melting of the glass-ceramics begins at temperatures >860°C.
Based on these results, the crystallization and sintering cycle of the powder compacts was developed. It starts with a fast heating to 480°C, where the temperature is kept for 20 min in order to internally nucleate lithium silicate at a glassy amorphous P2O5-rich interface (Höland and Beall, 2012). After internal nucleation, the powder compacts were heated to 860°C with a reduced heating rate of 10 K/min. Crystallization and sintering takes place in this temperature range. To obtain a dense glass-ceramic with minimal crystal phase dissolution and viscous deformation of the specimen, the sintering cycle stops after a dwell time of 5 min at 860°C. The micrographs of the glass-ceramics in Figure 6 reveal pores in the glass-ceramics B and C. Glass-ceramics A and D show a more homogeneous and dense microstructure than the other glass-ceramics. Based on the quantitative crystal phase analysis of the glass-ceramics (Table 4) and assuming that the entire ratio of amorphous P2O5–Li2O phase precipitates to Li3PO4, one can roughly estimate the composition of the residual glass phases. The estimated composition of residual glass phase is shown in Table 6. According to this estimation, the residual glass phase of the glass-ceramic D comprises a considerably higher concentration of network modifiers such as Li2O, MgO, and CaO. This would result in a reduced viscosity of the residual glass phases compared to glass-ceramics A–C and therefore displays an interesting point of discussion regarding the sintering properties of the glass-ceramics. Comparing the sintering properties of glass-ceramic A to B and C, the restraining influence of crystallization processes on the sintering process becomes obvious, since glass-ceramic A shows no diopside formation in contrast to B and C. A more dedicated investigation is necessary to resolve the different results of the sintering process. Furthermore, enhancing the powder processing, e.g., by granulation of the glass powder prior to uniaxial pressing, could improve the densification and sintering of the powder compacts.
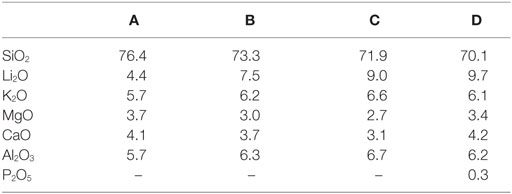
Table 6. Estimated chemical compositions of the residual glass phases in mol%.
The influence of the chemical composition of the base glasses on the quantitative crystal phase composition of the final glass-ceramics is shown in Table 4. Obviously, a concentration of ≤1.4 or 1.6 mol% of MgO and CaO in the base glasses is insufficient for the precipitation of CaMgSi2O6 in the present base glass system. However, as mentioned in the first paragraph of the discussion, this finding is only true for the activation of the glass powders surface by means of ball milling down to a mean grain size of about 20 μm. Increasing the concentration of MgO and CaO from 2.9 or 3.1 to 3.6 or 3.7 mol%, respectively, resulted in a considerable increase of the diopside content by 3.4 wt.%, while further increasing to 3.8 or 4 mol%, respectively, did not significantly change the concentration of diopside in the glass ceramic. The reduced content of SiO2 together with the consumption of SiO2 during the crystallization of Li2Si2O5 could explain the stagnant precipitation of CaMgSi2O6, although higher concentrations of CaO and MgO are provided in the base glass.
Based on the preliminary study of crystallization (Figure 4) and comparing the micrographs B–D with the micrograph of the diopside-free glass-ceramic A (Figure 6), the crystals forming the microstructure of the glass-ceramics can be identified. Large (5–10 μm) bar-like diopside crystals are embedded in a matrix of rather small (<1 μm) plate-like Li2Si2O5 crystals (Figure 6).
High concentrations of crystalline phases (>55 wt.%) (Table 4) as well as the arrangement of Li2Si2O5 and CaMgSi2O6 crystals in an interlocking microstructure (Figure 6) contribute to the biaxial strength of the glass-ceramics (Dittmer et al., 2014). However, pores in the microstructure, as they were found particularly in glass-ceramics B and C (Figure 6), can decrease the biaxial strength of the materials. In a previous study on tailoring, the thermal expansion of Li2Si2O5 glass-ceramics by precipitating a high CTE minor phase significant effects on the biaxial strength of the glass-ceramics due to the considerable mismatch in the CTEs of the involved crystal phases were reported (Rampf et al., 2015, 2016). However, in the glass-ceramic system investigated in the present study, such effects are unlikely since there is no such huge difference in the thermal expansion of the involved crystal phases. The CTE200–800°C of diopside crystals was investigated to be approximately 7.4 × 10−6 K−1 (Rigby and Green, 1942) and therefore very close to polycrystalline lithium disilicate (CTE25–600°C = 11.4 × 10−6 K−1) compared to the investigations by Rampf et al. (2015, 2016).
The precipitation of diopside as a minor phase in Li2Si2O5 glass-ceramics reduces the translucency of the glass-ceramic materials due to the introduction of a further interface where scattering of light occurs (Nassau, 2001). The influence of a minor crystal phase Ca5(PO4)3F on the optical properties of Li2Si2O5 glass-ceramics was previously investigated by Rampf et al. (2015, 2016). They reported the optical properties, in particular the translucency, to be dominated by the Li2Si2O5 crystals, which appeared to be significantly larger than the submicron sized apatite crystals. However, the apatite crystals having dimensions in the range of visible light did affect the opalescence of the materials. In the present system, the diopside crystals are significantly larger than the approximately 0.5–1 μm-sized Li2Si2O5 crystals. Hence, the precipitation of diopside displays an effective tool to influence the translucency of Li2Si2O5 glass-ceramics without affecting its opalescence properties.
Machining of the crystallized glass-ceramics using conventional CAD/CAM technology was feasible without problems. The minor diopside phase enhanced the machinability of the Li2Si2O5 glass-ceramics probably by interfering with the high-strength interlocking Li2Si2O5 microstructure.
Conclusion
Glass-ceramics comprising Li2Si2O5 as main crystal phase and CaMgSi2O6 as minor crystal phase can be produced from base glasses in the SiO2–Li2O–P2O5–Al2O3–K2O–MgO–CaO system. The parallel precipitation of a layer silicate (Li2Si2O5) on the one side and crystals of the chain silicate type (CaMgSi2O6) on the other side require the control of two separate nucleation and crystallization mechanisms. Li2Si2O5 can be nucleated internally at an amorphous glassy P2O5–Li2O-rich interphase. The activation of the glass surface, e.g., by means of ball milling, is mandatory to efficiently nucleate the crystallization of CaMgSi2O6.
The chemical composition, in particular a sufficient concentration of MgO and CaO, of the base glass is essential for the precipitation of the CaMgSi2O6 minor phase. However, the mandatory concentration of MgO and CaO can deviate with the extent or method applied for surface activation.
A reproducible powder compact process, including uniaxial pressing and sintering, was developed to produce dense Li2Si2O5–CaMgSi2O6 glass ceramic materials.
Li2Si2O5–CaMgSi2O6 glass-ceramics could be machined by means of conventional CAD/CAM technology and reach a biaxial strength of 391 ± 98 MPa based on the interlocking microstructure and possible additional compressive stress formation.
The large (5–10 μm) bar-like CaMgSi2O6 crystals embedded in a matrix of interlocking (≤1 μm) Li2Si2O5 crystals reduce the translucency of the material and hence display a further potential tool to create new combinations of material properties within Li2Si2O5-based glass-ceramics.
Author Contributions
MR: planning, organization, and execution of experiments; interpretation and discussion of results; and compilation of manuscript and artwork. MD: execution of Rietveld refinement; interpretation and discussion of results on XRD analysis; and assistance in compilation of the manuscript. CR: consulting in planning of experiments; interpretation and discussion of HMA and DSC; and assistance in compilation of the manuscript. WH: consulting regarding the state of the art literature on the topic; interpretation and discussion of results; and assistance in the compilation and proofreading of the manuscript.
Conflict of Interest Statement
The authors are employees of the company Ivoclar Vivadent AG. A patent was filed on the present subject.
The reviewer LM and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
References
Beall, G. H. (1971). “Structure, properties, and nucleation of glass-ceramics,” in Advances in Nucleation and Crystallization in Glasses, 8 Edn, Vol. Special Publ. 5, eds L. L. Hench and S. S. Freiman (Columbus, OH: The Am. Ceram. Soc.), 251–261.
Beall, G. H. (1992). Design and properties of glass-ceramics. Annu. Rev. Mater. Sci. 22, 91–119. doi:10.1146/annurev.ms.22.080192.000515
Beall, G. H. (2014). Milestones in glass-ceramics – a personal perspective. Int. J. Appl. Glass-Sci. 5, 1–11. doi:10.1111/ijag.12063
Dittmer, M., Ritzberger, C., Schweiger, M., Rheinberger, V., Wörle, M., and Höland, W. (2014). Phase and microstructure formation and their influence on the strength of two types of glass-ceramics. J. Non-Cryst. Solids 384, 55–60. doi:10.1016/j.jnoncrysol.2013.03.009
Höland, W., Frank, M., Schweiger, M., and Rheinberger, V. (1994). “Development of translucent glass-ceramics for dental applications,” in Proceedings of the 5th International Otto Schott Colloquium, ed. C. Rüssel (Frankfurt: Verlag der Deutschen Glastechnischen Gesellschaft), 117–121.
Höland, W., Rheinberger, V., Apel, E., and van’t Hoen, C. (2007). Principles and phenomena of bioengineering with glass-ceramics for dental restorations. J. Eur. Ceram. Soc. 27, 1521–1526. doi:10.1016/j.jeurceramsoc.2006.04.101
Höland, W., Rheinberger, V., van’t Hoen, C., and Apel, E. (2005). P2O5 as an effective nucleating agent of lithium disilicate glass-ceramics. Phosphorus Res. Bull. 19, 36–41. doi:10.3363/prb1992.19.0_36
Höland, W., Schweiger, M., Rheinberger, V. M., and Kappert, H. (2009). Bioceramics and their application for dental restoration. Adv. Appl. Ceram. 108, 373–380. doi:10.1179/174367609X414099
Höland, W., Schweiger, M., Watzke, R., Peschke, A., and Kappert, H. (2008). Ceramics as biomaterials for dental restoration. Expert Rev. Med. Devices 5, 729–745. doi:10.1586/17434440.5.6.729
Kokubo, T., Sakka, S., Sako, W., and Ikejiri, S. (1989). Preparation of glass-ceramic containing crystalline apatite and magnesium titanate for dental crown. J. Ceram. Soc. Jpn. Ed. 97, 236–240.
Mauro, J. C., Ellison, A. J., and Pye, L. D. (2013). Glass: the nanotechnology connection. Int. J. Appl. Glass-Sci. 4, 64–75. doi:10.1111/ijag.12030
Müller, R., Zanotto, E. D., and Fokin, V. M. (2000). Surface crystallization of silicate glasses: nucleation sites and kinetics. J. Non-Cryst. Solids 274, 208–213. doi:10.1016/S0022-3093(00)00214-3
Rampf, M., Dittmer, M., Ritzberger, C., Schweiger, M., and Höland, W. (2015). Properties and crystallization phenomena in Li2Si2O5–Ca5(PO4)3F and Li2Si2O5–Sr5(PO4)3F glass–ceramics via twofold internal crystallization. Front. Bioeng. Biotechnol. 3:122. doi:10.3389/fbioe.2015.00122
Rampf, M., Fisch, M., Dittmer, M., Ritzberger, C., and Höland, W. (2016). Tailoring the thermal expansion of glass-ceramics by controlled twofold crystallization of Li2Si2O5 and CsAlSi5O12. Int. J. Appl. Glass Sci. 7, 285–294. doi:10.1111/ijag.12180
Reinsch, S., Nascimento, M. L. F., Müller, R., and Zanotto, E. D. (2008). Crystal growth kinetics in cordierite and diopside glasses in wide temperature ranges. J. Non-Cryst. Solids 354, 5386–5394. doi:10.1016/j.jnoncrysol.2008.09.007
Rigby, G. R., and Green, A. T. (1942). The thermal expansion characteristics of some calcareous and magnesium minerals. Trans. Br. Ceram. Soc. 41, 123–143.
Stookey, S. D. (1959). Catalyzed crystallization of glass in theory and practice. Ind. Eng. Chem. 51, 805–808. doi:10.1021/ie50595a022
Keywords: glass-ceramics, lithium disilicate, diopside, twofold crystallization, nucleation, surface activation, sintering
Citation: Rampf M, Dittmer M, Ritzberger C and Höland W (2016) Controlled Parallel Crystallization of Lithium Disilicate and Diopside Using a Combination of Internal and Surface Nucleation. Front. Mater. 3:47. doi: 10.3389/fmats.2016.00047
Received: 29 July 2016; Accepted: 26 September 2016;
Published: 17 October 2016
Edited by:
John C. Mauro, Corning Incorporated, USAReviewed by:
Takayuki Komatsu, Nagaoka University of Technology, JapanLina Ma, Corning Incorporated, USA
Copyright: © 2016 Rampf, Dittmer, Ritzberger and Höland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Rampf, markus.rampf@ivoclarvivadent.com