A Cross-sectional Study of KLKB1 and PRCP Polymorphisms in Patient Samples with Cardiovascular Disease
 Haley R. Gittleman1†
Haley R. Gittleman1†
 Alona Merkulova2†
Omar Alhalabi2†
Evi X. Stavrou2,3
Martina L. Veigl1
Alona Merkulova2†
Omar Alhalabi2†
Evi X. Stavrou2,3
Martina L. Veigl1
 Jill S. Barnholtz-Sloan1
Jill S. Barnholtz-Sloan1  Alvin H. Schmaier1,2,4*
Alvin H. Schmaier1,2,4*
- 1Case Comprehensive Cancer Center, Case Western Reserve University, Cleveland, OH, USA
- 2Department of Medicine, Division of Hematology-Oncology, Case Western Reserve University, Cleveland, OH, USA
- 3Louis Stokes Veterans Administration Hospital, Cleveland, OH, USA
- 4University Hospitals Case Medical Center, Cleveland, OH, USA
Plasma kallikrein formed from prekallikrein (PK) produces bradykinin from kininogens and activates factor XII. Plasma PK is activated by factors αXIIa, βXIIa, or prolylcarboxypeptidase (PRCP). A cross-sectional investigation determined if there is an association of PRCP and KLKB1 polymorphisms with cardiovascular disease (CVD). DNA was obtained from 2243 individuals from the Prevention of Events with Angiotensin Converting Enzyme trial. Two PRCP SNPs, rs7104980 and rs2298668, and two KLKB1 SNPs, rs3733402 and rs3087505, were genotyped. Logistic regression models were performed for history of diabetes, myocardial infarction, stroke, angina, angiographic coronary disease, CABG, intermittent claudication, percutaneous transluminal coronary angioplasty (PTCA), and transient ischemic attack. The PRCP SNP rs7104980 increased the odds of having a history of PTCA by 21% [odds ratio (OR) = 1.211; 95% confidence intervals (CI) = (1.008, 1.454)]; P = 0.041, but was non-significant after Bonferroni correction. Alternatively, having the G allele for rs3733402 (KLKB1 gene) decreased the odds of having a history of angiographic coronary disease by 24% [OR = 0.759; 95% CI = (0.622, 0.927)]; P = 0.007 that was statistically significant (P < 0.01) after Bonferroni correction for multiple hypothesis testing. When the best-fit model based on the Akaike information criterion controlled for age, weight, gender, hypertension, and history of angina, the G allele of KLKB1 rs3733402 that is associated with less plasma kallikrein activity correlated with reduced history of CVD.
Introduction
Plasma prekallikrein (PK) is a zymogen whose enzyme plasma kallikrein is known to initiate and amplify factor XII activation on collagen and polyphosphates in plasma in the intravascular compartment to activate intrinsic blood coagulation. Formed plasma kallikrein is also the major enzyme to generate bradykinin from high molecular weight kininogen (HK) and to convert plasminogen to plasmin and single-chain urokinase to two-chain urokinase for fibrinolysis, C3 and C5 to C3a and C5a, respectively, in the complement system and prorenin to renin in the renin angiotensin system. There are several SNPs that have been recognized to impact PK function. KLKB1 rs3087505 is associated with venous thrombosis and this SNP is in linkage disequilibrium (LD) with two F11 SNPs (rs2036914, rs3756008) also associated with thromboembolism (1, 2). Also an exon 5 N124S polymorphism (rs3733402) in Apple domain 2 results in reduced PK complex formation to HK, its plasma, and cell membrane-binding protein (3–5), and this interferes with optimal PK activation and BK formation. Recent studies with klkb1−/− mice indicate that animals lacking klkb1 have delayed arterial thrombosis not due to reduced contact activation but due to reduced vessel wall tissue factor (6, 7). These combined studies suggest that PK contributes to vascular homeostasis. Infact, previous work by our own group demonstrated that elevated plasma PK correlates with accelerated vascular disease and proteinuria in a large population of diabetic patients (8, 9).
Prolylcarboxypeptidase (PRCP) is a membrane-associated serine protease that cleaves peptides where the penultimate position is a Pro-X bond (10, 11). As an exopeptidase, it has been recognized to degrade desArg9-bradykinin, angiotensin II and III, and α-melanocyte-stimulating hormone1–13 (10–12). We also have shown that it is a plasma PK activator when PK assembles on HK on endothelial and other cell membranes, suggesting that it is an endopeptidase as well (13–15). Genetic studies have identified an association between PRCP and essential hypertension and metabolic syndrome (16, 17). An exon SNP of PRCP (rs2298668, E112D) indicates risk for chronic hypertension and preeclampsia in an African-American patient population (18). In a Han Chinese population, it is associated with treatment resistance to an ACE inhibitor (19). In another study of Han Chinese subjects, the G allele of intron SNP rs7104980, but not rs2298668, is associated with essential hypertension [odds ratio (OR) = 1.98, 95% confidence intervals (CI) = 1.62–2.43, P = 0.3 × 10−10] (20). PRCP-deficient gene-trap (prcpgt/gt) mice that have 25% PRCP normal protein levels in their organs are constitutively hypertensive and have a heightened risk for arterial thrombosis (21). Likewise, spontaneous hypertensive rats (SHR) compared to their control Wistar rats also have reduced PRCP in cardiac tissue (22). PRCP deficiency in mice is associated with increased vascular inflammation and reduced cell proliferation, angiogenesis, and wound repair from physical injury and ischemia reperfusion (23). Opposite to PK, the presence of PRCP reduces vascular inflammation and arterial thrombosis.
In the present investigation, we asked if polymorphisms of PRCP and PK are associated with cardiovascular disease (CVD). In a single cross-sectional study, we show that the G allele for PRCP intronic rs7014980 associates with CVD but does not meet statistical significance. Alternatively, the minor G allele of an exonic SNP rs3733402 (N124S) of KLKB1 is significantly associated with reduced CVD. These data suggest that a selected KLKB1 SNP (rs3733402) mirrors the phenotype observed in patient studies with diabetes and on murine deletions of klkb1 (7–9).
Materials and Methods
Data Collection
Plasma samples for our cross-sectional investigation below were provided from the Prevention of Events with Angiotensin Converting Enzyme (PEACE) Inhibition Trial that were in the NIH Biorepository BioLincc (https://biolincc.nhlbi.nih.gov/home/) (24). In the original PEACE trial, all participants signed informed consent that was approved by their local IRBs for blood collection (24). The goal of the PEACE trial was to test whether ACE-inhibitor therapy would reduce the rate of non-fatal myocardial infarction (MI), death from cardiovascular causes, or coronary revascularization in low-risk patients with stable coronary artery disease and normal or slightly reduced left ventricular function (ejection fraction >40%). The criteria for selection in the PEACE trial were individuals >50 years old with evidence of coronary artery disease documented by one of the following three criteria: (1) MI >3 months from enrollment, (2) coronary artery bypass grafting (CABG) or percutaneous transluminal coronary angioplasty (PTCA) >3months from enrollment, and (3) obstruction of at least one luminal diameter >50% of at least one native vessel on coronary angiography, and toleration of the medication and >80% compliance with the medication (24). The cardiovascular phenotypes were defined in the PEACE trial and obtained from its data dictionary (24). The outcomes of the present analysis were compared with the baseline, pretreatment cardiovascular phenotype of the subjects. We determined if the present selected SNPs correlated with the mild cardiovascular phenotypes of the subjects of the PEACE trial.
The subsample of subject plasmas from the repository were de-identified from the individual donors but were linked by code to clinical information in the data dictionary (23). The samples that were provided by NHLBI BioLincc were chosen by random availability of 1 ml plasma aliquots for shipment. We obtained an exempt approval from the University Hospitals Case Medical Center IRB NHR-12-13. Two thousand two hundred forty-three (N = 2243) plasma samples from the PEACE trial were used for DNA isolation. DNA was extracted by the NucleoSpin Plasma XS technique (Macherey-Nagel, Inc.) yielding 0.03–0.1 μg/sample. The extracted DNA had whole-genome amplification using REPLI-g Mini Kit (Cat# 150023) (QIAGEN) to yield 10 mg/ml. DNA samples were quantified using Qubit DNA quantification assay and a Qubit 2.0 Fluorometer (Life Technologies).
As described above, two SNPs from PRCP that have been associated with hypertension, intronic rs7104980 (11:82864153) (20) and exonic rs2298668 (11:82853252) (E112D) (18, 19) were studied. Also, two SNPs from KLKB1 that have been associated with vascular function were chosen for study; rs3087505 (Chromosome location, 4:186258332) (1, 2) from the 3′UT and exonic rs3733402 (4:186236880) (N124S) (3). All SNPs studied were listed as Illumina Golden Gate validated and ABI TaqMan genotyping assays were available for them. ABI TaqMan chemistry consists of two probes labeled with either VIC or FAM and a primer pair to detect the specific SNP. Genotyping these samples was performed according to the manufacturer’s protocol. Specifically, 3–5 μl aliquots, containing 5–10 ng of DNA were transferred from 96-well reservoir plates to 384-well assay plates for each individual being genotyped. Multiple 384-well assay plates were generated; the DNA was dried down, the plates then sealed, and frozen until assayed. A 5 μl aliquot of Master Mix, Probe and Primer was robotically added to each well of a 384-well plate previously plated with DNA. PCR (40 cycles) was carried out on an ABI GeneAmp PCR System 9700 Dual Head Instrument and endpoint reads were carried out using the ABI 7900 Sequence Detection System (SDS). All assay mixtures were prepared in an amplicon-free room to avoid contamination. Data from each SNP were clustered using ABI’s SDS version 2.3 Software and Genotyping Calls (Vic/Vic; Fam/Fam; Vic/Fam) were automatically made by the software. Each file was manually inspected to remove outliers. The error rate of each SNP not identifying the genotype of eight control DNA samples examined 15 times over the course of the study was 6.9% for rs7104980, 6.2% for rs3087505, 13% for rs2298668, and 7.4% for rs3733402. These rates are similar to other large-scale genetic epidemiology studies.
Statistical Analysis
Genotyping rates were calculated for each SNP across all individuals. Allele and genotype frequencies were calculated for each SNP. Hardy–Weinberg equilibrium (HWE) and pairwise LD were assessed using standard methods. A heatmap was used to determine if SNP rs3733402 is in LD with F11 SNPs rs2036914 and rs3756008, respectively. Unconditional logistic regression models assessed the association between candidate SNPs and various disease outcomes calculating ORs and 95% CI. For each candidate SNP, having the rarer allele was compared to not having the rarer allele. Other covariates included in the logistic regression models were age, weight, gender, hypertension status, history of diabetes, history of angina, and/or cigarette use. Hypertension in the PEACE trial was defined as having systolic blood pressure greater or equal to 140 mmHg and/or having diastolic blood pressure greater or equal to 90 mmHg (24). This variable is user-defined specific for this investigation and different from the history of hypertension variable used in the PEACE trial. For cigarette use, the “rarely” and “never” categories were combined and compared against “frequent” cigarette use. Logistic regression models were then compared using specific −2 log likelihood values in order to calculate the Akaike information criterion (AIC) (25). The result with the smallest AIC was chosen as the best-fitting model. Multiple hypothesis testing was corrected by the Bonferroni correction method. All statistical analyses were performed using SAS version 9.3 and R.
Results
Frequencies
Patient characteristics are presented in Table 1. Ninety-two percent of the subjects in the control and treated groups of the PEACE trial were Caucasians. The mean age of the 2243 successfully genotyped participants was 64 ± 8 years, female (18%), frequently smoked cigarettes (14%), and had a history of transient ischemic attack (TIA) (3%), stroke (4%), intermittent claudication (8%), diabetes (15%), and CABG (36%). The majority of patients did have a history of angina (71%) and angiographic coronary disease (66%). These patient characteristics were similar to the overall PEACE study population (24). This subset study that represents 2243 out of the total 8290 patients in the overall PEACE study population (27% of total) had similar clinical characteristics as the PEACE trial with exception of the incidence of hypertension (current trial 25 vs. 45% in PEACE), history of PTCA (current trial 43 vs. 72% in PEACE), and history of MI (current trial 42 vs. 55% in PEACE) (24).
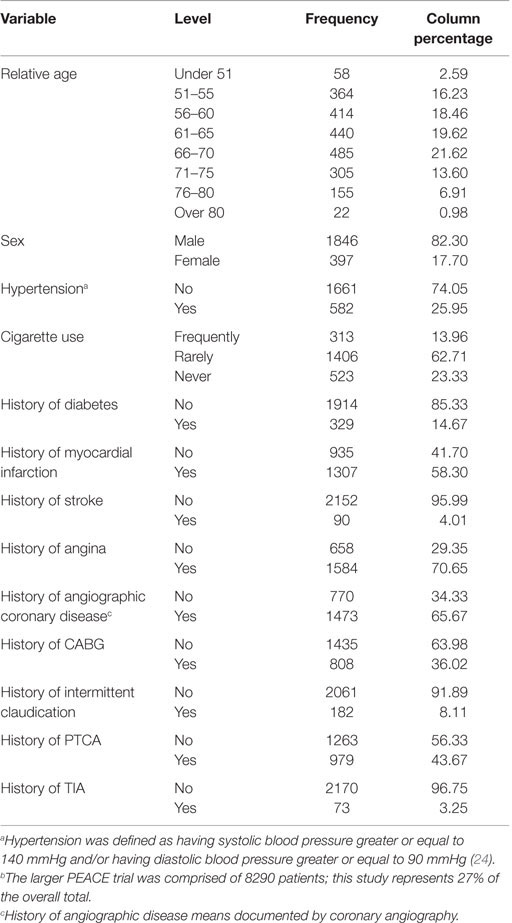
Table 1. Individual characteristics of the studied population (N = 2243 from the PEACE trialb).
Allele and genotype frequencies are presented in Table 2 for the candidate SNPs in the PRCP and KLKB1 genes. Across all four SNPs, the frequency of undetermined genotypes ranged from 5.1 to 8.4%. For rs7104980 in the PRCP gene, the less common allele was G (44.7%) and the rarer genotype was the G homozygote (G/G) (29%). For rs2298668 in the PRCP gene, the less common allele was the G allele (8.3%) and the rarer genotype was the G homozygote (G/G) (5.8%). For rs3087505 in the KLKB1 gene, the less common allele was the A allele (10.29%) and the rarer genotype was the A homozygote (A/A) (3.3%). For rs3733402 in the KLKB1 gene, the less common allele was the G allele (48.1%) and the rarer genotype was G/G (31.4%).
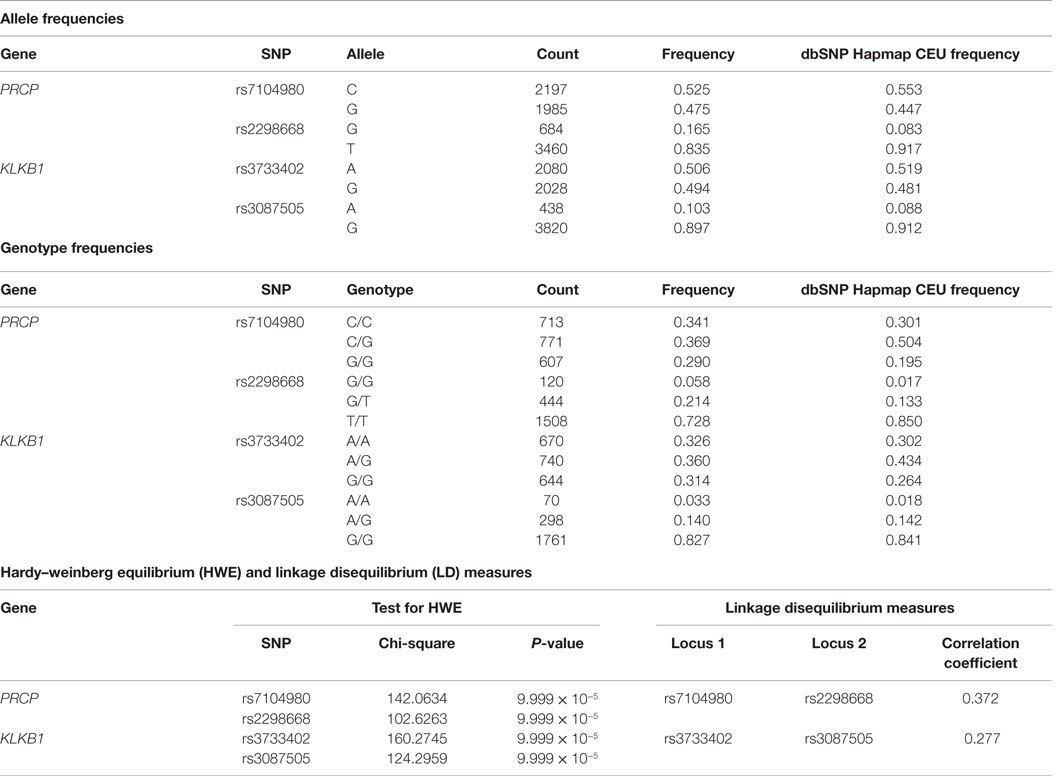
Table 2. Allele and genotype frequencies for the PRCP and KLKB1 SNPs.
Assessment of Hardy–Weinberg Equilibrium and Linkage Disequilibrium
The Hardy–Weinberg chi-square values for each SNP (HWE), as well as the LD correlation coefficients between each SNP per gene, were computed. Low P-values associated with the chi-square statistic for the HWE tests were observed that were the result of a large sample size, suggesting that the SNPs were in violation of HWE. This finding was due to having a statistical power large enough to detect small differences in this study that were not truly biologically meaningful. Hence, no SNPs were removed. In addition, both the allele and genotype frequencies for all tested SNPs were very similar to those from the dbSNP Hapmap CEU frequencies (Table 2). There were no SNPs in significant LD. The correlation coefficient between the two PRCP SNPs was 0.372, and the correlation coefficient between the two KLKB1 SNPs was 0.277, which were both smaller than the cutoff value of 0.8 for assessing significant LD. In a previous study examining SNPs associated with venous thrombosis, KLKB1 SNP rs3087505 was found to be in LD with two F11 SNPs rs2036914 and rs3756008 (1). We determined that KLKB1 SNP rs3733402 is not in LD with F11 SNPs rs2036914 and rs3756008, although the two F11 SNPs are in LD with each other (Figure 1).
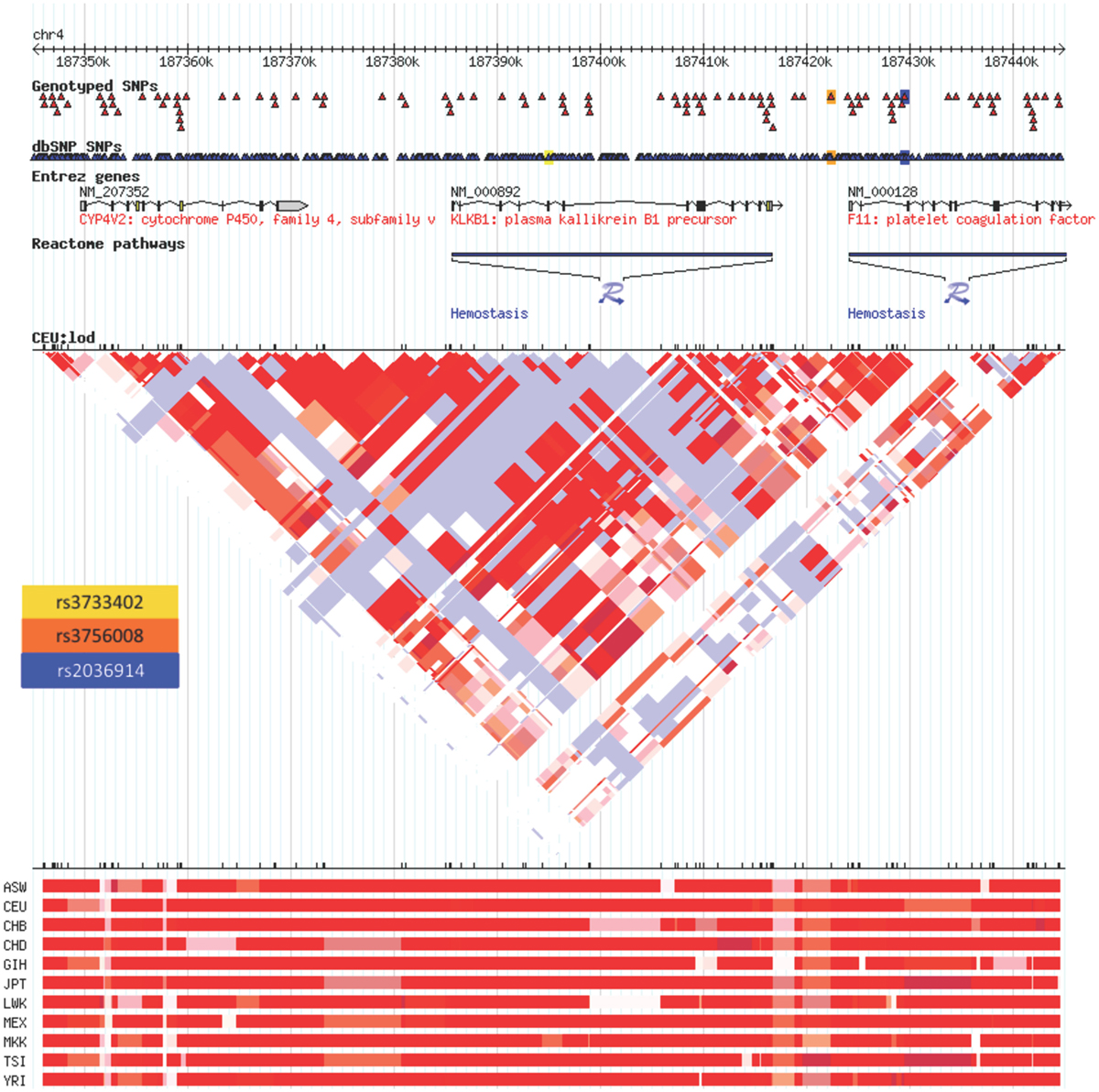
Figure 1. Linkage disequilibrium heatmap for rs3733402 vs. F11 SNPs. KLKB1 SNP rs3733402 is not in LD with F11 SNPs rs2036914 and rs3756008, although the two F11 SNPs are in LD with each other. The LD heatmap is gray between the KLKB1 and F11 genes and red within the F11 gene.
Logistic Regression Models and Model Selection
Initially, logistic regression models were run for various CVD-associated outcomes, including history of diabetes, MI, stroke, angina, angiographic coronary disease, CABG, intermittent claudication, PTCA, and TIA. The “baseline” model controlled for patient age, weight, and gender (Table 3). No associations were found for the PRCP rs2298668 and KLKB1 rs3087505 SNPs. Having the rare G allele compared to not having the G allele for rs7104980 (PRCP gene) increased the odds of having a history of PTCA by 21% [OR = 1.211; 95% CI = (1.008, 1.454); P = 0.041]. Alternatively, having the rarer G allele compared to not having the G allele for rs3733402 (KLKB1 gene) decreased the odds of having a history of angiographic coronary disease by 24% [OR = 0.759; 95% CI = (0.622, 0.927); P = 0.007]. After correcting for testing multiple SNPs by the Bonferroni correction method, only SNP rs3733402 (KLKB1 gene) was significantly associated with an outcome of having a history of angiographic coronary disease (adjusted P < 0.0125) (Table 3).
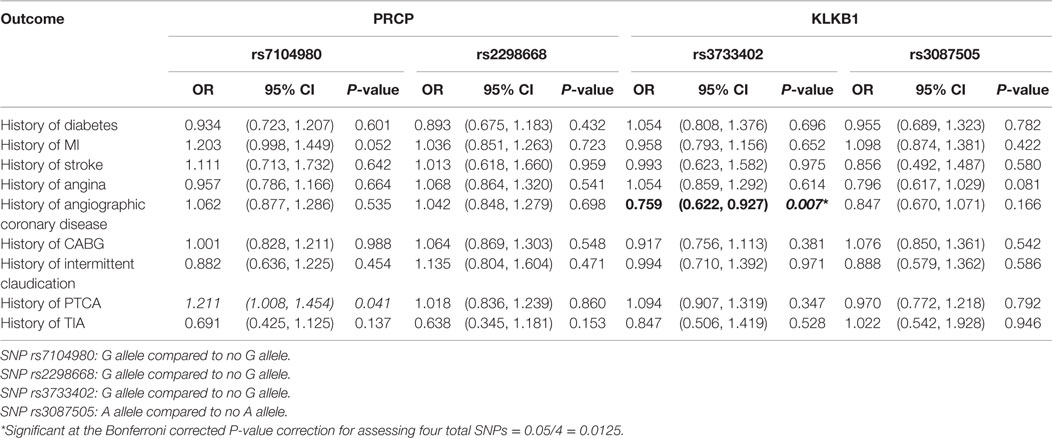
Table 3. Association of cardiovascular disease outcomes with SNPs in the PRCP and KLKB1 genes controlling for age, weight, and gender.
In order to better understand the potential association between having the G alleles for rs7104980 and rs3733402 and CVD, we calculated the −2 log-likelihood values in order to calculate the AIC to determine the best-fitting model (25). As shown in Table 4, the final model chosen for rs7104980, which had the lowest AIC, controlled for the baseline covariates (age, gender, and weight), hypertension, and history of angina. Having the G allele compared to not having the G allele for rs7014980 (PRCP gene) increased the odds of having hypertension and a history of angina by 22% [OR = 1.220; 95% CI = (1.016, 1.467)]; P = 0.034, but this analysis was not statistically significant (P > 0.01) after Bonferroni correction. Alternatively, having the G allele compared to not having the G allele for rs3733402 (KLKB1 gene) decreased the odds of the baseline covariates, hypertension, and history of angina by 24% [OR = 0.762; 95% CI = (0.623, 0.931); P = 0.008], which was statistically significant (P ≤ 0.01) for multiple testing after Bonferroni correction.
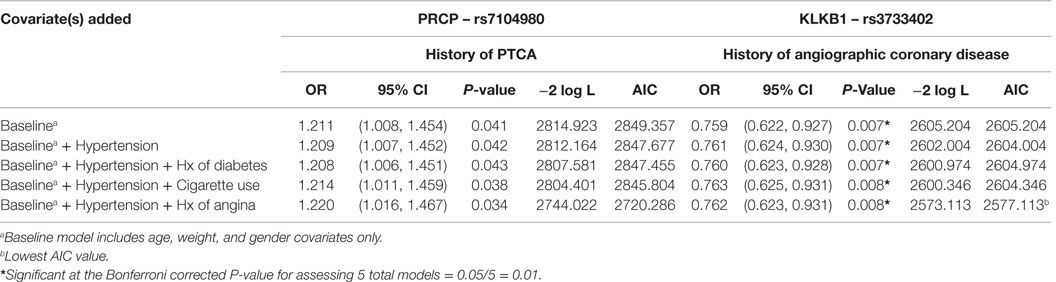
Table 4. Model selection for the best fit for the association between SNP rs7104980 (PRCP gene) and SNP rs3733402 (KLKB1 gene) with hypertension and a history of diabetes, cigarette use, or angina.
Discussion
The present cross-sectional study suggests that a polymorphism in KLKB1 associated with reduced PK binding to HK leading to a less actable zymogen is associated with reduced CVD. However, we are cautious in over interpreting the results of this investigation because it has several limitations. The PEACE trial was performed on low-risk individuals with CVD who had stable coronary artery disease and normal or slightly reduced left ventricular function. The patient population was 82% male with the majority of patients between 51 and 70 years of age with only 14% frequent smokers. The majority of patients did not have TIA, stroke, claudication, diabetes, hypertension, CABG, or PTCA. The present subset analysis of samples from the PEACE trial was similar in most ways, but actually had subjects with significantly reduced incidence of hypertension, PTCA, and MI. Since these three indicators are important disease markers of CVD, their reduction may have influenced the outcomes of the present studies. Overall, the present study examined a healthier subset of individuals with lower risk CVD than in the original PEACE trial itself.
Although there were no associations with the non-coding SNP associated with venous thrombosis, it is of interest that the KLKB1 exonic SNP, rs3733402 (N124S), was shown to be protective in the studied subjects for having a history of angiographic coronary artery disease (Table 3). Rs3733402 is not in LD with two F11 SNPs associated with venous thrombosis even though KLKB1 SNP rs3087505 from the 3′prime UT region is (1). The meaning of SNPs of different genes being in LD and associated with one disorder, such as venous thrombosis, is not known. The N124S polymorphism is in the Apple domain 2 of PK where HK binds (3, 26). Since the majority of plasma PK circulates in complex with HK and HK is a PK receptor on endothelial cells, the polymorphism of rs3733402 may be functionally important in vivo (4, 5). Rs3733402, a common polymorphism, is associated with reduced PK binding to HK (3). The polymorphism alters a PK glycosylation site that reduces its ability to bind to HK (3, 26). PK’s ability to bind to HK is important to protect plasma kallikrein from inhibition by C1 inhibitor and for PK’s activation by PRCP on cells (5, 27). This polymorphism may promote C1 inhibition of plasma kallikrein and reduce PRCP activation of PK to plasma kallikrein, thus limiting plasma kallikrein activity in two ways in vivo (27, 28).
Previous studies by us have shown that elevated plasma PK is associated with accelerated diabetic vascular disease (8, 9). Elevations of plasma kallikrein may be associated with increased prorenin conversion to renin and renin formation leading to elevated angiotensin II. Recent genome-wide meta-analysis studies indicate that the KLKB1 SNP rs3733402 has a positive correlation with circulating B-type natriuretic peptide, a recognized cardioprotective vasodilator in African Americans (29). Other recent studies show that PK deficient mice (klkb1−/− mice) have reduced arterial thrombosis providing support in mouse models to enhance the observation made in human association studies (6–9). It is of interest that mechanism for delayed thrombosis in klkb1−/− mice is mediated in part through elevated prostacyclin and vasculo-protective transcription factors Sirt1 and KLF4 with reduction of vessel wall tissue factor and not reduced contact activation as is commonly believed (7). The results of the present investigation showing that rs3733402 is associated with decreased CVD are consistent with these other studies (8, 9). Combined these investigations suggest that lowered PK may improve vascular health indirectly by reducing vessel thrombosis risk.
In the present investigation, it might be construed that significance of rs3733402 with a history of angiographic coronary disease is not consistent with the results observed for intermittent claudication, PTCA, and angina (Table 3). The lack of association with intermittent claudication is not surprising because there was a very small group of individuals in the study population with this clinical problem. In a similar manner, one may argue that the population of study subjects with PTCA was also a smaller group than the angiographic coronary disease group. When a logistic regression study was performed with a baseline model, including age, weight, gender, hypertension, and history of angina, history of angina did contribute to the best-fitting model (25) (Table 4).
The studies on PRCP SNPs were performed because PK is a substrate of PRCP. Furthermore, investigations show that an absence of PRCP in gene-trap mice is associated with CVD manifested as vascular inflammation, including increased vessel leukocytosis, reactive oxygen species, and apoptosis with reduced cellular proliferation, constitutive anticoagulation, and vessel angiogenesis and repair after mechanical and ischemia/reperfusion injury (21, 23). The current study suggests that the G allele of rs7104980 of PRCP may be a cardiovascular risk factor for PTCA, controlling for age, weight, and gender (P < 0.041), but the Bonferroni correction of the P-value for the total number of SNPs (P < 0.0125) shows non-significance (Table 3). Likewise the use of the AIC to choose the best-fitting result did not identify additional subsets such as hypertension or history of angina that increased the significance of this association (Table 4). In this population of patient with less severe CVD, we did not observe PRCP SNPs as risk factors. Our investigations were consistent with the investigations of Wu et al. who showed that the G allele of SNP rs710980 was associated with susceptibility for essential hypertension in 1020 Han Chinese (20). However, our present study has results that do not meet the significance level set by our investigations’ parameters. The implication of these data may be that polymorphisms in PRCP may be significant in patients with more serious hypertensive CVD than those investigated in the present study.
Furthermore, our studies like others did not show any significant association with the rs2298668 PRCP polymorphism (E112D PRCP) that was associated with chronic hypertension with or without increased risk of preeclampsia or resistance to an ACE-inhibitor therapy for hypertension (18, 19). First, the studies of Wang et al. and Zhang et al. were with patients who had more advanced hypertension and signs of deteriorating cardiovascular health than the subjects examined in the present report (18, 19). This difference also accounts for the different results with SNP rs2298668 between these two studies and the one of Wu et al. (18–20). The investigation of Wu et al. used subjects who just had essential hypertension without history of diabetes mellitus, MI, cerebrovascular accident, or other serious diseases (20). Finally, since previous studies with SNPs rs7104980 and rs2298668 were performed on Asian and African-Americans subjects, respectively, the non-statistically significant results observed in the present study also may be related to the mostly male Caucasian population examined.
When we performed this investigation, we were concerned whether we were able to get adequate DNA from stored plasma samples. We avoided serum samples for DNA preparation in this investigation since it has been associated with degraded DNA (30). We reasoned that well-collected plasma samples properly stored preserves soluble DNA for adequate preparation for assay like it does for study of blood coagulation zymogens. Our studies show that the genotyping error rate of 5–8.4% in the DNA prepared from subject plasma samples is less than that of 6.2–13% error rate in control DNA prepared from tissue across the SNPs.
Another caution in interpreting these findings is that this investigation is based only on a single cross-sectional study of samples. It needs to be confirmed by similar investigations on additional sample sets. Furthermore, the results of this cross-sectional study should be confirmed in a case-controlled study to obviate any unintended selection bias.
In sum, we postulate that the G allele of a KLKB1 SNP, rs3733402, is associated with reduced hypertension and coronary artery disease. These data suggest that reduction of PK activity may reduce CVD consistent with our previous studies in diabetic patients (8, 9). Our current findings also are consistent with our murine models. In our animal models, PK and PRCP may have opposing activities in the cardiovascular system regulating our risks for arterial disease. Reduction of PK or elevation of PRCP may be future targets to promote vascular health to reduce CVD.
Conclusion
This paper is the first cross-sectional investigation on two KLKB1 SNPs on a group of patients with mild CVD. When subject age, male gender, and weight are taken into account, KLKB1 SNP rs3733402 significantly associates with those with a reduced history of angiographic coronary artery disease. When subjects have covariates of age, weight, gender, hypertension, rs3733402 best associates with those individuals who both have a reduced history of angina. Rs3733402, an exonic SNP that is associated with reduced PK activation, also is associated with reduced CVD.
Author Contributions
JB-S and AS designed the project. AM, OA, MV, and ES performed experimental work. HG, JB-S, MV, and AS performed data analysis. HG, JB-S, MV, ES, and AS performed manuscript drafting and editing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by NIH HL052779-17 and HL112666-02 to AS and NIH/NCI 2P30 CA043703-23 to JB-S and HG. NIH/NCI P30 CA43703 also supports the Gene Expression and Genotyping Facility of the Case Comprehensive Cancer Center.
Abbreviations
AIC, akaike information criterion; CVD, cardiovascular disease; KLKB1, plasma prekallikrein gene; PEACE, prevention of Events with Angiotensin Converting Enzyme Inhibition Trial; PK, plasma prekallikrein protein; PRCP, prolylcarboxypeptidase gene; PRCP, prolylcarboxypeptidase protein.
References
1. Austin H, De Staercke C, Lally C, Bezemer ID, Rosendaal FR, Hooper WC. New gene variants associated with venous thrombosis: a replication study in White and Black Americans. J Thromb Haemost (2011) 9:489–95. doi: 10.1111/j.1538-7836.2011.04185.x
2. Heit JA, Cunningham JM, Petterson TM, Armasu SM, Rider DN, De Andrade M. Genetic variation within the anticoagulant, procoagulant, fibrinolytic and innate immunity pathways as risk factors for venous thromboembolism. J Thromb Haemost (2011) 9:1133–42. doi:10.1111/j.1538-7836.2011.04272.x
3. Katsuda I, Maruyama F, Ezaki K, Sawamura T, Ichihara Y. A new type of plasma prekallikein deficiency associated with homozygosity for Gly104Arg and Asn124Ser in apple domain 2 of the heavy-chain region. Eur J Haematol (2007) 79:59–68. doi:10.1111/j.1600-0609.2007.00871.x
4. Scott CF, Colman RW. Function and immunochemistry of prekallikrein-high molecular weight kininogen complex in plasma. J Clin Invest (1980) 65:413–21. doi:10.1172/JCI109684
5. Motta G, Rojkjaer R, Hasan AAK, Cines DB, Schmaier AH. High molecular weight kininogen regulates prekallikrein assembly activation on endothelial cells: a novel mechanism for contact activation. Blood (1998) 91:516–28.
6. Bird JE, Smith PL, Wang X, Schumacher WA, Barbera F, Revelli JP, et al. Effects of plasma kallikrein deficiency on haemostasis and thrombosis in mice: murine ortholog of the Fletcher trait. Thromb Haemost (2012) 107:1141–50. doi:10.1160/TH-11-10-0682
7. Stavrou EX, Fang C, Merkulova A, Alhalabi O, Grobe N, Antoniak S, et al. Reduced thrombosis in KLKB1-/- mice is mediated by increased Mas receptor, prostacyclin, Sirt1 and KLF4 and decreased tissue factor. Blood (2015) 125:710–9. doi:10.1182/blood-2014-01-550285
8. Jaffa AA, Durazo-Arvizu R, Zheng D, Lackland DT, Srikanth S, Garvey WT, et al. A risk factor for hypertension and nephropathy in type 1 diabetes. Diabetes (2003) 52:1215–21. doi:10.2337/diabetes.52.5.1215
9. Jaffa MA, Luttrell D, Schmaier AH, Klein RL, Lopes-Virella M, Luttrell LM, et al. Plasma prekallikrein is associated with carotid intima-media throickness in typoe 1 diabetes. Diabetes (2016) 65:498–502. doi:10.2337/db15-0930
10. Erdos EG, Yang HY. An enzyme in microsomal fraction of kidney that inactivates bradykinin. Life Sci (1967) 6:569–74. doi:10.1016/0024-3205(67)90090-2
11. Odya CE, Marcinkovic DV, Hammon KJ, Stewart TA, Erdos EG. Purification and properties of prolylcarboxypeptidase (angiotensinase C) from human kidney. J Biol Chem (1978) 253:5927–31.
12. Wallingford N, Perroud B, Gao Q, Coppola A, Gao X-B, Diament A, et al. Prolylcarboxypeptidase influences food intake by promoting breakdown of α-MSH. J Clin Invest (2009) 119:2291–302. doi:10.1172/JCI37209
13. Shariat-Madar Z, Mahdi F, Schmaier AH. Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J Biol Chem (2002) 277:17962–9. doi:10.1074/jbc.M106101200
14. Shariat-Madar Z, Mahdi F, Schmaier AH. Recombinant prolylcarboxypeptidase activates plasma prekallikrein. Blood (2004) 103:4554–61. doi:10.1182/blood-2003-07-2510
15. Shariat-Madar Z, Rahimi E, Mahdi F, Schmaier AH. Over-expression of prolylcarboxypeptidase enhances plasma prekallikrein activation on Chinese hamster ovary cell. Am J Physiol Heart Circ Physiol (2005) 289:H2697–703. doi:10.1152/ajpheart.00715.2005
16. Watson B Jr, Nowak NJ, Myracle AD, Shows TB, Warnock DG. The human angiotensinase C gene (HUMPCP) maps to 11q14 within 700 kb of D11S901: a candidate gene for essential hypertension. Genomics (1997) 15:365–7. doi:10.1006/geno.1997.4883
17. McCarthy JJ, Meyer J, Moliterno DJ, Newby LK, Rogers WJ, Topol EJ, et al. Evidence for substrantial effect modification by gender in a large scale genetic association study of the metabolic syndrome among coronary heart disease patients. Hum Genet (2003) 114:87–98. doi:10.1007/s00439-003-1026-1
18. Wang L, Feng Y, Zhang Y, Zhou H, Jiang S, Niu T, et al. Prolylcarboxypeptidase gene, chronic hypertension, and risk of preeclampsia. Am J Obstet Gynecol (2006) 195:162–71. doi:10.1016/j.ajog.2006.01.079
19. Zhang Y, Hong X-M, Xing H-X, Li J-P, Huo Y, Xu X-P. E112D polymorphism in the prolylcarboxypeptidase gene is associated with blood pressure response to benazepril in Chinese hypertensive patients. Chin Med J (2009) 122:2461–5.
20. Wu Y, Yang H, Yang B, Yang K, Xiao C. Association of polymorphisms in prolylcarboxypeptidase and chymase genes with essential hypertension in the Chinese Han population. J Renin Angiotensin Aldosterone Syst (2013) 14:263–70. doi:10.1177/1470320312448949
21. Adams GN, LaRusch GA, Stavrou E, Zhou Y, Nieman MT, Jacobs GH, et al. Murine prolylcarboxypeptidase depletion induces vascular dysfunction with hypertension and faster arterial thrombosis. Blood (2011) 117:3929–37. doi:10.1182/blood-2010-11-318527
22. Marangoni RA, Santos RA, Piccolo C. Deficient prolylcarboxypeptidase gene and protein expression in left ventricles of spontaneously hypertensive rats (SHR). Peptides (2014) 61:69–74. doi:10.1016/j.peptides.2014.08.016
23. Adams GN, Stavrou EX, Fang C, Merkulov A, Alaiti AM, Nakajima K, et al. Prolylcarboxypeptidase promotes angiogenesis and vascular repair. Blood (2013) 122:1522–31. doi:10.1182/blood-2012-10-460360
24. Braunwald E, Domanski MJ, Fowler SE, Geller NL, Gersh BJ, Hsia J, et al. Angiotensin-converting-enzyme inhibition in stable coronary artery disease. N Engl J Med (2004) 351:2058–68. doi:10.1056/NEJMoa042739
25. Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr (1974) AC-19(6):716–23. doi:10.1109/TAC.1974.1100705
26. Girolami A, Vidal J, Salagh M, Gervan N, Parody M, Peroni E, et al. The old and the new in prekallikrein deficiency: historical context and a family from Argentina with PK deficiency due to a new mutation (Arg541Gln) in exon 14 associated with a common polymorphism (Asn124Ser) in exon 5. Semin Thromb Hemost (2014) 40:592–9. doi:10.1055/s-0034-1384767
27. Schapira M, Scott CF, Colman RW. Protection of human plasma kallikrein from inactivation by C1 inhibitor and other protease inhibitors. The role of high molecular weight kininogen. Biochemistry (1981) 20:2738–43. doi:10.1021/bi00513a006
28. Wang J, Matafonov A, Madkhali H, Mahdi F, Watson D, Schmaier AH, et al. Prolylcarboxypeptidase independently activates plasma prekallikrein (Fletcher Factor). Curr Mol Med (2014) 14:1173–85. doi:10.2174/1566524014666141015153519
29. Musani SK, Fox ER, Kraja A, Bidulescu A, Lieb W, Lin H, et al. Genome-wide association analysis of plasma B-type natriuretic peptide in Blacks. The Jackson Heart Study. Circ Cardiovasc Genet (2015) 8:122–30. doi:10.1161/CIRCGENETICS.114.000900
Keywords: cardiovascular disease, prekallikrein, KLKB1, prolylcarboxypeptidase, PRCP, high molecular weight kininogen
Citation: Gittleman HR, Merkulova A, Alhalabi O, Stavrou EX, Veigl ML, Barnholtz-Sloan JS and Schmaier AH (2016) A Cross-sectional Study of KLKB1 and PRCP Polymorphisms in Patient Samples with Cardiovascular Disease. Front. Med. 3:17. doi: 10.3389/fmed.2016.00017
Received: 08 March 2016; Accepted: 14 April 2016;
Published: 29 April 2016
Edited by:
Jorge Di Paola, University of Colorado School of Medicine, USAReviewed by:
Owen McCarty, Oregon Health & Science University, USAMatthew Thomas Rondina, University of Utah, USA
Copyright: © 2016 Gittleman, Merkulova, Alhalabi, Stavrou, Veigl, Barnholtz-Sloan and Schmaier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alvin H. Schmaier, schmaier@case.edu
†Haley R. Gittleman, Alona Merkulova, and Omar Alhalabi contributed equally to this work.