Corrigendum: Chlamydia pneumoniae CdsL regulates CdsN ATPase activity, and disruption with a peptide mimetic prevents bacterial invasion
 Chris B. Stone1 David C. Bulir1 Connor A. Emdin1 Ryan M. Pirie1 Elisa A. Porfilio1 Jerry W. Slootstra2 James B. Mahony1*
Chris B. Stone1 David C. Bulir1 Connor A. Emdin1 Ryan M. Pirie1 Elisa A. Porfilio1 Jerry W. Slootstra2 James B. Mahony1*
- 1 Michael G. DeGroote Institute for Infectious Disease Research, Faculty of Health Sciences, Department of Pathology and Molecular Medicine, McMaster University, Father Sean O’Sullivan Research Centre, St. Joseph’s Healthcare, Hamilton, ON, Canada
- 2 PepScan Presto, Epitope Mapping, Lelystad, Netherlands
Chlamydiae are obligate intracellular pathogens that likely require type III secretion (T3S) to invade cells and replicate intracellularly within a cytoplasmic vacuole called an inclusion body. Chlamydia pneumoniae possess a YscL ortholog, CdsL, that has been shown to interact with the T3S ATPase (CdsN). In this report we demonstrate that CdsL down-regulates CdsN enzymatic activity in a dose-dependent manner. Using Pepscan epitope mapping we identified two separate binding domains to which CdsL binds viz. CdsN221–229 and CdsN265–270. We confirmed the binding domains using a pull-down assay and showed that GST–CdsN221–270, which encompasses these peptides, co-purified with His–CdsL. Next, we used orthology modeling based on the crystal structure of a T3S ATPase ortholog from Escherichia coli, EscN, to map the binding domains on the predicted 3D structure of CdsN. The CdsL binding domains mapped to the catalytic domain of the ATPase, one in the central channel of the ATPase hexamer and one on the outer face. Since peptide mimetics have been used to disrupt essential protein interactions of the chlamydial T3S system and inhibit T3S-mediated invasion of HeLa cells, we hypothesized that if CdsL–CdsN binding is essential for regulating T3S then a CdsN peptide mimetic could be used to potentially block T3S and chlamydial invasion. Treatment of elementary body with a CdsN peptide mimetic inhibited C. pneumoniae invasion into HeLa cells in a dose-dependent fashion. This report represents the first use of Pepscan technology to identify binding domains for specific T3S proteins viz. CdsL on the ATPase, CdsN, and demonstrates that peptide mimetics can be used as anti-virulence factors to block bacterial invasion.
Introduction
Chlamydia pneumoniae is an obligate, intracellular Gram-negative bacteria that has been associated with community acquired pneumonia (Clifton et al., 2004), atherosclerosis (Grayston, 2000), arthritis (Ardeniz et al., 2005), and Alzheimer’s disease (Balin et al., 2008). The members of the genus Chlamydia all share a unique, biphasic life-cycle that is initiated by attachment of the metabolically quiescent elementary body (EB) to the host cell. The association between the EB and the host cell membrane is poorly understood, but glycosaminoglycans may be involved (Chen and Stephens, 1997). Once attached to the host cell, type III secretion (T3S) is utilized to inject the translocated actin recruitment protein (TARP) to facilitate bacterial internalization into a plasma-membrane derived vacuole, termed an inclusion (Clifton et al., 2004; Lane et al., 2008). The MEK–ERK and PI 3-kinase pathways of the host cell are also involved in bacterial uptake, and are possibly targets of other T3S effectors translocated across the cytoplasmic membrane upon EB contact and binding to host cells (Coombes and Mahony, 2002; Carabeo et al., 2004; Subtil et al., 2004). Once inside the host cell the remainder of the life-cycle occurs inside the inclusion body where EBs differentiate into the metabolically active, non-infectious reticulate bodies (RB). The RB remains in close association with the inclusion membrane, suggesting a need for active T3S in RBs. This interaction with the inclusion membrane allows the RB to communicate with the host cell via T3S, allowing the Chlamydia to commandeer host cell pathways to obtain lipids, cholesterol, and other nutrients crucial for its growth and replication, and also to prevent phagosome endosome fusion (Wylie et al., 1997; Hoare et al., 2008; Scidmore and Hackstadt, 2008). Following inclusion body growth to accommodate replicating RBs, an unknown signal (possibly quorum sensing) triggers the detachment of RBs from the membrane and subsequent re-assortment into infectious EBs. The EBs then exit the host cell either by cell lysis or through extrusion, a packaged release mechanism which leaves the host cell intact (Hybiske and Stephens, 2007).
Type III secretion is a virulence mechanism commonly used by Gram-negative bacteria to directly translocate effector proteins from the bacterial cytoplasm to the host cell cytoplasm in a single step, through the use of a syringe-like apparatus termed an injectisome (Galan and Collmer, 1999; Ghosh, 2004; Galan and Wolf-Watz, 2006). The injectisome is constructed of 20–25 proteins spanning the inner membrane, periplasm, and outer membrane, extending into the extracellular milieu to allow for host cell sensing and contact. Upon host cell contact, the T3S injectisome apparatus injects two translocator proteins into the host cell membrane to form the translocon, a molecular pore through which secreted proteins can enter the host cell (Goure et al., 2004). Despite the fact that Chlamydia possesses a full repertoire of T3S genes scattered throughout the genome on at least 10 distinct operons, a systematic study of the injectisome has yet to be undertaken as chlamydial species are genetically intractable (Hefty and Stephens, 2007). Recent reports have identified a few T3S structural and effector proteins, providing some understanding of the Chlamydia T3SS. Our laboratory has shown that CdsD, a unique protein orthologs to YscD that contains two fork-head associated domains, interacts with the predicted C. pneumoniae ATPase tethering protein, CdsL, as well as CdsQ, a putative multi-cargo transport protein (Johnson et al., 2008; Spaeth et al., 2009). We extended these findings to show that CdsN also binds to this complex, and interacts independently with CdsD, CdsL, and CdsQ (Stone et al., 2008). Betts et al. (2008) have also recently identified the C. trachomatis CT666 gene encoding CdsF, the needle filament protein of the injectisome. The role of a few effector proteins of C. pneumoniae have been recently discovered. Cpn0585 is found in the inclusion membrane and interacts with Rab 1, 10, and 11, possibly recruiting these ATPases to the inclusion membrane (Cortes et al., 2007). Cpn0827 is a second effector protein that is detectable in the inclusion membrane at 20 hpi (Hermann et al., 2006). The Chlamydia protein associating with death domains (CADD) is a conserved effector known to interact with TNF family receptors (Stenner-Liewen et al., 2002). Recent biophysical evidence has also been presented by Markham et al. (2009) suggesting that the C. trachomatis protein CT584 could function as the needle tip protein, which is imperative for host cell sensing and translocator insertion into the host cell membrane.
Type III secretion ATPases are believed to be assembled as a hexameric ring at the basal body of the injectisome and play a role in delivery of effector proteins through the injectisome (Zarivach et al., 2007). Several T3S ATPases have been partially characterized, including EscN from Escherichia coli (Andrade et al., 2007), YscN from Yersinia (Blaylock et al., 2006), and InvC from Salmonella (Akeda and Galan, 2004), although the ability of the T3S ATPase to coordinate numerous protein interactions has not been explored. These ATPases have been shown to have significant sequence orthology to the β subunit of the F0F1 ATPase, and to hydrolyze ATP. Not only are these ATPases important for providing energy for protein transport, but they are believed to play a role in unfolding the effector proteins before translocation, which may be accomplished by releasing the chaperone from its cognate effector protein (Akeda and Galan, 2005). In Yersinia, the ATPase tethering protein YscL localizes the ATPase to the inner membrane, potentially by performing a function similar to that of the gamma-stalk of the F0F1 ATPase (Pallen et al., 2006). YscL has also been shown to play a role in regulation of the ATPase activity by down-regulating enzymatic activity (Blaylock et al., 2006). The quaternary structure of ATPase, viz. the hexamer, has been associated with enhanced enzymatic activity and most likely reflects the native conformation of this protein.
In this report we show that CdsL down-regulates the enzymatic activity of CdsN. We utilized Pepscan epitope mapping to determine where CdsL binds to CdsN, and used an orthology modeling approach to map this domain onto the predicted 3D structure of CdsN (Bernard et al., 2004; Teeling et al., 2006; Timmerman et al., 2007). We found that CdsN possesses two distinct binding domains for CdsL, one to potentially mediate the tethering function and the other to mediate the down-regulation function. We also show that a CdsN peptide acts as a peptide mimetic preventing bacterial invasion. This is the first report to identify binding domains for a T3S ATPase regulating protein and the use of a peptide mimetic as a novel antimicrobial agent to potentially target T3S.
Materials and Methods
Expression Plasmids
Chlamydia pneumoniae CWL029 (VR1310:ATCC; GenBank accession No. AE001363) was the strain used to isolate genomic DNA for cloning and protein expression. Full length cdsL and cdsN were amplified from CWL029 using AttB-containing primers (Gateway; Invitrogen). The amplified products were cloned into pDONR201 (Gateway; Invitrogen) to generate pENT vectors. The pENT vectors were then used in LR reactions (Gateway; Invitrogen) to produce pEX vectors containing the genes of interest. We used either pEX17 (N-terminal His-tag) or pEX15 (N-terminal GST-tag) vectors for our protein expression. All constructs were confirmed by sequencing at the Molecular Biology Facility at McMaster University.
Protein Expression
All constructs were expressed in E. coli Rosetta pLysS. Expression plasmids were used to transform E. coli Rosetta pLysS and plated on LB plates containing 100 μg/mL ampicillin. LB broth (750 mL), containing antibiotics, was then inoculated with 12 mL of an overnight culture and grown at 37°C until they reached an optical density (OD)600 of approximately 0.8. Cultures were then cooled on ice to 20°C and induced with 0.2 mM of isopropyl β-D-thiogalactopyranoside (IPTG). Cultures were then incubated at 23°C for 2 h and bacteria were harvested by centrifugation at 6500×g for 10 min in a Sorvall RC-5B centrifuge and washed with ice-cold phosphate-buffered saline (PBS). Bacteria containing His-tagged protein were resuspended in either Binding Buffer (50 mM potassium phosphate pH 7.2, 150 mM KCl, 1 mM MgCl2) when used in GST pull-downs, or Nickel A buffer (20 mM Tris pH 7.0, 0.02% β-mercaptoethanol, 400 mM KCl, 1% Triton X-100) when used in ATPase activity assays, while the bacteria containing GST-tagged protein were resuspended in PBS and stored at −20°C until further use.
Purification of Recombinant Proteins
Escherichia coli pellets containing over-expressed His- or GST-tagged proteins were thawed on ice and sonicated using a Fischer Scientific Sonic Dismembrator Model 100, followed by centrifugation at 20,000×g for 40 min to remove insoluble material. Supernatants containing His-tagged protein for use in GST pull-down assays were stored at 4°C. GST-tagged protein for use in GST pull-down assays were bound to 300 μL of glutathione beads overnight at 4°C, then blocked overnight in Tris Buffered Saline with 0.1% Tween-20 and 4% BSA and stored at 4°C until use. His- and GST-tagged protein supernatants for use in ATPase activity assays were filtered through 0.45 μm acrodisc filters (Pall Corporation) and purified on either a 1-mL GSTrap FF column (GE Healthcare) or a 1-mL HisTrap HP column (GE Healthcare). For GST-tagged protein, columns were washed with PBS + 0.1% Tween until the flow-through had an OD280 of less than 0.005. GST-tagged protein was then eluted off the beads using 1.5 μg/μL reduced glutathione (Sigma) and dialyzed against activity buffer (50 mM Tris–HCl pH 7.0, 5 mM MgCl2, 10 mM KCl). Purity was confirmed using SDS–PAGE and Coomassie blue staining. For His-tagged proteins, columns were washed with increasing imidazole concentrations and eluted with 300 mM imidazole then dialyzed into activity buffer. Purity was confirmed using SDS–PAGE and Coomassie blue staining.
CdsN Enzymatic Activity Down-Regulation Experiments
ATP hydrolysis by GST–CdsN purified from GSTrap columns was measured using a malachite green assay (R&D Systems) which measures released inorganic phosphate from ATP. For all experiments, the specific activity was determined using the equation of a standard line generated using phosphate standard (R&D Systems). Reaction mixtures contained 150 ng of GST–CdsN, 40 μM ATP, 50 mM Tris–HCl pH 7.0, 5 mM MgCl2, and 10 mM KCl. The reaction mixture (1 mL) was incubated at 37°C for 1 h and 50 μL of the mixture was taken for inorganic phosphate determination at various time points. The reaction was stopped by the addition of 10 μL of Malachite Green Reagent A followed by 10 μL of Malachite Green Reagent B and incubated at room temperature for 1 min before an OD610 reading was taken, according to the manufacturer’s instructions. For the negative control, purified CdsN was digested for 10 min at 37°C using Proteinase K (Invitrogen). Also, as a negative control, another GST-tagged protein (CopN) that lacks ATPase activity was purified in the same manner and tested for activity. ATPase activity was expressed as nmol phosphate released min−1, and all experiments were performed in triplicate. His–CdsL was purified from Ni-NTA agarose beads and tested for any contaminating ATPase activity using the Malachite Green Assay. It was then added in varying amounts into the reaction mixture with CdsN prior to addition of ATP and incubated for 20 min on ice. The activity assay was then performed as described above.
GST Pull-Down Assays
To examine the interaction of CdsN221–270 with CdsL, GST pull-down assays were performed as described previously by Johnson et al. (2008), with the following modifications. Briefly, glutathione–agarose beads (30 μL) bound to 50 ng of GST-tagged CdsN221–270 protein was used in the assay. The beads were incubated overnight at 4°C with the E. coli lysate expressing the His-tagged proteins. The beads were collected by centrifugation and washed with 0.1% Triton X-100 and increasing concentrations of NaCl to eliminate spurious protein interactions. The presence of the GST- and His-tagged proteins were confirmed by both Coomassie blue and Western blot. All proteins were eluted from the glutathione beads and electrophoresed on an 11% SDS–PAGE gel before being probed for His-tagged protein. GST alone bound to glutathione beads was used as a negative control for the pull-down.
Peptide Library Synthesis, Screening Assays, and 3D Modeling
The linear and/or CLIPS peptides are synthesized based on the amino acid sequence of the target protein using standard Fmoc-chemistry and de-protected using trifluoric acid with scavengers. The constrained peptides are synthesized on chemical scaffolds in order to reconstruct conformational epitopes, using Chemically Linked Peptides on Scaffolds (CLIPS) technology (Timmerman et al., 2007). For example, the single looped peptides are synthesized containing a dicysteine, which was cyclized by treating with alpha, α′-dibromoxylene, and the size of the loop is varied by introducing cysteine residues at variable spacing. If other cysteines besides the newly introduced cysteines are present, they are replaced by alanine. The side-chains of the multiple cysteines in the peptides are coupled to CLIPS templates by reacting onto credit-card format polypropylene Pepscan cards (455 peptide formats/card) with a 0.5-mM solution of CLIPS template such as 1,3-bis (bromomethyl) benzene in ammonium bicarbonate (20 mM, pH 7.9)/acetonitrile [1:1(v/v)]. The cards are gently shaken in the solution for 30–60 min while completely covered in solution. Finally, the cards are washed extensively with excess of H2O and sonicated in disrupt-buffer containing 1% SDS/0.1% β-mercaptoethanol in PBS (pH 7.2) at 70°C for 30 min, followed by sonication in H2O for another 45 min. The binding of antibody or labeled protein/peptide to each peptide is tested in a Pepscan-based ELISA. The 455-well credit-card format polypropylene cards containing the covalently linked peptides are incubated with peptide solution for example consisting of 1 μg/mL diluted in blocking solution, for example 4% horse serum, 5% ovalbumin (w/v) in PBS/1% Tween. After washing, the peptides are incubated with a monoclonal mouse anti-His-tag antibody (1/1000, Novagen, 70796-3) and subsequently after washing with a rabbit-anti-mouse antibody peroxidase conjugate (1/1000, Southern Biotech, 6175-05) for 1 h at 25°C. After washing, the peroxidase substrate 2,2′-azino-di-3-ethylbenzthiazoline sulfonate (ABTS) and 2 μL of 3% H2O2 are added. After 1 h, the color development is measured. The color development is quantified with a charge coupled device (CCD)-camera and an image processing system. The raw-data are optical values obtained by a CCD-camera. The values mostly range from 0 to 3000, a log scale similar to 1–3 of a standard 96-well plate ELISA-reader. First the CCD-camera makes a picture of the card before peroxidase coloring and then again a picture after the peroxidase coloring. These two pictures are subtracted from each other which results in the data which is called raw-data. This is copied into the Peplab™ database. Then the values are copied to excel and this file is labeled as raw-data file. One follow-up manipulation is allowed. Sometimes a well contains an air-bubble resulting in a false-positive value, the cards are manually inspected, and any values caused by an air-bubble are scored as 0. 3D structure prediction was performed using 3D-JIGSAW (http://www.bmm.icnet.uk/∼3djigsaw/). Modeling was then performed using PYMOL software.
Peptide Design and Entry into EBs
Based on the Pepscan epitope mapping of CdsN, two domains were found that mediate the interaction between CdsN and CdsL. The smaller of the two domains (residues 265–279), corresponding to TRFARA, was chosen to create a peptide mimetic compound. We synthesized the TRFARA peptide with a N-terminal membrane transport sequence (MTS), YGRKKRRQRRR (Efthymiadis et al., 1998). We also extended the CdsN sequence flanking the TRFARA peptide to ensure the entire binding domain was included, and flanked the peptide with cysteine residues to assist with any disulfide bonding that may be required for binding. The final peptide utilized was the 28-amino acid sequence YGRKKRRQRRRCVVLMMDSVTRFARALC and was used in its linear form for inhibition experiments. Peptides were synthesized by Peptide 2.0.
GST–MTS Uptake by EBs
Chlamydial EBs were purified using a discontinuous gastrografin gradient, and resuspended in PBS. Either GST or GST–MTS was incubated at varying concentrations (0, 100, 500 μm) or for varying times (5, 30, or 60 min) with the purified EBs at 4°C. The EBs were then pelleted and washed three times with PBS, followed by trypsinization for 30 min to ensure that no protein was bound to the outside of the EB. EBs were then boiled in loading dye and probed for the presence of GST using Western blot.
Peptide Cytotoxicity
The cytotoxicity was performed as described by Johnson et al. (2009). The effect of the CdsN TRFARA peptide on HeLa cell viability was determined. Briefly, 50 or 100 μM of the CdsN peptide, control peptides, or the positive control (1% SDS plus 0.1% Triton X-100) were added to sub-confluent HeLa cells in six-well plates. At 1 h supernatants were harvested and tested for the presence of adenylate kinase using a cytotoxicity assay (Lonza ToxiLight® BioAssay, Rockland). The cytotoxicity assay was performed as per the manufacturer’s protocol. Briefly, supernatants from HeLa cell cultures incubated with the CdsN TRFARA peptide (in MEM containing cycloheximide) were tested for evidence of eukaryotic cell cytotoxicity. Aliquots (5 μL) of each supernatant were mixed with 25 μL of Adenylate Kinase Detection Reagent and samples were incubated at room temperature for 5 min. Relative light units (RLUs) were measured using a 20/20-n Single Tube Luminometer from Turner BioSystems (Sunnyvale). Assays were conducted in triplicate for each condition. Cell monolayers were washed with warm PBS. 0.75 mL of trypsin was added to each well, and 0.75 mL of MEM was added after complete trypsinization (trypsinization was monitored by light microscopy). Each sample was thoroughly resuspended and aliquoted into a plastic cuvette and the cell number immediately quantitated by determining the optical density at 800 nM using a spectrophotometer.
Electron Microscopy and Immunofluorescence
HeLa cells grown to 90% confluency in 6-well plates were infected with C. pneumoniae, pre-incubated with linear CdsN peptide for 30 min or with linear random peptide (MTS–MFAVNAQ) for 30 min at various concentrations, centrifuged for 45 min at room temperature, followed by incubation at 37°C for 1 h. The inoculum and peptide were removed and the medium replaced with MEM containing 1 μg/mL cycloheximide to inhibit host cell protein synthesis. Cells were incubated at 37°C for 72 h, collected by trypsinization and centrifugation at 1000 rpm for 10 min then fixed overnight at 4°C with 2% glutaraldehyde (v/v) in cacodylate buffer, 7.2. The cell pellets were post-fixed in 2% osmium tetroxide (v/v), embedded in Spurs medium, and thin sections were cut on a Reichert Ultracut E microtome. Sections were examined in a Jeol 1200 electron microscope at 80 kV and pictures were captured with an AMT digital camera. For immunofluorescent staining of inclusions, HeLa cells were infected as above using cell monolayers on glass cover slips in shell vials, fixed in methanol, and stained using the Pathfinder kit, which uses a FITC-conjugated major outer membrane protein (MOMP)-monoclonal antibody specific to Chlamydia, as per the manufacturer’s instructions.
Results
Regulation of CdsN by CdsL
Type III secretion ATPases are known to be enzymatically down-regulated by YscL orthologs (Blaylock et al., 2006). C. pneumoniae encodes a YscL ortholog, CdsL (Cpn0826), which led us to explore whether CdsL down-regulates CdsN in C. pneumoniae. We have previously confirmed that CdsN interacted with CdsL using a GST pull-down assay, showing that CdsL co-purified with CdsN in 500 mM NaCl (Stone et al., 2008). We also showed previously that a CdsN-GST fusion protein possessed enzymatic activity consistent with other T3S ATPases (Stone et al., 2008). We found that CdsN hydrolyzed ATP at a rate of 0.0734 ± 0.0053 nmol/min at the concentration used in this assay (150 ng total). Prior to testing the ability of CdsL to regulate CdsN activity, we confirmed that CdsL lacked any ATPase activity by assaying it alone in the malachite green assay to control for possible contamination and found that CdsL possessed no ATPase activity. To test for regulation of CdsN by CdsL we added increasing amounts of CdsL to the CdsN reaction mixture. We found that addition of CdsL reduced CdsN enzymatic activity at a 0.3 CdsL:1 CdsN molar ratio, but the effect was most dominant at a 2.1 CdsL:1 CdsN molar ratio of CdsL: CdsN, where enzymatic activity was reduced by 82% (Figure 1). Further increasing the CdsL concentration only marginally reduced activity. The addition of a control His-tagged protein (His-CopN, a T3S structural component) had no effect on CdsN activity. Also as a control, His–CdsL had no effect on the enzymatic activity of a second C. pneumoniae ATPase, GspE (data not shown). GspE is a type II secretion ATPase which possesses enzymatic activity.
FIGURE 1
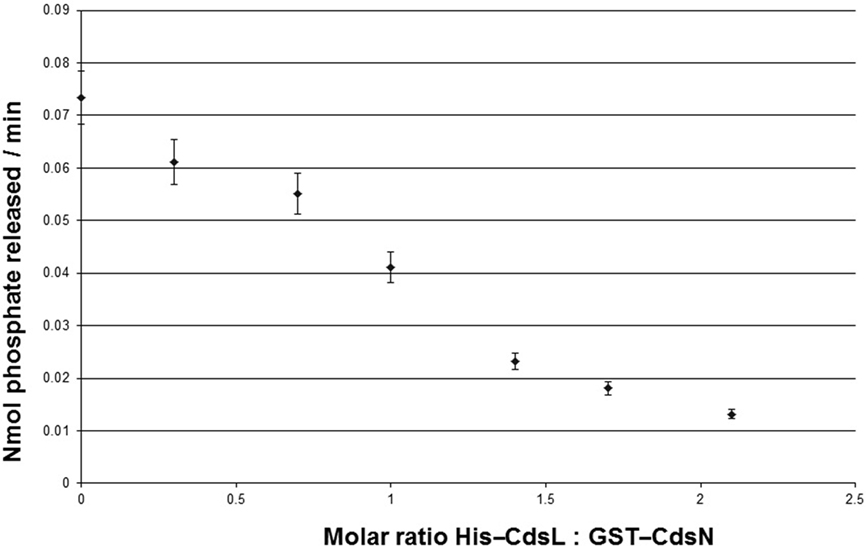
Figure 1. CdsL down-regulations CdsN enzymatic activity in a dose-dependent fashion. Aliquots (150 ng) of GST–CdsN were assayed for enzymatic activity using the malachite green assay to detect released inorganic phosphate from ATP. His–CdsL was added to the reaction mixture at molar ratios between 0.3 and 2.1, and ATPase activity of GST–CdsN was measured. Increasing amounts of CdsL resulted in a decrease in CdsN enzymatic activity. The maximum reduction of activity was seen at a 2.1: CdsL:1 CdsN molar ratio (82% reduction in CdsN enzymatic activity). All experiments were performed in triplicate and data is presented as the mean with error bars representing 1 SD of the mean.
Pepscan Analysis of CdsN–CdsL Interaction
We have shown that CdsN is enzymatically down-regulated by CdsL, but the mechanism by which this occurs is unknown. In an attempt to expand our understanding of how CdsL may function to down-regulate CdsN, we have utilized Pepscan epitope mapping to identify the domains within CdsN responsible for the CdsL interaction. Purified His–CdsL was added to an over-lapping peptide library consisting of 4000 over-lapping linear and conformational peptides of the CdsN protein in search for the peptide domain mediating protein binding (Timmerman et al., 2007). CdsN contained two binding regions that mediated the interaction with CdsL. The two CdsL binding sequences spanned residues 221–229, and residues 265–270 corresponding to the amino acids “RSVIVVSTS” and “TRFARA,” respectively, both of which are present within the catalytic domain (Figure 2). To corroborate the Pepscan data, we cloned and expressed a fragment of CdsN spanning residues 221–270 containing both of the CdsL predicted binding domains to test whether this CdsN fragment bound to His–CdsL. We found that the CdsN221–270 co-purified with His–CdsL in the presence of 500 mM NaCl while GST alone did not co-purify with CdsL under any salt conditions (Figure 3).
FIGURE 2
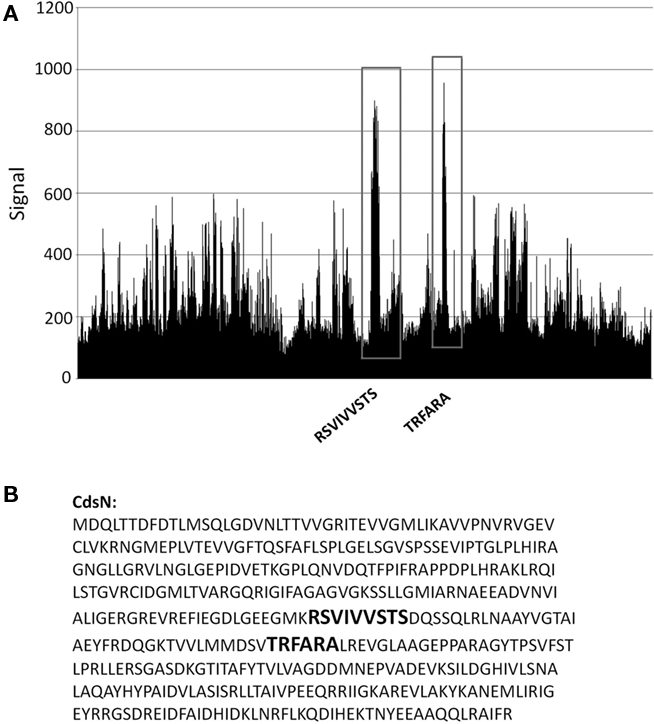
Figure 2. Pepscan analysis of the CdsL binding domain on CdsN. An over-lapping peptide library for full length CdsN consisting of 4000 linear and looped peptides was constructed by Pepscan Presto (The Netherlands). His–CdsL was screened against the peptide library to identify CdsN peptides that interact with CdsL. (A) Raw Pepscan data obtained by screening full length His–CdsL against the over-lapping peptide library of 4000 linear and looped CdsN peptides (x-axis). The two distinct CdsL binding domains are highlighted in black within the vertical boxes, and the signal strength of the interaction is visualized on the y-axis. The two peaks correspond to the sequences RSVIVVSTS and TRFARA. (B) The two amino acid sequences to which CdsL binds (RSVIVVSTS and TRFARA) are bolded within the full length CdsN amino acid sequence.
FIGURE 3
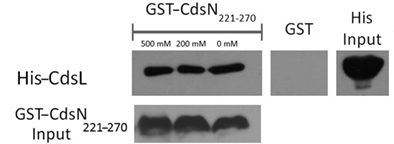
Figure 3. The CdsN221–270 peptide, containing both the predicted CdsN–CdsL binding domains, co-purifies with CdsL. GST–CdsN221–270 was bound to glutathione beads and used to pull-down His–CdsL from Escherichia coli lysates. Beads were collected by centrifugation, washed with 0, 200, and 500 mM NaCl and probed for His-tagged protein by Western blot using anti-His antibody. GST–CdsN221–270 co-purified with CdsL while GST alone did not.
3D Modeling of CdsN–CdsL Binding Domain
The crystal structure of the E. coli T3S ATPase, EscN, has recently been elucidated, allowing us to use an orthology modeling approach to obtain a predicted 3D structure of CdsN and to visualize the CdsL binding domains (Zarivach et al., 2007). CdsN and EscN possess a large amount of sequence orthology within the catalytic domain, where the CdsL binding domains are located (Stone et al., 2008). Using 3D-JIGSAW, a 3D structure of CdsN was generated and used to model the binding domains for CdsL. Modeling of the binding regions of CdsL on the predicted CdsN monomer revealed that both of the binding domains for CdsL are present in the central region of CdsN (Figure 4A). A more revealing mapping of the binding domains is shown when hexameric CdsN is modeled from the EscN crystal structure (Zarivach et al., 2007). For clarity, we have shown only one half of the predicted hexameric structure of CdsN (a CdsN trimeric complex). Residues 221–229 are exposed toward the outer face of the hexamer (Figure 4B, blue), while residues 265–270 are exposed in the central channel of the hexameric ring (Figure 4C, red). The location of the binding domains is consistent with a proposed mechanism of CdsL down-regulation of CdsN sterically hindering access of ATP to the P-loop (Figures 4B,C, magenta).
FIGURE 4
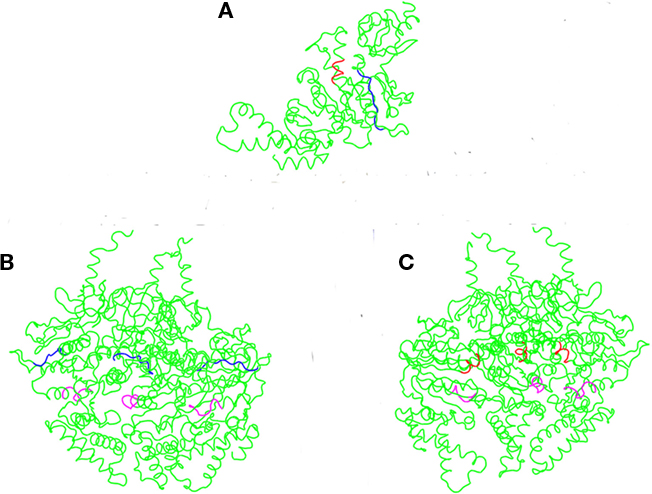
Figure 4. CdsL binding domains mapped onto the predicted 3D structure of CdsN reveals two distinct domains. A 3D structure of CdsN was generated based on orthology modeling using 3D-JIGSAW based on the structure of EscN, and used to visualize the CdsL binding domains identified from – 30 – the peptide library binding data. The two CdsL binding domains on CdsN are in blue (residues 221–229) or red (residues 265–270), and the ATP coordinating P-loop region of CdsN is highlighted in magenta. (A) Representation of the predicted CdsN monomeric structure indicating both CdsL binding domains (RSVIVVSTS in blue, TRFARA in red). (B) Structural model showing the location of residues 221–229 (RSVIVVSTS), displayed on one half of the predicted hexameric structure and viewed from the outer face, revealing that the binding domains are exposed to the outside of the hexamer. The P-loop is represented in magenta. (C) Structural model viewed from outside looking toward the central channel showing the location of residues 265–270 (TRFARA), displayed on one half of the predicted hexameric structure revealing that the domain is exposed within the central channel. The P-loop is represented in magenta.
Inhibition of C. pneumoniae Growth and Replication by CdsN Cognate Peptide
The Pepscan binding data revealed two distinct domains of CdsN that mediate the interaction between CdsN and CdsL. Using this information we sought to design and utilize a peptide mimetic targeting the interaction between CdsN and CdsL and examine its effect on chlamydial replication. As discussed above, CdsN possessed two different binding domains for CdsL; we selected the six amino acid sequence TRFARA to create a potential peptide mimetic. We synthesized the TRFARA peptide with a N-terminal MTS, YGRKKRRQRRR (Efthymiadis et al., 1998). We also extended the CdsN sequence flanking the TRFARA peptide to ensure the entire binding domain was included, and flanked the peptide with cysteine residues to assist with any disulfide bonding that may be required for binding. The final peptide utilized was the 28-amino acid sequence YGRKKRRQRRRCVVLMMDSVTRFARALC. To examine whether the peptide-MTS was entering EBs, we first incubated either GST alone or GST–MTS at 50, 100, and 500 μM with gastrografin purified EBs. We saw that the amount of GST–MTS entering EBs increased in a dose-dependent manner (Figure 5A). The GST control (500 μm) was taken up poorly by Chlamydia. We then added either GST or GST–MTS for increasing time periods (5, 30, or 60 min) which showed increasing uptake with time, starting as early as 5 min (Figure 5B). Again, the GST control showed very poor uptake into Chlamydia, suggesting that the MTS facilitates peptide entry in a time- and dose-dependent manner. HeLa cells were then infected with C. pneumoniae at an MOI of 2 and stained for inclusions at 72 h using a FITC-conjugated anti-MOMP monoclonal antibody. The CdsN peptide was added to EBs at a 0, 20, 50, and 100-μM concentrations for 30 min prior to infection and we saw that infectivity was reduced in a dose–response fashion (Figure 6A). In the absence of the peptide, inclusions are clearly visible at 72 h (Figure 7). Pre-treatment of the cells with 50 μM peptide resulted in a drastic reduction in the number of inclusions (>95%), while at 100 μM there was toxicity for HeLa cells. To rule out the possibility that the inhibitory activity at 50 μM might be due to toxicity we tested the effect of the peptide at 50 μM on HeLa cells using an adenylate kinase release assay and found little or no toxicity (Figure 6B). We next examined infected cells with the CdsN or the random peptide by transmission electron microscopy. In infected cells exposed to the random peptide large inclusion bodies containing a mixture of RBs and EBs were readily visible while infected cells incubated with 50 μM CdsN peptide lacked any detectable inclusions when over 200 cells were examined and showed no evidence of cytotoxicity (Figure 7).
FIGURE 5
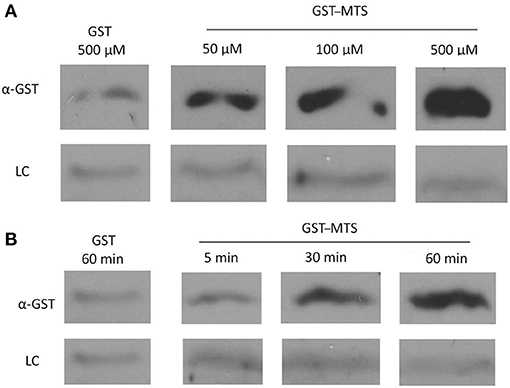
Figure 5. Uptake of a GST-Membrane transport signal (MTS) fusion protein into EBs. EBs were purified on a discontinuous gastrografin gradient and collected by centrifugation to ensure the use of a homogenous EB mixture. The GST alone and GST–MTS protein was expressed in Escherichia coli cells and purified using glutathione beads. The EBs were then incubated with either GST or GST–MTS, trypsinized for 30 min to ensure that all extracellular GST or GST–MTS was degraded, and examined by α-GST Western blot for the presence of intracellular GST-tagged protein. CdsL, an intracellular type III secretion protein, was used as a loading control (LC). (A) Time-course for incubation of EBs with GST–MTS for 5, 30, or 60 min. GST control at 60 min displayed very little protein while GST–MTS at 5, 30, and 60 min had increasing amounts of GST–MTS present within the EBs. (B) Dose–response of EBs incubated with GST–MTS at 50, 100, and 500 μm. GST control at 500 μm displayed very little protein while GST–MTS accumulated within the EBs as the concentration increased. CdsL is shown as a LC.
FIGURE 6
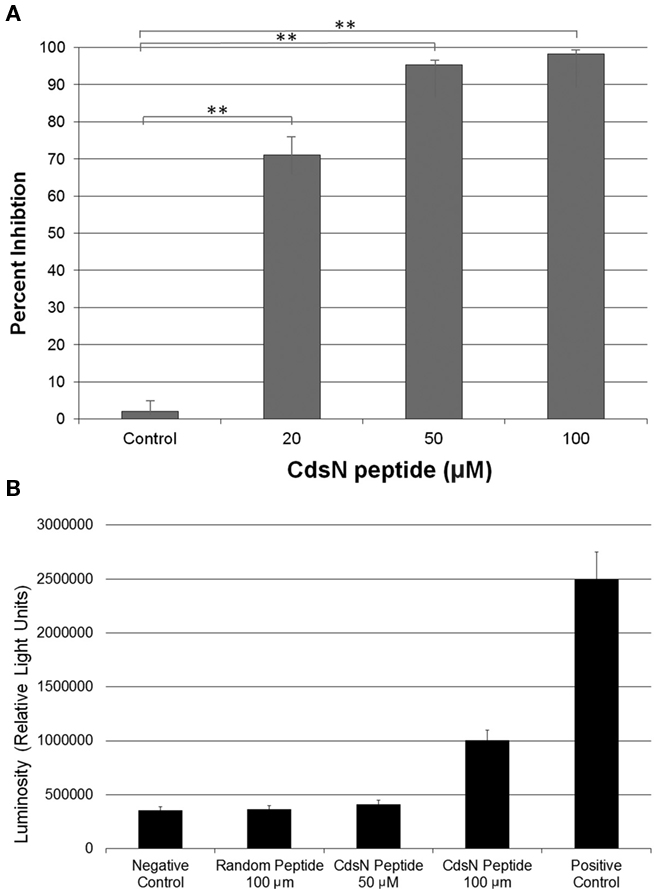
Figure 6. CdsN peptide containing the CdsL binding sequence TRFARA inhibits the infection of HeLa cells in a dose-dependent fashion and shows low toxicity at 50 μM. Shell vials grown to 85% confluency were infected with Chlamydia pneumoniae at an MOI of 3 after incubation with the CdsN or control peptide. Infected cells were counted by staining with a Chlamydia specific major outer membrane protein (MOMP)-monoclonal antibody (Pathfinder kit). Toxicity was measured using an adenylate kinase release assay for HeLa cells in the presence of either the CdsN or control peptide. (A) CdsN peptide was added at increasing concentrations (20, 50, and 100 μM) and demonstrated a dose–response in reduction of C. pneumoniae infectivity of HeLa cells. The control represents a random peptide with the sequence MFAVNAQ–MTS that had no effect on C. pneumoniae infectivity. All experiments were performed in triplicate and data is presented as the mean with error bars representing the mean + 1 SD with **representing p < 0.01. (B) Toxicity of the CdsN peptides toward HeLa cells was evaluated using the Lonza adenylate kinase release assay. The CdsN peptide was added to HeLa cells for 1 h at 50 and 100 μM, and the random peptide was added for 1 h at 100 μM. We saw that the CdsN peptide at 50 μM had no toxicity toward HeLa cells, and only minor toxicity at 100 μM.
FIGURE 7
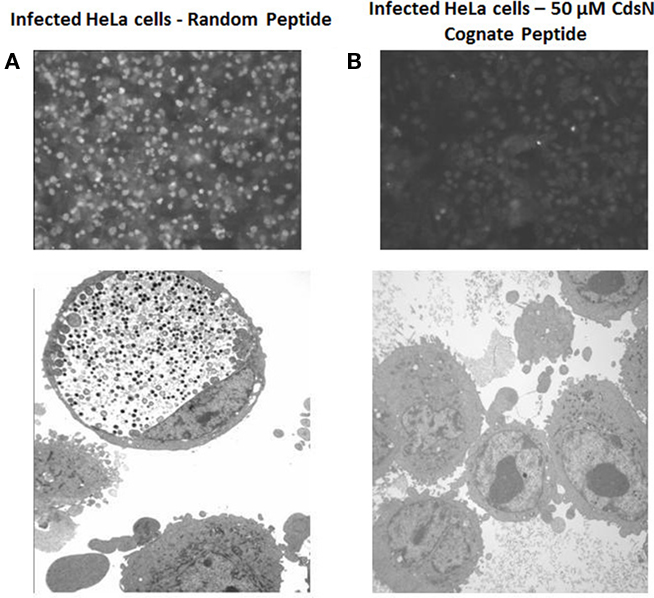
Figure 7. CdsN peptide reduces chlamydial infectivity of HeLa cells by IF staining and EM. HeLa cells were pretreated with CdsN or a control peptide (MTS–MFAVNAQ), then infected with Chlamydia pneumoniae at an MOI of 3 and examined by either IF staining using anti-MOMP monoclonal antibody treatment with the pathfinder kit (top) or by electron microscopy (bottom). (A) HeLa cells treated with the control peptide (100 μM) and infected with C. pneumoniae. Immunofluorescent staining reveals numerous infected cells (top) while electron microscopy reveals a large inclusion body with both EBs and RBs (bottom). (B) HeLa cells treated with the CdsN peptide (50 μM) and infected with C. pneumoniae. Evaluation of over 200 cells treated with the CdsN peptide by IF staining and EM examination revealed a drastic reduction in C. pneumoniae infected cells and a complete lack of detectable inclusions by EM.
Discussion
Although C. pneumoniae contains all the genes coding for a T3SS, only a small number of these have been characterized. We have previously shown that CdsN possesses enzymatic activity and interacts with the putative type III secretion protein, CdsL (Stone et al., 2008). We have now extended these observations to show that CdsL functions to down-regulate CdsN enzymatic activity. We have also used a novel approach to map the exact region within CdsN that mediates the CdsN–CdsL interaction, and used this information to design a peptide mimetic that disrupts the chlamydial replication cycle. Combined, this data provides new insights into the regulation of the T3S system and its importance in the chlamydial life-cycle.
YscL orthologs have been shown to down-regulate T3S ATPase enzymatic activity in Yersinia, and C. pneumoniae encodes a YscL ortholog (CdsL; Blaylock et al., 2006; Stone et al., 2008). We found that CdsN treated with increasing concentrations of CdsL reduced enzymatic activity up to 82%. To show that the regulation by CdsL was specific we tested the ability of CdsL to down-regulate the type II secretion ATPase of Chlamydia, GspE, and found that it had no effect on its enzymatic activity. This suggests that CdsL is specific in its down-regulation of CdsN. CdsL has previously been shown to be expressed at all time points throughout the replication cycle with an apparent accumulation during the final 48 h (Slepenkin et al., 2003). This is consistent with a role for CdsL regulating T3S during invasion and intracellular replication phases of the life-cycle in EBs and RBs, respectively.
Type III secretion ATPases are known to mediate numerous protein interactions between both structural components and chaperone/effector complexes, and play a critical role in construction of the injectisome and secretion of effectors (Gauthier and Finlay, 2003; Akeda and Galan, 2005). We used Pepscan Analysis to determine the CdsN domain that mediates the interaction with CdsL. We found that there were two distinct domains within CdsN that interacted with CdsL; one exposed in the central channel of the hexameric structure of CdsN and the other exposed on the outer face. Using GST pull-downs, we corroborated the Pepscan data by demonstrating that a 49-amino acid fragment of CdsN containing both of the predicted CdsL binding domains was able to co-purify with full length CdsL. We have shown previously that CdsL forms dimers, and it is likely that the two CdsL molecules interact with different regions of CdsN (Pallen et al., 2006; Stone et al., 2008). This would be consistent with the two binding domains on CdsN. Along with regulating ATPase activity, CdsL is believed to tether the ATPase to the inner membrane, functioning in a similar manner as the gamma-stalk of the F0F1 ATPase which runs through the central channel of the F0F1 hexamer and may interact with the membrane (Pallen et al., 2006). The location of the CdsL binding domain within the central channel of CdsN supports the concept that CdsL functions in a similar manner as the gamma-stalk domain for CdsN. Both binding domains are found in close proximity to the P-loop within the catalytic domain which coordinates ATP binding and cleavage (Stone et al., 2008). It is possible that when CdsL is bound to either of these domains it could sterically hinder access of ATP to the P-loop, reducing enzymatic function of CdsN. The tethering function of CdsL would be crucial for T3S function, and if this tethering is mediated by the binding domain within the central channel of the CdsN hexamer then likely this interaction is maintained throughout the entire developmental cycle. The exposed binding domain on the outer surface of the CdsN hexamer, however, could be responsible for the down-regulation of CdsN by CdsL. CdsL could bind and dissociate from the outer CdsN–CdsL binding domain upon a conformational change in CdsN, thereby activating ATPase activity upon CdsL release by allowing ATP to access the P-loop. It is tempting to speculate that delivery of effector/chaperone complexes to CdsN, possibly by a multi-cargo transport protein such as CdsQ, could be the trigger for CdsL release and ATPase activation, allowing for the dissociation of the effector/chaperone complex and subsequent secretion of the effector (Spaeth et al., 2009).
Using Pepscan epitope mapping we have shown where CdsL binds to CdsN. Using this binding data we designed a cognate CdsN peptide containing a membrane transport signal to allow the peptide to enter Chlamydia and showed that this peptide blocked chlamydial invasion of HeLa cells. Membrane transport signals are known to interact hydrophobically with eukaryotic membranes, triggering entry by endocytosis across the membrane (Efthymiadis et al., 1998). Our working hypothesis is that this transport signal would facilitate entry of the CdsN peptide into EBs, and that this could potentially disrupt the CdsN–CdsL interaction and inhibit T3S, and thus invasion, although we have not shown inhibition of T3S per se. This could be achieved by either disrupting the regulation of the ATPase or interfering with the tethering function of the ATPase, displacing its peripheral association with the inner membrane. Either of these two scenarios could disrupt the T3SS, preventing chlamydial infection and cellular invasion. We first confirmed that the MTS facilitated uptake of the peptide into EBs in a time- and dose-dependent manner. Afterward, we treated the chlamydial EBs for 30 min with the CdsN peptide which reduced chlamydial infection in a dose–response fashion, and at a concentration of 50 μM reduced Chlamydia infectivity by >95%, as determined by IF staining and counting of inclusions at 72 h. An alternative explanation of this data could be that the peptide was bactericidal toward Chlamydia, but we feel that this is unlikely since a random peptide failed to inhibit chlamydial replication. We have also shown that two additional peptide mimetics targeting other essential protein interactions can also inhibit chlamydial replication (Mahony et al., 2010). We are currently investigating whether the CdsN peptide mimetic can inhibit chlamydial replication if administered during the infection cycle, once infection has occurred, which if successful would indicate that the peptide can disrupt CdsN–CdsL complexes within RBs and block T3S across the inclusion membrane. This would suggest that ATPase activity is carefully regulated by CdsL in both EBs and RBs, and the CdsN–CdsL interaction is critical for chlamydial replication.
The discovery of unique CdsN binding domains for CdsL coupled with structural modeling of the binding domains has allowed us to postulate how CdsL functions to down-regulate CdsN. This has allowed us to create a cognate CdsN peptide that functions as a novel antimicrobial agent, preventing C. pneumoniae from invading host cells. One of the major difficulties in working with Chlamydia is that they are genetically intractable. Peptide mimetics such as this CdsN peptide could effectively create chemical knockouts by disrupting protein complexes, allowing us to mimic genetic knockouts and explore specific functions of various proteins in the T3SS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Chris B. Stone is a recipient of a Father Sean O’Sullivan Research Center Studentship. This research was funded by a Canadian Institute of Health Research grant to James B. Mahony.
References
Akeda, Y., and Galan, J. (2004). Genetic analysis of the Salmonella enterica type III secretion-associated ATPase InvC defines discrete functional domains. J. Bacteriol. 186, 2402–2412.
Akeda, Y., and Galan, J. (2005). Chaperone release an unfolding of substrates in type III secretion. Nature 437, 911–915.
Andrade, A., Pardo, J., Espinosa, N., Perez-Hernandez, G., and Gonzalez-Pedrajo, B. (2007). Enzymatic characterization of the enteropathogenic Escherichia coli type III secretion ATPase EscN. Arch. Biochem. Biophys. 468, 121–7.
Ardeniz, O., Gulbahar, O., Mete, N., Cicek, C., Basoglu, O., Sin, A., and Kokuludag, A. (2005). Chlamydia pneumoniae arthritis in a patient with common variable immunodeficiency. Ann. Allergy Asthma Immunol. 94, 504–508.
Balin, B., Little, C., Hammond, C., Appelt, D., Whittum-Hudson, J., Gerard, H., and Hudson, A. (2008). Chlamydophila pneumoniae and the etiology of late-onset Alzheimer’s disease. J. Alzheimers Dis. 13, 371–380.
Bernard, J., Harb, C., Mortier, E., Quéméner, A., Meloen, R. H., Vermot-Desroches, C., Wijdeness, J., van Dijken, P., Grötzinger, J., Slootstra, J. W., Plet, A., and Jacques, Y. (2004). Identification of an interleukin-15 receptor-binding site on human interleukin-15. J. Biol. Chem. 279, 24313–24322.
Betts, H., Twiggs, L., Sal, M., Wyrick, P., and Fields, K. (2008). Bioinformatic and biochemical evidence for the identification of the type III secretion system needle protein of Chlamydia trachomatis. J. Bacteriol. 190, 1680–1690.
Blaylock, B., Riordan, K., Missiakas, D., and Schneewind, O. (2006). Characterization of the Yersinia enterocolitica type III secretion ATPase YscN and its regulator, YscL. J. Bacteriol. 188, 3525–3534.
Carabeo, R., Grieshaber, S., Hasenkrug, A., Dooley, C., and Hackstadt, T. (2004). Requirement for the Rac GTPase in Chlamydia trachomatis invasion of non-phagocytic cells. Traffic 5, 418–425.
Chen, J., and Stephens, R. (1997). Chlamydia trachomatis glycosaminoglycan-dependent and independent attachment to eukaryotic cells. Microb. Pathog. 22, 23–30.
Clifton, D., Fields, K., Grieshaber, S., Dooley, C., Fischer, E., Mead, D., Carabeo, R., and Hackstadt, T. (2004). A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc. Natl. Acad. Sci. U.S.A. 101, 10166–10171.
Coombes, B., and Mahony, J. (2002). Identification of MEK- and phosphoinositide-3-kinase-dependent signaling as essential events during Chlamydia pneumoniae invasion of HEp2 cells. Cell. Microbiol. 4, 447–460.
Cortes, C., Rzomp, K., Tvinnereim, A., Scidmore, M., and Wizel, B. (2007). Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple Rab GTPases. Infect. Immun. 75, 5586–5595.
Efthymiadis, A., Briggs, L., and Jans, D. (1998). The HIV-1 Tat nuclear localization sequences confers novel nuclear import properties. J. Biol. Chem. 273, 1623–1628.
Galan, J., and Collmer, A. (1999). Type III secretion machines: bacterial devices for protein delivery into host cells. Science 284, 1322–1328.
Galan, J., and Wolf-Watz, H. (2006). Protein delivery into eukaryotic cells by type III secretion machines. Nature 444, 567–573.
Gauthier, A., and Finlay, B. (2003). Translocated intimin receptor and its chaperone interact with ATPase of the type III secretion apparatus of enteropathogenic Escherichia coli. J. Bacteriol. 185, 6747–6755.
Ghosh, P. (2004). Process of protein transport by the type III secretion system. Microbiol. Mol. Biol. Rev. 68, 771–795.
Goure, J., Pastor, A., Faudry, E., Chabert, J., Dessen, A., and Attree, I. (2004). The V antigen of Pseudomonas aeruginosa is required for assembly of the function PopB/PopD translocation pore in host cell membranes. Infect. Immun. 72, 4741–4750.
Grayston, J. (2000). Background and current knowledge of Chlamydia pneumoniae and atherosclerosis. J. Infect. Dis. 181, S402–S410.
Hefty, P., and Stephens, R. (2007). Chlamydial type III secretion system is encoded on ten operons preceded by sigma 70-like promoter elements. J. Bacteriol. 189, 198–206.
Hermann, M., Schuhmacher, A., Muhldorfer, I., Melchers, K., Prothmann, C., and Dammeier, S. (2006). Identification and characterization of secreted effector proteins of Chlamydophila pneumoniae TW183. Res. Microbiol. 157, 513–524.
Hoare, A., Timms, P., Bavoil, P., and Wilson, D. (2008). Spatial constraints within the chlamydial host cell inclusion predict interrupted development and persistence. BMC Microbiol. 8, 5. doi: 10.1186/1471-2180-8-5
Hybiske, K., and Stephens, R. (2007). Mechanisms of host cell exit by the intracellular bacterium. Chlamydia. Proc. Natl. Acad. Sci. U.S.A. 104, 11430–11435.
Johnson, D., Stone, C., Bulir, D., Coombes, B., and Mahony, J. (2009). A novel inhibitor of Chlamydophila pneumoniae protein kinase D (PknD) inhibits phosphorylation of CdsD and suppresses bacterial replication. BMC Microbiol. 9, 218. doi: 10.1186/1471-2180-9-218
Johnson, D., Stone, C., and Mahony, J. (2008). Interactions between CdsD, CdsQ, and CdsL, three putative Chlamydophila pneumoniae type III secretion proteins. J. Bacteriol. 190, 2972–2980.
Lane, B., Mutchler, C., Khodor, S., Grieshaber, S., and Carabeo, R. (2008). Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog. 4, e1000014. doi: 10.1371/journal.ppat.1000014
Mahony, J., Stone, C., Liu, L., Pyrie, R., Chong, S., and Whittum-Hudson, J. (2010). “Peptide mimetics inhibiting type III secretion decrease host cell invasion and reduce infection levels in the mouse genital tract,” in Chlamydial Infections, Proceedings of the 12th International Symposium on Human Chlamydial Infections, June 20–25, eds J. Schachter, G. Byrne, H. Caldwell, M. Chernesky, I. Clarke, D. Mabey, J. Paavonen, P. Saikku, M. Starnbach, A. Stary, R. Stephens, P. Timms, and P. Wyrick. Hof bei Salzburg, 325–328.
Markham, A., Jaafar, Z., Kernege, K., Middaugh, C., and Hefty, P. (2009). Biophysical characterization of Chlamydia trachomatis CT584 supports its potential role as the type III secretion needle tip protein. Biochemistry 48, 10353–10361.
Pallen, M., Bailey, C., and Beatson, S. (2006). Evolutionary links between FliH/YscL-like proteins from bacterial type III secretion systems and second-stalk components of the F0F1 and vacuolar ATPases. Protein Sci. 15, 935–940.
Scidmore, M., and Hackstadt, T. (2008). Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol. Microbiol. 39, 1638–1650.
Slepenkin, A., Motin, V., de la Maza, L., and Peterson, E. (2003). Temporal expression of type III secretion genes of Chlamydia pneumoniae. Infect. Immun. 71, 2555–2562.
Spaeth, K., Chen, Y., and Valdivia, R. (2009). The Chlamydia type III secretion system c-ring engages chaperone-effector protein complexes. PLoS Pathog. 5, e1000579. doi: 10.1371/journal.ppat.1000579
Stenner-Liewen, F., Liewen, H., Zapata, J., Pawlowski, K., Godzik, A., and Reed, J. (2002). CADD, a Chlamydia protein that interacts with death receptors. J. Biol. Chem. 277, 9633–9636.
Stone, C., Johnson, D., Bulir, D., and Mahony, J. (2008). Characterization of the putative type III secretion ATPase CdsN (Cpn0707) of Chlamydophila pneumoniae. J. Bacteriol. 190, 6580–6588.
Subtil, A., Wyplosz, B., Balana, M., and Dautry-Varsat, A. (2004). Analysis of Chlamydia caviae entry sites and involvement of Cdc42 and Rac activity. J. Cell Sci. 117, 3923–3933.
Teeling, J. L., Mackus, W. J. M., Wiegman, L. J. J. M., van den Brakel, J. H. N., Beers, S. A., French, R. R., van Meerten, T., Ebeling, S., Vink, T., Slootstra, J. W., Parren, P. W. H. I., Glennie, M. J., and van de Winkel, J. G. J. (2006). The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J. Immunol. 177, 362–371.
Timmerman, P., Puijk, W. C., and Meloen, R. H. (2007). Functional reconstruction and synthetic mimicry of a conformational epitope using CLIPSTM technology. J. Mol. Recognit. 20, 283–299.
Wylie, J., Hatch, G., and McClarty, G. (1997). Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. J. Bacteriol. 179, 7233–7242.
Keywords: type III secretion, peptide mimetic, Chlamydia, ATPase, Pepscan
Citation: Stone CB, Bulir DC, Emdin CA, Pirie RM, Porfilio EA, Slootstra JW and Mahony JB (2011) Chlamydia pneumoniae CdsL regulates CdsN ATPase activity, and disruption with a peptide mimetic prevents bacterial invasion. Front. Microbio. 2:21. doi: 10.3389/fmicb.2011.00021
Received: 01 November 2010;
Accepted: 30 January 2011;
Published online: 14 February 2011.
Edited by:
Rey Carabeo, Imperial College London, UKCopyright: © 2011 Stone, Bulir, Emdin, Pirie, Porfilio, Slootstra and Mahony. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: James B. Mahony, Regional Virology and Chlamydiology Laboratory, St. Joseph’s Hospital, L305, 50 Charlton Avenue East, Hamilton, ON, Canada L8N 4A6. e-mail: mahonyj@mcmaster.ca