
- 1 Institute of Microbiology and Genetics, Technische Universität Darmstadt, Darmstadt, Germany
- 2 Structural Biology Laboratory, Macromolecular Crystallography Unit, ITQB-UNL, Oeiras, Portugal
Background: The sulfur oxygenase reductase (SOR) is the initial enzyme of the sulfur oxidation pathway in the thermoacidophilic Archaeon Acidianus ambivalens. The SOR catalyzes an oxygen-dependent sulfur disproportionation to H2S, sulfite and thiosulfate. The spherical, hollow, cytoplasmic enzyme is composed of 24 identical subunits with an active site pocket each comprising a mononuclear non-heme iron site and a cysteine persulfide. Substrate access and product exit occur via apolar chimney-like protrusions at the fourfold symmetry axes, via narrow polar pores at the threefold symmetry axes and via narrow apolar pores within in each subunit. In order to investigate the function of the pores we performed site-directed mutagenesis and inhibitor studies. Results: Truncation of the chimney-like protrusions resulted in an up to sevenfold increase in specific enzyme activity compared to the wild type. Replacement of the salt bridge-forming Arg99 residue by Ala at the threefold symmetry axes doubled the activity and introduced a bias toward reduced reaction products. Replacement of Met296 and Met297, which form the active site pore, lowered the specific activities by 25–55% with the exception of an M296V mutant. X-ray crystallography of SOR wild type crystals soaked with inhibitors showed that Hg2+ and iodoacetamide (IAA) bind to cysteines within the active site, whereas Zn2+ binds to a histidine in a side channel of the enzyme. The Zn2+ inhibition was partially alleviated by mutation of the His residue. Conclusions: The expansion of the pores in the outer shell led to an increased enzyme activity while the integrity of the active site pore seems to be important. Hg2+ and IAA block cysteines in the active site pocket, while Zn2+ interferes over a distance, possibly by restriction of protein flexibility or substrate access or product exit.
Introduction
A large number of microorganisms oxidize sulfur and reduced inorganic sulfur compounds (ISC) for energy conservation (for review, see for example Friedrich et al., 2005; Kletzin, 2007; Frigaard and Dahl, 2009; Ghosh and Dam, 2009). Most studies on ISC oxidation were performed with soluble sulfur species like thiosulfate, sulfite and sulfide. Their mechanisms of activation and oxidation are reasonably well understood (Friedrich et al., 2005; Ghosh and Dam, 2009). Less is known how the barely soluble elemental sulfur is mobilized and oxidized (19–30 nmol/l α-S8 at 25°C, 478 nmol/l at 80°C; Kamyshny, 2009). Different enzymes and enzyme activities were described but few were analyzed in molecular detail (Rohwerder and Sand, 2003; Kletzin, 2007; Ghosh and Dam, 2009; also see Protze et al., 2011, this volume).
The best-known sulfur-oxidizing enzymes are sulfur oxygenase reductases (SOR), which were purified from two different thermoacidophilic Acidianus species (Emmel et al., 1986; Kletzin, 1989). In addition, SORs obtained by heterologous gene expression were studied from Ac. ambivalens and Ac. tengchongensis, from the hyperthermophilic bacterium Aquifex aeolicus, and from a moderately thermophilic bacterium from a bioleaching reactor (Sun et al., 2003; Urich et al., 2004; Chen et al., 2007; Pelletier et al., 2008). The SOR is the initial sulfur-oxidizing enzyme in the Archaeon Ac. ambivalens, which is our model organism for sulfur metabolism, and which grows optimally at 80°C and pH 1–3. The SOR or sor genes do not occur frequently; so far they are restricted to some thermoacidophilic Archaea and to some mesophilic and thermophilic Bacteria (Figure 1). The SORs catalyze an oxygen-dependent sulfur disproportionation reaction with sulfite, thiosulfate and sulfide as products (Eqs 1–3; Kletzin, 1989; Sun et al., 2003; Pelletier et al., 2008).

Figure 1. Multiple alignments of known and complete SOR amino acid sequences from isolated proteins and from genome sequences. PCR-generated fragments with high similarity to the Acidianus enzymes (Chen et al., 2007) were omitted. Genbank identification (GI) numbers are given at the end; *derived from the genome sequence available at JGI1. The horizontal line separates Archaea from Bacteria. Abbreviations: ASP, active site pore residues (Figure 5); AS, active site cysteines; Css, cysteine persulfide; Fe, iron-coordinating residues; Chimney, chimney-like protrusions at the fourfold symmetry axes, identical to the DelL deletion (Figure 2); DelK, short deletion of the residues around the outer Phe ring (Figure 2); Zn, 2-His motif around the zinc-coordinating His277.
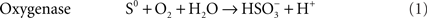
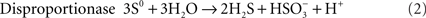
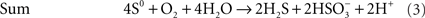
External cofactors or electron donors are not required and the two enzyme activities could not be separated. Zn2+, Hg2+ and thiol-modifying organic agents like iodoacetamide (IAA) and N-ethylmaleimide (NEM) inhibit activity (Kletzin, 1989). The SORs do not catalyze sulfur disproportionation in the absence of oxygen and they have neutral or slightly acidic pH optima (5–7.4; Emmel et al., 1986; Kletzin, 1989; Sun et al., 2003).
X-ray crystallographic structures were determined of two of these enzymes, from Ac. ambivalens and from Ac. tengchongensis. Both showed that the spherical, hollow oligomers are composed of 24 identical subunits arranged in a 432 point-group symmetry (Figure 2; Urich et al., 2006; Li et al., 2008). The outer surfaces of the spheres are apparently smooth and impervious with the exception of very narrow pores at the four- and threefold rotational axes of the oligomers (Figure 2). Each subunit contains a low-potential mononuclear non-heme iron site as the putative redox-active cofactor (Urich et al., 2004, 2006). We had shown using site-directed mutagenesis that the three Fe-coordinating residues (H86, H90 and E114 in Ac. ambivalens numbering) and a persulfurated cysteine (C31) are essential for catalysis. Most likely, the cysteine persulfide is involved in sulfur binding. Mutation of the other two cysteine residues did not abolish activity, not even in a double mutant (Urich et al., 2005b). Similar results had been obtained for the Ac. tengchongensis SOR (Chen et al., 2005). Our current hypothesis about the reaction mechanism of the SOR predicts that the catalytic cycle is initiated by covalent sulfur binding to the active site C31 as a polysulfide chain (R–Sn–SH), followed by hydrolytic cleavage of the cysteine polysulfide to sulfide and a polysulfenyl moiety (R–Sn–SOH). Either Fe2+ or the sulfenyl group would subsequently activate oxygen (Kletzin, 2008).
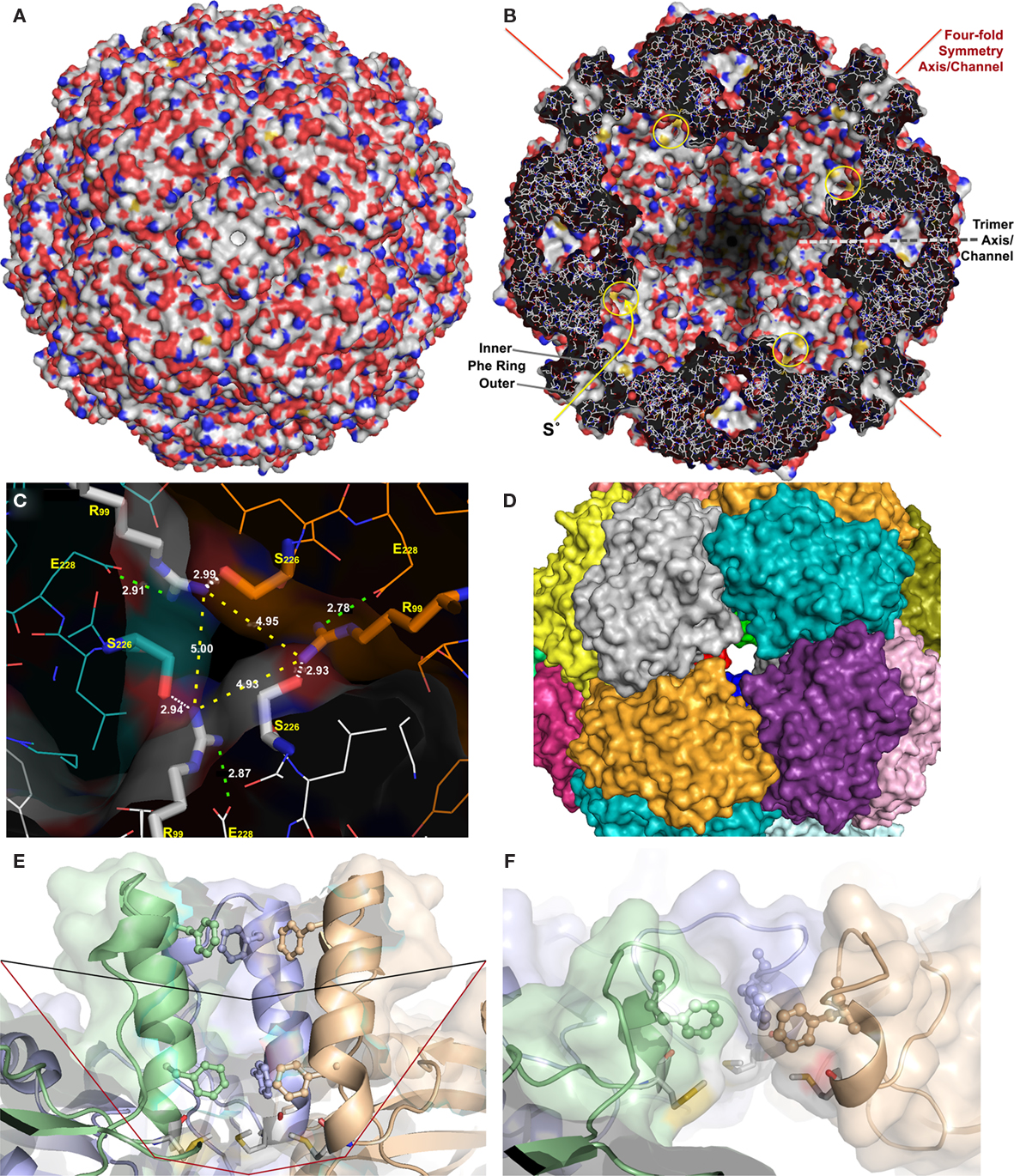
Figure 2. X-ray crystallography and modeling of the SOR and its pores. (A) Surface representation of the holoenzyme centered at the chimney-like structure at the fourfold symmetry axis. (B) Representation of the protein structure and the inner surface of the holoenzyme sliced at the center of the fourfold symmetry axes; the position of the inner and outer phenylalanine rings are indicated, also the active site pores (yellow circles), and the approximate position of the trimer symmetry axis, which is tilted out of plane. (C) Channel at the threefold symmetry axis formed by R99 and S226; distances are given between the Nη atoms of the arginines (yellow dashes), for the salt bridges to E228 (green dashes), and for the putative hydrogen bond to the Oγ of the S226 of the neighboring subunit (white dashes). (D) Subunit representation of the large deletion mutant at the fourfold symmetry axis DelL (Figure 1) modeled at the SwissModel server (Arnold et al., 2006). (E) Side view of the channel at the fourfold symmetry axis with outer (F141) and inner phenylalanine rings (F133; from top, showing three out of four subunits; wild type) and the methionine ring at its base (M130); black line, approximate position of small deletion (DelK); red line, large deletion of entire chimney (DelL). (F) Model of the same three subunits as in panel E of the short DelK deletion, modeled at the Phyre server (Kelley and Sternberg, 2009).
The iron site and the three conserved cysteine residues (Figure 1) are located in an active site pocket that is connected to the inner cavity of the sphere by a narrow pore formed by two adjacent methionines and a phenylalanine (3–4 Å diameter; M296/M297, F23; Figures 1 and 2). In consequence, substrate and products must pass first the outer shell of the holoenzyme into the inner cavity and then the active site pore into the pocket or vice versa. We had postulated that only linear polysulfane species but not circular S8 could pass both barriers (Urich et al., 2006). Therefore, S8-sulfur would require an initial activation step for catalysis, either by binding to a thiol group or by sulfide. The polysulfide sulfur formed in this reaction should be stable at the near-neutral pH of the cellular cytoplasm (6.5; Moll and Schäfer, 1988). In contrast, polysulfides are not stable in the outside medium (pH 1–3) and disproportionate into sulfur and sulfide (Schauder and Kröger, 1993).
The question now arises how the sulfur gets inside of the enzyme and how the products get out. Two successive rings of four phenylalanine residues each form the hydrophobic pore at the fourfold symmetry axis, followed at the inside by four methionines (Figure 2). In the crystallized protein, the pore is not fully open with C–C distances down to 5.0 Å in the inner Phe ring (Figure 2; Urich et al., 2006). Nevertheless, it is feasible that apolar molecules like the linear polysulfane species should be able to pass the pore when the protein is heated. In contrast, the polar reaction products hydrogen sulfide, sulfite and thiosulfate were proposed to exit the sphere via channels located at the threefold symmetry axes (Li et al., 2008). The narrow channel outlets are lined with hydrophilic residues such as R99 and S226. The shortest distances between the Nη of the three arginines are ≈5 Å, or ≈6.3 Å between the Oγ of the Ser and Nη of the Arg (Figure 2). In addition, the arginines form salt bridges to the gamma-carboxylate group of a glutamate within the same subunit and potential hydrogen bonds to the S226 residues of the neighboring subunit. Nevertheless, the arginines should be able to bind transiently the anionic reaction products sulfide, sulfite and thiosulfate allowing their exit.
Here, we show by site-directed mutagenesis that opening the putative substrate and product pathways in the outer shell leads to a significant increase in specific activity and to a shift in the stoichiometry of the products. In contrast, the integrity of the inner pore seems to be important. We also show by mutagenesis studies and crystallographic analysis of inhibitor derivatives that Hg2+ and IAA bind in the active site as expected, whereas Zn2+ does not and could interfere with the movement of substrates and products.
Materials and Methods
Construction of Site-Directed Mutants and Heterologous Gene Expression in Escherichia Coli
The sor gene (EMBL accession number X56616) was expressed heterologously using the pASK75 vector and a C-terminal Strep-tag fusion as described elsewhere (pASK-SOR.05 plasmid; Skerra, 1994; Urich et al., 2004). The site-directed mutants of several codons were constructed by using the Quikchange method with pASK-SOR.05 as a template (Stratagene; now Agilent Technologies, Böblingen, Germany; see Table 1 for a list of the mutants and oligonucleotides used).

Table 1. Forward oligonucleotides used in this study for the mutagenesis of the sor gene; the corresponding reverse-complimentary oligonucleotides required for the Quikchange method (Stratagene) are not shown.
The PCR product was digested for 4 h with 10 U DpnI (Fermentas; St. Leon-Rot, Germany), subsequently purified via the PCR Clean-Up Kit (Sigma-Aldrich; Steinheim, Germany) and eluted with 25 μl of elution buffer. After transformation of E. coli TOP 10’ cells (Invitrogen, Darmstadt, Germany) with 6–7 μl of the purified PCR product, the resulting constructs were analyzed by restriction digestion and by sequencing. Plasmid minipreparations of 25 colonies were sequenced using the degenerated MM296/297 primers, which resulted in the identification of the four mutants M297A, M296V, MM296/7VT, and MM296/7TT. The double mutant F133A/F141 was constructed using the F141A oligonucleotides with the previously constructed F133A mutant plasmid. In the DelL mutant (deletion, l = long), 23 chimney-forming amino acid residues were replaced by three glycines (Figure 1; Table 1). In the DelK derivative (deletion, K = kurz; German for short), 10 residues were replaced by two glycines.
Escherichia coli BL 21 Codon plus (DE3) RIL cells (Stratagene) were transformed with the mutant plasmids and the original pASK-SOR.05. The expression of the sor genes was induced by addition of anhydrotetracycline (200 μg/l of culture; IBA; Göttingen, Germany) to either 0.5 or 15 l cultures growing at 37°C in 2× LB medium at an OD600 between 0.6 and 0.8. The cultures were incubated for 20 h after induction with either vigorous shaking (0.5 l) or with vigorous aeration and stirring (15 l). In order to ensure sufficient iron incorporation, 100 μM ferric citrate was added to the media at the time of induction.
Protein Purification
The harvested cells were washed once in approximately 10 volumes of 100 mM Tris–HCl/150 mM NaCl buffer pH 8 and then re-suspended in five volumes of the same buffer. Cells were disrupted with a High Pressure Homogenizer (Constant Systems; 0.18 mm nozzle and 1.35 MPa pressure). After a first centrifugation step (10,000 × g for 30 min, Sorvall, SLA-3000; Thermo Fisher Scientific, Schwerte, Germany), the soluble protein-containing supernatant was centrifuged in an ultracentrifuge (100,000 × g for 45 min, Beckman Instruments, 45Ti). The particle-free protein extracts from 5 to 50 g of cells (wet mass) were applied to an 8 ml Strep-Tactin super-flow column (IBA, Göttingen, Germany) connected to an ÄKTApurifier 10 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). The elution step was performed with three column volumes of washing buffer containing 2.5 mM desthiobiotin (IBA). The column was washed and regenerated according to the manufacturer’s recommendations. Alternatively, the column was regenerated with three column volumes each of ddH2O, 0.5 M NaOH, and ddH2O instead of the regular HABA solution (IBA).
Analytical Procedures
Specific activities of the wild type and mutant proteins were determined by incubation of 2–5 μg of purified enzyme/ml of Tris–HCl buffer pH 7.2 containing 2% sulfur and 0.1% of Tween 20 as described previously (Kletzin, 1989; Urich et al., 2004, 2005b). Samples were taken at appropriate time points (usually after 0, 2, 4, 6, 8, and 10 min) and the concentration of the products was determined colorimetrically. Specific activities were calculated from the linear increase of product concentrations (Kletzin, 1989). At least two activity measurements were performed for each mutant using independently purified protein preparations (see below, Table 2). Iron quantification was performed with pure protein preparations using the 2,4,6-tripyridyl-1,3,5-triazine method (TPTZ; Fischer and Price, 1964). Protein concentrations were determined by the Coomassie Blue method (Bradford, 1976) or using the BCA Protein Assay Kit (Novagen/Merck, Darmstadt, Germany) according to the manufacturer’s instructions. Samples of purified proteins were separated by SDS-PAGE (Schägger and von Jagow, 1987) and visualized with colloidal Coomassie Blue staining (Roti-Blue; Roth, Karlsruhe, Germany).
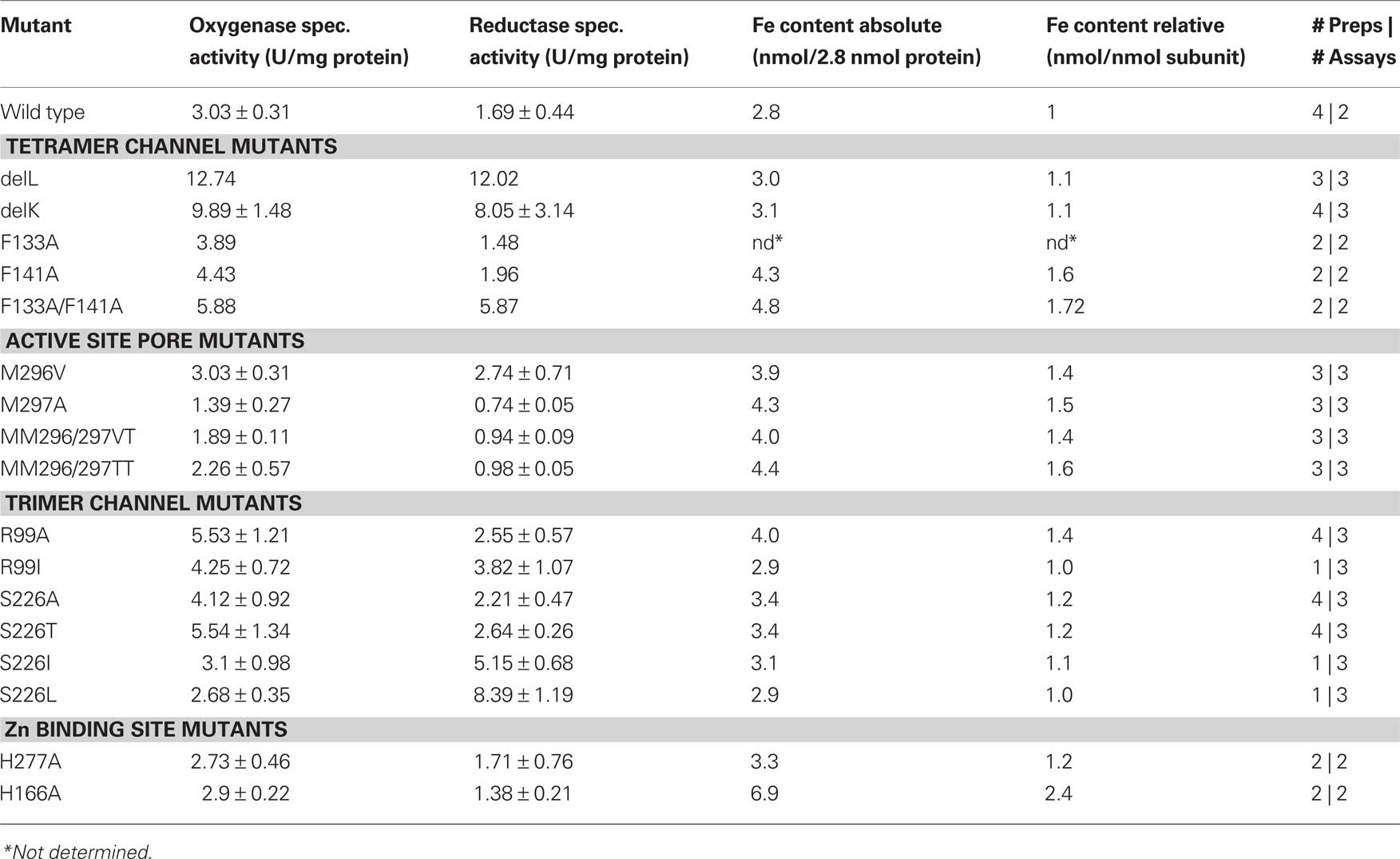
Table 2. Specific activities of wild type and mutant SOR, iron content, numbers (#) of preparations and assays per preparation.
Inhibition Assays
Zinc inhibition assays of the wild type and mutant proteins were performed by addition of a freshly prepared zinc chloride solution to the reaction buffer in concentrations ranging from 0.01 to 1 mM (Kletzin, 1989). After addition of the enzyme, the mixture was incubated for 30 min at room temperature before starting the reaction by heating to 85°C and measurement of the specific activities. A zinc/buffer mixture without protein was used as a negative control.
Crystallization, Data Collection, Alignment and Modeling
Crystallization of the SOR protein was performed as described previously (Urich et al., 2005a). The mercury derivative was prepared by addition of 5 mM sodium 4-(hydroxymercury)-benzoic acid (or p-chloromercuribenzoic acid, p-CMB) for 5 days at 22°C to crystallization drops containing grown crystals. The zinc-complexed SOR was prepared by co-crystallization of the SOR in presence of 10 mM zinc acetate for 2 days at 32°C. SOR was also co-crystallized with 10 mM IAA for 2 days. After washing in a cryoprotectant solution without the reagents used for soaking, the crystals were flash-frozen in liquid nitrogen (Urich et al., 2005a). The diffraction data sets of the SOR co-crystallized with zinc and IAA were collected with a resolution of 1.7 and 1.9 Å, respectively, at the ESRF beam line 14-1. The Hg2+ dataset was collected with a resolution of 2.5 Å at the ESRF beam line 14-3.
Images from the three diffraction experiments were processed with DENZO and the observed intensities merged and scaled with SCALEPACK of the HKL Suite (Otwinowski and Minor, 1997). Structure factor amplitudes were then calculated with TRUNCATE and the three sets scaled together against the native data (PDB entry 2CB2-SF; Urich et al., 2006) with SCALEIT of the CCP4.2.2 suite (Collaborative Computational Project, 1994). The final model of the native structure including cysteine persulfide, Fe ions and waters molecules was used as template for structure refinement with REFMAC (Murshudov et al., 1997) or PHENIX (Adams et al., 2010). The stereochemistry of the Zn and Hg centers was refined without target geometrical restraints. After an initial rigid body refinement the models were iteratively refined, analyzed and edited with XTALVIEW-XFIT (McRee, 1999) against electron density maps, until convergence of R-values was achieved (Urich et al., 2006).
The sequences used for the multiple alignment (Figure 1) were obtained following a BLAST search at NCBI2 with the Ac. ambivalens SOR sequence as input. The Sulfobacillus acidophilus SOR sequence was identified in the almost complete genome sequence available at the JGI genome server3 (January 2011). The alignment was made with MAFFT using the default parameter at the Kyushu server4. The DelL and DelK deletion mutants were modeled using the Phyre6 (Kelley and Sternberg, 2009) and SwissModel servers5 (Arnold et al., 2006) with the wild type 3D structure as template. The figures were prepared in Pymol, including the qualitative surface electrostatic distribution (DeLano, 2002).
Data Deposition
The atomic coordinates of the SOR derivatives were deposited at the Protein Data Bank with identification numbers 2yav (Zn derivative), 2yaw (Hg) and 2yax (IAA).
Results
Properties of the SOR Mutants
SOR mutant plasmids generated via site-directed mutagenesis were sequenced and were introduced, if correct, into E. coli BL21 Codon Plus cells. Wild type or mutant SOR protein was obtained after overnight incubation of the induced cultures. After breaking of the cells, 10–30% of the protein was present in soluble form while ≥70% precipitated in inclusion bodies as observed previously (Urich et al., 2004). The recombinant wild type and mutant SOR proteins were purified from the soluble E. coli fractions in a one-step procedure using the attached Strep-tag and Strep-Tactin columns. The yields of purified protein ranged from 0.6 to 1.2 mg/l of culture volume. When analyzed with SDS-PAGE, all SOR preparations were more than 90% homogeneous. In each protein preparation one larger band with a molecular mass of 70 kDa was seen and several smaller bands (Figure 3), the latter of which had been shown to represent cleavage products (Urich et al., 2004). Wild type and mutant proteins were analyzed for specific enzyme activity and iron content in order to verify that altered enzyme activities reflect true mutagenesis effects and not low iron incorporation into the active site. The iron content of the purified proteins was in the range of 1–3 Fe/subunit (Table 3). The average values of specific wild type SOR activities were 3.03 ± 0.31 U/mg (oxygenase) and 1.69 ± 0.44 U/mg (reductase).
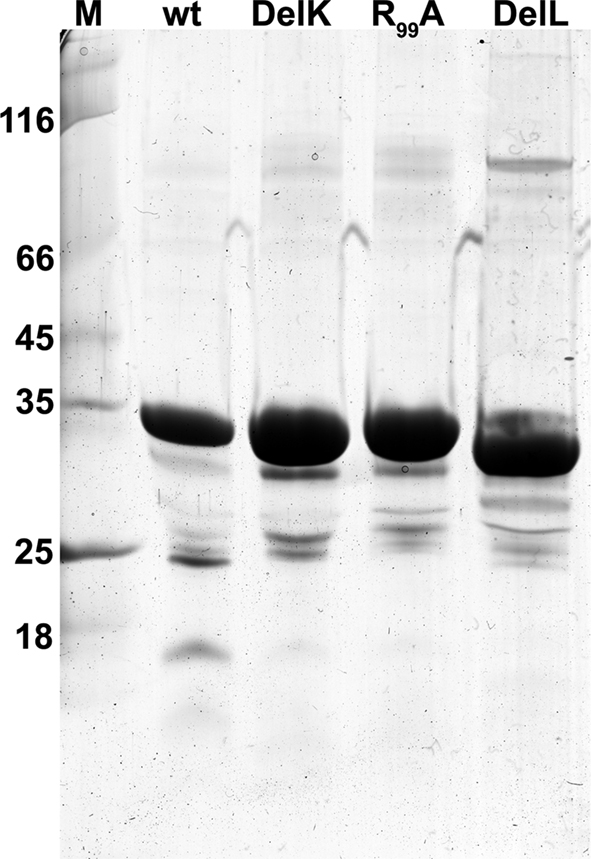
Figure 3. Coomassie-stained SDS gel of SOR wild type and three of the mutants; 5 μg of protein per lane; M, molecular weight marker.
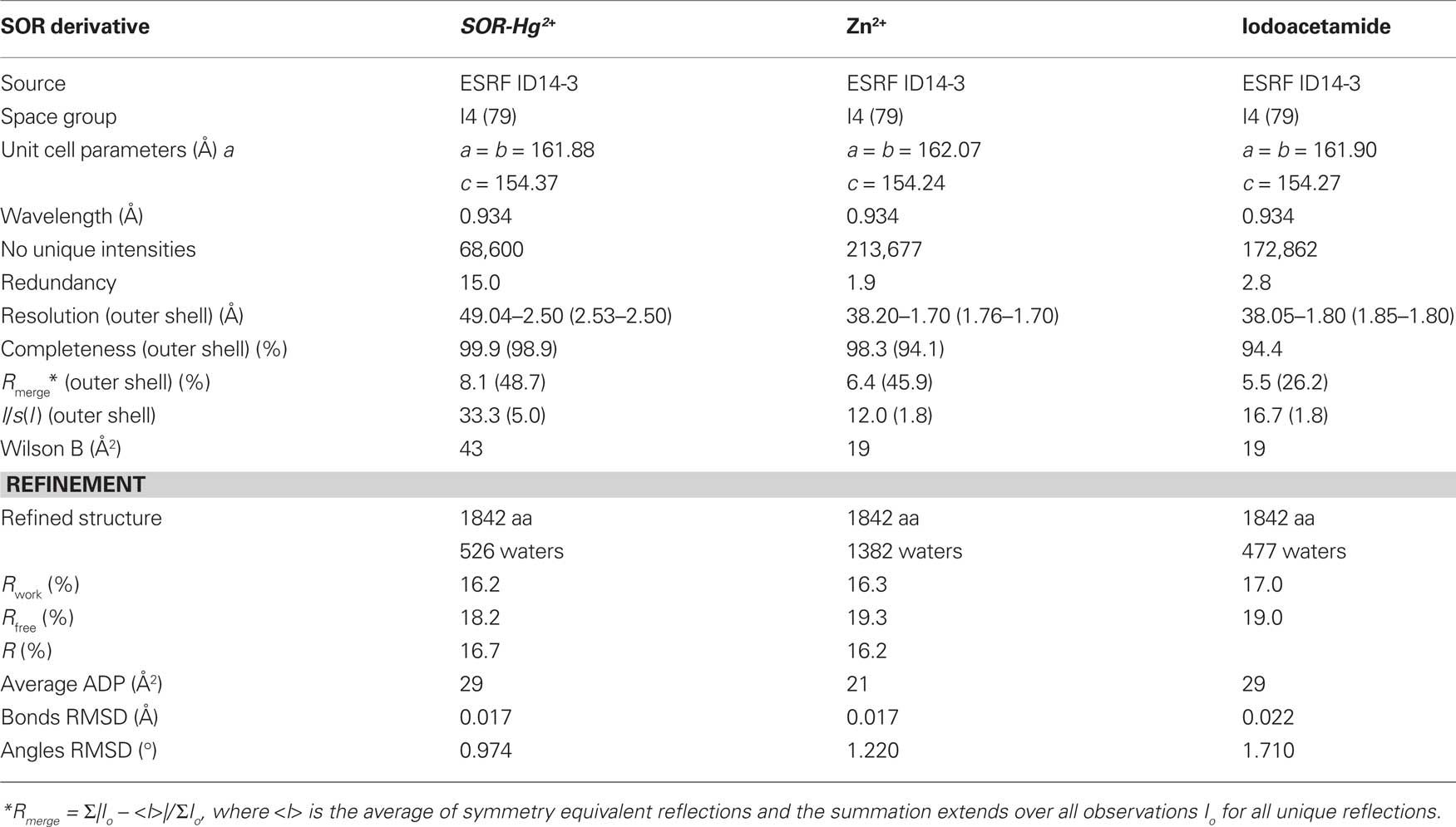
Table 3. Diffraction data processing and refinement statistics.
Opening of the Outer Sphere Increases Activity
Two mutants were constructed, which feature truncated versions of the chimney-like structures located at the fourfold symmetry axes. In the DelL mutant (deletion, L = long) the 23 amino acid residues that form the protrusions including both phenylalanine rings (Figures 1 and 2D) are replaced by three glycines. In the DelK derivative (deletion, K = kurz; German for short) 10 residues including the outer phenylalanine ring were replaced by two glycines. When modeled into the SOR holoenzyme structure, the pore opened to a diameter of 9–10 Å in the DelL mutant. The atomic distances of the inner phenylalanine ring did not change significantly in the model of the DelK mutant, so that it does not display an open pore (from 5.0 Å in the wild type protein to 5.4 Å in the mutant; Figure 2). As expected, the apparent molecular masses of the DelL and DelK mutants were slightly smaller in SDS gels compared to the wild type enzyme (Figure 3).
Several-fold increased enzyme activities were observed in both cases. DelL showed 420% of the oxygenase and up to 771% of the reductase activities, while DelK showed an increase up to 326% (oxygenase) and 476% (reductase), respectively (Figure 4; Table 2). The phenylalanine residues were mutated into alanine independently (F133A, F141A) and as a double mutant (F133A/F141A). All three different mutants showed increased activities. The double mutant showed the highest activities (194% of the oxygenase and 347% of the reductase). Mutation of M130A located at the base of the channel (Figure 2) did not alter the catalytic properties of the enzyme (not shown).
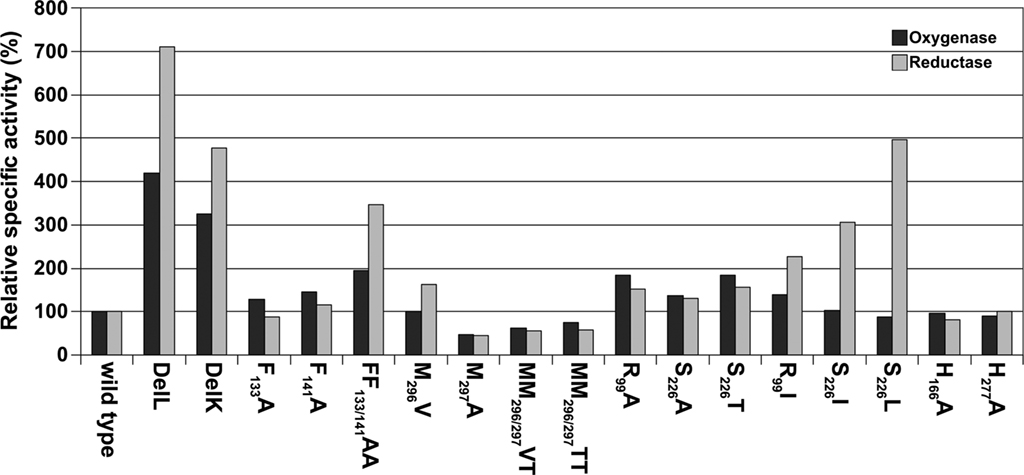
Figure 4. Histogram of relative activities of wild type SOR and mutants, the wild type was set to 100% according to the values in Table 3.
R99 and S226, both located at the postulated channel outlet at the threefold symmetry axis, were substituted for alanines independently. Together with an S226T variant, all three mutants showed elevated enzyme activities. R99A and S226T were comparable having both about 182% oxygenase and 156% reductase activities. Isoleucine and leucine variants of the pore-forming channel outlet residues were comparable to the wild type in oxygenase activity but showed a significantly increased reductase activity of up to 496% in case of S226L (Figure 2; Table 2).
The Integrity of the Active Site Pore is Essential
The active site pore entrance, which provides access to the reaction center, is formed by two adjacent methionines (M296/M297) and one phenylalanine F23 (Figure 5). We substituted the two methionines via site-directed mutagenesis using degenerated primers that allowed for 16 variations. Twenty-five different plasmids were screened and four different mutants were obtained, two double mutants, MM296/297VT, MM296/297TT and two single mutants, M297A and M296V. Mutagenesis led to an opening of the active site pore as compared to the wild type with the exception of the M296V mutant (Figure 5). Both double mutants MM296/297VT, MM296/297TT and also M297A showed a decrease to approximately 50% of wild type activity (Figure 4; Table 2). The M296V mutant showed an increase in reductase activity (162%) but not in oxygenase activity. The latter residue is also present in several of the naturally occurring SORs from other species (Figure 1).
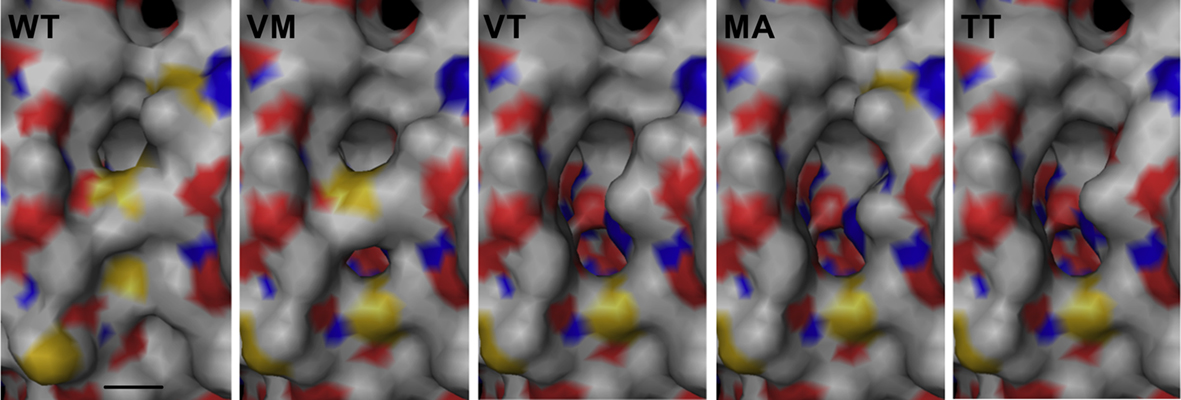
Figure 5. Modeling of active site pore mutants based on the wild type (WT) structure at the SwissModel server (Arnold et al., 2006). Abbreviations and symbols: VM, M296V; VT, MM296/297VT; MA, M297A; TT, MM296/297TT; bar, 4 Å.
Zinc Binds Far From the Active Site
Zn2+ had been shown to be a potent inhibitor of SOR activity as long as it is free in solution and not complexed by ligands such as EDTA (Kletzin, 1989; Urich et al., 2004). Crystals were soaked with Zn2+ in order to determine its binding site within the SOR. One Zn2+ ion was detected in the Zn-complexed structure (with 0.8 occupancy) refined at 1.7 Å resolution (Table 3). It was located on the opposite side of the beta barrel core of the monomer (not shown; see Urich et al., 2006) but not in close vicinity to the active site (Figure 6). The zinc ion was bound in a distance of 2.1 Å to His277 imidazole (interatomic distances from the here-presented structures are averages over of the six crystallographically independent monomers; Figure 6). An acetate and a chloride ion together with two water molecules completed the coordination sphere of Zn2+ (Figure 6). His166, which constitutes a conserved 2-His motif together with His277 (Figure 1), had its Nε2 in hydrogen-bonding distance to the two water molecules that coordinate the zinc (Figure 6). Both histidines are located at the bottom of a long, semi-closed channel that opens to the inner cavity of the holoenzyme next to the active site entrance of a neighboring monomer. The Fe–Zn distances were about 27 Å, both within the same subunit and with the neighboring subunit. The side chains of Css31 and Met297 separate the entrances to the active site pocket and the zinc-binding channel. We had postulated that Css31 is the sulfur-binding residue in the active site pocket (Urich et al., 2006). The distance between Zn2+ and the Sδ atom of the cysteine persulfide Css31 was 18 Å, so that direct interference of zinc in the catalysis is improbable.
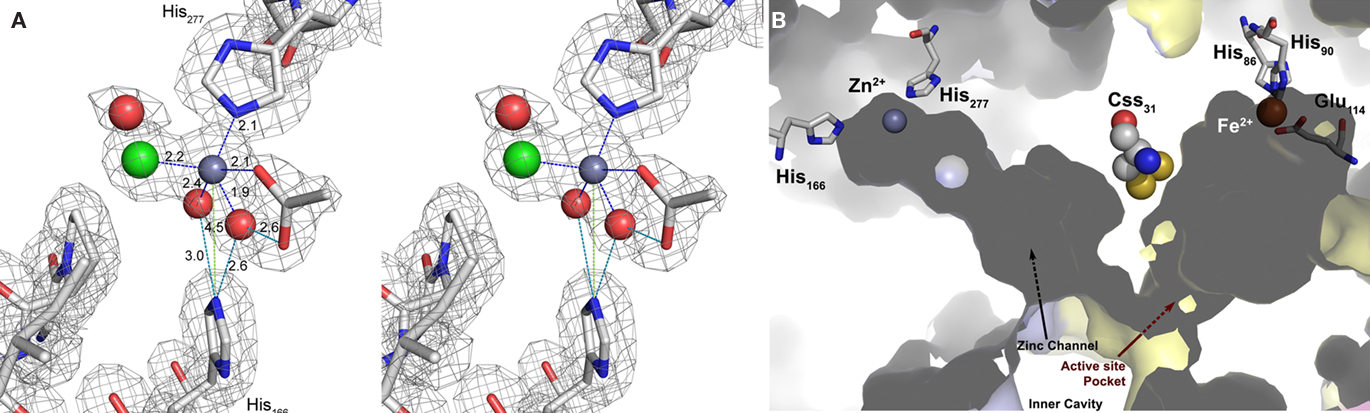
Figure 6. The Zn2+-binding site. (A) Electron density at the Zn2+-binding site (gray mesh representing 2m|Fo| − D|Fc| map at 1 σ). Cross-eyes stereo image showing the Zn2+ ion coordination (dark gray sphere with blue dashed lines) with the His277 side chain, one acetate ion (ACT), one chloride ion (green sphere), and two waters (red spheres) that are also at hydrogen-bonding distances (cyan dashed lines) from Nε2 of the His166 side chain. The other acetate oxygen is at hydrogen-bonding distance (cyan dashed line) from one of the Zn2+ coordinating waters; light green dashes, non-bonding distances; sticks representation: carbon in gray, nitrogen in blue, oxygen in red. (B) Surface representation of two neighboring monomers (light blue and yellow) highlighting the positions of the Zn and Fe sites with their respective ligands and channels and the bridging cysteine persulfide. The zinc site is located at the end of a tunnel leading to the active site Css31 of the neighboring monomer.
When His166 and His277 were substituted independently for alanine, the specific activities were similar to the wild type enzyme (Figure 4; Table 2). Ki-values (half-maximal inhibitory concentration) of the wild type enzyme were 45 μM zinc chloride for the oxygenase activity and 39 μM for the reductase activity. Ki-values for H166A were 121 μM (oxygenase) and 150 μM (reductase). The mutant H277A showed comparable Ki-values of 157 μM (oxygenase) and 144 μM (reductase).
Mercury and Iodoacetamide Bind at Active Site Cysteines
Analysis of the crystallographic model of the p-CMB treated crystal refined at 2.5 Å resolution (Table 3) showed in the active site pocket two partially occupied Hg2+ ions per monomer. One Hg2+, with 0.5 occupancy, was bound at a distance of 2.1 Å to the Sγ atom of the cysteine persulfide (Css31; Figure 7). A putative acetate ion refined reasonably well, also with 0.5 occupancy, with one of his carboxylic oxygens coordinating Hg2+ at 2.0 Å distance, although the limited resolution maps also show some density noise in its neighborhood. The second mercury ion, with 0.3 occupancy, refined at 2.6 Å distance from Sγ of non-essential cysteine Cys101, with the side chain in trans conformation for torsional angle chi1, and with occupancy 0.3. The other side chain conformation, chi1gauche(−) (with occupancy 0.7) corresponds to that of the native structure. This Hg2+ is not too far away from the Sδ atom of Met108, at 3.7 Å distance. Thus, the inhibition of the enzyme by mercury compounds (Kletzin, 1989) is caused by modification of the active site cysteine(s).
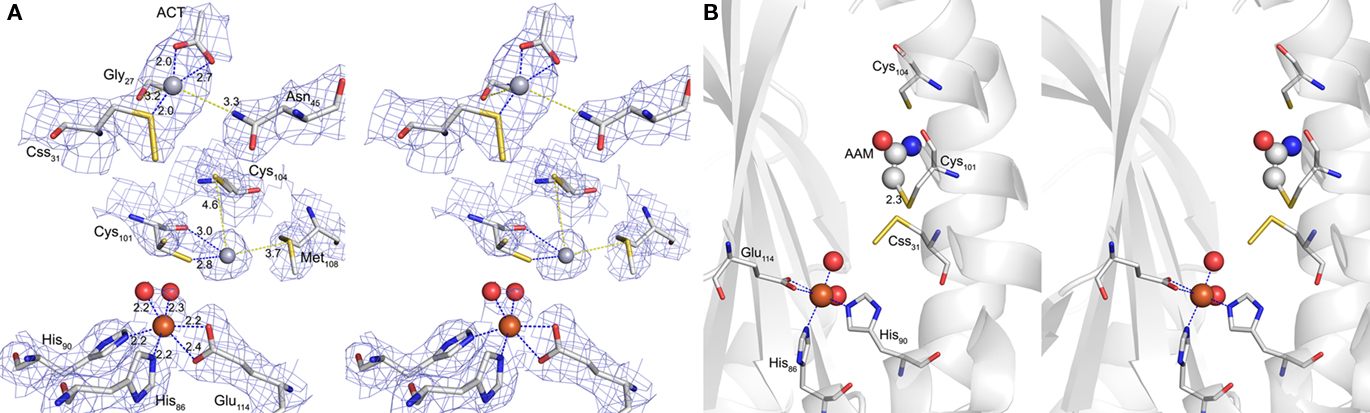
Figure 7. Hg2+ and iodoacetamide-binding sites. (A) Electron density at the Hg2+-binding sites (blue mesh representing 2m|Fo| − D|Fc| map at 1 σ). Cross-eyes stereo image of the coordination spheres of the two Hg2+ ions (gray spheres) and the active site Fe3+ (brown sphere); Hg1 at Sγ of Css31 with additional distances to one acetate (ACT) and the backbone oxygen of Gly27 (blue dashed lines) and to Nδ of Asn45 (yellow dashed line); Hg2 at Sγ and the backbone oxygen of Cys101 (blue dashed lines) and with the distances to Sγ of Cys104 and to Sδ of Met108 (yellow dashed lines); Fe3+ with two water ligands (red spheres), two histidine side chains and a bidentate glutamate (all blue dashed lines); sticks representation: carbon in gray, nitrogen in blue, sulfur in yellow and oxygen in red; ACT, acetate. (B) Cross-eyes stereo image of the acetamide (AAM) binding site with secondary structure representation of the subunit backbone; Fe site and sticks representation as above; AAM in sticks and spheres.
The co-crystallization with the thiol-modifying reagent IAA resulted in additional electron density resembling acetamide in covalent distance to Cys101 (Figure 7). The electron density was present with low occupancy in three out of six monomers in the asymmetric unit. No additional density was observed at Css31 or Cys104. In consequence, it appears that Cys101 is the primary target for alkylation with IAA, although this conclusion is tentative due to the low reactivity between enzyme and inhibitor in this experiment.
Discussion
Sulfur oxygenase reductases are uncommon sulfur-disproportionating enzymes restricted to a few species of sulfur-oxidizing Archaea and Bacteria, which are either facultative or obligatory chemolithoautotrophs (Figure 1). Determination the 3D structure of two of these enzymes showed that they form large hollow homomultimers with more or less secluded inner chambers. Each subunit contains a reaction pocket with conserved sites consisting of a mononuclear iron and a cysteine residue (Urich et al., 2004, 2005b, 2006; Chen et al., 2005; Li et al., 2008). The SORs thus provide enclosed reaction and/or storage compartments physically separated from the cytoplasm (Figure 2). Despite previous biochemical and mutagenesis studies it is still unclear how the proteins work in detail. Here, we focus on the pores in the protein and on the mode of action of important inhibitors of enzyme activity.
The Pores at the Fourfold Symmetry Axis Restrict Enzyme Activity
Narrow pores padded with apolar amino acids are localized at the chimney-like structures at the fourfold symmetry axes (Figure 2). They were already considered to be the substrate entrance points to the inner cavity of SOR (Urich et al., 2006; Li et al., 2008). Two rings of four phenylalanine side chains each define these pores in the present 3D model (Figure 2). Here we show that mutation of the Phe residues into Ala, which induces enlargement of the pores, gave a moderate, less than twofold increase in specific enzyme activity (Figure 4). A full deletion of the loop building the chimneys (Figures 1 and 2) resulted in a more than sevenfold increase in enzyme activity showing that substrate access to the active site and/or product exit is indeed limited by the outer shell of the protein. Therefore, it can be concluded that the specific activity of the wild type SOR is curbed to a lower level than optimally possible. The reasons for making the enzyme slow might be speculated upon. One option (among others) would predict that the reactive reaction products are not released uncontrollably into the cytoplasm but that they are delivered directly to the downstream oxidoreductases (Kletzin, 2008). One experimental indication for this interpretation – in the absence of known interaction partners – came from antibody/immunogold electron microscopy results of the Ac. tengchongensis SOR: the enzyme seemed to be attached to the inside of the cytoplasmic membrane (Chen et al., 2005). In addition, these pores might provide highly controlled access points to the active sites, preventing the oxidation of “unwanted substrates.”
Mutagenesis at the Threefold Symmetry Axis and Reaction Mechanism
The amino acids Arg99 and Ser226 are central elements of the subunit interface at the threefold symmetry axis. Arg99 forms an intra-subunit salt bridge to Glu228 (Figure 2). The other η-nitrogen atom is in hydrogen-bonding distance (2.8 Å) to the Oγ of Ser226 of the neighboring subunit. Alternative hydrogen-bonding networks are also possible: Arg99 might link to the α-carbonyl oxygen atoms of Ser226 of two subunits. In addition, a hypothetical salt bridge between the ε-nitrogen of the Arg99 and Glu228 is possible, provided that an extensive charge delocalization exists in the guanidinium group. We did not obtain protein of an E228A mutant from E. coli cells, so that we cold not analyze its effects on salt bridge formation (data not shown). In contrast, mutation of Arg99 and Ser226 into Ala gave a modest, less than 1.5-fold increase in specific activity (Figure 4). Mutation into more hydrophobic residues changed the ratio between oxidized and reduced reaction products in favor of sulfide (Figure 4; Table 2). The same had happened in a less pronounced way in the chimney mutants DelK and DelL, suggesting that opening of the closed reaction chamber in the interior of the protein changes the ratio between the oxygenase and disproportionase partial reactions (Eqs 1 and 2).
The interpretation of the activity data is complicated by non-enzymatic reactions occurring with sulfur and ISC in aqueous solutions, which depend on the incubation temperature and the pH of the buffer. For example, sulfite reacts rapidly with excess sulfur to thiosulfate at pH ≥ 5 and 85°C so that it is still unclear whether thiosulfate is a primary or secondary reaction product of the SOR (Kletzin, 1989). The sulfur disproportionation depicted in Eq. 2 and with H2S, polysulfides and thiosulfate (instead of sulfite) as products occurs non-enzymatically at alkaline pH (detectable above pH 7.5 at 75°C; Roy and Trudinger, 1970; Kletzin, 1989). We had concluded from previous activity assays in the presence of chemically complexed zinc (to overcome zinc inhibition and to precipitate sulfide in situ) that an approximate 1:1 ratio between oxidized and reduced reaction products is maintained (Eq. 3) as opposed to the 1:2 ratio for the oxygen-independent disproportionation shown in Eq. 2. We also had assumed that the sub-stoichiometric hydrogen sulfide detection in the standard assay is due to the rapid non-enzymatic re-oxidation under the aerobic assay conditions (see Table 2, wild type enzyme; Kletzin, 1989, 2008; Kletzin et al., 2004; Urich et al., 2004). This picture however changes with the channel mutants, which prove that the ratio between oxidized and reduced reaction products is not constant and that it depends on the integrity of the protein shell.
The Integrity of the Active Site Pore Seems to be Important
A different story resulted from the mutations at the active site pore, made from two adjacent methionines, M296/M297 and a phenylalanine, F23. M297 is conserved in all SORs, while M296 is exchanged for another hydrophobic amino acid in several naturally occurring SOR sequences (Figure 1). F23 is conserved in Archaea while Bacteria mostly use methionine at this position. The pore supposedly represents the entrance of substrate and the exit of the products; at least, we were unable to find a “back exit” in the active site pocket of the Ac. ambivalens enzyme as suggested for the Ac. tengchongensis SOR (Li et al., 2008). Expansion of the active site pore did not increase the enzyme activity, which is contrasting our observations on the tetramer and trimer channels (Figures 4 and 5). Replacement of the hydrophobic Met residues with the smaller and in the case of the threonine more hydrophilic residues diminished the specific activity by half, suggesting that the hydrophobic barrier and/or at least one of the methionines are essential. One could speculate that non-covalent nucleophilic interaction between the atoms of the sulfur substrate and the Sδ atom of the gate-keeping methionine residues could direct the sulfur substrate toward the active site cavity.
Different Mechanisms of Inhibitor Action
Zinc and the thiol-binding agents p-CMB, IAA and maleimide were previously shown to act as inhibitors (Kletzin, 1989). No activity had been observed in the presence of 1 mM Hg2+ or Zn2+ ions, whereas 0.1 mM resulted in 37 and 74% residual activity, respectively, for the oxidized reaction products (Kletzin, 1989). Inhibition by Zn2+ was neutralized by addition of at least equimolar concentrations of EDTA (Urich et al., 2004). Both Hg2+ and Zn2+ ions can theoretically bind to sulfur atoms of cysteine(s), to histidine and/or carboxylate ligands. The alkylating agents IAA and NEM were less potent inhibitors: 27% of oxygenase activity remained in the presence of 1 mM IAA (Kletzin, 1989), while 47% remained with 1 mM NEM. In order to determine the mechanisms of inhibition, we resolved the 3D structures of wild type SOR crystals soaked in p-CMB, IAA, and Zn2+.
Two Hg2+ ions were found in the active site pocket of p-CMB treated crystals, one of them in bonding distance (2.1 Å) to the Sγ atom of the essential cysteine persulfide Css31, thus explaining the inhibitory effect of Hg2+ (Figure 7). A second ligand was provided by the oxygen (at 2.0 Å distance) of a putative acetate from the crystallization media. The second Hg2+ ion is bound (2.6 Å) to Sγ of conserved cysteine Cys101. Although unresolved, water molecules might provide additional ligands as both mercury ions face the lumen of the water-filled active site pocket. To conclude, the main mechanism of inhibition seems to be the result of mercury binding to Css31, an amino acid that cannot be mutated without total loss of enzyme activity (Urich et al., 2005b). Mercury binding to Cys101 seems to play a less important role.
In contrast, the effect of IAA is more difficult to explain. Acetamide bound to Cys101 was observed only in three out of six subunits of the asymmetric unit and only with low occupancy (Figure 7). This observation has two implications. First it shows that the 24 subunits are not equal, minor distortions could be the result of inhibitor and/or substrate binding. Distortions were so far not seen at all because of the sheer number of subunits and the rotational symmetry of the holoenzyme. Both would average out minor distortions in the electron density and make subtle changes undetectable. We had tried to soak crystals with many different ISC without seeing additional electron density anywhere in the molecule (Urich, 2005), presumably because substrate or product binding occur only in a minority of subunits at a given time. Bound sulfur species would be averaged out within the holoenzyme. This situation is slightly different with IAA, where the low occupancy points into the same direction, supported by the observation that not all of the potential binding sites are occupied. However, the stoichiometry of inhibitor binding is high enough to facilitate the positive identification of the bound molecule. A second implication came from the binding site: Cys101 could be mutated to Ala with partial loss of activity (≈20%; Urich et al., 2005b). A Cys101S mutant had very low residual activity (≈1%) concomitant with a very low iron content (<0.1/subunit; Urich et al., 2005b). This observation showed that the chemical nature of the amino acid side chain at this position directly affects enzyme activity and active site integrity, although the cysteine itself is not essential.
Inhibition by zinc seems to follow a completely different mechanism. The Zn2+ ion was found in a dead-end channel, which opens next to the active site pore of the adjacent subunit. The active site Css31 separates the lumina of both channels (Figure 6). The question arises how zinc inhibits the enzyme activity over a distance of approximately 27 Å to the next iron. The zinc ion is coordinated by histidine residue H277, one acetate, one chloride and two water molecules that in turn are hydrogen-bonding His166 (Figure 6). Mutation of either histidine into alanine did not alter enzyme activity significantly. However, their Ki-values for Zn2+ increased two- to threefold, compared to the wild type. The 2-His motif is conserved in the SOR sequences (Figure 1). It cannot be answered at present which role the obviously conserved zinc channel plays. In contrast, several options exist about the mechanism of inhibition at a distance. The Zn2+ ion might make the protein less flexible and thus block the substrate entry or product exit from the active site. In addition, it might block the important Css31 residue, which is located at the interface of both channels, in its movements during the catalytic cycle. Stiffening of the protein seems to be the most probable mechanism of inhibition because no further connection between the zinc channel and the active site pocket is present in the enzyme.
Conclusion
From these and previous findings we can propose a hypothetical model of the sulfur pathway in the SOR and the different modes of enzyme inhibition. The chimney-like structures at the fourfold symmetry axes formed by two phenylalanine rings are not essential for activity, but presumably act as restrictive elements for the access of the hydrophobic sulfur substrate to the inner hollow. Product exit might occur via hydrophilic channels, which have their outlets at the threefold symmetry axes. Enlargement of both openings increased the enzyme activity several-fold and also affected the formation of H2S and the stoichiometry of reaction products. In contrast, the integrity of the active site pore, which provides a passage to the catalytic center, cannot be opened significantly without decreasing the specific activity of the enzyme. The inhibition of the SOR activity by Hg2+ and IAA occurs by binding of the compounds to different cysteine residues within the active site. In contrast, Zn2+ does not bind anywhere in the active site but in a separate channel. It might restrict protein flexibility and/or substrate and product movement within the protein subunits. It might also effect movements of the active site-cysteine persulfide (Css31) side chain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Felicitas Pfeifer (Darmstadt, Germany) for her generosity and encouragement. A. Veith and A. Kletzin were supported by a grant of the Deutsche Forschungsgemeinschaft (Az Kl885-5/1). T. Urich was supported by a Marie Curie Host Fellowship (HPMT-CT-2000-00045).
Footnotes
References
Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L. W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C., and Zwart, P. H. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221.
Arnold, K., Bordoli, L., Kopp, J., and Schwede, T. (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254.
Chen, Z. W., Jiang, C. Y., She, Q., Liu, S. J., and Zhou, P. J. (2005). Key role of cysteine residues in catalysis and subcellular localization of sulfur oxygenase-reductase of Acidianus tengchongensis. Appl. Environ. Microbiol. 71, 621–628.
Chen, Z. W., Liu, Y. Y., Wu, J. F., She, Q., Jiang, C. Y., and Liu, S. J. (2007). Novel bacterial sulfur oxygenase reductases from bioreactors treating gold-bearing concentrates. Appl. Microbiol. Biotechnol. 74, 688–698.
Collaborative Computational Project. (1994). The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763.
DeLano, W. L. (2002). The PyMOL Molecular Graphics System, 0.97 Edn. San Carlos, CA: DeLano Scientific.
Emmel, T., Sand, W., König, W. A., and Bock, E. (1986). Evidence for the existence of a sulfur oxygenase in Sulfolobus brierleyi. J. Gen. Microbiol. 132, 3415–3420.
Fischer, D. S., and Price, D. C. (1964). Simple serum iron method using new sensitive chromogen tripyridyl-s-triazine. Clin. Chem. 10, 21–25.
Friedrich, C. G., Bardischewsky, F., Rother, D., Quentmeier, A., and Fischer, J. (2005). Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8, 253–259.
Frigaard, N. U., and Dahl, C. (2009). Sulfur metabolism in phototrophic sulfur bacteria. Adv. Microb. Physiol. 54, 103–200.
Ghosh, W., and Dam, B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 33, 999–1043.
Kamyshny, A. (2009). Solubility of cyclooctasulfur in pure water and sea water at different temperatures. Geochim. Cosmochim. Acta 73, 6022–6028.
Kelley, L. A., and Sternberg, M. J. (2009). Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371.
Kletzin, A. (1989). Coupled enzymatic production of sulfite, thiosulfate, and hydrogen sulfide from sulfur: purification and properties of a sulfur oxygenase reductase from the facultatively anaerobic archaebacterium Desulfurolobus ambivalens. J. Bacteriol. 171, 1638–1643.
Kletzin, A. (2007). “23: Metabolism of inorganic sulfur compounds in archaea,” in Archaea. Evolution, Physiology, and Molecular Biology, eds R. A. Garrett and H.-P. Klenk (Oxford: Blackwell Publishing), 261–274.
Kletzin, A. (2008). “15. Oxidation of sulfur and inorganic sulfur compounds in Acidianus ambivalens,” in Microbial Sulfur Metabolism, eds C. Dahl and C. G. Friedrich (Berlin: Springer), 184–201.
Kletzin, A., Urich, T., Müller, F., Bandeiras, T. M., and Gomes, C. M. (2004). Dissimilatory oxidation and reduction of elemental sulfur in thermophilic archaea. J. Bioenerg. Biomembr. 36, 77–91.
Li, M., Chen, Z., Zhang, P., Pan, X., Jiang, C., An, X., Liu, S., and Chang, W. (2008). Crystal structure studies on sulfur oxygenase reductase from Acidianus tengchongensis. Biochem. Biophys. Res. Commun. 369, 919–923.
McRee, D. E. (1999). XtalView Xfit – a versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125, 156–165.
Moll, R., and Schäfer, G. (1988). Chemiosmotic H+ cycling across the plasma membrane of the thermoacidophilic archaebacterium Sulfolobus acidocaldarius. FEBS Lett. 232, 359–363.
Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255.
Otwinowski, Z., and Minor, W. (1997). “Processing of X-ray diffraction data collected in oscillation mode,” in Methods in Enzymology, Vol. 276: Macromolecular Crystallography, Part A, eds C. W. Carter, Jr. and R. M. Sweet (New York: Academic Press), 307–326.
Pelletier, N., Leroy, G., Guiral, M., Giudici-Orticoni, M. T., and Aubert, C. (2008). First characterisation of the active oligomer form of sulfur oxygenase reductase from the bacterium Aquifex aeolicus. Extremophiles 12, 205–215.
Read, R. J. (1986). Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A 42, 140–149.
Rohwerder, T., and Sand, W. (2003). The sulfane sulfur of persulfides is the actual substrate of the sulfur-oxidizing enzymes from Acidithiobacillus and Acidiphilium spp. Microbiology 149, 1699–1710.
Roy, A. B., and Trudinger, P. A. (1970). “The chemistry of some sulphur compounds,” in The Biochemistry of Inorganic Compounds of Sulphur, eds A. B. Roy and P. A. Trudinger (Cambridge: Cambridge University Press), 7–29.
Schägger, H., and von Jagow, G. (1987). Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379.
Skerra, A. (1994). Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene 151, 131–135.
Sun, C. W., Chen, Z. W., He, Z. G., Zhou, P. J., and Liu, S. J. (2003). Purification and properties of the sulfur oxygenase/reductase from the acidothermophilic archaeon, Acidianus strain S5. Extremophiles 7, 131–134.
Urich, T. (2005). The Sulfur Oxygenase Reductase from Acidianus ambivalens; Functional and Structural Characterization of a Sulfur-Disproportionating Enzyme. Ph.D. dissertation, Technische Universität, Darmstadt.
Urich, T., Bandeiras, T. M., Leal, S. S., Rachel, R., Albrecht, T., Zimmermann, P., Scholz, C., Teixeira, M., Gomes, C. M., and Kletzin, A. (2004). The sulphur oxygenase reductase from Acidianus ambivalens is a multimeric protein containing a low-potential mononuclear non-haem iron centre. Biochem. J. 381, 137–146.
Urich, T., Coelho, R., Kletzin, A., and Frazao, C. (2005a). The sulfur oxygenase reductase from Acidianus ambivalens is an icosatetramer as shown by crystallization and Patterson analysis. Biochim. Biophys. Acta 1747, 267–270.
Urich, T., Kroke, A., Bauer, C., Seyfarth, K., Reuff, M., and Kletzin, A. (2005b). Identification of core active site residues of the sulfur oxygenase reductase from Acidianus ambivalens by site-directed mutagenesis. FEMS Microbiol. Lett. 248, 171–176.
Keywords: Archaea, sulfur metabolism, site-directed mutagenesis, structural biology, X-ray crystallography
Citation: Veith A, Urich T, Seyfarth K, Protze J, Frazão C and Kletzin A (2011) Substrate pathways and mechanisms of inhibition in the sulfur oxygenase reductase of Acidianus ambivalens. Front. Microbio. 2:37. doi: 10.3389/fmicb.2011.00037
Received: 29 December 2010;
Paper pending published: 10 January 2011;
Accepted: 17 February 2011;
Published online: 07 March 2011.
Edited by:
Martin G. Klotz, University of Louisville, USAReviewed by:
Kathleen Scott, University of South Florida, USAUlrike Kappler, University of Queensland, Australia
Copyright: © 2011 Veith, Urich, Seyfarth, Protze, Frazão and Kletzin. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Arnulf Kletzin, Institute of Microbiology and Genetics, Technische Universität Darmstadt, Schnittspahnstraße 10, 64287 Darmstadt, Germany. e-mail: kletzin@bio.tu-darmstadt.de