
- 1 Coxiella Pathogenesis Section, Laboratory of Intracellular Parasites, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Hamilton, MT, USA
- 2 Department of Biomedical Sciences, Oregon State University, Corvallis, OR, USA
Infections by obligate intracellular bacterial pathogens result in significant morbidity and mortality worldwide. These bacteria include Chlamydia spp., which causes millions of cases of sexually transmitted disease and blinding trachoma annually, and members of the α-proteobacterial genera Anaplasma, Ehrlichia, Orientia, and Rickettsia, agents of serious human illnesses including epidemic typhus. Coxiella burnetii, the agent of human Q fever, has also been considered a prototypical obligate intracellular bacterium, but recent host cell-free (axenic) growth has rescued it from obligatism. The historic genetic intractability of obligate intracellular bacteria has severely limited molecular dissection of their unique lifestyles and virulence factors involved in pathogenesis. Host cell restricted growth is a significant barrier to genetic transformation that can make simple procedures for free-living bacteria, such as cloning, exceedingly difficult. Low transformation efficiency requiring long-term culture in host cells to expand small transformant populations is another obstacle. Despite numerous technical limitations, the last decade has witnessed significant gains in genetic manipulation of obligate intracellular bacteria including allelic exchange. Continued development of genetic tools should soon enable routine mutation and complementation strategies for virulence factor discovery and stimulate renewed interest in these refractory pathogens. In this review, we discuss the technical challenges associated with genetic transformation of obligate intracellular bacteria and highlight advances made with individual genera.
Introduction
Obligate intracellular bacterial pathogens are an understudied but significant group of human disease agents. These bacteria are thought to have emerged from ancestral non-pathogens through a pathoadaptive evolutionary process that involves significant genome reduction (Pallen and Wren, 2007). Ongoing gene loss in obligate intracellular bacteria is indicated by the presence of pseudogenes whose functions are often compensated for by the host cell. Some obligate intracellular bacteria, such as Rickettsia prowazekii and Chlamydia trachomatis, appear in the final stages of host cell adaptation, having cleared most pseudogenes from their respective genomes (Andersson and Andersson, 1999). Relative to these bacteria, Coxiella burnetii has a sophisticated genome with central metabolic pathways largely intact, suggesting a more recent adaptation to an obligate intracellular lifestyle (Seshadri et al., 2003).
Despite dramatically reduced genomes relative to most free-living bacterial pathogens, obligate intracellular bacterial pathogens still retain potent pathogenetic potential that can manifest in infections ranging from asymptomatic to fulminating and deadly (Walker, 1989; Maurin and Raoult, 1999). Unfortunately, the historic lack of genetic tools for obligates has severely limited molecular dissection of mechanisms associated with intracellular parasitism and animal pathogenesis. Many genes encoding putative virulence factors have been revealed by pathogen genome sequences (Andersson et al., 1998; Seshadri et al., 2003). However, because methods for site-specific gene inactivation and complementation are lacking, molecular Koch’s postulates (Falkow, 2004) have been impossible to fulfill for these genes. Consequently, gene function and regulation have often been explored using heterologous expression in surrogate hosts (Whitworth et al., 2005; Raghavan et al., 2008; Voth et al., 2009).
In this review, we discuss the experimental hurdles associated with developing genetic transformation systems for obligate intracellular bacteria and review the genetic tools that are currently available.
Technical Considerations in Transforming Obligate Intracellular Bacteria
A pathogen’s obligate reliance on a eukaryotic host cell for growth complicates several steps in genetic transformation that are easily conducted with free-living bacteria. Nonetheless, by employing tenacity and attention to detail, several investigators have overcome technical hurdles to establish at least rudimentary genetic systems for most pathogenic obligate intracellular bacteria. In this section, we highlight the special experimental considerations associated with genetic transformation systems of these bacteria.
Bacterial Purification
Before any genetic transformation procedure, obligate intracellular bacteria must be purified to some extent from host cells and concentrated to high density in a viable form. Depending on the degree of purity, the procedure can involve several centrifugation steps that take nearly a full day to complete (Shannon and Heinzen, 2007). For organisms that grow to low density in host cells, such as spotted fever group (SFG) rickettsia, yields can be poor and allow for only a few electroporation experiments (Kleba et al., 2010). To ensure utmost viability, some obligate intracellular bacteria are electroporated immediately after purification (Qin et al., 2004), thereby eliminating the convenience of storing purified bacteria for subsequent transformation experiments. Several low ionic strength electroporation buffers have been used, ranging from distilled water (Binet and Maurelli, 2009) to buffers containing osmoprotectants such as sucrose and glycerol (Beare et al., 2009). Organisms are washed several times in buffers and resuspended at high density (approx. 1010 bacteria per ml) prior to electroporation.
A consideration when purifying obligate intracellular bacteria for transformation experiments is that many display developmental forms that may be differentially infective and/or receptive to electroporation. For example, the large reticulate cell (RC) of Anaplasma phagocytophilum may be more amenable to electroporation than the smaller dense-cored cell (DC) with its characteristic condensed chromatin. However, the RC is poorly infective relative to the DC (Troese and Carlyon, 2009). A similar and more extreme example involves reticulate bodies (RB) of chlamydia that may be quite receptive to electroporation but are difficult to purify and considered non-infectious (Bavoil et al., 2000). Large cell variant (LCV) and small cell variant (SCV) development forms of C. burnetii appear equally infectious for host cells (Coleman et al., 2004). However, because the permissiveness of SCV and LCV to electroporation is unknown, bacteria used in transformation experiments are purified when host cells contain roughly equal numbers of cell forms (Beare et al., 2009).
Antibiotic Selection and Construct Optimization
Thus far, positive selection of transformed obligate intracellular bacteria has been conducted exclusively by selecting for antibiotic resistance. Restrictions based on antibiotic clinical efficacy in treating human infections significantly reduces the set of antibiotic resistance genes suitable for transformation studies. Furthermore, in the United States, the Centers for Disease Control and Prevention, Division of Select Agents and Toxins, ultimately approves the use of antibiotic resistance genes in select agent pathogens. R. prowazekii, Rickettsia rickettsii, and virulent phase I strains of C. burnetii fall into this category (Atlas, 2003). Work with these organisms also requires stringent biosafety level 3 procedures.
The minimal inhibitory concentrations (MIC) of approved antibiotics must first be established in a relevant host cell model system. Complicating the establishment of MICs are issues related to permeability and subcellular pharmacological activity. With the exception of cells infected with cytoplasmically localized Rickettsia or Orientia spp., antibiotics used in selection must permeate at least two host cell lipid bilayers: the plasma membrane and the membrane of the pathogen-occupied vacuole. To overcome these diffusion barriers, the concentration of antibiotics required for selection may be several fold higher than typically used with free-living bacteria. A high MIC may be toxic to host cells. For instance, high levels of chloramphenicol can inhibit mitochondrial function (Li et al., 2010). Moreover, the microenvironment of intracellular compartments may inhibit antibiotic activity. For example, the acidic parasitophorous vacuole of C. burnetii clearly inhibits the bactericidal effect of certain antibiotics (Maurin et al., 1992). Raising vacuolar pH with alkalizing agents, such as hydroxychloroquine, can dramatically increase antibiotic killing of C. burnetii (Maurin et al., 1992). Indeed, long-term combination doxycycline/hydroxychloroquine therapy is now recommended for treatment of chronic Q fever endocarditis (Maurin and Raoult, 1999). Finally, high rates of spontaneous mutation to resistance is a problem with some pathogen–antibiotic pairs such as C. burnetii–ampicillin (Suhan et al., 1996) and R. prowazekii–rifampin (Rachek et al., 1998). Expansion of non-transformed resistant mutants is exacerbated by the several week selection procedures used in many transformation protocols for obligates.
Constructs used in transformation generally have antibiotic resistance genes and supplementary screenable markers, such as genes encoding fluorescent proteins, under control of a characterized pathogen promoter. Again, developmental biology needs to be considered when picking a promoter to ensure constitutive expression of marker genes throughout the infectious cycle of a pathogen. Optimal gene expression may require use of chemically synthesized genes that more closely match the codon usage of a particular organism. This is a particular issue with pathogens having unusual genomic G + C content, such as Rickettsia spp. (Qin et al., 2004). Finally, active pathogen restriction/modification systems may significantly lower transformation frequency. Propagation of transformation constructs in methylation defective E. coli strains may alleviate this problem (Binet and Maurelli, 2009).
Introduction of DNA
Electroporation has been universally effective in introducing DNA into obligate intracellular bacteria. Because of their relatively small particle size, high electrical field strengths (e.g., 16–24 kV/cm) are generally used, with organisms typically maintaining high viability after the procedure. Indeed, two pulses of 16 kV/cm were found optimal for DNA uptake by the ∼0.3 μm in diameter elementary bodies (EB) of Chlamydia psittaci (Binet and Maurelli, 2009). Also facilitating DNA uptake is the use of relatively large amounts of DNA (e.g., 5–10 μg; Rachek et al., 1998; Beare et al., 2009; Binet and Maurelli, 2009). Prior to infection of host cells, DNaseI protection assays can be conducted to confirm uptake of target DNA by microorganisms. Electroporated organisms are treated with DNaseI to remove extracellular plasmid, and the plasmid DNA protected by pathogen uptake is detected by Q-PCR (Tam et al., 1994; Rachek et al., 1998; Binet and Maurelli, 2009).
Scoring and Expansion of Transformants
Obligate intracellular bacteria have slow generation times relative to most free-living bacteria. For example, C. burnetii has a generation time of approximately 11 h during exponential phase in Vero cells (Coleman et al., 2004). Thus, to allow adequate expression of resistance markers, antibiotics are generally added to tissue culture media at 12–24 h post-infection with electroporated organisms (Rachek et al., 1998; Baldridge et al., 2005; Felsheim et al., 2006; Beare et al., 2009; Binet and Maurelli, 2009). Delayed addition of antibiotic may also be required to allow differentiation and metabolic activation of developmental forms (Bavoil et al., 2000).
A preliminary score of successful transformation is obvious growth of organisms in tissue culture containing an inhibitory concentration of antibiotic. Low transformation frequencies combined with slow growth often require incubations over several weeks to expand transformants to a level detectable by microscopy (Rachek et al., 1998; Baldridge et al., 2005, 2010b; Beare et al., 2009; Felsheim et al., 2010). Expansion of organisms that do not actively lyse host cells, such as C. burnetii, is aided by sequential rounds of infection, mechanical host cell lysis, and re-infection of fresh host cells (Beare et al., 2009). Transformants of R. rickettsii and C. psittaci have been detected in 7 and 10 days, respectively, by using plaque assays (described below; Binet and Maurelli, 2009; Kleba et al., 2010; Clark et al., 2011).
Because spontaneous mutation to antibiotic resistance can result in significant background numbers of non-transformed bacteria (Suhan et al., 1996; Rachek et al., 1998), additional screens are required to confirm genetic transformation. Molecular typing of transformants, either as clones or pools, by Southern blotting is arguably the gold standard and most commonly used method for confirming stable maintenance of target DNA (Suhan et al., 1996; Rachek et al., 1998; Qin et al., 2004; Felsheim et al., 2006; Baldridge et al., 2007a, 2010b; Liu et al., 2007; Beare et al., 2009; Binet and Maurelli, 2009; Driskell et al., 2009; Chen et al., 2010; Kleba et al., 2010). Often complementing Southern blotting is rescue cloning of transposon (Tn)-encoded antibiotic resistance genes and sequencing to map integration sites (Qin et al., 2004; Baldridge et al., 2005, 2007a, 2010b; Felsheim et al., 2006, 2010; Liu et al., 2007; Beare et al., 2009). Direct sequencing of transformant chromosomal DNA has also mapped Tn integration sites (Kleba et al., 2010). Sequencing of recombination sites using PCR and primers flanking target genes has confirmed allelic exchange experiments (Rachek et al., 1998; Binet and Maurelli, 2009). Assessment of transformation by PCR with primers specific to only transformation DNA should be avoided as false positive amplicons can arise from the persistence of free transformation DNA in cell culture (P. A. Beare and R. A. Heinzen, unpublished data).
Direct detection of reporter/antibiotic resistance gene expression has frequently supplemented genome analysis in scoring transformation. A commonly used method is microscopic visualization of green or red fluorescent proteins encoded by introduced transgenes (Lukacova et al., 1999; Troyer et al., 1999; Renesto et al., 2002; Baldridge et al., 2005, 2010b; Felsheim et al., 2006, 2010; Liu et al., 2007; Beare et al., 2009; Chen et al., 2010). RT-PCR, northern blotting and immunoblotting have been employed by several investigators to confirm expression of fluorescent and antibiotic resistance proteins (Suhan and Thompson, 2000; Baldridge et al., 2005, 2010b; Felsheim et al., 2010).
Cloning of Transformants
Generation of clones from a mixed population of transformants is essential for downstream phenotypic analyses. Cloning derives organisms with single mutational events and eliminates potential viable but non-transformed carryover bacteria that, depending on the experiment, may allow removal of antibiotic selection. Cloning of free-living bacteria, but not obligate intracellular bacteria, is easily done by colony formation on agar plates. A convenient method for some obligate intracellular bacteria is the plaque assay. This procedure relies on pathogen destruction of a localized region of the host cell monolayer that is visible to the naked eye. Monolayer plaques are picked through an agarose overlay and the harvested clonal populations of bacteria further expanded. The assay is generally applicable to organisms with active mechanisms of cell-to-cell spread and/or host cell lysis, such as SFG rickettsia (Wike et al., 1972) and some Chlamydia spp. (Matsumoto et al., 1998). Indeed, the assay was recently employed to clone transformants of R. rickettsii (Kleba et al., 2010) and C. psittaci (Binet and Maurelli, 2009). An alternate method of cloning involves extraction of bacteria from individual infected cells by micromanipulation (Beare et al., 2007). Infections conducted at a low multiplicity of infection (e.g., 0.1) initially result in cells that contain the biological equivalent of a very small bacterial colony. A micromanipulator is used to place a glass micropipette, attached to a hydraulically driven microinjector, directly into an isolated infected cell. The clonal bacterial contents are then removed by running the microinjector in reverse. This procedure is applicable to pathogens where infected monolayer-forming host cells are easily identifiable by light microscopy. The method was recently used to clone a Tn mutant of C. burnetii (Beare et al., 2009). Finally, transformant clones of C. burnetii (Suhan et al., 1996) and R. prowazekii (Rachek et al., 2000; Qin et al., 2004) have been derived by the tedious and time-consuming method of end-point limiting dilution.
Successes in Transformation of Obligate Intracellular Bacterial Pathogens
Despite considerable technical barriers, significant advances in genetic manipulation of obligate intracellular bacteria have occurred in the last decade. These include (1) Tn mutagenesis, which has identified a key virulence determinant in rickettsia and allowed generation of fluorescent bacteria useful in host–pathogen interaction studies, (2) discovery of a C. burnetii shuttle vector used in identifying secreted effector proteins, and (3) the first successful site-specific inactivation of a virulence gene by allelic exchange. Achievements specific to bacterial genera are outlined below.
Rickettsia Spp.
Efforts with R. prowazekii, the cause of epidemic typhus, have led the way in genetic manipulation of obligate intracellular bacteria. The organism was originally transformed to rifampin resistance by using a portion of the R. prowazekii rpoB gene from a strain containing a mutation conferring resistance to rifampin (Rachek et al., 1998). Following electroporation with a ColE1-based suicide plasmid carrying the allele, recombination occurred between the plasmid and chromosomal rpoB locus to result in allelic exchange and genetic transformation to rifampin resistance (Rachek et al., 1998).
The described rifampin resistance allele is recessive in a merodiploid strain containing a wild-type gene, thereby limiting its use in R. prowazekii as a selectable marker (Wood and Azad, 2000). Therefore, two alternate methods of positive selection were tested that employed genes that are dominant by producing enzymes that inactivate antibiotics. The first system utilized ereB, which encodes an esterase that hydrolyzes the lactone ring of erythromycin (Rachek et al., 2000). Successful transformation to erythromycin resistance resulted from chromosomal integration of a suicide plasmid carrying ereB and rickettsial gltA, encoding citrate synthase. Homologous recombination by a single crossover event was mediated by gltA to result in two chromosomal copies of gltA flanking heterologous ereB (Rachek et al., 2000). The second system used arr-2 which encodes an enzyme that inactivates rifampin by ADP-ribosylation (Qin et al., 2004; Liu et al., 2007). For optimal expression, arr-2 codons were engineered to mimic rickettsial codon usage and cloned downstream from a strong R. prowazekii ribosomal subunit promoter (rpsLP). The arr-2 cassette was incorporated into two Tn systems that were used to randomly mutagenize R. prowazekii. The first system employed an Epicentre EZ::Tn5 transposon system (Qin et al., 2004). Experiments inconsistently yielded transformants, and when successful, only a few unique Tn insertion sites were identified (Qin et al., 2004). Inconsistency in generating active transposome complexes (i.e., Tn plus transposase) was speculated as contributing to low transformation efficiency (Qin et al., 2004; Liu et al., 2007). Seeking a more robust system of random mutagenesis, a mariner-based Himar1 Tn system was tested (Lampe et al., 1999; Liu et al., 2007). Transposition of Himar1 only requires expression of the Himar1 transposase and no additional host factors (Lampe et al., 1999), and was previously used by Felsheim et al. (2006) in successful Tn mutagenesis of the obligate intracellular bacterium A. phagocytophilum. R. prowazekii was electroporated with a suicide plasmid (pMW1650) encoding Himar1 with an integral arr-2 cassette, a green fluorescent protein (GFP) gene driven by the strong rickettsial outer membrane protein A (OmpA) promoter (Baldridge et al., 2005), and a ColE1 origin of replication for rescue cloning. Transformation resulted in green fluorescent rickettsiae and numerous transposition events. The improved transformation efficiency of Himar1 over the transposome system was attributed, in part, to the 2-bp TA recognition sequence of Himar1 and the A + T richness of rickettsiae (∼71%).
Transformation using pMW1650 has recently provided novel information on virulence mechanisms of R. rickettsii, the cause of Rocky Mountain spotted fever. In one study, transformation yielded an unusually small rifampin-resistant plaque (Kleba et al., 2010). Unlike typhus group rickettsia, SFG rickettsia readily form plaques in epithelial cell and fibroblast monolayers (Wike et al., 1972). Mapping of the Himar1 insertion site of this clone revealed that sca2, encoding an autotransporter protein, had been insertionally inactivated. The mutation and small plaque phenotype correlated with defective actin-based motility, a mechanism used by SFG rickettsia for cell-to-cell spread (Figure 1; Heinzen et al., 1993). Sca2 has since been defined as a bacterial actin nucleator that functionally mimics eukaryotic formin proteins (Haglund et al., 2010). A definitive virulence role for sca2 was demonstrated by showing attenuated virulence of the mutant in a guinea pig challenge model (Kleba et al., 2010). A second study elucidated the mechanism of plaque clarity in SFG rickettsia and resolved a running debate on whether the phenotype is associated with virulence. Lytic strains that produce clear plaques have historically been considered more virulent than non-lytic strains that form opaque or “turbid” plaques (Hackstadt, 1996). Genome sequencing of spontaneously arising non-lytic and lytic variants of normally lytic and non-lytic strains, respectively, revealed that lytic strains of R. rickettsii have mutations in a gene annotated as relA/spoT-like that are predicted to disrupt protein function. Transformation of a lytic variant with a pMW1650 construct that contains Himar1 with a wild-type version of relA/spoT, complemented the mutant to result in a non-lytic, turbid plaque phenotype. Thus, RelA/SpoT activity is responsible for the turbid plaque phenotype, presumably manifested by the protein’s putative role in rickettsial alarmone physiology. Interestingly, isogenic lytic and non-lytic strains were equally virulent for guinea pigs, indicating plaque clarity is not a correlate of virulence in SFG rickettsia (Clark et al., 2011).
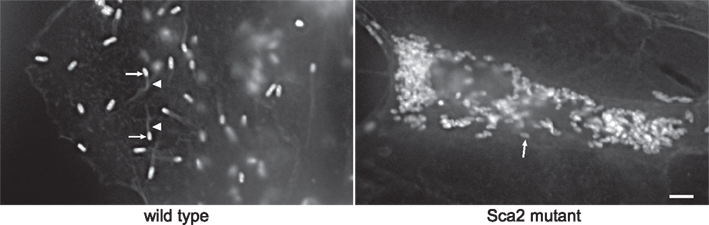
Figure 1. A Himar1 insertion in R. rickettsii sca2 eliminates actin-based motility. The right micrograph shows wild-type R. rickettsii (arrows) with typical filamentous actin tails (arrowhead). The left micrograph depicts a small plaque-forming mutant of R. rickettsii with a Himar1 insertion in sca2, which encodes an autotransporter protein (Kleba et al., 2010). The mutant rickettsia (arrow) lack actin tails. Rickettsia were stained by immunofluorescence using a specific monoclonal antibody and filamentous actin was stained with Alex Fluor 598 phalloidin. Bar, 3 μm. (Micrographs courtesy of Ted Hackstadt, Rocky Mountain Laboratories).
Finally, in the first and currently only example of targeted disruption of a putative virulence gene encoded by an obligate intracellular bacterium, the R. prowazekii pld gene, encoding phospholipase D, was disrupted by a double crossover event with a linear 2698 bp DNA fragment containing pld disrupted with the arr-2 cassette (Driskell et al., 2009). Like the R. rickettsii sca2 mutant, the pld mutant showed attenuated virulence in a guinea pig challenge model (Driskell et al., 2009).
Achievements with R. prowazekii were followed by transformation of non-pathogenic Rickettsia monacensis and Rickettsia montanensis to chloramphenicol resistance using the EZ::Tn5 system (Baldridge et al., 2005, 2010b). In transformation of R. monacensis, the Tn carried genes encoding chloramphenicol acetyltransferase (CAT) and GFP independently driven by the rickettsial OmpA promoter. Following extensive tissue culture passage, a clonal population of green fluorescent rickettsia with a single Tn insertion was obtained. The transformant proved useful in visualizing infection dynamics of R. monacensis in ticks (Baldridge et al., 2007b). Mapping of a Tn5 insertion site in a later transformation experiment revealed a hitherto unknown 23.5 kb low copy-number plasmid in R. monacensis (Baldridge et al., 2007a). Termed pRM, the plasmid is highly related to pRF, 39.2 kb plasmid previously identified in Rickettsia felis by genome sequencing (Ogata et al., 2005). Related plasmids have now been discovered in multiple Rickettsia spp. (Baldridge et al., 2010a). A valuable shuttle vector for rickettsial transformation could be derived by subcloning regions necessary for pRM/pRF replication into a standard E. coli cloning vector. For transformation of R. montanensis, the EZ::Tn5 construct was altered to contain GFP and CAT genes under control of the rickettsial outer membrane protein B (OmpB) promoter (Baldridge et al., 2010a). Because OmpB is more abundant than OmpA in SFG rickettsia (Policastro and Hackstadt, 1994), enhanced expression of GFP was anticipated. Again, a clone containing a single Tn insertion was isolated after cell culture passage; however, GFP expression was poor, leading the authors to conclude that the ompA promoter is a better choice for driving expression of foreign genes in SFG rickettsia (Baldridge et al., 2010a).
Single reports have been published on transformation of Rickettsia typhi (Troyer et al., 1999) and Rickettsia conorii (Renesto et al., 2002), the etiologic agents of murine typhus and Mediterranean spotted fever, respectively. Both studies employed suicide vectors containing gfp cloned immediately downstream of the 3′ end of a wild-type version of the pathogen’s rpoB gene. PCR demonstrated homologous recombination between chromosomal and plasmid rpoB sequences, and flow cytometry showed elevated levels of green fluorescence in cells infected with transformant populations. While demonstrating the feasibility of transforming these rickettsial species, the lack of a selectable marker limits the utility of the described transformation strategies.
A. phagocytophilum and Anaplasma marginale
Anaplasma phagocytophilum and A. marginale, which cause human and bovine anaplasmosis, respectively, have been transformed with Himar1 (Felsheim et al., 2006, 2010). In these cases, the Himar1 transposase and Tn were carried on separate suicide plasmids, thereby eliminating promiscuous transposition events that can occur during plasmid propagation in E. coli when both elements are carried on a single plasmid (P. A. Beare, personal observation). Expression of Tn-encoded GFP and spectinomycin resistance genes was driven by the A. marginale tr promoter (Barbet et al., 2005) as single transcriptional unit. Transposase expression was also directed by the tr promoter. Bright green fluorescent bacteria resulted from co-transformation with both suicide plasmids. Multiple Tn integration sites were mapped in A. phagocytophilum transformants (Felsheim et al., 2006). Conversely, after 2 months of antibiotic selection, only a single Tn insertion site was identified in A. marginale. Surprisingly, in this instance, transformation did not result from Tn transposition but rather from integration of plasmid DNA by single crossover at the tr promoter region (Felsheim et al., 2010). Nonetheless, this transformant has proven useful in monitoring transmission of A. marginale between cattle and ticks (Noh et al., 2011).
Coxiella Burnetii
The most sophisticated genetic tools are currently available for C. burnetii, which incidentally, was the first obligate intracellular bacterium to be stably genetically transformed in 1996 (Suhan et al., 1996). As discussed in more detail below, recent advances in C. burnetii genetic transformation have been dramatically aided by the recent discovery of a medium that supports host cell-free (axenic) growth of the organism (Omsland et al., 2009, 2011).
The first successful genetic transformation of C. burnetii used a chimeric plasmid termed pSKO(+)1000 comprised of a previously defined C. burnetii 5.8 kb autonomous replication sequence (ars) (Suhan et al., 1994) and the E. coli cloning vector pBluescript, which carries a β-lactamase (blaM) gene and a ColE1 ori (Suhan et al., 1996). Ampicillin-resistant C. burnetii were recovered from host cells after 2–3 months of selection. Southern blotting of transformant DNA with a blaM probe showed both chromosomal integration and ars-dependent autonomous replication of pSKO(+)1000. Integration occurred via homologous recombination between the plasmid ars and the corresponding region in the C. burnetii chromosome (Suhan et al., 1996). The lengthy selection process also yielded ampicillin-resistant organisms without blaM, indicating spontaneous mutation to antibiotic resistance had occurred. In a subsequent transformation report, pSKO(+)1000 was modified to contain a GFP reporter gene that, like the blaM gene, was driven by an E. coli promoter (Lukacova et al., 1999). Weakly green fluorescent organisms were shown, but molecular data confirming transformation was not provided (Lukacova et al., 1999).
Over a decade elapsed before the next successful transformation of C. burnetii, an accomplishment that employed the Himar1 Tn (Beare et al., 2009). Similar to the Anaplasma spp. Himar1 system (Felsheim et al., 2006), a two-plasmid system was used with a pathogen promoter, in this case 1169P, driving expression of transposase, CAT, and mCherry red fluorescent protein genes. CBU1169 encodes the small heat shock protein Hsp20 and is constitutively expressed at a moderate level throughout the C. burnetii infectious cycle (P. A. Beare and R. A. Heinzen, unpublished data). After 5 weeks of incubation in the presence of antibiotic and multiple passages, Vero cells infected with C. burnetii co-electroporated with both Himar1 plasmids displayed obvious bacteria-filled intracellular vacuoles. Subsequent rescue cloning of the ColE1 ori revealed 35 unique Tn insertion sites, with two located in the 37.4-kb QpH1 plasmid. A transformant termed B2c was cloned by micromanipulation (Beare et al., 2007), expanded for 5 weeks in cell culture, and shown to contain a Tn insertion in ftsZ, a gene required for bacterial cell division. The FtsZ mutant grew in long filaments having incomplete division septae and represented the first example of a clonal population of C. burnetii harboring a defined gene mutation generated by genetic transformation.
Random mutagenesis of C. burnetii using a host cell-based transformation protocol was a significant advance in genetic manipulation of the organism. However, the lengthy selection and clonal expansion processes severely limits the number of clones that can be phenotyped as well as the testing of new transformation technologies. Moreover, obligatism eliminates Tn mutants that cannot infect and subsequently grow in host cells, thereby precluding identification of many genes required for virulence. However, the recent breakthrough in axenic growth of C. burnetii has now removed the substantial experimental constraints associated with an obligate intracellular lifestyle (Omsland et al., 2009, 2011). An axenic growth medium called acidified citrate cysteine medium (ACCM) was developed that supports roughly 3 logs (log10) of growth of C. burnetii over 6 days in a microaerobic environment. Importantly, axenically grown organisms plate with high efficiency in semi-solid ACCM agarose and the minute colonies that arise contain clonal populations (Omsland et al., 2009, 2011).
An entirely axenic transformation protocol is now accelerating development of C. burnetii genetic tools. Bacteria for electroporation are grown in ACCM for 6 days to early stationary phase. Following electroporation, organisms are incubated in ACCM for 24 h prior to addition of antibiotic. Three days later, organisms are plated in semi-solid ACCM agarose containing antibiotic and plates incubated for 6 days to allow colony formation. Colonies are picked, added to ACCM, and cultures incubated for 6 days to expand the clone. Thus, expanded transformant clones can now be derived in 2–3 weeks using ACCM as opposed to 2–3 months using cell culture. Moreover, both CAT and Kan genes work well as selectable markers in ACCM, allowing two transformation procedures in a single strain (Omsland et al., 2011).
Using ACCM, an optimized Himar1-based Tn mutagenesis system was developed for C. burnetii. 1169P-driven expression of CAT and mCherry as a single transcriptional unit in the original Himar1 system resulted in sufficient chloramphenicol resistance, but disappointing mCherry fluorescence (Beare et al., 2009). Therefore, a second generation Himar1 construct employed the CBU0311 (outer membrane porin P1) promoter (311P) to drive expression of mCherry independent of 1169P-driven CAT (Figure 2A). CBU0311 is constitutively expressed at high levels during the C. burnetii infectious cycle (P. A. Beare and R. A. Heinzen, unpublished data). A cloned transformant termed MC1 with an intergenic Himar1 insertion between CBU0316 and CBU0317 was obtained that shows no growth defect in Vero cells relative to wild-type C. burnetii (Beare et al., 2010). By both fluorescence microscopy and flow cytometry, mCherry protein fluorescence was significantly higher in MC1 than the previously described B2c clone (Beare et al., 2009; Figures 2B,C).
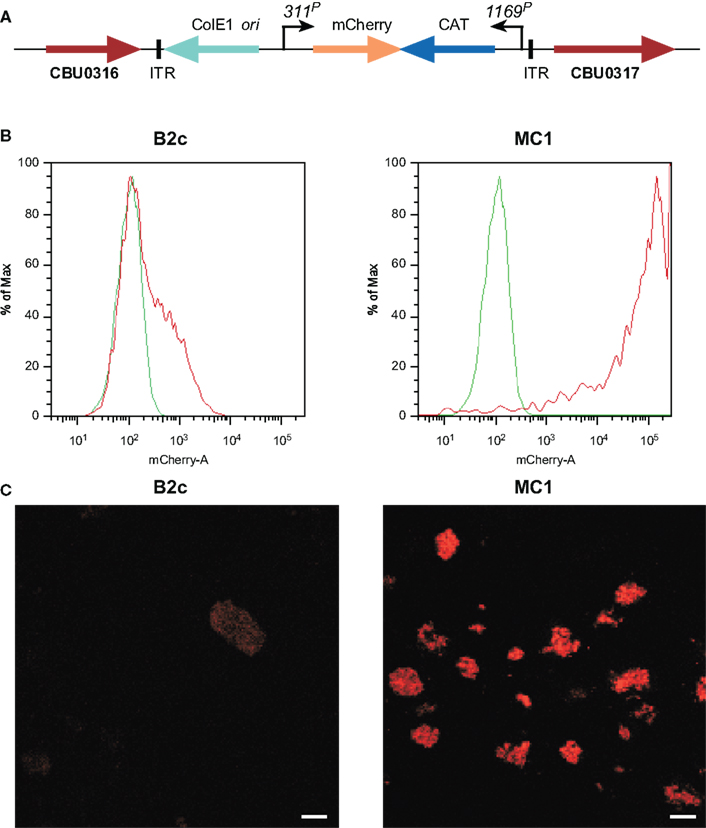
Figure 2. Characterization C. burnetii Nine Mile (phase II) MC1 and B2c Himar1 transformants. (A) Schematic of the Himar1 chromosomal integration site in C. burnetii MC1. The Himar1 transposon is flanked by inverted terminal repeat (ITR) elements and inserted into an intergenic region between CBU0316 and CBU0317. Flow cytometry (B) and confocal fluorescence microscopy (C) of live Vero cells infected with MC1 or B2c for 5 days. Both assays revealed considerably more mCherry fluorescence from MC1-containing vacuoles, where expression is driven by 311P, then from B2c-containing vacuoles, where expression is driven by 1169P (Beare et al., 2009). The green trace in flow cytometry histograms shows autofluorescence of Vero cells infected with wild-type C. burnetii for 5 days. Bars, 5 μm.
Acidified citrate cysteine medium facilitated development of two new C. burnetii transformation systems: (1) a shuttle vector that is compatible with the endogenous plasmid of the organism, and (2) a Tn7 system for single-copy, site-specific chromosomal gene integration. Plasmids with an RSF1010 ori were recently shown to autonomously replicate in C. burnetii (Chen et al., 2010; Voth et al., 2011) with a copy number of roughly 3–6 (P. A. Beare and R. A. Heinzen, unpublished data). Chen et al. (2010) constructed the vector pKM230 by inserting a cassette encoding mCherry and CAT genes, under control of C. burnetii groES (CBU1719) and com1 (CBU1910) promoters, respectively, into the Legionella pneumophila plasmid pJB908 (Sexton et al., 2004). Transformation with pKM230 yielded chloramphenicol-resistant, mCherry-fluorescing C. burnetii as assessed by growth in both ACCM and L929 cells. Subsequently, a second plasmid called pCBTEM was constructed that allows identification of C. burnetii secreted proteins that are putative Dot/Icm type IV secretion system (T4SS) substrates. The secretion assay relies on cytosolic delivery of BlaM C-terminally fused to a C. burnetii protein containing a T4SS translocation signal. Once secreted, the BlaM moiety of the protein chimera cleaves the β-lactam ring of a cell loaded fluorescent compound (CCF4/AM), resulting in blue cytosolic fluorescence (Campbell, 2004; de Felipe et al., 2008). The mCherry/CAT cassette of pKM230 was cloned into the BlaM fusion vector pXDC61 (de Felipe et al., 2008) where BlaM expression is under control of IPTG-inducible tacp, to generate pCBTEM. Infection of THP-1 cells with pCBTEM transformants confirmed translocation of six suspected T4SS effector proteins by C. burnetii. In a similar study, Voth et al. (2011) used both BlaM and adenylate cyclase (CyaA) translocation assays to show that the cryptic QpH1 plasmid of C. burnetii encodes several T4SS substrates. Two C. burnetii–E. coli shuttle vectors, termed pJB-CAT and pJB-KAN were first derived by modifying the L. pneumophila plasmid pJB2581 (Bardill et al., 2005) to encode CAT and Kan genes, respectively, under control of 1169P. These plasmids were then modified to become T4SS reporter plasmids by adding 1169P-driven blaM or cyaA (Figure 3A). Like the BlaM assay, the CyaA assay is an enzymatic reporter assay that relies on cytosol delivery of CyaA C-terminally fused to a C. burnetii T4SS effector protein. Once in the cytosol, the CyaA moiety is activated by binding cytosolic calmodulin, resulting in elevated levels of cyclic AMP (Sory and Cornelis, 1994). Both assays revealed that CBUA0015 is one of six QpH1 plasmid-encoded proteins that are secreted (Voth et al., 2011; Figure 3B). A transformation frequency (transformants/protocol) of approximately 5 × 10−5 has since been established for C. burnetii using pJB-CAT or pJB-KAN and ACCM culture techniques (Omsland et al., 2011). These vectors have multiple applications including in trans complementation of mutants generated by Tn or other mutagenesis techniques, expression of dominant/negative proteins, and expression of epitope tagged proteins for intracellular trafficking studies.
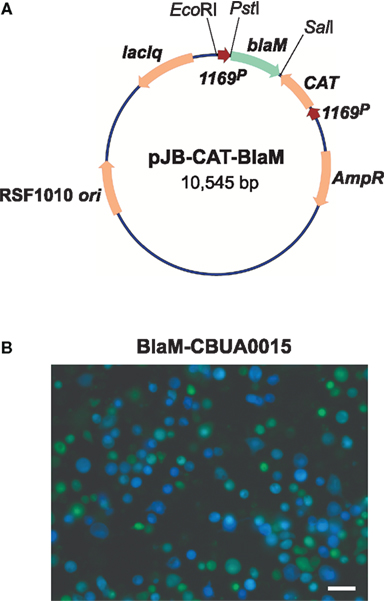
Figure 3. A C. burnetii BlaM translocation assay shuttle vector. (A) Map of the reporter plasmid pJB-CAT-BlaM which contains an RSF1010 ori that functions in both E. coli and C. burnetii. Genes encoding suspected secreted proteins are cloned downstream and in-frame with blaM using the unique SalI site. (B) BlaM translocation assay showing cytosolic delivery of a BlaM-CBUA0015 fusion protein. THP-1 cells were infected with C. burnetii Nine Mile (phase II) containing pJB-CAT-BlaM::CBUA0015 for 48 h, then incubated for 1 h with CCF4/AM. Cleavage of CCF4/AM by cytosolic BlaM results in blue fluorescent cells and indicates secretion of the fusion protein. Bar, 30 μm.
Gene dosage effects that skew phenotypes are a potential concern with the plasmid copy number of pJB-CAT/KAN vectors. As discussed above, Himar1 can be used for in cis complementation and single-copy expression of a heterologous protein (e.g., mCherry; Beare et al., 2010; Clark et al., 2011). However, the highly random nature of Himar1 genomic integration necessitates mapping and phenotyping of clones to ensure an insertion event has not generated an unintended phenotype. An alternative system of single-copy chromosomal transgene expression was developed in C. burnetii that employs the transposon Tn7. The Tn7 system utilizes a transposase encoded by tnsABCD that recognize a specific 30 bp site termed attTn7 that, in Pseudomonas aeruginosa and other Gram-negative bacteria, is located in the extreme 3′ end of glmS encoding glucosamine-6-phosphate synthetase (Choi et al., 2005). Transposition is orientation specific and occurs 36 bp downstream of attTn7 in an intergenic region (Choi et al., 2005). Analysis of C. burnetii glmS (CBU1787) revealed 24 nucleotide identity with the P. aeruginosa attTn7 site, suggesting this region comprises a functional Tn7 recognition site. To test this possibility, a two-plasmid Tn7 system was constructed (Choi et al., 2005). One suicide plasmid, termed pTNS2::1169P-tnsABCD, carries the transposase genes tnsABCD under control of 1169P. The second suicide plasmid, called pMiniTn7T-CAT::GFP, carries Tn7 containing CAT and GFP genes driven independently by 1169P and 311P, respectively (Figure 4A). Co-electroporation of C. burnetii with pTNS2::1169P-tnsABCD and pMiniTn7T-CAT::GFP, followed by chloramphenicol selection of transformants in ACCM, yielded clones with Tn7 inserted 36 bp downstream of the attTn7 site in an intergenic region between glmS (CBU1787) and the hypothetical protein encoding gene CBU1788 (Figure 4B). Vero cells infected with expanded C. burnetii Tn7 transformants display high levels of GFP expression when visualized by fluorescence microscopy at 5 days post-infection (Figure 4C).
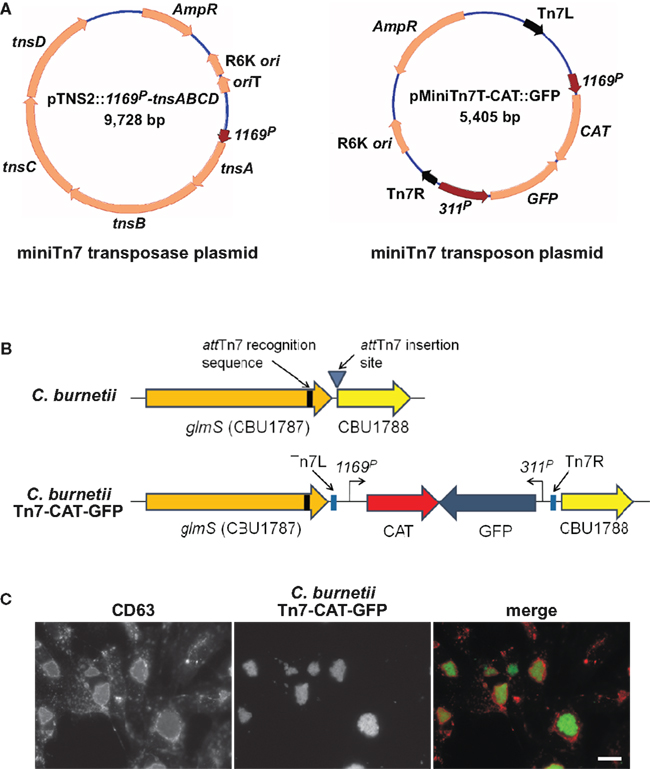
Figure 4. Coxiella burnetii Tn7 transformation system. (A) Maps of two-plasmid C. burnetii miniTn7 transposon system. The suicide plasmid pTNS2::1169P-tnsABCD encodes the tnsABCD operon under control of 1169P. The suicide plasmid pMiniTn7T-CAT::GFP encodes CAT and GFP genes driven independently by 1169P and 311P, respectively. The CAT/GFP cassette is flanked by Tn7L and Tn7R elements. (B) Schematic of the glmS regions in C. burnetii Nine Mile (phase II) and C. burnetii Nine Mile (phase II)/Tn7-CAT-GFP. (C) Fluorescence microscopy of Vero cells infected for 5 days with C. burnetii Nine Mile (phase II)/Tn7-CAT-GFP (green). Cells were fixed with 4% paraformaldehyde, then immunostained for the lysosomal protein CD63 (red). Bar, 5 μm.
Chlamydia trachomatis and Chlamydia psittaci
A perhaps underappreciated fact is that Tam et al. (1994) published the first description of genetic transformation, albeit transient in nature, of an obligate intracellular bacterium in 1994. C. trachomatis was transformed to chloramphenicol resistance by electroporation with the plasmid pPBW100. The plasmid contains 3.6 kb of an E. coli ColE1 ori cloning vector and 7 kb of pCTE1, the 7.5-kb endogenous plasmid of C. trachomatis serovar E. For antibiotic selection, a CAT gene was also cloned immediately downstream from a native chlamydial promoter. Electroporated EB, the infectious developmental form of chlamydiae, were used to infect McCoy cells and inclusions containing chloramphenicol-resistant chlamydia were initially detected. However, these were lost following several tissue culture passes. Suggested reasons for the transient nature of transformants included incompatibility of pPBW100 with pCTE1, activation of DNA restriction/modification systems that degraded pPBW100, and developmental regulation of the promoter driving CAT gene expression that resulted in insufficient levels of expression during the chlamydial developmental cycle (Tam et al., 1994). Nonetheless, this study was a technological triumph by showing that DNA could be introduced into metabolically inert EB by electroporation, that expression of heterologous selectable markers could be achieved, and that transformants could be detected in cell culture.
Fifteen years elapsed before the next successful transformation of Chlamydia spp. C. psittaci was transformed by allelic exchange using both linear and circular DNA containing an allele of the 16s rRNA gene of the organism engineered to contain mutations conferring resistance to kasugamycin and spectinomycin (Binet and Maurelli, 2009). EB were electroporated with different amounts of DNA that contained up to 8.1 kb of the C. psittaci rRNA chromosomal region plus the 16s rRNA mutations. Molecular typing of antibiotic resistant plaques picked from L929 mouse fibroblast monolayers revealed that transformation had occurred by homologous recombination, mediated by a minimum of two crossover events, with both linear and circular DNA substrates. The highest transformation frequencies (approximately 3 × 10−6) were observed using 10–20 μg of circular DNA propagated in a methylase-deficient E. coli strain. Recombination frequencies using plasmid templates dropped almost 10-fold when rRNA homologous DNA was reduced from 8.1 to 2.5 kb. Similar to the situation with the rifampin resistance allele used in rickettsial transformation (Wood and Azad, 2000), kasugamycin, and spectinomycin resistance are recessive in a merodiploid strain. Thus, their use as selectable markers necessitates elimination of the wild-type gene, thereby limiting their utility in chlamydial transformation. Nonetheless, this study provided considerable information on optimal electroporation conditions and the state of DNA substrates that favor homologous recombination.
Chlamydial natural competence?
A growing body of evidence from genomic data and in vitro experiments indicates that chlamydiae actively engage in horizontal gene transfer and that the process drives genomic diversification and acquisition of antibiotic resistance genes (DeMars et al., 2007; DeMars and Weinfurter, 2008; Suchland et al., 2009). These observations are surprising as the chlamydial genomes are largely syntenous and contain a limited number of strain-specific genes. Our understanding of natural genetic exchange in chlamydia is based on several studies. Comprehensive genetic linkage analyses of C. trachomatis clinical isolates and genome wide comparative studies identified possible recombination hot spots flanking highly polymorphic loci (Gomes et al., 2007). OmpA and the Pmp family of proteins are surface exposed in chlamydiae and are among the most polymorphic gene products in this system (Lampe et al., 1993; Millman et al., 2001; Gomes et al., 2007; Nunes et al., 2010). The genetic variability of these genes suggests that recombination drives functional and/or antigenic variation that might be important for immune evasion. Individual patients are commonly infected with multiple strains; thus, it is logical to suspect that co-infections may provide the necessary reservoir of genetic material to drive intraspecies recombination and genome diversification (Suchland et al., 2003).
An artificial system was recently developed to study in vitro lateral gene transfer and recombination in chlamydia (DeMars et al., 2007; DeMars and Weinfurter, 2008; Suchland et al., 2009). Transfer of tetracycline, rifampin, ofloxacin, and/or clindamycin resistance is routinely detected between C. trachomatis, Chlamydia suis, or Chlamydia muridarum strains following coinfection of cells with differently resistant parental strains and selection for doubly resistant progeny. A dynamic range of DNA fragment sizes incorporate into recipient strains ranging from 100 bp up to 790 kbp (DeMars and Weinfurter, 2008; B. M. Jeffrey and D. D. Rockey, unpublished data) with recombination occurring throughout the genome without apparent site specificity. Each of the three Chlamydia species mentioned above occupy and grow within the same vacuole, but whether or not co-occupancy of an inclusion is a requirement for horizontal gene transfer in chlamydia is unknown.
It is difficult to determine how chlamydial DNA exchange occurs in the absence of identifiable conjugative machinery, competence inducing genes or pathways, restriction modification systems, or marked genomic differences among strains. However, it is possible that DNA is transferred between chlamydiae via uptake of extracellular DNA released by lysed organisms. If so, perhaps this “natural competence” could be exploited to develop a transformation system for chlamydiae, an exciting possibility for these organisms that are still genetically intractable relative to other obligate intracellular bacteria.
A reverse genetic approach to genetic manipulation of C. trachomatis
An exciting new development in genetic manipulation of Chlamydia spp. that circumvents difficulties in genetic transformation was recently described by Kari et al. (2011) who used a reverse genetic approach to generate isogenic mutants of C. trachomatis. The strategy is based on an approach frequently used in plant genetics called Targeting Induced Local Lesions in Genomes (TILLING; McCallum et al., 2000). C. trachomatis was first chemically mutagenized with ethyl methanesulfonate in a manner that generated an estimated 0.5 mutations per genome. Mutagenized chlamydiae in pools of roughly 10 organisms were then expanded in McCoy cells in individual wells of 96 well plates. Genomic DNA was isolated from a duplicate library for use in PCR. To provide proof of principle, a PCR-based screen of pool DNA was conducted to detect mutations in trpB, encoding tryptophane synthase beta chain. PCR amplicons derived from pools were digested with the mismatch specific endonuclease CEL I. Amplicons containing mismatch mutations as detected by agarose gel electrophoresis were nucleotide sequenced to identify single nucleotide polymorphisms (SNPs). Of the 2,800 pools screened, 13 contained non-synonymous SNPs in trpB, with one being a nonsense mutation predicted to disrupt TrpB function. Unlike wild-type bacteria, the plaque-cloned mutant could not be rescued with indole during interferon-γ induced tryptophane depletion. While labor intensive, this approach represents the only technology currently available for generating isogenic null mutants of chlamydiae. As mentioned above, and in keeping with other obligate intracellular bacteria, null mutations in genes required for growth in cell culture will not be attainable using this method. However, knockouts in genes that function primarily in animal infection and pathogenesis, such as TrpB, implicated in strain-specific tissue tropism (Caldwell et al., 2003), can be accomplished.
Perspective
Successful transformation of several obligate intracellular bacteria proves that barriers to genetic manipulation, while often significant, are not insurmountable. Electroporation is universally effective in introducing heterologous DNA into these bacteria. Positive selection of transformants using resistance to antibiotics is feasible despite restricted use of antibiotics based on clinical efficacy. Homologous recombination and transposition occur at some frequency.
While genetic methods for obligates are still rudimentary compared to those available for most free-living bacterial pathogens, several milestones have been reached. The Himar1 transposon has proven particularly useful with Coxiella, Rickettsia, and Anaplasma in generating transformants expressing fluorescent proteins with applications in pathogen–host interaction studies. Himar1-mediated gene inactivation also revealed a novel virulence gene (sca2) in R. rickettsii and, by in cis complementation of a naturally occurring mutant, identified a relA/spoT-like gene as involved in the lytic plaque phenotype of the organism. The first targeted gene disruption using transformation in any obligate intracellular bacterium was achieved with the knockout of pld in R. prowazekii. Recent axenic growth of C. burnetii is significantly aiding development of genetic tools that now include Himar1 Tn mutagenesis, RSF1010 ori-based shuttle vectors, and a Tn7 system for single-copy, site-specific gene delivery. Finally, progress has been made in understanding requirements for transformation of the most recalcitrant of obligates, Chlamydia spp. A chronological overview of achievements in genetic transformation of obligate intracellular bacterial pathogens is detailed in Table 1.
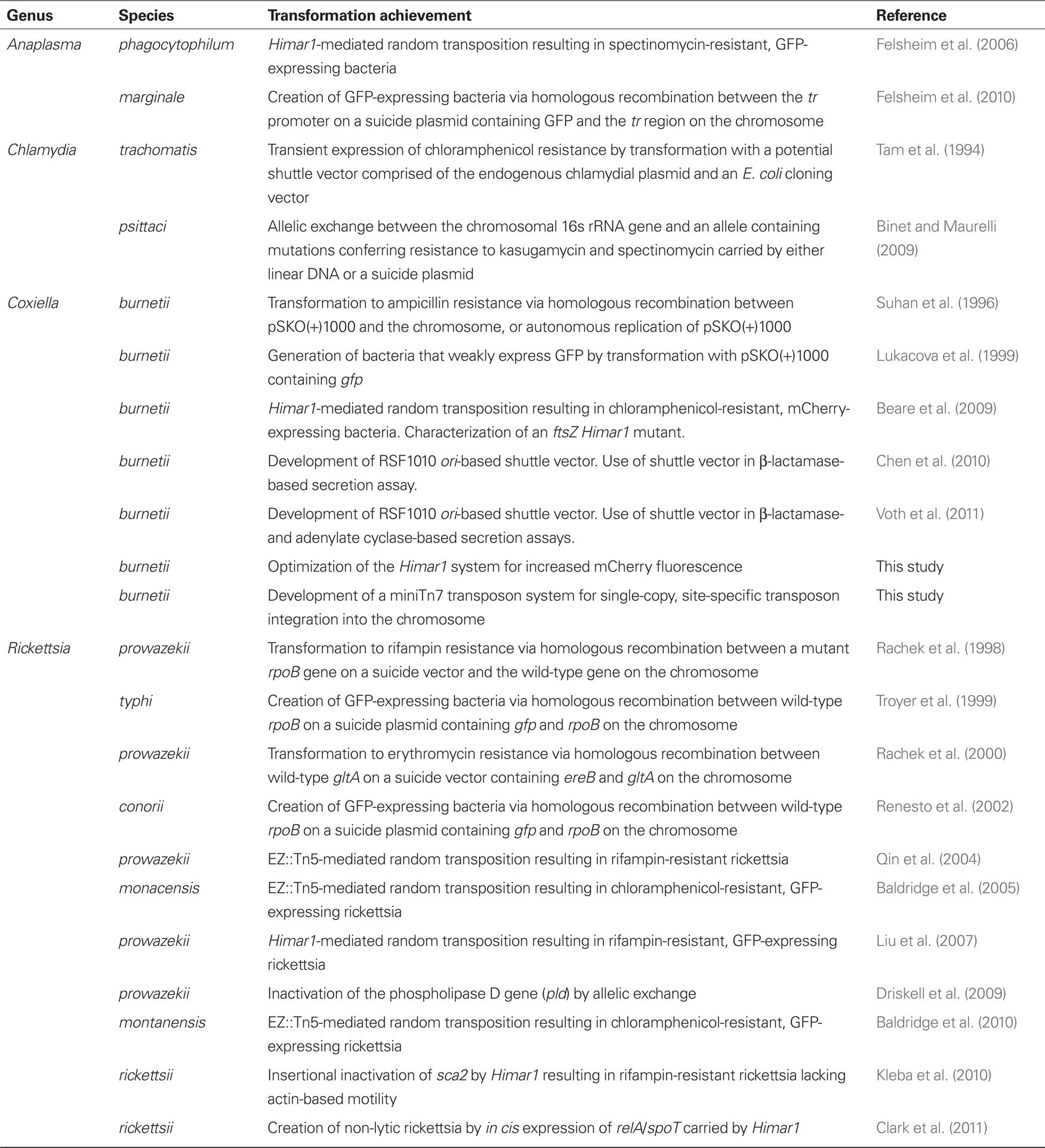
Table 1. History of genetic transformation of obligate intracellular bacterial pathogens.
Notwithstanding these notable achievements, additional advances in genetic manipulation of obligates are still required before molecular dissection of virulence factors can become routine. First and foremost, efficient methods of targeted gene disruption and complementation are needed. Traditional methods of allelic exchange rely upon double crossover events between a mutated allele of a gene, carried by a suicide plasmid or linear fragment, and the wild-type copy on the chromosome. The low frequency of double crossover events, combined with the poor transformation efficiencies of obligates (discussed in more detail below), makes this approach difficult. Counterselectable markers, such as sacB conferring sucrose sensitivity, can dramatically aid recovery of gene knockout mutants (Reyrat et al., 1998). In this strategy, chromosomal integration of a suicide plasmid by a single crossover event between a plasmid-coded mutant allele and the chromosomal wild-type gene is first achieved by positive selection for antibiotic resistance. A second crossover event that resolves the co-integrant and generates the desired mutation is then selected for by growth under counterselective conditions (e.g., media containing sucrose). While widely used in free-living bacteria to generate both marked and unmarked mutants, novel counterselective strategies may be necessary for obligate intracellular bacteria. Another possibility for targeted gene disruption in obligates involves mobile group II introns (targetrons). This technique is effective in other fastidious bacteria (Rodriguez et al., 2008) and relies on retargeting of the intron to a gene of interest by a process referred to as “retrohoming” (Karberg et al., 2001). Finally, recombineering using bacteriophage λ Red recombination functions should be considered (Thomason et al., 2007). λ Red functions promote high efficiency in vivo homologous recombination of sequences with homologies as short as 40 bases. Site-directed gene disruption might be accomplished by co-transforming with a linear targeting sequence and a suicide plasmid expressing of the λ Red recombination enzymes. With the exception of C. burnetii, plasmid shuttle vectors are needed for in trans complementation and gene expression studies using reporters such as lacZ and destabilized forms of GFP (Barysheva et al., 2008). Systems for inducible gene expression are also desirable for understanding protein function. For example, gene induction during a specific stage of the infectious cycle can be useful for determining the temporal requirement of a specific virulence factor.
Although robust gene knockout technologies are clearly needed for obligate intracellular bacteria, any gene inactivation that lowers fitness for intracellular growth may preclude isolation of the desired mutant. Indeed, to date, just a handful of genes have been inactivated in obligate intracellular bacteria. Isolation of null mutants of obligate intracellular bacteria may be particularly problematic due to their extensively downsized genomes. Nonetheless, there are clearly a subset of genes that are not essential for growth in cultured cells but play important roles in animal colonization and virulence. Rickettsial pld and sca2 (Driskell et al., 2009; Kleba et al., 2010), and chlamydial trpB (Kari et al., 2011), are examples. A system for inducible in trans expression may allow inactivation of genes essential for intracellular growth (Molofsky and Swanson, 2003). These procedures typically involve creation of a strain carrying a plasmid with an inducible copy of a chromosomal gene targeted for inactivation. IPTG induction of lacI-tacp works with intracellular Legionella pneumophila (Roy et al., 1998) and may be similarly effective with obligates.
Low transformation efficiencies remain an obstacle to further development of genetic tools for obligates. Many reasons could account for poor efficiency including suboptimal electroporation conditions/buffers, purity of host cell-derived organisms, and restriction/modification systems that degrade heterologous DNA. Improvements in transformation efficiencies that allow near saturation-level Tn mutagenesis might permit identification of genes required for host cell/animal infection by deep sequencing technologies (Gawronski et al., 2009). Cloning also remains a challenge for non-plaque-forming organisms, but limiting dilution and micromanipulation are alternatives (Suhan et al., 1996; Rachek et al., 2000; Qin et al., 2004; Beare et al., 2007).
The lack of methods to genetically manipulate obligate intracellular bacteria has historically impeded progress in understanding the genetic basis of their unique lifestyles and virulence. However, recent achievements in genetic transformation of nearly all medically relevant genera of this group will undoubtedly fuel new interest in these understudied but fascinating bacterial pathogens. Greater genetic tractability is inevitable, and will lead to novel insight into intracellular parasitism and disease pathogenesis, in addition to enabling development of new pathogen countermeasures, such as rationally designed attenuated or subunit vaccines.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Shelly Robertson for critical reading of the manuscript and Anita Mora for graphic support. This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases.
References
Andersson, J. O., and Andersson, S. G. (1999). Insights into the evolutionary process of genome degradation. Curr. Opin. Genet. Dev. 9, 664–671.
Andersson, S. G., Zomorodipour, A., Andersson, J. O., Sicheritz-Ponten, T., Alsmark, U. C., Podowski, R. M., Naslund, A. K., Eriksson, A. S., Winkler, H. H., and Kurland, C. G. (1998). The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396, 133–140.
Atlas, R. M. (2003). Bioterrorism and biodefence research: changing the focus of microbiology. Nat. Rev. Microbiol. 1, 70–74.
Baldridge, G. D., Burkhardt, N., Herron, M. J., Kurtti, T. J., and Munderloh, U. G. (2005). Analysis of fluorescent protein expression in transformants of Rickettsia monacensis, an obligate intracellular tick symbiont. Appl. Environ. Microbiol. 71, 2095–2105.
Baldridge, G. D., Burkhardt, N. Y., Felsheim, R. F., Kurtti, T. J., and Munderloh, U. G. (2007a). Transposon insertion reveals pRM, a plasmid of Rickettsia monacensis. Appl. Environ. Microbiol. 73, 4984–4995.
Baldridge, G. D., Kurtti, T. J., Burkhardt, N., Baldridge, A. S., Nelson, C. M., Oliva, A. S., and Munderloh, U. G. (2007b). Infection of Ixodes scapularis ticks with Rickettsia monacensis expressing green fluorescent protein: a model system. J. Invertebr. Pathol. 94, 163–174.
Baldridge, G. D., Burkhardt, N. Y., Labruna, M. B., Pacheco, R. C., Paddock, C. D., Williamson, P. C., Billingsley, P. M., Felsheim, R. F., Kurtti, T. J., and Munderloh, U. G. (2010a). Wide dispersal and possible multiple origins of low-copy-number plasmids in Rickettsia species associated with blood-feeding arthropods. Appl. Environ. Microbiol. 76, 1718–1731.
Baldridge, G. D., Burkhardt, N. Y., Oliva, A. S., Kurtti, T. J., and Munderloh, U. G. (2010b). Rickettsial ompB promoter regulated expression of GFPuv in transformed Rickettsia montanensis. PLoS ONE 5, e8965. doi: 10.1371/journal.pone.0008965
Barbet, A. F., Agnes, J. T., Moreland, A. L., Lundgren, A. M., Alleman, A. R., Noh, S. M., Brayton, K. A., Munderloh, U. G., and Palmer, G. H. (2005). Identification of functional promoters in the msp2 expression loci of Anaplasma marginale and Anaplasma phagocytophilum. Gene 353, 89–97.
Bardill, J. P., Miller, J. L., and Vogel, J. P. (2005). IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol. Microbiol. 56, 90–103.
Barysheva, O. V., Fujii, J., Takaesu, G., and Yoshida, S. (2008). Application of unstable Gfp variants to the kinetic study of Legionella pneumophila icm gene expression during infection. Microbiology 154, 1015–1025.
Bavoil, P. M., Hsia, R., and Ojcius, D. M. (2000). Closing in on Chlamydia and its intracellular bag of tricks. Microbiology 146, 2723–2731.
Beare, P. A., Howe, D., Cockrell, D. C., and Heinzen, R. A. (2007). Efficient method of cloning the obligate intracellular bacterium Coxiella burnetii. Appl. Environ. Microbiol. 73, 4048–4054.
Beare, P. A., Howe, D., Cockrell, D. C., Omsland, A., Hansen, B., and Heinzen, R. A. (2009). Characterization of a Coxiella burnetii ftsZ mutant generated by Himar1 transposon mutagenesis. J. Bacteriol. 191, 1369–1381.
Beare, P. A., Omsland, A., Cockrell, D. C., Howe, D., and Heinzen, R. A. (2010). Expansion of the Coxiella burnetii genetics tool box using axenic culture. Abstr. 24th Meet. Am. Soc. Rickettsiol. Abstr. 94.
Binet, R., and Maurelli, A. T. (2009). Transformation and isolation of allelic exchange mutants of Chlamydia psittaci using recombinant DNA introduced by electroporation. Proc. Natl. Acad. Sci. U.S.A. 106, 292–297.
Caldwell, H. D., Wood, H., Crane, D., Bailey, R., Jones, R. B., Mabey, D., Maclean, I., Mohammed, Z., Peeling, R., Roshick, C., Schachter, J., Solomon, A. W., Stamm, W. E., Suchland, R. J., Taylor, L., West, S. K., Quinn, T. C., Belland, R. J., and McClarty, G. (2003). Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J. Clin. Invest. 111, 1757–1769.
Campbell, R. E. (2004). Realization of beta-lactamase as a versatile fluorogenic reporter. Trends Biotechnol. 22, 208–211.
Chen, C., Banga, S., Mertens, K., Weber, M. M., Gorbaslieva, I., Tan, Y., Luo, Z. Q., and Samuel, J. E. (2010). Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc. Natl. Acad. Sci. U.S.A. 107, 21755–21760.
Choi, K. H., Gaynor, J. B., White, K. G., Lopez, C., Bosio, C. M., Karkhoff-Schweizer, R. R., and Schweizer, H. P. (2005). A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2, 443–448.
Clark, T. R., Ellison, D. W., Kleba, B., and Hackstadt, T. (2011). Complementation of Rickettsia rickettsii RelA/SpoT restores a non-lytic plaque phenotype. Infect. Immun. 79, 1631–1637.
Coleman, S. A., Fischer, E. R., Howe, D., Mead, D. J., and Heinzen, R. A. (2004). Temporal analysis of Coxiella burnetii morphological differentiation. J. Bacteriol. 186, 7344–7352.
de Felipe, K. S., Glover, R. T., Charpentier, X., Anderson, O. R., Reyes, M., Pericone, C. D., and Shuman, H. A. (2008). Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 4, e1000117. doi: 10.1371/journal.ppat.1000117
DeMars, R., and Weinfurter, J. (2008). Interstrain gene transfer in Chlamydia trachomatis in vitro: mechanism and significance. J. Bacteriol. 190, 1605–1614.
DeMars, R., Weinfurter, J., Guex, E., Lin, J., and Potucek, Y. (2007). Lateral gene transfer in vitro in the intracellular pathogen Chlamydia trachomatis. J. Bacteriol. 189, 991–1003.
Driskell, L. O., Yu, X. J., Zhang, L., Liu, Y., Popov, V. L., Walker, D. H., Tucker, A. M., and Wood, D. O. (2009). Directed mutagenesis of the Rickettsia prowazekii pld gene encoding phospholipase D. Infect. Immun. 77, 3244–3248.
Falkow, S. (2004). Molecular Koch’s postulates applied to bacterial pathogenicity – a personal recollection 15 years later. Nat. Rev. Microbiol. 2, 67–72.
Felsheim, R. F., Chavez, A. S., Palmer, G. H., Crosby, L., Barbet, A. F., Kurtti, T. J., and Munderloh, U. G. (2010). Transformation of Anaplasma marginale. Vet. Parasitol. 167, 161–174.
Felsheim, R. F., Herron, M. J., Nelson, C. M., Burkhardt, N. Y., Barbet, A. F., Kurtti, T. J., and Munderloh, U. G. (2006). Transformation of Anaplasma phagocytophilum. BMC Biotechnol. 6, 42. doi: 10.1186/1472-6750-6-42
Gawronski, J. D., Wong, S. M., Giannoukos, G., Ward, D. V., and Akerley, B. J. (2009). Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc. Natl. Acad. Sci. U.S.A. 106, 16422–16427.
Gomes, J. P., Bruno, W. J., Nunes, A., Santos, N., Florindo, C., Borrego, M. J., and Dean, D. (2007). Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res. 17, 50–60.
Haglund, C. M., Choe, J. E., Skau, C. T., Kovar, D. R., and Welch, M. D. (2010). Rickettsia Sca2 is a bacterial formin-like mediator of actin-based motility. Nat. Cell Biol. 12, 1057–1063.
Heinzen, R. A., Hayes, S. F., Peacock, M. G., and Hackstadt, T. (1993). Directional actin polymerization associated with spotted fever group Rickettsia infection of Vero cells. Infect. Immun. 61, 1926–1935.
Karberg, M., Guo, H., Zhong, J., Coon, R., Perutka, J., and Lambowitz, A. M. (2001). Group II introns as controllable gene targeting vectors for genetic manipulation of bacteria. Nat. Biotechnol. 19, 1162–1167.
Kari, L., Goheen, M. M., Randall, L. B., Taylor, L. D., Carlson, J. H., Whitmire, W. M., Virok, D., Rajaram, K., Endresz, V., McClarty, G., Nelson, D. E., and Caldwell, H. D. (2011). Generation of targeted Chlamydia trachomatis null mutants. Proc. Natl. Acad. Sci. U.S.A. (in press).
Kleba, B., Clark, T. R., Lutter, E. I., Ellison, D. W., and Hackstadt, T. (2010). Disruption of the Rickettsia rickettsii Sca2 autotransporter inhibits actin-based motility. Infect. Immun. 78, 2240–2247.
Lampe, D. J., Akerley, B. J., Rubin, E. J., Mekalanos, J. J., and Robertson, H. M. (1999). Hyperactive transposase mutants of the Himar1 mariner transposon. Proc. Natl. Acad. Sci. U.S.A. 96, 11428–11433.
Lampe, M. F., Suchland, R. J., and Stamm, W. E. (1993). Nucleotide sequence of the variable domains within the major outer membrane protein gene from serovariants of Chlamydia trachomatis. Infect. Immun. 61, 213–219.
Li, C. H., Cheng, Y. W., Liao, P. L., Yang, Y. T., and Kang, J. J. (2010). Chloramphenicol causes mitochondrial stress, decreases ATP biosynthesis, induces matrix metalloproteinase-13 expression, and solid-tumor cell invasion. Toxicol. Sci. 116, 140–150.
Liu, Z. M., Tucker, A. M., Driskell, L. O., and Wood, D. O. (2007). Mariner-based transposon mutagenesis of Rickettsia prowazekii. Appl. Environ. Microbiol. 73, 6644–6649.
Lukacova, M., Valkova, D., Quevedo Diaz, M., Perecko, D., and Barak, I. (1999). Green fluorescent protein as a detection marker for Coxiella burnetii transformation. FEMS Microbiol. Lett. 175, 255–260.
Matsumoto, A., Izutsu, H., Miyashita, N., and Ohuchi, M. (1998). Plaque formation by and plaque cloning of Chlamydia trachomatis biovar trachoma. J. Clin. Microbiol. 36, 3013–3019.
Maurin, M., Benoliel, A. M., Bongrand, P., and Raoult, D. (1992). Phagolysosomal alkalinization and the bactericidal effect of antibiotics: the Coxiella burnetii paradigm. J. Infect. Dis. 166, 1097–1102.
McCallum, C. M., Comai, L., Greene, E. A., and Henikoff, S. (2000). Targeted screening for induced mutations. Nat. Biotechnol. 18, 455–457.
Millman, K. L., Tavare, S., and Dean, D. (2001). Recombination in the ompA gene but not the omcB gene of Chlamydia contributes to serovar-specific differences in tissue tropism, immune surveillance, and persistence of the organism. J. Bacteriol. 183, 5997–6008.
Molofsky, A. B., and Swanson, M. S. (2003). Legionella pneumophila CsrA is a pivotal repressor of transmission traits and activator of replication. Mol. Microbiol. 50, 445–461.
Noh, S. M., Ueti, M. W., Palmer, G. H., Munderloh, U. G., Felsheim, R. F., and Brayton, K. A. (2011). Stability and tick transmission phenotype of gfp-transformed Anaplasma marginale through a complete in vivo infection cycle. Appl. Environ. Microbiol. 77, 330–334.
Nunes, A., Nogueira, P. J., Borrego, M. J., and Gomes, J. P. (2010). Adaptive evolution of the Chlamydia trachomatis dominant antigen reveals distinct evolutionary scenarios for B- and T-cell epitopes: worldwide survey. PLoS ONE 5, e13171. doi: 10.1371/journal.pone.0013171
Ogata, H., Renesto, P., Audic, S., Robert, C., Blanc, G., Fournier, P. E., Parinello, H., Claverie, J. M., and Raoult, D. (2005). The genome sequence of Rickettsia felis identifies the first putative conjugative plasmid in an obligate intracellular parasite. PLoS Biol. 3, e248. doi: 10.1371/journal.pbio.0030248
Omsland, A., Beare, P. A., Hill, J., Cockrell, D. C., Howe, D., Hansen, B., Samuel, J. E., and Heinzen, R. A. (2011). Isolation from animal tissue and genetic transformation of Coxiella burnetii facilitated by an improved axenic growth medium. Appl. Environ. Microbiol. (in press).
Omsland, A., Cockrell, D. C., Howe, D., Fischer, E. R., Virtaneva, K., Sturdevant, D. E., Porcella, S. F., and Heinzen, R. A. (2009). Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc. Natl. Acad. Sci. U.S.A. 106, 4430–4434.
Policastro, P. F., and Hackstadt, T. (1994). Differential activity of Rickettsia rickettsii ompA and ompB promoter regions in a heterologous reporter gene system. Microbiology 140, 2941–2949.
Qin, A., Tucker, A. M., Hines, A., and Wood, D. O. (2004). Transposon mutagenesis of the obligate intracellular pathogen Rickettsia prowazekii. Appl. Environ. Microbiol. 70, 2816–2822.
Rachek, L. I., Hines, A., Tucker, A. M., Winkler, H. H., and Wood, D. O. (2000). Transformation of Rickettsia prowazekii to erythromycin resistance encoded by the Escherichia coli ereB gene. J. Bacteriol. 182, 3289–3291.
Rachek, L. I., Tucker, A. M., Winkler, H. H., and Wood, D. O. (1998). Transformation of Rickettsia prowazekii to rifampin resistance. J. Bacteriol. 180, 2118–2124.
Raghavan, R., Hicks, L. D., and Minnick, M. F. (2008). Toxic introns and parasitic intein in Coxiella burnetii: legacies of a promiscuous past. J. Bacteriol. 190, 5934–5943.
Renesto, P., Gouin, E., and Raoult, D. (2002). Expression of green fluorescent protein in Rickettsia conorii. Microb. Pathog. 33, 17–21.
Reyrat, J. M., Pelicic, V., Gicquel, B., and Rappuoli, R. (1998). Counterselectable markers: untapped tools for bacterial genetics and pathogenesis. Infect. Immun. 66, 4011–4017.
Rodriguez, S. A., Yu, J. J., Davis, G., Arulanandam, B. P., and Klose, K. E. (2008). Targeted inactivation of Francisella tularensis genes by group II introns. Appl. Environ. Microbiol. 74, 2619–2626.
Roy, C. R., Berger, K. H., and Isberg, R. R. (1998). Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol. Microbiol. 28, 663–674.
Seshadri, R., Paulsen, I. T., Eisen, J. A., Read, T. D., Nelson, K. E., Nelson, W. C., Ward, N. L., Tettelin, H., Davidsen, T. M., Beanan, M. J., Deboy, R. T., Daugherty, S. C., Brinkac, L. M., Madupu, R., Dodson, R. J., Khouri, H. M., Lee, K. H., Carty, H. A., Scanlan, D., Heinzen, R. A., Thompson, H. A., Samuel, J. E., Fraser, C. M., and Heidelberg, J. F. (2003). Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc. Natl. Acad. Sci. U.S.A. 100, 5455–5460.
Sexton, J. A., Miller, J. L., Yoneda, A., Kehl-Fie, T. E., and Vogel, J. P. (2004). Legionella pneumophila DotU and IcmF are required for stability of the Dot/Icm complex. Infect. Immun. 72, 5983–5992.
Shannon, J. G., and Heinzen, R. A. (2007). Infection of human monocyte-derived macrophages with Coxiella burnetii. Methods Mol. Biol. 431, 189–200.
Sory, M. P., and Cornelis, G. R. (1994). Translocation of a hybrid YopE-adenylate cyclase from Yersinia enterocolitica into HeLa cells. Mol. Microbiol. 14, 583–594.
Suchland, R. J., Eckert, L. O., Hawes, S. E., and Stamm, W. E. (2003). Longitudinal assessment of infecting serovars of Chlamydia trachomatis in Seattle public health clinics: 1988–1996. Sex. Transm. Dis. 30, 357–361.
Suchland, R. J., Sandoz, K. M., Jeffrey, B. M., Stamm, W. E., and Rockey, D. D. (2009). Horizontal transfer of tetracycline resistance among Chlamydia spp. in vitro. Antimicrob. Agents Chemother. 53, 4604–4611.
Suhan, M., Chen, S. Y., Thompson, H. A., Hoover, T. A., Hill, A., and Williams, J. C. (1994). Cloning and characterization of an autonomous replication sequence from Coxiella burnetii. J. Bacteriol. 176, 5233–5243.
Suhan, M. L., Chen, S. Y., and Thompson, H. A. (1996). Transformation of Coxiella burnetii to ampicillin resistance. J. Bacteriol. 178, 2701–2708.
Suhan, M. L., and Thompson, H. A. (2000). Expression of beta-lactamase in Coxiella burnetii transformants. FEMS Microbiol. Lett. 184, 303–306.
Tam, J. E., Davis, C. H., and Wyrick, P. B. (1994). Expression of recombinant DNA introduced into Chlamydia trachomatis by electroporation. Can. J. Microbiol. 40, 583–591.
Thomason, L. C., Costantino, N., Shaw, D. V., and Court, D. L. (2007). Multicopy plasmid modification with phage lambda Red recombineering. Plasmid 58, 148–158.
Troese, M. J., and Carlyon, J. A. (2009). Anaplasma phagocytophilum dense-cored organisms mediate cellular adherence through recognition of human P-selectin glycoprotein ligand 1. Infect. Immun. 77, 4018–4027.
Troyer, J. M., Radulovic, S., and Azad, A. F. (1999). Green fluorescent protein as a marker in Rickettsia typhi transformation. Infect. Immun. 67, 3308–3311.
Voth, D. E., Beare, P. A., Howe, D., Sharma, U. M., Samoilis, G., Cockrell, D. C., Omsland, A., and Heinzen, R. A. (2011). The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV secretion system substrates. J. Bacteriol. 193, 1493–1503.
Voth, D. E., Howe, D., Beare, P. A., Vogel, J. P., Unsworth, N., Samuel, J. E., and Heinzen, R. A. (2009). The Coxiella burnetii ankyrin repeat domain-containing protein family is heterogeneous, with C-terminal truncations that influence Dot/Icm-mediated secretion. J. Bacteriol. 191, 4232–4242.
Walker, D. H. (1989). Rocky Mountain spotted fever: a disease in need of microbiological concern. Clin. Microbiol. Rev. 2, 227–240.
Whitworth, T., Popov, V. L., Yu, X. J., Walker, D. H., and Bouyer, D. H. (2005). Expression of the Rickettsia prowazekii pld or tlyC gene in Salmonella enterica serovar Typhimurium mediates phagosomal escape. Infect. Immun. 73, 6668–6673.
Wike, D. A., Tallent, G., Peacock, M. G., and Ormsbee, R. A. (1972). Studies of the rickettsial plaque assay technique. Infect. Immun. 5, 715–722.
Keywords: transposon mutagenesis, electroporation, antibiotic selection, allelic exchange, genetic transformation, virulence factor, shuttle vector, complementation
Citation: Beare PA, Sandoz KM, Omsland A, Rockey DD and Heinzen RA (2011) Advances in genetic manipulation of obligate intracellular bacterial pathogens. Front. Microbio. 2:97. doi: 10.3389/fmicb.2011.00097
Received: 24 February 2011;
Paper pending published: 14 March 2011;
Accepted: 19 April 2011;
Published online: 02 May 2011.
Edited by:
Rey Carabeo, Imperial College London, UKReviewed by:
Mikhail A. Gavrilin, Ohio State University, USAAndres Vazquez-Torres, University of Colorado Medical School, USA
Copyright: © 2011 Beare, Sandoz, Omsland, Rockey and Heinzen. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Robert A. Heinzen, Rocky Mountain Laboratories, Laboratory of Intracellular Parasites, 903 South Fourth Street, Hamilton, MT 59840, USA. e-mail: rheinzen@niaid.nih.gov