 Weiyan Zhu1 Ching-Ju Chen2 Christopher E. Thomas1 James E. Anderson1 Ann E. Jerse3 P. Frederick Sparling1*
Weiyan Zhu1 Ching-Ju Chen2 Christopher E. Thomas1 James E. Anderson1 Ann E. Jerse3 P. Frederick Sparling1*- 1 Department of Medicine, University of North Carolina, Chapel Hill, NC, USA
- 2 Human Vaccine Institute, Duke University, Durham, NC, USA
- 3 Department of Microbiology, Uniformed University of the Health Sciences, Bethesda, MD, USA
Immune responses to the gonococcus after natural infection ordinarily result in little immunity to reinfection, due to antigenic variation of the gonococcus, and redirection or suppression of immune responses. Brinton and colleagues demonstrated that parenteral immunization of male human volunteers with a purified pilus vaccine gave partial protection against infection by the homologous strain. However, the vaccine failed in a clinical trial. Recent vaccine development efforts have focused on the female mouse model of genital gonococcal infection. Here we discuss the state of the field, including our unpublished data regarding efficacy in the mouse model of either viral replicon particle (VRP) vaccines, or outer membrane vesicle (OMV) vaccines. The OMV vaccines failed, despite excellent serum and mucosal antibody responses. Protection after a regimen consisting of a PorB-VRP prime plus recombinant PorB boost was correlated with apparent Th1, but not with antibody, responses. Protection probably was due to powerful adjuvant effects of the VRP vector. New tools including novel transgenic mice expressing human genes required for gonococcal infection should enable future research. Surrogates for immunity are needed. Increasing antimicrobial resistance trends among gonococci makes development of a vaccine more urgent.
Introduction: Need for a Vaccine for Gonorrhea
Neisseria gonorrhoeae (the gonococcus, or GC) remains an important disease. Still relatively common in the US, with over 300,000 reported cases annually, and probably as many that are not reported, it is much more common in Africa and in many other parts of the less-developed world. Untreated gonococcal infection in women may progress to pelvic inflammatory disease, increasing the risk of ectopic pregnancy and infertility. Calculations of attributable risk show that GC is one of the significant cofactors for HIV transmission (Fleming and Wasserheit, 1999), increasing risks of HIV transmission and acquisition about threefold. These factors alone should promote interest in a vaccine for this ancient disease.
Another reason to urge development of a gonococcal vaccine is emergence of antibiotic resistant GC. In many parts of the world, fluorinated quinolones are no longer recommended because of the prevalence of resistance (Newman et al., 2007; Schultz et al., 2001; Lewis, 2010). Resistance to cephalosporins also is emerging, rendering oral formulations such as cefixime less effective (Lo et al., 2008; Golparian et al., 2010; Lewis, 2010). A steady creep toward decreased in vitro susceptibility to ceftriaxone in SE Asia and elsewhere (Chisholm et al., 2010) will threaten utility of ceftriaxone, the principal remaining parenteral therapy for GC, if the trend continues. Current U.S. gonorrhea treatment guidelines increased the dose of parenteral ceftriaxone from 125 to 250 mg to attempt to counteract the slow increases in cephalosporin resistance (Centers for Disease Control, 2010). Resistance to quinolones and beta lactams is due to chromosomal mutations, including alterations of the target sites for these antimicrobials, and also alterations of porin entry channels and/or efflux pumps (reviewed in Lewis, 2010). Most of the resistant mutants appear to be fit, in the sense that they persist and spread in the natural world.
Although plasmid-mediated resistance to penicillins and tetracyclines has leveled off (Lewis, 2010), history teaches that continued selection of more resistant GC variants is to be expected, either by additional chromosomal mutations or acquisition of plasmids. We expect that in a decade, GC resistance to antimicrobials will increase, and therapy with existing cephalosporins will no longer be useful. There are few if any promising new antimicrobials for GC on the horizon. It takes at least a decade or more to create vaccines. Now is the time to be serious about the problem.
The increasing threat of difficult-to-treat GC should make a gonococcal vaccine an important objective, but there is relatively little work at present on such a vaccine. A PubMed search on 12.27/2010 under “gonococcal vaccine” yielded 247 entries, whereas a similar search under “meningococcal vaccine” yielded 3326 entries. The reasons for this glaring discrepancy are not obvious, but may include such commercial factors as estimated market size for vaccines, the probability that the public would accept and utilize the vaccine, and who would pay for the vaccine. It is possible that the shock of witnessing meningococcal (MC) disease and death in innocent infants and young adults helps to energize the MC vaccine programs. By contrast, GC disease is a silent killer of the unborn, due to salpingitis and ectopic pregnancy. Some might view GC as just a minor infection, and one that is acquired by personal choice. If it can be treated, why bother with a vaccine? Another obvious consideration is the likelihood that a vaccine can be developed, based solely on the available science. In the absence of evidence for naturally acquired immunity after infection, and correlates for protective immunity, it is difficult for commercial operations to push forward with vaccine development. The result has been discontinuance of the former US military GC vaccine program, and in termination of related programs in virtually every large-scale manufacturers of vaccines in the US and Europe.
In this review, we examine the natural history of infection with emphasis on immune response and immunity to infection, and certain new evidence regarding the pathogenesis of infection. Other comprehensive reviews of GC pathogenesis and animal models are included elsewhere in this volume. Previous reviews of gonococcal vaccines covered pathogenesis and antigens that might be used for a vaccine (Blake and Wetzler, 1995; Sparling et al., 2003; Edwards and Apicella, 2004; Virji, 2009). We discuss previous vaccine attempts, as well as lessons learned from vaccines for comparable diseases, especially the close sibling of GC, the MC. We then present previously unpublished experiments from our laboratories, which were “negative” in the sense that they did not show evidence for protection in the female mouse model of genital GC infection. Nevertheless, they inform future investigations, and one surprising result in particular suggests a path forward for additional research.
Is there Evidence for Naturally Acquired Immunity to Reinfection?
In the preantibiotic era, symptomatic infections resolved spontaneously over months, although the basis for apparent immune resolution was not studied (Hill, 1942). In the current era of readily available antibiotics, persons commonly acquire gonorrhea, are treated, but are soon reinfected. Mild or asymptomatic infection may be carried for months without treatment, both in men and women (Handsfield et al., 1974). A community outbreak due to a highly resistant strain demonstrated that reinfections with the same strain were common after treatment of uncomplicated genital infection (Faruki et al., 1985). There was no evidence for increased resistance to a second infection in studies of experimental GC of male human volunteers, even when the repeat infection was initiated only 2 weeks after treatment of the first infection by the same MS11 gonococcal strain (Schmidt et al., 2001) The infections of experimental subjects had to be terminated at onset of symptoms for ethical reasons, undoubtedly before a full immune response was initiated.
A study of highly experienced female commercial sex workers in Africa showed apparent immunity to recurrent infection; protection was specific to particular outer membrane porin protein B (PorB) serovars, and was not generally protective against other strains (Plummer et al., 1989). However, a similar study in a small US community, involving men and women, who certainly had fewer previous episodes of GC than the African female cohort, actually found an increased rate of reinfection by the same PorB serovar (Fox et al., 1999). Increased rates of second infections by the same strain could have been due to the greater likelihood of sexual reexposure to still-infected partners. Other data from this same study showed there was some antigenic variation in exposed PorB surface loops during repeated passage of this strain through the community (Hobbs et al., 1999). A small study claimed PorB serovar specific immunity to reinfection in women who had salpingitis (Buchanan et al., 1980). Conceivably, repeated infections elicited immune memory responses, providing the basis for protective immunity on reexposure.
A somewhat more optimistic view of immunity after local mucosal infection was provided by studies of experimental GC in chimpanzees. Certain GC strains were capable of infecting the chimp urethra, pharynx, and cervix; the infectious dose was high for the pharynx and cervix, but for the male urethra the required inoculum was about 1 × 104 colony forming units (CFU), essentially the same as for human urethral infection (Kraus et al., 1975). Infection persisted for weeks, but then an uncharacterized immune response cleared infection, as defined by two successive negative cultures. Initiation of a second infection by the same strain 1 week after termination of first infection required an infectious inoculum about 1000-fold greater. Repeat challenge 2 years after the first infection showed that immunity had completely waned (Kraus et al., 1975). These experiments also showed that bactericidal serum antibodies protected against urethral infection in the chimp: the only GC capable of infection were of the PorB1B serovar class, which were able to bind chimp complement four binding protein (C4bp), rendering them phenotypically serum resistant (Ngampasutadol et al., 2005). PorB1 strains were not able to bind C4bp and were unable to infect at the highest inocula tested. Chimps are expensive, difficult to work with, and are now virtually unavailable for similar studies. That is unfortunate, since they are the only non-human primate capable of mucosal infection by GC.
In sum, the evidence does not provide much support for naturally acquired immunity, and outside of chimps, almost none for immunity after uncomplicated infection. What evidence exists suggests immunity is strain specific, which is not helpful in the clinical arena. Regardless, failure of natural immunity after infection can have many causes, and does not in itself prove that a vaccine is not possible. A vaccine might induce a much greater and different immune response than occurs after mucosal infection.
Human Immune Response to GC
Uncomplicated mucosal GC in humans results in an immune response, but in many studies the response was weak and brief. Serum antibodies have been documented against many different GC antigens (reviewed in Sparling et al., 2003). Men tend to have a poorer response than women (Tapchaisri and Srinisinha, 1976; Miettinen et al., 1989; Hedges et al., 1999). In almost all studies detectable serum antibodies were gone within a few months, or less. In cervical mucus, IgG antibodies were at least as prevalent as IgA (Tapchaisri and Srinisinha, 1976). In keeping with an overall modest immune response to GC, Hedges et al. (1998) found elevations only of lL-6 in serum of infected women; none of the measured cytokines was elevated in cervical mucus. Only transient elevations of IL-1β, IL-6, IL-8, and TNFα in urine and serum occurred in experimentally infected men (Ramsey et al., 1995).
Kasper et al. (1977) found bactericidal responses in about one-third of uncomplicated infections, but in only 5% of women with salpingitis at the time of their presentation for care, suggesting that bactericidal antibodies might protect against salpingitis. Convalescent sera showed a fourfold rise in bactericidal titers in 70% of women with severe salpingitis, as compared to only 11% with mild disease (Kasper et al., 1977). Bactericidal antibodies were common in patients with mucosal infection, but did not prevent infection (Brooks and Ingwer, 1978).
Hedges et al. (1999) provided the best study in many ways of immune responses in uncomplicated GC infection of men and women. Sera and genital secretions were tested by ELISA for isotype-specific responses to whole formaldehyde-fixed GC strains, both their own infecting strain, and a standard MS11 strain. Strain-specific IgA1 but not IgA2 antibodies were observed in both serum and cervical mucus in some subjects. A weak secondary IgG response was seen in some individuals. Women had greater responses than men, but responses were brief and temporary, declining within weeks of treatment. Previous infection did not result in immunological memory; titers and duration of antibody responses were identical in first infections and in those with previous infections. The authors’ suggested that GC might somehow suppress the immune response of the host, although no mechanism was offered.
The concept of immune suppression or redirection by commensal and pathogenic bacteria is now a very active area of research. There is evidence that GC are able to suppress either human CD4 T cell responses (Boulton and Gray-Owen, 2002; Lee et al., 2008) or human B cell antibody responses (Pantelic et al., 2005) in vitro. These effects of GC on human immune cells are regulated by binding of GC opacity (Opa) proteins to human carcinoembryonic antigen cellular adhesion molecule 1 (CEACAM1) on lymphocytes (Boulton and Gray-Owen, 2002; Pantelic et al., 2005). Binding CEACAM1 initiates a broad-range of inhibitory effects on T cells (Nagashi et al., 2008). In mice, GC infection stimulates a Th-17 response, with influx of mucosal polymorphonuclear leukocytes (PMNs; Feinen et al., 2010). This apparently is beneficial, since GC are able to substantially resist PMN-mediated killing (Simons et al., 2005, 2006; Criss and Seifert, 2008; Criss et al., 2009). From the view of a vaccine development, evidence that GC suppresses or redirects immune responses is good news, since it opens the possibility that a vaccine might circumvent such immune manipulation by GC.
The most promising approach has been to examine targets for biologically relevant immune responses. Much work has focused on a complex of three antigens that cluster together in the outer membrane (OM): lipooligosaccharide (LOS); Reduction modifiable protein (RMP), formerly designated PIII; and porin protein (PorB). The structures and biology of these molecules have been reviewed many times (Sparling et al., 2003; Virji, 2009). LOS undergoes frequent phase variation in the structure of its polysaccharide side chains, and also can be sialylated, rendering strains more serum bactericidal resistant but less invasive. Sialylated LOS also partially masks adjacent PorB trimers in the OM, reducing binding of monoclonal antibodies (mAbs) to PorB by about 50% (Elkins et al., 1992). RMP is a potent immunogen and elicits blocking antibodies that subvert the killing effects of bactericidal antibodies directed against PorB or LOS (Blake et al., 1989). PorB exists in two main classes, designated PorB1 and PorB1B, each the product of the porB gene. The PorB1 and PorB1B classes vary in certain domains of the eight exposed loops that project out from the OM, and within each class there are many minor variants that differ in sequence and antigenicity (serovars). PorB is the major protein in the outer membrane, and is essential; porB knockouts are lethal. PorB is crucial for entry of low molecular weight anions, and is involved with Pil and LOS in cooperative binding to complement receptor 3 (CR3) in primary cervical cells (Edwards et al., 2002). PorB also is capable of penetrating epithelial cells, facilitating invasion (Weel and Putten, 1991; see also reviews by Massari et al., 2003; Sparling et al., 2003; Edwards and Apicella, 2004; Virji, 2009), and leading to apoptosis, by means of entry into mitochondrial membranes within the cell (Massari et al., 2003; Kozjak-Pavlovic et al., 2009; Rudel et al., 2010). PorB does not undergo phase variation, unlike many important cell surface molecules involved in GC pathogenesis including LOS, pili (Pil), and opacity proteins (Opa). Thus, it is no surprise that PorB has evolved many mechanism for protection from host defenses, including binding the complement regulatory proteins C4bp and factor H (fH; Ram et al., 1998; Jarva et al., 2007; Madico et al., 2007; Lewis et al., 2008; Ngampasutadol et al., 2008), in addition to the protections offered by sialylation of LOS and the elicitation of blocking antibodies by RMP.
The immediate importance to this discussion is evidence that differences in immune responses to the RMP, LOS, and PorB triad affects susceptibility to infection. Data presented in summary form suggested that susceptibility to infection after sexual exposure to an infected partner was determined by an equation: antibodies against (PorB + LOS)/antibodies against Rmp equals susceptibility; the higher the ratio, the lower the risk (Rice et al., 1974; Blake and Wetzler, 1995). Unfortunately, complete details of these experiments have not been published, to our knowledge. Other efforts for a vaccine based on PorB or certain LOS epitopes are discussed below. Avoidance of Rmp antigen is one goal of current vaccine design.
Gonococcal Vaccines for Humans
Only two vaccines for GC have entered into clinical trials. The first was a crude killed whole cell vaccine, which was studied in a controlled experiment in a population of Inuit in northern Canada with high incidence and prevalence of GC infection (Greenberg et al., 1974; Greenberg, 1975). There was no evidence for protection, even though the vaccine was said to be well tolerated and induced an antibody response in over 90% of vaccine recipients. Since it was a crude whole cell vaccine, the alleged good tolerance, with only mild reactions, was surprising, since the vaccine certainly contained highly inflammatory LOS. Similar efforts with MC vaccines have utilized either detergent delipidated OMs to remove toxic lipid A, or mutants of LOS that no longer produce toxic forms of lipid A (reviewed by Granoff, 2010). The whole cell GC vaccine was not developed further. An analogous vaccine made from a piliated GC strain was tested in chimps by investigators from the CDC in Atlanta, with good serum bactericidal and immunofluorescent antibody responses. This vaccine resulted in protection very similar to that observed after natural immune clearance of infection in chimps, requiring a 1000-fold larger inoculum to infect vaccinated animals (Arko et al., 1976).
The most significant effort focused on a purified Pil vaccine. Pil are crucial for initial attachment to a variety of human cells. Either the pilus fibril, the product of the pilE gene, or a minor pilus-associated protein designated PilC, or both, mediate attachment to a still uncertain receptor. On primary cervical epithelial cells, Pil mediate attachment to CR3 in conjunction with PorB (Edwards et al., 2002; Edwards and Apicella, 2004). A series of elegant experiments proved that isolated and purified Pil containing an uncertain amount, but probably not much, of contaminating PilC, was able to protect human volunteers from experimental urethral infection after parenteral immunization (Brinton et al., 1982). The vaccine elicited a broad antibody response in serum and in genital secretions, including secretory IgA. Mucosal antibodies blocked adherence in vitro of piliated GC to various cells (McChesney et al., 1982). A subsequent trial that was never published in detail showed no protection against a heterologous strain expressing antigenically variant Pil (Tramont and Boslego, 1985). Nevertheless, a large-scale filed trial of the single-antigen Pil vaccine was carried out in high-risk US military personnel stationed in Korea, using a smaller dose and a different route (intradermal) than that used in the initial proof-of-principle experiment. The result was not even a hint of protection (Boslego et al., 1991). The probable reason for failure was antigenic variation of expressed Pil in the naturally acquired infections (Criss et al., 2005). Since then, there have been relatively small efforts to discover and develop a common Pil epitope for use in a next-generation vaccine, but so far, no useful data have been forthcoming.
Other vaccine candidates have been and are being considered, as will be discussed below, but none has advanced to clinical trial.
What Can be Learned from Meningococcal Vaccines?
An examination of the state of MC vaccines may be instructive. Vaccines for MC disease traditionally used capsular polysaccharides, which induced bactericidal responses that were protective against invasive disease including bacteremia and meningitis. These developments built upon classic studies that showed that epidemic MC disease was the result of colonization of persons by epidemic strains to which the subjects had no pre-existing bactericidal antibodies (Goldschneider et al., 1969a, b). Serum bactericidal antibody (SBA) titers of at least 1:4 were strongly correlated with protection from disease, whether induced by naturally occurring infections with bacteria that share similar capsular antigens with encapsulated MC, or by the MC capsular antigen vaccines (Frasch et al., 2009; Granoff, 2009). SBA is a better measure of protection than ELISA titers against capsular antigen (Frasch et al., 2009). SBA also is well correlated with protection from invasive disease by several other encapsulated bacteria, including pneumococci and Hemophilus influenzae (Conference Report, 2010). A variety of efficacious capsular vaccines were developed, including capsular polysaccharides conjugated to protein antigens to increase immunogenicity in the young. Capsular vaccines against serogroup C MC result in decreased carriage as well as protection from invasive disease, yielding a significant effect on transmission within the community (Maiden et al., 2008). A conjugate vaccine for group A MC designated “MenAfriVac” is now being introduced in the meningitis belt in Africa, under the aegis of a consortium known as the Meningitis Vaccine Project (Butler, 2010), and conjugate vaccines for other MC capsular types are now widely deployed in practice.
MC disease differs from GC in two crucial respects: GC do not make capsular polysaccharides, and GC only occasionally invade the blood stream, typically restricting damage to the lower and upper genital tracts. Thus, correlates for protection against MC disease such as SBA may not predict correlates for protection against GC. Moreover, SBA is not the only predictor of protection for MC disease; Granoff argues that the SBA level of at least 1:4 is sufficient for protection, but is not a sensitive measure of protection, since many are protected with lower levels of measurable SBA (Granoff, 2009). Opsonophagoyctic activity (OPA) with human serum and PMNs appears to be a good predictor of efficacy for experimental group B MC vaccines (Granoff, 2009).
Progress in development of a vaccine for group B MC might help inform development of a GC vaccine. Non-capsulate GC and MC are not identical, but share many outer membrane proteins, and their LOS are similar. For decades, serogroup B MC have been an elusive target for a vaccine because the capsule of group B MC is identical to human central nervous system (CNS) antigens, causing fear of cross-reactive anti-CNS immune responses. For this reason, all efforts for a group B MC vaccine are now focused on non-capsular antigens. Detergent-extracted outer membrane vesicles (OMV) appear to be safe, and have been used successfully to control local epidemics. The LPS also can be modified genetically so as to express a lipid A bearing either four or five acyl residues instead of the usual six, reducing the reactogenicity of OMV vaccines significantly (Donnelly et al., 2010; Keiser et al., 2010). However, the immunodominant OMV protein PorA is antigenically variable (reviewed in Granoff, 2010). OMV vaccines prepared from multiple PorA type strains have been tested and are efficacious, but are not highly immunogenic in infants (Granoff, 2010). (GC contain a porA pseudogene, but do not express PorA).
By a variety of methods including whole genome sequencing, in silico predictions of which genes express novel outer membrane proteins, proteomics, genetic engineering, and immunology, a small set of candidate novel protein immunogens were selected for further study. SBA served as the surrogate for protection, and enabled more rapid development than would have been possible without such a reliable simple assay for protective immunity. Progress required a large investment of resources by industry, the FDA and academic laboratories. Two new vaccines are in late stages of clinical trials. In one, three protein immunogens, consisting of two fusion proteins and a third single protein, are coupled with an OMV vaccine expressing PorA (Keiser et al., 2010). The vaccine appears to be more effective when the recombinant proteins are coupled with OMV rather than being administered alone (Findlow et al., 2010). The key novel proteins are NadA, an adhesin; a heparin binding protein formerly designated 2132, now designated NHBP; and a protein that binds fH, designated fHbp (Beerink and Granoff, 2008; Donnelly et al., 2010). Antibodies against fHbp are directly bactericidal, and also prevent binding of fH, further promoting bactericidal activity. All of these proteins are widely expressed among MC strains, and are relatively conserved, although fHbp exists in three major antigenically distinct families (Beerink and Granoff, 2008; Donnelly et al., 2010). Clinical trials have been conducted in adults and infants, and results are sufficiently promising to support large phase three trials, which are underway (Findlow et al., 2010; Snape et al., 2010). Meanwhile, others are investigating construction of additional novel immunogens (Lewis et al., 2010), including OMV expressing hybrids of the fHbp family that result in antibodies that cross react widely against MC strains (Beerink and Granoff, 2008; Koeberling et al., 2009). With any luck, we soon will witness the clinical deployment of efficacious vaccines in adults and infants for all MC strains, a triumph.
What are the lessons for a gonococcal vaccine? Many of the key protein antigens identified in MC are not present or are not functional in GC. PorA is not expressed in GC. NadA is absent from all of the sequenced GC genomes (Comanducci et al., 2002; and unpublished observations). GC has a fHbp homologue but it is not predicted to be localized to the surface as in MC, since it lacks a functional signal sequence. Deletion of this gene does not decrease factor H binding or alter serum resistance (Welsch and Ram, 2008). NHBP is present in several of the sequenced GC genomes and remains a viable target. Development of a vaccine similar to the MC group B vaccine should be possible, but will require a much larger effort than that currently underway. Use of OMVs with engineered LOS structures that are less reactogenic should be easy. OMVs can be constructed to express a variety of antigens, including hybrid PorB. Reliance on a single protein antigen is problematic. All the tools to do this are available, with one major exception: there is no reliable surrogate such as the SBA to guide development.
Choice of the Mouse Model for Development a GC Vaccine
Development of the female genital tract mouse model for studies of pathogenesis of GC (Jerse, 1999) opened up new possibilities for early phase studies of possible vaccines for GC. Infection in mice persists for about 2 weeks after the initial inoculation, and rates of decline can be used to measure resistance to infection. Mixed infections also can be used to test relative fitness of strains, and presumably could be used to test relative abilities to resist specific immune responses to particular antigens. The most attractive aspect of the mouse for vaccine development, apart from availability and cost, is the advanced state of development of mouse genetics, and the superb tools for monitoring immune responses in the mouse. Similar to observations in humans, mice develop an inflammatory immune response, but no resistance to repeat infection by the same strain, and no immune memory responses, after genital tract infection (Song et al., 2008).
The mouse model has many deficiencies, including (at the time our studies were performed, see below) lack of: receptors for binding Pil and Opa; human C4bp or fH; and iron binding proteins other than hemoglobin and heme that can be used by GC for growth. Each of these is a problem, and in aggregate, it is uncertain how findings in a mouse can be translated to human infection. Nevertheless, confidence that the mouse model is relevant to studies of GC biology is enhanced because of the correlations in roles of some virulence factors for both mice and humans. Sialylation of LOS appears to be requisite for maximal infectivity in mice, just as in humans (Wu and Jerse, 2006). Surprisingly, Opa expression is selected in the mouse, even in the absence of the CEACAM receptors for Opa (Cole et al., 2010). In humans, there also is strong selection for Opa expression in male volunteers (Jerse et al., 1994). Drug resistance efflux pumps involved in antibiotic resistance are required for maximal infectivity in the mouse, apparently by means of exporting cationic innate immune defense proteins (Warner et al., 2008). The mouse model, although imperfect, seemed the best choice for our efforts to investigate protective immune responses, aiming to discover which antigens might be useful in a vaccine, and correlates of protection.
But which antigens? Because of lack of their receptors, expression of phase-variable GC pili is not maintained in mice, making study of Pil vaccines impossible. Moreover, the mouse model is not particularly robust, making it very difficult to utilize a shotgun of antigens, either as genetic constructs or as proteins. We decided to focus on intranasal (IN) vaccination with outer membrane preparations, because IN immunization results in strong genital mucosal responses, and because of the success of the MC OMV vaccines. We also chose to closely examine a few outer membrane proteins. For initial studies of immunogenicity, we included the GC transferrin receptor protein TbpB, because it is an immunogenic lipoprotein, and is essential for gonococcal infection of male humans using strain FA1090 (Cornelissen et al., 1998; Cornelissen, 2008). The other candidate selected was PorB, because it plays many essential roles in infection, including binding to CR3 on female epithelial genital cells in a complex with Pil and LOS (Edwards et al., 2002); binding of PorB1 to Gp96 and SREC on epithelial cells, facilitating both adherence and invasion (Rechner et al., 2007); binding of both C4bp and fH to discrete and different loops on either PorB1 or PorB1B, enabling resistance to complement-mediated killing (summarized above); and evidence that some polyclonal and mAbs to PorB are bactericidal (Virji et al., 1986; Heckels et al., 1989; Butt et al., 1990). Moreover, PorB is an adjuvant, through binding to TLR2 (Wetzler, 2010).
OMV Vaccines Not Uniformly Successful in Mice
Immunization with gonococcal OMV is an attractive vaccine strategy due to the potential of OMV to elicit an immune response against several different conformationally correct components on the bacterial surface. There was reason to believe OMV vaccines were likely to be effective, by analogy to success with MC OMV vaccines, and because of one report of a successful trial of a GC OMV vaccine in the mouse model. Plante et al. (2000) reported that IN immunization of female mice with gonococcal OMV reduced colonization following vaginal challenge with the homologous wild-type strain MS11. Significant protection was observed in the two experiments that were reported in this study, and in an unreported experiment that was performed prior to these experiments. However, no protection was observed in similar experiments testing either MS 11or FA1090 OMV that were performed subsequent to these published experiments. The OMV preparations that showed protection were prepared by shaking whole bacteria with glass beads for 2 h at 45°C, whereas the failed experiments used OMVs prepared by passing bacteria through a needle. It is possible that differences in the preservation of conformational epitopes might be responsible for inability to reproduce these data. Additional differences in the experiments included a slightly higher dose of estradiol in the mice that did not show protection by OMV vaccine.
Failure to reproduce the initial OMV vaccine results could have been due to presence of Rmp in outer membranes from wild-type MS11. Anti-Rmp blocking antibodies could have reduced effectiveness of antibodies against either LOS or PorB. To test this, we immunized mice with OMV isolated from an rmpA insertion mutant of another PorB1B strain, strain FA1090, grown under iron-restricted conditions so as to express iron-repressed proteins. OMVs were prepared by passing bacteria through a small bore needle. Mice developed substantial anti-PorB serum IgG antibodies, a rise in SBAs, vaginal PorB-specific IgG, and very high levels of vaginal PorB-specific IgA antibodies (Zhu et al., 2005). Despite what one might have assumed were the types of responses that would correlate with protection, there was no protection after IN OMV immunization (Table 1). We concluded that anti-Rmp blocking antibodies were not responsible for the lack of effectiveness of OMV vaccine antigens in vivo. These results also showed that induction of mucosal IgG and IgA antibodies, and a bactericidal serum response, did not predict protection.
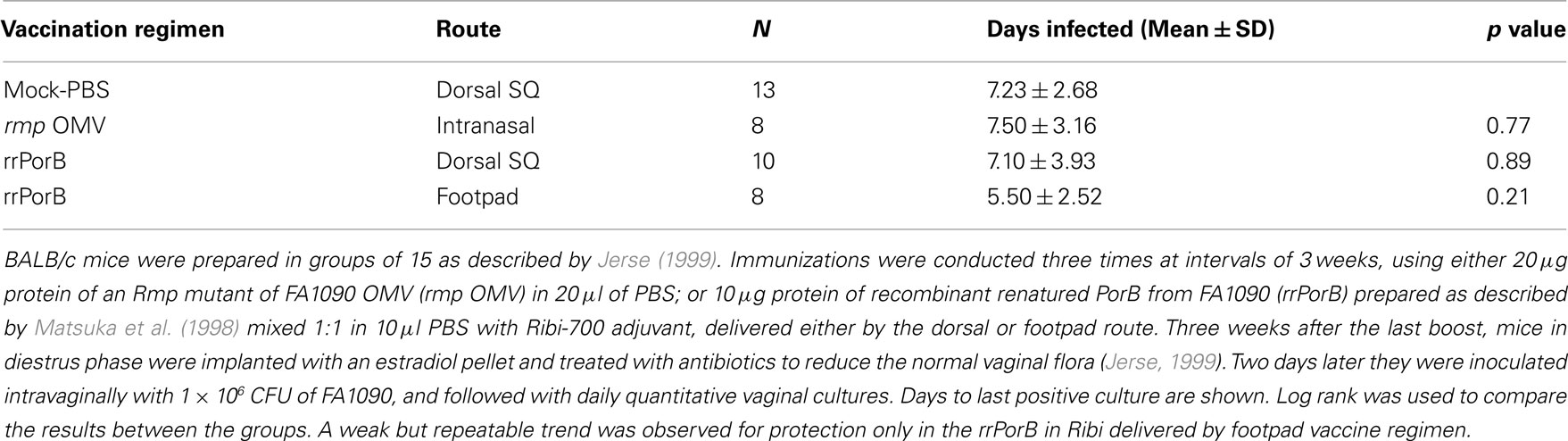
Table 1. Immunization with OMV or rrPorB fails to protect female mice from genital infection by FA1090.
Another reason for failure of the vaccines could be in vivo sialylation of LOS, leading to resistance to mucosal antibodies and complement. Increased resistance to complement-mediated defenses in humans occurs via the covalent linkage of sialic acid to LOS molecules with a lactose-N-tetraose moiety, which reduces activation of the alternative pathway of complement activation via the binding of fH. Although mouse fH does not bind to sialylated LOS (Ngampasutadol et al., 2008), sialylation does reduce opsonophagocytosis following incubation of GC in mouse serum, and an lst mutant (which lacks sialyltransferase, and can not sialylate LOS) was attenuated for murine infection (Wu and Jerse, 2006). To test the effect of sialylation in vivo on possible escape from OMV-induced immune protection, we used wild-type F62 and an isogenic lst mutant as the challenge strains, after IN immunization with F62 OMVs. Ovariectomized BALB/c mice were used to circumvent the need for mice to be in anestrus or in the diestrus stage of the estrous cycle during the challenge phase of the experiment. High titers of OMV-specific serum IgG and vaginal IgG and IgA were detected following immunization. Mice were challenged 3 weeks after the final immunization. There was no difference in the duration of recovery or number of wild-type or lst mutant bacteria recovered (Figure 1). Similar results were obtained after IN immunization with the strain MS11 OMVs, followed by challenge with either MS11 or its isogenic MS11lst mutant. We concluded that sialylation was not responsible for gonococcal evasion of an OMV vaccine-induced host response in the mouse model.
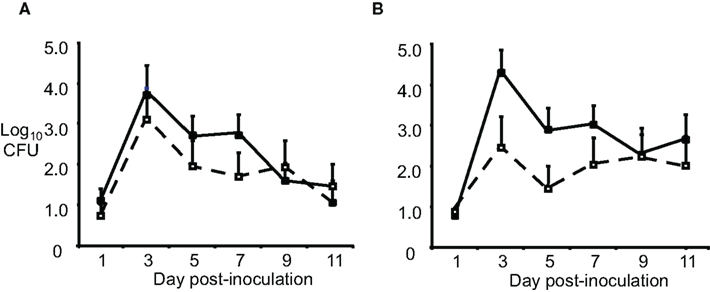
Figure 1. Sialylation does not protect GC from OMV vaccine-induced immune responses. Groups of BALB/c mice (n = 10–12 per group) were immunized with 20 μg of OMV (■) from strain F62 via the IN, subcutaneous, and IN routes separated by 3-week intervals or given PBS (□) by the same routes. Three weeks after the final immunization, mice were challenged vaginally with (A) wild-type F62 bacteria or the (B) F62 lst::aphA3 mutant (GP330). Vaginal mucus was cultured every other day for 11 days. The average log10CFU per 100 μl vaginal swab suspension over time is shown with standard error bars. There was no difference in the number of GC recovered (repeated measures ANOVA) or duration of colonization (Log rank test) for any experimental group. This experiment was repeated using three IN immunizations and another experiment was performed with strain MS11 and an MS11 lst mutant, and the results were similar.
Thus, three different outer membrane vaccines delivered IN resulted in high titers of serum antibodies, excellent serum bactericidal activity, and robust mucosal responses as assessed by analysis of vaginal wash and fecal pellet antibodies (Zhu et al., 2005; and data not shown), yet were uniformly completely ineffective in either accelerating clearance or preventing GC infection in the female mouse genital tract. OMV may still play some role as a component of a GC vaccine, but we shifted our attention to individual outer membrane protein antigens.
Recombinant Protein Vaccines
The first consideration was how to prepare purified protein antigens in a conformationally correct form, in sufficient amounts, and in the absence of contaminants that might be a problem, including LOS and the blocking antigen Rmp. Problems due to the blocking effects of antibodies to Rmp could be avoided by use of cloned recombinant preparations of PorB. Considerable effort by commercial partners went into preparing a refolded recombinant FA1090 PorB1B (rrPorB) from E. coli, in a conformation that closely mimicked that of native PorB extracted from GC (Matsuka et al., 1998). A phase 1 human trial was undertaken, but was not carried forward to test for protection of male volunteers, apparently because of problems with adverse rates of local inflammation due to the vaccine. The rrPorB served as an excellent immunogen for studies in mice. Preparing batches of the much less hydrophobic TbpB was less problematic (Thomas et al., 2006).
VRP and DNA Vaccines
Another very attractive option was to use genetic vaccines expressing either PorB or TbpB. Such an approach would allow much greater flexibility in exploring combinations and variations in the antigens. DNA vaccines have the advantage of great simplicity. There has been a surge in interest in viral derivatives that deliver antigens either as a viral like particle (VLP) that do not replicate, or as viral replicon particles that have a single cycle of replication (VRP). One of these VRP systems, based on the alphavirus Venezuelan encephalitis virus (VEE), provides both systemic and mucosal immune responses, and is being used for a variety of novel vaccines Davis et al., 1996; Hubby et al., 2007). We prepared both DNA and VEE-VRP vaccines for each of FA1090 TbpB and PorB (Zhu et al., 2004, 2005; Thomas et al., 2006).
The PorB-DNA vaccine appeared promising in terms of immune responses (Zhu et al., 2004). PorB-DNA was particularly immunogenic in BALB/c mice when boosted either by rrPorB in Ribi-R700 adjuvant, or by PorB-VRP. Delivery of PorB-DNA by the IM route resulted in a polarized Th1 response, but delivery by a gene gun to the epidermis, resulted in a predominantly Th2 response (Zhu et al., 2004). Boosting with PorB-VRP drove the response to a more dominant Th1 response. Serum antibodies with opsonophagocytic activity against FA1090 were observed after DNA immunization. Ability to control the direction of the immune response might be useful for vaccine development in the mouse, but because of the relatively greater immune responses elicited by PorB-VRPs boosted by rrPorB (below), and by other practical considerations, our vaccine efficacy experiments were limited to the PorB-VRP and rrPorB antigens.
Based on the ratio of PorB-specific IgG1/IgG2 antibody responses, there was an apparent Th1 bias with either the PorB-VRP vaccine, delivered without additional adjuvants into a rear footpad (FP), or with the rrPorB vaccine delivered with Ribi-R700 adjuvant into a rear FP. In contrast, there was an apparent Th2 bias when the rrPorB vaccine with Ribi was delivered into the dorsal SQ region. ELISPOT assays of harvested splenocytes showed that there was a significant interferon-γ (IFN-γ) response to PorB peptides only after immunization with the PorB-VRP vaccine (Zhu et al., 2005). Highest serum IgG responses were observed with the rrPorB immunization, accompanied by vaginal mucosal IgG but not IgA responses. The best mucosal IgG responses were observed after rrPorB immunization, whereas the highest mucosal IgA responses were after IN OMV immunization (Zhu et al., 2005).
Mice were Protected by a VRP Vaccine
Initial vaccination/protection experiments utilized rrPorB with Ribi-R700 adjuvant. Delivery of the vaccine in the dorsal SQ route resulted in no protection, but delivery into the FP resulted in a weak, statistically insignificant, but repeatable trend toward protection, manifest by about a 2-day reduction in colonization (Table 1 and data not shown). We designed additional experiments to test whether a prime-boost regimen utilizing the rrPorB and PorB-VRP vaccines might yield improved results. Four groups of 24 animals each were immunized three times at 2-week intervals with either mock control, rrPorB-R700 in the FP × 3, PorB-VRP in FP × 3, or PorB-VRP in FP × 2 plus a boost with rrPorB-R700 in FP (Figure 2). There was a trend toward reduced duration of infection in the PorB-VRP in FP × 3 group (p = 0.11), and in significant protection (p < 0.01) in the PorB-VRP plus rrPorB-R700 boost group.
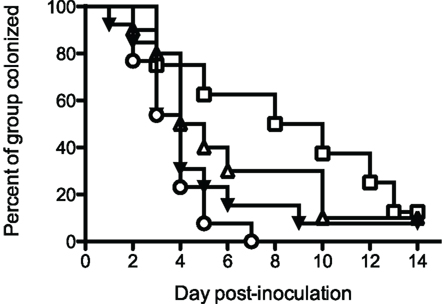
Figure 2. PorB-VRP × 2 in FP plus boost with rrPorB in FP protects against vaginal infection in the mouse model. Three- to four-week old female BALB/c mice were divided randomly into four groups of 24 and vaccinated three times at 2 weeks interval. Mice were immunized with three doses of PBS (□, mock, n = 8), rrPorB (△, n = 10), PorB-VRP (▼, n = 13) or 2 doses of PorB-VRP with 1 boost of rrPorB (○, n = 13). All doses of immunogens, in a volume of 20 μl, were given through the left footpad after the animals had been anesthetized. Two weeks after the last boost, mice in the diestrus phase were implanted with estradiol pellets and treated with antibiotic. They were inoculated intravaginally with 1 × 106 CFU of FA1090 2 days later. Daily vaginal cultures were processed for total recovery from the infection. Days to last positive culture are shown. Log-rank test for equality to the Mock group yielded the following p values: PorB-VRP + rrPorB boost, p = <0.01; PorB-VRP without rrPorB boost, p = 0.11; rrPorB alone, p = 0.40.
These results were encouraging, even though the vaccines did not prevent infection. The experiment was repeated, adding an additional control consisting of an irrelevant influenza virus hemagglutinin antigen VRP construct (HA-VRP). Because the most effective vaccine in the previous experiment was PorB-VRP × 2 boosted with rrPorB, the control HA-VRP vaccine was given twice, and boosted with rrPorB. This tested for the possible effects of the VRP vector. The mock PBS control also was given twice and boosted once with rrPorB, to control for the effects of a single dose of rrPorB. The results (Figure 3) were interesting. Both the PorB-VRP vaccine, boosted once with rrPorB, and the HA-VRP vaccine boosted once with rrPorB resulted in significant (p < 0.05) reduction in duration of infection compared to the PBS control boosted once with rrPorB. The single booster dose of rrPorB given after PBS × 2 had no effect, compared to PBS without rrPorB. There was no difference in colonization between the HA-VRP × 2 + rrPorB boost and the PorB-VRP × 2 + rrPorB boost vaccines. We tentatively concluded that the VRP vector was the important component of the vaccine, and that PorB expressed by the VRP vector might not be necessary for protection. Note that infectious challenge was initiated 4 weeks after the last dose of PorB-VRP in the PorB-VRP × 2 plus rrPorB boost group, so the effects of the VRP vector were durable.
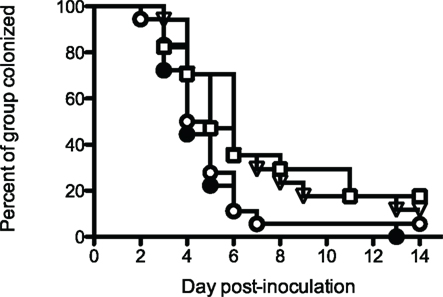
Figure 3. A PorB-VRP vaccine for gonorrhea in the mouse is no more effective than a vaccine composed of the same vector expressing an irrelevant peptide. BALB/c female mice were immunized in FP with either PBS three times (□, mock, n = 17); PBS twice with a single boost of rrPorB (▽, n = 17); FA1090 PorB-VRP twice with a single boost of rrPorB (○, n = 18); or influenza HA-VRP twice, also boosted once with rrPorB (●, n = 18). The rrPorB boosts consisted of 10 μg of FA1090 rrPorB in 10 μl PBS mixed 1:1 with Ribi-700 adjuvant. Immunizations were at 2-week intervals. The VRP preparations were administered in a dose of about 1 × 106 particles without additional adjuvant. The volume of all immunizations was 20 μl. Two weeks after the final immunization, mice in diestrus were recruited for the challenge with FA1090, as in Table 1. Days to last positive culture are shown. Log rank was used to compare all four groups; results were significantly different among the groups (p = 0.03). The only pairwise comparisons which were significantly different (p < 0.05) were PBS alone vs HA-VRP + rrPorB (p = 0.03), PBS + rrPorB vs HA-VRP + rrPorB (p = 0.01), and PBS + rrPorB vs PorB-VRP + rrPorB (p = 0.04).
How could a vector expressing an irrelevant peptide provide protection against GC infection? Protection was not correlated with serum PorB-specific IgG, since there was no more PorB IgG in sera after immunization with the effective HA-VRP + rrPorB boost vaccine than the ineffective control PBS + rrPorB boost vaccine (Figure 4). Serum levels of specific PorB IgG were at least 10-fold higher after immunization with the ineffective rrPorB × 3 vaccine (Zhu et al., 2005, and data not shown). Surface-bound antibodies to whole GC also were much higher after the rrPorB × 3 in FP vaccine than after any of the VRP-based vaccines (data not shown). Vaginal levels of PorB-specific IgA and IgG were much higher after immunization with the ineffective rrPorB and OMV vaccines than after the effective VRP-based vaccines (Zhu et al., 2005). The only bactericidal antibody responses were observed after the totally ineffective OMV vaccine. Thus, protection was not correlated with measured antibody responses. Since we did not measure specific IgG2b and IgG3 responses, conclusions about the roles of isotypes in protection were not possible.
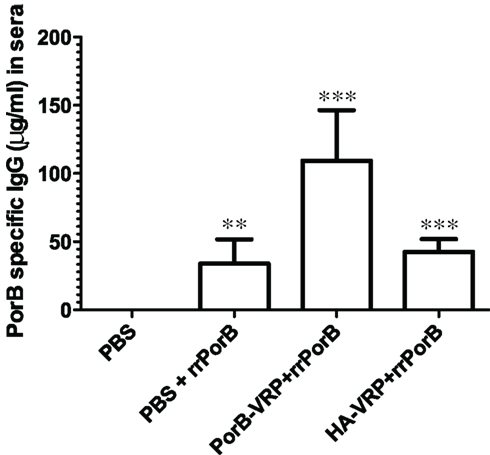
Figure 4. PorB specific IgG levels in sera after different vaccination regimens. BALB/c mice were immunized with HA-VRP, PorB-VRP, or PBS for two times at 2 weeks intervals and then boosted with rrPorB as the last immunization. Another control group was immunized three times with PBS at 2 weeks intervals. Four weeks after the last immunization, sera were collected and IgG responses against a pool of synthetic PorB peptides were measured by quantitative ELISA. Each column represents mean ± SD. Data were analyzed by one-way ANOVA followed by unpaired Student’s t-test. Symbols: **p < 0.01; ***p < 0.001 as compared to PBS control. PBS + rrPorB is different from PorB-VRP + rrPorB (p = 0.002); PorB-VRP + rrPorB is different from HA-VRP + rrPorB (p = 0.003); but PBS + rrPorB is not different from HA-VRP + rrPorB (p = 0.32).
Although quantities of serum PorB-specific IgG were the same in the ineffective PBS + rrPorB boost group and the effective HA-VRP + rrPorB boost group (Figure 4), there could have been unmeasured qualitative differences that accounted for the observed differences in protection. Responses in Figure 4 were measured to synthetic peptides; PorB-conformation-specific antibodies were not measured, nor were antibody avidities. Moreover, there was a minimal serum IgG response to PorB peptides after HA-VRP immunization × 2, without the rrPorB boost (520 ng/ml PorB-reactive IgG as compared to 4135 ng/ml after PorB-VRP × 2 without rrrPorB boost, and no detectable response after immunization with PBS × 2, data not shown). Therefore, the rrPorB boost after the HA-VRP regimen could have resulted in a secondary immune response with different antibody characteristics as compared to a primary immunization with rrPorB after immunization with PBS. Reasons for the small serum IgG response to HA-VRP are not understood; no linear amino acid (aa) sequence identities longer than 4 aa were identified between FA1090 PorB, and either HA, the VEE capsid protein, or the VEE surface glycoproteins E1, E2, or E3 (data not shown). Since experiments were not conducted with an empty VRP vector, the observed protection could have had something to do with HA rather than the VRP vector.
We considered that an IFN-γ Th1 response might be the best correlate of protection, since PorB peptide-specific spenocyte IFN-γ responses were significantly greater after the VRP vaccines than after either the rrPorB or OMV vaccines (Zhu et al., 2005). However, the IFN-γ responses measured 4 weeks after the immunizations were completed, and 2 weeks after infection, were identical in the ineffective rrPorB × 3 and the PorB-VRP × 2 + rrPorB boost vaccines (Zhu et al., 2005, Figures 5A,B), suggesting that it might not have been the IFN-γ responses per se, but something else triggered after vaccination with the HA-VRP vaccine that was most important. Although IFN-γ responses are important for immune responses to antigens expressed by VRP vectors, they are not important for the adjuvant effect of empty VRPs (Thompson et al., 2008a).
What is Special about VRPs?
The VEE VRP system has been shown to stimulate a strong adjuvant effect, especially when empty VRPs are delivered simultaneously with antigen (Thompson et al., 2006; Tonkin et al., 2010; Carroll et al., 2011). The effect is at least as great as that driven by other adjuvants such as CpG, and depends on VRP RNA replication (Thompson et al., 2006). Systemic and mucosal responses are enhanced, involving both B and T cells; a robust antigen-specific CD8+ T cell response is generated (Thompson et al., 2008b). The effect does not depend on delivery of VRP into the mouse FP, but is seen after IM injection as well (Tonkin et al., 2010). After VRP delivery, dendritic cells as well as macrophages and NK cells are stimulated to release a variety of cytokines and chemokines, with increased cellularity in involved lymph nodes. The list of cytokines and chemokines is interesting both by what is stimulated, and what is not: IL-12 and IL-17 are not stimulated, but IL-6, G-CSF, GM-CSF, IFN-γ, MIP-2, and MCP-1 are stimulated (Tonkin et al., 2010). We did not try to dissect the various components of innate immune responses after the VRP immunizations, and did not measure specific T cell responses. The purported effects of the VRP vector could have been mediated by a variety of mechanisms, including increased production of defensins or other innate responses, or by conformation or avidity differences in antibodies produced in the different vaccination regimens. Future exploration of the basis of the apparent protective effects of VRP-based immunization as a means to enhance GC vaccines should be a fertile field of study.
Future Directions
If there is to be a vaccine for GC, a renewed effort is needed in several areas. First is in the search for correlates of immune protection in humans. It is not obvious how to accomplish this in the absence of acquired immunity in humans. One way forward might be a prospective examination of sexual partners of infected subjects, inquiring as to the immune parameters (especial at mucosal surfaces, including innate responses) in those who, and those who do not, become infected. About two-thirds of exposed women become infected; there may be differences in immune responses at first exposure, or a week after exposure, that would provide leads. Similar studies in male volunteers are indicated as well; about 50% of volunteers exposed to 1 × 105 CFU of strain FA1090 develop urethral infection. There may be differences in early innate responses in those who do, and those who do not, become infected.
The mouse model also offers opportunities, especially as new transgenic mice become available, that express one or several human gene products essential for GC infection. Transgenic mice expressing receptors for Opa proteins including CEACAM1 (Gu et al., 2010), human fH (Ufret-Vincenty et al., 2010), and transferrin (Li et al., 2010) are available already. Humanized mouse models are being generated in multiple laboratories (Zhang et al., 2007; Denton and Garcia, 2009), and with further improvement, may be helpful in assessing human-like immune responses in mice. A mouse model that allowed substantial growth of the inoculum, and longer persistence of infection, would assist in developing correlates of protection in the mouse. Among other parameters, mice can be monitored for the quality of the immune response to experimental vaccines, focusing on generation of cells that are positive for each of IL-2, IFN-γ, and TNF. Substantial experience shows that such T cell responses are correlated with a variety of effective vaccines (Seder et al., 2008).
There are multiple candidate proteins that can be considered for a subunit vaccine, with or without lipid A-detoxified OMVs. These include PorB, which remains attractive even though extensive studies cited above have not been fruitful. An approach aimed specifically at PorB loop domains involved in binding fH, C4bp, and/or host receptor binding to CR3 might yield better results than whole PorB. Such a directed response might block binding to and invasion of host cells, and could be bactericidal either directly, or by blocking fH or C4b binding. One way to do this is being explored by one of us (Ann E. Jerse), utilizing circular loop peptides as the immmunogens, based on evidence that analagous circular Opa peptides are able to elicit potentially protective immune responses (Cole and Jerse, 2009). Intranasal immunization with the GC transferrin-binding proteins TbpA or TbpB, or both, elicited bactericidal immune responses; TbpA stimulated more broadly cross-reactive antibodies than did TbpB (Price et al., 2005). Immunization of mice with genetic chimeras that fused parts of TbpA and TbpB stimulated production of vaginal antibodies that inhibited growth in vitro (Price et al., 2007). A novel adhesin designated OmpA is yet another candidate (Serino et al., 2007).
Extensive studies over the past 15 years have shown the potential of vaccines directed at a conserved LOS epitope, which is defined by binding the mAb 2C7 (Gulati et al., 1996). About 95% of GC strains express the 2C7 epitope, composed principally of a lactose residue attached to heptose-2 on GC LOS (Yamasaki et al., 1999). Synthetic peptides that mimic the 2C7 epitope are immunogenic, and result in bactericidal activity, even against strains that are resistant to killing by normal human serum (Ngampasutadol et al., 2006).
There may be other effective immunogens. Perhaps improved mouse models will permit a discovery approach that utilizes pools of antigens, delivered as DNA constructs. Priming with VRP-based vaccines, or a variety of other novel adjuvants (Gwinn et al., 2010; Pulendran et al., 2010), may enable more effective immune responses. The pace of discovery in mucosal immunology is rapid; efforts to develop more effective genital immune responses, including nasal immunization strategies (Gwinn et al., 2010), may pay dividends. Progress will require a substantial commitment of effort and resources. It is not too early to renew the effort.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Author’s unpublished experiments were supported by research grants RO1AI142053 and U19AI031496. Studies on sialylation as a protection from vaccines were performed by Sandra Veit, Afrin Bequm, and Hong Wu. Experiments with vaccines composed of VRPs were aided in many ways by discussions with Nancy Davis and Bob Johnston. Martha Collier prepared the VRP vaccines and Rashina Cholera provided statistical support.
References
Arko, R. J., Duncan, W. P., Brown, W. J., Peacock, W. L., and Tomizawa, T. (1976). Immunity in infection with Neisseria gonorrhoeae: duration and serological response in the chimpanzee. J. Infect. Dis. 133, 441–447.
Beerink, P. T., and Granoff, D. M. (2008). Bactericidal antibody responses induced by meningococcal recombinant chimeric factor H-binding protein vaccines. Infect. Immun. 76, 2568–2575.
Blake, M. S., and Wetzler, L. M. (1995). Vaccines for gonorrhea: where are we on the curve? Trends Microbiol. 3, 469–474.
Blake, M. S., Wetzler, L. M., Gotschlich, E. C., and Rice, P. A. (1989). Protein III structure, function, and genetics. Clin. Microbiol. Rev. 2S, S60–S63.
Boslego, J. W., Tramont, E. C., Chung, R. C., Mcchesney, D. G., Ciak, J., Sadoff, J. C., Piziak, M. V., Brown, J. D., Brinton, C. C. J., Wood, S. W., and Bryan, J. R. (1991). Efficacy trial of a parenteral gonococcal pilus vaccine in men. Vaccine 9, 154–162.
Boulton, I. C., and Gray-Owen, S. D. (2002). Neisserial binding to CEACAM1 arrests the activation and proliferation of CD4+ T lymphocytes. Nat. Immunol. 3, 229–236.
Brinton, C. C., Wood, S. W., Brown, A., Labik, A. M., Bryan, J. R., Lee, S. W., Polen, S. E., Tramont, E. C., Sadoff, J., and Zollinger, W. (1982). “The development of a Neisserial pilus vaccine for gonorrhea and meningococcal meningitis,” in Seminars in Infectious Diseases, Vol. IV: Bacterial Vaccines, eds J. B. Robbins, J. C. Hill, and J. C. Sadoff (New York, NY: Thieme-Stratton), 140–159.
Brooks, G. F., and Ingwer, I. (1978). Studies on the relationships between serum bactericidal activity and uncomplicated genital infections due to Neisseria gonorrhoeae. J. Infect. Dis. 138, 333–339.
Buchanan, T. M., Eschenbach, D. A., Knapp, J. S., and Holmes, K. K. (1980). Gonococcal salpingitis is less likely to recur with Neisseria gonorrhoeae of the same principal outer membrane protein antigenic type. Am. J. Obstet. Gynecol. 138, 978–980.
Butt, N. J., Virji, M., Vayreda, F., Lambden, P. R., and Heckels, J. E. (1990). Gonococcal outer membrane-protein P1B: comparative sequence analysis and localization of epitopes which are recognized by type-specific and cross-reacting monoclonal antibodies. J. Gen. Microbiol. 136, 2165–2172.
Carroll, T. D., Matzinger, S. R., Barro, M., Fritts, L., McChesney, M. B., Miller, C. J., and Johnston, R. E. (2011). Alphavirus replicon-based adjuvants enhance the immunogenicity and effectiveness of FluzoneR in rhesus macaques. Vaccine 931–940.
Centers for Disease Control. (2010). Sexually transmitted diseases treatment guidelines. Morbidity and Mortality Weekly Report 59, December 17, 1–114.
Chisholm, S. A., Mouton, J. W., Lewis, D. A., Nichols, T., Ison, C. A., and Livermore, D. M. (2010). Cephalosporin creep among gonococci: time for a pharmacodynamic rethink? J. Antimicrob. Chemother. 65, 2141–2148.
Cole, J. G., Fulcher, N. B., and Jerse, A. E. (2010). Opacity proteins increase fitness in the female genital tract due to a factor under ovarian control. Infect. Immun. 78, 1629–1641.
Cole, J. G., and Jerse, A. E. (2009). Functional characterization of antibodies against Neisseria gonorrhoeae opacity protein loops. PLoS ONE 4, e8108. doi:10.1371/journal.pone.0008108
Comanducci, M., Bambini, S., Brunelli, B., Adu-Bobie, J., Aricò, B., Capecchi, B., Giuliani, M. M., Masignani, V., Santini, L., Savino, S., Granoff, D. M., Caugant, D. A., Pizza, M., Rappuoli, R., and Mora, M. (2002). NadA, a novel vaccine candidate of Neisseria meningitidis. J. Exp. Med. 195, 1445–1454.
Conference Report. (2010) . Utilization of serologic assays to support efficacy of vaccines in nonclinical and clinical trials: meeting at the crossroads. Vaccine 28, 4539–4547.
Cornelissen, C. N. (2008). Identification and characterization of gonococcal iron transport systems as potential vaccine antigens. Future Microbiol. 3, 287–298.
Cornelissen, C. N., Kelley, M., Hobbs, M. M., Anderson, J. E., Cannon, J. G., Cohen, M. S., and Sparling, P. F. (1998). The gonococcal transferrin receptor is required for human infection. Mol. Microbiol. 27, 611–616.
Criss, A. K., Katz, B. Z., and Seifert, H. S. (2009). Resistance of Neisseria gonorrhoeae to non-oxidative killing by adherent human polymorphonuclear leukocytes. Cell. Microbiol. 11, 1074–1087.
Criss, A. K., Kline, K. A., and Seifert, H. S. (2005). The frequency and rate of pilin antigenic variation in Neisseria gonorrhoeae. Mol. Microbiol. 58, 510–519.
Criss, A. K., and Seifert, H. S. (2008). Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell. Microbiol. 10, 2257–2270.
Davis, N. L., Brown, K. W., and Johnston, R. E. (1996). A viral vaccine vector that expresses foreign genes in lymph nodes and protects against mucosal challenge. J. Virol. 70, 3781–3787.
Denton, P. W., and Garcia, J. V. (2009). Novel humanized murine models for HIV research. Curr. HIV/AIDS Rep. 6, 13–19.
Donnelly, J., Medini, D., Boccadifuoco, G., Biolchi, A., Ward, J., Frasch, C., Moxon, E. R., Stella, M., Comanducci, M., Bambini, S., Muzzi, A., Andrews, W., Chen, J., Santos, G., Santini, L., Boucher, P., Serruto, D., Pizza, M., Rappuoli, R., and Giuliani, M. M. (2010). Qualitative and quantitative assessment of meningococcal antigens to evaluate potential strain coverage of protein-based vaccines. Proc. Natl. Acad. Sci. 107, 19490–19495.
Edwards, J. L., and Apicella, M. A. (2004). The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin. Microbiol. Rev. 17, 965–981.
Edwards, J. L., Brown, E. J., Uk-Nham, S., Cannon, J. G., Blake, M. S., and Apicella, M. A. (2002). A co-operative interaction between Neisseria gonorrhoeae and complement receptor 3 mediates infection of primary cervical epithelial cells. Cell. Microbiol. 4, 571–584.
Elkins, C., Carbonetti, N. H., Varela, V. A., Stirewalt, D., Klapper, D. G., and Sparling, P. F. (1992). Antibodies to N terminal peptides of gonococcal porin are bactericidal when gonococcal lipopolysaccharide is not sialylated. Mol. Microbiol. 6, 2617–2630.
Faruki, H., Kohmescher, R. N., McKinney, W. P., and Sparling, P. F. (1985). A community-based outbreak of a penicillin-resistant Neisseria gonorrhoeae not producing penicillinase (chromosomally mediated resistance). N. Engl. J. Med. 313, 607–611.
Feinen, B., Jerse, A. E., Gaffen, S. L., and Russel, M. W. (2010). Critical role of Th17 responses in a murine model of Neisseria gonorrhoeae genital infection. Mucosal Immunol. 3, 312–321.
Findlow, J., Borrow, R., Snape, M. D., Dawson, T., Holland, A., John, T. M., Evans, A., Telford, K. L., Ypma, E., Toneatto, D., Oster, P., Miller, E., and Pollard, A. J. (2010). Multicenter, open-label, randomized phase II controlled trial of an investigational recombinant meningococcal serogroupB vaccine with and without outer membrane vesicles, administered in infancy. Clin. Infect. Dis. 51, 1127–1137.
Fleming, D. T., and Wasserheit, J. N. (1999). From epidemiological synergy to public health policy and practice: the contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sex. Transm. Infect. 75, 3–17.
Fox, K.K. Thomas J.C., Weiner, D. H., Davis, R. H., Sparling, P. F., and Cohen, M. S. (1999). Longitudinal evaluation of serovar-specific immunity to Neisseria gonorrhoeae. Am. J. Epidemiol. 149, 353–358.
Frasch, C. A., Borrow, R., and Donnelly, J. (2009). Bactericidal antibody is the immunological surrogate of protection against meningococcal disease. Vaccine 27S, B112–B116.
Goldschneider, I., Gotschlich, E. C., and Artenstein, M. S. (1969a). Human immunity to the meningococcus. I. The role of humoral antibodies. J. Exp. Med. 129, 1307–1326.
Goldschneider, I., Gotschlich, E. C., and Artenstein, M. S. (1969b). Human immunity to the meningococcus. II. Development of natural immunity. J. Exp. Med. 129, 1327–1348.
Golparian, D., Hellmark, B., Fredlund, H., and Unemo, M. (2010). Emergence, spread, and characteristics of Neisseria gonorrhoeae isolates with in vitro decreased susceptibility and resistance to extended-spectrum cephalosporins in Sweden. Sex. Transm. Infect. 86, 454–460.
Granoff, D. (2009). Relative importance of complement-mediated bactericidal and opsonic activity for protection against meningococcal disease. Vaccine 27S, B117–B125.
Greenberg, L., Diena, B. B., Ashtron, F. A., Wallace, R., Kenney, C. P., Znamirowski, R., Ferrari, H., and Atkinson, J. (1974). Gonococcal vaccine studies in Inuvik. Can. J. Public Health 65, 29–33.
Gu, A., Zhang, Z., Chang, N., Tsark, W., and Shively, J. E. (2010). Generation of human CEACAM1 transgenic mice and binding of Opa protein to their neutrophils. PLoS ONE 5, e10067. doi:10.1371/journal.pone.0010067
Gulati, S., McQuillen, D. P., Mandrell, R. E., Jani, D. B., and Rice, P. A. (1996). Immunogenicity of Neisseria gonorrhoeae lipooligosaccharide epitope, 2C7, widely expressed in vivo with no immunochemical similarity human glycosphingolipids. J. Infect. Dis. 174, 1223–1237.
Gwinn, W. M., Kirwan, S. M., Wang, S. H., Ashcraft, K. A., Sparks, N. L., Doil, C. R., Tlusty, T. G., Casey, L. S., Hollingshead, S. K., Briles, D. E., Dondero, R. S., Hickey, A. J., Foster, W. M., and Staats, H. F. (2010). Effective induction of protective systemic immunity with nasally administered vaccines adjuvanted with IL-1. Vaccine 28, 6901–6914.
Handsfield, H. H., Lipman, T. O., Harnish, J. P., Tronca, E., and Holmes, K. K. (1974). Asymptomatic gonorrhea in men. Diagnosis natural course, prevalence and significance. N. Engl. J. Med. 290, 117–123.
Heckels, J. E., Fletcher, J. N., and Virji, M. (1989). The potential protective effect of immunization with outer membrane protein 1 from Neisseria gonorrhoeae. J. Gen. Microbiol. 135, 2269–2276.
Hedges, S. R., Mayo, M. S., Mestecky, J., Hook, E. W. III, and Russell, M. W. (1999). Limited local and systemic antibody responses to Neisseria gonorrhoeae during uncomplicated genital infections. Infect. Immun. 67, 3937–3946.
Hedges, S. R., Sibley, D. A., Mayo, M. S., Hook, E. W., and Russell, M. W. (1998). Cytokine and antibody responses in women infected with Neisseria gonorhoeae: effects of concomitant infections. J. Infect. Dis. 178, 742–751.
Hill, J. (1942). Experimental infection with Neisseria gonorrhoeae. Am. J. Syph. Gonorrhea Vener. Dis. 27, 733–771.
Hobbs, M. M., Alcorn, T. M., Davis, R. H., Fischer, W., Thomas, J. C., Martin, I., Ison, C., Sparling, P. F., and Cohen, M. S. (1999). Molecular typing of Neisseria gonorrhoeae causing repeated infections: evolution of porin during passage within a community. J. Infect. Dis. 179, 371–381.
Hubby, B., Talarico, T., Maughan, K. W., Reap, E. A., Berglund, P., Kamrud, K. L., Copp, L., Lewis, W., Cecil, C., Norberg, P., Wagner, J., Watson, A., Negri, S., Burnett, B. K., Graham, A., Smith, J. F., and Chulay, J. D. (2007). Development and preclinical evaluation of an alphavirus replicon vaccine for influenza. Vaccine 25, 8180–8189.
Jarva, H., Ngampasutadol, J., Ram, S., Rice, P. A., Villoutreix, B. O., and Blom, A. M. (2007). Molecular characterization of the interaction between porins of Neisseria gonorrhoeae and C4b-binding protein. J. Immunol. 179, 540–547.
Jerse, A. E. (1999). Experimental gonococcal genital tract infection and opacity protein expression in estradiol-treated mice. Infect. Immun. 67, 5699–5708.
Jerse, A. E., Cohen, M. S., Drown, P. M., Whicker, L. G., Isbey, S. F., Seifert, H. S., and Cannon, J. G. (1994). Multiple gonococcal opacity proteins are expressed during experimental urethral infection in the male. J. Exp. Med. 179, 911–920.
Kasper, D. L., Rice, P. A., and McCormick, W. M. (1977). Bactericidal antibody in genital infection due to Neisseria gonorrhoeae. J. Infect. Dis. 135, 243–251.
Keiser, P. B., Gibbs, B. T., Coster, T. S., Moran, E. E., Stoddard, M. B., Labrie, J. E. III, Schmiel, D. H., Pinto, V., Chen, P., and Zollinger, W. D. (2010). A phase 1 study of a group B meningococcal native outer membrane vesicle vaccine made from a strain with deleted lpxL2 and synX and stable expression of opcA. Vaccine 28, 6970–6976.
Koeberling, O., Giuntini, S., Seubert, A., and Granoff, D. M. (2009). Meningococcal outer membrane vesicle vaccines derived from mutant strains engineered to express factor H binding proteins from antigenic variant groups 1 and 2. Clin. Vaccine Immunol. 16, 156–162.
Kozjak-Pavlovic, V., Dian-Lothrop, E. A., Meincke, M., Kepp, O., Ross, K., Rajalingam, K., Harsman, A., Hauf, E., Brinkmann, V., Gunther, D., Herrmann, I., Hurwitz, R., Rassow, J., Wagner, R., and Rudel, T. (2009). Bacterial porin disrupts mitochondrial membrane potential and sensitizes host cells to apoptosis. PLoS Pathog. 5, e1000629. doi: 10.1371/journal.ppat.1000629
Kraus, S. J., Brown, J. W., and Arko, R. J. (1975). Acquired and natural immunity to gonococcal infection in chimpanzees. J. Clin. Invest. 55, 1349–1356.
Lee, H. S., Ostrowski, M. A., and Gray-Owen, S. D. (2008). CEACAM1 dynamics during Neisseria gonorrhoeae suppression of CD4+ T lymphocyte activation. J. Immunol. 180, 6827–6835.
Lewis, D. A. (2010). The gonococcus fights back: is this time a knock-out? Sex. Transm. Infect. 86, 415–421.
Lewis, L. A., Ngampasutadol, J., Wallace, P., Reid, J. E., Vogel, U., and Ram, S. (2010). the meningococcal vaccine candidate neisserial surface proteinA (NspA). binds to factor H and enhances meningococcal resistance to complement. PLoS Pathog. 6, e1001027. doi: 10.1371/journal.ppat.1001027
Lewis, L. A., Ram, S., Prasad, A., Gulati, S., Getzlaff, S., Blom, A. M., Vogel, U., and Rice, P. A. (2008). Defining targets for complement components C4b and C3b on the pathogenic neisseriae. Infect. Immun. 76, 339–350.
Li, S., Guo, X., Gong, X., Xu, M., Yan, J., Huang, Y., and Ren, Z. (2010). Bovine prolactin promotes the expression of human transferrin in the milk of transgenic mice. Biotechnol. Lett. 32, 787–793.
Lo, J. Y., Ho, K. M., Leung, A. O., Tiu, F. S., Tsang, G. K., Lo, A. C., and Tapsall, J. W. (2008). Cefibuten resistance and treatment failure of Neisseria gonorrhoeae infection. Antimicrob. Agents Chemother. 52, 3564–3567.
Madico, G., Ngampasutadol, J., Gulati, S., Vogel, U., Rice, P. A., and Ram, S. (2007). Factor H binding and function in sialylated pathogenic neisseriae is influenced by gonococcal, but not meningococcal, porin. J. Immunol. 178, 4489–4497.
Maiden, M. C., Ibarz-Pavón, A. B., Urwin, R., Gray, S. J., Andrews, N. J., Clarke, S. C., Walker, A. M., Evans, M. R., Kroll, J. S., Neal, K. R., Ala’aldeen, D. A., Crook, D. W., Cann, K., Harrison, S., Cunningham, R., Baxter, D., Kaczmarski, E., Maclennan, J., Cameron, J. C., and Stuart, J. M. (2008). Impact of meningococcal serogroup C conjugate vaccines on carriage and herd immunity. J. Infect. Dis. 197, 737–743.
Massari, P., Ram, S., Macleod, H., and Wetzler, L. M. (2003). The role of porins in neisserial pathogenesis and immunity. Trends Microbiol. 11, 87–93.
Matsuka, Y. V., Dilts, D. A., Hoiseth, S., and Arumugham, R. (1998). Characterization of a subunit structure and stability of the recombinant porin from Neisseria gonorrhoeae. J. Protein Chem. 17, 719–728.
McChesney, D., Tramont, E. C., Boslego, J. W., Ciak, J., Sadoff, J. C., and Brinton, C. C. (1982). Genital antibody response to a parenteral gonococcal pilus vaccine. Infect. Immun. 36, 1006–1012.
Miettinen, SA., Hakkarainen, K., Gronroos, P., Heinonen, P., Teisala, K., Aine, R., Sillantaka, I., Saarenmaa, K., Lehtinen, M., Punnonen, R., and Paavonen, J. (1989). Class specific antibody response to gonococcal infection. J. Clin. Pathol. 42, 72–76.
Nagashi, T., Chen, Z., Chen, L., Iijima, H., Nakajima, A., and Blumberg, R. S. (2008). CEACAM1 and the regulation of mucosal inflammation. Mucosal Immunol. 1(Suppl. 1), S39–S42.
Newman, L. M., Moran, J. S., and Workowski, K. A. (2007). Update on the management of gonorrhea in adults in the United States. Clin. Infect. Dis. 44S, S84–S101.
Ngampasutadol, J., Ram, S., Blom, A. M., Jarva, H., Jerse, A. E., Lien, E., Goguen, J., Gulati, S., and Rice, P. A. (2005). Human C4b-binding protein selectively interacts with Neisseria gonorrhoeae and results in species-specific infection. Proc. Natl. Acad. Sci. 102, 17142–17147.
Ngampasutadol, J., Ram, S., Gulati, S., Agarwal, S, Li, C., Visintin, A., Monks, B., Madico, G., and Rice, P. A. (2008). Human factor H interacts selectively with Neisseria gonorrhoeae and results in species-specific complement evasion. J. Immunol. 180, 3426–3435.
Ngampasutadol, J., Rice, P. A., Walsh, M. T., and Gulati, S. (2006). Characterization of a peptide vaccine candidate mimicking an oligosaccharide epitope of Neisseria gonorrhoeae and resultant immune responses and function. Vaccine 24, 157–170.
Pantelic, M., Kim, Y. J., Bolland, S., Chen, I., Shively, J., and Chen, T. (2005). Neisseria gonorrhoeae kills carcinoembryonic antigen-related cellular adhesion molecule 1 (CD66a)-expressing human B cells and inhibits antibody production. Infect. Immun. 73, 4171–4179.
Plante, M., Jerse, A. E., Hamel, J., Couture, F., Rioux, C. R., Brodeur, B. R., and Martin, D. (2000). Intranasal immunization with gonococcal outer membrane preparations reduces the duration of vaginal colonization of mice by Neisseria gonorrhoae. J. Infect. Dis. 182, 848–855.
Plummer, F. A., Simonsen, N. J., Chubb, H., Slaney, L., Kimata, J., Bosire, M., Ndinya-Achola, J. O., and Ngugi, E. N. (1989). Epidemiologic evidence for the development of serovar-specific immunity after gonococcal infection. J. Clin. Invest. 83, 1472–1476.
Price, G. A., Masri, H. P., Hollander, A. M., Russell, M. W., and Cornelissen, C. N. (2007). Gonococcal transferrin binding protein chimeras induce bactericidal and growth inhibitory antibodies in mice. Vaccine 25, 7247–7260.
Price, G. A., Russell, M. W., and Cornelissen, C. N. (2005). Intranasal administration of recombinant Neisseria gonorrhoeae transferrin binding proteins A and B conjugated to the cholera toxin B subunit induces systemic and vaginal antibodies in mice. Infect. Immun. 73, 3945–2953.
Ram, S., Mcquillen, D. P., Gulati, S., Elkins, C., Pangburn, M. K., and Rice, P. A. (1998). Binding of complement factor H to loop 5 of porin protein 1A: a molecular mechanism of serum resistance of nonsialylated Neisseria gonorrhoeae. J. Exp. Med. 188, 671–680.
Ramsey, K. H., Schneider, H., Cross, A. S., Boslego, J. W., Hoover, D. L., Staley, T. L., Kuschner, R. A., and Deal, C. D. (1995). Inflammatory cytokines produced in response to experimental human gonorrhea. J. Infect. Dis. 172, 186–191.
Rechner, C., Kuhlewein, C., Muller, A., Schild, H., and Rudel, T. (2007). Host glycoprotein Gp96 and scavenger receptor SREC interact with PorB of disseminating Neisseria gonorrhoeae in an epithelial invasion pathway. Cell Host Microbe 2, 393–403.
Rice, P. A., McQuillen, D. P., Gulati, S., Jani, D. B., Wetzler, L. M., Blake, M. S., and Gotschlich, E. C. (1974). Serum resistance of Neisseria gonorhoeae. Does it thwart the inflammatory response and facilitate the transmission of infection? Ann. N. Y. Acad. Sci. 730, 7–14.
Rudel, T., Kepp, O., and Kozjak-Pavlovic, V. (2010). Interactions between bacterial pathogens and the mitochondrial cell death pathways. Nat. Rev. Microbiol. 8, 693–705.
Schmidt, K. A., Schneider, H., Lindstrom, J. A., Boslego, J. W., Warren, R. A., Van de Berg, L., Deal, C. D., Mcclain, J. B., and Griffiss, J. M. (2001). Experimental gonococcal urethritis and reinfection with homologous gonococci in male volunteers. Sex. Transm. Dis. 28, 555–564.
Schultz, T. R., Tapsall, J. W., and White, P. A. (2001). Correlation of in vitro susceptibilities to newer quinolones of naturally occurring quinolone-resistant Neisseria gonorrhoeae strains with changes in GyrA and ParC. Antimicrob. Agents Chemother. 45, 734–738.
Seder, R., Darrah, P. A., and Roederer, M. (2008). T-cell quality in memory and protection: implications for vaccine design. Nat. Rev. Immunol. 8, 247–258.
Serino, L., Nesta, B., Leuzzi, R., Fontana, M. R., Monaci, E., Mocca, B. T., Cartocci, E., Masignani, V., Jerse, A. E., Rappuoli, R., and Pizza, M. (2007). Identification of a new OmpA-like protein in Neisseria gonorrhoeae involved in the binding to human epithelial cells and in vivo colonization. Mol. Microbiol. 64, 1391–1403.
Simons, M. P., Nauseef, W. M., and Apicella, M. A. (2005). The interactions of Neisseria gonorrhoeae with adherent polymorphonuclear leukocytes. Infect. Immun. 73, 1971–1977.
Simons, M. P., Nauseef, W. M., Griffith, T. S., and Apicella, M. A. (2006). Neisseria gonorrhoeae delays the onset of apoptosis in polymorphonuclear leukocytes. Cell. Microbiol. 8, 1780–1790.
Snape, M. D., Dawson, T., Oster, P., Evans, A., John, T. M., Ohene-Kena, B., Findlow, J., Yu, L. M., Borrow, R., Ypma, E., Toneatto, D., and Pollard, A. J. (2010). Immunogenicity of two investigational serogroup B meningococcal vaccines in the first year of life: a randomized comparative trial. Pediatr. Infect. Dis. J. 11, e71–e79.
Song, W., Condron, S., Mocca, B. T., Veit, S. J., Hill, D., Abbas, A., and Jerse, A. E. (2008). Local and humoral immune responses against primary and repeat Neisseria gonorrhoeae genital tract infections of 17 beta-estradiol-treated mice. Vaccine 26, 5741–5751.
Sparling, P. F., Thomas, C. E., and Zhu, W. (2003). “A vaccine for gonorrhea,” in Bacterial Vaccines, eds R. W. Ellis and R. W. Brodeur (Georgetown, TX: Landes Bioscience), 128–154.
Tapchaisri, P., and Srinisinha, S. (1976). Serum and secretory antibody responses to Neisseria gonorrhoeae in patients with gonococcal infections. Brit. J. Vener. Dis. 52, 374–380.
Thomas, C. E., Zhu, W., Van Dam, C. N., Davis, N. L., Johnston, R. E., and Sparling, P. F. (2006). Vaccination of mice with gonococcal TbpB expressed in vivo from Venezuelan encephalitis viral replicon particles. Infect. Immun. 74, 1612–1620.
Thompson, J. M., Whitmore, A. C., Konopka, J. L., Collier, M. L., Richmond, E. M. B., Davis, N. L., Staats, H. F., and Johnston, R. E. (2006). Mucosal and systemic adjuvant activity of alphavirus replicon particles. Proc. Natl. Acad. Sci. 103, 3722–3727.
Thompson, J. M., Whitmore, A. C., Staats, H. F., and Johnston, R. E. (2008a). The contribution of type I interferon signaling to immunity induced by alphavirus replion vaccines. Vaccine 26, 4998–5003.
Thompson, J. M., Whitmore, A. C., Staats, H. F., and Johnston, R. E. (2008b). Alpha virus replicon particles acting as adjuvants promote CD8+ T cell responses to co-delivered antigen. Vaccine 26, 4267–4275.
Tonkin, D. R., Jocquera, P., Todd, T., Beard, C. W., Johnston, R. E., and Barro, M. (2010). Alphavirus replicon-based enhancement of mucosal and systemic immunity is linked to the innate immune response generated by primary immunization. Vaccine 28, s3238–s3246.
Ufret-Vincenty, R. L., Aredo, B., Liu, X., McMahan, A., Chen, P. W., Sun, H., Niederkorn, J. Y., and Kedzierski, W. (2010). Transgenic mice expressing variants of complement factor H develop AMD-like retinal findngs. Invest. Ophthalmol. Vis. Sci. 11, 5878–5887.
Virji, M. (2009). Pathogenic neisseria: surface modulation, pathogenesis and infection control. Nat. Rev. Microbiol. 7, 274–286.
Virji, M., Zak, K., and Heckels, J. E. (1986). Monoclonal antibodies to gonococcal membrane protein 1B; use in investigation of the potential protective effect of antibodies directed against conserved and type-specific epitopes. J. Gen. Microbiol. 132, 1621–1629.
Warner, D. M., Shafer, W. M., and Jerse, A. E. (2008). Clinically relevant mutations that cause derepression of the Neisseria gonorhoeae MtrC-MtrD-MtrE efflux pump system confer different levels of antimicrobial resistance and in vivo fitness. Mol. Microbiol. 70, 462–478.
Weel, J. F. L., and Putten, J. P. M. (1991). Fate of the major outer membrane protein P.1A in early and late events of gonococcal infection of epithelial cells. Res. Microbiol. 142, 985–993.
Wetzler, L. M. (2010). Innate immune functions of the neisserial porins and the relationship to vaccine adjuvant activity. Future Microbiol. 5, 749–758.
Wu, H., and Jerse, A. E. (2006). Sialyltransferase enhances Neisseria gonorrhoeae survival during experimental murine genital tract infection. Infect. Immun. 74, 4094–4103.
Yamasaki, R., Koshino, H., Kurono, S., Nishinaka, Y., McQuillen, D. P., Kume, A., Gulati, S., and Rice, P. A. (1999). Structural and immunochemical characterization of a Neisseria gonorrhoeae epitope defined by a monoclonal antibody 2C7; the antibody recognizes a conserved epitope on specific lipooligosaccharides in spite of presence of human carbohydrate epitopes. J. Biol. Chem. 51, 36550–36558.
Zhang, L., Kovalev, G. I., and Su, L. (2007). HIV-1 infection and pathogenesis in a novel humanized mouse model. Blood 109, 2978–2981.
Zhu, W., Thomas, C. E., Chen, C. -J., Van Dam, C. N., Johnston, R. E., Davis, N. L., and Sparling, P. F. (2005). Comparison of immune responses to gonococcal PorB delivered as outer membrane vesicles, recombinant protein, or Venezuelan equine encephalitis viral replicon particles. Infect. Immun. 73, 7558–7568.
Keywords: Neisseria gonorrhoeae, vaccines, mouse models, immune responses, viral replicon particles, outer membranes, recombinant proteins
Citation: Zhu W, Chen C-J, Thomas CE, Anderson JE, Jerse AE and Sparling PF (2011) Vaccines for gonorrhea: can we rise to the challenge?. Front. Microbio. 2:124. doi: 10.3389/fmicb.2011.00124
Received: 31 January 2011; Paper pending published: 07 March 2011;
Accepted: 19 May 2011; Published online: 03 June 2011.
Edited by:
Cynthia N. Cornelissen, Virginia Commonwealth University School of Medicine, USAReviewed by:
Magdalene So, University of Arizona, USAPeter A. Rice, University of Massachusetts Medical School/UMass Memorial Medical Center, USA
Copyright: © 2011 Zhu, Chen, Thomas, Anderson, Jerse and Sparling. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: P. Frederick Sparling, Department of Medicine, University of North Carolina, 5113 Bioinformatics Building, 130 Mason Farm Road, Chapel Hill, NC 27599-7537, USA. e-mail: zman@med.unc.edu