 Geoffrey L. Rogers1 Ashley T. Martino1 George V. Aslanidi1 Giridhara R. Jayandharan2 Arun Srivastava1,3,4,5,6* Roland W. Herzog1,3,4,5,6*
Geoffrey L. Rogers1 Ashley T. Martino1 George V. Aslanidi1 Giridhara R. Jayandharan2 Arun Srivastava1,3,4,5,6* Roland W. Herzog1,3,4,5,6*- 1 Division of Cellular and Molecular Therapy, Department of Pediatrics, University of Florida College of Medicine, Gainesville, FL, USA
- 2 Department of Haematology, Centre for Stem Cell Research, Christian Medical College, Vellore, Tamil Nadu, India
- 3 Department of Molecular Genetics and Microbiology, University of Florida College of Medicine, Gainesville, FL, USA
- 4 Powell Gene Therapy Center, University of Florida College of Medicine, Gainesville, FL, USA
- 5 Genetics Institute, University of Florida College of Medicine, Gainesville, FL, USA
- 6 Shands Cancer Center, University of Florida College of Medicine, Gainesville, FL, USA
Gene replacement therapy by in vivo delivery of adeno-associated virus (AAV) is attractive as a potential treatment for a variety of genetic disorders. However, while AAV has been used successfully in many models, other experiments in clinical trials and in animal models have been hampered by undesired responses from the immune system. Recent studies of AAV immunology have focused on the elimination of transgene-expressing cells by the adaptive immune system, yet the innate immune system also has a critical role, both in the initial response to the vector and in prompting a deleterious adaptive immune response. Responses to AAV vectors are primarily mediated by the TLR9–MyD88 pathway, which induces the production of pro-inflammatory cytokines by activating the NF-κB pathways and inducing type I IFN production; self-complementary AAV vectors enhance these inflammatory processes. Additionally, the alternative NF-κB pathway influences transgene expression in cells transduced by AAV. This review highlights these recent discoveries regarding innate immune responses to AAV and discusses strategies to ablate these potentially detrimental signaling pathways.
Introduction
Adeno-associated virus (AAV) has emerged as a promising vector for viral gene therapy over the past 20 years (Mueller and Flotte, 2008; Mays and Wilson, 2011; Mingozzi and High, 2011). AAV is a parvovirus, which is a family of small, non-enveloped viruses containing a single-stranded linear DNA genome of about 5 kb; the wild-type virus is replication-deficient, requiring a helper virus in order to reproduce (Srivastava et al., 1983). In humans, AAV has not been found to be pathogenic. This fact, along with the tendency for the genomes of recombinant AAV (rAAV) vectors to remain as episomal concatemers rather than integrating into the host genome (reducing the risk for insertional mutagenesis), makes AAV a relatively safe gene therapy vector for testing in the clinic (Nakai et al., 2001). Indeed, AAV has become a preferred choice by many investigators for in vivo viral gene transfer, and due to its wide tissue tropism, it has been used in over 20 clinical trials to treat a wide variety of monogenetic diseases, including but not limited to: hemophilia B, α1 antitrypsin deficiency, cystic fibrosis, Parkinson’s disease, and Leber’s congenital amaurosis (LCA; Zhao et al., 2006; Nathwani et al., 2011).
It is widely accepted that the trial for LCA represents the first example of successful AAV gene therapy in humans without immune consequences. LCA is a genetic disease characterized by severe vision deficits due to a mutation in RPE65. Patients who received one subretinal injection of rAAV encoding RPE65 tolerated the transgene well and showed improved visual capability both in psychophysical (e.g., visual acuity) and functional (e.g., ability to navigate an obstacle course) measures (Maguire et al., 2009). So far, the improvements have persisted for over 3 years (Mingozzi and High, 2011). However, other clinical trials using AAV have not been as successful due to interference from the immune system (Manno et al., 2006). The success of the LCA trials is likely due to unique features of the target tissue that make it ideally suited for gene transfer. The retina is easy to access without potential exposure of vector to other tissues, and the relatively small number of target cells allow a small volume and low vector titer to result in a high multiplicity of infection (Mccarty, 2008). Additionally, the eye is generally regarded as an immunoprivileged site, greatly reducing the likelihood of developing a deleterious immune response (Hauswirth et al., 2008).
Conversely, other target tissues do not possess the near complete immunological ignorance displayed by the retina. The potential effects of an immune response on gene transfer are illustrated by the 2006 trial using AAV serotype 2 (AAV2) to transfer human blood coagulation factor IX (hF.IX) to the livers of hemophilia B patients deficient in factor IX. In this trial, one patient showed elevated circulating F.IX at 2 weeks, followed by a decline of transgene with a concomitant rise in liver enzymes, indicative of the destruction of hepatocytes; this damage was most likely mediated by a CD8+ T cell response against the AAV capsid (Manno et al., 2006; Mingozzi et al., 2007). Subsequently, it was shown that input capsid-derived peptides are presented by MHC class I molecules on the surface of hepatocytes following transduction with AAV2 vectors (Pien et al., 2009). Therefore, transduced cells may become targets for AAV capsid-specific CTLs. Another subject with a higher neutralizing antibody (NAB) titer against the capsid did not show appreciable levels of circulating F.IX, likely because the antibodies prevented transduction of hepatocytes (Mingozzi and High, 2007). These results illustrate a major problem with using AAV for in vivo gene therapy – pre-existing immunity. Since AAV is a naturally occurring infection in the human population, it is not surprising that reports have indicated that CD8+ memory T cells as well as NAB to AAV are common (Mingozzi and High, 2007; Calcedo et al., 2009; Boutin et al., 2010).
Studies in animal models have also revealed concerns beyond pre-existing immune responses to AAV. Without a memory response against the capsid developed due to natural infection, it is easier to successfully transduce wild-type mice with hF.IX via hepatic gene transfer; the resulting induction of tolerance to the transgene is thought to be mediated by hF.IX-specific regulatory T cells (Tregs; Dobrzynski et al., 2006; Cao et al., 2007). However, even in animal models, sustained transgene expression is not guaranteed. Hemophilic mice with missense mutations in transgenically expressed hF.IX genes are more tolerant to hF.IX gene transfer than total deletion mutants. The target tissue for transgene expression can also affect the outcome of gene transfer. In the same hemophilic mouse strains, hF.IX was less tolerated when expressed in skeletal muscle than when expressed in hepatocytes (Cao et al., 2009). Furthermore, tolerance can be affected by the serotype of AAV that is used; increased transduction efficiency in the liver is more likely to lead to tolerance to the transgene. In this regard, AAV8 is more tolerogenic than AAV2 (Cooper et al., 2009). Transduction efficiency can be also be increased by mutating surface exposed tyrosine residues on the capsid, which is thought to reduce proteasomal degradation, increasing trafficking to the nucleus (Zhong et al., 2008; Markusic et al., 2010). Though a variety of mechanisms are involved in these studies, they, along with other studies in animals, are united by a common theme: in current murine models, functional CD8+ T cell infiltrates in AAV transduced tissues are primary directed against the transgene product rather than the capsid, while an antibody response is often observed to both potential immunogens (Siders et al., 2009).
With these concerns in mind, many investigators have focused more on the adaptive immune response to AAV2. Additionally, a previous study comparing adenoviral vectors and AAV2 found that the innate immune response to AAV was weak and transient relative to the potent and prolonged response to adenovirus, suggesting that innate immunity to AAV2 may be insignificant (Zaiss et al., 2002). It is commonly accepted that innate responses provide activation signals critical for subsequent adaptive immunity. Even though the adaptive immune system has the effector functions that impact viral gene transfer, signals provided by the innate immune system can recruit and activate antigen presenting cells, T cells, and B cells (Hensley and Amalfitano, 2007). In the absence of proper activation signals, lymphocytes may be unresponsive to the presence of antigen. In this article, we will review the mechanisms that the innate immune system uses to respond to viruses, and then specifically consider how responses to rAAV vectors are mediated and how they affect successful transgene expression.
Overview of Innate Immune Responses to Viruses
As with other pathogens, in order to respond to viruses, the innate immune system needs to identify the particle as foreign and potentially dangerous. This occurs by recognizing structural motifs unique to non-self organisms, commonly referred to as pathogen-associated molecular patterns (PAMPs), via pattern recognition receptors (PRRs). The innate immune system distinguishes the unique characteristics of viruses via PRRs that can recognize both viral nucleic acids and membrane glycoproteins (Akira et al., 2006).
The most studied family of PRRs are the toll-like receptors (TLRs), of which 13 have been described so far (Huang and Yang, 2009). These transmembrane proteins are characterized by an extracellular domain that binds to the receptor’s cognate PAMP and an intracellular Toll/IL-1R homology (TIR) domain. TLR2 (glycoproteins and lipoproteins), TLR3 (dsRNA), TLR4 (glycoproteins and bacterial LPS), TLR7 (ssRNA), TLR8 (ssRNA), and TLR9 (unmethylated CpG DNA) have all been implicated in initiating inflammatory responses to viruses (Kawai and Akira, 2011). The glycoprotein-recognizing receptors are generally found on the cell surface, while the nucleic acid specific TLRs are most commonly located in endosomal compartments. TLRs are mostly found in immune cells – including DCs, macrophages, B cells, and some T cells – but also in some non-immune cells such as fibroblasts and epithelial cells (Akira et al., 2006).
Upon receptor engagement, most TLRs recruit MyD88, which causes phosphorylation of IRAK4 and IRAK1. Subsequent interaction of the IRAKs with TRAF6 and NEMO leads to ubiquitination of the latter molecules. Once ubiquitinated, TRAF6 and NEMO recruit TAK1, which activates the MAP kinase and classical NF-κB pathways (via ubiquitin-mediated degradation of IκB, freeing the RelA-p50 heterodimer to enter the nucleus). This results in the production of pro-inflammatory cytokines such as TNF-α and IL-6. TLR3, on the other hand, uses TRIF as an adaptor molecule instead of MyD88, but this pathway also leads to TRAF6-mediated activation of TAK1 and the same downstream inflammatory pathways. TLR2 and TLR4 can also induce an inflammatory response by signaling through TRIF (Kawai and Akira, 2007).
TLR7 and TLR9 signaling can induce the production of type I interferons (IFNs) in plasmacytoid dendritic cells (pDCs), which are a subset of DCs specifically designed to sense and respond to viral infections by producing large amounts of IFN. This signaling pathway is also dependent on MyD88; however, in order to stimulate IFN production, TLR9 must interact with AP-3 and traffic to the LAMP2+ lysosome-related organelle (LRO), where it can engage TRAF3 and signal IRF7 (Gilliet et al., 2008; Sasai et al., 2010). It is important to note that this pathway need not be activated in the same cell as the classical NF-κB pathway. IFNs are a class of soluble cytokines uniquely suited to combat intracellular infections by stimulating the production of over 100 interferon response genes (ISGs). The combined actions of these genes induce an antiviral state that renders the cell resistant to viral infection. Although the individual effects of most of these genes are still unknown, the family includes proteins such as Mx, which sequesters viral ribonucleoproteins, PKR, which leads to phosphorylation of eIF2α, inhibiting translation, and OAS, which activates RNAse L to degrade cellular and viral RNA (Garcia-Sastre and Biron, 2006). Furthermore, type I IFNs play a role in augmenting the innate immune response and stimulating the adaptive immune response to a viral infection by activating NK cells, inducing short-term proliferation of memory CD8+ T cells, aiding expansion and functionality of naïve T cell populations, and acting on CD4+ T cells and B cells to induce antibody isotype switching (Garcia-Sastre and Biron, 2006; Zhu et al., 2007, 2008).
In addition to the transmembrane TLRs, soluble PRRs can be found in the cytoplasm. As with the TLRs, these receptors mostly respond to viral nucleic acids. RIG-I and MDA5 detect dsRNA formed during the life cycle of most RNA viruses, while DAI has been implicated in cytoplasmic responses to DNA (Akira et al., 2006). There is also evidence that NLR proteins in association with mitochondrial antiviral signaling protein (MAVS; also known as IPS-1) are capable of regulating type I IFN and NF-κB production in response to viral single-stranded or double-stranded RNA (Ting et al., 2010). Finally, the NLRP3 inflammasome, a cytoplasmic complex that consists of NLRP3, ASC, and procaspase-1, can cleave pro-IL-1β and pro-IL-18 into their active and secreted forms in response to viral DNA (Muruve et al., 2008).
In summary, the innate immune system has a wide variety of membrane-bound and cytoplasmic mechanisms to detect viral components and initiate inflammatory responses that are primarily mediated by NF-κB and type I IFNs. Only some of these pathways and mechanisms are involved in innate immunity to AAV, which elicits a limited response. Our current knowledge of the interactions between the innate immune system and AAV vectors is summarized in the following.
Innate Responses to AAV
As previously noted, the innate immune responses to single-stranded AAV vectors (ssAAV) are typically low, particularly when compared with adenoviral vectors (Zaiss et al., 2002). In some situations, these vectors appear to lack the inflammatory signals that allow transduced cells such as hepatocytes to be targeted by a CTL response (Somanathan et al., 2010). Nevertheless, other studies have indicated that innate immune signaling through TLR9 can have a crucial effect on the development of a CD8+ T cell response against a transgene product (Zhu et al., 2009). The authors showed that AAV can induce the production of type I IFNs in pDCs [but not cDCs, Kupffer cells (KCs), or macrophages] in vitro, and that this induction is dependent on the TLR9–MyD88 pathway. In order to be exposed to this endosomal PRR, AAV must enter the endosomal pathway. There are two potential mechanisms by which AAV particles may enter APCs and traffic to endosomes. Binding to a specific receptor and co-receptor cause the particle to be endocytosed into a clathrin-coated pit (Figure 1A). Receptor specificity varies among the AAV serotypes. Examples of primary receptors utilized by some serotypes include heparin sulfate proteoglycan (HSPG) or sialic acid. AAV2 can utilize several different co-receptors, including αVβ5 integrin and platelet-derived growth factor receptor (PDGFR; Ding et al., 2005; Muzyczka, 2010). Alternatively, the virus may enter APCs via a clathrin-independent mechanism, such as pinocytosis (Figure 1A; Harbison et al., 2008). Once in the endosome, degradation of the viral capsid can expose the genome to TLR9, which then signals through MyD88 to activate NF-κB and ISGs (Figures 1B–F; Zhu et al., 2009).
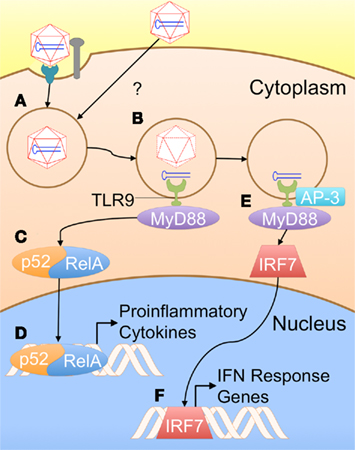
Figure 1. A proposed model for innate immune recognition of AAV vectors (based on TLR9 dependence of the response and general knowledge of TLR9 signaling). (A) AAV particles are taken up by APCs; the exact mechanism of uptake in these cell types is not presently known. Typically, AAV is taken up via receptor/co-receptor interactions that lead to internalization in a clathrin-coated pit. However, viral particles might also enter APCs through other pathways such as pinocytosis. (B) Some capsid breaks down in the endosome, exposing the genome to TLR9. (C) MyD88 initiates a signaling cascade, resulting in activation of NF-κB1 (p52–RelA heterodimer). (D) NF-κB enters the nucleus, initiating transcription of pro-inflammatory cytokines. (E) Alternatively, TLR9 may be rerouted to a LAMP2+ compartment by AP-3, allowing it to activate IRF7. (F) IRF7 translocates into the nucleus, inducing transcription of interferon response genes, including IFNα/β.
Zhu et al. (2009) also demonstrated that this pathway is critical for the development of an anti-transgene CTL response and NAB to both the transgene product and the AAV capsid. This study represented a significant breakthrough in attempts to elucidate the source of innate immune responses to AAV and their effects on the formation of an adaptive immune response that can lead to transgene rejection. A recent study by Martino et al. (2011) explored the innate immune responses to ssAAV and self-complementary (scAAV vectors) during hepatic gene transfer. Such scAAV vectors could potentially display altered responses due to their unique genome organization (dsDNA rather than single-stranded) and transduction kinetics (Mccarty, 2008).
TLR9 Facilitates Enhanced Responses to ScAAV Vectors during Hepatic Gene Transfer
Overall, the study by Martino et al. (2011) found that the innate responses to scAAV vectors were increased relative to ssAAV (Figure 2). Consistent with previous findings, the response to single-stranded vectors was rapid yet transient. Within 2 h after administration, a small increase in type 1 IFNs, TLR9, MyD88, and TNF-α RNA levels could be observed, which faded within 6 h. Conversely, the scAAV vectors induced much higher expression of these genes, plus increases in IL-6 (which now could be observed systemically by 2 h), CCL5, MIP-1, and TLR2. Cytokine responses varied with vector dose for scAAV but not ssAAV. Other pro-inflammatory genes, such IL-1α/1β, TLR1, and TLR3-8, were not upregulated following injection with either single-stranded or self-complementary AAV. Unlike adenoviral vectors, neither type of AAV vector could activate the inflammasome (Martino et al., 2011).
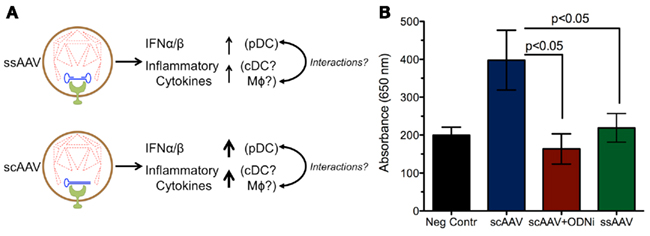
Figure 2. Increased innate responses are induced by scAAV relative to ssAAV. (A) Degradation of the capsid within the endosome can expose the vector genome to TLR9, which subsequently induces production of inflammatory cytokines and type I IFNs. The stronger response to self-complementary genomes may be due to reduced capsid stability of scAAV, leading to more genome exposure to TLR9. Alternatively, the structure of the double-stranded DNA may lead to increased TLR9 signaling and heightened inflammation relative to single-stranded DNA. (B) Experiments in a TLR9 reporter cell line demonstrate that sensing of scAAV results in stronger TLR9 signaling compared to ssAAV; this response can be blocked by TLR9 antagonists. Secreted embryonic alkaline phosphatase (SEAP) levels were measured by absorbance at 650 nm following TLR9 activation in a 293 reporter cell line (Invivogen, San Diego, CA, USA) after infection at an MOI of 104 with ssAAV, scAAV, or scAAV with a TLR9 inhibitory oligonucleotide (ODNi).
In accordance with the differential induction of chemokines by ssAAV and scAAV, the vectors provoked dissimilar levels of innate immune cell infiltrates 2 h after vector administration. The ssAAV vectors caused a slight increase in neutrophil and macrophage infiltration, whereas scAAV injection led to more substantial infiltration by neutrophils, macrophages, and NK cells.
The production of pro-inflammatory cytokines in response to scAAV is almost entirely dependent on TLR9. TLR9−/− mice injected with scAAV displayed no increases in RNA transcripts for any of the markers previously upregulated, except for a marginal increase in TLR2. The same effect can be achieved using an inhibitory oligodeoxynucleotide (ODN); these molecules binds strongly to TLR9 without inducing signaling, allowing them to serve as antagonists and block normal TLR9 signals (Stunz et al., 2002). Co-administration of inhibitory ODN with scAAV broadly blocked the cytokine response in a similar manner to the TLR9 knockout mice. It was also able to effectively blunt infiltration by macrophages, NK cells, and neutrophils, demonstrating that the recruitment of those cells depends on chemokines induced by TLR9 signaling. The rapid activation of pro-inflammatory cytokines suggests that the classical NF-κB pathway initiates their production, which is supported by other studies (Jayandharan et al., 2011).
At the cellular level, innate immune responses to scAAV in the liver were found to be partially dependent on KCs, concurring with previous data from ssAAV (Zaiss et al., 2002). Inactivation of KCs via gadolinium chloride (GdCl3) injection prior to scAAV administration significantly altered the profile of the immune response. Transcripts for IFNα/β, IP-10, and IL-6 were not increased by scAAV injection following GdCl3 treatment. More minor changes in TLR9, MyD88, MCP-1, and MIP-1 were observed, while transcripts for TNF-α and TLR2 were not significantly affected by KC inactivation. This treatment was also able to diminish recruitment of neutrophils and NK cells, but only slightly reduced macrophage accumulation. The underlying mechanism of these results is not yet clear. However, since changing the scAAV capsid to serotype 8, which more effectively transduces hepatocytes, did not increase the innate immune response, the hepatocytes themselves may not respond to the vector genomes; it is therefore more likely that the response is due to an uptake of AAV particles by APCs. Additionally, inactivation of KCs in the presence of scAAV vector prevented the type I IFN response (Martino et al., 2011).
Not surprisingly, the disparity found in innate immune responses between ssAAV and scAAV correlates with differences observed in adaptive responses. At equal doses, scAAV vectors induced higher CD8+ T cell and antibody responses against the vector capsid compared to ssAAV. Prevention of TLR9 signaling either via injection into TLR9−/− mice or co-administration of an inhibitory ODN caused a significant reduction in CTL responses and delayed the formation of anti-AAV antibodies. This suggests that the increased adaptive response to scAAV, like the innate response, depends on TLR9 signaling. However, following liver-directed delivery, the adaptive response to the F.IX transgene was unchanged between ssAAV and scAAV, most likely due to the temporal separation between the innate signals and transgene expression. The pro-inflammatory cytokine response induced by scAAV occurs primarily during the first 9 h after vector administration, while the transgene is only expressed after viral uncoating and transcriptional activation. Thus, the immune system would not encounter hF.IX in the presence of inflammatory signals that could induce an effector response (Martino et al., 2011). In contrast to hepatic gene transfer, there is some evidence that, in skeletal muscle, scAAV vectors can direct a stronger response to the transgene product (H. C. Ertl, personal communication). Still, despite the lack of anti-transgene response after scAAV liver gene transfer, inhibition of TLR9 signaling increases systemic F.IX expression. This would imply that factors other than an anti-transgene adaptive response, such as NK cells or type I IFN activity, could affect the overall levels of transgene expression in animal models for AAV, as is known to be the case for adenovirus (Zhu et al., 2008).
Interestingly, the heightened responses observed for scAAV vectors appear to be entirely dependent on their unique genome conformation. Single-stranded AAV8 and ssAAV–tyrosine mutant (TM; an altered AAV2 vector with three surface tyrosines changes to phenylalanine) displayed a comparable cytokine response to ssAAV2 vectors, while scAAV8–GFP was similar to scAAV2–hF.IX. Thus, the capsid serotype and transgene do not appear to affect the TLR9-mediated innate responses to scAAV during hepatic gene transfer (Martino et al., 2011).
The highly transient nature of the innate response further explains why hepatic gene transfer with AAV vectors has been successful for tolerance induction to transgene products, which is further facilitated by the anti-inflammatory microenvironment of the liver (Breous et al., 2009; Loduca et al., 2009; Martino et al., 2009; Somanathan et al., 2010; Hoffman et al., 2011).
Role of the AAV Capsid in Immune Responses
Although capsid alterations do not modify the TLR9 dependent innate response, variable immune responses to different AAV capsids has been demonstrated. Some reports have suggested that certain serotypes, specifically AAV1, are capable of transducing DCs (Lu and Song, 2009). After transduction and maturation, DCs may initiate an immune response to the foreign transgene product; thus, the report by Lu and Song found that transgene delivered to NOD mice in an AAV1 capsid was more immunogenic and less tolerated than when it was delivered with AAV8, which does not effectively transduce DCs.
Other AAV capsid types have also been shown to have variable immunogenicity. AAVrh32.33, an evolutionarily divergent serotype isolated from rhesus macaques, yields reduced transgene expression and stronger anti-vector and anti-transgene CD8+ T cell responses than AAV8 when injected into skeletal muscle (Mays et al., 2009). This enhanced response was shown to depend on CD4+ helper T cells, plus CD40L and CD28, both of which are co-stimulatory molecules involved in immune activation. Interestingly, anti-CD40 antibody, which is usually able to restore immune responsiveness in CD40L−/− mice, was ineffective in this case.
Interactions between the viral capsid and complement have also been shown to play a role in the innate immune response to AAV (Zaiss et al., 2008). Immunoprecipitation studies showed that iC3b can bind to the AAV capsid, and that, in vitro, complement binding to AAV2 can increase capsid uptake and pro-inflammatory cytokine production by macrophages. This effect was also observed in vivo. Mice deficient in complement receptor 1/2 or C3 were less able to mount a humoral immune response to AAV than wild-type mice. However, AAV was not found to activate the alternative complement pathway. The capsid appears to bind factor H, which can then inhibit factor I-mediated degradation of C3b to iC3b, providing the virus some protection against complement.
The observation that scAAV vectors can slightly activate TLR2 transcription raises the question of whether this PRR may play a minor role in responses to AAV (Martino et al., 2011). While TLR2 is more commonly associated with sensing surface glycoproteins of enveloped viruses, leading to the production of type I IFNs in inflammatory monocytes, it has also been demonstrated to be involved in sensing adenoviral particles, which, like AAV, are non-enveloped (Appledorn et al., 2008; Barbalat et al., 2009; Quigley et al., 2009). Further, it has been reported that empty AAV capsids can induce innate immune responses (Hoesel et al., 2010). Though that mechanism is unclear, it is possible that the capsid-based inflammatory response could involve TLR2; further study will be required to elucidate this molecule’s role in innate immune responses to AAV.
Effects of Target Tissue on Innate Immune Responsiveness
As has been mentioned previously, the target tissue transduced by AAV can significantly impact the resulting immune response. Preclinical and clinical studies of gene therapy for hemophilia have focused on two target tissues: the liver and skeletal muscle (Matrai et al., 2010). Generally, the tolerogenic nature of the liver renders hepatic gene transfer more conducive to long-term transgene expression than gene transfer to skeletal muscle, as demonstrated by an 8-year study in hemophilic dogs and a detailed analysis in hemophilia B mice (Cao et al., 2009; Niemeyer et al., 2009). The divergent outcomes between muscle and liver gene transfer do not appear to be related to differences in innate responses due to TLR3 signaling (Cao and Herzog, 2008). Despite this, skeletal muscle gene transfer remains a useful tool for investigators. The more pronounced immune responses generated in muscle make it easier to elucidate the mechanisms of immune responsiveness to AAV. Muscular injection of AAV–CMV–OVA vectors demonstrated that a strong, transgene specific immune response could cause a loss of transgene expression (Wang et al., 2005). Other studies in muscle have demonstrated that the nature of the transgene can affect the development of an immune response. Membrane-bound β-gal delivered via AAV was found to be more immunogenic than cytoplasmic transgene, presumably because it was more accessible for uptake by DCs and presentation to T cells on MHC class I and II (Sarukhan et al., 2001). Hopefully, expanding studies of scAAV to muscle can further our understanding of the immune responses to these vectors and help determine organ-specific effects on innate immunity.
The Alternative NF-κB Pathway and AAV
So far, this article has focused on traditional innate responses to AAV initiated through the classical NF-κB pathway. These responses use NF-κB1 (p105), which is constitutively cleaved into p50 and complexes with RelA; this heterodimer can be disinhibited through ubiquitin-mediated degradation of IκB by IKKβ (Kawai and Akira, 2007). In contrast, the alternative pathway uses NF-κB2 (p100). NIK activates IKKα, which induces proteolytic cleavage of p100 into p52, allowing p52–RelB dimers to translocate to the nucleus and initiate transcription (Senftleben et al., 2001; Lawrence and Bebien, 2007). While the classical pathway is strongly involved in the production of pro-inflammatory cytokines during the innate immune response, the alternative pathway is considered important for adaptive immunity (Lawrence and Bebien, 2007).
However, a recent study has also described a role for the alternative pathway in AAV transduction (Jayandharan et al., 2011). The authors report that, in addition to classical NF-κB activation by 2 h (Figure 3A), infection with AAV is capable of inducing alternative pathway activation after 9 h, as demonstrated by increased levels of nuclear p52 both in vitro and in vivo. Expression of an EGFP transgene was also increased by 20–25 fold following alternative pathway activation. VP16, an activator of both pathways, increased fluorescence in HeLa cells, while Bay11, which can inhibit both the classical and alternative pathways, ablated transgene expression. PDTC (an inhibitor of the classical pathway), on the other hand, did not affect transgene expression. This result concurs with previous studies demonstrating that the AAV genome can be bound and transcriptionally inhibited by cellular proteins, one of which was later shown to be an NF-κB-repressing factor (Qing et al., 1997; Jayandharan et al., 2011).
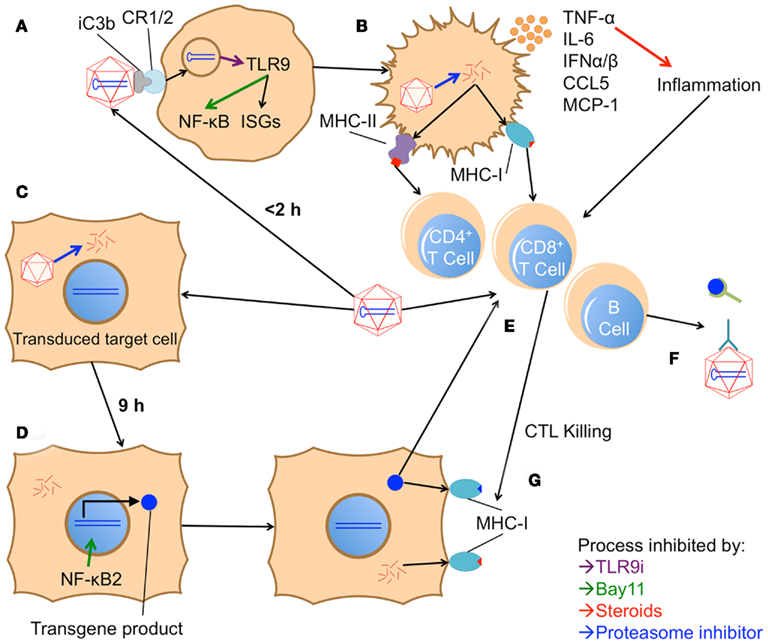
Figure 3. Model of proposed in vivo innate and adaptive immune responses to AAV vectors. (A) Within 2 h after vector injection, AAV is phagocytosed by an APC; it is unclear whether initial contact is through a cDC, pDC, KC, MΦ, or other cell type. Phagocytosis may be aided by opsonization through iC3b and complement receptor 1/2 (CR1/2). Once in the endosome, the virus’s genome is detected by TLR9, which subsequently activates the classical NF-κB pathway and interferon response genes (ISGs). TLR9i can inhibit both these responses, while Bay11 can block the NF-κB pathway. (B) Initiation of these pathways causes the APC to mature, up-regulating costimulatory molecules and producing pro-inflammatory cytokines like TNF-α, IL-6, CCL5, MCP-1, and type I IFNs. Steroids can inhibit the inflammatory activity of these cytokines. Mature APCs also present capsid fragments on MHC class I and II, unless protease inhibitors are utilized to hinder degradation of the capsid. (C) Concurrently, AAV productively transduces target cells. (D) Within 9 h, the alternative NF-κB pathway is triggered in these cells, enhancing expression of the transgene product. Bay11 can also blockade this NF-κB pathway. (E) Over the course of several days, the inflammatory signals from internalized AAV particles and the local environment induce maturation of APCs. Activation of the alternative NF-κB pathway in these APCs may enhance transgene expression and cross-presentation, augmenting their ability to prime an adaptive response (involving B cells, CD4+ T cells, and CD8+ T cells) to both the AAV capsid and the transgene product. (F) B cells subsequently produce antibodies against the capsid, inhibiting further transduction, and against the transgene product, inhibiting its efficacy. (G) Capsid or transgene specific CD8+ T cells recognize peptide fragments of either protein in class I MHC on transduced cells. Recognition prompts the CTLs to attack and kill the target cell. Death of a significant number of transduced cells can lead to a reduction or complete elimination of transgene expression. Capsid-specific CTL killing can be reduced by inhibiting capsid degradation in target cells with proteasome inhibitors.
Adeno-associated virus was also able to induce alternative pathway signaling in vivo (Jayandharan et al., 2011). Liver homogenates from 9 h after injection showed increased nuclear p52, suggesting NF-κB2 signaling; p52 elevation could be ablated if Bay11 was administered before the vector (Figure 3D). That alternative pathway signaling could be detected from whole liver suggests the transduced hepatocytes, rather than tissue-resident APCs, are responsible for this expression. Indeed, DCs exposed in vitro to AAV did not active the classical or alternative NF-κB pathways and were not effectively transduced to produce EGFP. Addition of VP16 or pro-inflammatory cytokines, which did activate both pathways, caused the DCs to become permissive to AAV transduction. Addition of cytokines and Bay11 resulted in reduced NF-κB activation and transduction relative to non-inhibited cells.
Thus, the alternative pathway seems to support AAV transduction in multiple cell types. While increased transduction efficiency is desired in hepatocytes, expression in APCs is more likely to lead to an enhanced immune response against the transgene. Activation of the alternative pathway can also enhance the ability of DCs to cross-present exogenous antigen to CD8+ T cells, so even in the absence of APC transduction, NF-κB2 activation could induce a stronger immune response (Lind et al., 2008). The authors therefore investigated the effects of transient NF-κB suppression via Bay11 on long-term transgene expression (Jayandharan et al., 2011). Administration of Bay11 prior to gene transfer abrogated the rapid production of pro-inflammatory cytokines mediated by the classical pathway and reduced the anti-capsid antibody response. Additionally, 2 weeks post injection, both Bay11 treated and untreated mice showed comparable EGFP expression, suggesting that temporary inhibition of the alternative pathway does not interfere with sustained transgene expression. The mechanism by which AAV infection activates the alternative pathway is unclear and requires further study.
However, preliminary studies to test the mechanism of NF-κB-mediated increases in transgene expression from AAV vectors suggest that activation of the classical NF-κB p65 component in the cytosol may require the vector capsid, while the activation and function of alternative NF-κB p52 is subsequently amplified by its binding to AAV2 inverted terminal repeats (ITRs; Jayandharan et al., 2011; Jayandharan, unpublished results). Transfection in HeLa cells with plasmids containing increasing numbers of functional ITRs + D-sequence in the presence of the NF-κB activator VP-16 showed an increase in EGFP expression in an ITR + D-sequence-dependent fashion, whereas Bay11 ablated this effect. This suggested that putative NF-κB-responsive transcription factor binding sites exist in the AAV–ITRs. Further in silico analysis with a human transcription factor database demonstrated the presence of several binding sites for NF-κB binding co-factors, such as p300, TFIIB, and SplI (Jayandharan et al., 2011). One of these is the p300/CREB transcription factor that has been recently shown to be associated with the AAV genome (Dean et al., 2009).
Therapeutic Potential of Blocking Innate Responses to AAV
After examining what is known about the innate immune responses to AAV, it is prudent to discuss how these findings could be translated into improved therapies in the clinic. Given the brief nature of these responses, it seems that transient immunosuppression will down-regulate immune responses that are potentially deleterious to successful gene therapy. The recent reports reviewed here have, through preclinical studies, highlighted the potential for this sort of protocol to succeed (Figure 3). Inhibition of TLR9 signaling through an ODN antagonist and suppression of both NF-κB pathways were each able to reduce, but not completely eliminate, immune responses to the vector (Jayandharan et al., 2011; Martino et al., 2011). Bay11 alone was probably only partially successful because, as an NF-κB inhibitor, it does not block production of type I IFNs, which are critical mediators of responses to viruses (Figures 3A,D). Obstructing TLR9 activation (Figure 3A), on the other hand, seemed mostly successful, but an anti-capsid antibody response began to develop at 4 weeks post injection. Although either alone was not completely effective, perhaps a combination of these two therapies would more successfully abolish the immune response to AAV.
While primary adaptive immune responses rely heavily on activation by inflammatory signals such as those derived from TLR signaling, a recent report has indicated that secondary expansion of memory CD8+ T cells occurs independently of MyD88 signaling (Rahman et al., 2011). Hence, therapeutics directed against specific innate pathways such as TLR–MyD88 may not be capable of preventing the loss of transduced cells due to capsid-specific memory CD8+ T cells.
Other groups are also exploring different transient treatments to co-administer with vectors to increase the success of the therapy. Treatment with glucocorticoids prior to systemic delivery of adenoviral vectors could ablate immunotoxicity without negatively impacting transduction efficiency (Figure 3B; Seregin et al., 2009). This strategy may also be applicable to AAV vectors. Adenoviral research has also indicated that complement inhibition may reduce the immune response to viral vectors. Fusion of decay-activating factor (DAF), a complement inhibitor, to the capsid of adenovirus 5 reduced the antibody and CD8+ T cell response to Ad-encoded HIV-Gag; antibody responses to the adenovirus capsid were also diminished by this strategy (Seregin et al., 2011). Thus, complement inhibition may be another strategy to increase the efficacy of AAV-mediated gene transfer.
Instead of artificially blocking the immune response with drugs, the other way to reduce inflammation would be to reduce vector load. To that end, as mentioned previously, tyrosine mutant AAV vectors have been developed that enhance transduction efficiency by resisting proteasomal degradation; this allows fewer vector particles to achieve the same expression levels as significantly higher titers of wild-type AAV (Li et al., 2010; Markusic et al., 2010). A similar effect could also potentially be achieved using proteasome inhibitors at the time of vector administration to enhance transduction efficiency (Figures 3B,C).
These two treatment modalities do not have to be mutually exclusive. It is possible that a combination of methods that enhance vector efficiency and suppress the immune response could lead to safer and more effective AAV gene therapy in humans.
Conclusion
It is clear that, despite its low immunogenicity, AAV is detected and can cause innate immune responses. These responses can interfere with transgene expression, negatively impacting the outcome of gene therapy. Among the variety of mechanisms to respond to viruses, sensing of vector genomes by TLR9 appears to be the key mediator of these responses. Through MyD88, TLR9 induces the production of pro-inflammatory cytokines and chemokines (via NF-κB) and type I IFNs (Figure 1). These recruit additional pro-inflammatory cells, interfere with transduction, and lead to the development of an adaptive immune response (Figures 3E–G). Preventing signaling through TLR9 completely blocks the innate response and reduces adaptive responses, suggesting that it is the most important innate response element to AAV. However, there is also evidence that the capsid can be involved in these responses, by dictating which cell types are transduced, by binding complement, and perhaps by some signaling through TLR2. Additionally, the alternative NF-κB pathway has a role in immune responses and transgene expression.
Though we have a much greater understanding of AAV immunology due to recent findings, a number of questions remain unanswered. For instance, it is unclear why the response to AAV disappears so rapidly compared to other viral vectors. Is the initial inflammatory signal too weak to continue to self-propagate, is the duration of active signaling following pathogen recognition reduced compared to other vectors, or could the vector trigger a pathway that down-regulates the response? Also, the mechanisms behind the enhanced responses observed to scAAV vectors need further investigation. Although there is evidence that self-complementary TLR9 agonists can signal more strongly than single-stranded agonists, other factors maybe involved (Struthers et al., 2010). The capsid of self-complementary vectors may be less stable, causing more DNA to be released in the endosome, or perhaps the self-complementary genome is not completely encapsulated (Figure 2A). The vector cassette does not affect the response, and it is therefore unlikely that specific sequences in the DNA are responsible.
Furthermore, the cell–cell interactions during in vivo responses to AAV are still uncertain. In vitro data indicate that pDCs are responsible for type I IFN production, yet in vivo results from hepatic gene transfer demonstrate that KCs are required (Zhu et al., 2009; Martino et al., 2011). These cell types, and perhaps others, likely cooperate within in the host to mount an immune response. Blocking innate responses noticeably reduces the adaptive response to capsid, yet it is uncertain whether this effect extends to the transgene product. Nonetheless, in TLR9−/− mice, AAV2–influenza hemagglutinin (HA) gene transfer to skeletal muscle exhibits diminished responses against HA (Zhu et al., 2009). On the other hand, in the liver, increasing signaling through TLR9 with scAAV vectors did not increase the adaptive response to hF.IX (Martino et al., 2011). Whether adaptive immune responses to the transgene product can be effectively prevented by blocking innate immunity can be addressed in future studies.
Clearly, a number of additional studies will be required in order to better understand the innate immunology of AAV. Hopefully, in answering these questions, we can define interventions that will allow us to improve AAV gene therapy and make it a practical treatment for a variety of genetic diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Appledorn, D. M., Patial, S., Mcbride, A., Godbehere, S., Van Rooijen, N., Parameswaran, N., and Amalfitano, A. (2008). Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J. Immunol. 181, 2134–2144.
Barbalat, R., Lau, L., Locksley, R. M., and Barton, G. M. (2009). Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat. Immunol. 10, 1200–1207.
Boutin, S., Monteilhet, V., Veron, P., Leborgne, C., Benveniste, O., Montus, M. F., and Masurier, C. (2010). Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 21, 704–712.
Breous, E., Somanathan, S., Vandenberghe, L. H., and Wilson, J. M. (2009). Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology 50, 612–621.
Calcedo, R., Vandenberghe, L. H., Gao, G., Lin, J., and Wilson, J. M. (2009). Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 199, 381–390.
Cao, O., Dobrzynski, E., Wang, L., Nayak, S., Mingle, B., Terhorst, C., and Herzog, R. W. (2007). Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood 110, 1132–1140.
Cao, O., and Herzog, R. W. (2008). TLR3 signaling does not affect organ-specific immune responses to factor IX in AAV gene therapy. Blood 112, 910–911.
Cao, O., Hoffman, B. E., Moghimi, B., Nayak, S., Cooper, M., Zhou, S., Ertl, H. C., High, K. A., and Herzog, R. W. (2009). Impact of the underlying mutation and the route of vector administration on immune responses to factor IX in gene therapy for hemophilia B. Mol. Ther. 17, 1733–1742.
Cooper, M., Nayak, S., Hoffman, B. E., Terhorst, C., Cao, O., and Herzog, R. W. (2009). Improved induction of immune tolerance to factor IX by hepatic AAV-8 gene transfer. Hum. Gene Ther. 20, 767–776.
Dean, J., Plante, J., Huggins, G. S., Snyder, R. O., and Aikawa, R. (2009). Role of cyclic AMP-dependent kinase response element-binding protein in recombinant adeno-associated virus-mediated transduction of heart muscle cells. Hum. Gene Ther. 20, 1005–1012.
Ding, W., Zhang, L., Yan, Z., and Engelhardt, J. F. (2005). Intracellular trafficking of adeno-associated viral vectors. Gene Ther. 12, 873–880.
Dobrzynski, E., Fitzgerald, J. C., Cao, O., Mingozzi, F., Wang, L., and Herzog, R. W. (2006). Prevention of cytotoxic T lymphocyte responses to factor IX-expressing hepatocytes by gene transfer-induced regulatory T cells. Proc. Natl. Acad. Sci. U.S.A. 103, 4592–4597.
Garcia-Sastre, A., and Biron, C. A. (2006). Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312, 879–882.
Gilliet, M., Cao, W., and Liu, Y. J. (2008). Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 8, 594–606.
Harbison, C. E., Chiorini, J. A., and Parrish, C. R. (2008). The parvovirus capsid odyssey: from the cell surface to the nucleus. Trends Microbiol. 16, 208–214.
Hauswirth, W. W., Aleman, T. S., Kaushal, S., Cideciyan, A. V., Schwartz, S. B., Wang, L., Conlon, T. J., Boye, S. L., Flotte, T. R., Byrne, B. J., and Jacobson, S. G. (2008). Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum. Gene Ther. 19, 979–990.
Hensley, S. E., and Amalfitano, A. (2007). Toll-like receptors impact on safety and efficacy of gene transfer vectors. Mol. Ther. 15, 1417–1422.
Hoesel, M., Broxtermann, M., Janicki, H., Esser, K., Arzberger, S., Von Freyend, M.J., Stabenow, D., Knolle, P., Hallek, M., Protzer, U., and Buening, H. (2010). Innate immune response towards adeno-associated viral (AAV) vectors in human liver non-parenchymal cells. Mol. Ther. 18, S120.
Hoffman, B. E., Martino, A. T., Sack, B. K., Cao, O., Liao, G., Terhorst, C., and Herzog, R. W. (2011). Nonredundant roles of IL-10 and TGF-beta in suppression of immune responses to hepatic AAV-Factor IX gene transfer. Mol. Ther. 19, 1263–1272.
Huang, X., and Yang, Y. (2009). Innate immune recognition of viruses and viral vectors. Hum. Gene Ther. 20, 293–301.
Jayandharan, G. R., Aslanidi, G., Martino, A. T., Jahn, S. C., Perrin, G. Q., Herzog, R. W., and Srivastava, A. (2011). Activation of the NF-kappaB pathway by adeno-associated virus (AAV) vectors and its implications in immune response and gene therapy. Proc. Natl. Acad. Sci. U.S.A. 108, 3743–3748.
Kawai, T., and Akira, S. (2007). Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 13, 460–469.
Kawai, T., and Akira, S. (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650.
Lawrence, T., and Bebien, M. (2007). IKKalpha in the regulation of inflammation and adaptive immunity. Biochem. Soc. Trans. 35, 270–272.
Li, M., Jayandharan, G. R., Li, B., Ling, C., Ma, W., Srivastava, A., and Zhong, L. (2010). High-efficiency transduction of fibroblasts and mesenchymal stem cells by tyrosine-mutant AAV2 vectors for their potential use in cellular therapy. Hum. Gene Ther. 21, 1527–1543.
Lind, E. F., Ahonen, C. L., Wasiuk, A., Kosaka, Y., Becher, B., Bennett, K. A., and Noelle, R. J. (2008). Dendritic cells require the NF-kappaB2 pathway for cross-presentation of soluble antigens. J. Immunol. 181, 354–363.
Loduca, P. A., Hoffman, B. E., and Herzog, R. W. (2009). Hepatic gene transfer as a means of tolerance induction to transgene products. Curr. Gene Ther. 9, 104–114.
Lu, Y., and Song, S. (2009). Distinct immune responses to transgene products from rAAV1 and rAAV8 vectors. Proc. Natl. Acad. Sci. U.S.A. 106, 17158–17162.
Maguire, A. M., High, K. A., Auricchio, A., Wright, J. F., Pierce, E. A., Testa, F., Mingozzi, F., Bennicelli, J. L., Ying, G. S., Rossi, S., Fulton, A., Marshall, K. A., Banfi, S., Chung, D. C., Morgan, J. I., Hauck, B., Zelenaia, O., Zhu, X., Raffini, L., Coppieters, F., De Baere, E., Shindler, K. S., Volpe, N. J., Surace, E. M., Acerra, C., Lyubarsky, A., Redmond, T. M., Stone, E., Sun, J., Mcdonnell, J. W., Leroy, B. P., Simonelli, F., and Bennett, J. (2009). Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 374, 1597–1605.
Manno, C. S., Pierce, G. F., Arruda, V. R., Glader, B., Ragni, M., Rasko, J. J., Ozelo, M. C., Hoots, K., Blatt, P., Konkle, B., Dake, M., Kaye, R., Razavi, M., Zajko, A., Zehnder, J., Rustagi, P. K., Nakai, H., Chew, A., Leonard, D., Wright, J. F., Lessard, R. R., Sommer, J. M., Tigges, M., Sabatino, D., Luk, A., Jiang, H., Mingozzi, F., Couto, L., Ertl, H. C., High, K. A., and Kay, M. A. (2006). Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 12, 342–347.
Markusic, D. M., Herzog, R. W., Aslanidi, G. V., Hoffman, B. E., Li, B., Li, M., Jayandharan, G. R., Ling, C., Zolotukhin, I., Ma, W., Zolotukhin, S., Srivastava, A., and Zhong, L. (2010). High-efficiency transduction and correction of murine hemophilia B using AAV2 vectors devoid of multiple surface-exposed tyrosines. Mol. Ther. 18, 2048–2056.
Martino, A. T., Nayak, S., Hoffman, B. E., Cooper, M., Liao, G., Markusic, D. M., Byrne, B. J., Terhorst, C., and Herzog, R. W. (2009). Tolerance induction to cytoplasmic beta-galactosidase by hepatic AAV gene transfer: implications for antigen presentation and immunotoxicity. PLoS ONE 4, e6376. doi: 10.1371/journal.pone.0006376
Martino, A. T., Suzuki, M., Markusic, D. M., Zolotukhin, I., Ryals, R. C., Moghimi, B., Ertl, H. C., Muruve, D. A., Lee, B., and Herzog, R. W. (2011). The genome of self-complementary AAV vectors increases TLR9-dependent innate immune responses in the liver. Blood 117, 6459–6468.
Matrai, J., Chuah, M. K., and Vandendriessche, T. (2010). Preclinical and clinical progress in hemophilia gene therapy. Curr. Opin. Hematol. 17, 387–392.
Mays, L. E., Vandenberghe, L. H., Xiao, R., Bell, P., Nam, H. J., Agbandje-Mckenna, M., and Wilson, J. M. (2009). Adeno-associated virus capsid structure drives CD4-dependent CD8+ T cell response to vector encoded proteins. J. Immunol. 182, 6051–6060.
Mays, L. E., and Wilson, J. M. (2011). The complex and evolving story of T cell activation to AAV vector-encoded transgene products. Mol. Ther. 19, 16–27.
Mccarty, D. M. (2008). Self-complementary AAV vectors; advances and applications. Mol. Ther. 16, 1648–1656.
Mingozzi, F., and High, K. A. (2007). Immune responses to AAV in clinical trials. Curr. Gene Ther. 7, 316–324.
Mingozzi, F., and High, K. A. (2011). Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 12, 341–355.
Mingozzi, F., Maus, M. V., Hui, D. J., Sabatino, D. E., Murphy, S. L., Rasko, J. E., Ragni, M. V., Manno, C. S., Sommer, J., Jiang, H., Pierce, G. F., Ertl, H. C., and High, K. A. (2007). CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 13, 419–422.
Mueller, C., and Flotte, T. R. (2008). Clinical gene therapy using recombinant adeno-associated virus vectors. Gene Ther. 15, 858–863.
Muruve, D. A., Petrilli, V., Zaiss, A. K., White, L. R., Clark, S. A., Ross, P. J., Parks, R. J., and Tschopp, J. (2008). The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107.
Muzyczka, N. (2010). “Adeno-associated viral (AAV) vectors,” in A Guide to Human Gene Therapy, eds R. W. Herzog and S. Zolotukhin (Hackensack, NJ: World Scientific Publishing), 87–102.
Nakai, H., Yant, S. R., Storm, T. A., Fuess, S., Meuse, L., and Kay, M. A. (2001). Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 75, 6969–6976.
Nathwani, A. C., Rosales, C., Mcintosh, J., Rastegarlari, G., Nathwani, D., Raj, D., Nawathe, S., Waddington, S. N., Bronson, R., Jackson, S., Donahue, R. E., High, K. A., Mingozzi, F., Ng, C. Y., Zhou, J., Spence, Y., Mccarville, M. B., Valentine, M., Allay, J., Coleman, J., Sleep, S., Gray, J. T., Nienhuis, A. W., and Davidoff, A. M. (2011). Long-term safety and efficacy following systemic administration of a self-complementary aav vector encoding human fix pseudotyped with serotype 5 and 8 capsid proteins. Mol. Ther. 19, 876–885.
Niemeyer, G. P., Herzog, R. W., Mount, J., Arruda, V. R., Tillson, D. M., Hathcock, J., Van Ginkel, F. W., High, K. A., and Lothrop, C. D. Jr. (2009). Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood 113, 797–806.
Pien, G. C., Basner-Tschakarjan, E., Hui, D. J., Mentlik, A. N., Finn, J. D., Hasbrouck, N. C., Zhou, S., Murphy, S. L., Maus, M. V., Mingozzi, F., Orange, J. S., and High, K. A. (2009). Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J. Clin. Invest. 119, 1688–1695.
Qing, K., Wang, X. S., Kube, D. M., Ponnazhagan, S., Bajpai, A., and Srivastava, A. (1997). Role of tyrosine phosphorylation of a cellular protein in adeno-associated virus 2-mediated transgene expression. Proc. Natl. Acad. Sci. U.S.A. 94, 10879–10884.
Quigley, M., Martinez, J., Huang, X., and Yang, Y. (2009). A critical role for direct TLR2-MyD88 signaling in CD8 T-cell clonal expansion and memory formation following vaccinia viral infection. Blood 113, 2256–2264.
Rahman, A. H., Zhang, R., Blosser, C. D., Hou, B., Defranco, A. L., Maltzman, J. S., Wherry, E. J., and Turka, L. A. (2011). Antiviral memory CD8 T-cell differentiation, maintenance, and secondary expansion occur independently of MyD88. Blood 117, 3123–3130.
Sarukhan, A., Soudais, C., Danos, O., and Jooss, K. (2001). Factors influencing cross-presentation of non-self antigens expressed from recombinant adeno-associated virus vectors. J. Gene Med. 3, 260–270.
Sasai, M., Linehan, M. M., and Iwasaki, A. (2010). Bifurcation of toll-like receptor 9 signaling by adaptor protein 3. Science 329, 1530–1534.
Senftleben, U., Cao, Y., Xiao, G., Greten, F. R., Krahn, G., Bonizzi, G., Chen, Y., Hu, Y., Fong, A., Sun, S. C., and Karin, M. (2001). Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 293, 1495–1499.
Seregin, S. S., Aldhamen, Y. A., Appledorn, D. M., Zehnder, J., Voss, T., Godbehere, S., and Amalfitano, A. (2011). Use of DAF-displaying adenovirus vectors reduces induction of transgene- and vector-specific adaptive immune responses in mice. Hum. Gene Ther. doi: 10.1089/hum.2010.218. [Epub ahead of print].
Seregin, S. S., Appledorn, D. M., Mcbride, A. J., Schuldt, N. J., Aldhamen, Y. A., Voss, T., Wei, J., Bujold, M., Nance, W., Godbehere, S., and Amalfitano, A. (2009). Transient pretreatment with glucocorticoid ablates innate toxicity of systemically delivered adenoviral vectors without reducing efficacy. Mol. Ther. 17, 685–696.
Siders, W. M., Shields, J., Kaplan, J., Lukason, M., Woodworth, L., Wadsworth, S., and Scaria, A. (2009). Cytotoxic T lymphocyte responses to transgene product, not adeno-associated viral capsid protein, limit transgene expression in mice. Hum. Gene Ther. 20, 11–20.
Somanathan, S., Breous, E., Bell, P., and Wilson, J. M. (2010). AAV vectors avoid inflammatory signals necessary to render transduced hepatocyte targets for destructive T cells. Mol. Ther. 18, 977–982.
Srivastava, A., Lusby, E. W., and Berns, K. I. (1983). Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol. 45, 555–564.
Struthers, M., Bett, A. J., Wisniewski, T., Dubey, S. A., Precopio, M., Jiang, W., Sun, Z., Wang, H., Nowak, I., Putta, M. R., Yu, D., Tang, J. X., Kandimalla, E. R., Agrawal, S., and Casimiro, D. R. (2010). Synthesis and immunological activities of novel agonists of toll-like receptor 9. Cell Immunol. 263, 105–113.
Stunz, L. L., Lenert, P., Peckham, D., Yi, A. K., Haxhinasto, S., Chang, M., Krieg, A. M., and Ashman, R. F. (2002). Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur. J. Immunol. 32, 1212–1222.
Ting, J. P., Duncan, J. A., and Lei, Y. (2010). How the noninflammasome NLRs function in the innate immune system. Science 327, 286–290.
Wang, L., Dobrzynski, E., Schlachterman, A., Cao, O., and Herzog, R. W. (2005). Systemic protein delivery by muscle-gene transfer is limited by a local immune response. Blood 105, 4226–4234.
Zaiss, A. K., Cotter, M. J., White, L. R., Clark, S. A., Wong, N. C., Holers, V. M., Bartlett, J. S., and Muruve, D. A. (2008). Complement is an essential component of the immune response to adeno-associated virus vectors. J. Virol. 82, 2727–2740.
Zaiss, A. K., Liu, Q., Bowen, G. P., Wong, N. C., Bartlett, J. S., and Muruve, D. A. (2002). Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 76, 4580–4590.
Zhao, W., Zhong, L., Wu, J., Chen, L., Qing, K., Weigel-Kelley, K. A., Larsen, S. H., Shou, W., Warrington, K. H. Jr., and Srivastava, A. (2006). Role of cellular FKBP52 protein in intracellular trafficking of recombinant adeno-associated virus 2 vectors. Virology 353, 283–293.
Zhong, L., Li, B., Mah, C. S., Govindasamy, L., Agbandje-Mckenna, M., Cooper, M., Herzog, R. W., Zolotukhin, I., Warrington, K. H. Jr., Weigel-Van Aken, K. A., Hobbs, J. A., Zolotukhin, S., Muzyczka, N., and Srivastava, A. (2008). Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. U.S.A. 105, 7827–7832.
Zhu, J., Huang, X., and Yang, Y. (2007). Type I IFN signaling on both B and CD4 T cells is required for protective antibody response to adenovirus. J. Immunol. 178, 3505–3510.
Zhu, J., Huang, X., and Yang, Y. (2008). A critical role for type I IFN-dependent NK cell activation in innate immune elimination of adenoviral vectors in vivo. Mol. Ther. 16, 1300–1307.
Keywords: AAV vectors, gene therapy, innate immunity
Citation: Rogers GL, Martino AT, Aslanidi GV, Jayandharan GR, Srivastava A and Herzog RW (2011) Innate immune responses to AAV vectors. Front. Microbio. 2:194. doi: 10.3389/fmicb.2011.00194
Received: 08 June 2011;
Accepted: 31 August 2011;
Published online: 19 September 2011.
Edited by:
Katherine High, Children’s Hospital of Philadelphia, USAReviewed by:
Dirk Dittmer, University of North Carolina at Chapel Hill, USAMelanie M. Brinkmann, Helmholtz Centre for Infection Research, Germany
Copyright: © 2011 Rogers, Martino, Aslanidi, Jayandharan, Srivastava and Herzog. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Arun Srivastava, University of Florida, 2033 Mowry Road, Room 492-A, Gainesville, FL 32610, USA. e-mail: aruns@peds.ufl.edu; Roland W. Herzog, Cancer and Genetics Research Complex, University of Florida, 2033 Mowry Road, Room 203, Gainesville, FL 32610, USA. e-mail: rherzog@ufl.edu