Erratum: Acquired antibiotic resistance genes: an overview
 Angela H. A. M. van Hoek1 Dik Mevius2,3 Beatriz Guerra4 Peter Mullany5 Adam Paul Roberts5 Henk J. M. Aarts1*
Angela H. A. M. van Hoek1 Dik Mevius2,3 Beatriz Guerra4 Peter Mullany5 Adam Paul Roberts5 Henk J. M. Aarts1*- 1 Laboratory for Zoonoses and Environmental Microbiology, Centre for Infectious Disease Control, National Institute of Public Health and the Environment, Utrecht, Netherlands
- 2 Central Veterinary Institute of Wageningen UR, Lelystad, Netherlands
- 3 Department of Infectious Diseases and Immunology, Utrecht University, Utrecht, Netherlands
- 4 National Salmonella Reference Laboratory, Federal Institute for Risk Assessment, Berlin, Germany
- 5 Department of Microbial Diseases, University College London Eastman Dental Institute, University College London, London, UK
In this review an overview is given on antibiotic resistance (AR) mechanisms with special attentions to the AR genes described so far preceded by a short introduction on the discovery and mode of action of the different classes of antibiotics. As this review is only dealing with acquired resistance, attention is also paid to mobile genetic elements such as plasmids, transposons, and integrons, which are associated with AR genes, and involved in the dispersal of antimicrobial determinants between different bacteria.
Introduction
The discovery and production of (synthetic) antibiotics in the first half of the previous century has been one of medicine’s greatest achievements. The use of antimicrobial agents has reduced morbidity and mortality of humans and contributed substantially to human’s increased life span. Antibiotics are, either as therapeutic or as prophylactic agents, also widely used in agricultural practices.
The first discovered antimicrobial compound was penicillin (Flemming, 1929) a β-lactam antibiotic. Soon after this very important discovery, antibiotics were used to treat human infections starting with sulfonamide and followed by the aminoglycoside streptomycin and streptothricin (Domagk, 1935; Schatz and Waksman, 1944). Nowadays numerous different classes of antimicrobial agents are known and they are classified based on their mechanisms of action (Neu, 1992). Antibiotics can for instance inhibit protein synthesis, like aminoglycoside, chloramphenicol, macrolide, streptothricin, and tetracycline or interact with the synthesis of DNA and RNA, such as quinolone and rifampin. Other groups inhibit the synthesis of, or damage the bacterial cell wall as β-lactam and glycopeptide do or modify, like sulfonamide and trimethoprim, the energy metabolism of a microbial cell.
Upon the introduction of antibiotics it was assumed that the evolution of antibiotic resistance (AR) was unlikely. This was based on the assumption that the frequency of mutations generating resistant bacteria was negligible (Davies, 1994). Unfortunately, time has proven the opposite. Nobody initially anticipated that microbes would react to this assault of various chemical poisons by adapting themselves to the changed environment by developing resistance to antibiotics using such a wide variety of mechanisms. Moreover, their ability of interchanging genes, which is now well known as horizontal gene transfer (HGT) was especially unexpected. Later on it was discovered that the emergence of resistance actually began before the first antibiotic, penicillin, was characterized. The first β-lactamase was identified in Escherichia coli prior to the release of penicillin for use in medical practice (Abraham and Chain, 1940). Besides β-lactams, the aminoglycoside–aminocyclitol family was also one of the first groups of antibiotics to encounter the challenges of resistance (Wright, 1999; Bradford, 2001). Over the years it has been shown by numerous ecological studies that (increased) antibiotic consumption contributes to the emergence of AR in various bacterial genera (MARAN, 2005, 2007; NethMap, 2008). Some examples of the link between antibiotic dosage and resistance development are the rise of methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE). The initial appearance of MRSA was in 1960 (Jevons et al., 1963), whereas VRE were first isolated about 20 years ago (Uttley et al., 1988). Over the last decades they have remained a reason for concern, but additional public health threats in relation to resistant microorganisms have also arisen (see for example Cantón et al., 2008; Goossens, 2009; Allen et al., 2010).
Bacteria have become resistant to antimicrobials through a number of mechanisms (Spratt, 1994; McDermott et al., 2003; Magnet and Blanchard, 2005; Wright, 2005):
I. Permeability changes in the bacterial cell wall which restricts antimicrobial access to target sites,
II. Active efflux of the antibiotic from the microbial cell,
III. Enzymatic modification of the antibiotic,
IV. Degradation of the antimicrobial agent,
V. Acquisition of alternative metabolic pathways to those inhibited by the drug,
VI. Modification of antibiotic targets,
VII. Overproduction of the target enzyme.
These AR phenotypes can be achieved in microorganisms by chromosomal DNA mutations, which alter existing bacterial proteins, through transformation which can create mosaic proteins and/or as a result of transfer and acquisition of new genetic material between bacteria of the same or different species or genera (Spratt, 1994; Maiden, 1998; Ochman et al., 2000).
There are numerous examples of mutation based resistance. For example, macrolide resistance can be due to nucleotide(s) base substitutions in the 23S rRNA gene. However, a similar resistance phenotype may also result from mutations within the ribosomal proteins L4 and L22 (Vester and Douthwaite, 2001). Single nucleotide polymorphisms (SNPs) can be the cause for resistance against the synthetic drugs quinolones, sulfonamides, and trimethoprim (Huovinen et al., 1995; Hooper, 2000; Ruiz, 2003) and mutations within the rpsL gene, which encodes the ribosomal protein S12, can result in a high-level streptomycin resistance (Nair et al., 1993). A frame shift mutation in the chromosomal ddl gene, encoding a cytoplasm enzyme D-Ala–D-Ala ligase, can account for glycopeptides resistance (Casadewall and Courvalin, 1999).
Acquired Resistance
This review deals with the description of acquired resistance against several classes of antibiotics. For each class the development of resistance is summarized along with the mechanisms of action. Furthermore an extensive summary is given of the resistance mechanisms and resistance genes involved.
Aminoglycoside
History and action mechanism
The aminoglycoside antibiotics initially known as aminoglycosidic aminocyclitols are over 60 years old (Siegenthaler et al., 1986; Begg and Barclay, 1995). In the early 1940s the first aminoglycoside discovered was streptomycin in Streptomyces griseus (Schatz and Waksman, 1944). Several years later, additional aminoglycosides were characterized from other Streptomyces species; neomycin and kanamycin in 1949 and 1957, respectively. Furthermore, in the 1960s gentamicin was recovered from the actinomycete Micromonospora purpurea. Because most aminoglycosides have been isolated from either Streptomyces or Micromonospora a nomenclature system has been set up based on their source. Aminoglycosides that are derived from bacteria of the Streptomyces genus are named with the suffix “-mycin,” while those which are derived from Micromonospora are named with the suffix “-micin.”
The first semi-synthetic derivatives were isolated in the 1970s. For example netilmicin is a derivative of sisomicin whereas amikacin is derived from kanamycin (Begg and Barclay, 1995; Davies and Wright, 1997).
Aminoglycosides are antimicrobials since they inhibit protein synthesis and/or alter the integrity of bacterial cell membranes (Vakulenko and Mobashery, 2003). They have a broad antimicrobial spectrum. Furthermore, they often act in synergy with other antibiotics as such it makes them valuable as anti-infectants.
Resistance mechanisms
Several aminoglycoside resistance mechanisms have been recognized; (I) Active efflux (Moore et al., 1999; Magnet et al., 2001), (II) Decreased permeability (Hancock, 1981; Taber et al., 1987), (III) Ribosome alteration (Poehlsgaard and Douthwaite, 2005), (IV) Inactivation of the drugs by aminoglycoside-modifying enzymes (Shaw et al., 1993). Intrinsic mechanisms, i.e., efflux pumps and 16S rRNA methylases but also chromosomal mutations can cause the first three resistance properties. In recent years acquired 16S rRNA methylases appear to have increased in importance (Galimand et al., 2005; Doi and Arakawa, 2007; Table 1). The first gene identified of a plasmid-mediated type of aminoglycoside resistance was armA (Galimand et al., 2003). To date five additional methylases have been reported, i.e., npmA, rmtA, rmtB, rmtC, and rmtD (Courvalin, 2008; Doi et al., 2008). Data regarding the 16S rRNA methylase genes are accumulated and provided at the website: www.nih.go.jp/niid/16s_database/index.html.
TABLE 1
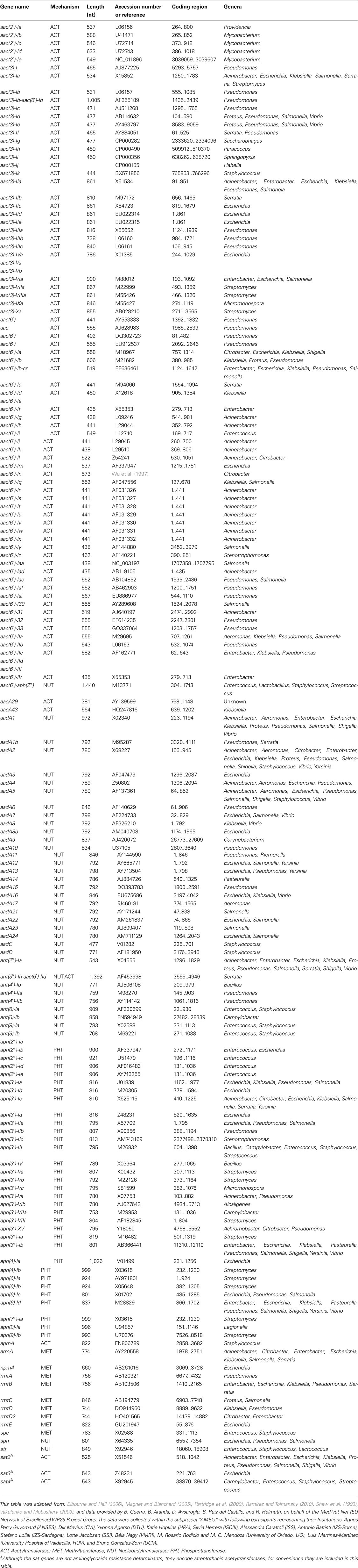
Table 1. Acquired Aminoglycoside resistance genes.
The major encountered aminoglycoside resistance mechanism is the modification of enzymes. These proteins are classified into three major classes according to the type of modification: AAC (acetyltransferases), ANT (nucleotidyltransferases or adenyltransferases), APH (phosphotransferases; Shaw et al., 1993; Wright and Thompson, 1999; Magnet and Blanchard, 2005; Wright, 2005; Ramirez and Tolmansky, 2010). Within these classes, an additional subdivision can be made based on the enzymes different region specificities for aminoglycoside modifications: i.e., there are four acetyltransferases: AAC(1), AAC(2′), AAC(3), and AAC(6′); five nucleotidyltransferases: ANT(2″), ANT(3″), ANT(4′), ANT(6), and ANT(9) and seven phosphotransferases: APH(2″), APH(3′), APH(3″), APH(4), APH(6), APH(7″), and APH(9). Furthermore, there also exists a bifunctional enzyme, AAC(6′)–APH(2″), that can acetylate and phosphorylate its substrates sequentially (Shaw et al., 1993; Kotra et al., 2000). Table 1 displays the currently known aminoglycoside resistance genes. The action mechanisms of the determinants, the variety in gene lengths, accession numbers, and the distribution are all indicated. As can be deduced from the second column of Table 1, inconsistencies arose in the nomenclature of genes for aminoglycoside-modifying enzymes (Vakulenko and Mobashery, 2003). In some cases, genes were named according to the site of modification, followed by a number to distinguish between genes. Using a different nomenclature, for example, the genes for AAC(6′)-Ia and AAC(3)-Ia are referred to as aacA1 and aacC1, respectively. The nomenclature proposed by Shaw et al. (1993), who utilize the identical names for the enzymes and the corresponding genes, but the names of genes are in lowercase letters and italicized will be used in this review (see Table 1). According to this more convenient nomenclature, the genes for the AAC(6′)-Ia and AAC(3)-Ia enzymes are termed aac(6′)-Ia and aac(3)-Ia, respectively.
β-Lactam
History and action mechanism
As already mentioned before, the first antibiotic discovered was a β-lactam, i.e., penicillin. The Scottish scientist Alexander Flemming accidentally noticed the production of a substance with antimicrobial properties by the mold Penicillium notatum (Flemming, 1929). Over the last 30 years, many new β-lactam antibiotics have been developed. By definition, all β-lactam antibiotics have a β-lactam nucleus in their molecular structure. The β-lactam antibiotic family includes penicillins and derivatives, cephalosporins, carbapenems, monobactams, and β-lactam inhibitors (Williams, 1987; Bush, 1989; Petri, 2006; Queenan and Bush, 2007).
The core compound of penicillin, 6-aminopenicillanic acid (6-APA) is used as the main starting point for the preparation of numerous semi-synthetic derivatives. Although the cephalosporins are often thought of as new and improved derivatives of penicillin, they were actually discovered as naturally occurring substances (Petri, 2006). They can be grouped in first, second, third, and forth generation cephalosporins according to their spectrum of activity and timing of the agent’s introduction. In general, first generation agents have good Gram-positive activity and relatively modest coverage for Gram-negative organisms; second generation cephalosporins have increased Gram-negative and somewhat less Gram-positive activity; third generation antimicrobials have improved Gram-negative and variable Gram-positive activity; forth generation β-lactams have good true broad spectrum activity against both Gram-negatives and Gram-positives (Williams, 1987; Marshall et al., 2006). The second generation cephamycins are sometimes also grouped among the cephalosporins.
Because carbapenems diffuse easily in bacteria they are considered as broad spectrum β-lactam antibiotic. Imipenem and meropenem are well known representative. Even though monobactams do not contain a nucleus with a fused ring attached, they still belong to the β-lactam antibiotics. The β-lactamase inhibitors, like clavulanic acid, do contain the β-lactam ring, but they exhibit negligible antimicrobial activity and are used in combination with β-lactam antibiotics to overcome resistance in bacteria that secrete β-lactamase, which otherwise inactivates most penicillins.
The β-lactam antibiotics work by inhibiting the cell wall synthesis by binding to so-called penicillin-binding proteins (PBPs) in bacteria and interfering with the structural cross linking of peptidoglycans and as such preventing terminal transpeptidation in the bacterial cell wall. As a consequence it weakens the cell wall of the bacterium and finally results in cytolysis or death due to osmotic pressure (Kotra and Mobashery, 1998; Andes and Craig, 2005).
The β-lactamase inhibitors can be classified as either reversible or irreversible and the latter are considered more effective in that they eventually result in the destruction of enzymatic activity. Not surprisingly the inhibitors in clinical use, i.e., clavulanic acid, sulbactam, and tazobactam are all examples of irreversible β-lactamase inhibitors (Bush, 1988; Drawz and Bonomo, 2010).
Resistance mechanisms
The first bacterial enzyme reported to destroy penicillin was an AmpC β-lactamase of E. coli (Abraham and Chain, 1940). Nowadays, bacterial resistance against β-lactam antibiotics is increasing at a significant rate and has become a common problem. There are several mechanisms of antimicrobial resistance to β-lactam antibiotics. The most common and important mechanism through which bacteria can become resistant against β-lactams is by expressing β-lactamases, for example extended-spectrum β-lactamases (ESBLs), plasmid-mediated AmpC enzymes, and carbapenem-hydrolyzing β-lactamases (carbapenemases; Bradford, 2001; Jacoby and Munoz-Price, 2005; Paterson and Bonomo, 2005; Poirel et al., 2007; Queenan and Bush, 2007; Jacoby, 2009).
The β-lactamase family has been subdivided either based on functionality or molecular characteristics. Initially, before genes were routinely sequenced various biochemical parameters were determined of the different β-lactamases which allowed classification of this AR determinants family into four groups (Bush et al., 1995; Wright, 2005). Groups 1, 2, and 4 are serine-β-lactamases, while members of group 3 are metallo-β-lactamases. Classification based on molecular characteristics, i.e., amino acid homology has also resulted in four major groups, the so-called Ambler classes A–D, which correlate well with the functional scheme but lack details concerning the enzymatic activity. Ambler classes A, C, and D include the β-lactamases with serine at their active site, whereas Ambler class B β-lactamases are all metallo-enzymes who require zinc as a metal cofactor for their catalytic activities (Ambler, 1980; Bradford, 2001; Paterson and Bonomo, 2005; Wright, 2005; Poirel et al., 2007, 2010; Bush and Jacoby, 2010; Drawz and Bonomo, 2010). In this review the Ambler classification will be used (Table 2).
TABLE 2
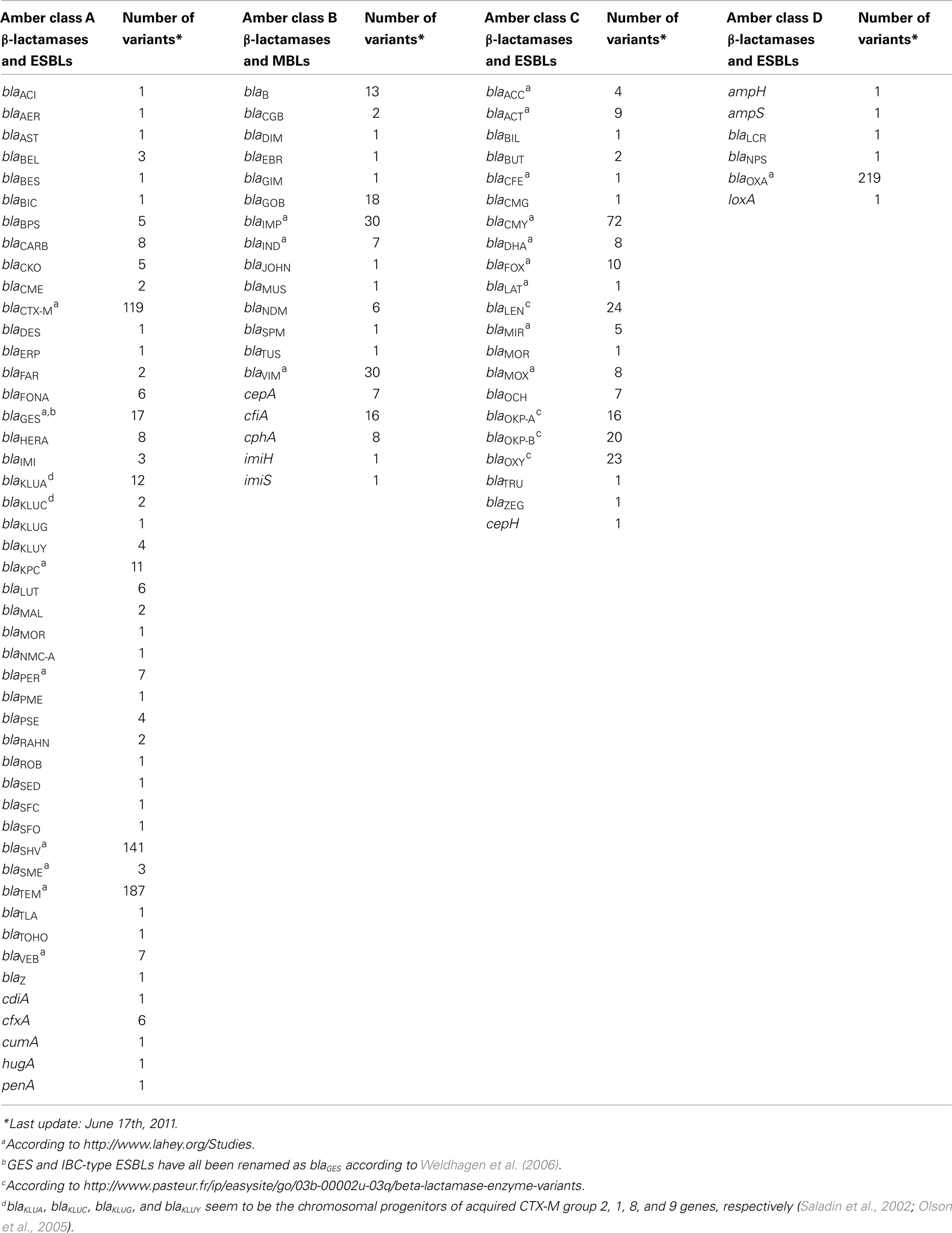
Table 2. β-Lactamases and ESBLs families.
In addition to the production of β-lactamases resistance can also be due to possession of altered PBPs. Since β-lactams cannot bind as effectively to these altered PBPs, the antibiotic is less effective at disrupting cell wall synthesis. The PBPs are thought to be the ancestors of the naturally occurring chromosomally mediated β-lactamase in many bacterial genera (Bradford, 2001).
Although plasmid-encoded penicillinase arose much earlier in Gram-positives in Staphylococcus aureus, due to the use of penicillin (Aarestrup and Jensen, 1998), the first plasmid-mediated β-lactamase, TEM-1, was described in the early 1960s in Gram-negatives (Datta and Kontomichalou, 1965). Currently over 1,150 chromosomal, plasmid, and transposon located β-lactamases are currently known (Bush and Jacoby, 2010; Drawz and Bonomo, 2010; Table 2).
Based on their activity to hydrolyze a small number or a variety of β-lactams the enzymes can be subdivided into narrow-, moderate-, broad-, and ESBLs. A commonly used definition specifies that broad spectrum β-lactamases are capable to provide resistance to the penicillins and cephalosporins and are not inhibited by inhibitors such as clavulanic acid and tazobactam. The ESBLs confer resistance to the penicillins, first-, second-, and third-generation cephalosporins and aztreonam, but not to carbapenems and are inhibited by β-lactamase inhibitors. In recent years acquired AR genes encoding ESBLs have become a major concern (Bradford, 2001). In time the parent enzymes blaTEM-1, blaTEM-2, and blaSHV-1 have undergone amino acid substitutions (point mutations) evolving to the ESBLs, starting with blaTEM-3 and blaSHV-2 (Bradford, 2001). Additional mutations at critical amino acids important for catalysis resulted in over 140 currently known SHV and TEM ESBL variants. In addition, plasmid-encoded class C β-lactamases or AmpC determinants, like blaCMY have also caught people’s awareness (Jacoby, 2009). Furthermore, in the past decade CTX-M enzymes have become very prevalent ESBLs, both in nosocomial and in community settings (Cantón and Coque, 2006).
Table 2 illustrates the size and diversity of the group of β-lactamases and ESBLs. The vast and still increasing number of (broad spectrum) β-lactamases and ESBLs has become a problem for the nomenclature for novel genes. Names have been assigned according to individual preference rather than according to systematic procedures (Bush, 1989). Fortunately, an authoritative website has been constructed on the nomenclature of ESBLs hosted by Jacoby and Bush1.
Chloramphenicol
History and action mechanism
In 1947, the first chloramphenicol, originally referred to as chloromycetin, was isolated from Streptomyces venezuelae (Ehrlich et al., 1947). Probably because chloramphenicol is a molecule with a rather simple structure only a small number of synthetic derivates have been synthesized without adverse effects on antimicrobial activity (Schwarz et al., 2004). In azidamfenicol two chlorine atoms (−Cl2) are replaced by an azide group. Substitution of the nitro group (−NO2), by a methyl–sulfonyl residue (−SO2CH3) resulted in the synthesis of thiamphenicol, whereas in the fluorinated thiamphenicol derivative florfenicol the hydroxyl group (−OH) is replaced with fluorine (−F).
Chloramphenicol is a highly specific and potent inhibitor of protein synthesis through its affinity for the peptidyltransferase of the 50S ribosomal subunit of 70S ribosomes (Schwarz et al., 2004). Due to its binding to this enzyme the antibiotic prevents peptide chain elongation. The substrate spectrum of chloramphenicol includes Gram-positive and Gram-negative, aerobic and anaerobic bacteria. Chloramphenicol analogs including the fluorinated derivative florfenicol have a similar spectrum of activity.
Resistance mechanism
The first and still most frequently encountered mechanism of bacterial resistance to chloramphenicol is enzymatic inactivation by acetylation of the drug via different types of chloramphenicol acetyltransferases (CATs; Murray and Shaw, 1997; Schwarz et al., 2004; Wright, 2005). CATs are able to inactivate chloramphenicol as well as thiamphenicol and azidamfenicol, however, due to its structural modification florfenicol is resistant to inactivation by these enzymes. Consequently, chloramphenicol resistant strains, in which resistance is exclusively based on the activity of CAT, are susceptible to florfenicol. There are two defined types of genes coding CATs which distinctly differ in their structure: i.e., the classical catA determinants and the novel, also known as xenobiotic CATs, encoded by catB variants (Table 3). Besides the inactivating enzymes, there are also reports on other chloramphenicol resistance systems, such as inactivation by phosphotransferases, mutations of the target site, permeability barriers, and efflux systems (Schwarz et al., 2004). Of the latter mechanism, cmlA and floR are the most commonly known (Bissonnette et al., 1991; Briggs and Fratamico, 1999). The presence of a cmlA gene will result in resistance to chloramphenicol, but susceptibility to florfenicol. In contrast, floR will give rise to a chloramphenicol and florfenicol resistance phenotype. Inconsistencies in the nomenclature arose, like with many other AR genes, due to the increasing number of chloramphenicol resistance determinants. Schwarz et al. (2004) suggested a unified nomenclature. Table 3 represents the currently known chloramphenicol/florfenicol resistance genes. Some characteristics which are mentioned in Table 3 are mechanism of action, diverse gene lengths, accession numbers, and the distribution.
TABLE 3
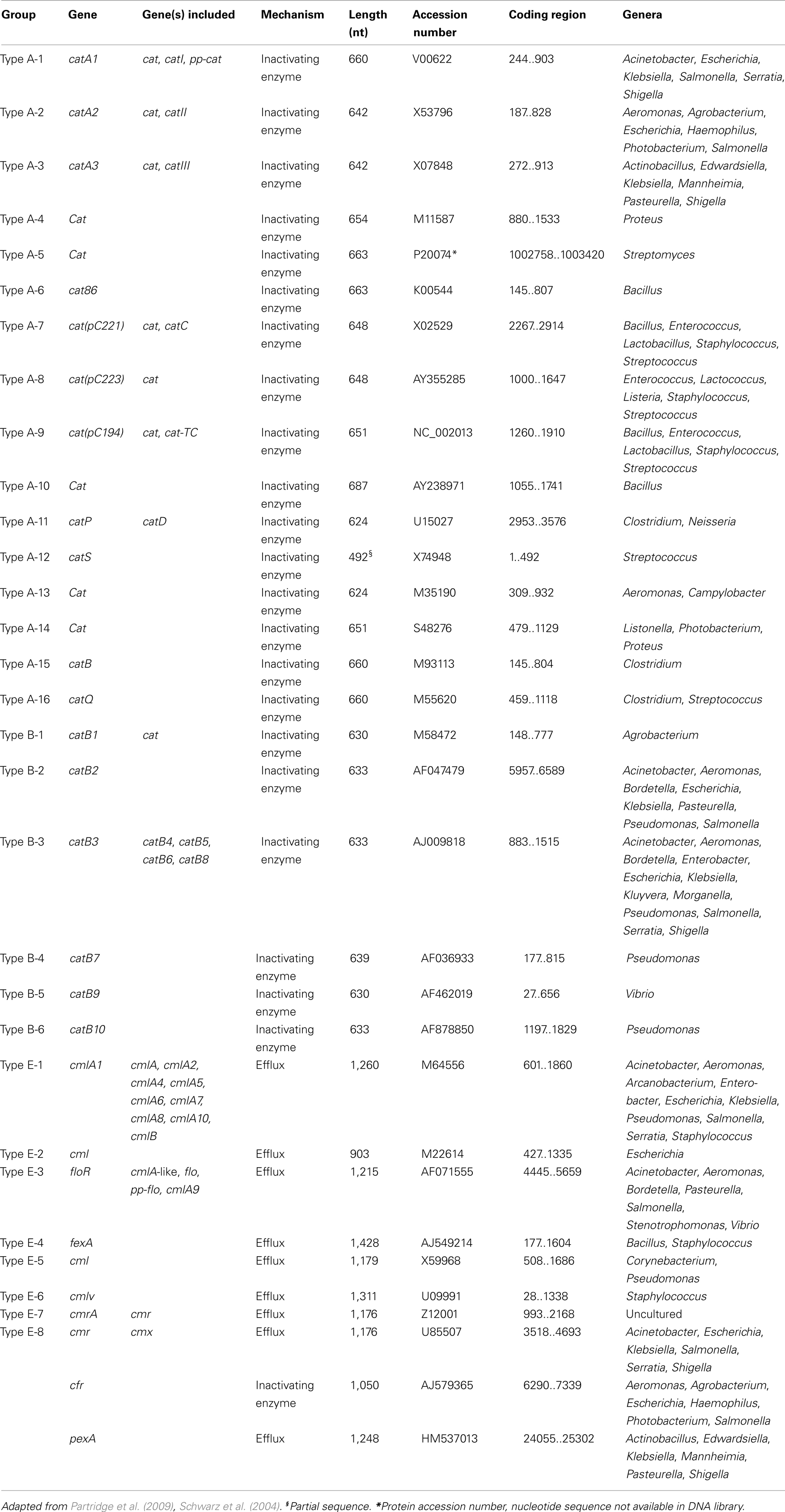
Table 3. Acquired chloramphenicol resistance genes.
Glycopeptide
History and action mechanism
In the late 1950s, the first glycopeptide, vancomycin was introduced in a clinical setting. Vancomycin was isolated as a fermentation product from a soil bacterium, Streptomyces orientalis, displaying antimicrobial activity (McCormick et al., 1956). Nearly 30 years later followed another glycopeptide antibiotic, teicoplanin (Parenti et al., 1978). Currently, four groups of glycopeptides are recognized, i.e., vancomycin type, avoparcin type, ristocetin type, and teicoplanin type. (Yao and Crandall, 1994). Among them, vancomycin and teicoplanin are the only two therapeutics currently used against Gram-positive microorganisms. During the 1990s, an association between the use of avoparcin and the occurrence of glycopeptide-resistant enterococci (GRE), more commonly designated VRE, in farm animals was demonstrated (Aarestrup, 1995; Klare et al., 1995). As a consequence avoparcin was banned as a growth promoter in all European Union countries in 1997.
Glycopeptides have an unusual mode of action. Instead of inhibiting an enzyme, they bind to a substrate. To be more specific, the molecular target of these glycopeptide antibiotics is the D-alanyl–D-alanine (D-Ala–D-Ala) terminus of the cell wall peptidoglycan precursor. After the glycopeptides are bound to their target, they inhibit the subsequent transglycosylation reaction by steric hindrance. (Gao, 2002; Klare et al., 2003).
Resistance mechanism
The introduction of antibiotics into clinical setting is usually followed by the fairly rapid emergence of resistant bacteria. In this respect, vancomycin was somewhat atypical, because for almost 30 years following its introduction, resistance to this glycopeptide was reported only rarely and appeared to have little clinical significance. However, in the late 1980s, the emergence of acquired glycopeptides resistance was recognized for the first time (Leclercq et al., 1988; Johnson et al., 1990). This vancomycin resistance resulted from the production of modified peptidoglycan precursors ending in D-Ala–D-Lac (VanA, VanB, and VanD) or D-Ala–D-Ser (VanC, VanE, and VanG), to which glycopeptides exhibit low binding affinities. Classification of glycopeptide resistance is based on the primary sequence of the structural genes for the resistance-mediating ligases. The vanA and vanB operons are located on plasmids or on the chromosome, whereas the vanC1, vanC2/3, vanD, vanE, and vanG have so far been found exclusively on the chromosome (Gao, 2002; Klare et al., 2003; Depardieu et al., 2007). Currently, resistance to the glycopeptides, vancomycin, and teicoplanin or both, has been detected in six, all Gram-positive bacterial genera: Enterococcus, Erysipelothrix, Lactobacillus, Leuconostoc, Pediococcus, and Staphylococcus (Woodford et al., 1995).
Macrolide–Lincosamide–Streptogramin B
History and action mechanism
The first macrolide, erythromycin A, was discovered in the early 1950s (McGuire et al., 1952). The main structural component of this molecule is a large lactone ring to which amino and/or neutral sugars are attached by glycosidic bonds. To address the limitations of erythromycin, like chemical instability, poor absorbance, and bitter taste, newer 14-, 15-, and 16-membered ring macrolides such as clarithromycin and the azalide, azithromycin, have been developed (Kirst, 2002; Roberts, 2002).
Macrolides have a similar mode of antibacterial action and comparable antibacterial spectra as two other antibiotic classes, i.e., lincosamides and streptogramins B. Consequently, these antibiotics, although chemically distinct, have been clustered together as Macrolide–Lincosamide–Streptogramin B (MLS) antibiotics (Roberts, 2002). Nowadays this class of antibiotics should even be extended due to the development of various synthetic drugs. The ketolides (Zhanel et al., 2002; Ackermann and Rodloff, 2003) and oxazolidinones (Diekema and Jones, 2000) can be grouped together with the MLS antimicrobial agents which results in the MLSKO family of antibiotics (Roberts, 2008).
Macrolides, lincosamides, and streptogramins B all inhibit protein synthesis by binding to the 50S ribosomal subunit of bacteria (Weisblum, 1995; Roberts, 2002).
Resistance mechanism
Shortly after the introduction of erythromycin into clinical setting in the 1950s, bacterial resistance to this antibiotic was reported for the first time in staphylococci (Weisblum, 1995). Since then a large number of bacteria have been identified that are resistant to MLS due to the presence of various different genes. The AR determinants responsible include rRNA methylases, efflux, and inactivating genes (Roberts et al., 1999; Roberts, 2008). The latter group can be further subdivided in esterases, lyases, phosphorylases, and transferases (Table 4).
TABLE 4
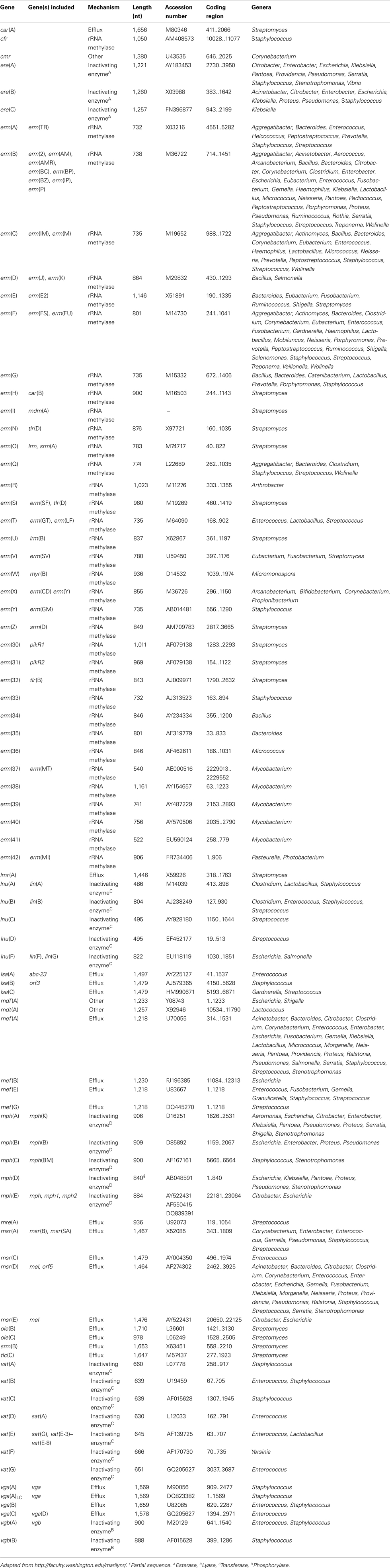
Table 4. Acquired macrolide–lincosamide–streptogramin B (MLS) resistance genes.
The most common mechanism of MLS resistance is due to the presence of rRNA methylases, encoded by the erm genes. These enzymes methylate the adenine residue(s) resulting in MLS resistance. The methylated adenine prevents the binding of the drugs from binding to the 50S ribosomal subunit. The other two mechanisms efflux pumps and inactivating genes are encoded by msr and ere determinants, respectively.
Because currently over 60 MLS resistance genes are recognized a nomenclature for naming these genes has been proposed that considers the same rules developed for identifying and naming new tetracycline resistance genes (see below; Roberts et al., 1999; Roberts, 2008). Table 4 represents the MLS acquired resistance genes. The genes included, the resistance mechanism, diverse gene lengths and accession number, and their distribution are displayed in this table.
Quinolone
History and action mechanism
In 1962, during the process of synthesis and purification of chloroquine (an antimalarial agent), a quinolone derivative, nalidixic acid, was discovered which possessed bactericidal activity against Gram-negatives (Lescher et al., 1962). The second generation quinolones arose when it became clear that the addition of a fluoride atom at position 6 of a quinolone molecule, creating a fluoroquinolone, greatly enhanced its biological activity. During the 1980s, various fluoroquinolones were developed, e.g., ciprofloxacin, norfloxacin, and ofloxacin. These fluoroquinolones demonstrated a broadened antimicrobial spectrum, including some Gram-positives (Wolfson and Hooper, 1989; Hooper, 2000; King et al., 2000).
In the 1990s, further alterations resulted in the third-generation (fluoro)quinolones, e.g., levofloxacin and sparfloxacin, showing potent activity against both Gram-negative and Gram-positive microbes. The new compounds, such as trovafloxacin, also show promising activity against anaerobic bacteria (Hooper, 2000; King et al., 2000).
Quinolones inhibit the action of DNA gyrase and topoisomerase IV, two enzymes essential for bacterial DNA replication and as a result the microbes are killed. (Hooper, 1995, 2000). DNA gyrase is a tetrameric enzyme composed of 2 GyrA and 2 GyrB subunits. The topoisomerase IV has a similar structure, comprised of 2 A and 2 B subunits, encoded by parC and parE, respectively. The four genes coding for the subunits of these enzymes are the targets for resistance mutations (see below).
Resistance mechanism
For decades, the mechanisms of resistance to quinolones were believed to be only chromosome-encoded, however, recently three plasmid-mediated resistance mechanisms have been reported (Robicsek et al., 2006a; Courvalin, 2008; Martínez-Martínez et al., 2008). The chromosome-encoded resistance result in either a decreased outer-membrane permeability related to porin loss, to the (over)expression of naturally occurring efflux pumps or mutations of the molecular targets DNA gyrase and topoisomerase IV (Hooper, 2000; Ruiz, 2003; Jacoby, 2005). In the latter case mutations occur at specific “quinolone resistance determining regions” (QRDR) in the genes gyrA, gyrB, parC, and parE encoding the subunits of DNA gyrase and topoisomerase IV. Not surprisingly this QRDR is situated on the DNA-binding surface of the enzymes (Jacoby, 2005).
Although the possibility of the existence of plasmid-mediated resistance was already suggested in 1990 (Courvalin, 1990), the first actually identified plasmid-mediated quinolone resistance gene, a qnr determinant, which encodes for a protein that protects DNA gyrase and type IV topoisomerase from quinolone inhibition, was reported nearly a decade later (Martínez-Martínez et al., 1998).
Currently five families of qnr genes have been reported; qnrA (7), qnrB (39), qnrC (1), qnrD (1), and qnrS (4). The number in between brackets indicates the variants known of each type (Jacoby et al., 2008; Cattoir and Nordmann, 2009; Cavaco et al., 2009; Strahilevitz et al., 2009; Torpdahl et al., 2009). Because of the increasing number of qnr genes a database has been constructed and will be maintained to assign further allele numbers to novel variants2. Very recently an additional family has been described, qnrAS in the fish pathogen Aliivibrio salmonicida (Sun et al., 2010). Table 5 describes all known qnr families and their variants, together with the gene lengths, accession numbers, and in which bacterial genera they have been identified so far.
TABLE 5
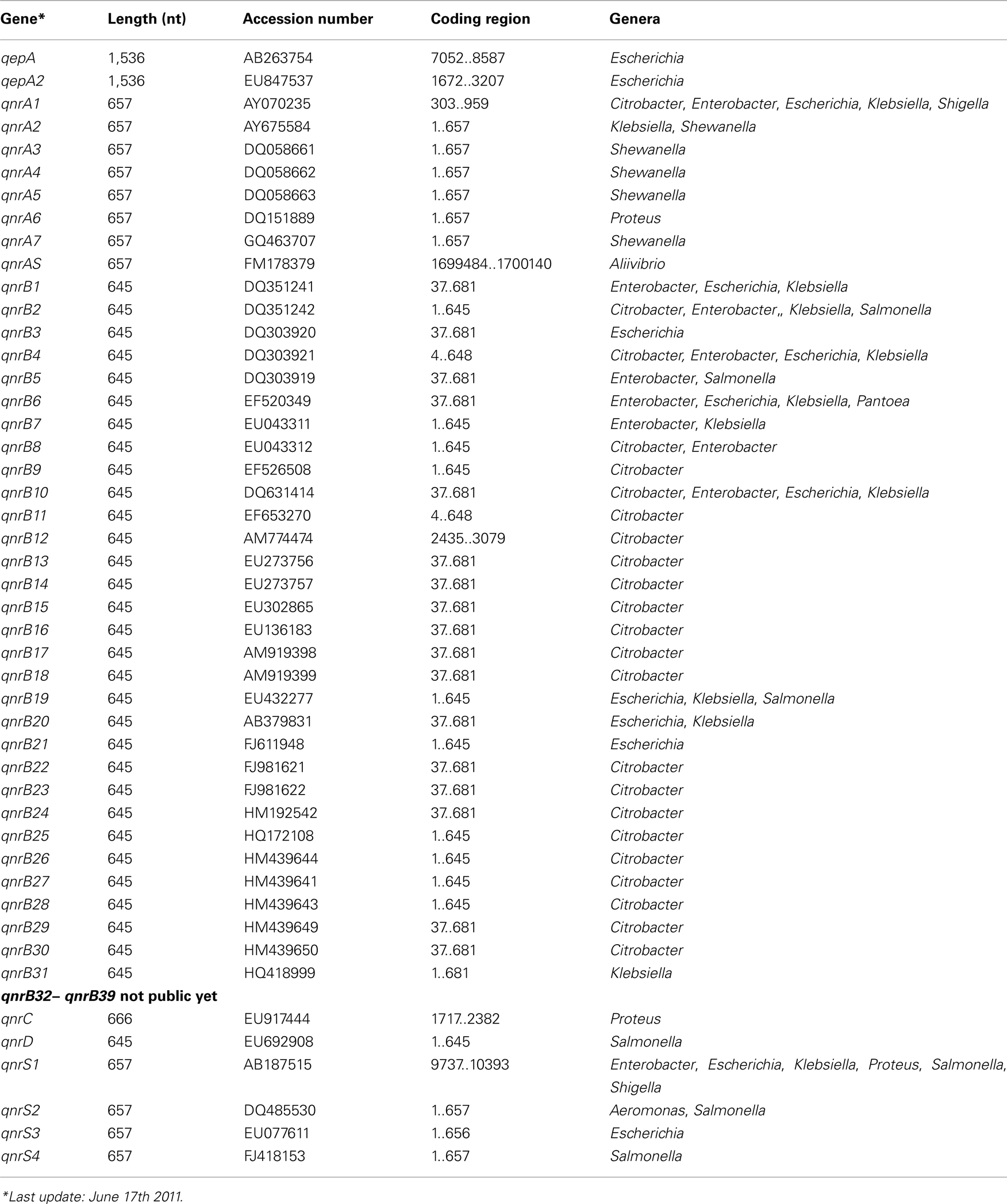
Table 5. Acquired quinolone resistance genes.
The second type of plasmid located quinolone resistant gene is a cr variant of aac(6′)-Ib, aac(6′)-Ib-cr, responsible for low-level ciprofloxacin resistance. It encodes an aminoglycoside acetyltransferase, called AAC(6′)-Ib-cr which has two amino acid changes, Trp102Arg and Asp179Tyr. These substitutions are responsible for the enzyme’s ability to acetylate ciprofloxacin (Park et al., 2006; Robicsek et al., 2006b; Strahilevitz et al., 2009).
The third mechanism is qepA, a plasmid-mediated efflux pump which can extrude hydrophilic fluoroquinolones, e.g., ciprofloxacin and enrofloxacin (Périchon et al., 2007; Yamane et al., 2007). A variant of this resistance pump, QepA2, was identified in an E. coli isolate from France (Cattoir et al., 2008).
Streptothricin
History and action mechanism
In the early days of the antibiotics era screening for new compound resulted in the discovery of a Streptomyces lavendulae isolate which inhibited growth of Gram-negative as well as Gram-positive bacteria. Isolation of the active antimicrobial substance resulted in the identification of streptothricin (Waksman and Woodruff, 1942). Delayed toxicity prevents streptothricin’s use in man, but it is effective in preventing animal infections.
Streptothricins consist of three moieties: gulosamine, streptolidin, and a β-lysine peptide chain. Since, the discovery of the streptothricin, six analogs have been reported, streptothricin A–F. The analogs differ from the parent molecule in the number of β-lysine residues (Keeratipibul et al., 1983; Tschäpe et al., 1984).
The streptothricins are potent inhibitors of bacterial protein synthesis, via direct binding to ribosomes. They also cause misreading of mRNA codons, although they are unrelated to other drugs that cause translational ambiguity, like the aminoglycosides (Tschäpe et al., 1984).
Resistance mechanism
Since streptothricin is inactivated by acetylation in its producer it is not surprising that the identified resistance mechanisms are acetyltransferases. The first streptothricin resistant bacterium identified was an E. coli isolate from a rectal swab of pigs under streptothricin F treatment. The AR gene was localized on a transferable plasmid (Tschäpe et al., 1984). Currently three different streptothricin acetyltransferases are recognized, sat2–sat4 (Partridge and Hall, 2005; see Table 1).
Sulfonamide
History and action mechanism
Sulfonamides belong to the oldest introduced synthetic drugs. They were first used in 1932 (Domagk, 1935; Sköld, 2001). A number of different sulfonamides have been developed of which the most commonly used nowadays is sulfamethoxazole. Moreover, since 1968, the combination of trimethoprim and sulfamethoxazole (called co-trimoxazole) has been used extensively because a combination of both drugs at certain concentrations has a synergetic bactericidal effect, it reduces selection of AR to either drug and associated costs (Roberts, 2002; Grape, 2006).
A sulfonamide, with its structural analogy to p-aminobenzoic acid, which is involved in the biosynthetic pathway leading to folic acid, competitively inhibits the enzyme dihydropteroate synthase (DHPS). This protein is part of the next to last step of the folate biosynthetic pathway that is required for thymine production and bacterial cell growth (Sköld, 2000, 2001; Roberts, 2002).
Resistance mechanism
Resistance to sulfonamide among pathogenic bacteria appeared quite soon after its introduction into clinical practice in the 1930s (Sköld, 2001). Since sulfonamides are synthetic antibacterial agents, naturally occurring enzymes degrading, or modifying this drug were not to be expected. However, chromosomal sulfonamide resistance occurs, mostly low level, by mutations in the folP gene encoding DHPS (Huovinen et al., 1995; Sköld, 2000, 2001; Grape, 2006).
Acquired sulfonamide resistance was discovered in the 1960s, but the plasmid-mediated genes were characterized later on in the 1980s as sul1 and sul2 (Swedberg and Sköld, 1983; Rådström and Swedberg, 1988; Sundström et al., 1988). Currently three plasmid-borne drug-resistant variants of the DHPS enzymes are known; besides the two genes mentioned above also sul3 has been identified (Perreten and Boerlin, 2003).
Tetracycline
History and action mechanism
The first tetracycline antibiotic was characterized in 1948 as chlortetracycline from Streptomyces aureofaciens (Chopra et al., 1992; Chopra and Roberts, 2001). In consecutive decades additional tetracyclines were identified either as naturally occurring molecules mostly in Streptomyces species (e.g., oxytetracycline, tetracycline) or products of semi-synthetic approaches (e.g., doxycycline, minocycline; Chopra et al., 1992; Hunter and Hill, 1997; Chopra and Roberts, 2001).
Tetracyclines were the first major group to which the term “broad spectrum” was applied (Chopra and Roberts, 2001). Because of this spectrum of activity, their relative safety, and low cost, tetracyclines have been used widely throughout the world and are second after penicillin in world consumption. This class of antibiotic can be separated into two groups, typical, (e.g., chlortetracycline, doxycycline, minocycline, oxytetracycline, and tetracycline) and atypical tetracyclines (e.g., anhydrotetracycline and 6-thiatetracycline), see below (Rasmussen et al., 1991; Oliva and Chopra, 1992; Chopra and Roberts, 2001).
Initially, it was thought that tetracyclines and most of its derivatives are antimicrobial agents only because they inhibit the growth of microbes by entering the bacterial cell, interacting with the ribosomes, and consequently blocking protein synthesis, the so-called typical tetracyclines (Speer et al., 1992; Roberts, 2002). However, Oliva and Chopra (1992) suggested an additional mode of action. Certain tetracycline derivatives are poor inhibitors of protein synthesis and appear to bind ribosomes inefficiently or not at all, in stead they interact with the bacterial membrane (Rasmussen et al., 1991; Chopra, 1994).
Resistance mechanism
Prior to the mid-1950s, the majority of commensals and pathogens were susceptible to tetracycline. However, in 1953 the first tetracycline resistant bacteria were isolated (Watanabe, 1963). The resistance mechanisms for the tetracycline class of antibiotics fall in three categories; energy-dependent efflux pumps, ribosomal protection proteins (RPPs), or enzymatic inactivation.
A novel tetracycline resistance determinant is identified as unique if it shares <79% amino sequence identity with all previously described genes. Initially, letters of the Roman alphabet have been used to name tetracycline resistance determinants. However, the number of tet genes has reached the end of the alphabet and to accommodate new genes, a nomenclature employing numerals for future determinants was introduced (Levy et al., 1999). Moreover, also naturally occurring hybrid tetracycline resistance genes exist. A simple, descriptive nomenclature for these mosaic tet determinants has been proposed incorporating the designations of the known tet genes classes forming the hybrid, e.g., tet(O/W) and tet(O/W/O; Levy et al., 2005; Stanton et al., 2005; van Hoek et al., 2008).
There are currently over 40 different acquired tetracycline resistance determinants recognized, i.e., 38 tet (tetracycline resistance) and 3 otr (oxytetracycline resistance) genes, additionally 1 tcr gene has been identified (Roberts, 1996, 2005; Brown et al., 2008; see Table 6). Among these 25 of the tet, 2 of the otr genes and the only tcr determinant code for efflux pumps, whereas 10 tet and 1 otr code for a RPP. The enzymatic inactivation mechanism can be attributed to 3 tet genes. The tet(U) determinant represents an unknown tetracycline resistance mechanism since its sequence does not appear to be related to either efflux or RPPs, nor to the inactivation enzymes (Table 6). The efflux and RPP encoding genes are found in members of Gram-positive, Gram-negative, aerobic, as well as anaerobic bacterial species. In contrast the enzymatic tetracycline inactivation mechanism has so far only been identified in Gram-negatives (Table 6). The tet(M) has the broadest host range of all tetracycline resistance genes, whereas tet(B) gene has the widest range among the Gram-negative microbes. In recent years published data indicate that there are increasing numbers of Gram-negative bacteria that carry “Gram-positive tet genes” (Roberts, 2002).
TABLE 6
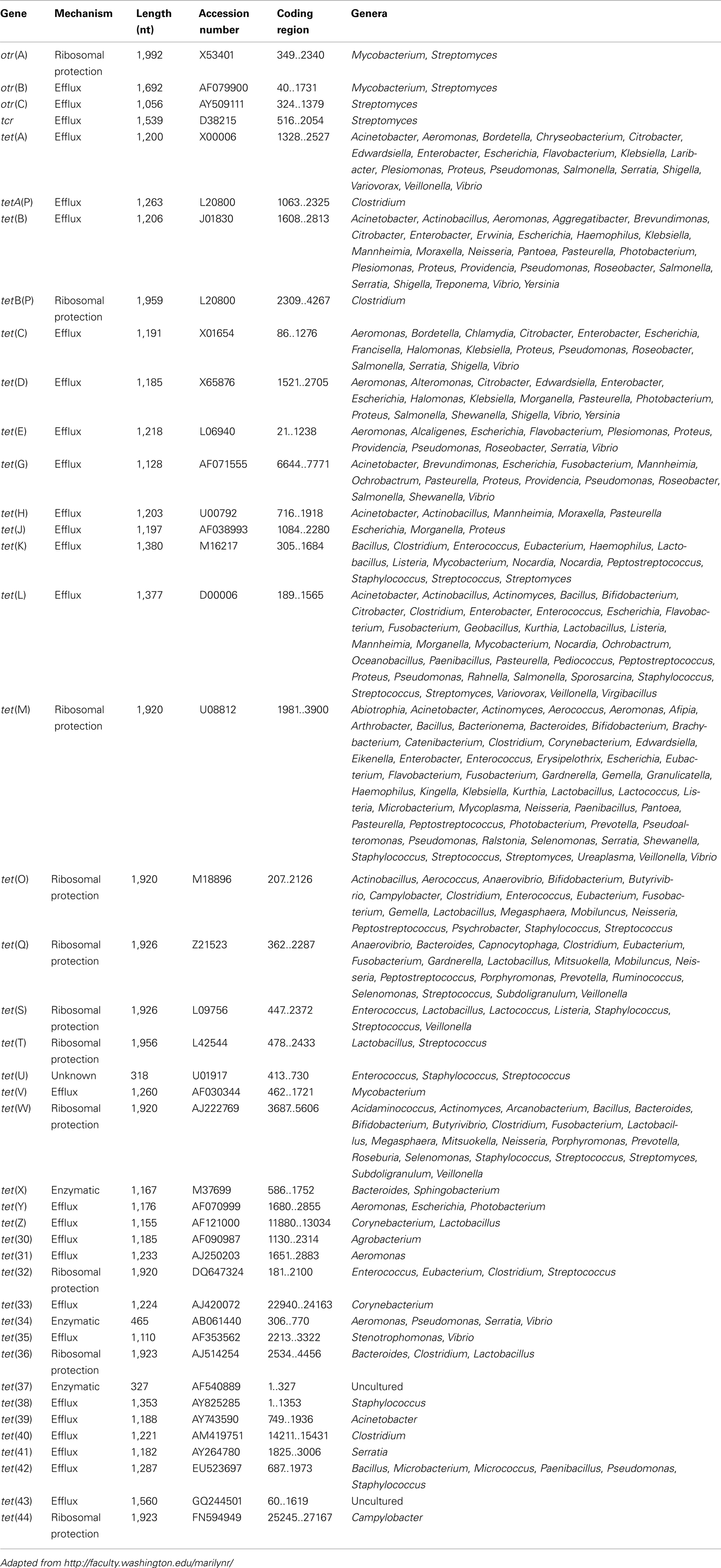
Table 6. Acquired tetracycline resistance genes.
Trimethoprim
History and action mechanism
Trimethoprim has been available since 1962 and is considered the last truly new antibacterial agent introduced into clinical practice (Roth et al., 1962). All later developed agents have been variations of older antibiotics, that is, belonging to families of agents, within which cross-resistance is common (Sköld, 2001; Roberts, 2002). Trimethoprim is a completely synthetic drug, belonging to the diaminopyrimidine group of compounds, i.e., 5-benzyl-2,4-diamino-pyrimidine (Huovinen, 1987).
Trimethoprim inhibits the enzyme dihydrofolate reductase (DHFR) by competitively binding to its active site. DHFR catalysis the NAHPH-dependent reduction of dihydrofolate acid to the active co-enzyme tetrahydrofolate. As such trimethoprim can be regarded as an antifolate, a structural analog of folic acid. DHFR, like DHPS is part of the folate biosynthetic pathway (Sköld, 2001; Grape, 2006; see section Sulfonamides).
Resistance mechanism
Because trimethoprim like sulfonamide is a synthetic antibacterial agent, naturally occurring enzymes degrading, or modifying it are unlikely. However, resistance, mostly low level, can for example occur via non-allelic and drug-resistant variants of the chromosomal folA gene encoding the bacterial DHFR (Huovinen et al., 1995; Sköld, 2001; Grape, 2006).
High-level resistance is generally achieved by a bypass mechanism through the action of an acquired gene which is a non-allelic and drug-insusceptible variant of a chromosomal DHFR. These plasmid-mediated DHFRs emerged in Gram-negative bacteria within several years of the clinical introduction of the drug (Fleming et al., 1972; Huovinen and Toivanen, 1980; Amyes and Towner, 1990).
Initially, the acquired DHFRs fell into two quite distinct families, dfrA and dfrB genes (Howell, 2005). Members of the dfrA group are at least 474 nucleotides (nt) long (157 amino acids, aa), whereas the dfrB genes are 237 nt in length (78 aa). Currently six plasmid-mediated families can be distinguished with relatively few dfr determinants originating from Gram-positive bacteria. (Table 7). The dfrK gene is the newest addition to the trimethoprim resistance determinant family (Kadlec and Schwarz, 2009). In contrast to the latest reported DHFRs, the oldest families, dfrA and dfrB, each contain several members (Roberts, 2002; Levings et al., 2006). For example, the dfrA group accommodates over 30 genes. Determinant dfrA27 is the newest reported DHFR gene among Gram-negatives (Wei et al., 2009), although a newer, however unpublished, dfrA variant is present in the public DNA library and some genes apparently have changed nomenclature (Table 7). Among this family two sub-families can be distinguished (Adrian et al., 2000). The dfrA1-group with 12 different genes share 64–90% identity on amino acids level. The dfrA12-group, with five members, display 84% amino acid identity and similar trimethoprim-inhibition profiles. The additional dfrA genes are less related to each other, some have even less than 25% amino acid sequence identity. In contrast to the dfrA family, the dfrB group is somewhat smaller, with only eight reported genes (Levings et al., 2006; Partridge et al., 2009).
TABLE 7
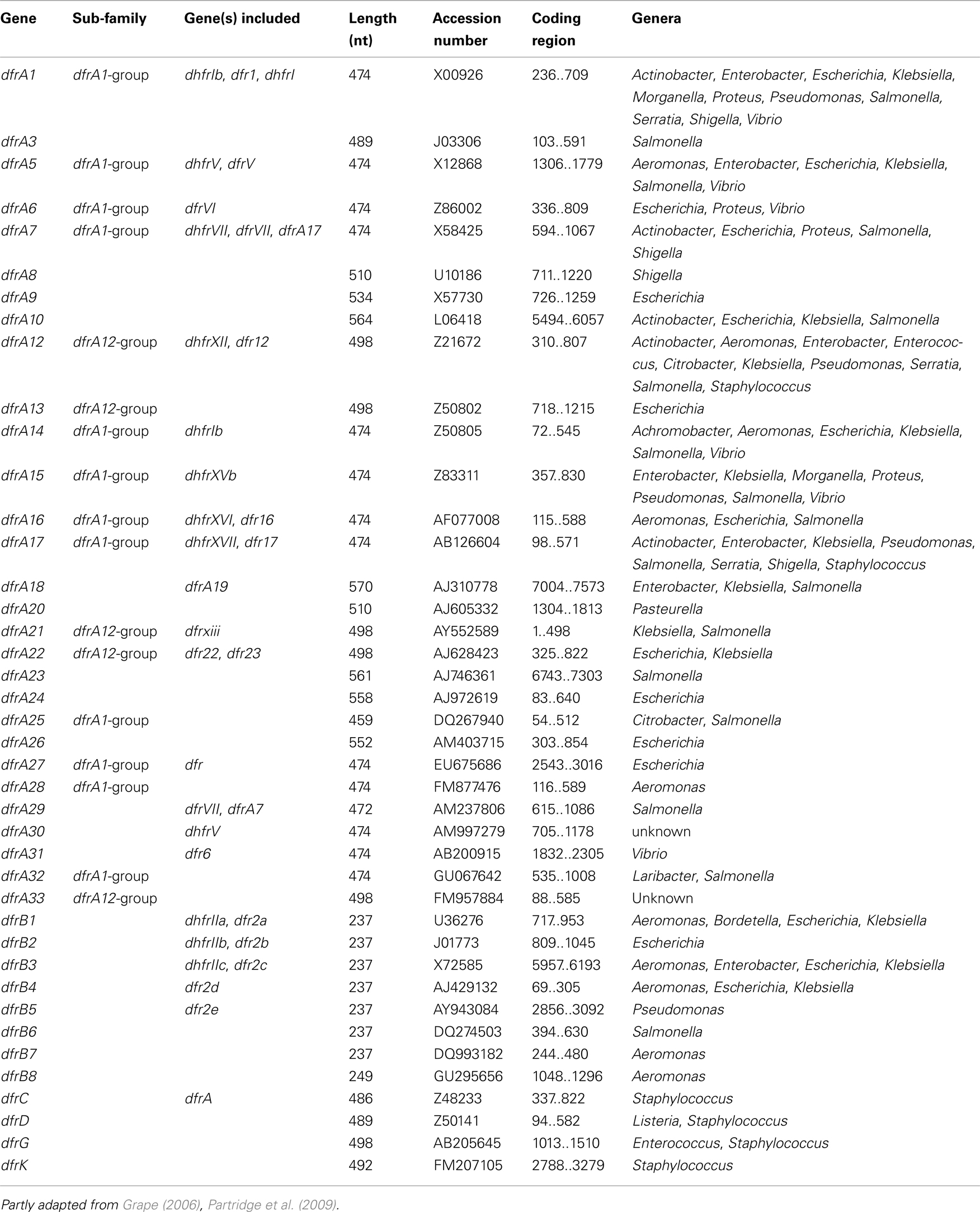
Table 7. Acquired trimethoprim resistance genes.
Mobile Genetic Elements
Acquired AR genes are frequently contained within mobile DNA which can be loosely defined as any segment of DNA that is capable of translocation from one part of a genome to another or between genomes. This definition includes a wide range of distinct mobile elements. The major players in HGT are the conjugative and mobilizable elements, the former contain all the genetic information required to transfer from one bacterium to another whilst the latter use the conjugation functions of co-resident conjugative elements (conjugative plasmids or conjugative transposons) to transfer to another host. Bacteriophages also play a role in the spread of DNA between bacteria, they do this by a process called transduction in which bacterial DNA, rather than phage DNA, is packaged into the phage head and injected into the recipient bacterium. There are also elements which are capable of translocation to new sites in the genome but are not themselves capable of transfer to a new host (of course if they transpose to a conjugative element they can be moved to new hosts). These include the transposons and the mobile introns.
Bacteria can also acquire AR genes by transformation. The process occurs in both Gram-positive and Gram-negative bacteria. Bacteria capable of taking up DNA from the environment are termed “competent.” Some microorganisms, such as many streptococci, are competent at a specific stage in their growth whilst others have no obvious competence window. Some bacteria have specific sequence requirements to successfully take up DNA such as Neisseria (Smith et al., 1999), while others like Bacillus subtilis have no obvious such requirements. In this process naked DNA is taken up by the recipient bacteria and either incorporated into the host genome by homologous recombination or transposition. Alternatively the DNA molecule may be able to replicate autonomously, e.g., plasmids. Mobile genetic elements are often acquired by transformation as well as by conjugation. For a recent review of the mechanisms of transformation see (Kovács et al., 2009; Aune and Aachmann, 2010; Burton and Dubnau, 2010).
Conjugative Elements (Plasmids)
Typically plasmids are extra chromosomal elements that contain their own origin of replication. They have been found in almost all bacterial genera and the simplest of these elements just contain an origin of replication and genes encoding replication functions, e.g., see Chambers et al. (1988). Plasmids also commonly contain an origin of transfer and genes encoding functions that allow them to transfer to new hosts via conjugation (Smillie et al., 2010). Plasmids that harbor conjugation genes are called conjugative and plasmids that only contain an origin of transfer (oriT) but no conjugation genes are called mobilizable as they can make use of the conjugation functions of conjugative plasmids to transfer to a new host.
In addition to functions involved in replication and transfer plasmids commonly encode resistance to antibiotics. If a resistance gene is on a conjugative or mobilizable plasmid then it has the potential to transfer to new hosts. Some plasmids have a broad host range and can transfer between different species whereas others have a much narrower host range and are confined to one genus or species. There are also plasmids that have the capability of transferring to a particular host but cannot replicate in the new host or do not replicate well. In these circumstances the plasmid may be lost, however if it contains a resistance gene on a transposon this genetic element can translocate to the bacterial chromosome and be maintained in the absence of the plasmid. Therefore a plasmid does not necessarily need to be maintained in a particular host in order to contribute to the spread of resistance.
Both circular and linear plasmids have been described. Circular plasmids have in general been more intensively investigated then linear plasmids. This probably reflects the relative ease which they can be separated from the bacterial chromosome. Nonetheless linear plasmids have now been relatively well characterized and have been shown to convey advantageous phenotypes on the host. Like circular plasmids linear plasmids are often capable of conjugation (Meinhart et al., 1997; Chaconas and Kobryn, 2010).
Some (resistance) plasmid types cannot coexist in a microbial cell and this fact gave rise to the division into incompatibility groups (Couturier et al., 1988). Four major groups have been defined on the basis of genetic relatedness and pilus structure: IncF group (containing IncC, IncD, IncF, IncJ, and IncS), IncI group (including IncB, IncI, and IncK), IncP group (consisting of IncM, IncP, IncU, and IncW), and Ti.
Conjugative Elements (Integrative)
The integrative conjugative elements (ICE), also called conjugative transposons (Roberts et al., 2008), like the conjugative plasmids contain an origin of transfer and the genes required to make the conjugation apparatus. Unlike plasmids these elements do not contain an origin of replication and have to integrate into a replicon in order to be maintained. This replicon can be either plasmid or chromosome. This gives them an advantage over plasmids as they do not have to have replication machinery that is compatible with the host so tend to have a larger host range than plasmids.
Integrative conjugative elements are a highly heterogeneous group of genetic elements with different properties and host ranges. However in general they do have a modular organization, i.e., a conjugation, recombination, regulation, and accessory modules. The latter commonly contains genes encoding AR.
There are also integrative elements that do not contain the conjugation region but can by mobilized by co-resident conjugative ICE or conjugative plasmids. Again these can mediate the spread of AR. There have been a number of comprehensive reviews in this area (Roberts and Mullany, 2009; Frost and Koraimann, 2010; Wozniak and Waldor, 2010).
Transduction
There have been examples of AR genes, and even entire mobile genetic elements, being mobilized by transduction (Willi et al., 1997; Del Grosso et al., 2011). Transduction is a process in which the phage particles are packaged with bacterial DNA instead of phage. There are two type of transduction, generalized in which any segment of bacterial DNA can be packaged into the phage head, and specialized, in which the DNA adjacent to the phage insertion site is packaged.
Translocation within Genomes
The simplest of the mobile genetic elements are insertion sequence (IS). These elements just consist of the gene required for element mobility and the inverted repeat at the ends of the element. IS elements can be as short as 1Kb (Siguier et al., 2006). When these elements contain accessory genes not involved in element translocation they are called transposons. A simple transposon will contain an accessory gene (often encoding AR) together with the transposase (for examples of each type of element see Roberts et al., 2008). There are more complex classes of transposons that move using different mechanisms including class II transposons.
The transposons mentioned above are not capable of conjugal transfer to other bacteria and in order for them to be disseminated they need to be contained within a conjugative element. However some of ICE elements as well as being able to transfer to new hosts (see above) are also able to transpose to new genomic sites. Their ability to use different integration sites in the chromosomes depends on the type of recombinases they contain. For example Tn916 can use a large number of different integration sites in most hosts (reviewed in Roberts and Mullany, 2009). However some elements are highly site-specific such as Tn916 (Wozniak and Waldor, 2010). Presumably elements like Tn916 have evolved to use different integration sites in order to increase their host range. Elements that can only use a particular number of insertion sites are limited in the hosts they can use if the site is mutated or occupied.
Gene Capture Elements
Integrons are genetic elements that include components of a site-specific recombination system enabling them to capture and mobilize genes, in particular AR determinants (Stokes and Hall, 1989; Rechia and Hall, 1995; Fluit and Schmitz, 1999; Depardieu et al., 2007). They harbor an intI gene, encoding a site-specific integrase of the tyrosine recombinase family that carries out recombination between two distinct target sites, i.e., an attI recombination site and a 59-base element (attC site) where attI is the target site for cassette integration and a promoter (Hall and Stokes, 1993; Hall and Collis, 1995; Rechia and Hall, 1997; Mazel, 2006). In contrast to transposons integrons are not flanked by repeat sequences, in addition they do not include any genes encoding proteins that catalyze their movement. HGT of integrons to other bacteria is mostly mediated by plasmids or transposons.
The intI genes have been used as a basis for grouping integrons into ”classes.” Currently, four classes are recognized; those carrying intI1 are defined as class 1, intI2 as class 2, intI3 as class 3, and intI4 as class 4 (Carattoli, 2001; Partridge et al., 2009).
Factors Influencing Acquisition of Mobile Genetic Elements
The ability of mobile genetic elements containing AR genes to spread is modulated by a range of factors including, selective pressures in the environment, host factors, and properties of the genetic elements themselves. Each of these factors will be examined in turn in the next sections.
Specific host encoded factors
Bacteria have a number of systems that protect them from incoming DNA, including restriction/modification systems and CRISPR-Cas systems (Makarova et al., 2011). These systems although mechanistically very different have the same end point of identifying and destroying foreign DNA. Restriction systems work by identifying particular sequences in the incoming DNA that have not been protected by methylation and digesting them. CRISPRs act as a memory of past infection by a mobile element and can destroy that element if the bacterium encounters it again. Both these systems can be effective in stopping the spread of phage, ICE, and plasmids.
A specific host factor that attracts mobile elements has been documented in the pheromone responsive systems, in which a plasmid less recipient secrets a pheromone to which plasmids containing strains respond and transfer their plasmid to the recipients (Palmer et al., 2010).
None specific host factors
Some none specific factors that can act as barriers to HGT have been eluded to above such as not having the target site for a particular ICE or having incompatible replication systems that stop plasmids replicating in a particular host. Also the architecture of the cell surface my not allow the conjugation systems of all mobile elements to work productively. Additionally one member of a mating pair may produce inhibitory substances. Bacteria produce a number of antimicrobial products the most common being the peptide antibiotics. The best understood are the colicins produced by E. coli. Gram-positive bacteria also produce a diverse array of antimicrobial peptides (Riley and Wertz, 2002).
Genetic element encoded factors
Mobile genetic elements have a plethora of ways to overcome bacterial defense systems. Many plasmids and ICE encode anti-restriction proteins that as the name suggests inactivate the host restriction system allowing the element to enter the new host and survive. Also many mobile genetic elements do not have many restriction enzyme recognition sites so that they avoid the attention of the restriction enzymes. Some, including the common Tn916-like family of conjugative transposons, encode anti-restriction proteins which have been shown to mimic DNA and are recognized by the restriction enzyme. The anti-restriction protein ArdA from Tn916 is one of the best characterized (McMahon et al., 2009).
Many transposons and ICE can transpose into essential genes. If this happens the host will die, to get around this some mobile elements are site-specific or preferentially target inter-genic regions (Cookson et al., 2011). Also most transposable elements (including ICE) are tightly regulated so that they only transpose at low frequency or transpose when the bacteria are stressed, such as antibiotics in their environment (reviewed in Roberts and Mullany, 2009; Wozniak and Waldor, 2010). For example members of the CTndot family of ICE transfer at a much higher frequency in the presence of tetracycline (the antibiotic to which they encode resistance). This is an advantageous response for both the element and the host bacteria (Moon et al., 2005).
Environmental factors
All the factors outlined in the previous sections are important in modulating the spread of AR but obviously if antibiotics are present in the environment there is strong selective pressure for spread of resistance and those factors that promote the spread of resistance will be selected for and those stopping the spread of mobile elements selected against.
Gene transfer is also more likely in environments where bacteria are in close proximity to each other and in relatively high density such as the gut and oral cavity. In order to control the spread of resistance it is important to have an understanding of the molecular biology of the different mobile genetic elements and of the ecology of the environments in which spread is likely.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors (Peter Mullany and Adam Paul Roberts) have received financial support from the Commission of the European Communities, specifically the Infectious Diseases research domain of the Health theme of the 7th Framework Programme, contract 241446, “The effects of antibiotic administration on the emergence and persistence of antibiotic-resistant bacteria in humans and on the composition of the indigenous microbiotas at various body sites.”
Footnotes
References
Aarestrup, F. M. (1995). Occurrence of glycopeptide resistance among Enterococcus faecium isolates from conventional and ecological poultry farms. Microb. Drug Resist. 1, 255–257.
Aarestrup, F. M., and Jensen, N. E. (1998). Development of penicillin resistance among Staphylococcus aureus isolated from bovine mastitis in Denmark and other countries. Microb. Drug Resist. 4, 247–256.
Abraham, E. P., and Chain, E. (1940). An enzyme from bacteria able to destroy penicillin. Nature 146, 837.
Ackermann, G., and Rodloff, A. C. (2003). Drugs of the 21st century: telithromycin (HMR 3647) – the first ketolide. J. Antimicrob. Chemother. 51, 497–511.
Adrian, P. V., Thomson, C. J., Klugman, K. P., and Amyes, S. G. B. (2000). New gene cassettes for trimethoprim resistance, dfr13, and streptomycin-spectinomycin resistance, aadA4, inserted on a class 1 integron. Antimicrob. Agents Chemother. 44, 355–361.
Allen, H. K., Donato, J., Wang, H. H., Cloud-Hansen, K. A., Davies, J., and Handelsman, J. (2010). Call of the wild: antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 8, 251–259.
Ambler, R. P. (1980). The structure of β-lactamases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 289, 321–331.
Amyes, S. G. B., and Towner, K. J. (1990). Trimethoprim resistance; epidemiology and molecular aspects. J. Med. Microbiol. 31, 1–19.
Andes, D. R., and Craig, W. A. (2005). “Cephalosporins,” in Principles and Practice of Infectious Diseases, eds G. L. Mandell, J. E. Bennett, and R. Dolin (Philadelphia, PA: Churchill Livingstone), 294–311.
Aune, T. E. V., and Aachmann, F. L. (2010). Methodologies to increase the transformation efficiencies and the range of bacteria that can be transformed. Appl. Microbiol. Biotechnol. 85, 1301–1313.
Begg, E. J., and Barclay, M. L. (1995). Aminoglycosides – 50 years on. Br. J. Clin. Pharmacol. 39, 597–603.
Bissonnette, L., Champetier, S., Buisson, J.-P., and Roy, P. H. (1991). Characterization of the non-enzymatic chloramphenicol resistance (cmlA) gene of the In4 integron of Tn1696: similarity of the product to transmembrane transport proteins. J. Bacteriol. 173, 4493–4502.
Bradford, P. A. (2001). Extended-spectrum β-lactamase in the 21st century: characterization, epidemiology, and detection of this important resistance threat. Clin. Microbiol. Rev. 14, 933–951.
Briggs, C. E., and Fratamico, P. M. (1999). Molecular characterization of an antibiotic resistance gene cluster of Salmonella typhimurium DT104. Antimicrob. Agents Chemother. 43, 846–849.
Brown, M. G., Mitchell, E. H., and Balkwill, D. L. (2008). Tet 42, a novel tetracycline resistance determinant isolated from deep terrestrial subsurface bacteria. Antimicrob. Agents Chemother. 52, 4518–4521.
Burton, B., and Dubnau, D. (2010). Membrane-associated DNA transport machines. Cold Spring Harb. Perspect. Biol. 2, a000406.
Bush, K. (1988). β-Lactamase inhibitors from laboratory to clinic. Clin. Microbiol. Rev. 1, 109–123.
Bush, K., and Jacoby, G. A. (2010). Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 54, 969–976.
Bush, K., Jacoby, G. A., and Medeiros, A. A. (1995). A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 39, 1211–1233.
Cantón, R., and Coque, T. M. (2006). The CTX-M beta-lactamase pandemic. Curr. Opin. Microbiol. 9, 466–475.
Cantón, R., Novais, A., Valverde, A., Machado, E., Peixe, L., Baquero, F., and Coque, T. M. (2008). Prevalence and spread of extended-spectrum β-lactamase-producing Enterobacteriaceae in Europe. Clin. Microbiol. Infect. 14, 144–153.
Carattoli, A. (2001). Importance of integrons in the diffusion of resistance. Vet. Res. 32, 243–259.
Carattoli, A. (2003). Plasmid-mediated antimicrobial resistance in Salmonella enterica. Curr. Issues Mol. Biol. 5, 113–122.
Carattoli, A. (2009). Resistance plasmid families in Enterobactericeae. Antimicrob. Agents Chemother. 53, 2227–2238.
Casadewall, B., and Courvalin, P. (1999). Characterization of the vanD glycopeptide resistance gene cluster from Enterococcus faecium BM4339. J. Bacteriol. 181, 3644–3648.
Cattoir, V., and Nordmann, P. (2009). Plasmid-mediated quinolone resistance in gram-negative bacterial species: an update. Curr. Med. Chem. 16, 1028–1046.
Cattoir, V., Poirel, L., and Nordmann, P. (2008). Plasmid-mediated quinolone resistance pump QepA2 in an Escherichia coli isolate from France. Antimicrob. Agents Chemother. 52, 3801–3804.
Cavaco, L. M., Hasman, H., Xia, S., and Aarestrup, F. M. (2009). qnrD, a novel gene conferring transferable quinolone resistance in Salmonella enterica serovar Kentucky and Bovismorbificans strains of human origin. Antimicrob. Agents Chemother. 53, 603–608.
Chaconas, G., and Kobryn, K. (2010). Structure, function, and evolution of linear replicons in Borrelia. Annu. Rev. Microbiol. 64, 185–202.
Chambers, S. P., Prior, S. E., Barstow, D. A., and Minton, N. P. (1988). The pMTL nic- cloning vectors. I. Improved pUC polylinker regions to facilitate the use of sonicated DNA for nucleotide sequencing. Gene 68, 139–149.
Chopra, I. (1994). Tetracycline analogs whose primary target is not the bacterial ribosome. Antimicrob. Agents Chemother. 38, 637–640.
Chopra, I., Hawkey, P. M., and Hinton, M. (1992). Tetracyclines, molecular and clinical aspects. J. Antimicrob. Chemother. 29, 245–277.
Chopra, I., and Roberts, M. C. (2001). Tetracycline antibiotics: mode of action, applications, molecular biology and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 65, 232–260.
Cookson, A. L., Noel, S., Hussein, H., Perry, R., Sang, C., Moon, C. D., Leahy, S. C., Altermann, E., Kelly, W. J., and Attwood, G. T. (2011). Transposition of Tn916 in the four replicons of the Butyrivibrio proteoclasticus B316T genome. FEMS Microbiol. Lett. 316, 144–151.
Courvalin, P. (1990). Plasmid-mediated 4-quinolone resistance: real or apparent absence? Antimicrob. Agents Chemother. 34, 681–684.
Courvalin, P. (2008). New plasmid-mediated resistances to antimicrobial agents. Arch. Microbiol. 189, 289–291.
Couturier, M., Bex, F., Bergquist, P. L., and Maas, W. K. (1988). Identification and classification of bacterial plasmids. Microbiol. Rev. 52, 375–395.
Datta, N., and Kontomichalou, P. (1965). Penicillinase synthesis controlled by infectious R factors in enterobacteriaceae. Nature 208, 239–241.
Davies, J. (1994). Inactivation of antibiotics and the dissemination of resistance genes. Science 64, 375–382.
Davies, J., and Wright, G. D. (1997). Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol. 5, 234–240.
Del Grosso, M., Camilli, R., Barbabella, G., Blackman Northwood, J., Farrell, D. J., and Pantosti, A. (2011). Genetic resistance elements carrying mef subclasses other than mef(A) in Streptococcus pyogenes. Antimicrob. Agents Chemother. 55, 3226–3230.
Depardieu, F., Podglajen, I., Leclercq, R., Collatz, E., and Courvalin, P. (2007). Modes and modulations of antibiotic resistance gene expression. Clin. Microbiol. Rev. 20, 79–114.
Doi, Y., and Arakawa, Y. (2007). 16S ribosomal RNA methylation: emerging resistance mechanism against aminoglycosides. Clin. Infect. Dis. 45, 88–94.
Doi, Y., Wachino, J.-I., and Arakawa, Y. (2008). Nomenclature of plasmid-mediated 16S rRNA methylases responsible for panaminoglycoside resistance. Antimicrob. Agents Chemother. 52, 2287–2288.
Domagk, G. J. (1935). Ein beitrag zur chemotherapie der bakteriellen infektionen. Dtsch. Med. Wochenschr. 61, 250–253.
Drawz, S. M., and Bonomo, R. A. (2010). Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 23, 160–201.
Ehrlich, J., Bartz, Q. R., Smith, R. M., Joslyn, D. A., and Burkholder, P. R. (1947). Chloromycetin a new antibiotic from a soil actinomycete. Science 106, 417.
Elbourne, L. D. H., and Hall, R. M. (2006). Gene cassette encoding a 3-N-aminoglycoside acetyltransferase in a chromosomal integron. Antimicrob. Agents Chemother. 50, 2270–2271.
Fleming, M. P., Datta, N., and Grüneberg, R. N. (1972). Trimethoprim resistance determined by R factors. Br. Med. J. 1, 726–728.
Flemming, A. (1929). Classics in infectious diseases: on the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. Brit. J. Exp. Pathol. 10, 226–236.
Fluit, A. C., and Schmitz, F. J. (1999). Class 1 integrons, gene cassettes, mobility, and epidemiology. Eur. J. Clin. Microbiol. Infect. Dis. 18, 761–770.
Frost, L. S., and Koraimann, G. (2010). Regulation of bacterial conjugation: balancing opportunity with adversity. Future Microbiol. 5, 1057–1071.
Galimand, M., Courvalin, P., and Lambert, T. (2003). Plasmid-mediated high level resistance to aminoglycosides in Enterobacteriaceae due to 16S rRNA methylation. Antimicrob. Agents Chemother. 47, 2565–2571.
Galimand, M., Sabtcheva, S., Courvalin, P., and Lambert, T. (2005). Worldwide disseminated armA aminoglycoside resistance methylase gene is borne by composite transposon Tn1548. Antimicrob. Agents Chemother. 49, 2949–2953.
Gao, Y. (2002). Glycopeptide antibiotics and development of inhibitors to overcome vancomycin resistance. Nat. Prod. Rep. 19, 100–107.
Goossens, H. (2009). Antibiotic consumption and link to resistance. Clin. Microbiol. Infect. 15, 12–15.
Grape, M. (2006). Molecular Basis for Trimethoprim and Sulphonamide Resistance in Gram Negative Pathogens. Ph.D. thesis, Karolinska Institutet, Stockholm.
Hall, R. M., and Collis, C. M. (1995). Mobile gene cassettes and integrons: capture and spread of genes by site-specific recombination. Mol. Microbiol. 15, 593–600.
Hall, R. M., and Stokes, H. W. (1993). Integrons: novel DNA elements which capture genes by site-specific recombination. Genetica 90, 115–132.
Hancock, R. E. W. (1981). Aminoglycoside uptake and mode of action–with special reference to streptomycin and gentamicin. J. Antimicrob. Chemother. 8, 249–276.
Hooper, D. C. (2000). Mechanisms of action and resistance of older and newer fluoroquinolones. Clin. Infect. Dis. 31, S24–S28.
Howell, E. E. (2005). Searching sequence space: two different approaches to dihydrofolate reductase catalysis. Chembiochem 6, 590–600.
Hunter, I. S., and Hill, R. A. (1997). “Tetracyclines, chemistry and molecular genetics of their formation,” in Biotechnology of Antibiotics (Drugs and the Pharmaceutical Sciences), ed. W. R. Strohl (London, UK: Informa HealthCare), 659–682.
Huovinen, P., Sundström, L., Sewdberg, G., and Sköld, O. (1995). Trimethoprim and sulfonamide resistance. Antimicrob. Agents Chemother. 39, 279–289.
Huovinen, P., and Toivanen, P. (1980). Trimethoprim resistance in Finland after five years’ use of plain trimethoprim. Br. Med. J. 280, 72–74.
Jacoby, G., Cattoir, V., Hooper, D., Martínez-Martínez, L., Nordmann, P., Pascual, A., Poirel, L., and Wang, M. (2008). qnr Gene nomenclature. Antimicrob. Agents Chemother. 52, 2297–2299.
Jevons, M. P., Coe, A. W., and Parker, M. T. (1963). Methicillin resistance in staphylococci. Lancet 281, 904–907.
Johnson, A. P., Uttley, A. H. C., Woodford, N., and George, R. C. (1990). Resistance to vancomycin and teicoplanin: an emerging clinical problem. Clin. Microbiol. Rev. 3, 280–291.
Kadlec, K., and Schwarz, S. (2009). Identification of a novel trimethoprim resistance gene, dfrK, in a methicillin-resistant Staphylococcus aureus ST398 strain and its physical linkage to the tetracycline resistance gene tet(L). Antimicrob. Agents Chemother. 53, 776–778.
Keeratipibul, S., Sugiyama, M., and Nomi, R. (1983). Mechanism of resistance to streptothricin of a producing microorganism. Biotechnol. Lett. 5, 441–446.
King, D. E., Malone, R., and Lilley, S. H. (2000). New classification and update on the quinolone antibiotics. Am. Fam. Physician 61, 2741–2748.
Kirst, H. A. (2002). “Introduction to the macrolide antibiotics,” in Macrolide Antibiotics, eds W. Schönfeld and H. A. Kirst (Basel: Birkhäuser Verlag), 1–14.
Klare, I., Heier, H., Claus, H., Reissbrodt, R., and Witte, W. (1995). vanA-mediated high-level glycopeptide resistance in Enterococcus faecium from animal husbandry. FEMS Microbiol. Lett. 125, 165–171.
Klare, I., Konstabel, C., Badstübner, D., Werner, G., and Witte, W. (2003). Occurrence and spread of antibiotic resistances in Enterococcus faecium. Int. J. Food Microbiol. 88, 269–290.
Kotra, L. P., Haddad, J., and Mobashery, S. (2000). Aminoglycosides: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob. Agents Chemother. 44, 3249–3256.
Kotra, L. P., and Mobashery, S. (1998). β-Lactam antibiotics, β-lactamases and bacterial resistance. Bull. Inst. Pasteur 96, 139–150.
Kovács, A. T., Smits, W. K., Mironczuk, A. M., and Kuipers, O. P. (2009). Ubiquitous late competence genes in Bacillus species indicate the presence of functional DNA uptake machineries. Environ. Microbiol. 11, 1911–1922.
Leclercq, R., Derlot, E., Duval, J., and Courvalin, P. (1988). Plasmid mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N. Engl. J. Med. 319, 157–161.
Lescher, G. Y., Froelich, E. J., Gruett, M. D., Bailey, J. H., and Brundage, R. P. (1962). 1,8-Naphthyridine derivatives: a new class of chemotherapy agents. J. Med. Pharm. Chem. 5, 1063–1068.
Levings, R. S., Lightfoot, D., Elbourne, L. D. H., Djordjevic, S. P., and Hall, R. M. (2006). New integron-associated gene cassette encoding a trimethoprim-resistant DfrB-type dihydrofolate reductase. Antimicrob. Agents Chemother. 50, 2863–2865.
Levy, S. B., McMurry, L. M., Barbosa, T. M., Burdett, V., Courvalin, P., Hillen, W., Roberts, M. C., Rood, J. I., and Taylor, D. E. (1999). Nomenclature for new tetracycline resistance determinants. Antimicrob. Agents Chemother. 43, 1523–1524.
Levy, S. B., McMurry, L. M., and Roberts, M. C. (2005). Tet protein hybrids. Antimicrob. Agents Chemother. 49, 3099.
Magnet, S., and Blanchard, J. S. (2005). Molecular insights into aminoglycoside action and resistance. Chem. Rev. 105, 477–497.
Magnet, S., Courvalin, P., and Lambert, T. (2001). Resistance-nodulation-cell division type effux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob. Agents Chemother. 45, 3375–3380.
Maiden, M. C. J. (1998). Horizontal genetic exchange, evolution, and spread of antibiotic resistance in bacteria. Clin. Infect. Dis. 27, S12–S20.
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J., Charpentier, E., Horvath, P., Moineau, S., Mojica, F. J., Wolf, Y. I., Yakunin, A. F., van der Oost, J., and Koonin, E. V. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477.
MARAN. (2005). Monitoring of Antimicrobial Resistance and Antibiotic Usage in Animals in the Netherlands in 2005. Lelystad: CIDC-Lelystad. Available at: www.cidc-lelystad.nl.
MARAN. (2007). Monitoring of Antimicrobial Resistance and Antibiotic Usage in Animals in the Netherlands in 2006/2007. Lelystad: CVI-Lelystad. Available at: www.cvi.wur.nl.
Marshall, W. F., Vink, A., Orenstein, R., Wilson, J. W., and Estes, L. L. (2006). “Infectious diseases,” in Mayo Clinic Internal Medicine Review 2006-2007, eds T. M. Habermann, A. K. Ghosh, and D. J. Rhodes (New York, NY: Taylor & Francis Group), 589.
Martínez-Martínez, L., Cano, M. E., Rodríguez-Martínez, R. M., Calvo, J., and Pascual, A. (2008). Plasmid-mediated quinolone resistance. Expert Rev. Anti Infect. Ther. 6, 685–711.
Martínez-Martínez, L., Pascual, A., and Jacoby, G. A. (1998). Quinolone resistance from a transferable plasmid. Lancet 351, 797–799.
McCormick, M. H., Stark, W. M., Pittenger, G. E., Pittenger, R. C., and McGuire, J. M. (1956). Vancomycin a new antibiotic. I. Chemical and biological properties. Antibiot. Annu. 1955–1956, 606–611.
McDermott, P. F., Walker, R. D., and White, D. G. (2003). Antimicrobials: modes of action and mechanisms of resistance. Int. J. Toxicol. 22, 135–143.
McGuire, J. M., Bunch, R. L., Anderson, R. C., Boaz, H. E., Flynn, E. H., Powell, H. M., and Smith, J. W. (1952). ‘Ilotycin’ a new antibiotic. Antibiot. Chemother. 2, 281–283.
McMahon, S. A., Roberts, G. A., Johnson, K. A., Cooper, L. P., Liu, H., White, J. H., Carter, L. G., Sanghvi, B., Oke, M., Walkinshaw, M. D., Blakely, G. W., Naismith, J. H., and Dryden, D. T. F. (2009). Extensive DNA mimicry by the ArdA anti-restriction protein and its role in the spread of antibiotic resistance. Nucleic Acids Res. 37, 4887–4897.
Meinhart, F., Schaffrath, R., and Larsen, M. (1997). Microbial linear plasmids. Appl. Microbiol. Biotechnol. 47, 329–336.
Moon, K., Shoemaker, N. B., Gardner, J. F., and Salyers, A. A. (2005). Regulation of excision genes of the Bacteroides conjugative transposon CTnDOT. J. Bacteriol. 187, 5732–5741.
Moore, R. A., DeShazer, D., Reckseidler, S., Weissman, A., and Woods, D. E. (1999). Effux-mediated aminoglycoside and macrolide resistance in Burkholderia pseudomallei. Antimicrob. Agents Chemother. 43, 465–470.
Murray, I. A., and Shaw, W. V. (1997). O-Acetyl-transferases for chloramphenicol and other natural products. Antimicrob. Agents Chemother. 41, 1–6.
Nair, J., Rouse, D. A., Bai, G.-H., and Morris, S. L. (1993). The rpsL gene and streptomycin resistance in single and multiple drug-resistant strains of Mycobacterium tuberculosis. Mol. Microbiol. 10, 521–527.
NethMap. (2008). Consumption of Antimicrobial Agents and Antimicrobial Resistance Among Medically Important Bacteria in the Netherlands. Bilthoven: RIVM. Available at: www.swab.nl.
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304.
Oliva, B., and Chopra, I. (1992). Tet determinants provide poor protection against some tetracyclines: further evidence for division of tetracyclines into two classes. Antimicrob. Agents Chemother. 36, 876–878.
Olson, A. B., Silverman, M., Boyd, D. A., McGeer, A., Willey, B. M., Pong-Porter, V., Daneman, N., and Mulvey, M. R. (2005). Identification of a progenitor of the CTX-M-9 group of extended-spectrum β-lactamases from Kluyvera georgiana isolated in Guyana. Antimicrob. Agents Chemother. 49, 2112–2115.
Palmer, K. L., Kos, V. N., and Gilmore, M. S. (2010). Horizontal gene transfer and the genomics of enterococcal antibiotic resistance. Curr. Opin. Microbiol. 13, 632–639.
Parenti, F., Beretta, G., Berti, M., and Arioli, V. (1978). Teichomycins, new antibiotics from Actinoplanes teichomyceticus Nov. Sp. I. Description of the producer strain, fermentation studies and biological properties. J. Antibiot. 31, 276–283.
Park, C. H., Robicsek, A., Jacoby, G. A., Sahm, D., and Hooper, D. C. (2006). Prevalence in the United States of aac(6′)-Ib-cr encoding a ciprofloxacin-modifying enzyme. Antimicrob. Agents Chemother. 50, 3953–3955.
Partridge, S. R., and Hall, R. M. (2005). Correctly identifying the streptothricin resistance gene cassette. J. Clin. Microbiol. 43, 4298–4300.
Partridge, S. R., Tsafnat, G., Coiera, E., and Iredell, J. R. (2009). Gene cassettes and cassette arrays in mobile resistance integrons. FEMS Microbiol. Rev. 33, 757–784.
Paterson, D. L., and Bonomo, R. A. (2005). Extended-spectrum β-Lactamases: a clinical update. Clin. Microbiol. Rev. 18, 657–686.
Périchon, B., Courvalin, P., and Galimand, M. (2007). Transferable resistance to aminoglycosides by methylation of G1405 in 16S rRNA and to hydrophilic fluoroquinolones by QepA-mediated efflux in Escherichia coli. Antimicrob. Agents Chemother. 51, 2464–2469.
Perreten, V., and Boerlin, P. (2003). A new sulfonamide resistance gene (sul3) in Escherichia coli is widespread in the pig population of Switzerland. Antimicrob. Agents Chemother. 47, 1169–1172.
Petri, W. A. (2006). “Penicillins, cephalosporins, and other β-lactam antibiotics,” in Goodman & Gilman’s, The Pharmacologic Basis of Therapeutics, eds L. L. Brunton, J. S. Lazo, and K. L. Parker (New York: The McGraw-Hill Companies), 1127–1154.
Poehlsgaard, J., and Douthwaite, S. (2005). The bacterial ribosome as a target for antibiotics. Nat. Rev. Microbiol. 3, 870–881.
Poirel, L., Naas, T., and Nordmann, P. (2010). Diversity, epidemiology, and genetics of class D β-lactamases. Antimicrob. Agents Chemother. 54, 24–38.
Poirel, L., Pitoud, J. D., and Nordmann, P. (2007). Carbapenemases: molecular diversity and clinical consequences. Food Microbiol. 2, 501–512.
Queenan, A. M., and Bush, K. (2007). Carbapenemases: the versatile β-lactamases. Clin. Microbiol. Rev. 20, 440–458.
Rådström, P., and Swedberg, G. (1988). RSF1010 and a conjugative plasmid contain sulII, one of two known genes for plasmid-borne sulfonamide resistance dihydropteroate synthase. Antimicrob. Agents Chemother. 32, 1684–1692.
Ramirez, M. S., and Tolmansky, M. E. (2010). Aminoglycoside modifying enzymes. Drug Resist. Updat. 13, 151–171.
Rasmussen, B., Noller, H. F., Daubresse, G., Oliva, B., Misulovin, Z., Rothstein, D. M., Ellestad, G. A., Gluzman, Y., Tally, F. P., and Chopra, I. (1991). Molecular basis of tetracycline action: identification of analogs whose primary target is not the bacterial ribosome. Antimicrob. Agents Chemother. 35, 2306–2311.
Rechia, G. D., and Hall, R. M. (1995). Gene cassettes: a new class of mobile element. Microbiology 141, 3015–3027.
Rechia, G. D., and Hall, R. M. (1997). Origins of the mobile gene cassettes found in integrons. Trends Microbiol. 5, 389–394.
Riley, M. A., and Wertz, J. E. (2002). Bacteriocins: evolution, ecology and application. Annu. Rev. Microbiol. 56, 117–137.
Roberts, A. P., Chandler, M., Courvalin, P., Guédon, G., Mullany, P., Pembroke, T., Rood, J. I., Jeffery Smith, C., Summers, A. O., Tsuda, M., and Berg, D. E. (2008). Revised nomenclature for transposable genetic elements. Plasmid 60, 167–173.
Roberts, A. P., and Mullany, P. (2009). A modular master on the move: the Tn916 family of mobile genetic elements. Trends Microbiol. 6, 251–258.
Roberts, M. C. (1996). Tetracycline resistance determinants: mechanisms of action, regulation of expression, genetic mobility, and distribution. FEMS Microbiol. Rev. 19, 1–24.
Roberts, M. C. (2002). Resistance to tetracycline, macrolide-lincosamide-streptogramin, trimethoprim, and sulfonamide drug classes. Mol. Biotechnol. 20, 261–284.
Roberts, M. C. (2005). Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 245, 195–203.
Roberts, M. C. (2008). Update on macrolide-lincosamide-streptogramin, ketolide, and oxazolidinone resistance genes. FEMS Microbiol. Lett. 282, 147–159.
Roberts, M. C., Sutcliffe, J., Courvalin, P., Jensen, L. B., Rood, J., and Seppala, H. (1999). Nomenclature for macrolide and macrolide-lincosamide streptogramin B antibiotic resistance determinants. Antimicrob. Agents Chemother. 43, 2823–2830.
Robicsek, A., Jacoby, G. A., and Hooper, D. C. (2006a). The worldwide emergence of plasmid-mediated quinolone resistance. Lancet Infect. Dis. 6, 629–640.
Robicsek, A., Strahilevitz, J., Jacoby, G. A., Macielag, M., Abbanat, D., Park, C. H., Bush, K., and Hooper, D. C. (2006b). Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat. Med. 12, 83–88.
Roth, B., Falco, E. A., Hitchings, G. H., and Bushby, S. R. M. (1962). 5-Benzyl-2,4-diaminopyrimidines as anti-bacterial agents. I. Synthesis and antibacterial activity in vitro. J. Med. Pharm. Chem. 5, 1103–1123.
Ruiz, J. (2003). Mechanisms of resistance to quinolones: target alterations, decreased accumulation and DNA gyrase protection. J. Antimicrob. Chemother. 51, 1109–1117.
Saladin, M., Cao, V. T. B., Lambert, T., Donay, J.-L., Herrmann, J.-L., Ould-Hocine, Z., Verdet, C., Delisle, F., Philippon, A., and Arlet, G. (2002). Diversity of CTX-M β-lactamases and their promoter regions from Enterobacteriaceae isolated in three Parisian hospitals. FEMS Microbiol. Lett. 209, 161–168.
Schatz, A., and Waksman, S. A. (1944). Effect of streptomycin and other antibiotic substances upon Mycobacterium tuberculosis and related organisms. Proc. Soc. Exp. Biol. Med. 57, 244–248.
Schwarz, S., Kehrenberg, C., Doublet, B., and Cloeckaert, A. (2004). Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev. 28, 519–542.
Shaw, K. J., Rather, P. N., Hare, R. S., and Miller, G. H. (1993). Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 57, 138–163.
Siegenthaler, W. E., Bonetti, A., and Luthy, R. (1986). Aminoglycoside antibiotics in infectious diseases. An overview. Am. J. Med. 80, 2–14.
Siguier, P., Filée, J., and Chandler, M. (2006). Insertion sequences in prokaryotic genomes. Curr. Opin. Microbiol. 9, 526–531.
Smillie, C., Garcillán-Barcia, M. P., Francia, M. V., Rocha, E. P. C., and de la Cruz, F. (2010). Mobility of plasmids. Microbiol. Mol. Biol. Rev. 74, 434–452.
Smith, H. O., Gwinn, M. L., and Salzberg, S. L. (1999). DNA uptake signal sequences in naturally transformable bacteria. Res. Microbiol. 150, 603–616.
Speer, B. S., Shoemaker, N. B., and Salyers, A. A. (1992). Bacterial resistance to tetracycline: mechanisms, transfer, and clinical significance. Clin. Microbiol. Rev. 5, 387–399.
Spratt, B. G. (1994). Resistance to antibiotics mediated by target alterations. Science 264, 388–393.
Stanton, T. B., Humphrey, S. B., Scott, K. P., and Flint, H. J. (2005). Hybrid tet genes and tet gene nomenclature: request for opinion. Antimicrob. Agents Chemother. 49, 1265–1266.
Stokes, H. W., and Hall, R. M. (1989). A novel family of potentially mobile DNA elements encoding site-specific gene-integration functions: integrons. Mol. Microbiol. 3, 1669–1683.
Strahilevitz, J., Jacoby, G. A., Hooper, D. C., and Robicsek, A. (2009). Plasmid-mediated quinolone resistance: a multifaceted threat. Clin. Microbiol. Rev. 22, 664–689.
Sun, H. I., Jeong, D. U., Lee, J. H., Wu, X., Park, K. S., Lee, J. J., Jeong, B. C., and Lee, S. H. (2010). A novel family (QnrAS) of plasmid-mediated quinolone resistance determinant. Int. J. Antimicrob. Agents 36, 573–580.
Sundström, L., Rådström, P., Swedberg, G., and Sköld, O. (1988). Site-specific recombination promotes linkage between trimethoprim- and sulfonamide-resistance genes. Sequence characterization of dhfrV and sulI and a recombination active locus of Tn21. Mol. Gen. Genet. 213, 191–201.
Swedberg, G., and Sköld, O. (1983). Plasmid-borne sulfonamide resistance determinants studied by restriction enzyme analysis. J. Bacteriol. 153, 1228–1237.
Taber, H. W., Mueller, J. P., Miller, P. F., and Arrow, A. S. (1987). Bacterial uptake of aminoglycoside antibiotics. Microbiol. Rev. 51, 439–457.
Torpdahl, M., Hammerum, A. M., Zachariasen, C., and Nielsen, E. M. (2009). Detection of qnr genes in Salmonella isolated from humans in Denmark. J. Antimicrob. Chemother. 63, 406–408.
Tschäpe, H., Tietze, E., Prager, R., Voigt, W., Wolter, E., and Seltmann, G. (1984). Plasmid-borne streptothricin resistance in gram-negative bacteria. Plasmid 12, 189–196.
Uttley, A. H. C., Collins, C. H., Naidoo, J., and George, R. C. (1988). Vancomycin-resistant enterococci. Lancet 331, 57–58.
Vakulenko, S. B., and Mobashery, S. (2003). Versatility of aminoglycosides and prospects for their future. Clin. Microbiol. Rev. 16, 430–450.
van Hoek, A. H. A. M., Mayrhofer, S., Domig, K. J., Flóres, A. B., Ammor, M. S., Mayo, B., and Aarts, H. J. M. (2008). Mosaic tetracycline resistance genes and their flanking regions in Bifidobacterium thermophilum and Lactobacillus johnsonii. Antimicrob. Agents Chemother. 52, 248–252.
Vester, B., and Douthwaite, S. (2001). Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob. Agents Chemother. 45, 1–12.
Waksman, S. A., and Woodruff, H. B. (1942). Strepto-thricin, a new selective bacteriostatic and bactericidal agent, primarily active against Gram-negative bacteria. Proc. Soc. Exp. Biol. Med. 49, 207–210.
Watanabe, T. (1963). Infective heredity of multiple drug resistance in bacteria. Bacteriol. Rev. 27, 87–115.
Wei, Q., Jiang, X., Yang, Z., Chen, N., Chen, X., Li, G., and Lu, Y. (2009). dfrA27, a new integron-associated trimethoprim resistance gene from Escherichia coli. J. Antimicrob. Chemother. 63, 405–406.
Weisblum, B. (1995). Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother. 39, 577–585.
Weldhagen, G. F., Kim, B., Cho, C.-H., and Lee, S. H. (2006). Definitive nomenclature of GES/IBC-type extended-spectrum β-lactamases. J. Microbiol. Biotechnol. 16, 1837–1840.
Willi, K., Sandmeier, H., Kulik, E. M., and Meyer, J. (1997). Transduction of antibiotic resistance markers among Actinobacillus actinomycetemcomitans strains by temperate bacteriophages Aaφ23. Cell. Mol. Life Sci. 53, 904–910.
Williams, J. D. (1987). “Classification of cephalosporins,” in The Cephalosporins, ed. J. D. Williams (Auckland: ADIS Press Ltd.), 15–22.
Wolfson, J. S., and Hooper, D. C. (1989). Fluoroquinolone antimicrobial agents. Clin. Microbiol. Rev. 2, 378–424.
Woodford, N., Johnson, A. P., Morrison, D., and Speller, D. C. E. (1995). Current perspectives on glycopeptide resistance. Clin. Microbiol. Rev. 8, 585–615.
Wozniak, R. A. F., and Waldor, M. K. (2010). Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 8, 552–563.
Wright, G. D. (2005). Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Deliv. Rev. 57, 1451–1470.
Wright, G. D., and Thompson, P. R. (1999). Aminoglycoside phosphotransferases: proteins, structure, and mechanism. Front. Biosci. 4, 9–21.
Wu, H. Y., Miller, G. H., Guzmán Blanco, M., Hare, R. S., and Shaw, K. J. (1997). Cloning and characterization of an aminoglycoside 6′-N-acetyltransferase gene from Citrobacter freundii which confers an altered resistance profile. Antimicrob. Agents Chemother. 41, 2439–2447.
Yamane, K., Wachino, J. I., Suzuki, S., Kimura, K., Shibata, N., Kato, H., Shibayama, K., Konda, T., and Arakawa, Y. (2007). New plasmid-mediated fluoroquinolone efflux pump, QepA, found in an Escherichia coli clinical isolate. Antimicrob. Agents Chemother. 51, 3354–3360.
Yao, R. C., and Crandall, L. W. (1994). “Glycopeptides: classification, occurrence and discovery,” in Glycopeptide Atibiotics, ed. R. Nagarajan (New York, NY: Taylor & Francis Group), 1–28.
Keywords: antimicrobial resistance mechanisms, acquired, antibiotics, mobile genetic elements
Citation: van Hoek AHAM, Mevius D, Guerra B, Mullany P, Roberts AP and Aarts HJM (2011) Acquired antibiotic resistance genes: an overview. Front. Microbio. 2:203. doi: 10.3389/fmicb.2011.00203
Received: 01 July 2011;
Accepted: 08 September 2011;
Published online: 28 September 2011.
Edited by:
Timothy Rutland Walsh, Cardiff University, UKReviewed by:
M. Pilar Francino, Center for Public Health Research, SpainJun Liu, Mount Sinai School of Medicine, USA
Copyright: © 2011 van Hoek, Mevius, Guerra, Mullany, Roberts and Aarts. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Henk J. M. Aarts, National Institute of Public Health and the Environment, Antonie van Leeuwenhoekla 9, 3721 MA Bilthoven, Utrecht, Netherlands. e-mail: henk.aarts@rivm.nl