
- Department I Protein Evolution, Max Planck Institute for Developmental Biology, Tübingen, Germany
The subcellular localization (SCL) of proteins provides important clues to their function in a cell. In our efforts to predict useful vaccine targets against Gram-negative bacteria, we noticed that misannotated start codons frequently lead to wrongly assigned SCLs. This and other problems in SCL prediction, such as the relatively high false-positive and false-negative rates of some tools, can be avoided by applying multiple prediction tools to groups of homologous proteins. Here we present ClubSub-P, an online database that combines existing SCL prediction tools into a consensus pipeline from more than 600 proteomes of fully sequenced microorganisms. On top of the consensus prediction at the level of single sequences, the tool uses clusters of homologous proteins from Gram-negative bacteria and from Archaea to eliminate false-positive and false-negative predictions. ClubSub-P can assign the SCL of proteins from Gram-negative bacteria and Archaea with high precision. The database is searchable, and can easily be expanded using either new bacterial genomes or new prediction tools as they become available. This will further improve the performance of the SCL prediction, as well as the detection of misannotated start codons and other annotation errors. ClubSub-P is available online at http://toolkit.tuebingen.mpg.de/clubsubp/
Introduction
Gram-negative bacteria have a multi-layered cell envelope, which consists of a symmetrical phospholipid bilayer (the cytoplasmic or inner membrane, IM) and an asymmetrical bilayer comprised of phospholipids and lipopolysaccharides (the outer membrane, OM). These membranes are separated by the periplasmic space, which contains a thin peptidoglycan layer as a cell wall (Gardy and Brinkman, 2006; Bos et al., 2007). The IM is the boundary for the cytosol; thus the Gram-negative cell consists of four compartments (cytosol, IM, periplasm, OM). Each subcellular compartment contains a defined set of proteins to fulfill distinct tasks.
To perform their functions at their native subcellular localization (SCL), newly synthesized proteins must be sorted and transported to their respective subcellular compartments. While most of the newly synthesized proteins remain in the cytoplasm, other proteins are inserted into the cytoplasmic membrane via the signal recognition particle (SRP) and YidC pathways. Proteins are targeted to the cytoplasmic membrane via the SRP pathway. YidC acts like an additional insertase to fold and assemble a defined subset of these proteins in the cytoplasmic membrane (Luirink et al., 2005). Proteins with native functions in the periplasmic space and in the OM are secreted across the cytoplasmic membrane into the periplasmic space by the Sec, TAT, or Holin (which secretes autolytic enzymes during cell death; Saier et al., 2008) secretory pathways. From the periplasm some proteins are further translocated to the OM or across the OM via Type II secretion systems (T2SS), T5SS, T7SS, and T8SS. Secretion systems such as T1SS, T3SS, T4SS, and T6SS span both membranes and can secrete proteins from the cytoplasm directly into the extracellular space or even into the host cytoplasm (Desvaux et al., 2009).
The general secretion system (Sec; Desvaux et al., 2009) is the most common pathway; it is conserved in all living organisms. In Gram-negative bacteria, it translocates unfolded proteins across the cytoplasmic membrane into the periplasmic space. The Sec translocon recognizes signal sequences present at the N-terminus of its substrate proteins. These general Sec signals are highly conserved and consist of a positively charged N-terminal region (n-region), a hydrophobic central region (h-region), and a polar C-terminal region (c-region; Nielsen et al., 1997). Alternatively, some folded proteins use the twin-arginine translocation (TAT) pathway for secretion across the cytoplasmic membrane, which recognizes its substrates through a modified general signal peptide with an additional RRXFL motif found between the n-region and h-region (Bendtsen et al., 2005). Typically, TAT signal peptides are longer than general signal peptides. The secretion of lipoproteins is accomplished by another modification of the general Sec signal peptide pathway. Here a cysteine residue follows immediately after the signal peptide cleavage site; this signal peptide is recognized and cleaved by lipoprotein signal peptidase (SPaseII or Lsp) after the N-terminal cysteine is modified with a lipid moiety, which anchors the protein to the membrane. Finally, an additional fatty acid is attached to the new N-terminus (Juncker et al., 2003). These proteins are then either retained at the cytoplasmic membrane or translocated to the OM by the Lol lipoprotein-sorting pathway (Lewenza et al., 2008). Although this sorting is assumed to be based on the residue at the +2 position after the cleavage site (Seydel et al., 1999), it has been shown that residues at +3 and +4 also play important roles in the sorting of these proteins in Pseudomonas aeruginosa (Lewenza et al., 2008). So far, the detailed patterns of lipoprotein-sorting remain unclear. A number of specialized secretion systems exist, each one typically translocating only a small subset of proteins.
The SCL of proteins provides important clues to their function in the cell. Determining the SCL of proteins by experimental means is accurate but time-consuming and expensive. As a result of new and more efficient sequencing technologies, the number of newly deposited sequences is increasing exponentially, while the number of proteins annotated with experimentally verified SCL stagnates. Thus, computational SCL prediction is important and has become indispensible in protein research, e.g., for genome-wide SCL studies. There are two types of SCL prediction tools. One type is predicting only the features specific to localizations, such as signal peptides (Nielsen et al., 1997; Rose et al., 2002; Juncker et al., 2003; Bendtsen et al., 2004, 2005; Hiller et al., 2004; Käll et al., 2004; Bos et al., 2007; Szabó et al., 2007; Arnold et al., 2009; Bagos et al., 2009; Löwer and Schneider, 2009), transmembrane helices (TMHs; Krogh et al., 2001; Tusnady and Simon, 2001; Käll et al., 2004), or transmembrane β-barrels (TMBBs; Berven et al., 2004; Remmert et al., 2009). The other type is predicting the exact localization of a protein by combining various localization-specific features (Su et al., 2007; Yu et al., 2010) or general features like amino acid composition (Yu et al., 2006), evolutionary information (Rashid et al., 2007), structure conservation information (Su et al., 2007), and gene ontology (Chou and Shen, 2006b).
It has been shown that the combination of different SCL prediction tools increases the quality of the overall prediction significantly (Shen and Burger, 2007; Horler et al., 2009; Giombini et al., 2010; Goudenège et al., 2010). Moreover, Imai and Nakai (2010) recently reported that homology-based methods perform better even on datasets with a low overall sequence identity cutoff, when compared to state-of-the-art single-sequence SCL predictors. Mah et al. (2010) used clustering information to optimize OM β-barrel protein predictions in seven proteomes of Mycobacteria.
Our interest is predominantly in surface-localized proteins of Gram-negative bacteria that could be exploited for vaccine development. We found most single SCL prediction methods to be either not useful or not sensitive enough for our bioinformatics pipeline. Moreover, we found many proteins with misannotated start codons. These are easily identified from the multiple sequence alignments of homologous proteins but are hard to find on the level of individual sequences. The differences in start codon predictions between orthologous sequences from closely related organisms are typically a result of using different automated gene prediction methods while annotating the sequenced genome (Overbeek et al., 2007). These misannotations are a common source of error in SCL prediction, especially since feature prediction tools based on N-terminal signal peptides depend essentially on accurate annotations of the translation start. Conversely, the TMBB prediction tool BOMP uses a C-terminal β-barrel motif for its predictions and thus relies on correctly sequenced stop codons (Berven et al., 2004).
In this work, we developed a method called cluster-based SCL prediction, or ClubSub-P, which combines different localization-specific features and SCL prediction tools, using rules based on the biology of protein sorting to annotate the SCL for Gram-negative bacterial proteins. In contrast to other general SCL prediction tools, it uses homology information taken from clusters of orthologous proteins from different species to further increase the confidence of the prediction. Since we use information from the whole cluster to increase the confidence, we overcome the problem of misannotation of start codons and thus increase the specificity of the method further. Performance measurements with ClubSub-P show that the additional use of homology information from simple clustering increases the precision of our tool over other state-of-the-art SCL prediction tools. Our tool relies on an expandable database. The constantly increasing number of sequenced genomes will, over time, allow us to cluster more sequences, which will further increase the quality of homology detection and thus, the precision of our predictions. To show how easily the tool can be expanded to whole new organism groups, we have included an additional module for the SCL prediction of archaeal proteins.
Materials and Methods
Datasets
To create the ClubSub-P database (see Database, below), 607 Gram-negative bacterial proteomes (2,331,935 sequences) were downloaded from the NCBI RefSeq genome database1 in July 2011. A non-redundant dataset was created using CD-HIT (Li and Godzik, 2006) from the above sequences at 40% local sequence identity, and at 80% sequence alignment coverage to the longest sequence in the cluster. The “accurate and slow” mode was used to ensure clustering of proteins into the most similar cluster, which is not given when using the fast mode. Shorter sequences (<40 amino acids) were removed from the dataset for two reasons. First, such short proteins are only annotated in very few bacterial genomes and frequently do not show significant homology to proteins with experimentally verified SCL (Warren et al., 2010). Second, even when there is available experimental data, small proteins are frequently considered fragments and are removed from datasets of many SCL prediction tools (Chou and Shen, 2006a), making a consensus prediction impossible. The final dataset, which we named DB_ClubSub-P, contained 1,911,760 proteins. The list of the downloaded proteomes and the accession numbers of the replicons are given in Data Sheet S1 in Supplementary Material.
We used the Gram-negative bacterial protein sequences from the training dataset of PSORTb v3.0.22 (Yu et al., 2010) to test the clustering parameters. This dataset contains 8,227 protein sequences with experimentally determined SCLs and we named it DB_ePSORT.
To obtain a test set for the evaluation of the performance of ClubSub-P, Gram-negative bacterial protein sequences with experimentally verified SCL annotation were extracted from UniProt Release 2011_07 (UniProt-Consortium, 2010). We wrote a parser to extract Gram-negative bacterial protein sequences with literature reference to their SCL annotations, but ignoring sequences with “potential,” “by similarity,” or “probable” annotations, sequences labeled as “Fragment,” or sequences with “chromatophore” localization.
Sequences with ≤40 aa length were removed from this dataset; we also removed sequences which have more than 40% sequence identity to the PSORTb v.3 training dataset to allow an objective comparison between the tools. Likewise, since the SCL tools used in performance measure do not separately annotate lipoproteins, we removed sequences with “lipid anchor” SCL annotation which leaves 171 sequences for our DB_Experimental dataset.
Subcellular Localization Prediction
Subcellular localization prediction using the DB_ClubSub-P dataset was done on two levels. First, we combined different prediction tools as listed in Table 1 for localization-specific features, and SCL prediction tools based on known biological rules as shown in Table 2, to annotate the SCL of each single protein in the DB_ClubSub-P dataset. Figure 1 displays the procedure in form of flow chart. Second, we clustered all protein sequences and combined their SCL annotations into a consensus SCL prediction for each protein cluster.
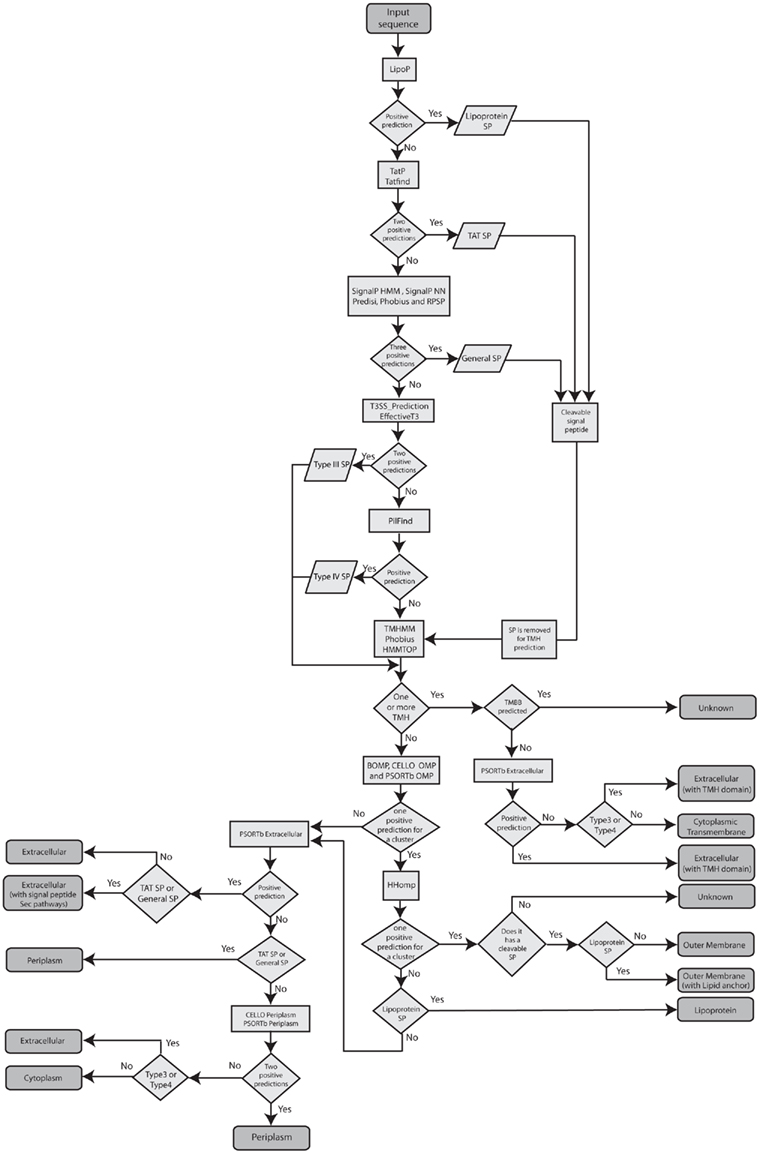
Figure 1. Flowchart for SCL prediction at the level of single proteins.
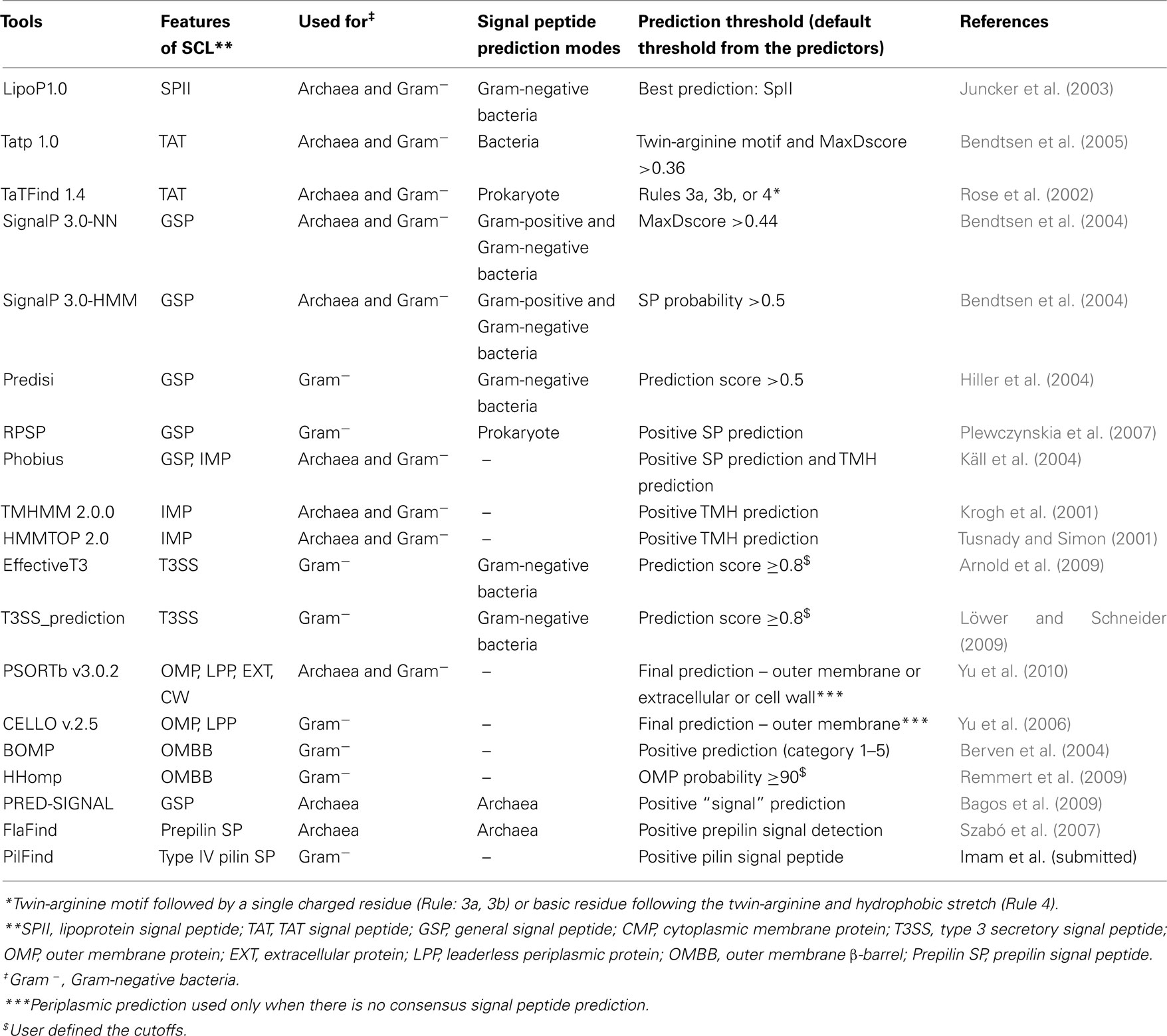
Table 1. List of SCL and feature specific tools used in the prediction pipeline.
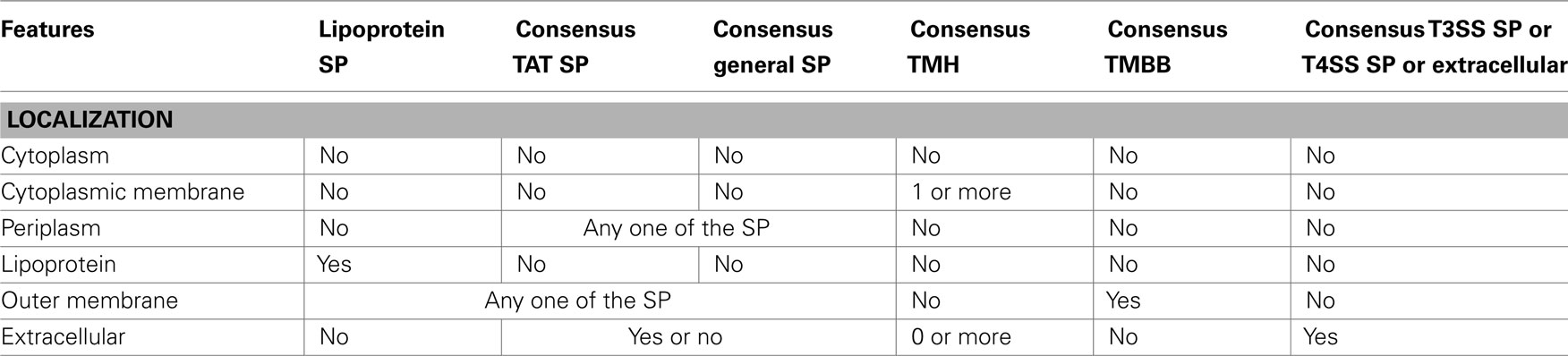
Table 2. Logic for SCL prediction at the protein level.
Consensus subcellular localization at the protein level
Consensus signal peptide prediction. Signal peptide predictions for Lipoprotein signals, TAT pathway signal peptides, general secretory signal peptides, T3SS signal peptides and T4SS signal peptides were done on all proteins in the DB_ClubSub-P dataset. A lipoprotein prediction was considered positive when the best prediction of LipoP 1.0 (Juncker et al., 2003) was for a signal peptidase II cleavage site. For TAT pathway signal peptide prediction in ClubSub-P, both TatP 1.0 (Bendtsen et al., 2005) and the rule-based predictor TatFind 1.4 (Rose et al., 2002) had to be positive; the cutoff for a positive TatP 1.0 prediction was a MaxD score above 0.36, while TatFind 1.4 requires the presence of the twin-arginine motif and additional sequence features.
Five tools were combined for the consensus prediction of general signal peptides: SignalP-HMM (with a default cutoff of p = 0.5), SignalP-NN (with MaxD value above 0.44), Predisi (with a default cutoff of p = 0.5), RPSP (with positive signal peptide), or Phobius (with positive signal peptide prediction; Bendtsen et al., 2004; Hiller et al., 2004; Käll et al., 2004). For a positive prediction, three out of five tools were required to be positive; in this case, a consensus SP cleavage site was predicted from the individual cleavage site predictions. Here, Phobius was also used to differentiate between the SP and TMH predictions (see below). If only two tools predict the presence of a signal peptide with zero or one consensus TMHs, the protein’s SCL is annotated as “Unknown” to avoid false-positive predictions.
To reduce the false-positive prediction rate of type III signal peptide prediction, positive predictions from both EffectiveT3 (Arnold et al., 2009) and T3SS_prediction (Löwer and Schneider, 2009) were required; predictions with scores ≥0.8 were considered as positive type III signal peptides (Burstein et al., 2009). We used a new, unpublished tool named PilFind (Imam et al., submitted) to predict type IV secretion system (T4SS) signals.
If one or more SP were predicted for a protein, it was classified based on the hierarchy described above (Figure 1), since there are cases where Lipoprotein or TAT SPs are also predicted as general SPs by general SP prediction tools, and taking into consideration that the accuracy of T3SS and T4SS SP prediction tools is still insufficient.
Consensus transmembrane helix prediction. TMHMM (Krogh et al., 2001), HMMTOP (Tusnady and Simon, 2001), and Phobius (Käll et al., 2004) were used for the prediction of TMHs. For the consensus TMH prediction, we ruled that a helix must be predicted independently by at least 2 of the tools used, over a length of at least 10 residues. Consensus TMH prediction was avoided over the length of previously predicted cleavable signal peptides, because signal peptides are known to be frequently misinterpreted as TMHs by TM prediction tools. The consensus TMH prediction is displayed in Figure 2.
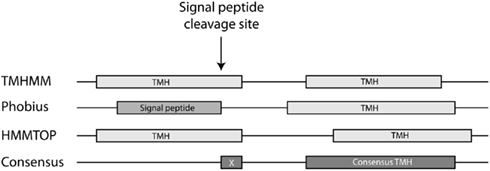
Figure 2. Consensus transmembrane helix prediction module. A consensus TMH should be predicted by at least two tools. Signal peptides frequently result in false-positive TMH predictions and are removed with consensus TMH predictions.
Consensus transmembrane β-barrel prediction. We used BOMP (Berven et al., 2004), CELLO (Yu et al., 2006), PSORTb (Yu et al., 2010), and HHomp (Remmert et al., 2009) to predict outer membrane proteins (OMPs). Since classifier-based predictions are faster than sensitive search methods such as HHomp, only BOMP, CELLO, and PSORTb were ran on all the sequences. If any one of BOMP, PSORTb, or CELLO had a positive prediction for OMPs in a cluster (see Subcellular Localization on the Level of Sequence Clusters for details on clustering), we selected a random sequence from the cluster and ran HHomp. When the sequence was predicted as OMP with probability above 90%, we annotated all the sequences in the cluster as OM-localized TMBBs.
Consensus subcellular localization prediction. For the consensus SCL prediction we applied rules based on the biology of protein sorting along with the previously predicted protein features as mentioned in the Table 2. The lipoprotein-sorting signal is based on the amino acids after the SPII cleavage site and species-specific (Juncker et al., 2003). Currently there is not sufficient experimental data to postulate a common sorting pattern for all species. Thus, we annotated proteins with lipoprotein signal peptides and without TMHs as “IM/OM lipoprotein.” Also, as there is insufficient experimental data available to annotate the extracellular presence of lipoproteins, we didn’t analyze the further destination of lipoproteins (Pugsley et al., 1990). Proteins featuring general Sec or TAT signal peptides and without TMHs and TMBBs were annotated as “periplasmic.” Proteins predicted to be periplasmic by PSORTb v3.0.2 (Yu et al., 2010) and CELLO v.2.5 (Yu et al., 2006) but without any signal peptide, TMHs and TMBBs were also predicted as periplasmic. Additionally, they were tagged with a note stating that they could be secreted via signal peptide-independent pathways (leaderless pathways). Proteins with one or more consensus TMHs were annotated as “cytoplasmic membrane.” Proteins with consensus TMBB prediction containing one of the previously predicted cleaved general, TAT, or lipoprotein signal peptides were annotated as “outer membrane protein,” as OMPs are typically secreted by SP-dependent pathways. The SCL of proteins with positive TMBB predictions, but without any signal peptide predictions were annotated as “Unknown”. Proteins predicted to be extracellular by PSORTb or predicted to have a T3SS or T4SS signal peptide were annotated as “extracellular.” Proteins without TMHs, TMBBs, signal peptide, or extracellular prediction were annotated as “cytoplasmic.”
Subcellular localization on the level of sequence clusters
To add homology information to single-sequence results in order to improve the overall prediction quality, all protein sequences from the DB_ClubSub-P dataset were clustered using CD-HIT (Li and Godzik, 2006); the clustering parameters are given in the Section “Datasets,” above.
Since we cannot infer homology from singletons, we skipped 291,727 singletons and used the remaining 1,620,033 sequences, which resulted in 174,028 clusters with sequence numbers ranging from 2 to 1,667. If a fraction of 0.7 or above of all proteins in the cluster have the same given SCL (i.e., 70% or more), this SCL is considered the SCL of the respective cluster. Clusters where no single SCL amounts to a protein fraction ≥0.7 (including “unknown”) were annotated as “uncertain,” and details of the predictions are kept available in the database for expert users to study further. Note that “uncertain” clusters are different from “unknown” clusters, as in the “unknown” ones most of the sequences show contradictory predictions to the rules described in the above section. Dual localization annotations were allowed only when two SCLs amounted to a fraction ≥0.7. The cutoff of 0.7 was chosen because any higher cutoff value leads to a steep increase in the number of “uncertain” clusters (see Figure 3).
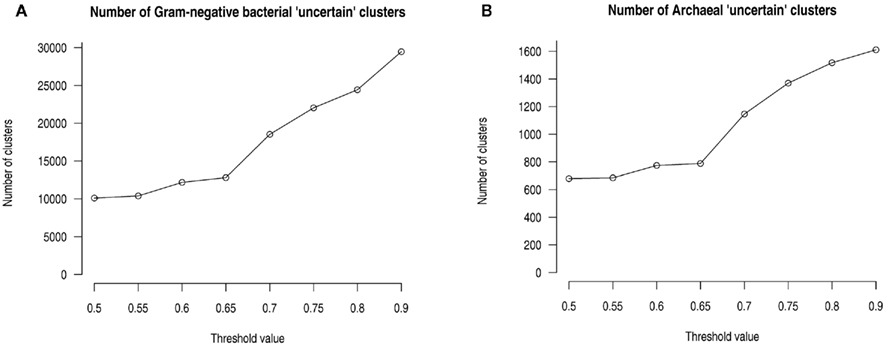
Figure 3. Threshold determination for the assignment of subcellular localizations to whole clusters. (A) For Gram-negative bacteria; (B) for Archaea. In both cases, the number of clusters annotated as “uncertain” (i.e., with no SCL prediction above the threshold) increases at 0.7 – to minimize the number of “uncertain” clusters, cluster SCLs were assigned when a fraction of 0.7 or above (=70%) of the proteins in a cluster are predicted to have a given SCL.
Subcellular Localization Prediction for Archaea
We created a similar protocol to expand ClubSub-P to archaeal proteins. To this end, we used proteins from 65 archaeal proteomes (shown in Data Sheet S1 in Supplementary Material). After removing 779 small proteins with length 40 and below, we obtained 151,553 proteins for clustering using CD-HIT (Li and Godzik, 2006) with the same parameters as above. This resulted in 22,184 clusters with cluster size two and above. We named this dataset as DB_ClubSub-P_Archaea.
We used a similar parser to obtain a test dataset for Archaea. We obtained all the reviewed archaeal sequences without any “potential,” “by similarity,” or “probable” annotations in their SCL. We thus obtained 744 archaeal sequences with SCL annotation from UniProt Release 2011_07 (UniProt-Consortium, 2010). Sequences with ≤40 aa length were removed from the dataset and a non-redundant dataset with 40% sequence identity was created using CD-HIT (Li and Godzik, 2006), resulting in 252 sequences for the performance test. We named this dataset DB_experimental_Archaea.
For archaeal proteins, Lipoprotein, and TAT signal peptides were predicted using the same tools (LipoP, TatP, TatFind) as for Gram-negative bacterial proteins. For general signal peptide prediction, SignalP in Gram-positive mode was used, and Predisi was replaced by the tool PRED-SIGNAL (Bagos et al., 2009), which is an archaeal signal peptide prediction program. Phobius was used in default mode for the predictions. FlaFind (Szabó et al., 2007) was used to predict archaeal prepilin signal peptides; here, a TMH follows the signal peptide, and Prepilin peptidase cleaves the signal peptide before the TMH (Szabó et al., 2007). Thus, the protein is anchored to the membrane.
When two or more SPs were predicted, a consensus SP was annotated using a similar hierarchy as described in Figure 1, with the exception that there is no T3SS SP prediction for Archaea. Consensus TMH prediction was performed the same way as for Gram-negative bacteria. Archaeal proteins with TAT, general, or prepilin signal peptides or with PSORTb extracellular predictions (Yu et al., 2010) were annotated as “secreted/extracellular.” Proteins with lipoprotein SP were annotated as “lipoproteins.” “Cell wall” binding proteins were predicted using PSORTb’s cell wall predictions (Yu et al., 2010). Proteins with one or more consensus TMH prediction were annotated as “cytoplasmic membrane” proteins. Proteins without any membrane domains or signal peptides or cell wall annotations were annotated as “cytoplasmic” proteins. Table 3 explains the rules for SCL prediction for Archaea.
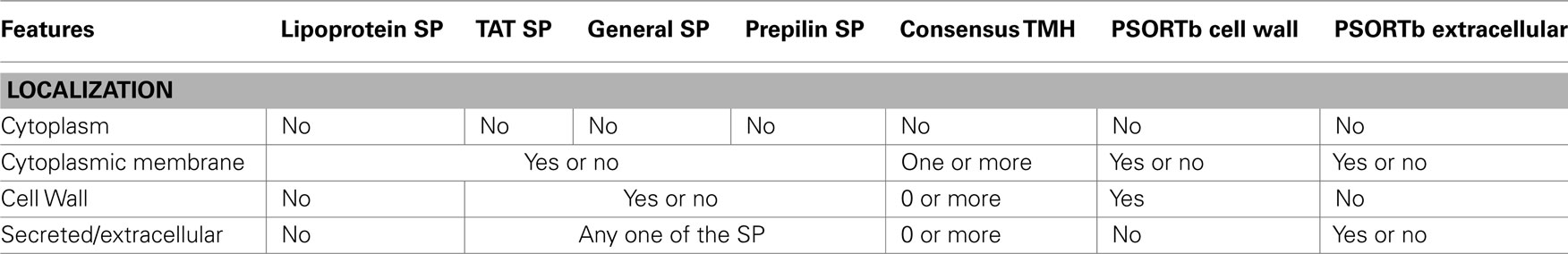
Table 3. Logical rules used for archaeal SCL predictions.
Database
We built a database from the above SCL annotations, which we named ClubSub-P, for “Cluster-based Subcellular localization Prediction.” Results and input features are stored in SQL tables. The database is integrated into the classification section of the MPI Bioinformatics Toolkit (Biegert et al., 2006). The database is fully searchable using keywords or GI identifiers; moreover, FASTA sequences can be entered and will be assigned to the appropriate cluster through an internal BLAST search at >75% sequence coverage and >40% identity cutoff.
Evaluation
We used the previously described DB_Experimental datasets to compare the performance of ClubSub-P with state-of-the-art SCL prediction tools. We calculated the precision, recall, accuracy, and the Mathew’s correlation coefficient (MCC) for performance measure. In the following equations TP stands for true positives, TN for true negatives, FP for false positives, and FN for false negatives.
Precision is a measure of the ability of the system to predict only the relevant data and it was calculated as the ratio between the number of predicted true positives against all positively predicted values, TP/(TP + FP).
Recall is a measure of the ability of the system to predict all the relevant data and was calculated as the ratio between the number of predicted true positives against all true values, TP/(TP + FN).
The accuracy of the system is defined by the closeness of its prediction toward the true values and was calculated by (TP + TN)/(TP + TN + FP + FN).
The MCC calculates the correlation between the prediction and the observation and was calculated by 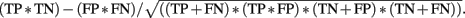
Results
Clustering Using the PSORTb V3 Gram-Negative Bacterial Training Dataset
As a first step, we had to make sure that the transfer of SCL information between homologous proteins is legitimate, and at which cutoffs for clustering (sequence identity and sequence coverage) this is still a valid procedure. To this end, we tested various clustering parameters using the 8,227 sequences in the DB_ePSORT dataset at decreasing cutoffs. To avoid problems with multi-domain proteins that might have different functions, and thus SCL, we decided to keep high sequence coverage. At 40% sequence identity and 80% sequence coverage, 6,136 sequences of the test set were clustered into 1,023 clusters with at least two sequences. 964 (94.2%) of these clusters had one common SCL for all of the proteins in the cluster. 47 (4.6%) of the clusters contained proteins with multiple SCL annotations which partially overlapped, and only 12 (1.2%) of the clusters had proteins with contradictory SCLs in them. Consequently, clustering done with the same parameters on the DB_ClubSub-P dataset can be expected to a have high number of clusters with homologous sequences that have a common SCL.
There are reports of orthologous proteins that have different SCLs in different organisms as a result of different evolutionary requirements. One prominent example is the glycerophosphoryl diester phosphodiesterase GlpQ, which is a periplasmic enzyme in E. coli, but is a surface-exposed lipoprotein in Haemophilus influenzae (Protein D; Janson et al., 1992). Such cases are rare, but they are easily missed when inferring their SCL from homology alone. In the case of Protein D/GlpQ, the two proteins are correctly predicted to have their respective – and different – SCL. Thus, one should always have a close look at the single-protein SCLs in cases where clustering leads to unclear or contradictory localization information. The ClubSub-P database allows for such manual inspection.
Cluster-Based Comparison of Signal Peptide and Transmembrane Prediction Tools
Applying a feature prediction tool such as a signal peptide predictor to sequences in a cluster of orthologous proteins should return similar results for all the proteins in the cluster (with very few but notable exceptions, see above). Inconsistency in such predictions will most probably be due to a lack of precision of the respective tool. Since different tools already use most of the proteins with experimentally verified SCL in their training sets, examining the performance of a tool at the cluster level is better suited to measure its sensitivity in a larger dataset, and to compare different tools.
Figure 4 shows the performance of different signal peptide prediction tools on our clusters produced from the DB_ClubSub-P dataset. We used only clusters with more than four sequences in this analysis; in detail, 66,716 clusters were included containing 1,341,180 sequences. If only <20% of the sequences in a cluster were positively predicted to contain a signal peptide, these predictions were assumed to be false positives; in a cluster with >80% of the sequences positively predicted, the remaining differing sequences were assumed to be false negatives.
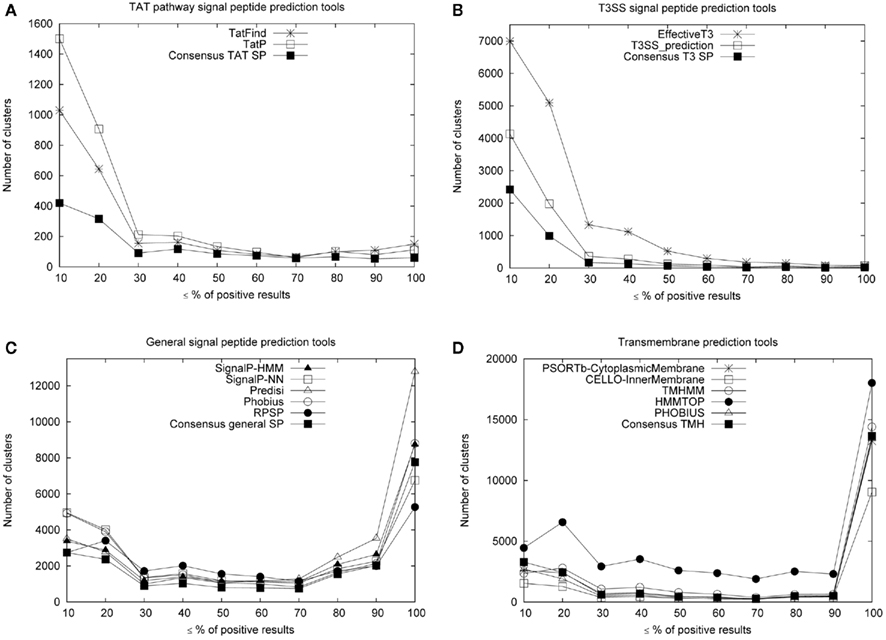
Figure 4. Cluster-based comparison of signal peptide prediction tools. Shown are the comparisons for TAT (A), T3SS (B), General signal peptide (C) and transmembrane (D) prediction tools. The x-axis describes how many sequences (in %) in a cluster are predicted to have a signal peptide, where 10% means 0.1–10%, 20 means 10.1–20% etc. The majority of clusters contains only sequences where no signal sequence is predicted (0% positive results) – for clarity, these are ignored in the graph.
Using these assumptions, we compared the TAT, type III, general signal peptide, and IM helix prediction tools along with their consensus predictions. Positive predictions from both TatP (Bendtsen et al., 2005) and TaTFind (Rose et al., 2002) tools were considered as a consensus TAT signal peptide. False-positive predictions were largely reduced by these consensus predictions (Figure 4A). This shows that most of the positive predictions in clusters with <20% positives are in fact false-positive predictions from the tools. A similar result can be seen with the consensus prediction for type III signal peptides (Figure 4B), where we considered positive predictions from both T3SS_prediction (Löwer and Schneider, 2009) and EffectiveT3 (Arnold et al., 2009) tools as a consensus type III signal peptide. For consensus general signal peptide prediction, we required at least three positive predictions from highly precise general signal peptide tools (Choo et al., 2009) like SignalP-HMM, SignalP-NN (Bendtsen et al., 2004), Predisi (Hiller et al., 2004), RPSP (Plewczynskia et al., 2007), and Phobius (Käll et al., 2004). Figure 4C shows the cluster-based comparison for general signal peptide tools and the consensus made from their prediction.
Similarly, we compared the performance of TMH prediction by CELLO v.2.5 (Yu et al., 2006), PSORTb v3.0.2 (Yu et al., 2010), Phobius (Käll et al., 2004), TMHMM 2.0 (Krogh et al., 2001), and HMMTOP v2.0 (Tusnady and Simon, 2001; Figure 4D), assuming that prediction of at least one TMH indicates that the protein is a transmembrane protein. The result clearly shows the high false-positive rate of HMMTOP predictions (Figure 4D), compared to the predictions of the other tools. However, the consensus TMH prediction of Phobius, TMHMM 2.0, and HMMTOP v2.0 (see Materials and Methods) eliminated most of these false-positive predictions.
Such comparisons of different prediction tools help in selecting the best tools for consensus predictions; alternatively, one could use this performance measure to weigh different tools, giving more importance to tools that performed better.
ClubSub-P Database Statistics
The core of the cluster-based SCL prediction is the ClubSub-P database. Of 2,331,935 retrieved sequences, 404,542 identical sequences and 15,633 sequences with less than 40 residues and were removed. The remaining 1,911,760 sequences were clustered using CD-HIT (Li and Godzik, 2006). We used 40% local sequence identity at 80% sequence coverage for clustering, When these settings were applied, 1,620,033 sequences (84.74%) were clustered into 174,028 clusters with size range from 2 to 1,677 and 291,727 proteins (15.26%) appeared to be singletons, meaning these sequences do not have any homolog among the sequences in the database at these settings. These singletons were not analyzed in detail, since no homology information can be inferred for them. Note though that with expansion of the database, these proteins might fall into newly formed clusters at a later time point as discussed below.
We were able to annotate the SCL of 1,500,778 of 1,620,033 sequences that are grouped in clusters of at least two sequences, which is 78.50% of the sequences used in clustering (1,911,760 sequences – note again that singletons, i.e., sequences that do not fall into clusters, are excluded from our predictions). For comparison, PSORTb v3.0.2 annotates 71.25% of all sequences used in our clustering approach (1,362,110 of 1,911,760 sequences). The details of the ClubSub-P prediction statistics for Gram-negative bacteria are shown in Table 4.
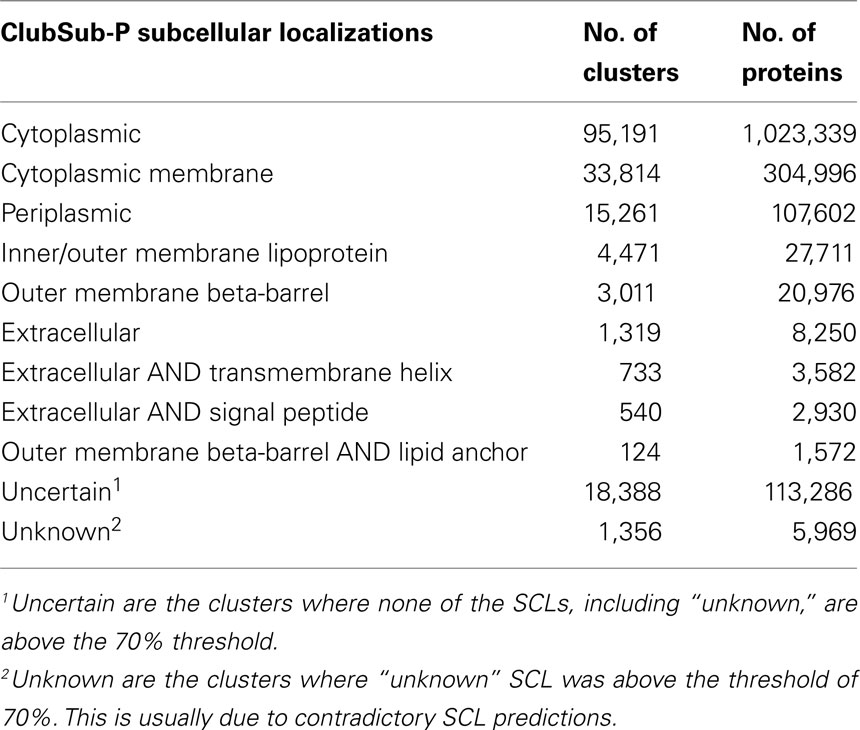
Table 4. Statistics of the ClubSub-P database.
Multiple Subcellular Localization Predictions
In addition to the common SCL classifications in Gram-negative bacteria, we found clusters of proteins with features that correspond to two different SCLs, e.g., “extracellular” proteins that have signal peptides for secretion to the “periplasm,” “extracellular” proteins with “TMHs” to get inserted into host membranes, and “OM β-barrel” proteins with a “lipoprotein” signal peptide. In many cases, experimental evidence for these double localizations exists, demonstrating that they are not artifacts of our SCL prediction pipeline. As an example, the “Pertussis toxin subunit 1” (UniProt ID – TOX1_BORPE/gi|33594638) is predicted by ClubSub-P to have an “extracellular” and a “periplasmic” localization; by experimental evidence (Farizo et al., 2002) it is an extracellular protein that is first secreted to the periplasm using the general signal peptide pathway, and only subsequently is secreted to the extracellular space. Moreover, the “Outer membrane protein oprM” (UniProt ID – OPRM_PSEAE/gi|116054158; Nakajima et al., 2000) has been shown experimentally to be attached to the OM via a lipid anchor, while it also spans the OM with a TMBB domain. ClubSub-P predicts OprM to be an OM β-barrel protein as well as a lipoprotein. A prominent example for “extracellular” and “transmembrane” localization are proteins secreted by pathogens to insert in to the host membrane, such as the needle tip components of the Type III secretion apparatus (Marlovits and Stebbins, 2010); and indeed, we find SipB from Salmonella (UniProt ID – SIPB_SALTY/gi|62181387) among the proteins with both extracellular and transmembrane localization. Thus, double localizations in our database, while sometimes counterintuitive, can reflect important information on complex secretion pathways.
Performance Measure
The performance of ClubSub-P was compared to PSORTb v.3.0.2 (Yu et al., 2010) and CELLO v.2.5 (Yu et al., 2006). We calculated the precision, recall, accuracy, and MCC.
Unfortunately, the Proteome Analyst prediction server (Lu et al., 2004) is not active any more, thus we could not compare ClubSub-P against it. A recently published database for SCL prediction of Gram-negative bacteria, CobaltDB v1 (Goudenège et al., 2010), provides meta predictions for different signal peptide and secondary structural features; however, it does not combine these results to annotate a final SCL for the proteins. For this reason we could not use CobaltDB in our performance measure.
Dual localization predictions were considered for all the tools compared in the performance measure, but only CELLO and the UniProt original annotations had proteins with dual annotations in our test dataset. However, proteins with more than two localization predictions in CELLO v.2.5 were not considered and annotated as unknown. In cases where two different SCLs for a single protein are either predicted by a tool or given from UniProt data in the test set, a hit is considered as “true positive” if at least one of the localizations matches. All the “Unknown” predictions were considered as false negatives in our performance measurements. Sequences from test datasets were used to search against the ClubSub-P database, in order to assign their SCL. Only hits with sequence identities above 40% and pairwise alignment coverage above 75% were annotated to the corresponding cluster and sequences with no hits or below this cutoff were assigned as “Unknown”. The hits with “Uncertain” localization (see Materials and Methods) were also considered as “Unknown” for the performance measurements.
The results of the performance measurement are shown in Table 5. With the DB_Experimental test dataset, ClubSub-P (83.85%) shows a higher precision than PSORTb v3.0.2 (80%) and CELLO v2.5 (66.67%). Since the recall value for periplasmic proteins (15.79%) is very low for PSORTb v3.0.2, the overall recall value of PSORTbv3.0.2 (54.55%) is lower than that of CELLO (70.18%) and ClubSub-P (62.64%). Overall, the accuracy of all tools is comparable. Since we considered any one of correct dual localization predictions as “true positive,” CELLO’s overall performance (0.6) in terms of MCC is comparable to PSORTb (0.59). ClubSub-P has a superior overall performance (MCC 0.67). In summary, ClubSub-P has a higher precision than PSORTb and CELLO, showing that its strength is a reduced false-positive rate through the use of homology information.
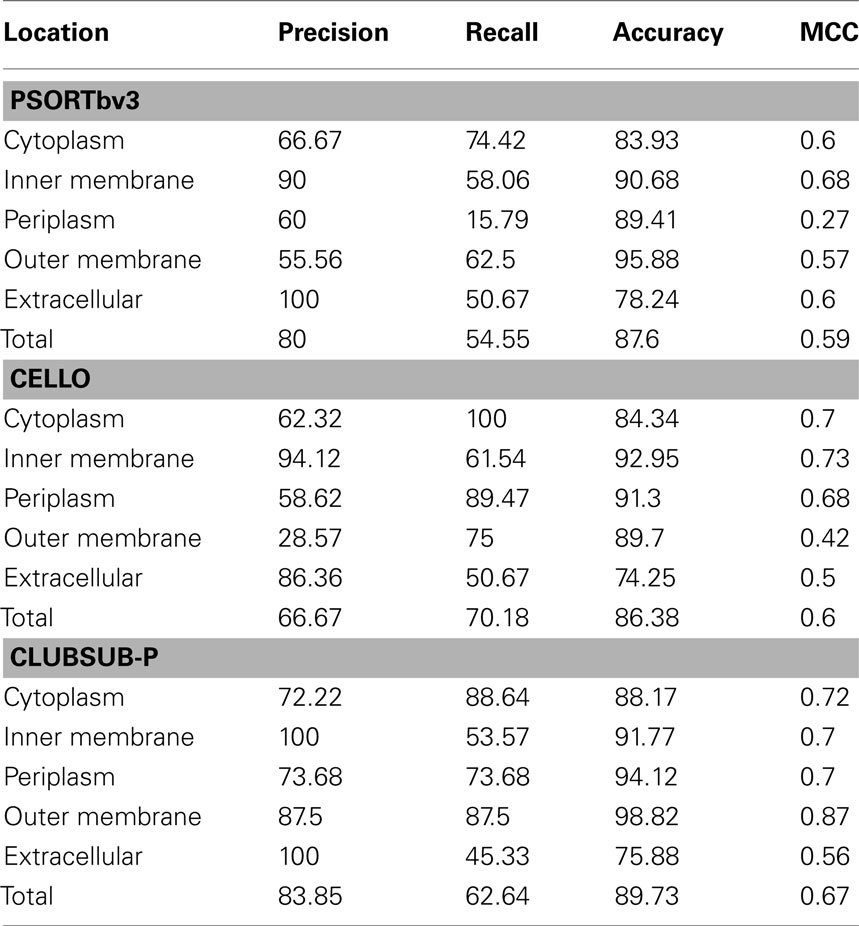
Table 5. Performance measurement for different Gram-negative bacterial subcellular localization prediction tools.
Incorrect Start Codons Resulting in Misannotated Signal Peptides
A known problem in SCL prediction is the quality of the input sequences; especially the exact start position for proteins with N-terminal signal peptides is essential. In the course of our analysis, we noted that in clusters where the majority of sequences are predicted to contain an N-terminal signal peptide, the false-negative results typically stem from misannotated start codons. When we corrected such gene annotation errors in the sequence, the signal peptides were correctly predicted in most cases. We found examples for both possible cases, where the misannotated start codons either extended or shortened the sequence N-terminally. Examples for these cases are shown in Figure 5; the sequences in this cluster that contained misannotated start codons were not predicted to contain a signal peptide, but had a OM beta-barrel annotation and thus were annotated as unknown, while the correctly annotated sequences in the cluster were predicted to be OM beta-barrel protein with a lipid anchor. The SCL annotation of the cluster reassigns them to OMP proteins with lipid anchor via the cluster consensus annotation, which shows one strength of ClubSub-P, the additional use of homology information on top of single-sequence predictions.

Figure 5. Cluster alignment and start codon mispredictions (sequences are labeled with gene identifiers). The alignment shows extended and shortened ends of orthologous sequences at the DNA level. Wrong extensions are colored in red, and shortened sequences are highlighted in yellow. Corrected sequences with alternative start codons are shown in bold. These corrections in most cases lead to corrected predictions of signal peptides. For clearer view, sequences are chopped.
Overall, we found 3,558 proteins with false-negative predictions in different clusters of proteins with signal peptides (annotated as periplasmic, OMP, OMP with lipid anchor, lipoproteins, or extracellular with signal peptide). These 3,558 proteins were spread across 547 of the 607 genomes that we used in this study, and were present in 2,222 different clusters (Data Sheet S2 in Supplementary Material). These errors were significantly accumulated in certain genomes compared with the rest of the genomes in the database (Table 6). This could be due to differences in the gene prediction and ORF finder methods used in the gene annotation process. But as we can easily find these mistakes only in the signal peptide-containing clusters, we cannot provide good statistical data on the performance of the different gene prediction pipelines – there might be additional misannotations in other proteins that do not have an N-terminal signal peptide.
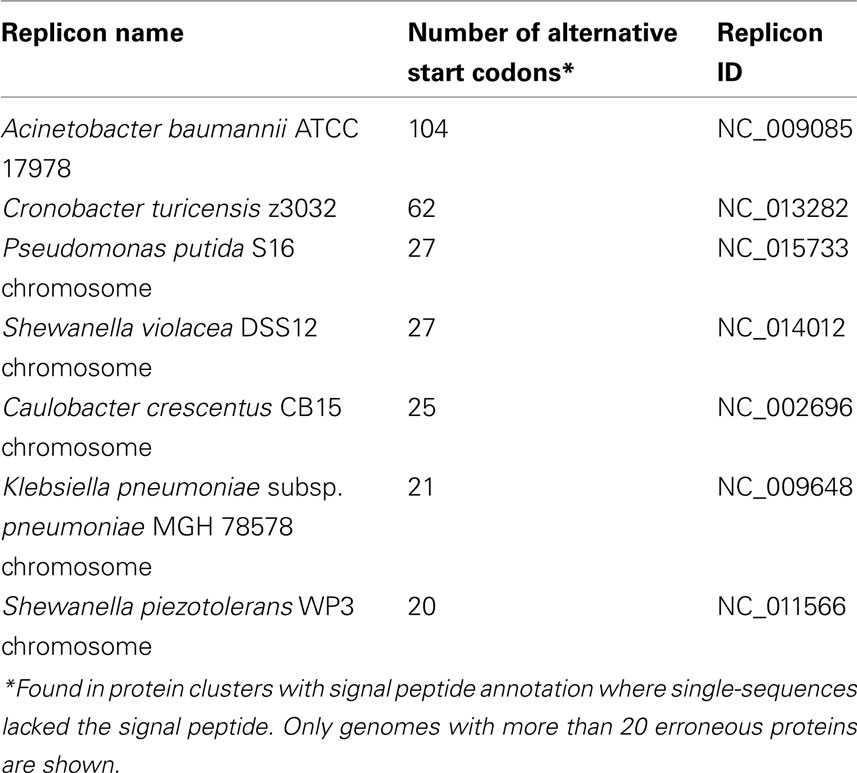
Table 6. Genomes with multiple signal peptide/start codon errors in secretory clusters.
Random manual checking revealed that most of the 3,558 protein sequences with false-negative signal peptide predictions have a mispredicted start codon on the DNA level. Studies have shown that the biased use of the uncommon start codons GUG and UUG over AUG is common among mispredicted start codons (Starmer et al., 2006; Pallejà et al., 2008). Confirming these findings, we also found a biased use of uncommon start codons among the above mentioned 3,558 proteins. The frequency of start codon usage in all bacterial coding sequences (3,690,458 sequences) used for this analysis is AUG (80.7%), GUG (12.6%), UUG (6.5%), and other start codons (0.2%). But the gene start codon frequencies of the 3,558 falsely predicted proteins are AUG (62.73%), GUG (21.61%), UUG (12.45%), and other start codons (3.2%), again showing that these gene predictions need revision.
We wanted to check if we could detect signal peptides from the genes with alternative start codons after re-annotation. ProTISA (Hu et al., 2008) is a database which combines translation initiation site (TIS) information from different sources, e.g., from experimental Swiss-Prot annotations, conserved domain hits and from alignments of orthologous sequences, to refine the RefSeq TIS annotations. Unfortunately, it doesn’t cover all the proteomes we used in our database; thus, we used the alternative start codons predicted by gene prediction programs instead (see above). The NCBI RefSeq FTP site provides updated gene predictions for all sequenced bacterial genomes, based on the latest version of four gene prediction programs [GeneMark-2.5m (Borodovsky and Mcininch, 1993), GeneMarkHMM-2.6r (Borodovsky and Lukashin, 1998), Glimmer3 (Delcher et al., 2007), and Prodigal-2.50 (Hyatt et al., 2010)]. To obtain more quantitative information on the phenomenon, we used this precomputed data to find an alternative start codon for the 3,558 proteins with false-negative signal peptide predictions (see methods), which translates into a protein with a signal peptide according to SignalP-HMM. Together, 2,290 sequences with an alternative start leading to a positive signal peptide prediction were found by one or several gene prediction programs. Of these 2,290 positive predictions, GeneMark-2.5m predicts 69.91% (1,601), GeneMarkHMM-2.6r predicts 72.79% (1,667), Glimmer3 predicts 66.86% (1,531), and Prodigal-2.50 predicts 84.93% (1,945). The numbers do not significantly change by using LipoP or Phobius instead of SignalP-HMM. The details of the alternative start codons with positive signal peptide predictions are given in Data Sheet S2 in Supplementary Material.
Subcellular Localization in Archaea
Archaea have a comparable cellular architecture to Gram-positive bacteria, except that instead of a peptidoglycan layer, different types of surface layers made from proteins, glycoproteins, or pseudo-murein are observed (Ellen et al., 2010). As there are no specialized SCL prediction programs available for Archaea other than the recently published PSORTb v3.0.2 program (Yu et al., 2010), we combined different feature prediction tools along with homology information in the same way as described above for Gram-negative bacteria.
As a result we were able to assign unambiguous SCLs to 69.21% of all proteins obtained from the 65 archaeal proteomes (104,896 of 151,553), where PSORTbV3.0.2 annotates 86.99% (131,839 of 151,553). When exclusively looking at proteins found in clusters with size two and above, i.e., where homology information is available, ClubSub-P can annotate 96.35% (104,896 out of 108,872) of proteins with an unambiguous SCL, where PSORTbv3.0.2 predicts only 89.42% (97,349 of 108,872).
ClubSub-P archaeal SCL annotation statistics are found in the Table 7. Just like in the Gram-negative SCL predictions, we also found clusters of archaeal proteins with multiple localizations, such as “Secreted/extracellular AND membrane anchor” and “Cell wall AND membrane anchor.” We annotated these combinations separately as we assume that, again as for Gram-negative bacteria, these double localizations have a biological significance. In detail, proteins with a predicted signal peptide and one consensus membrane helix prediction were annotated as “Secreted/extracellular AND membrane anchor.” This also includes the proteins with prepilin signal peptide. Proteins with a cell wall prediction and one or two consensus membrane helix predictions were annotated as “Cell wall AND membrane anchor.” Note that membrane anchor in this context means a single N-terminal transmembrane helix that anchors proteins to the cytoplasmic membrane.
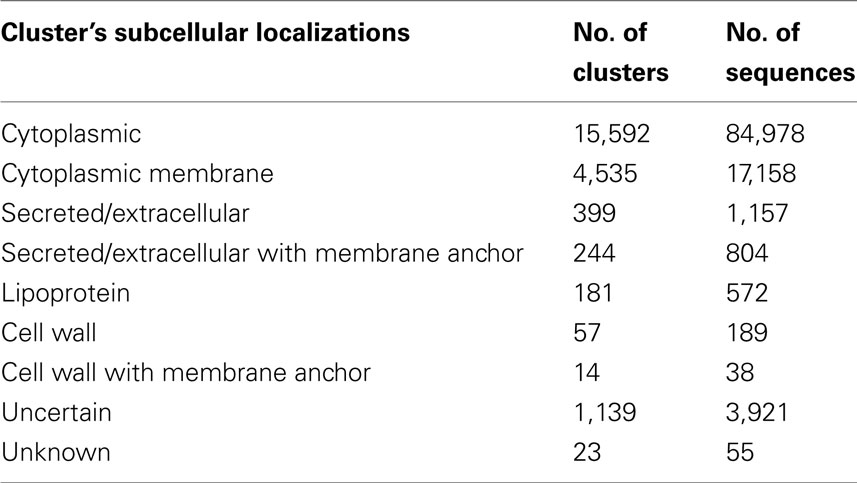
Table 7. ClubSub-P archaeal SCL prediction statistics.
Since there are very few archaeal proteins with experimentally annotated SCLs, most of these proteins are already included in the training sets of the tools we used in the consensus prediction, which makes the calculation of benchmarks very difficult. But, using the experimentally verified 252 archaeal sequences from UniProt, we were able to show that ClubSub-P has a slightly higher precision than PSORTbV3.0, but with a lower recall value. Overall, both tools are comparable in performance. Details on the performance of ClubSub-P with archaeal proteins are found in Table 8. With the addition of more archaeal proteomes from genomic data, and with the inclusion of further tools specialized on SCL prediction of archaeal proteins, ClubSub-P will be able to predict the archaeal SCLs more precisely in the future.
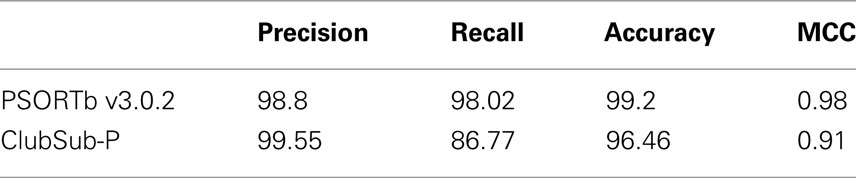
Table 8. Performance measurement of ClubSub-P archaeal predictions.
ClubSub-P Availability
We introduced the ClubSub-P database into the classification section of the MPI Bioinformatics Toolkit, a platform that integrates a great variety of tools for protein sequence analysis (Biegert et al., 2006). ClubSub-P can be found at http://toolkit.tuebingen.mpg.de/clubsubp. Users can browse the database to view the precomputed results, or they can annotate their query sequences by searching the database using BLAST. ClubSub-P is interconnected with other tools in the toolkit, so users can easily forward their results to other tools for further analysis. Screenshots of the ClubSub-P database are shown in Figure 6.
Figure 6. ClubSub-P database screenshots.
Discussion
Annotating the SCL of a protein is an important step in characterizing the native function of a protein. Thus, computational SCL predictions have gained importance in the post-genomic era, and various tools exist for this purpose. When combing different SCL predictors to create a meta-SCL predictor, it is important to select the best available individual predictors. We have developed a cluster-based meta-SCL prediction method for archaeal and Gram-negative bacterial proteins, by combining different published tools through consensus voting and protein sorting rules. In addition to the consensus SCL prediction for each single sequence, sequences are clustered according to their similarity. This homology information is exploited to eliminate false-positive and false-negative results. The performance of our tool is comparable with state-of-the-art SCL prediction methods, but with more precision (where precision is a measure of the ability of the system to predict only the relevant data, see Materials and Methods). In addition to the general SCLs, we were able to annotate more specific localizations, such as “OMP with lipid anchor,” “extracellular protein with transmembrane helix,” and “transmembrane with TAT or general signal peptide” for certain protein clusters, by combining different feature prediction tools. When more of such specific feature prediction tools become available we can include them into our prediction pipeline easily, and can annotate more specific localizations in a very precise way. In the cluster-based comparison of predictions for orthologous proteins, we have shown that there are inconsistencies between different prediction methods. We have demonstrated that by obtaining a consensus prediction from different tools, we can greatly reduce the number of false-positive predictions for single sequences. Furthermore, combining the single SCL predictions on the level of clusters further increases the precision of the predictions. The incorporation of additional proteomes from new sequencing projects will further decrease the number of singletons and will significantly increase the coverage and the precision of the SCL predictions of ClubSub-P in the future.
The pipeline can be expanded to other organism groups easily, as we show with the example of Archaea. Archaea are especially interesting in this context as comparably little experimental information is available for them. As only few reliable SCL prediction tools trained specifically on archaeal datasets are available, ClubSub-P is at an advantage as it combines different tools into a (more reliable) consensus prediction, and uses homology information where available to exclude most false-positive and false-negative predictions. Though the recall value is lower than that of PSORTb, the overall performance will increase dramatically by adding more sequenced archaeal genomes for clustering, and with new and Archaea-specific SCL prediction tools which can be incorporated into ClubSub-P easily.
The database can be used for a variety of applications. One obvious application is in genome annotation, where we show how misinterpreted start codons can be detected through SCL predictions and the use of homology information. We originally produced the database to screen for conserved immunogenic epitopes localized on the bacterial cell surface, in order to identify new vaccine candidates which would protect from diseases caused by Gram-negative human pathogens. Using the protein clusters with OM or extracellular localization, one can find conserved proteins which could be useful vaccine candidates or diagnosis markers specific to the bacterial species present in these clusters.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are very thankful to Christina Wassermann and Andre Noll for the useful discussions and technical assistance in integrating the database into the MPI toolkit, and to Vikram Alva, Thomas Arnold, Stanislaw Dunin-Horkawicz, Iwan Grin, Marcus Thein and others for helpful discussions, and to Andrei Lupas for continuing support. We are also thankful to the many authors that provided us with offline version of their tools. This work was funded by the Bill & Melinda Gates Foundation, Grand Challenges Explorations program.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/evolutionary_and_genomic_microbiology/10.3389/fmicb.2011.00218/abstract
Footnotes
References
Arnold, R., Brandmaier, S., Kleine, F., Tischler, P., Heinz, E., Behrens, S., Niinikoski, A., Mewes, H.-W., Horn, M., and Rattei, T. (2009). Sequence-based prediction of type III secreted proteins. PLoS Pathog. 5, e1000376. doi: 10.1371/journal.ppat.1000376
Bagos, P. G., Tsirigos, K. D., Plessas, S. K., Liakopoulos, T. D., and Hamodrakas, S. J. (2009). Prediction of signal peptides in Archaea. Protein Eng. Des. Sel. 22, 27–35.
Bendtsen, J., Nielsen, H., Widdick, D., Palmer, T., and Brunak, S. (2005). Prediction of twin-arginine signal peptides. BMC Bioinformatics 6, 167. doi: 10.1186/1471-2105-6-167
Bendtsen, J. D., Nielsen, H., Von Heijne, G., and Brunak, S. (2004). Improved prediction of signal peptides: signalP 3.0. J. Mol. Biol. 340, 783–795.
Berven, F. S., Flikka, K., Jensen, H. B., and Eidhammer, I. (2004). BOMP: a program to predict integral β-barrel outer membrane proteins encoded within genomes of Gram-negative bacteria. Nucleic Acids Res. 32, W394–W399.
Biegert, A., Mayer, C., Remmert, M., Söding, J., and Lupas, A. N. (2006). The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res. 34, W335–W339.
Borodovsky, M., and Lukashin, A. V. (1998). GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115.
Borodovsky, M., and Mcininch, J. (1993). GenMark: parallel gene recognition for both DNA strands. Comput. Chem. 17, 123–133.
Bos, M. P., Robert, V., and Tommassen, J. (2007). Biogenesis of the gram-negative bacterial outer membrane. Annu. Rev. Microbiol. 61, 191–214.
Burstein, D., Zusman, T., Degtyar, E., Viner, R., Segal, G., and Pupko, T. (2009). Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog. 5, e1000508. doi: 10.1371/journal.ppat.1000508
Choo, K., Tan, T., and Ranganathan, S. (2009). A comprehensive assessment of N-terminal signal peptides prediction methods. BMC Bioinformatics 10, S2. doi: 10.1186/1471-2105-10-S15-S2
Chou, K.-C., and Shen, H.-B. (2006a). Hum-PLoc: a novel ensemble classifier for predicting human protein subcellular localization. Biochem. Biophys. Res. Commun. 347, 150–157.
Chou, K.-C., and Shen, H.-B. (2006b). Large-scale predictions of gram-negative bacterial protein subcellular locations. J. Proteome Res. 5, 3420–3428.
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679.
Desvaux, M., Hébraud, M., Talon, R., and Henderson, I. R. (2009). Secretion and subcellular localizations of bacterial proteins: a semantic awareness issue. Trends Microbiol. 17, 139–145.
Ellen, A. F., Zolghadr, B., Driessen, A. M., and Albers, S. V. (2010). Shaping the archaeal cell envelope. Archaea 2010, 608243.
Farizo, K. M., Fiddner, S., Cheung, A. M., and Burns, D. L. (2002). Membrane localization of the S1 subunit of pertussis toxin in Bordetella pertussis and implications for pertussis toxin secretion. Infect. Immun. 70, 1193–1201.
Gardy, J. L., and Brinkman, F. S. L. (2006). Methods for predicting bacterial protein subcellular localization. Nat. Rev. Microbiol. 4, 741–751.
Giombini, E., Orsini, M., Carrabino, D., and Tramontano, A. (2010). An automatic method for identifying surface proteins in bacteria: {SLEP}. BMC Bioinformatics 11, 39. doi: 10.1186/1471-2105-11-39
Goudenège, D., Avner, S., Lucchetti-Miganeh, C., and Barloy-Hubler, F. (2010). CoBaltDB: complete bacterial and archaeal orfeomes subcellular localization database and associated resources. BMC Microbiol. 10, 88. doi: 10.1186/1471-2180-10-88
Hiller, K., Grote, A., Scheer, M., Münch, R., and Jahn, D. (2004). PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res. 32, W375–W379.
Horler, R. S. P., Butcher, A., Papangelopoulos, N., Ashton, P. D., and Thomas, G. H. (2009). EchoLOCATION: an in silico analysis of the subcellular locations of Escherichia coli proteins and comparison with experimentally derived locations. Bioinformatics 25, 163–166.
Hu, G.-Q., Zheng, X., Yang, Y.-F., Ortet, P., She, Z.-S., and Zhu, H. (2008). ProTISA: a comprehensive resource for translation initiation site annotation in prokaryotic genomes. Nucleic Acids Res. 36, D114–D119.
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. doi: 10.1186/1471-2105-11-119
Imai, K., and Nakai, K. (2010). Prediction of subcellular locations of proteins: where to proceed? Proteomics 10, 3970–3983.
Janson, H., Heden, L. O., and Forsgren, A. (1992). Protein D, the immunoglobulin D-binding protein of Haemophilus influenzae, is a lipoprotein. Infect. Immun. 60, 1336–1342.
Juncker, A. S., Willenbrock, H., Von Heijne, G., Brunak, S. R., Nielsen, H., and Krogh, A. (2003). Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12, 1652–1662.
Käll, L., Krogh, A., and Sonnhammer, E. L. L. (2004). A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036.
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J. Mol. Biol. 305, 567–580.
Lewenza, S., Mhlanga, M. M., and Pugsley, A. P. (2008). Novel inner membrane retention signals in Pseudomonas aeruginosa lipoproteins. J. Bacteriol. 190, 6119–6125.
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659.
Löwer, M., and Schneider, G. (2009). Prediction of type III secretion signals in genomes of Gram-negative bacteria. PLoS ONE 4, e5917. doi: 10.1371/journal.pone.0005917
Lu, Z., Szafron, D., Greiner, R., Lu, P., Wishart, D. S., Poulin, B., Anvik, J., Macdonell, C., and Eisner, R. (2004). Predicting subcellular localization of proteins using machine-learned classifiers. Bioinformatics 20, 547–556.
Luirink, J., Von Heijne, G., Houben, E., and De Gier, J.-W. (2005). Biogenesis of inner membrane proteins in Escherichia coli. Annu. Rev. Microbiol. 59, 329–355.
Mah, N., Perez-Iratxeta, C., and Andrade-Navarro, M. A. (2010). Outer membrane pore protein prediction in mycobacteria using genomic comparison. Microbiology 156, 2506–2515.
Marlovits, T. C., and Stebbins, C. E. (2010). Type III secretion systems shape up as they ship out. Curr. Opin. Microbiol. 13, 47–52.
Nakajima, A., Sugimoto, Y., Yoneyama, H., and Nakae, T. (2000). Localization of the outer membrane subunit OprM of resistance-nodulation-cell division family multicomponent efflux pump in Pseudomonas aeruginosa. J. Biol. Chem. 275, 30064–30068.
Nielsen, H., Engelbrecht, J., Brunak, S., and Von Heijne, G. (1997). Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. Des. Sel. 10, 1–6.
Overbeek, R., Bartels, D., Vonstein, V., and Meyer, F. (2007). Annotation of bacterial and archaeal genomes: improving accuracy and consistency. Chem. Rev. 107, 3431–3447.
Pallejà, A., Harrington, E. D., and Bork, P. (2008). Large gene overlaps in prokaryotic genomes: result of functional constraints or mispredictions? BMC Genomics 9, 335. doi: 10.1186/1471-2164-9-335
Plewczynski, D., Slabinski, L., Tkacz, A., Kajan, L., Holm, L., Ginalski, K., and Rychlewski, L. (2007). The RPSP: Web server for prediction of signal peptides. Polymer 48, 5493–5496.
Pugsley, A. P., Kornacker, M. G., and Ryter, A. (1990). Analysis of the subcellular location of pullulanase produced by Escherichia coli carrying the pulA gene from Klebsiella pneumoniae strain UNF5023. Mol. Microbiol. 4, 59–72.
Rashid, M., Saha, S., and Raghava, G. P. (2007). Support vector machine-based method for predicting subcellular localization of mycobacterial proteins using evolutionary information and motifs. BMC Bioinformatics 8, 337. doi: 10.1186/1471-2105-8-337
Remmert, M., Linke, D., Lupas, A. N., and Söding, J. (2009). HHomp – prediction and classification of outer membrane proteins. Nucleic Acids Res. 37, W446–W451.
Rose, R. W., Bruser, T., Kissinger, J. C., and Pohlschroder, M. (2002). Adaptation of protein secretion to extremely high-salt conditions by extensive use of the twin-arginine translocation pathway. Mol. Microbiol. 45, 943–950.
Saier, M. H., Ma, C. H., and Rodgers, L. (2008). Protein secretion and membrane insertion systems in bacteria and eukaryotic organelles. Adv. Appl. Microbiol. 65, 141–197.
Seydel, A., Gounon, P., and Pugsley, A. P. (1999). Testing the “+2 rule” for lipoprotein sorting in the Escherichia coli cell envelope with a new genetic selection. Mol. Microbiol. 34, 810–821.
Shen, Y. Q., and Burger, G. (2007). “Unite and conquer”: enhanced prediction of protein subcellular localization by integrating multiple specialized tools. BMC Bioinformatics 8, 420. doi: 10.1186/1471-2105-8-420
Starmer, J., Stomp, A., Vouk, M., and Bitzer, D. (2006). Predicting Shine-Dalgarno sequence locations exposes genome annotation errors. PLoS Comput. Biol. 2, e57. doi: 10.1371/journal.pcbi.0020057
Su, E. C.-Y., Chiu, H.-S., Lo, A., Hwang, J.-K., Sung, T.-Y., and Hsu, W.-L. (2007). Protein subcellular localization prediction based on compartment-specific features and structure conservation. BMC Bioinformatics 8, 330. doi: 10.1186/1471-2105-8-330
Szabó, Z., Stahl, A. O., Albers, S.-V., Kissinger, J. C., Driessen, A. J. M., and Pohlschröder, M. (2007). Identification of diverse archaeal proteins with class III signal peptides cleaved by distinct archaeal prepilin peptidases. J. Bacteriol. 189, 772–778.
Tusnady, G. E., and Simon, I. (2001). The HMMTOP transmembrane topology prediction server. Bioinformatics 17, 849–850.
UniProt-Consortium. (2010). The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 38, D142–D148.
Warren, A., Archuleta, J., Feng, W.-C., and Setubal, J. (2010). Missing genes in the annotation of prokaryotic genomes. BMC Bioinformatics 11, 131. doi: 10.1186/1471-2105-11-131
Yu, C.-S., Chen, Y.-C., Lu, C.-H., and Hwang, J.-K. (2006). Prediction of protein subcellular localization. Proteins 64, 643–651.
Yu, N. Y., Wagner, J. R., Laird, M. R., Melli, G., Rey, S., Lo, R., Dao, P., Sahinalp, S. C., Ester, M., Foster, L. J., and Brinkman, F. S. L. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615.
Keywords: subcellular localization prediction, signal peptide, clustering, protein homology, start codon prediction
Citation: Paramasivam N and Linke D (2011) ClubSub-P: cluster-based subcellular localization prediction for Gram-negative bacteria and archaea. Front. Microbio. 2:218. doi: 10.3389/fmicb.2011.00218
Received: 05 September 2011; Accepted: 12 October 2011;
Published online: 08 November 2011.
Edited by:
Martin G. Klotz, University of North Carolina at Charlotte, USAReviewed by:
Loren Hauser, Oak Ridge National Laboratory, USAUli Stingl, King Abdullah University of Science and Technology, Saudi Arabia
Copyright: © 2011 Paramasivam and Linke. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Dirk Linke, Department I Protein Evolution, Max Planck Institute for Developmental Biology, Spemannstr. 35, D-72076 Tübingen, Germany. e-mail: dirk.linke@tuebingen.mpg.de