
- 1 Laboratory of Bacterial Genomics, Department of Pathobiological Sciences, University of Wisconsin–Madison, Madison, WI, USA
- 2 Department of Food Hygiene, Cairo University, Giza, Egypt
Mycobacterium avium subspecies paratuberculosis (M. ap), the causative agent of Johne’s disease, infects many farmed ruminants, wild-life animals, and recently isolated from humans. To better understand the molecular pathogenesis of these infections, we analyzed the whole-genome sequences of several M. ap and M. avium subspecies avium (M. avium) isolates to gain insights into genomic diversity associated with variable hosts and environments. Using Next-generation sequencing technology, all six M. ap isolates showed a high percentage of similarity (98%) to the reference genome sequence of M. ap K-10 isolated from cattle. However, two M. avium isolates (DT 78 and Env 77) showed significant sequence diversity (only 87 and 40% similarity, respectively) compared to the reference strain M. avium 104, a reflection of the wide environmental niches of this group of mycobacteria. Within the M. ap isolates, genomic rearrangements (insertions/deletions) were not detected, and only unique single nucleotide polymorphisms (SNPs) were observed among M. ap isolates. While more of the SNPs (~100) in M. ap genomes were non-synonymous, a total of ~6,000 SNPs were detected among M. avium genomes, most of them were synonymous suggesting a differential selective pressure between M. ap and M. avium isolates. In addition, SNPs-based phylo-genomics had a enough discriminatory power to differentiate between isolates from different hosts but yet suggesting a bovine source of infection to other animals examined in this study. Interestingly, the human isolate (M. ap 4B) was closely related to a M. ap isolate from a dairy facility, suggesting a common source of infection. Overall, the identified phylo-genomes further supported the idea of a common ancestor to both M. ap and M. avium isolates. Genome-wide analysis described here could provide a strong foundation for a population genetic structure that could be useful for the analysis of mycobacterial evolution and for the tracking of Johne’s disease transmission among animals.
Introduction
Infection with Mycobacterium avium subspecies paratuberculosis (M. ap) causes Johne’s disease, or paratuberculosis, in a large number of ruminants and wild-life animals (Collins et al., 1994b). The combination of low milk yield and mortalities caused by Johne’s disease significantly impacts the economics of the dairy industry in the US and worldwide (Barrett et al., 2006; Raizman et al., 2009). Both infected and clinically ill animals can shed M. ap in their feces, a common source of infection, especially to young calves through a fecal–oral route (Collins et al., 1994a). The infected animals usually have a prolonged subclinical phase which eventually leads to severe gastroenteritis and death. The disease affects mainly ruminants with some evidence suggesting an association between M. ap and Crohn’s disease in humans (Naser et al., 2002, 2004; Over et al., 2011). As expected, M. ap isolates from variable hosts were the subject of several genetic analyses to decipher a potential role for genetic variations on M. ap virulence and pathogenesis. For example, approaches based on PCR amplification of specific targets (e.g., IS1311, IS900; Whittington et al., 2000) and short sequence repeats (SSR; Sevilla et al., 2008) revealed isolate variation on a genetic level. Recently, DNA microarrays were applied to examine variations on a whole-genome level (Paustian et al., 2005; Wu et al., 2006) which provided a comprehensive analysis of large scale differences among examined isolates. However, events of genomic rearrangements (insertions/deletions, Indels) were not easily identified. In this report, we resorted to a high throughput sequencing strategy to address our hypothesis linking genomic diversity to mycobacterial adaption to variable host and environments where they replicate and persist.
Several studies attempted to examine the genomic polymorphisms among M. avium complex (MAC) strains to better identify mycobacterial species associated with infections. In one study, dnaJ sequence revealed a limited genomic diversity among human and veterinary strains (Morita et al., 2004). However, comparative genomic hybridization revealed more diversity among M. avium, M. ap, and M. avium subsp. silvaticum (Semret et al., 2004; Paustian et al., 2005). Despite a 95% similarity at the nucleotide level between M. avium and M. ap, long oligonucleotide microarrays were able to assess genomic diversity among the genomes of MAC members (Semret et al., 2004; Wu et al., 2006). Fortunately, the complete genome sequence of M. ap K-10 was reported in 2005 and has been revised and re-sequenced recently (Li et al., 2005; Wu et al., 2009; Wynne et al., 2010) which allowed more detailed comparative analysis of several M. ap strains. For the M. tuberculosis complex, the presence of historical data and documented isolates collection helped in better understanding of the origin and evolution of members of this group (Behr and Small, 1999; Smith et al., 2009). Unfortunately, such records are not available for members of MAC.
With the advancements in next-generation sequencing technology (Bentley et al., 2008), we took advantage of Illumina-based technology to decipher the genome contents of M. ap isolates from various animals and their environments. This technology allows us to compare genomic sequences on unprecedented level, the nucleotide level, with high speed and accuracy. With the help of an array of bioinformatics tools, we were able to analyze the genomes of eight mycobacterial isolates including the ATCC 19698, a widely used M. ap isolate in several virulence and pathogenesis studies (Tanaka et al., 1994; Shin et al., 2006; Van et al., 2010). Most M. ap isolates had a high level of sequence similarity to the reference, M. ap K-10 strain on a genome-wide scale even when human isolates were analyzed (Wynne et al., 2011). However, the genomes of two M. avium isolates had lower level of sequence identity to the M. avium 104 genome, the reference genome for M. avium subsp. hominissuis (MAH). Overall, genomic rearrangements represented by large scale inversions and deletions were found between M. ap and M. avium genomes. However, single nucleotide polymorphisms (SNPs) were the most common variations observed among M. ap isolates from different animals despite the bovine origin for all of these isolates. The observed genomic polymorphism among MAC isolates provided us a better understanding for the evolutionary forces active on both closely related organisms with different characteristic phenotypes.
Materials and Methods
Bacterial Strains
Six strains of M. avium subspecies paratuberculosis (M. ap) from different hosts and environments and two M. avium subspecies avium (M. avium) were selected (Table 1) for whole-genome sequencing. Laboratory strain of M. ap ATCC 19698, JTC 1281, JTC 1285, and DT 3 were all obtained from Johne’s testing center (JTC) at the University of Wisconsin–Madison. M. ap 4B, a human isolate, was provided by Dr. Saleh Naser at the University of Central Florida (Wu et al., 2006). Environmental isolates were obtained from the soil and utensils of dairy farms and provided by National Veterinary Service Laboratories at Ames, IA. The identity of each strain was confirmed and genotyped as M. ap versus M. avium by PCR analysis of three genes (16S rRNA, IS1311, hsp65) and growth phenotype in presence/absence of mycobactin J as outlined before (Wu et al., 2006). Each strain was cultured in Middlebrook 7H9 broth media supplemented with 10% ADC (2% glucose, 5% bovine serum albumin factor V, and 0.85% NaCl), 0.05% Tween 80 at 37°C. For M. ap isolates, 2 μg/ml of Mycobactin J was added.

Table 1. A list of mycobacterial isolates used in this study.
Genomic DNA Extraction
Five to 10 ml of bacterial cultures at mid-log phase were used for DNA extraction and isolation. Briefly, M. ap cultures were centrifuged down at 10,000 rpm for 3 min and pellets were then resuspended in sterile Tris–EDTA buffer. Bacterial cells were killed at 80°C for 20 min before adding lysozyme (10 μl of 100 mg/ml) for an overnight incubation at 37°C. After cell lysis, 12 μl of 20 mg/ml proteinase K/pellet was added and incubated at 65°C for 2–3 h. For DNA isolation, 100 μl of 5 M NaCl/pellet was added and incubated at 65°C for 10 min, followed by adding 80 μl of CTAB/NaCl and then incubated for another 10 min at 65°C. In addition, equal volume of phenol–chloroform–isoamyl alcohol (25:24:1) was added to each tube and centrifuge at 10,000 rpm for 5 min at room temperature. The aqueous upper layers were collected and transferred to a fresh tube for washing with equal volume of chloroform–isoamyl alcohol (24:1) and then with isopropanol followed by incubation at −20°C for at least 1 h. Genomic DNA was precipitated by centrifugation at 10,000 rpm for 15 min followed by washing in 75% ethanol. After wash, DNA pellets were dried in a Speed-Vac and resuspended in nuclease-free sterile water.
Whole-Genome Sequencing
Purified genomic DNA (1–5 μg) samples isolated from each target strain were sent to the Genomic Resource Center (GRC) at the University of Maryland for Illumina GAIIx whole-genome sequencing with multiplexing using the sample preparation oligonucleotide kit from Illumina. The integrity and concentration of DNA was checked by GRC again and followed by fragmentation of DNA using nebulization. DNA ends were repaired and A-tails and adaptors were added using Illumina protocols. The desired size of DNA (around 200 bp) were selected and then amplified with adaptor specific primers that contained four nucleotide barcode tags. The amplified DNA libraries were analyzed by Agilent Bioanalyzer to determine the size and the concentration of DNA fragments. DNA libraries were loaded onto eight channel flow cell. DNA fragments were denatured and hybridized to the oligonucleotides in flow cells followed by and amplification step to form clusters. The flow cells were then transferred to Genome analyzer II for sequencing. For paired-end sequence reads, the amplicons were flipped on the flowcells so the other end can be read as described before (Quail et al., 2008). At the end of each run, four images were collected and used for base-calling.
Sequence Assembly and Alignments
Raw sequences obtained from the Illumina GAIIx were analyzed using the CLC Genomic Workbench software (version 4.0.3, CLC Bio, Cambridge, MA, USA) to perform both de novo and comparative reference assembly. For the M. ap genomes, all sequences were assembled in reference to the revised M. ap K-10 sequences (Wynne et al., 2010). The M. avium DT 78 genome was assembled using the genome of M. avium subsp. hominissuis 104 (NCBI accession NC 008595) as a reference. The de novo assembly was used for the genome sequence of Env 77 strain because of the lack of significant similarity to other genomes. Additionally, the MAUVE algorithm was used to align paired or multiple genomes for comparative purposes, as outlined before (Perna et al., 1998; Darling et al., 2010). The gapped consensus sequence of each strain was imported to MAUVE for sequence alignment at default seed weight setting.
Single Nucleotide Polymorphisms Analysis
For SNPs detection, we used algorithms implemented in CLC Bio Workstation. Criteria for identifying SNPs included a coverage range setting at 10–55 reads and a presence frequency in at least 50% of the reads before consideration for further analysis. A randomly selected number of SNPs were further analyzed using Sanger sequencing to confirm Next-Generation sequencing data. The primers were designed to cover 10 possible SNPs. The BigDye Terminator (Applied Biosystems, Foster City, CA, USA) version 3.1 cycle sequencing kit was used for sequencing. The sequencing PCR included an initial denaturation cycle at 95°C for 5 min followed by 35 cycles of 95°C for 20 s, 45°C for 30 s and 60°C for 2 min with a final extension at 72°C for 7 min. All samples were sent to the Biotechnology Center at the University of Wisconsin–Madison for sequencing on a ABI 3730XL machine (Applied Biosystems).
For the genome-wide phylogeny (phylo-genome analysis), the predicted SNPs from sequenced genomes (M. ap isolates) and the corresponding nucleotides in DT 78, M. ap K-10 and M. avium 104 were tabulated to create a concatenated sequences of each strain. The genome of M. avium Env 77 isolate was excluded from such analysis because of the low similarity to other genomes. The concatenated sequence of each strain was aligned using CLUSTALW, and phylogenetic trees were generated with MEGA version 5 using one of the following methods: maximum parsimony (MP), maximum likelihood (ML), maximum likelihood with molecular clock (MLK) assumption in addition to Neighbor-joining algorithm with a bootstrapping values of 1,000 replicates applied to all methods (Tamura et al., 2011).
Results
Characterization of Mycobacterial Isolates
Several mycobacterial species belonging to the M. avium complex are present in animal surroundings; each with different capacities to cause illness (e.g., M. ap, M. avium) and potential to spread to humans (Alvarez-Uria, 2010). Before initiating our genome analysis of members of the M. avium complex, we searched our collection of mycobacterial isolates originating from diverse hosts, diverse tissues as well as from environmental samples of dairy herds that might help in spreading the infection. Our selection scheme identified eight isolates that were subjected for further genotyping protocols to confirm their identity. Based on acid-fast staining and amplification of the 16S rRNA gene using mycobacteria-specific primers (Talaat et al., 1997), all eight isolates were shown to belong to the genus mycobacterium. Moreover, typing based on the hsp65 gene (Smole et al., 2002) confirmed the identity of two mycobacterial isolates, DT 78 and Env 77 as M. avium subspecies avium (M. avium) while the rest of the isolates were all M. ap. Identification of sheep or cattle types of M. ap was based on IS1311 amplification followed by HinfI digestion (data not shown). All of the six M. ap isolates belonged to the bovine origin (M. ap type II). A compiled list of all mycobacterial isolates used in this study and their origin is shown in Table 1.
Whole-Genome Sequencing of Mycobacterial Isolates
The Illumina sequencer generated an average read length of 50 nucleotides with an average coverage of 42–68× of each sequenced genome after reference assembly. The number of reads, mapped reads, and the length of consensus sequence are all listed in Table 2. The revised version of M. ap K-10 sequence (Wynne et al., 2010) and M. avium subspecies hominissuis (M. avium 104) were used as references for comparative genome assembly of the target isolates. As expected, all examined M. ap genomes showed a high sequence identity (up to 99%) to the M. ap K-10 genome. Lack of sequence coverage in some parts of the genome could explain some of the differences from the reference genome. Despite the presence of small deleted regions among M. ap genomes, only 2 gaps >1 kb had been seen among M. ap genomes, including the one isolated from human (M. ap 4B isolate), suggesting a high level of similarity to the M. ap K-10 strain isolated from cattle. On the other hand, the M. avium DT 78 strain had only 87% sequence identity to the M. avium 104 genome while it had a higher similarity (93%) to the M. ap K-10 genome, despite its established genotype as M. avium isolate. In the DT 78 genome, more gaps were present whether M. avium 104 or M. ap K-10 were used for reference alignment (Figure 1). The average gap size in this genome is ~4 kb.
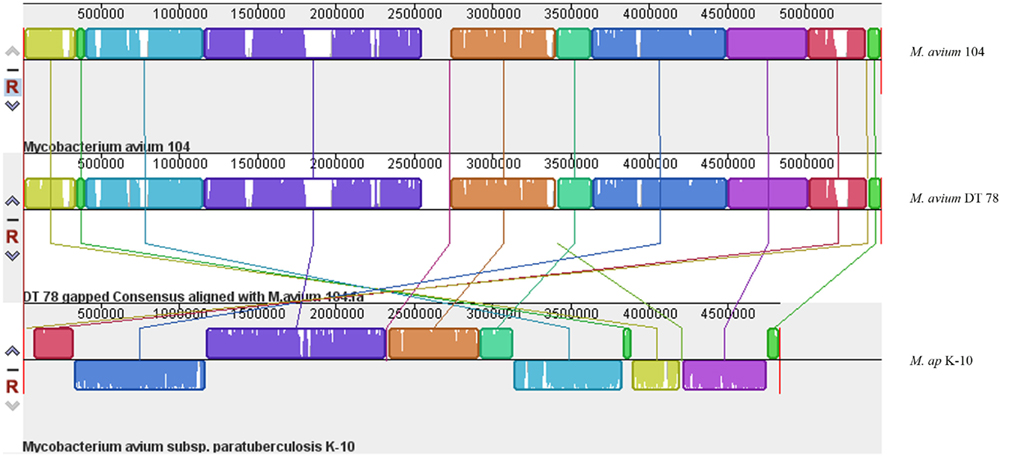
Figure 1. A whole-genome alignment of M. avium DT 78, M. avium 104 and M. ap DT 78. MAUVE algorithm (Darling et al., 2010) was used for the alignment of the three genomes where white areas indicate low coverage gaps in the sequence of M. avium DT 78 genome, and about seven large region Indels were identified in M. avium DT 78. Regions with the same color indicate high similarity and connected by same color bars. The genomes were drawn to scale based on the reference M. avium 104 genome.
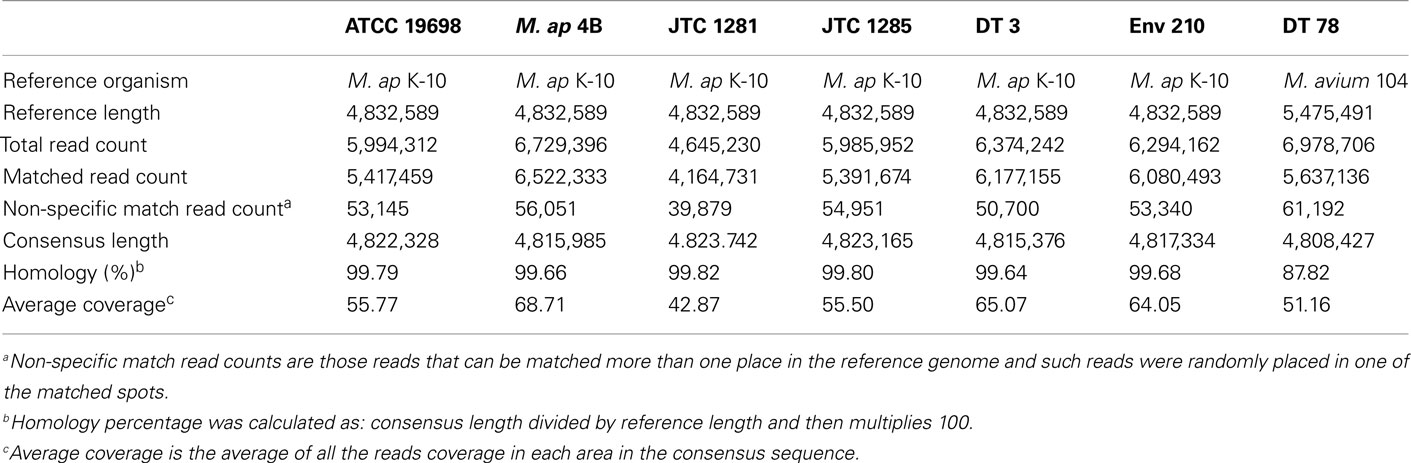
Table 2. A summary report for CLC Bio reference assembly of M. aviu m and M. ap isolates.
Among the sequenced genomes, the genome of M. avium Env 77 provided a significant challenge because of the low level of similarity to M. avium 104 genome during the reference assembly phase. Accordingly, we employed an algorithm for de novo assembly that generated 772 contigs. These contigs were used as queries in MegaBLAST search against the Mycobacteria genome database (blast.ncbi.nlm.nih.gov). The coverage of each contig is at least 20× and the average coverage of all contigs is around 30× for this strain. In fact, the Env 77 genome was sequenced twice with similar result for each sequencing run (data not shown). Interestingly, BLAST analysis showed only a third of the Env 77 genome with sequence similarity to the genomes of either the M. ap K-10 or M. avium 104 and to a lesser degree to other sequenced mycobacterial genomes, suggesting a mosaic genome structure (Figure 2). Detailed BLAST analysis of the Env 77 draft genome shared common conserved genes, mainly with four mycobacterium species, including ribosomal proteins, DNA polymerase, proteinase Clp, cell division protein Fts, and some transcription or translation regulatory factors. As indicated in Figure 2, the genome of M. avium Env 77 has higher similarity to M. avium 104 and M. ap K-10 than other mycobacterial species. Overall, the sequenced genomes from all strains, except Env 77, mapped to the reference genomes with a significantly high level of similarity. All sequenced genomes were deposited to GenBank database for download and further analysis. The accession numbers for the deposited sequences are listed in Table A1 in Appendix.
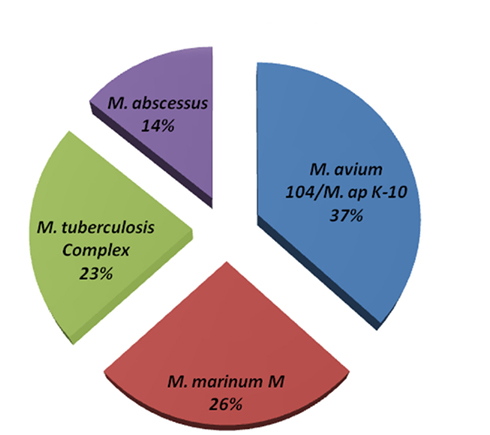
Figure 2. Genome composition of M. avium Env 77. MegaBLAST algorithm was used to identify closely related bacteria to all contig sequences from the M. avium Env 77 isolate. Genomes with <10% homology were excluded from representation. Members of the M. tuberculosis complex included M. tuberculosis and M. bovis with sequence divergence <5%. The same criteria was used to formulate M. avium and M. ap groups.
Genomic Rearrangements among M. ap Isolates
A major goal of our investigation was to delineate events of insertions and deletions among mycobacterial genomes to better understand their evolutionary relationships. To identify large scale events of insertions/deletions (Indels), we compared the assembled genomes of the six M. ap isolates to the standard M. ap K-10 genome using MAUVE software (version 2.3.1; Darling et al., 2010; Figure 3). Among the potential Indels that could exist among these genomes, we identified only gaps that are <1 kb. A common gap area located at reference position 3,767,550–3,767,870 which is part of MAPK 3350 gene encoding a hypothetical protein has been seen among all six strains with a gap size ~300 bp. At this region, low or zero read coverage has observed among all six strains suggesting a problematic region for Illumina sequencer. The sequence in this gap region appeared to have high GC contents (82%) but no repetitive elements involved.
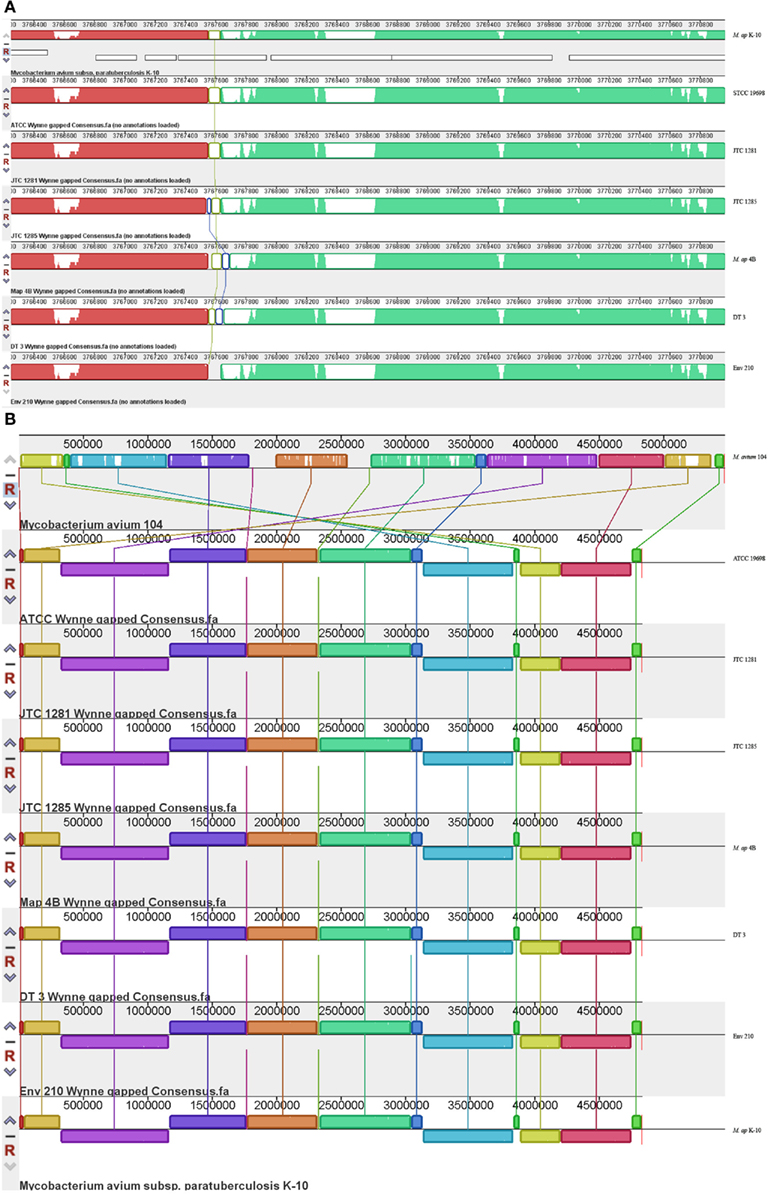
Figure 3. Comparative analysis of M. ap and M. avium from animals and environmental sources. The gapped consensus sequence of each strain was used for comparison by MAUVE version 2.3.1. (A) A close-up depiction of a breaking point in the alignment of six M. ap genomes in comparison to M. ap K-10 reference genome. The white areas indicated low or zero reads. In this example, the flanking sequences of the breaking point contain high GC percentage sequence but not repetitive sequences. (B) Indels among M. ap and M. avium genomes. Notice genome rearrangements are usually surrounding the genome origin of replication.
Based on the MAUVE comparison, the consensus sequences of these six strains are closely matched to the M. ap K-10 genome and no inversions were observed (Figure 3). On the other hand, when MAUVE was used to compare the genome of M. ap isolates to the M. avium 104 or M. avium DT78 genomes, about seven large regions of Indels were identified, confirming earlier findings by our group when DNA microarray was used (Wu et al., 2006). For example, one 11 kb Indel was found in all six M. ap strains at position 2,318,400–2,333,740 (MAPK 2038–MAPK 2050) but absent from M. avium. This 11 kb region encodes mostly hypothetical proteins in M. ap K-10 genome with two exceptions, MAPK 2040 and MAPK 2050. MAPK 2040 is a predicted hydrolase and earlier analysis (Santema et al., 2009) also showed the absence of this gene in M. avium 104, but present in other M. avium strain (Table 3). In addition, a total of six genomic inversions spanning ~2.4 Mb were identified among all M. ap strains when compared to M. avium 104 genome, similar to our earlier analysis of only M. ap K-10 and M. avium 104 genomes (Wu et al., 2006).
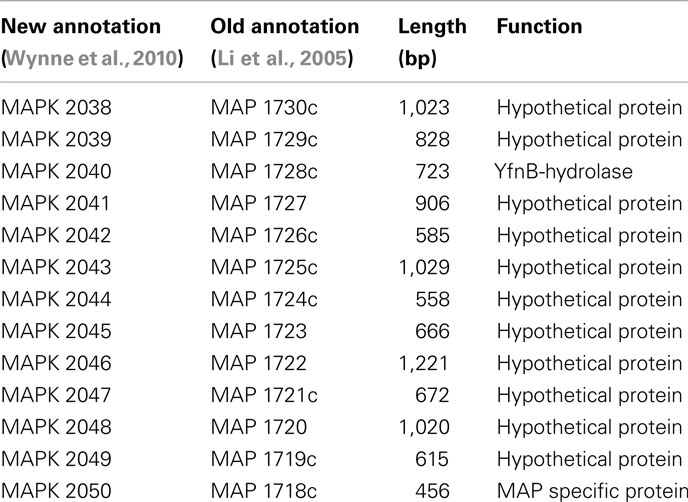
Table 3. A list of genes in the 11 kb island which is absent in M. aviu m 104.
SNPs among M. ap Isolates
To better analyze genomic diversity among M. ap isolates, we also examined genomic variations on the nucleotide level. For SNPs analysis, we set stringent criteria for SNP detection (see Materials and Methods). The total number of SNPs among six M. ap genomes ranged from 56 to 131 (Figure 4), among which 17 were found in >1 genome (Table 4). The number of non-synonymous SNPs (nSNPs) is slightly higher than synonymous SNPs (sSNPs), suggesting a positive selective pressure on the identified genes. In addition, most genes harbored one SNP with exceptions of 23 genes that contained two or three SNPs (Table A2 in Appendix). Interestingly, GlnE and MAPK 4304 contained three SNPs each, all are nSNPs, suggesting a high selective pressure on these two genes. Majority of genes contained >1 SNP are larger than 1 kb in size with an average SNP density of 1 SNP per 1.44 kb. Remaining 232 genes that harbored only one SNP represented a similar SNP density of one SNP per 1.44 kb that was identified in other mycobacterium (Qi et al., 2009). For the M. ap JTC 1281 and M. ap 4B, the percentage of nSNPs were 52.68 and 51.76% respectively, and the rest of M. ap strains with >60% of SNPs were nSNPs. Interestingly, genes encoding the Cytochrome P450 proteins harbored a high number of alleles in three of the six examined genomes (Table 5), similar to the same family of genes in M. tuberculosis (Cole, 1999). Intergenic SNPs were identified and counted for <10% of total SNPs.
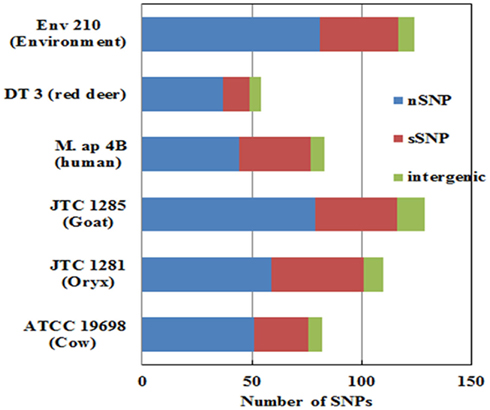
Figure 4. The total number of single nucleotide polymorphism (SNP) among M. ap isolates. The number of nSNP (non-synonymous) and sSNP (synonymous) and SNPs in the intergenic regions are color coded as indicated. SNPs were detected using reference assembled sequences of each strain. About 60–130 SNPs were detected M. ap isolates. Percentage of nSNP is generally higher than sSNP which indicates a high selective pressure in these strains.
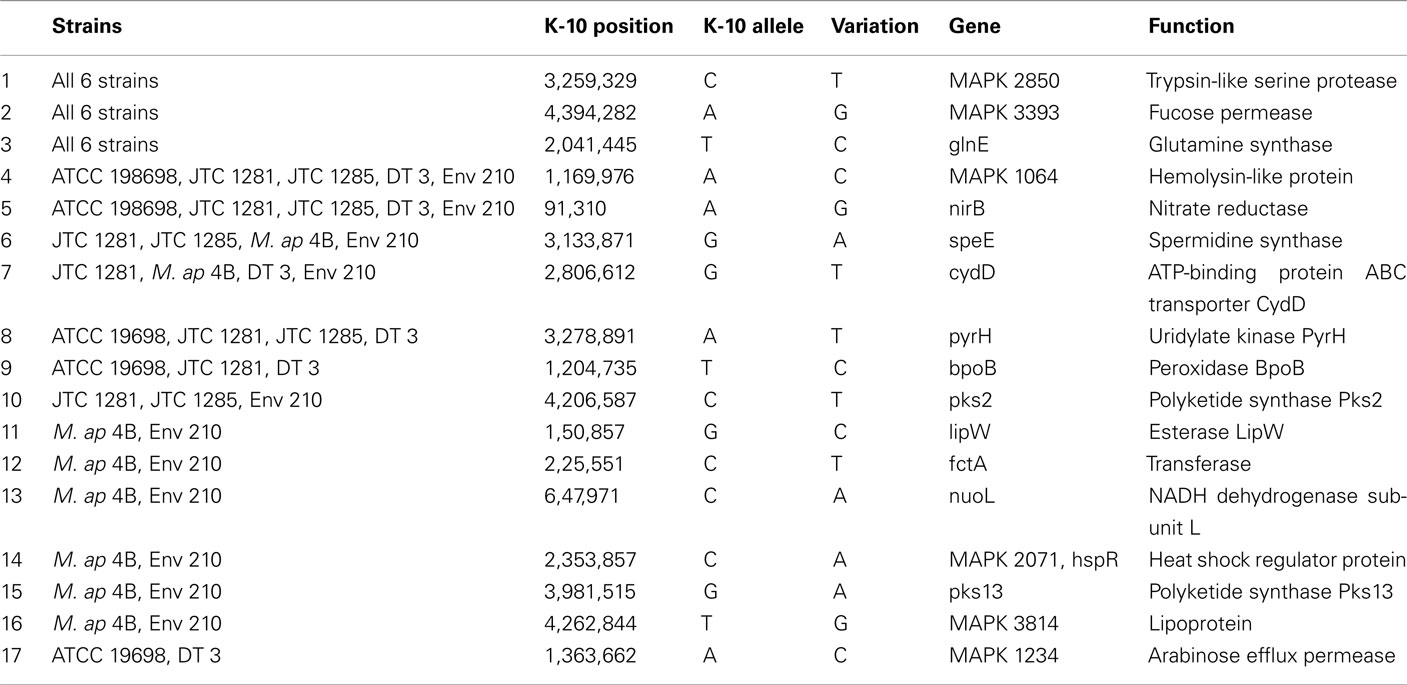
Table 4. A list of non-synonymous SNPs in M. ap genome resulted in more than one strain.
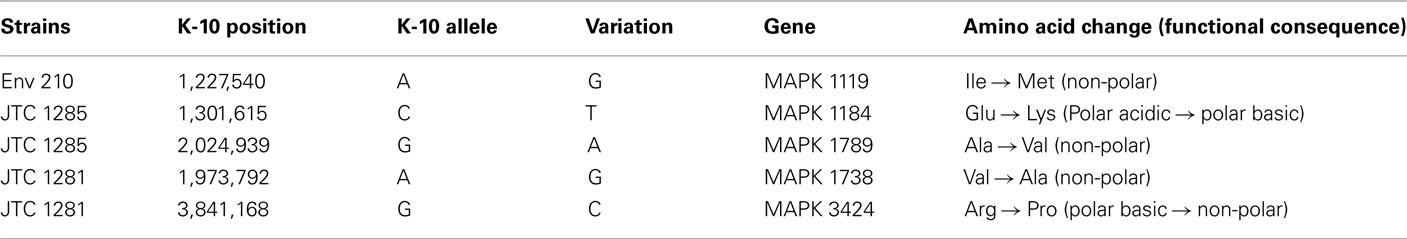
Table 5. A list of nSNP in cytochrome P450 proteins.
Generally, a modest number of SNPs were detected among genomes of M. ap isolates, unlike M. avium isolates. The M. avium DT 78 genome had a significantly high number of SNPs detected (6,278 SNPs) when compared to the standard M. avium 104 genome suggesting an earlier separation of this strain during its evolutionary pathway. In addition, >75% of the identified SNPs were synonymous, an indication of a higher stabilizing selective pressure for M. avium genes than those of M. ap. For the M. avium Env 77, SNP detection was not performed because the whole sequence aligned poorly with either M. ap K-10 or M. avium 104. Finally, 10 SNPs were randomly chosen for further confirmation using the Sanger sequencing method. The 10 SNPs were chosen based on the ATCC 19698 genome. The same 10 SNPs were also found in JTC 1281, while only 5 common SNPs were found in JTC 1285. All amplicons were sequenced from both forward and reverse strands (Table A3 in Appendix). Three SNPs were not detected in JTC 1285 based on the Sanger results, and is most likely caused by the Illumina sequencer error. Overall, Illumina sequencing was very beneficial in providing a high level of single nucleotide polymorphism in all examined genomes.
Phylo-Genomic Relationship among M. ap Isolates
Single nucleotide polymorphisms of six M. ap strains were concatenated and used for phylogenetic analysis on a genome-wide (phylo-genome) level. The two reference strains, M. ap K-10 and M. avium 104, were included in the analysis. A total of 301 SNPs present among the six M. ap strains as well as in M. avium 104 and M. avium DT 78 genomes were included in this analysis using the Neighbor-joining method (Tamura et al., 2011). The un-rooted tree showed a strong discriminatory power of SNP for all examined isolates based on their origin (Figure 5A) while maintained branches of M. avium genomes separate from genomes of M. ap isolates. Such discriminatory power was not possible when single-gene genotypes were tried (see above). Nonetheless, when the tree was rooted to M. avium 104 genome, two distinct major branches within the M. ap genomes were easily discerned (Figure 5B).
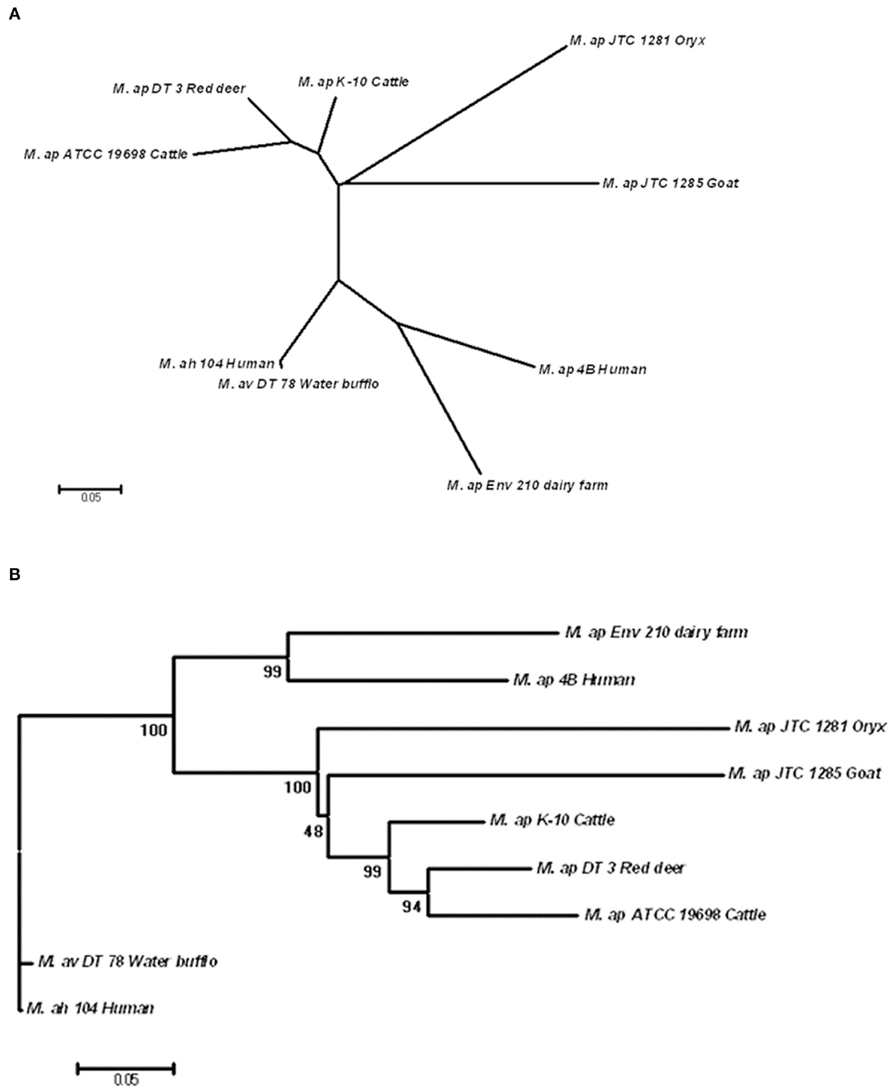
Figure 5. Phylogenomic analysis of M. ap and M. avium strains. (A) A dendrogram displaying an un-rooted, Neighbor-joining tree of the concatenated SNPs from all eight mycobacterial isolates under study. (B) A rooted Neighbor-joining tree using M. ah 104 genome as out group. The bootstrap consensus tree inferred from 1,000 replicates is taken to represent the evolutionary history of the taxa analyzed. The bootstrap replicates are marked on each branch and a less than 50% bootstrap replicates were collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test is shown next to the branches.
In one branch within M. ap genomes (Figure 5B), an isolate from red deer (M. ap DT 3) was closely related to the standard cattle strains (M. ap K-10 and ATCC 19698). On the other hand, isolates from goat and oryx (M. ap JTC 1281 and JTC 1285, respectively) were more closely related to the recently isolated cattle type strain (M. ap K-10) than to other laboratory strain (ATCC 19698), suggesting a cattle source of infection. In the other branch of the tree, M. ap 4B and M. ap Env 210 isolates from human and dairy farm, respectively, were closely related to each other. It is noteworthy to mention here that the association of M. avium DT 78 genome to the M. avium 104 strain based on phylo-genomic analysis confirmed our earlier identification of this isolate to belong M. avium group despites its overall higher similarity to the M. ap K-10. Finally, when we tried additional three methods for tree construction (MP, ML, MLK) on independent lists of sSNPs and nSNPs, a congregant topology was obtained for all trees with a high bootstrap support, similar to the one showed in Figure 5B. The Log Likelihood Ratio test for MLK consensus tree against ML tree indicated that the molecular clock assumption was not valid (p < 0.007). Overall, the identified tree topology suggests that M. avium 104 as a common ancestor from which M. ap likely emerged and diversified into two lineages: a lineage that clustered Env 210 with M. ap 4B (Human) while the second clustered all type II strains of M. ap. In both lineages, infected cows are the most likely reservoir for spreading the type II M. ap strains.
Discussion
Understanding the genome-wide variations among pathogenic Mycobacteria will improve our understanding of the pathogenesis and evolution of these important pathogens. Recently, Next-generation sequencing technologies provided us the opportunity to examine whole-genome variations on a much faster basis than traditional sequencing or DNA-microarray technologies. In this study, M. ap isolates were chosen from diverse hosts, sources, and locations to better assess the impact of these variations on pathogen genome composition. As expected, the six M. ap genomes sequenced in this study shared ~99% sequence similarity M. ap K-10 reference genome with a modest number of SNPs (~100) suggesting a stabilizing selective pressure. On the contrary, isolates of M. avium origin showed more diversity. For example, the M. avium DT 78 genome had a significant number of gaps and a large number of SNPs (~6,000) compared to M. avium 104 despite its significant similarity to M. ap K-10 (>90%) on a whole-genome level. This isolate is likely to represents an intermediate strain between M. avium 104 and M. ap K-10. Generally, M. avium replicate faster than M. ap and survive in a more diverse environments, those factors are likely to contribute to adaptive polymorphism. Previous analyses showed that M. avium has more diversity than M. ap strains (Turenne et al., 2006; Wu et al., 2006). Additionally, the M. avium Env 77 genome BLAST search indicated its complex and mosaic structure, another indication of diversity among M. avium isolates. Although standard genotyping protocols used here (based on hsp65 and IS1311) clearly typed DT78 and Env 77 isolates as M. avium, our genome sequencing approach question the validity of genotyping of Mycobacteria based on a single or a few genes and advocate for a whole-genome based approach.
Because of the close relatedness of M. ap genomes, SNPs from each strain provided valuable information on the divergence and evolutionary process that control members of MAC. A wide range of studies used SNPs for studying drug resistance mutations in organisms (Xu et al., 2008), analysis of genomic evolution (Filliol et al., 2006), and association of M. ap infection to Crohn’s disease patients (Wynne et al., 2011). In this study, SNPs were used for genome-wide typing of isolates to understand the dynamics of Johne’s disease transmission. Examining the modest number of SNPs detected in M. ap identified the presence of a higher percentage of nSNPs in all six M. ap isolates, suggesting a close relatedness among strains (Gutacker et al., 2002; Holden et al., 2004; Rocha et al., 2006). This close relationship among M. ap isolates could indicate a “spillover” infection from cattle to other animals (in this study red deer). However, the observed higher percentage of nSNPs could indicate adaptive evolution of M. ap to different hosts with positive selective pressure. A significant number of nSNPs were located in genes that encode hypothetical proteins while others in genes that encode proteins with enzymatic functions, some of them involved in metabolism and energy pathways, such as Pks proteins and NuoL protein. Interestingly, a SNP in the glnE gene were identified in all six M. ap genomes and additional SNPs in this gene were observed in ATCC 19698 and M. ap 4B separately. In M. tuberculosis, GlnE is an adenylyl transferase modulating glutamine synthetase activity and it is essential for bacterial growth under alternative nitrogen sources (Carroll et al., 2008). SNPs within this gene could likely be an indication of common evolutionary ancestor with environmental isolates. Similarity, SNPs in cytochrome P450 enzymes (Table 5) that catalyze mixed oxidation of hydrophobic compounds associated with free-living saprophyte (Arnold, 2007), another indication of a common environmental ancestor for M. ap. Finally, genes encoding cytochrome P450 were shown to play a role in the persistence of M. tuberculosis in tissues (McLean et al., 2010). SNPs found in M. ap counterpart could potentially contribute to the M. ap-host interactions.
An interesting outcome of the employed phylo-genomic analysis provided here is the further support provided to the hypothesis of presence of common origin to both subspecies of M. avium complex, namely, M. ap and M. avium subsp. avium. Such hypothesis was supported before based on large genomic regions of insertions/deletions (Wu et al., 2006; Alexander et al., 2009). This study provides further support to this hypothesis using SNPs on a genome-wide level. The whole-genome approach we employed here allowed us to explore the diversity among MAC isolates from different hosts and variable locations. It also provided more clues regarding the dynamic of mycobacterial transmission among animals. Sequencing the genome of more isolates will definitely enrich our understanding of the genome content and evolution of both environmental and pathogenic strains of mycobacteria and will eventually provide a comprehensive population genetic structure. Such knowledge base could elucidate the relationship between strains and host or some special environmental cues. In addition, sequencing more diverse isolates will help to evaluate the dynamic of disease transmission among animals or from animals to humans. Developing algorithms that can utilize the information gained from Next-generation sequencers will only improve the phylo-genomic analysis and is greatly needed to advance our understanding of microbe–host interactions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Meagan Cooney for reading the manuscript. We also like to thank Sarah Marcus and Eric Cabot for assisting with the CLC Bio assembling software. Illumina genome sequencing was performed by Genomic Resource center at University of Maryland, Baltimore. This project is supported by National Research Initiative of the U.S. Department of Agriculture Cooperative State Research, Education and Extension Service (grants #2007-35204-18400 and #2004-35605-14243 for Johne’s disease integrated program) and US-AID grant #1937.
References
Alexander, D. C., Turenne, C. Y., and Behr, M. A. (2009). Insertion and deletion events that define the pathogen Mycobacterium avium subsp. paratuberculosis. J. Bacteriol. 191, 1018–1025.
Alvarez-Uria, G. (2010). Lung disease caused by nontuberculous mycobacteria. Curr. Opin. Pulm. Med. 16, 251–256.
Arnold, C. (2007). Molecular evolution of Mycobacterium tuberculosis. Clin. Microbiol. Infect. 13, 120–128.
Barrett, D. J., Good, M., Hayes, M., and More, S. J. (2006). The economic impact of Johne’s disease in an Irish dairy herd: a case study. Ir. Vet. J. 59, 282.
Behr, M. A., and Small, P. M. (1999). A historical and molecular phylogeny of BCG strains. Vaccine 17, 915–922.
Bentley, D. R., Balasubramanian, S., Swerdlow, H. P., Smith, G. P., Milton, J., Brown, C. G., Hall, K. P., Evers, D. J., Barnes, C. L., Bignell, H. R., Boutell, J. M., Bryant, J., Carter, R. J., Keira, C. R., Cox, A. J., Ellis, D. J., Flatbush, M. R., Gormley, N. A., Humphray, S. J., Irving, L. J., Karbelashvili, M. S., Kirk, S. M., Li, H., Liu, X., Maisinger, K. S., Murray, L. J., Obradovic, B., Ost, T., Parkinson, M. L., Pratt, M. R., Rasolonjatovo, I. M., Reed, M. T., Rigatti, R., Rodighiero, C., Ross, M. T., Sabot, A., Sankar, S. V., Scally, A., Schroth, G. P., Smith, M. E., Smith, V. P., Spiridou, A., Torrance, P. E., Tzonev, S. S., Vermaas, E. H., Walter, K., Wu, X., Zhang, L., Alam, M. D., Anastasi, C., Aniebo, I. C., Bailey, D. M., Bancarz, I. R., Banerjee, S., Barbour, S. G., Baybayan, P. A., Benoit, V. A., Benson, K. F., Bevis, C., Black, P. J., Boodhun, A., Brennan, J. S., Bridgham, J. A., Brown, R. C., Brown, A. A., Buermann, D. H., Bundu, A. A., Burrows, J. C., Carter, N. P., Castillo, N., Chiara, E., Catenazzi, Chang, S., Neil, C. R., Crake, N. R., Dada, O. O., Diakoumakos, K. D., Dominguez-Fernandez, B., Earnshaw, D. J., Egbujor, U. C., Elmore, D. W., Etchin, S. S., Ewan, M. R., Fedurco, M., Fraser, L. J., Fuentes Fajardo, K. V., Scott, F. W., George, D., Gietzen, K. J., Goddard, C. P., Golda, G. S., Granieri, P. A., Green, D. E., Gustafson, D. L., Hansen, N. F., Harnish, K., Haudenschild, C. D., Heyer, N. I., Hims, M. M., Ho, J. T., Horgan, A. M., Hoschler, K., Hurwitz, S., Ivanov, D. V., Johnson, M. Q., James, T., Huw Jones, T. A., Kang, G. D., Kerelska, T. H., Kersey, A. D., Khrebtukova, I., Kindwall, A. P., Kingsbury, Z., Kokko-Gonzales, P. I., Kumar, A., Laurent, M. A., Lawley, C. T., Lee, S. E., Lee, X., Liao, A. K., Loch, J. A., Lok, M., Luo, S., Mammen, R. M., Martin, J. W., McCauley, P. G., McNitt, P., Mehta, P., Moon, K. W., Mullens, J. W., Newington, T., Ning, Z., Ling, N. B., Novo, S. M., O’Neill, M. J., Osborne, M. A., Osnowski, A., Ostadan, O., Paraschos, L. L., Pickering, L., Pike, A. C., Pike, A. C., Chris, P. D., Pliskin, D. P., Podhasky, J., Quijano, V. J., Raczy, C., Rae, V. H., Rawlings, S. R., Chiva, R. A., Roe, P. M., Rogers, J., Rogert Bacigalupo, M. C., Romanov, N., Romieu, A., Roth, R. K., Rourke, N. J., Ruediger, S. T., Rusman, E., Sanches-Kuiper, R. M., Schenker, M. R., Seoane, J. M., Shaw, R. J., Shiver, M. K., Short, S. W., Sizto, N. L., Sluis, J. P., Smith, M. A., Ernest Sohna, S. J., Spence, E. J., Stevens, K., Sutton, N., Szajkowski, L., Tregidgo, C. L., Turcatti, G., Vandevondele, S., Verhovsky, Y., Virk, S. M., Wakelin, S., Walcott, G. C., Wang, J., Worsley, G. J., Yan, J., Yau, L., Zuerlein, M., Rogers, J., Mullikin, J. C., Hurles, M. E., McCooke, N. J., West, J. S., Oaks, F. L., Lundberg, P. L., Klenerman, D., Durbin, R., and Smith, A. J. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59.
Carroll, P., Pashley, C. A., and Parish, T. (2008). Functional analysis of GlnE, an essential adenylyl transferase in Mycobacterium tuberculosis. J. Bacteriol. 190, 4894–4902.
Cole, S. T. (1999). Learning from the genome sequence of Mycobacterium tuberculosis H37Rv. FEBS Lett. 452, 7–10.
Collins, D. M., Radford, A. J., De Lisle, G. W., and Billman-Jacobe, H. (1994a). Diagnosis and epidemiology of bovine tuberculosis using molecular biological approaches. Vet. Microbiol. 40, 83–94.
Collins, M. T., Sockett, D. C., Goodger, W. J., Conrad, T. A., Thomas, C. B., and Carr, D. J. (1994b). Herd prevalence and geographic distribution of, and risk factors for, bovine paratuberculosis in Wisconsin. J. Am. Vet. Med. Assoc. 204, 636–641.
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5, e11147. doi:10.1371/journal.pone.0011147
Filliol, I., Motiwala, A. S., Cavatore, M., Qi, W., Hazbon, M. H., Bobadilla, d. V., Fyfe, J., Garcia-Garcia, L., Rastogi, N., Sola, C., Zozio, T., Guerrero, M. I., Leon, C. I., Crabtree, J., Angiuoli, S., Eisenach, K. D., Durmaz, R., Joloba, M. L., Rendon, A., Sifuentes-Osornio, J., Ponce de, L. A., Cave, M. D., Fleischmann, R., Whittam, T. S., and Alland, D. (2006). Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J. Bacteriol. 188, 759–772.
Gutacker, M. M., Smoot, J. C., Migliaccio, C. A. L., Ricklefs, S. M., Hua, S., Cousins, D.V., Graviss, E. A., Shashkina, E., Kreiswirth, B. N., and Musser, J. M. (2002). Genome-wide analysis of synonymous single nucleotide polymorphisms in Mycobacterium tuberculosis complex organisms: resolution of genetic relationships among closely related microbial strains. Genetics 162, 1533–1543.
Holden, M. T., Feil, E. J., Lindsay, J. A., Peacock, S. J., Day, N. P., Enright, M. C., Foster, T. J., Moore, C. E., Hurst, L., Atkin, R., Barron, A., Bason, N., Bentley, S. D., Chillingworth, C., Chillingworth, T., Churcher, C., Clark, L., Corton, C., Cronin, A., Doggett, J., Dowd, L., Feltwell, T., Hance, Z., Harris, B., Hauser, H., Holroyd, S., Jagels, K., James, K. D., Lennard, N., Line, A., Mayes, R., Moule, S., Mungall, K., Ormond, D., Quail, M. A., Rabbinowitsch, E., Rutherford, K., Sanders, M., Sharp, S., Simmonds, M., Stevens, K., Whitehead, S., Barrell, B. G., Spratt, B. G., and Parkhill, J. (2004). Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. U.S.A. 101, 9786–9791.
Li, L., Bannantine, J. P., Zhang, Q., Amonsin, A., May, B. J., Alt, D., Banerji, N., Kanjilal, S., and Kapur, V. (2005). The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc. Natl. Acad. Sci. U.S.A. 102, 12344–12349.
McLean, K. J., Belcher, J., Driscoll, M. D., Fernandez, C. C., Le, V. D., Bui, S., Golovanova, M., and Munro, A. W. (2010). The Mycobacterium tuberculosis cytochromes P450: physiology, biochemistry and molecular intervention. Future Med. Chem. 2, 1339–1353.
Morita, Y., Maruyama, S., Kabeya, H., Nagai, A., Kozawa, K., Kato, M., Nakajima, T., Mikami, T., Katsube, Y., and Kimura, H. (2004). Genetic diversity of the dnaJ gene in the Mycobacterium avium complex. J. Med. Microbiol. 53, 813–817.
Naser, S. A., Ghobrial, G., Romero, C., and Valentine, J. F. (2004). Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn’s disease. Lancet 364, 1039–1044.
Naser, S. A., Shafran, I., Schwartz, D., El Zaatari, F., and Biggerstaff, J. (2002). In situ identification of mycobacteria in Crohn’s disease patient tissue using confocal scanning laser microscopy. Mol. Cell. Probes 16, 41–48.
Over, K., Crandall, P. G., O’Bryan, C. A., and Ricke, S. C. (2011). Current perspectives on Mycobacterium avium subsp. paratuberculosis, Johne’s disease, and Crohn’s disease: a review. Crit. Rev. Microbiol. 37, 141–156.
Paustian, M. L., Kapur, V., and Bannantine, J. P. (2005). Comparative genomic hybridizations reveal genetic regions within the Mycobacterium avium complex that are divergent from Mycobacterium avium subsp. paratuberculosis isolates. J. Bacteriol. 187, 2406–2415.
Perna, N. T., Mayhew, G. F., Posfai, G., Elliott, S., Donnenberg, M. S., Kaper, J. B., and Blattner, F. R. (1998). Molecular evolution of a pathogenicity island from enterohemorrhagic Escherichia coli O157-H7. Infect. Immun. 66, 3810–3817.
Qi, W., Kaser, M., Roltgen, K., Yeboah-Manu, D., and Pluschke, G. (2009). Genomic diversity and evolution of Mycobacterium ulcerans revealed by next-generation sequencing. PLoS Pathog. 5, e1000580. doi:10.1371/journal.ppat.1000580
Quail, M. A., Kozarewa, I., Smith, F., Scally, A., Stephens, P. J., Durbin, R., Swerdlow, H., and Turner, D. J. (2008). A large genome center’s improvements to the Illumina sequencing system. Nat. Methods 5, 1005–1010.
Raizman, E. A., Fetrow, J. P., and Wells, S. J. (2009). Loss of income from cows shedding Mycobacterium avium subspecies paratuberculosis prior to calving compared with cows not shedding the organism on two Minnesota dairy farms. J. Dairy Sci. 92, 4929–4936.
Rocha, E. P., Smith, J. M., Hurst, L. D., Holden, M. T., Cooper, J. E., Smith, N. H., and Feil, E. J. (2006). Comparisons of dN/dS are time dependent for closely related bacterial genomes. J. Theor. Biol. 239, 226–235.
Santema, W., Overdijk, M., Barends, J., Krijgsveld, J., Rutten, V., and Koets, A. (2009). Searching for proteins of Mycobacterium avium subspecies paratuberculosis with diagnostic potential by comparative qualitative proteomic analysis of mycobacterial tuberculins. Vet. Microbiol. 138, 191–196.
Semret, M., Zhai, G., Mostowy, S., Cleto, C., Alexander, D., Cangelosi, G., Cousins, D., Collins, D. M., van Soolingen, D., and Behr, M. A. (2004). Extensive genomic polymorphism within Mycobacterium avium. J. Bacteriol. 186, 6332–6334.
Sevilla, I., Li, L., Amonsin, A., Garrido, J. M., Geijo, M. V., Kapur, V., and Juste, R. A. (2008). Comparative analysis of Mycobacterium avium subsp. paratuberculosis isolates from cattle, sheep and goats by short sequence repeat and pulsed-field gel electrophoresis typing. BMC Microbiol. 8, 204. doi:10.1186/1471-2180-8-204
Shin, S. J., Wu, C.-W., Steinberg, H., and Talaat, A. M. (2006). Identification of novel virulence determinants in Mycobacterium paratuberculosis by screening a library of insertional mutants. Infect. Immun. 7, 3825–3833.
Smith, N. H., Hewinson, R. G., Kremer, K., Brosch, R., and Gordon, S. V. (2009). Myths and misconceptions: the origin and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 7, 485–491.
Smole, S. C., McAleese, F., Ngampasutadol, J., von Reyn, C. F., and Arbeit, R. D. (2002). Clinical and epidemiological correlates of genotypes within the Mycobacterium avium complex defined by restriction and sequence analysis of hsp65. J. Clin. Microbiol. 40, 3374–3380.
Talaat, A. M., Reimschuessel, R., and Trucksis, M. (1997). Identification of mycobacteria infecting fish to the species level using polymerase chain reaction and restriction enzyme analysis. Vet. Microbiol. 58, 229–237.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739.
Tanaka, S., Sato, M., Taniguchi, T., and Yokomizo, Y. (1994). Histopathological and morphometrical comparison of granulomatous lesions in BALB/c and C3H/HeJ mice inoculated with Mycobacterium paratuberculosis. J. Comp. Pathol. 110, 381–388.
Turenne, C. Y., Semret, M., Cousins, D. V., Collins, D. M., and Behr, M. A. (2006). Sequencing of hsp65 distinguishes among subsets of the Mycobacterium avium complex. J. Clin. Microbiol. 44, 433–440.
Van, B. L., Coudijzer, K., Vlaemynck, G., Hendrickx, M., Michiels, C., Messens, W., Herman, L., and De, B. J. (2010). Localization of Mycobacterium avium subspecies paratuberculosis in artificially inoculated milk and colostrum by fractionation. J. Dairy Sci. 93, 4722–4729.
Whittington, R. J., Hope, A. F., Marshall, D. J., Taragel, C. A., and Marsh, I. (2000). Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis: IS900 restriction fragment length polymorphism and IS1311 polymorphism analyses of isolates from animals and a human in Australia. J. Clin. Microbiol. 38, 3240–3248.
Wu, C.-W., Glasner, J., Collins, M. T., Naser, S., and Talaat, A. M. (2006). Whole genome plasticity among Mycobacterium avium subspecies: insights from comparative genomic hybridizations. J. Bacteriol. 188, 711–723.
Wu, C. W., Schramm, T. M., Zhou, S., Schwartz, D. C., and Talaat, A. M. (2009). Optical mapping of the Mycobacterium avium subspecies paratuberculosis genome. BMC Genomics 10, 25. doi: 10.1186/1471-2164-10-25
Wynne, J. W., Bull, T. J., Seemann, T., Bulach, D. M., Wagner, J., Kirkwood, C. D., and Michalski, W. P. (2011). Exploring the zoonotic potential of Mycobacterium avium subspecies paratuberculosis through comparative genomics. PLoS ONE 6, e22171. doi:10.1371/journal.pone.0022171
Wynne, J. W., Seemann, T., Bulach, D. M., Coutts, S. A., Talaat, A. M., and Michalski, W. P. (2010). Resequencing the Mycobacterium avium subsp. paratuberculosis K10 genome: improved annotation and revised genome sequence. J. Bacteriol. 192, 6319–6320.
Xu, C., Zhou, Y. F., Deng, J. Y., Deng, X., Guo, Y. C., Cui, Z. Q., Zhang, Z. P., Wei, H. P., Bi, L. J., and Zhang, X. E. (2008). On-chip ligation of multiplexing probe-pairs for identifying point mutations out of dense SNP loci. Biosens. Bioelectron. 24, 818–824.
Appendix

Table A1. A list of accession numbers of genome deposited to GenBank.
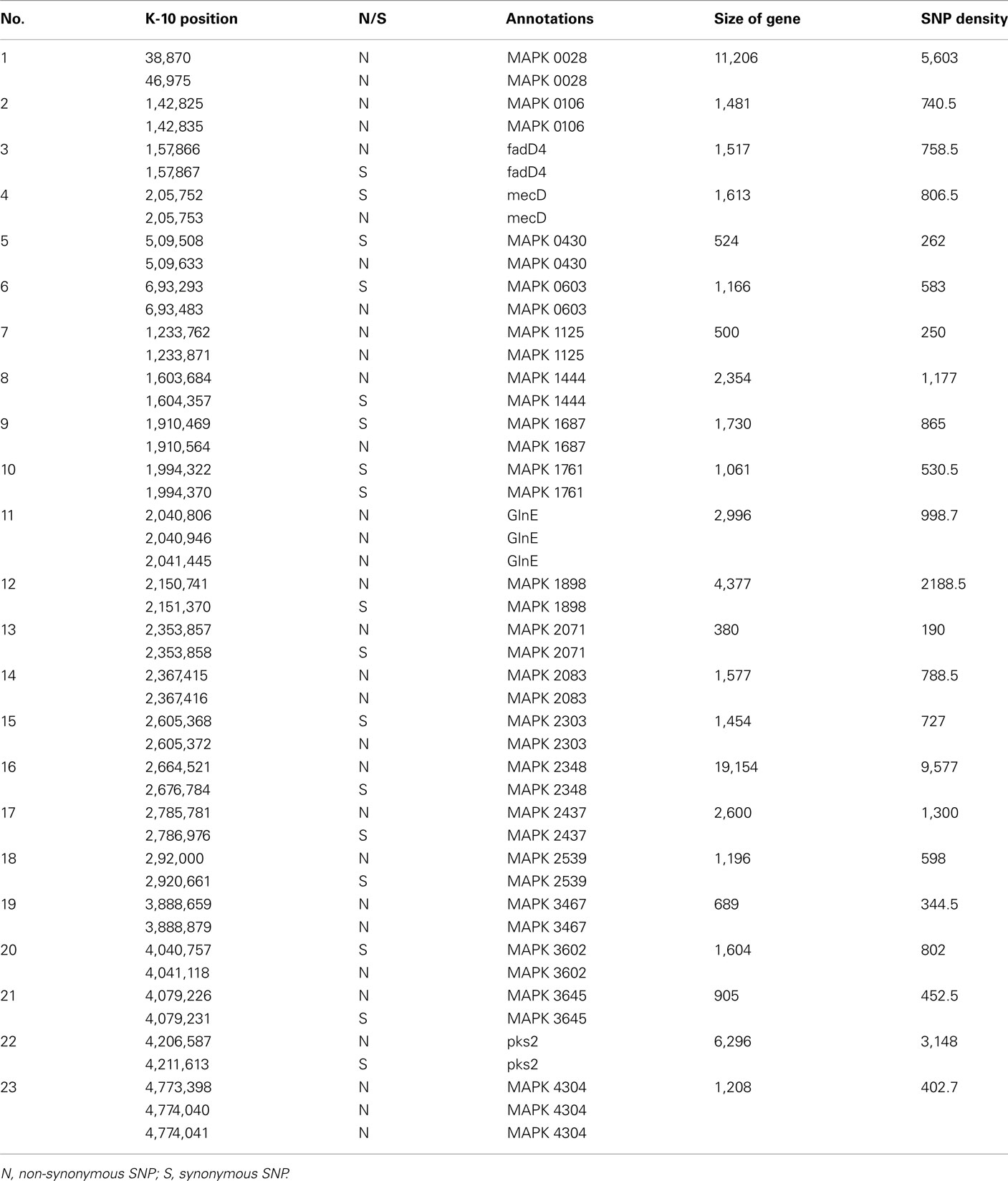
Table A2. A list of genes that harbored >1 SNPs and its SNPs density.
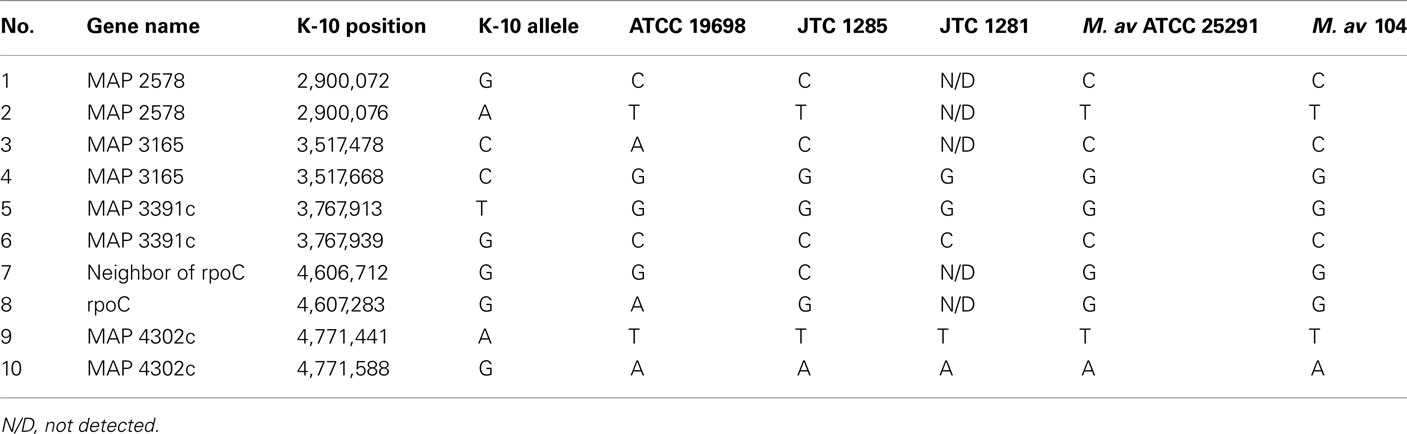
Table A3. A list of 10 SNPs picked for Sanger sequencing.
Keywords: Mycobacteria, paratuberculosis, Johne’s disease, whole-genome sequencing, genomics, pathogenesis
Citation: Hsu C-Y, Wu C-W and Talaat AM (2011) Genome-wide sequence variation among Mycobacterium avium subspecies paratuberculosis isolates: a better understanding of Johne’s disease transmission dynamics. Front. Microbio. 2:236. doi: 10.3389/fmicb.2011.00236
Received: 09 August 2011;
Paper pending published: 02 September 2011;
Accepted: 09 November 2011;
Published online: 09 December 2011.
Edited by:
Thomas A. Ficht, Texas A&M University, USAReviewed by:
Srinand Sreevastan, University of Minnesota, USAMichael L. Vasil, University of Colorado Medical School, USA
Copyright: © 2011 Hsu, Wu and Talaat. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Adel M. Talaat, Laboratory of Bacterial Genomics, Department of Pathobiological Sciences, University of Wisconsin–Madison, 1656 Linden Drive, Madison, WI 53706-1581, USA. e-mail: atalaat@wisc.edu