 Anna Blazejak1† Axel Schippers1,2*
Anna Blazejak1† Axel Schippers1,2*- 1 Geomicrobiology, Federal Institute for Geosciences and Natural Resources (BGR), Hannover, Germany
- 2 Faculty of Natural Sciences, Leibniz Universität Hannover, Hannover, Germany
Sulfate-reducing prokaryotes (SRP) are ubiquitous and quantitatively important members in many ecosystems, especially in marine sediments. However their abundance and diversity in subsurface marine sediments is poorly understood. In this study, the abundance and diversity of the functional genes for the enzymes adenosine 5′-phosphosulfate reductase (aprA) and dissimilatory sulfite reductase (dsrA) of SRP in marine sediments of the Peru continental margin and the Black Sea were analyzed, including samples from the deep biosphere (ODP site 1227). For aprA quantification a Q-PCR assay was designed and evaluated. Depth profiles of the aprA and dsrA copy numbers were almost equal for all sites. Gene copy numbers decreased concomitantly with depth from around 108/g sediment close to the sediment surface to less than 105/g sediment at 5 mbsf. The 16S rRNA gene copy numbers of total bacteria were much higher than those of the functional genes at all sediment depths and used to calculate the proportion of SRP to the total Bacteria. The aprA and dsrA copy numbers comprised in average 0.5–1% of the 16S rRNA gene copy numbers of total bacteria in the sediments up to a depth of ca. 40 mbsf. In the zone without detectable sulfate in the pore water from about 40–121 mbsf (Peru margin ODP site 1227), only dsrA (but not aprA) was detected with copy numbers of less than 104/g sediment, comprising ca. 14% of the 16S rRNA gene copy numbers of total bacteria. In this zone, sulfate might be provided for SRP by anaerobic sulfide oxidation. Clone libraries of aprA showed that all isolated sequences originate from SRP showing a close relationship to aprA of characterized species or form a new cluster with only distant relation to aprA of isolated SRP. For dsrA a high diversity was detected, even up to 121 m sediment depth in the deep biosphere.
Introduction
Sulfate reduction plays a crucial role in the past and present global sulfur cycle, and may be regarded as one of the oldest metabolic pathways on Earth (Castresana and Moreira, 1999; Schen et al., 2001). Therefore, sulfate-reducing prokaryotes (SRP) are biogeochemically important organisms in the environment, especially for the degradation of organic matter in coastal but also in deeply buried marine sediments in the open ocean (Jørgensen, 1982; Ferdelman et al., 1997; Knoblauch et al., 1999; Sahm et al., 1999; Thamdrup et al., 2000; Jørgensen et al., 2001; D’Hondt et al., 2004; Parkes et al., 2005; Schippers et al., 2005, 2010). Despite their importance in subsurface marine sediments the abundance and diversity of SRP in this environment is poorly understood. Global surveys of SRP cell numbers and gene sequencing data are missing and thus, more primary data for particular sediment sites are necessary. This includes the development of new methods for the detection of SRP in environmental samples.
The abundance of SRP in marine sediments has been determined by a variety of methods including MPN-cultivation (Knoblauch et al., 1999), 16S rRNA slot-blot hybridization (Sahm et al., 1999), or FISH and CARD-FISH with 16S rRNA gene probes (Ravenschlag et al., 2000; Gittel et al., 2008). Since SRP are phylogenetically diverse (Stahl et al., 2002), 16S rRNA approaches require a comprehensive set of 16S rRNA probes for a full, quantitative coverage of all SRP in an environmental sample (Ravenschlag et al., 2000). The functional gene encoding for dissimilatory sulfite reductase (dsrA) of SRP shows a high similarity in different SRP (Wagner et al., 1998), thus a dsrA specific PCR primer set targeting both, Gram-positive and Gram-negative SRP species, was developed for competitive PCR quantification (Kondo et al., 2004). These primers were also used to design a quantitative, real-time PCR (Q-PCR) assay for dsrA for SRP quantification in subsurface marine sediments (Schippers and Neretin, 2006; Leloup et al., 2007, 2009; Nunoura et al., 2009; Webster et al., 2009; Schippers et al., 2010) and the Black Sea water column (Neretin et al., 2007). Other Q-PCR assays for dsrA based on other primers (Wagner et al., 1998; Dhillon et al., 2003; Geets et al., 2006) were also applied to marine sediments (Wilms et al., 2007; Engelen et al., 2008), oil (Agrawal and Lal, 2009), and wastewater (Ben-Dov et al., 2007). Furthermore, RT-Q-PCR was applied to quantify mRNA of dsrA (Neretin et al., 2003).
Due to PCR bias or mismatches of the dsrA of not yet discovered SRP with the available dsrA primers, important SRP might have been overlooked in environmental samples. This might have happened in studies of deeply buried marine sediments (e.g., Peru continental margin, ODP Leg 201) in which sulfate reduction was identified as an important biogeochemical process, but dsrA or 16S rRNA genes of SRP were scarcely detected (D’Hondt et al., 2004; Parkes et al., 2005; Schippers et al., 2005; Inagaki et al., 2006; Schippers and Neretin, 2006; Teske, 2006; Webster et al., 2006, 2009; Fry et al., 2008; Nunoura et al., 2009). For this reason, another independent SRP quantification method is useful to reveal dsrA data and to confirm the full quantitative coverage of SRP in environmental sample analyses, especially for the deep biosphere.
A second functional gene of SRP is the adenosine 5′-phosphosulfate reductase gene aprA. In sulfate reducers, APS reductase catalyzes the two-electron reduction of APS to sulfite and AMP. APS reductase consists of an alpha and beta subunit, encoded by the genes aprA and aprB, respectively. The aprA gene has been thoroughly studied in SRP, and specific PCR and Q-PCR amplification of aprA was shown (Friedrich, 2002; Blazejak et al., 2005; Ben-Dov et al., 2007; Meyer and Kuever, 2007).
The objective of this study was a better understanding of the abundance and diversity of SRP in subsurface marine sediments. A Q-PCR assay specific for aprA of SRP was designed and applied to samples from different marine sediments together with the published Q-PCR assay for dsrA quantification (Schippers and Neretin, 2006). The diversity of SRP was analyzed based on cloning and sequencing of their functional genes aprA and dsrA. Marine sediments of the Peru continental margin, including samples from the deep biosphere (ODP site 1227), and the Black Sea were chosen because previous studies indicate that sulfate reduction is an important biogeochemical process in these sediments (Jørgensen et al., 2001; D’Hondt et al., 2004; Schippers et al., 2005). In addition, the abundance of sulfate reducers and other microorganisms was already determined using different assays, allowing comparisons with our newly developed method (Schippers et al., 2005; Inagaki et al., 2006; Schippers and Neretin, 2006; Leloup et al., 2007; Blazejak and Schippers, 2010).
Materials and Methods
Sample Collection
Samples were collected from different sediment depths at three marine sites during three research vessel expeditions. Site 1227 (8°59.5′S, 79°57.4′W) at a water depth of 427 m on the Peru margin was sampled with advanced piston coring up to 121 mbsf during Ocean Drilling Program (ODP) Leg 201 in March 2002 (D’Hondt et al., 2003; Jørgensen et al., 2005). Site 2MC (11°35.0′S, 77°33.1′W) at a water depth of 86 m on the Peru continental margin was sampled with a multicorer up to 0.34 mbsf during the cruise SO147 of R/V Sonne in June 2000. Site 20 (43°57.25′N, 35°38.46′E) at a water depth of 2048 m in the Black Sea was sampled with a gravity corer up to 5.8 mbsf during cruise M72-5 of R/V Meteor in May 2007.
Samples for molecular analysis were taken aseptically from the center of the cores at all stations and were stored at −20°C until further processing in the laboratory. For the recovery of deeply buried sediments from site 1227 on the Peru margin seawater based drilling fluid was used. Thus a potential contamination with seawater microorganisms was routinely checked by application of fluorescent beads of prokaryotic cell size and a chemical tracer (D’Hondt et al., 2003). Only uncontaminated samples were used for further analysis.
DNA Extraction
DNA was isolated from 0.5–4 g sediment of various depths using a FastDNA®Spin for Soil Kit (MP Biomedicals, Solon, OH, USA) with the following modification: to increase the yield of isolated DNA from clayish sediments 200 μg polyadenylic acid (Roche Diagnostics GmbH, Mannheim, Germany) dissolved in sterile water was added to the sample at the first step of the extraction procedure (Webster et al., 2003). DNA extracts from blank tubes (no sediment added) were used as procedural contamination control in later PCR analyses. Isolated DNA was stored in aliquots to avoid multiple defrosting and freezing and was thawed for Q-PCR measurements not more than twice.
Q-PCR Measurements
Quantitative PCR measurements were run in triplicate on an ABI Prism 7000 detection system (Applied Biosystems, Foster City, CA, USA). Quantification of Bacteria in total was performed using a Q-PCR assay based on the detection of the 16S rRNA gene (Nadkarni et al., 2002). The dissimilatory sulfite reductase gene dsrA of SRP was quantified using a published protocol (Schippers and Neretin, 2006) and primers (Kondo et al., 2004). The size of the amplified fragments was 219 bp. To quantify the adenosine 5′-phosphosulfate reductase gene aprA of SRP, a novel Q-PCR assay was designed. For specific amplification of this gene the primers APS1F (5-TGGCAGATCATGATYMAYGG-3) and APS4R (5-GCGCCAACYGGRCCRTA-3) were used (Blazejak et al., 2005; Meyer and Kuever, 2007). The size of the amplified fragments was 384–396 bp. The Q-PCR assay was performed with Platinum® SYBR® Green Q-PCR SuperMix-UDG with ROX (Invitrogen, Carlsberg, CA, USA), a primer concentration of 300 nM, and the following amplification conditions: 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. Two microliters sample DNA were added to a PCR reaction assay with a total volume of 25 μL. Melting curve analyses were run after each assay to check PCR specificity. For amplification of standards, DNA was extracted, amplified, and purified from minipreps of cloned aprA gene sequences from sulfate-reducing endosymbiotic bacteria with the accession numbers AM234052 and AM234053.
Q-PCR Data Analysis
Relative standards were prepared by serial dilution (1:10) of the PCR product. For each standard, the concentration was plotted against the cycle number at which the fluorescence signal increased above the background or cycle threshold (Ct value). The slope of each calibration curve was included into the following equation to determine the efficiency of the PCR reaction: efficiency = 10(−1/slope) − 1. According to this formula, an efficiency of 100% means a doubling of the product in each cycle. Data evaluation was performed with the software StepOne™ v2.0 (Applied Biosystems, Foster City, CA, USA).
PCR Amplification, Cloning, and Sequencing of the dsrA and aprA Genes
DNA was isolated from sediment samples of the Peru margin from three depths, 3.6, 65.3, and 121.4 mbsf (site 1227, ODP Leg 201) and in the Black Sea from four depths, 0.15, 2.7, 4.5, and 5.8 mbsf (site 20 GC, M72-5). Except for the number of cycles, amplification of the dsrA and aprA genes was carried out at the same conditions as for the Q-PCR assays (see above). For amplification of the dsrA gene, 30 cycles of PCR were required for the sediment sample from 3.6 mbsf depth of the Peru margin, and 35 cycles for the other samples. To amplify the aprA gene, 25 cycles of PCR were applied to the sediment samples from 0.15 and 2.7 mbsf depth in the Black Sea, and up to 35 cycles for the remaining samples. Three parallel PCR products obtained from each depth were combined, purified using a QIAquick PCR purification kit (Qiagen, Hilden, Germany), and subsequently cloned using the pGEM®-T Easy vector system (Promega, Madison, WI, USA) and TOP10 chemically competent cells (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Because of the high number of PCR cycles also the yield of the negative controls, although no visible amplification was observed, was purified, and cloned. Clones were randomly picked, suspended in PCR grade water and selected for the correct insert size by PCR with vector primers. Approximately 50 positive clones per depth were sequenced with the vector primer M13 Forward. Sequencing reactions were run using ABI BigDye on an ABI Prism 3100 genetic analyzer (Applied Biosystems, Foster City, CA, USA).
Phylogenetic Analysis
For sequence alignment and phylogenetic tree reconstruction sequences were analyzed with the BioEdit program1 and the software ARB2. Briefly, after removal of the vector sequence, sequences were aligned and clustered. Phylogenetic calculations for the partial aprA genes were generated from 128–132 deduced amino acids sequences using maximum-likelihood analyses with a 25% positional conservation filter. For the phylogenetic analysis of the partial dsrA sequences first a maximum-likelihood tree was generated from dsrAB sequences of full length (approximately 650 amino acids), than successively single partial dsrA sequences (73 amino acids) were added to the tree using a 25% positional conservation filter.
Nucleotide Accession Numbers
The dsrA and aprA gene sequences obtained in this study were submitted to the DDBJ/EMBL/GenBank nucleotide databases under the accession numbers HE575209–HE575212 and HE575674–HE575681 for aprA sequences and HE575682–HE575732 for dsrA sequences.
Results and Discussion
In this study the abundance and diversity of the functional genes for adenosine 5′-phosphosulfate reductase (aprA) and dissimilatory sulfite reductase (dsrA) of SRP were analyzed in marine sediments from the Black Sea, and the Peru continental margin, including deep biosphere sediments (ODP site 1227). For aprA quantification a Q-PCR assay was designed. The evaluation results for this assay are followed by data on the abundance and diversity of aprA and dsrA in sediments. For comparison and interpretation, 16S rRNA gene copy numbers of total bacteria from a previous study (Blazejak and Schippers, 2010) have been included here.
Evaluation of the Q-PCR Assay for aprA
Amplification quantities of the standard ranged from 1.0 × 101 to 1.0 × 107 molecules with a correlation coefficient of 0.996. The efficiency of the PCR reactions was 96%. Detection of contaminant DNA in the negative control was not observed. In our experiments the detection limit was set to 1.0 × 102 molecules. This could be lowered to 1.0 × 101 still ensuring reliable detection values since no contaminant DNA in the negative controls was identified. Detection limits for gene quantification by PCR for functional genes can range up to 10 copies per reaction (Vaerman et al., 2004; Bustin et al., 2009). However one critical limitation of PCR-based methods is their sensitivity to compounds that are co-extracted with the DNA from environmental samples, in particular from sediments and soils, that may influence and inhibit the real-time PCR-process. For example humic acids can hamper the PCR reaction and impair fluorescence, and metal ions can inhibit DNA polymerases (Lindberg et al., 2007) whereby the detection limit is lowered. The maximum fluorescence signal of the melting curve occurred at a temperature of 87°C. Melting curves were analyzed after each assay and always showed a single peak, verifying the specificity of the PCR amplification.
Quantification of the Functional Genes aprA and dsrA of SRP and 16S rRNA of Total Bacteria in Marine Sediment Samples
Depth profiles of DNA copy numbers of the functional genes aprA and dsrA as marker for sulfate-reducing prokaryotes (SRP) and the 16S rRNA gene of total Bacteria are shown in Figure 1 for three sediment sites, surface (site 2MC, 0–0.35 mbsf) and deep (site 1227, 0–121.4 mbsf) sediments on the Peru margin, and in the Black Sea (site 20, 0–5.8 mbsf). The copy numbers of all genes decreased with sediment depth in different depth gradients. An important finding of this study was that the depth profiles of copy numbers of both functional genes, aprA and dsrA, were almost equal for all sediment sites expect for the ODP site 1227 below 40 mbsf. Congruent SRP quantification profiles based on independent Q-PCR analysis of two functional genes imply that no SRP have been overlooked, and that the results are close to the actual SRP gene density in the subsurface. Two independent Q-PCR assays with different primers are very unlikely to generate identical PCR biases and quantification profiles by chance.
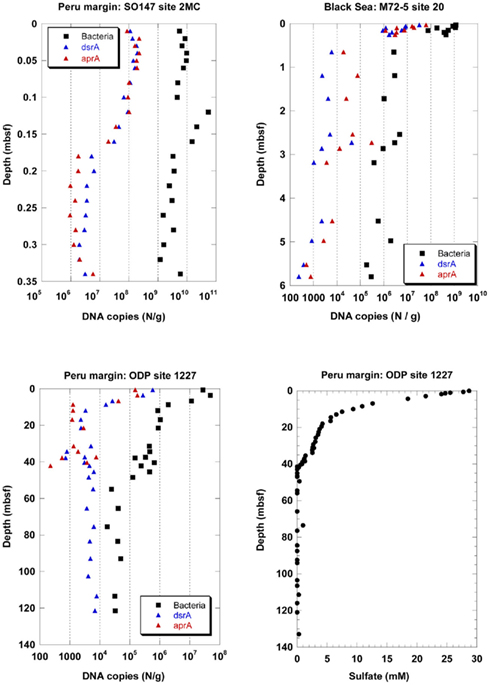
Figure 1. Depth profiles of DNA copy numbers of the functional genes aprA and dsrA as marker for sulfate-reducing prokaryotes (SRP) and the 16S rRNA gene of total Bacteria at three sediment sites, surface (site 2MC, 0–0.35 mbsf) and deep (site 1227, 0–121.4 mbsf) sediments on the Peru margin, and in the Black Sea (site 20, 0–5.8 mbsf), and depth profile of pore water sulfate concentrations at site 1227 (0–135 mbsf, D’Hondt et al., 2004) on the Peru margin. ■, Bacteria;  , dsrA;
, dsrA;  , aprA.
, aprA.
In the Black Sea at site 20, all gene copy numbers decreased rapidly within 65 cm from the sediment surface. The dsrA and aprA copy numbers decreased from 107–108 copies/g at the sediment surface to less than 105 copies/g below 0.6 mbsf. They decreased further to less than 104 copies/g below 3 mbsf. The dsrA copy numbers close to the sediment surface were similar to those for another sediment site of the Black Sea (Leloup et al., 2007). Downcore, the numbers in our study decreased toward lower counts than those in the previous study. Similar differences between these two sites were also found for the 16S rRNA gene copy numbers of total Bacteria. While site 20 was located in the central basin of the Black Sea southeast of the peninsula Crimea at 2048 m water depth, the site of the previous study was located west of the peninsula Crimea on the slope at 1024 m water depth. Thus, different organic matter availability may explain the different gene copy numbers in the two studies.
In the Peru continental margin near-surface sediments (site 2MC) the dsrA and aprA copy numbers were very close to each other and exhibited a more pronounced depth gradient than the 16S rRNA gene copy numbers of total Bacteria (Figure 1). The dsrA and aprA copy numbers decreased from more than 108 copies/g at the sediment surface to 106–107 copies/g between 0.18 and 0.34 mbsf. In a previous Q-PCR study of the same site (Schippers and Neretin, 2006), the dsrA and 16S rRNA gene copy numbers of total Bacteria copy numbers were similar to those of this new study.
In the deeply buried Peru margin sediment (site 1227) the dsrA and aprA copy numbers decreased from 105–106/g sediment at the top of the core at 0.6 mbsf to less than 104/g sediment at 10 mbsf. These numbers for both genes stay steady up to 35 mbsf. Below 35 mbsf the run of the curves are different. After a slight increase of the aprA gene copy numbers between 37–40 mbsf they drop to less than 103/g sediment at 42 mbsf and are not more detectable underneath this depth. In contrast, dsrA copy numbers below 104 copies/g sediment are still observed up to the depth of 121 mbsf. For all samples between 10–121 mbsf, dsrA copy numbers remained consistent in this range. In contrast, dsrA was only patchily detected (5 out of 19 samples) in the previous study (Schippers and Neretin, 2006). The dsrA values in the deeper sediment are close to the detection limit of the Q-PCR method. Thus, slight differences in the efficiency of DNA extraction from the sediment or differences in the total amount of sediment used for DNA extraction may explain this discrepancy.
The 16S rRNA gene copy numbers of total Bacteria exceeded those of the functional genes at all sediment depths, and allowed to calculate the proportion of SRP to total Bacteria. The aprA and dsrA copy numbers comprised in average 0.5–1% of the 16S rRNA gene copy numbers of total Bacteria in the sediments of the Black Sea and those from the Peru continental margin up to a depth of ca. 40 mbsf. Below, only dsrA (but not aprA) was detected with copy numbers of less than 104/g sediment, comprising ca. 14% of the 16S rRNA gene copy numbers of total Bacteria. In other marine sediments sulfate reducers contributed to <1–30% to the prokaryotic community based on Q-PCR, FISH, or rRNA slot blot hybridization analyses (Sahm et al., 1999; Ravenschlag et al., 2000; Knittel et al., 2003; Schippers and Neretin, 2006; Leloup et al., 2007, 2009; Wilms et al., 2007; Gittel et al., 2008; Julies et al., 2010; Schippers et al., 2010). Overall our Q-PCR analysis of the functional genes revealed that SRP are a minor part of the prokaryotic community in the Peru margin sediments, in agreement with clone library data (Parkes et al., 2005; Inagaki et al., 2006; Webster et al., 2006). Based on Q-PCR analysis of the same sediment samples especially the bacterial groups Chloroflexi and/or candidate division JS-1 were shown to be dominant (Blazejak and Schippers, 2010), while Archaea, Eukarya, and the Fe(III)- and Mn(IV)-reducing bacteria of the family Geobacteraceae (Inagaki et al., 2006; Schippers and Neretin, 2006) were of minor abundance.
Active sulfate reduction for the two Peru margin sites up to a depth of ca. 40 mbsf was confirmed by pore water sulfate profiles and sulfate reduction rate measurements (Böning et al., 2004; D’Hondt et al., 2004; Schippers et al., 2005). At ca. 40 mbsf sulfate is reduced by methane oxidation (sulfate–methane transition zone) and a slight maximum of 16S rRNA genes was detected (Schippers et al., 2005; Schippers and Neretin, 2006; Sørensen and Teske, 2006; Teske and Sørensen, 2008). This maximum is not reflected by higher copy numbers of the functional genes dsrA or aprA of SRP indicating that sulfate-dependent anaerobic methane oxidation is not linked to a SRP population peak.
The detection of dsrA of SRP below ca. 40 mbsf was surprising because sulfate as the electron acceptor for active SRP was not detectable in the pore water from ca. 40–121 mbsf of site 1227 (Figure 1). There are three possibilities to explain this finding: 1. The detected dsrA was not extracted from living cells but is part of fossil DNA, persisting adsorbed to sediment particles over geological time scales as previously discussed (Inagaki et al., 2005; Schippers and Neretin, 2006; Schippers et al., 2010); 2. The dsrA originated from living SRP which use another electron acceptor than sulfate, e.g., Fe(III) as shown for several genera of SRP (Vandieken et al., 2006); 3. Low amounts of sulfate might be provided by anoxic oxidation of sulfides. The sulfate formed by this process is constantly consumed by SRP, thus it remained undetectable in the pore water.
We believe that the third possibility is most relevant. On the one hand very low rates of sulfate reduction have been measured with sulfate radiotracer for site 1227 even below 40 mbsf (Schippers et al., 2005). On the other hand, stable isotope data of oxygen and sulfate for sediment and experimental studies support a deep anoxic sulfur cycle. Sulfide oxidation occurs with reactive iron or manganese oxides as oxidant in deeply buried sediments (Bottrell et al., 2000, 2008; Schippers and Jørgensen, 2001; Riedinger et al., 2010; Holmkvist et al., 2011a,b).
Diversity of the Functional Genes aprA and dsrA in Sediment Samples
To analyze not only the abundance but also the diversity of SRP, their metabolic key genes, aprA and dsrA genes, were cloned. Sediment samples of three depths at the Peru margin (site 1227), 3.6, 65.3, and 121.4 mbsf, and four depths in the Black Sea (site 20), 0.15, 2.7, 4.5, and 5.8 mbsf were selected for the study. Sequence analysis of the isolated aprA and dsrA sequences showed their relationship to aprA or dsrA genes from characterized SRP indicating that they also originate from SRP. Although high numbers of PCR cycles were required (up to 35 cycles for dsrA amplification), no visible amplification was observed in the negative controls. Sequences of a few clones obtained from the negative controls showed that only primer sequences were inserted into the cloning vectors. Thus, despite the high PCR cycle number no contamination was noted.
For aprA analysis, 50 clones from a Peru margin sediment at 3.6 mbsf, and 24–45 clones from each Black Sea sediment depth were sequenced. Sequences were grouped into distinct clone families based on their sequence similarities and their allocation within the phylogenetic tree after algorithmic calculations. Sequence similarities within a clone family as well as the similarity to the next relative sequence of a cultivated bacterium are shown in Table 1. For phylogenetic tree reconstruction all sequences were used; however only one representative sequence of each clone family is presented (Figure 2).
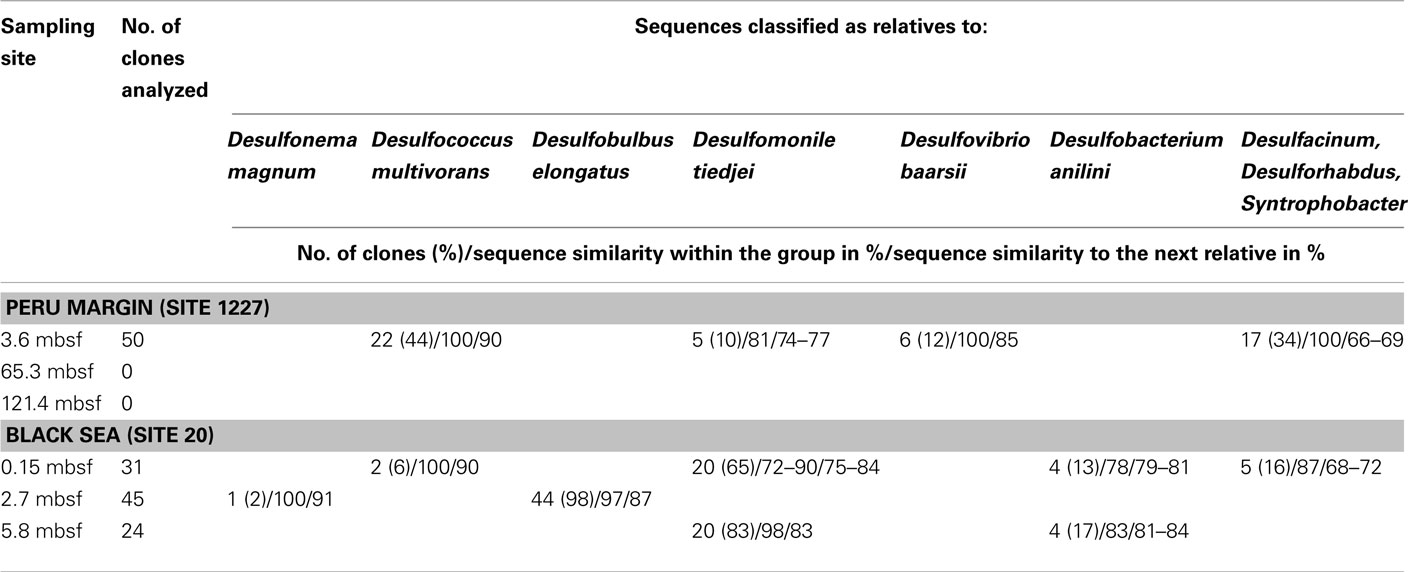
Table 1. Gene aprA clone library data for three sediment samples of the Peru margin and the Black Sea each.
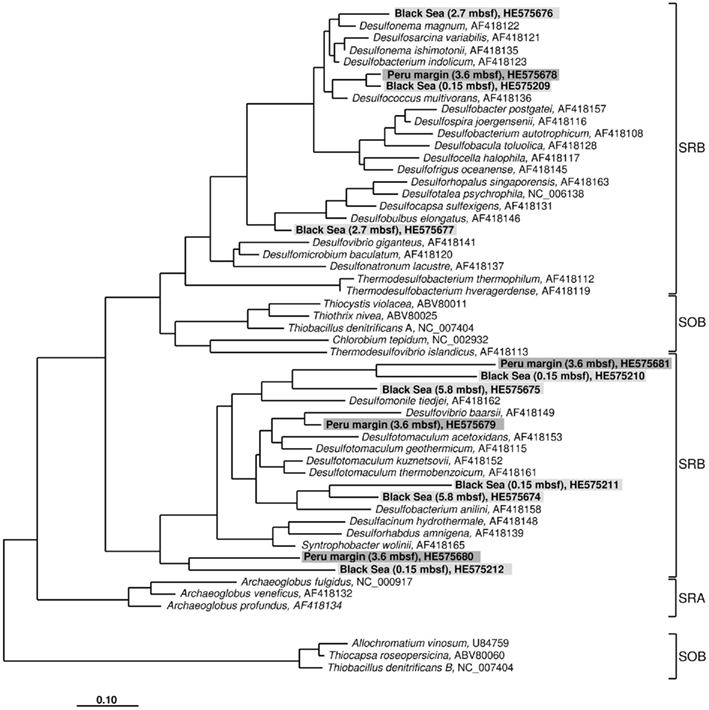
Figure 2. Gene aprA phylogeny based on deduced amino acid sequences of the aprA gene coding for the alpha subunit of the adenosine 5′ -phosphosulfate reductase. Sequences from this study are highlighted in light gray (Black Sea sediments) and dark gray (Peru margin sediments). Scale bar = 0.10 estimated substitutions per site. SOB, sulfide-oxidizing bacteria; SRB, sulfate-reducing bacteria; SRA, sulfate-reducing archaea.
The aprA sequences isolated from 3.6 mbsf depth on the Peru margin were classified into four distinct clone families showing a close relationship to the sulfate-reducing bacteria (SRB) Desulfococcus multivorans, Desulfomonile tiedjei, Desulfovibrio baarsii, and a cluster including the genera Desulfacinum, Desulforhabdus, and Syntrophobacter (Figure 2; Table 1). In deeper sediment layers, at 65.3 and 121.4 mbsf, the aprA gene was not amplified although a high number of cycles (up to 35) was applied. This result corresponds to the absence of quantitative data of the aprA using the newly designed Q-PCR assay (Figure 1).
In the Black Sea sediment a slight decline of diversity with depth could be observed. Near-surface (0.15 mbsf) aprA sequences were assigned into four distinct clone families: D. multivorans, D. tiedjei, Desulfobacterium anilini, and a cluster including the genera Desulfacinum, Desulforhabdus, and Syntrophobacter. In contrast, sequences from 2.7 and 5.8 mbsf formed only two clone groups each, related to Desulfonema magnum and Desulfobulbus elongatus, and Desulfomonile tiedjei and Desulfobacterium anilini, respectively (Figure 2; Table 1).
Almost all isolated aprA sequences showed a close relationship to aprA sequences of cultivated, well characterized SRB of the Deltaproteobacteria indicating that they also originate from bacteria with a same metabolism. For two sequences (accession numbers HE575680 and HE575212), their relationship to known organisms is difficult to predict because they form a separate branch and are only distantly related to aprA sequences of characterized SRB of the Deltaproteobacteria and Gram-positive SRB of the genus Desulfotomaculum (Figure 2; Table 1).
The dsrA sequences could be amplified from sediment samples of all analyzed depths from the Peru margin and the Black Sea (Table 2). Up to 51 clone sequences per depth were included in the phylogenetic analysis. Because of the high PCR cycle number of up to 35 cycles, the negative controls was also cloned although no PCR bands were observed. Sequences obtained from these negative controls showed that only primer sequences were inserted into the cloning vector, thus despite the high PCR cycle number no contamination was found. Phylogenetic analysis showed that all isolated dsrA sequences were closely related to the metabolic gene dsrA. The dsrA sequences isolated from the Peru margin and the Black Sea sediments were classified into eight clone families, showing overall a higher diversity than the isolated aprA sequences. Sequence similarities within a clone family as well as the similarity to the next relative sequence of cultivated prokaryote are shown in Table 2. For phylogenetic tree reconstruction all sequences were used however only one representative sequence of each clone family is presented in Figure 3. Except for three clone families, sequences belonging to all other groups are closely related to dsrA sequences isolated from SRP of Deltaproteobacteria showing for some sequences habitat specificity. For example: dsrA sequences related to Desulfovibrio acrylicus, Desulfohalobium utahense, and to the genera Desulfobulbus, Desulfacinum, and Syntrophobacter were only found in sediments from the Peru margin, whereas sequences related to the genus Desulfomicrobium and to Desulfoarculus baarsii were found exclusively in sediments from the Black Sea. A comparatively high proportion of dsrA sequences (20%) related to D. acrylicus were found in Peru margin deeply buried sediments at 121 mbsf. A specific feature of this anoxic, sulfate-reducing bacterium is the ability to switch from sulfate to acrylate reduction once this is energetically more favorable (van der Maarel et al., 1996). In contrast to dsrA sequences showing habitat specificity, sequences related to Desulfococcus oleovorans, Desulfobacterium autotrophicum, and Desulfotalea psychrophila were detected in sediments at both sites. Members of the genera Desulfococcus and Desulfobacterium belong to the family of Desulfobacteraceae that are known to be able to oxidize a great variety of different electron donors completely to CO2. Thus, they successfully inhabit anoxic marine environments such as Black Sea and Peru margin sediments (Ravenschlag et al., 2000; Liu et al., 2003; Mußmann et al., 2005; Kondo et al., 2007; Leloup et al., 2007, 2009) or the anoxic water column of the Black Sea (Vetriani et al., 2003; Neretin et al., 2007) and other marine habitats (Kondo et al., 2007). Besides dsrA sequences affiliated to Desulfobacteraceae, also numerous aprA sequences belonging to this family were detected in samples from the Black Sea and Peru margin sediments indicating that bacteria of this community play an important role in sulfate reduction in these sediments.
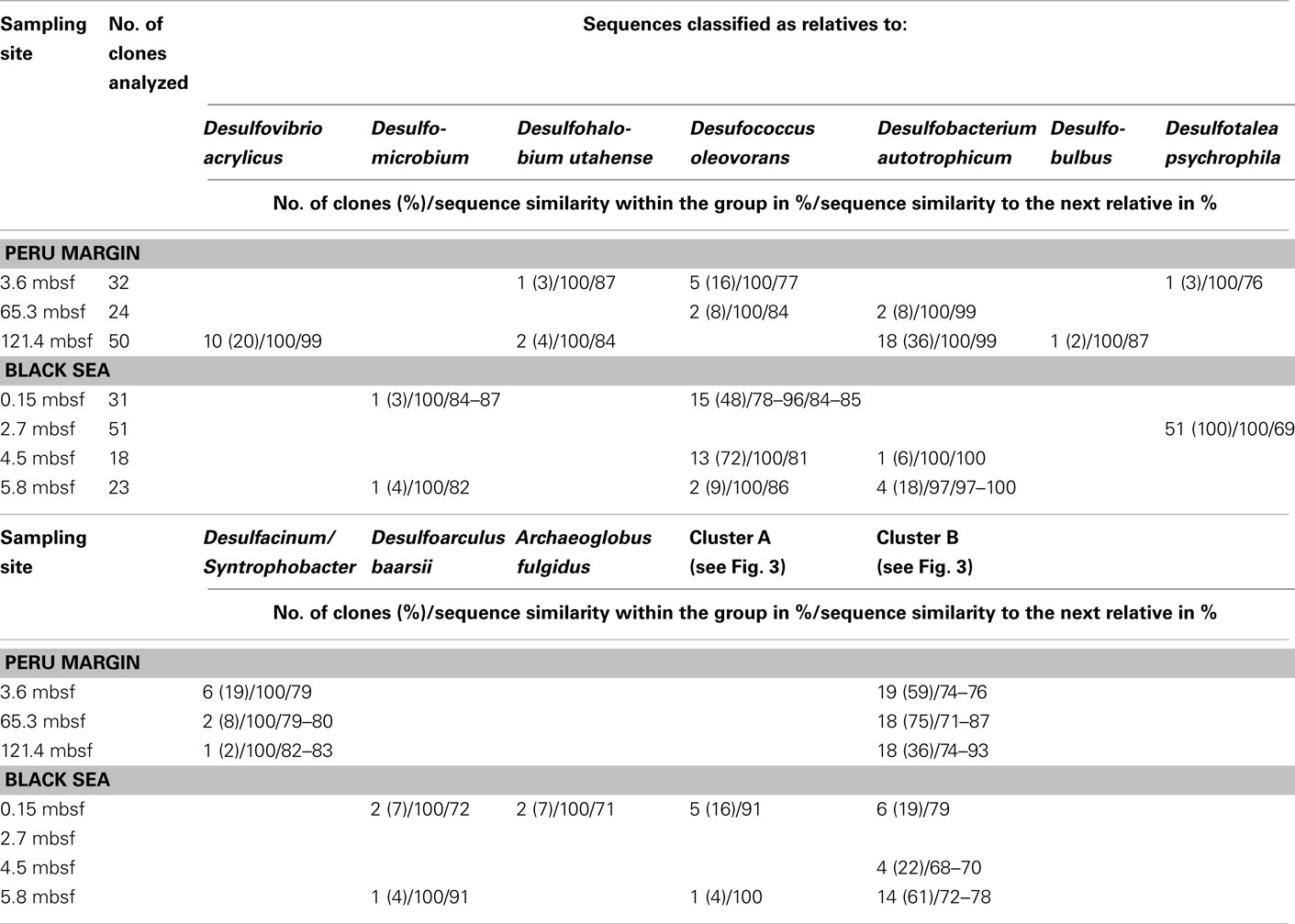
Table 2 Gene dsrA clone library data for three sediment samples of the Peru margin and four sediment samples of the Black Sea.
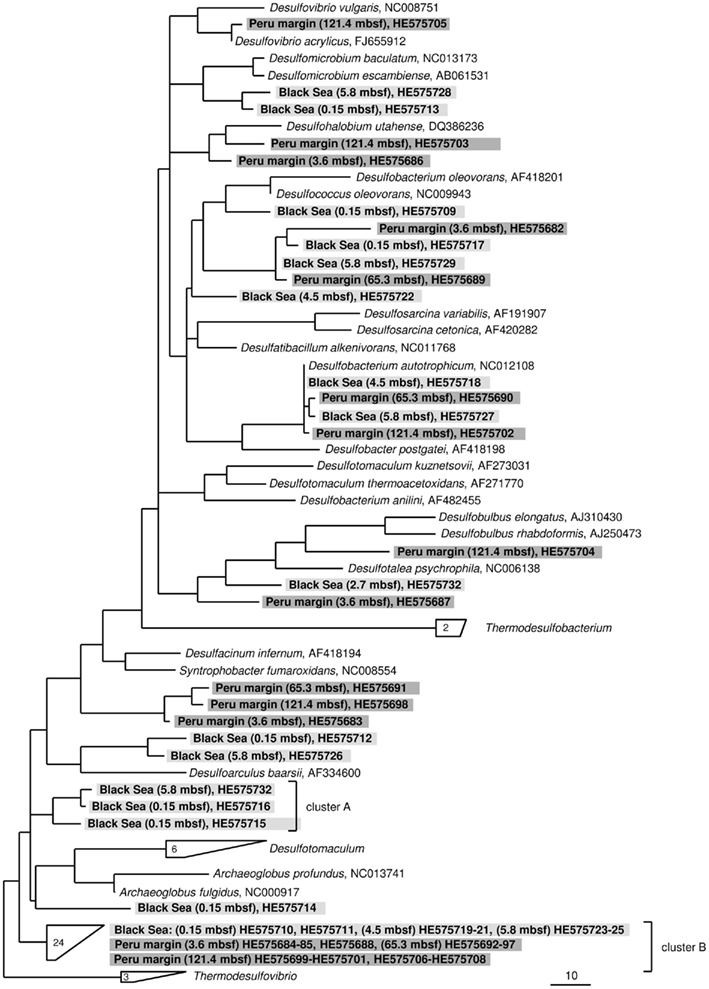
Figure 3. Gene dsrA phylogeny based on deduced amino acid sequences of the dsrA gene coding for the alpha subunit of dissimilatory (bi)sulfite reductase. Sequences from this study are highlighted in light gray (Black Sea sediments) and dark gray (Peru margin sediments). Scale bar = 0.10 estimated substitutions per site.
Only two dsrA sequences, isolated from sediment of the Black Sea, showed a distant relationship (71% amino acid similarity) to the sulfate-reducing archaeon (SRA) Archaeoglobus fulgidus. Primarily SRA of the genus Archaeoglobus were isolated from marine hydrothermal systems, North Sea oil fields, and from petroleum hydrocarbon-rich Guaymas Basin sediments off the coast of Mexico (Hartzell and Reed, 2006). A few dsrA sequences, allocated within the same cluster, could be isolated from other habitats as from the Nankai Trough deep-sea and Black Sea sediments (Kaneko et al., 2007; Leloup et al., 2007), and showed however also only a distant relationship to dsrA sequences of the genus Archaeoglobus. Because of this distant relationship and the fact that Black Sea sediments are not a typical habitat for SRA, the affiliation of these sequences to the genus Archaeoglobus is questionable. SRA may play only a minor role in these sediments because of the low numbers of detected dsrA sequences, and the lack of aprA clones related to SRA.
The affiliation of dsrA sequences of two clone families, named cluster A and B, could not be clearly identified. Sequences within the cluster A form a clearly separated branch based on their unique sequence signature and showed only distant similarities to aprA of A. fulgidus (70–71% amino acid similarity). The cluster B was generated by 24 different dsrA sequences isolated from sediments of both habitats from each depth. Within this cluster numerous dsrA clone sequences isolated from different marine sediments as from deep-sea sediments from the Nankai Trough (Kaneko et al., 2007) and the Guaymas Basin (Dhillon et al., 2003), but also from salt marsh sediments (Bahr et al., 2005), and fen soil (Loy et al., 2004) are represented (data not showed). The closest described relatives based on amino acid similarity searches are dsrA sequences from the archaeon A. fulgidus (65–76% amino acid sequence similarity), the SRB of the genus Thermodesulfovibrio of the class Nitrospira (63–76% amino acid sequence similarity), and gram-positive SRB of the genus Desulfotomaculum (61–73% amino acid sequence similarity). Because of the distant relationship to dsrA sequences from characterized microorganisms, the conclusion about the affiliation of the dsrA sequences of the cluster B to either SRA or SRB remains speculative. The common distribution of these dsrA sequences along the depth profiles of the two habitats and their high proportion within the clone libraries, up to 75%, argue for a significant role in sulfate/sulfite reduction of these microorganisms in marine sediments. They seem to be generalists and can adapt to a wide range of sulfate concentrations and electron donors, and it is tempting to suppose that they are a dominant group within the community of sulfate reducers in the anoxic sediments from the Black Sea and the Peru margin. Another unlike possibility could be that these dsrA sequences originate from microorganisms, which still contain this gene but have lost their ancestral ability of dissimilatory sulfate/sulfite reduction as shown for some members of Desulfotomaculum subcluster Ih (Imachi et al., 2006). This scenario would explain the unsuccessful cultivation of sulfate reducers related to the cluster B so far, because of incorrect selection of electron acceptors.
In summary of the dsrA diversity study, it can be concluded that this metabolic gene for the sulfate/sulfite reduction could be detected in all analyzed sediment layers demonstrating a high diversity. Even in deeply buried sediments from the Peru margin in depths of 65 and 121 mbsf dsrA sequences were identified. Surprisingly, also in such an extreme habitat these sequences showed a high diversity belonging to well characterized genera as Desulfobacterium and Desulfococcus of the family Desulfobacteraceae, and Desulfovibrio, but also to novel deep-branching clusters A and B which phylogenetic affiliation and thus metabolism remain hidden. The presence of aprA and dsrA sequences affiliated to the same phylogenetic clusters, for example genera of the family Desulfobacteraceae or the genera Syntrophobacter and Desulfacinum, showed that sulfate reducers of equal phylogenesis were detected. This is particularly demonstrative for aprA and dsrA clone libraries from the Black Sea sediment of 2.7 mbsf depth showing that almost all aprA sequences (98%) and all dsrA sequences (100%) are affiliated either to D. elongatus or to D. psychrophila, respectively. The aprA and dsrA phylogeny of these two SRP of the family Desulfobulbaceae showed a close relationship to each other indicating that aprA and dsrA sequences detected in the Black Sea sediments could originate from the same bacterium.
Conclusively, although SRP do not belong to the most abundant prokaryotic groups, this study shows that they represent an inherent, diverse part of the microbial community in Black Sea sediments and the deep biosphere of Peru margin.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the ODP Leg 201, RV Meteor M72-5 personnel, and especially Gerrit Köweker for sediment sampling. Special thanks to Rudolf Amann for providing laboratory space at MPI Bremen for cloning. This research used samples and data provided by the ODP which is sponsored by the US National Science Foundation (NSF) and participating countries under management of Joint Oceanographic Institutions (JOI), Inc. This work was supported by the German Research Foundation (DFG) priority program IODP/ODP grant SCHI 535/5 to Axel Schippers.
Footnotes
References
Agrawal, A., and Lal, B. (2009). Rapid detection and quantification of bisulfite reductase genes in oil field samples using real-time PCR. FEMS Microbiol. Ecol. 69, 301–312.
Bahr, M., Crump, B. C., Klepac-Ceraj, V., Teske, A., Sogin, M. L., and Hobbie, J. E. (2005). Molecular characterization of sulfate-reducing bacteria in a New England salt marsh. Environ. Microbiol. 7, 1175–1185.
Ben-Dov, E., Brenner, A., and Kushmaro, A. (2007). Quantification of sulfate-reducing bacteria in industrial wastewater, by real-time polymerase chain reaction (PCR) using dsrA and apsA genes. Microbiol. Ecol. 54, 439–451.
Blazejak, A., Erseus, C., Amann, R., and Dubilier, N. (2005). Coexistence of bacterial sulfide oxidizers, sulfate reducers, and spirochetes in a gutless worm (Oligochaeta) from the Peru margin. Appl. Environ. Microbiol. 71, 1553–1561.
Blazejak, A., and Schippers, A. (2010). High abundance of JS-1- and Chloroflexi-related Bacteria in deeply buried marine sediments revealed by quantitative, real-time PCR. FEMS Microbiol. Ecol. 72, 198–207.
Böning, P., Brumsack, H.-J., Böttcher, M. E., Schnetger, B., Kriete, C., Kallmeyer, J., and Borchers, S. L. (2004). Geochemistry of Peruvian near-surface sediments. Geochim Cosmochim Acta 68, 4429–4451.
Bottrell, S. H., Böttcher, M. E., Schippers, A., Parkes, R. J., Jørgensen, B. B, Raiswell, R., Telling, J., and Gehre, M. (2008). Abiotic sulfide oxidation via manganese fuels the deep biosphere. Goldschmidt Conference Abstracts. Geochim. Cosmochim. Acta 72(Suppl. 1), :A102.
Bottrell, S. H., Parkes, R. J., Cragg, B. A., and Raiswell, R. (2000). Isotopic evidence for anoxic pyrite oxidation and stimulation of bacterial sulphate reduction in marine sediments. J. Geol. Soc. London 157, 711–714.
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., Mueller, R., Nolan, T., Pfaffl, M. W., Shipley, G. L., Vandesompele, J., and Wittwer, C. T. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622.
Castresana, J., and Moreira, D. (1999). Respiratory chains in the last common ancestor of living organisms. J. Mol. Evol. 49, 453–460.
Dhillon, A., Teske, A., Dillon, J., Stahl, D. A., and Sogin, M. L. (2003). Molecular characterization of sulfate-reducing bacteria in the Guaymas Basin. Appl. Environ. Microbiol. 69, 2765–2772.
D’Hondt, S. L., Jørgensen, B. B., Miller, D. J., Batzke, A., Blake, R., Cragg, B. A., Cypionka, H., Dickens, G. R., Ferdelman, T. G., Hinrichs, K. U., Holm, N. G., Mitterer, R., Spivack, A., Wang, G., Bekins, B., Engelen, B., Ford, K., Gettemy, G., Rutherford, S. D., Sass, H., Skilbeck, C. G., Aiello, I. W., Guèrin, G., House, C. H., Inagaki, F., Meister, P., Naehr, T., Niitsuma, S., Parkes, R. J., Schippers, A., Smith, D. C., Teske, A., Wiegel, J., Padilla, C. N., and Solis Acosta, J. L. (2004). Distributions of microbial activities in deep subseafloor sediments. Science 306, 2216–2221.
D’Hondt, S. L., Jørgensen, B, B., Miller, D. J., Shipboard Scientific Party. (2003). Leg 201 Initial report – controls on microbial communities in deeply buried sediments, Eastern Equatorial Pacific and Peru margin. Proc. ODP, Init. Repts., 201: College Station, TX (Ocean 131 Drilling Program)..
Engelen, B., Ziegelmüller, K., Wolf, L., Köpke, B., Gittel, A., Cypionka, H., Treude, T., Nakagawa, S., Inagaki, F., Lever, M. A., and Steinsbu, B. O. (2008). Fluids from the oceanic crust support microbial activities within the deep biosphere. Geomicrobiol. J. 25, 56–66.
Ferdelman, T. G., Lee, C., Pantoja, S., Harder, J., Bebout, B. M., and Fossing, H. (1997). Sulfate reduction and methanogenesis in a Thioploca-dominated sediment off the coast of Chile. Geochim. Cosmochim. Acta 61, 3065–3079.
Friedrich, M. W. (2002). Phylogenetic analysis reveals multiple lateral transfers of adenosine-5′-phosphosulfate reductase genes among sulfate-reducing microorganisms. J. Bacteriol. 184, 278–289.
Fry, J. C., Parkes, J. R., Cragg, B. A., Weightman, A. J., and Webster, G. (2008). Prokaryotic biodiversity and activity in the deep subseafloor biosphere. FEMS Microbiol. Ecol. 66, 181–196.
Geets, J., Borremans, B., Diels, L., Springael, D., Vangronsveld, J., van der Lelie, D., and Vanbroekhoven, K. (2006). DsrB gene-based DGGE for community and diversity surveys of sulfate-reducing bacteria. J. Microbiol. Methods 66, 194–205.
Gittel, A., Mußmann, M., Sass, H., Cypionka, H., and Könneke, M. (2008). Identity and abundance of active sulfate-reducing bacteria in deep tidal flat sediments determined by directed cultivation and CARD-FISH analysis. Environ. Microbiol. 10, 2645–2658.
Holmkvist, L., Kamyshny, A. Jr., Vogt, C., Vamvakopoulos, K., Ferdelman, T. G., and Jørgensen, B. B. (2011a). Sulfate reduction below the sulfate–methane transition in Black Sea sediments. Deep Sea Res. Part I Oceanogr. Res. Pap. 58, 493–504.
Holmkvist, L., Ferdelman, T. G., and Jørgensen, B. B. (2011b). A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark). Geochim. Cosmochim. Acta 75, 3581–3599.
Imachi, H., Sekiguchi, Y., Kamagata, Y., Loy, A., Qiu, Y. L., Hugenholtz, P., Kimura, N., Wagner, M., Ohashi, A., and Harada, H. (2006). Non-sulfate-reducing, syntrophic bacteria affiliated with Desulfotomaculum cluster I are widely distributed in methanogenic environments. Appl. Environ. Microbiol. 72, 2080–2091.
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., Suzuki, M., Takai, K., Delwiche, M., Colwell, F. S., Nealson, K. H., Horikoshi, K., D’Hondt, S., and Jørgensen, B. B. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific ocean margin. Proc. Natl. Acad. Sci. U.S.A. 103, 2815–2820.
Inagaki, F., Okada, H., Tsapin, A. I., and Nealson, K. H. (2005). The paleome: a sedimentary genetic record of past microbial communities. Astrobiology 5, 141–153.
Jørgensen, B. B. (1982). Mineralization of organic matter in the sea bed; the role of sulphate reduction. Nature 296, 643–645.
Jørgensen, B. B., D’Hondt, S., and Miller, D. J. (eds). (2005). Proc. ODP, Sci. Results, 201. Available at: http://www-odp.tamu.edu/publications/201_SR/synth/synth.htm
Jørgensen, B. B., Weber, A., and Zopfi, J. (2001). Sulfate reduction and anaerobic methane oxidation in Black Sea sediments. Deep Sea Res. Part 1 Oceanogr. Res. Pap. 48, 2097–2120.
Julies, E. M., Fuchs, B. M., Arnosti, C., and Brüchert, V. (2010). Organic carbon degradation in anoxic organic-rich shelf sediments: biogeochemical rates and microbial abundance. Geomicrobiol. J. 27, 303–314.
Kaneko, R., Hayashi, T., Tanahashi, M., and Naganuma, T. (2007). Phylogenetic diversity and distribution of dissimila- tory sulfite reductase genes from deep-sea sediment cores. Mar. Biotechnol. 9, 429–436.
Knittel, K., Boetius, A., Eilers, H., Lochte, K., Pfannkuche, O., Linke, P., and Amann, R. (2003). Activity, distribution, and diversity of sulfate reducers and other bacteria in sediments above gas hydrate (Cascadia margin, Oregon). Geomicrobiol. J. 20, 269–294.
Knoblauch, C., Jørgensen, B. B., and Harder, J. (1999). Community size and metabolic rates of psychrophilic sulfate-reducing bacteria in Arctic marine sediments. Appl. Environ. Microbiol. 65, 4230–4233.
Kondo, R., Purdy, K. J., Silva, S. Q., and Nedwell, D. B. (2007). Spatial dynamics of sulphate-reducing bacterial compositions in sediment along a salinity gradient in a UK estuary. Microbes Environ. 22, 11–19.
Kondo, R., Shigematsu, K., and Butani, J. (2004). Rapid enumeration of sulphate-reducing bacteria from aquatic environments using real-time PCR. Plankton Benthos Res. 3, 180–183.
Leloup, J., Fossing, H., Kohls, K., Holmkvist, L., Borowski, C., and Jørgensen, B. B. (2009). Sulfate-reducing bacteria in marine sediment (Aarhus Bay, Denmark): abundance and diversity related to geochemical zonation. Environ. Microbiol. 11, 1278–1291.
Leloup, J., Loy, A., Knab, N. J., Borowski, C., Wagner, M., and Jorgensen, B. B. (2007). Diversity and abundance of sulfate-reducing microorganisms in the sulfate and methane zones of a marine sediment, Black Sea. Environ. Microbiol. 9, 131–142.
Lindberg, E., Albrechtsen, H. J., and Jacobsen, C. S. (2007). Inhibition of real-time PCR in DNA extracts from aquifer sediment. Geomicrobiol. J. 24, 343–352.
Liu, X., Bagwell, C. E., Wu, L., Devol, A. H., and Zhou, J. (2003). Molecular diversity of sulfate-reducing bacteria from two different continental margin habitats. Appl. Environ. Microbiol. 69, 6073–6081.
Loy, A., Kusel, K., Lehner, A., Drake, H. L., and Wagner, M. (2004). Microarray and functional gene analyses of sulfate-reducing prokaryotes in low-sulfate, acidic fens reveal cooccurrence of recognized genera and novel lineages. Appl. Environ. Microbiol. 70, 6998–7009.
Meyer, B., and Kuever, J. (2007). Phylogeny of the alpha and beta subunits of the dissimilatory adenosine-5′- phosphosulfate (APS) reductase from sulfate-reducing prokaryotes – origin and evolution of the dissimilatory sulfate-reduction pathway. Microbiology 153, 2026–2044.
Mußmann, M., Ishii, K, Rabus, R., and Amann, R. (2005). Diversity and vertical distribution of cultured and uncultured deltaproteobacteria in an intertidal mud flat of the Wadden Sea. Environ. Microbiol. 7, 405–418.
Nadkarni, M. A., Martin, F. E., Jacques, N. A., and Hunter, N. (2002). Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148, 257–266.
Neretin, L. N., Abed, R. M. M., Schippers, A., Schubert, C. J., Kohls, K., and Kuypers, M. M. M. (2007). Inorganic carbon fixation by sulfate-reducing bacteria in the Black Sea water column. Environ. Microbiol. 9, 3019–3024.
Neretin, L. N., Schippers, A., Pernthaler, A., Hamann, K., Amann, R., and Jørgensen, B. B. (2003). Quantification of dissimilatory (bi)sulphite reductase gene expression in Desulfobacterium autotrophicum using real-time RT-PCR. Environ. Microbiol. 5, 660–671.
Nunoura, T., Soffientino, B., Blazejak, A., Kakuta, J., Oida, H., Schippers, A., and Takai, K. (2009). Subseafloor microbial communities associated with rapid turbidite deposition in the Gulf of Mexico continental slope (IODP Expedition 308). FEMS Microbiol. Ecol. 69, 410–424.
Parkes, R. J., Webster, G., Cragg, B. A., Weightman, A. J., Newberry, C. J., Ferdelman, T. G., Kallmeyer, J., Jørgensen, B. B., Aiello, I. W., and Fry, J. C (2005). Deep sub-seafloor prokaryotes stimulated at interfaces over geological time. Nature 436, 390–394.
Ravenschlag, K., Sahm, K., Knoblauch, C., Jørgensen, B. B., and Amann, R. (2000). Community structure, cellular rRNA content and activity of sulfate-reducing bacteria in marine Arctic sediments. Appl. Environ. Microbiol. 66, 3592–3602.
Riedinger, N., Brunner, B., Formolo, M. J., Solomon, E., Kasten, S., Strasser, M., and Ferdelman, T. G. (2010). Oxidative sulfur cycling in the deep biosphere of the Nankai trough, Japan. Geology 38, 851–854.
Sahm, K., Knoblauch, C., and Amann, R. (1999). Phylogenetic affiliation and quantification of psychrophilic sulfate-reducing isolates in marine Arctic sediments. Appl. Environ. Microbiol. 65, 3976–3981.
Schen, Y., Buick, R., and Canfield, D. E. (2001). Isotopic evidence for microbial sulphate reduction in the early Archaean era. Nature 410, 77–81.
Schippers, A., and Jørgensen, B. B. (2001). Oxidation of pyrite and iron sulfide by manganese dioxide in marine sediments. Geochim. Cosmochim. Acta 65, 915–922.
Schippers, A., Köweker, G., Höft, C., and Teichert, B. (2010). Quantification of microbial communities in three forearc sediment basins off Sumatra. Geomicrobiol. J. 27, 170–182.
Schippers, A., and Neretin, L. N. (2006). Quantification of microbial communities in near-surface and deeply buried marine sediments on the Peru continental margin using real-time PCR. Environ. Microbiol. 8, 1251–1260.
Schippers, A., Neretin, L. N., Kallmeyer, J., Ferdelman, T. G., Cragg, B. A., Parkes, R. J., and Jørgensen, B. B. (2005). Prokaryotic cells of the deep sub-seafloor biosphere identified as living bacteria. Nature 433, 861–864.
Sørensen, K. B., and Teske, A. (2006). Stratified communities of active archaea in deep marine subsurface sediments. Appl. Environ. Microbiol. 72, 4596–4603.
Stahl, D. A., Fishbain, S., Klein, M., Baker, B. J., and Wagner, M. (2002). Origins and diversification of sulfate-respiring microorganisms. Antonie Van Leeuwenhoek 81, 189–195.
Teske, A. P. (2006). Microbial community composition in deep marine subsurface sediments of ODP Leg 201: sequencing surveys and cultivations. Proc. Ocean Drill. Prog. Sci. Results 201, Available at: http://www-odp.tamu.edu/publications/201_SR/120/120.htm
Teske, A. P., and Sørensen, K. B. (2008). Uncultured archaea in deep marine marine subsurface sediments: have we caught them all? The ISME J. 2, 3–18.
Thamdrup, B., Rosselló-Mora, R., and Amann, R. (2000). Microbial manganese and sulfate reduction in Black Sea shelf sediments. Appl. Environ. Microbiol. 66, 2888–2897.
Vaerman, J. L., Saussoy, P., and Ingargiola, I. (2004). Evaluation of real-time PCR data. J. Biol. Regul. Homeost. Agents 18, 212–214.
van der Maarel, M. J. E. C., van Bergeijk, S., van Werkhoven, A. F., Laverman, A. M., Meijer, W. G., Stam, W. T., and Hansen, T. A. (1996). Cleavage of dimethylsulfoniopropionate and reduction of acrylate by Desulfovibrio acrylicus sp. nov. Arch. Microbiol. 166, 109–115.
Vandieken, V., Finke, N., and Jørgensen, B. B. (2006). Pathways of carbon oxidation in an Arctic fjord sediment (Svalbard) and isolation of psychrophilic and psychrotolerant Fe(III)-reducing bacteria. Mar. Ecol. Prog. Ser. 322, 29–41.
Vetriani, C., Tran, H. V., and Kerkhof, L. J. (2003). Fingerprinting microbial assemblages from the oxic/anoxic chemocline of the Black Sea. Appl. Environ. Microbiol. 69, 6481–6488.
Wagner, M., Roger, A. J., Flax, J. L., Brusseau, G. A., and Stahl, D. A. (1998). Phylogeny of dissimilatory sulfite reductases supports an early origin of sulfate respiration. J. Bacteriol. 180, 2975–2982.
Webster, G., Blazejak, A., Cragg, B. A., Schippers, A., Sass, H., Rinna, J., Tang, X., Mathes, F., Ferdelman, T., Fry, J. C., Weightman, A. J., and Parkes, R. J. (2009). Subsurface microbiology and biogeochemistry of a deep, cold-water carbonate mound from the Porcupine Seabight (IODP Expedition 307). Environ. Microbiol. 11, 239–257.
Webster, G., Newberry, C. J., Fry, J. C., and Weightman, A. J. (2003). Assessment of bacterial community structure in the deep sub-seafloor biosphere by 16S rDNA-based techniques: a cautionary tale. J. Microbiol. Methods 55, 155–164.
Webster, G., Watt, L. C., Rinna, J., Fry, J. C., Evershed, R. P., Parkes, J., and Weightman, A, J. (2006). A comparison of stable-isotope probing of DNA and phospholipid fatty acids to study prokaryotic functional diversity in sulfate-reducing marine sediment enrichment slurries. Environ. Microbiol. 8, 1575–1589.
Keywords: deep biosphere, real-time PCR, subsurface, ODP, sulfate-reducing prokaryotes, aprA, dsrA
Citation: Blazejak A and Schippers A (2011) Real-Time PCR Quantification and Diversity Analysis of the Functional Genes aprA and dsrA of Sulfate-Reducing Prokaryotes in Marine Sediments of the Peru Continental Margin and the Black Sea. Front. Microbio. 2:253. doi: 10.3389/fmicb.2011.00253
Received: 11 August 2011; Accepted: 29 November 2011;
Published online: 22 December 2011.
Edited by:
Andreas Teske, University of North Carolina at Chapel Hill, USAReviewed by:
Julie A. Huber, Marine Biological Laboratory, USAKasthuri Venkateswaran, NASA-Jet Propulsion Laboratory, USA
Copyright: © 2011 Blazejak and Schippers. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Axel Schippers, Geomicrobiology, Federal Institute for Geosciences and Natural Resources (BGR), Stilleweg 2, 30655 Hannover, Germany. e-mail: axel.schippers@bgr.de
†Present address: Anna Blazejak, Max Planck Institute for Marine Microbiology, Bremen, Germany.