 Amanda J. Martino1 Matthew E. Rhodes1 Jennifer F. Biddle2 Leah D. Brandt1 Lynn P. Tomsho3 Christopher H. House1*
Amanda J. Martino1 Matthew E. Rhodes1 Jennifer F. Biddle2 Leah D. Brandt1 Lynn P. Tomsho3 Christopher H. House1*- 1 Department of Geosciences, Penn State Astrobiology Research Center, Pennsylvania State University, University Park, PA, USA
- 2 School of Marine Science and Policy, College of Earth Ocean and Environment, University of Delaware, Lewes, DE, USA
- 3 Department of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, PA, USA
A degenerate polymerase chain reaction (PCR)-based method of whole-genome amplification, designed to work fluidly with 454 sequencing technology, was developed and tested for use on deep marine subsurface DNA samples. While optimized here for use with Roche 454 technology, the general framework presented may be applicable to other next generation sequencing systems as well (e.g., Illumina, Ion Torrent). The method, which we have called random amplification metagenomic PCR (RAMP), involves the use of specific primers from Roche 454 amplicon sequencing, modified by the addition of a degenerate region at the 3′ end. It utilizes a PCR reaction, which resulted in no amplification from blanks, even after 50 cycles of PCR. After efforts to optimize experimental conditions, the method was tested with DNA extracted from cultured E. coli cells, and genome coverage was estimated after sequencing on three different occasions. Coverage did not vary greatly with the different experimental conditions tested, and was around 62% with a sequencing effort equivalent to a theoretical genome coverage of 14.10×. The GC content of the sequenced amplification product was within 2% of the predicted values for this strain of E. coli. The method was also applied to DNA extracted from marine subsurface samples from ODP Leg 201 site 1229 (Peru Margin), and results of a taxonomic analysis revealed microbial communities dominated by Proteobacteria, Chloroflexi, Firmicutes, Euryarchaeota, and Crenarchaeota, among others. These results were similar to those obtained previously for those samples; however, variations in the proportions of taxa identified illustrates well the generally accepted view that community analysis is sensitive to both the amplification technique used and the method of assigning sequences to taxonomic groups. Overall, we find that RAMP represents a valid methodology for amplifying metagenomes from low-biomass samples.
Introduction
The deep subseafloor biosphere represents a frontier for the discovery of new microbial life, and for investigations of the extent, versatility, and perseverance of life on earth. However, there are many challenges in studying this community of microorganisms, and the past 20 years of study have only begun to produce an understanding of this vast and complex ecosystem. Marine subsurface microorganisms are isolated from the direct energy of sunlight, receive limited nutrients, and sometimes experience extreme pressures and challenging temperatures. Investigations to date suggest that many of these microbes appear to be only distantly related to those we know from the study of surface environments (Sørensen et al., 2004; Inagaki et al., 2006; Lipp et al., 2008). Cultivation studies have produced some useful results (Bale et al., 1997; Mikucki et al., 2003; Toffin et al., 2004), but the majority of microbes in this environment (as well as most microbes on Earth) still evade cultivation efforts. Cultivation-independent methods such as polymerase chain reaction (PCR) amplification and subsequent sequencing directly from environmental DNA hold great promise, and have provided the majority of the information obtained to date (Jørgensen and Boetius, 2007; Orcutt et al., 2011); however, there are still many challenges to overcome in utilizing these methods to their full potential.
Among the available cultivation-independent methods, automated metagenomic sequencing via platforms such as Roche 454, is one of the most promising tools for probing the depths of diversity and exploring metabolic capabilities of subsurface microbes. However, even with recent advances in technology, this type of high-throughput sequencing requires, ideally, at least 500 ng of sample DNA (as per Roche 454 protocol). The relatively low concentrations of cells in the marine subsurface coupled with the difficulties of extracting DNA from marine sediment (Webster et al., 2003) results in quantities of extracted DNA which are often too low for direct, unamplified, metagenomic sequencing.
Over the past couple of decades, researchers have been experimenting with different methods of amplifying genomic DNA. Some of the resulting whole-genome amplification (WGA) methods are modifications to the standard PCR, which reduce its specificity, allowing for a general amplification of DNA. These methods include interspersed repetitive sequence PCR (IRS-PCR; Nelson et al., 1989), primer-extension-preamplification PCR (PEP-PCR; Zhang et al., 1992), improved primer-extension-preamplification PCR (I-PEP-PCR; Dietmaier et al., 1999), degenerate oligonucleotide-primed PCR (DOP-PCR; Telenius et al., 1992), and long products from low DNA quantities DOP-PCR (LL-DOP-PCR; Kittler et al., 2002). In addition to the PCR-based methods, a non-PCR method called multiple displacement amplification (MDA) was developed in attempt to overcome problems with the PCR methods, which included incomplete coverage, amplification artifacts, and DNA too short for some applications (Dean et al., 2002). MDA is an isothermal, strand-displacing reaction employing the phi29 DNA polymerase and random hexamer primers. Several commercial versions of MDA now exist, including the REPLI-g Whole-Genome Amplification Kit (Qiagen) and the GenomiPhi DNA Amplification Kit (GE Healthcare). Two commercial MDA kits, along with PEP-PCR and DOP-PCR, were analyzed for genome coverage bias in a 454 metagenomic sequencing study and all were found to induce significant bias (Pinard et al., 2006). For both microbial genomes utilized in that study, the MDA reactions resulted in the least bias, followed by PEP-PCR, and lastly, DOP-PCR. These results were consistent with those obtained through a TaqMan quantitative PCR analysis of eight genes after amplification of human genomic DNA using MDA, DOP-PCR, and PEP-PCR (Dean et al., 2002).
As a result, MDA-based techniques are most often the method of choice in applications where bias and coverage are significant concerns. For metagenomic sequencing of subseafloor environmental DNA samples, however, the use of MDA as an amplification technique is often problematic. In particular, the tendency of MDA to synthesize a DNA product even in the absence of added cells, means that a reliable negative control for the amplification reaction is very difficult to achieve (Raghunathan et al., 2005). These products that form in the negative controls can be as large as 15 kb and greater (Biddle, 2006). While they have not been sequenced, failure of attempts to PCR amplify specific genes from the products suggests that they are more likely primer-dimer type formations rather than microbial contamination (Biddle, 2006). This may be a consequence of the competitive nature of the reaction, whereby in the absence of anything else of which to anneal, the random hexamers anneal to each other. If this were the case, the problem would be of greatest significance when sample DNA template is very low, and unable to compete with the hexamers for annealing and amplification. It has been shown that the DNA product formed in negative controls can be avoided if the reaction is monitored closely with a qPCR protocol and stopped before amplification in the negative control begins (Biddle et al., 2011). However, in many cases, this occurs after less than 2 h reaction time, and stopping the reaction at this length of time may hinder the amplification of sample DNA as well, particularly when the sample had very small amounts of DNA to begin with (Biddle et al., 2011), as is the case with most deep subseafloor samples.
Due to the difficulties with using available WGA methods for amplifying subseafloor DNA samples, we undertook efforts to develop an alternative method of WGA that may be of use when sequencing low-biomass environmental samples. We hypothesized that the production of DNA in reaction negatives of MDA was a result of the high level of degeneracy of the primers. We recognized, however, that this high level of degeneracy was critical in obtaining amplified DNA with the least amount of bias, which appears to be the reason that the PEP-PCR and MDA methods produce less bias than the PCR methods with more specific primers. Consequently, our strategy was to design a PCR method with primers that would be more degenerate than IRS-PCR and DOP-PCR – and thus applicable to environmental DNA samples, but less degenerate than PEP-PCR and MDA. In addition, with the rapidly increasing use of next generation metagenomic sequencing technology, we aimed to have our new method aid in streamlining the process of preparing samples for metagenomic sequencing using these new technologies. The result was a PCR-based method of WGA utilizing 454 amplicon primers with an attached degenerate region at the 3′ end. We refer to this method as random amplification metagenomic PCR (RAMP). While optimized here for use with Roche 454 sequencing, the developed method may be viewed as a general framework for using PCR for WGA of low-biomass environmental DNA samples in preparation for metagenomic sequencing using next generation sequencing technologies.
Materials and Methods
DNA Extraction
E. coli DNA was extracted from a culture of E. coli Mach1™-T1R cells from a TOPO TA Cloning® Kit (Invitrogen, Inc.). The parental strain of Mach1™-T1R E. coli is the non-K-12, wild-type W strain (ATCC #9637, S. A. Waksman), and the cells contain slight modifications to the genome necessary for the cloning process. The cells were grown overnight in Luria–Bertani (LB) broth at 37°C. Cells for DNA extraction were pelleted using a centrifuge from 5 mL of the liquid culture. Cell pellets were added to bead tubes of a Mo Bio UltraClean DNA Isolation kit (MO BIO Laboratories). The kit protocol was followed except for the substitution of 30 s of vortexing in place of bead beating.
The environmental DNA samples used in this study were aliquots remaining from a previous study (Biddle et al., 2008), frozen at −80°C after the original extraction. The method of DNA extraction is described therein and summarized as follows: Frozen sediment cores from ODP Leg 201 Site 1229 were processed by removal of the top ∼1 cm of potentially contaminated sediment. Aliquots of the remaining sediment were homogenized and DNA was extracted using the Mo Bio UltraClean Microbial DNA kit (MO BIO Laboratories) with some modifications, including the addition of a 65°C water bath incubation step and a decrease in the recommended amount of bead beating time to 1 min (Biddle et al., 2008).
Primer Design
The non-degenerate 5′ ends of the RAMP primers were chosen from the primers utilized in Roche 454 sequencing technology. These included a set of primers used for amplicon sequencing, and a set of adaptors used for metagenomic sequencing. Both the amplicon primers and metagenomic adaptors were updated with the switch to “Titanium” sequencing reagents, providing a total of four sets of available primers to be tested. These sequencing primers were then altered by the addition of degenerate bases to the 3′ ends, with the goal of maximizing degeneracy while limiting the possibility of primer-dimers and DNA hairpins. A total of 12 unique primers were tested on culture DNA as well as environmental DNA for use in the RAMP protocol (Table 1). Success of the primers was evaluated by visualization of the amplified products after electrophoretic separation on 1% agarose gels. All primers were ordered from Integrated DNA Technologies, Inc. “B” primers included a 5′ biotin tag (for sequencing preparations) and HPLC purification, while “A” primers were ordered with standard desalting purification.

Table 1. All primer designs tested during method development.
PCR Optimization
The following PCR conditions were chosen for RAMP reactions of samples sequenced in this study: Reagents (Per 25 μL reaction): 1.6 μM each FLXampA + 5N and FLXampB + 5N primers (Table 1), 1.25 units SpeedSTAR™ HS DNA Polymerase (Takara Bio, Inc.) with 2.5 μL accompanying FB1 buffer containing 30 mM MgCl2, and 2.0 μL accompanying dNTP mixture containing 2.5 mM each dNTP. Cycling conditions: 5 min at 94°C, followed by 25 cycles of 10 s at 94°C, 15 s at 47 (or 25)°C, 20 s at 72°C, followed by a final extension of 10 min at 72°C. The amount of DNA template used per reaction varied by sample, but the sensitivity of the RAMP reaction to concentration of DNA template was tested using a dilution series of E. coli DNA ranging from about 10 ng/μL down to about 0.0625 ng/μL. Multiple ranges of other PCR parameters were tested as well during protocol optimization. For primers, concentrations tested included 1–7 μM in increments of 1, plus lower concentrations of 0.0016–1.6 μM by powers of 10, and higher concentrations of 8 and 36.5 μM (data not shown). Annealing temperature tested ranged from 25 to 70°C, and number of cycles ranged from 25 to 60. Another DNA Taq polymerase was tested, TaKaRa Ex Taq™ (Takara Bio, Inc.), as well as a Pfu DNA polymerase, Stratagene PfuUltra™ High-Fidelity DNA Polymerase (Agilent Technologies, Inc.), with PCR reagents and conditions as recommended by those manufacturers. Optimal values for each parameter were assessed, prior to sequencing, by visualization of product after electrophoretic separation on 1% agarose gels. All amplifications were performed in either an MJ Research PTC-100 Thermal Cycler or an Eppendorf Mastercycler Gradient Thermal Cycler. Only products amplified with SpeedSTAR polymerase were used for sequencing.
454 Sequencing
Amplified DNA products were purified via a gel extraction, to remove excess primer and to select for a size range of DNA fragments that worked well with the current 454 sequencing technology. Products were subject to electrophoretic separation on a 1% agarose gel, along with DNA markers to estimate the size of the fragments. The portion of the gel containing amplified products ranging in size from about 650 to 850 base pairs (bp) long for E. coli and environmental DNA samples sequenced with “Titanium” reagents (or 250–500 bp long for E. coli samples sequenced with FLX reagents) was excised, and the DNA was purified using a QIAEX II Gel Extraction Kit (Qiagen). After gel purification, DNA was quantified using PicoGreen, on a handheld fluorometer (Turner Biosystems, TBS-380). Because the RAMP primers incorporated the 454 sequencing primers, the library preparation step usually needed to ligate the sequencing primers to the DNA fragments was unnecessary. Isolation of only those DNA fragments with both “A” and “B” primers was accomplished using a biotin–streptavidin selection protocol, employing the biotin label incorporated onto the RAMP “B” primers. Sequencing was carried out by the Schuster Lab at the Pennsylvania State University on a Roche 454 Genome Sequencer FLX sequencing system (454 Life Sciences) as described (Poinar et al., 2006). For E. coli coverage tests, ¼ of a picotiter plate was sequenced on each of three separate occasions using FLX chemistry for tests 1 and 2, and the newer Titanium chemistry for test 3 due to upgrades in 454 technology. For sequencing of RAMP-amplified environmental samples, ¼ of a picotiter plate was sequenced for each sample using the Titanium chemistry. The two new RAMP-amplified environmental metagenomes were uploaded to MG-RAST (metagenomics.anl.gov) as job numbers 37975 and 37977.
Processing of Sequence Data
Raw metagenomic sequence datasets from three samples sequenced in 2008 (Biddle et al., 2008) were downloaded from the National Center for Biotechnology Information (NCBI) GenBank archive (SRA001015) and converted into FASTA format using the online resource, Galaxy (Giardine et al., 2005; Blankenberg et al., 2010; Goecks et al., 2010). These three metagenomes, along with all metagenomes generated in the present study, were screened to remove replicate sequences and sequences containing ambiguous bases.
Random amplification metagenomic PCR-amplified metagenomes from E. coli genomic DNA were compared to the reference E. coli W genome (downloaded from the NCBI genome database) in order to estimate genome coverage. Comparisons were carried out via a BLASTN search with an e-value of 10−15. Genomic locations of the respective top BLAST results were recorded, and the total number of matched base pairs was tabulated using a 98% identity cut-off value. This approach may have overlooked some highly repetitive regions and thus should serve as a slight underestimate. Genome coverage for test 3 of E. coli sequencing (carried out with newer sequencing technology) was estimated both for the whole dataset and for a random sub-sampling of the dataset, equivalent in size to those available from E. coli tests 1 and 2. The GC content of the metagenomic datasets was assessed via simple mathematical calculations performed with a home written Python script.
Environmental metagenomes from the 2008 study as well as the present study were further processed to include only those sequence reads greater than 150 bp in length. The environmental metagenomes were compared to the NCBI database of non-redundant (nr) protein sequences (downloaded August 2010) via BLASTX with an e-value of 10−2, to the Silva database of 16S rRNA nucleotide sequences (downloaded July 2010) via BLASTN with an e-value of 10−9, and to a compiled database containing all available sequences of the RNA polymerase beta-subunit encoding gene (rpoB; downloaded September 2011, DOE Joint Genome Institute) via BLASTX with an e-value of 10−2. For matches to the nr and rpoB databases detected by BLASTX, the software program MEGAN v.4.60.2 (Huson et al., 2007) was used to assign the sequences to phylogenetic groups, using the following parameter settings: minscore: 35.0, toppercent: 10.0, minsupport: 5, and winscore: 0.0. In short, MEGAN assigns BLAST-hit sequences to phylogenetic groups using bit-score to retain only significant hits, based on the parameters chosen. 16S matches detected by BLASTN were assigned to phylogenetic groups based on the most similar sequence found via the BLAST algorithm.
Results
Method Assessment
Of the 12 primers tested in the development of the RAMP protocol (some as pairs and some singly), two sets and one single primer resulted in some amplification of DNA (Table 1). Only one set, however, amplified environmental DNA consistently and robustly. That set was the FLXampA + 5N/FLXampB + 5N combination. These primers worked best at concentrations from 1 to 3 μM, although amplification of template DNA was observed for all concentrations up to 7 μM (Figure 1A). Lower and higher concentrations of primers did not result in amplification of DNA template (data not shown). Further, amplification of DNA was observed over a wide range of annealing temperatures (25–70°C). The most concentrated amplified product occurred in the reaction at 47°C, and concentrations appeared to decline at both lower and higher annealing temperatures (Figure 1B). Amplification was more consistent when using the SpeedSTAR DNA polymerase than when using the Ex Taq DNA polymerase, and the Pfu DNA polymerase failed to produce amplification at all. Using a primer concentration of 1.6 μM, an annealing temperature of 47°C, and SpeedSTAR DNA polymerase, the sensitivity of the method to initial concentration of DNA in a sample was tested using a dilution series of E. coli DNA ranging from 10 to 0.0625 ng/μL. Amplification, as viewed on an agarose gel, was seen for all but the lowest concentration and the negative control (Figure 1C). Therefore, the threshold starting DNA concentration for visible amplification by RAMP under these reaction conditions is somewhere between 0.125 and 0.0625 ng/μL. However, when testing the method on environmental DNA, it was observed that increasing the number of PCR cycles and/or increasing the volume of sample added to the reaction resulted in greater amplification in some samples, which suggests that the method’s sensitivity could be improved in some cases. Experimentation revealed that the number of PCR cycles for RAMP could be increased to as much as 50, but increasing to 60 cycles resulted in high molecular weight amplification products in the negative controls (Figure 1D). Attempts to use qPCR to better quantify a minimum threshold of DNA needed for RAMP amplification and to monitor possible formation of products in negative controls were unsuccessful. Further experimentation with the qPCR reaction suggested that the PicoGreen added to the qPCR reaction for quantification of the product may have been interfering in some way with the RAMP reaction. As an alternative means of quantification of amplified samples and negatives, future applications of RAMP could be coupled with other highly precise DNA quantification methods such as the use of a bioanalyzer or fluorometer.
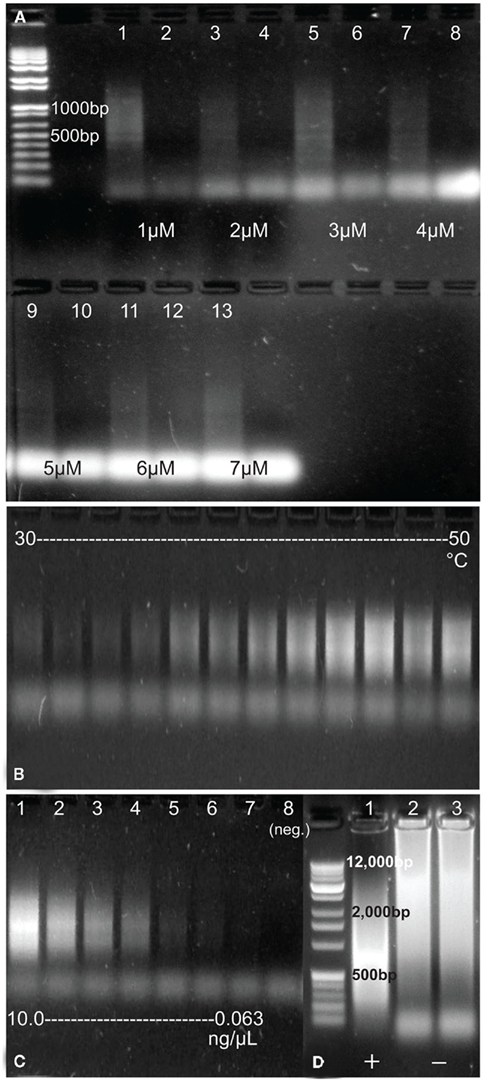
Figure 1. (A) Products of RAMP-amplified E. coli DNA using a range of primer concentrations from 1 to 7 μM and their corresponding negative controls (no DNA added). Sizes of two of the DNA marker bands (far left) are included on the image. (B) Products of RAMP-amplified E. coli DNA using a range of annealing temperatures, from 30 to 50°C. (C) Products of RAMP-amplified E. coli DNA of a series of concentrations [∼10, 2, 1, 0.5, 0.25, 0.125, 0.0625, and 0 (negative control) ng/μL]. (D) Products of amplification of environmental DNA (lane 1) and two negative controls (lanes 2 and 3) after 60 cycles of RAMP. Sizes of a few DNA marker bands (far left) are included on the image.
The method was further tested by sequencing amplified DNA products extracted from E. coli, chosen because its complete sequenced genome was available for reference in estimating coverage bias. The first sequencing test was carried out on E. coli DNA amplified with the chosen primer set, with an annealing temperature of 47°C, and 25 cycles of RAMP. Based on the amount of sequence data received, a theoretical genome coverage of 3.64× was calculated, while the actual genome coverage for this sequencing test was estimated at 0.30×, or 30% of the genome (Table 2). The GC content of the sequenced metagenome was calculated as 50.9%. For the second sequencing test, all PCR conditions remained the same; however, 15 separate amplification reactions were carried out on the same DNA sample and pooled together prior to sequencing. The results were nearly the same as those from the first test – perhaps slightly poorer, with a theoretical coverage of 3.86× and again, actual coverage of 30%. GC content was 52.15% for this metagenome. For test three, only one reaction was carried out, but the annealing temperature during PCR cycling was lowered from 47 to 25°C. Because this third test was sequenced with newer technology, the amount of data was far greater than the first two, but after accounting for that difference by randomly analyzing only a portion of the metagenome equivalent in size to the first two tests, the results showed a slight increase in genome coverage with sequencing effort, with 37% actual coverage resulting from a 3.51× sequencing effort.
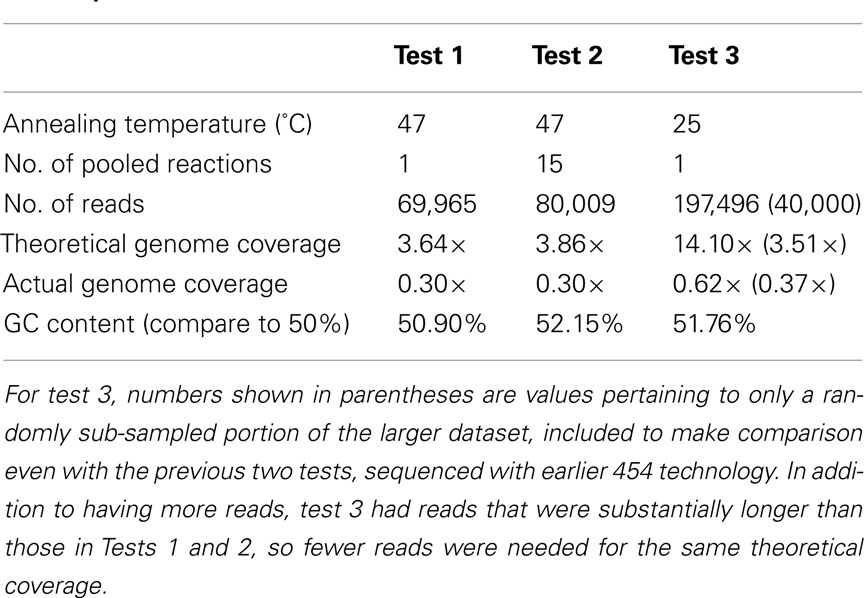
Table 2. Results of sequencing tests of RAMP-amplified E. coli DNA.
When analyzing the entire third metagenome, theoretical genome coverage was 14.10×, and actual genome coverage was about 62%. Given this significant increase, an estimate of genome coverage in relation to sequencing effort was performed, by analyzing sequentially larger and larger portions of this third metagenome (Figure 2). Using a logarithmic fit relationship as an estimate [y = 16.33Ln(x) − 9.4203], complete coverage would be achieved with 813 million base pairs of sequence data, or about two whole plates of 454 sequencing with current Titanium chemistry.
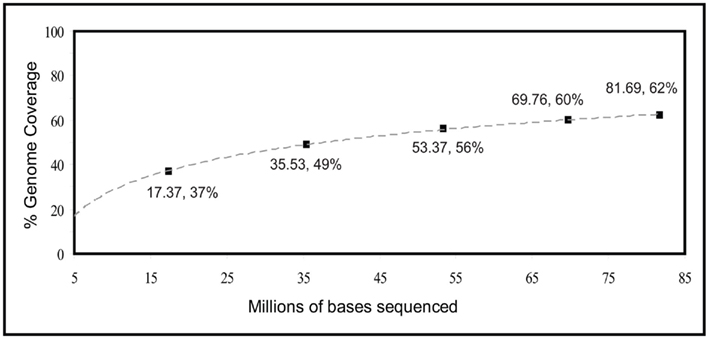
Figure 2. Estimated percentage of genome coverage in relation to sequencing effort of RAMP-amplified E. coli genomic DNA.
In all sequencing tests, the initial concentration of E. coli DNA prior to amplification with RAMP was ∼10 ng/μL. The concentration of DNA after amplification was not measured until after gel purification, which consumes a large portion of sample DNA, so absolute values are not available for the amount of DNA produced by the various RAMP conditions. However, by examining the values obtained for concentration after gel purification, we can still make relative comparisons. The concentrations for E. coli tests 1, 2, and 3 after gel purification were 8.2 ng/μL, 5.5 ng/μL, and 51 pg/μL, respectively. The concentrations of the 1- and 32-m below seafloor (mbsf) environmental samples after gel purification were 54 and 12.5 pg/μL, respectively. From this data, we see that DNA concentrations are orders of magnitude higher after RAMP at 47°C annealing (E. coli tests 1 and 2) than after RAMP at 25°C annealing (E. coli test 3 and both environmental samples).
Environmental Application
The RAMP method utilized for “test 3” of sequencing in method development was also applied to two environmental DNA samples extracted from marine subsurface sediment. The marine sediment was from two depths – 1 and 32 mbsf – of a Peru Margin subsurface location (ODP Leg 201 site 1229). Aliquots of the DNA extracted from these two samples for an earlier study (Biddle et al., 2008) were available for re-analysis using the new method. In this way, RAMP could be compared to the WGA method used in the earlier study without concerns of DNA extraction bias influencing results. In the Biddle et al. study, the 1-mbsf sample was sequenced with no prior WGA, as well as with WGA via the REPLI-g kit (Qiagen) using the available 454 GS 20 technology. The 32-mbsf sample was sequenced only after WGA using REPLI-g as concentration of DNA was too low for unamplified sequencing. In the present study, both the 1- and 32-mbsf samples were sequenced after amplification with RAMP.
In all, five metagenomes were analyzed, three old and two new: 1 mbsf, unamplified; 1 mbsf amplified with REPLI-g; 32 mbsf amplified with REPLI-g; 1 mbsf amplified with RAMP; and 32 mbsf amplified with RAMP. After comparing all five metagenomes to the nr, 16S rRNA, and rpoB databases, RAMP metagenomes were found to have higher percentages of sequences with identifiable homologs in all cases (Table 1). Further, within each amplification method, matches to the databases decreased with increasing depth of the sample.
Matches to the nr, 16S rRNA, and rpoB databases were used to analyze community composition of the sample metagenomes and to compare the results obtained via the different amplification methods (Figure 3). Examining first the results from phylogeny assigned via the nr dataset, the different amplification methods appear to give very similar results to each other as well as to the unamplified control, with a few notable differences. Most apparent is the over-representation of the Chloroflexi sequences in the RAMP 1 mbsf metagenome (17.9% higher for RAMP than unamplified). In the REPLI-g 1 mbsf metagenome, the Euryarchaeota appear to be over-represented, although to a lesser degree (6.4% higher for REPLI-g than unamplified). Though there is no unamplified control for the 32-mbsf sample, comparison of the REPLI-g and RAMP samples to each other, appears to confirm these two patterns of over-representation, with the RAMP 32 mbsf metagenome displaying a higher percentage of Chloroflexi sequences by 17.2%, and the REPLI-g metagenome having more Euryarchaeota sequences by 10.1%. An additional trend is the under-representation of the Bacteroidetes/Chlorobi group in the RAMP samples, as compared with the unamplified and REPLI-g metagenomes, at both depths.
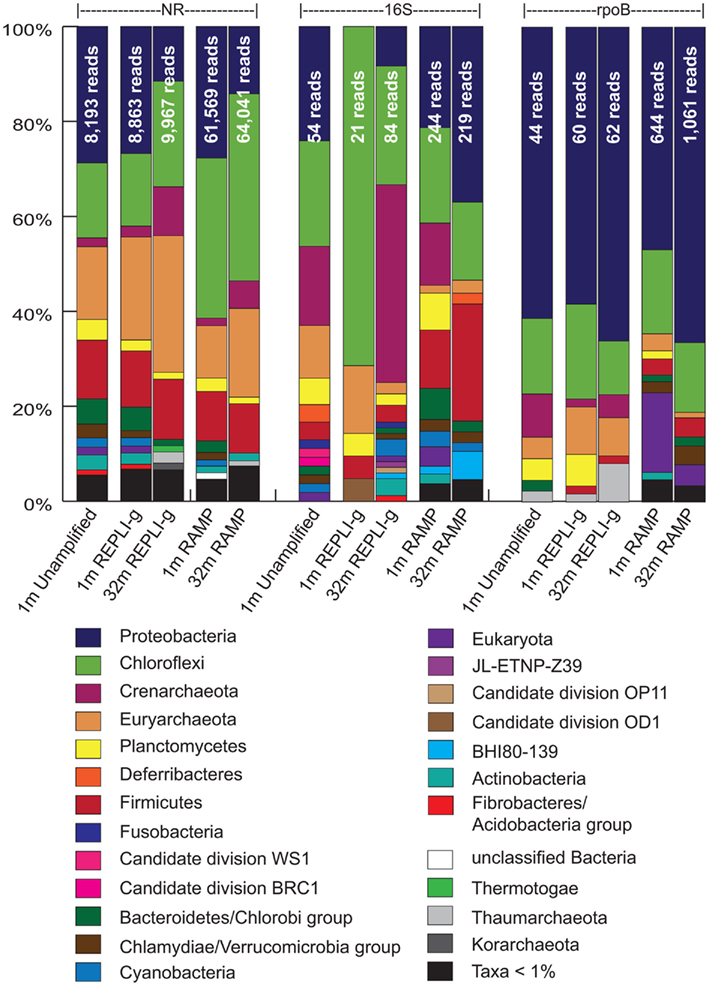
Figure 3. Phylogenetic identities of sequences in all five metagenomes as revealed through comparison to the nr, 16S, and rpoB databases. Shown are the percentages of total identifiable hits. Total number of identifiable hits for each condition is listed at the top of the bars.
In comparing the community composition of the 1-mbsf sample to the 32-mbsf sample using the nr results, both REPLI-g and RAMP samples reveal the same trends of increasing or decreasing of certain taxa with depth, with only the degree of these changes differing slightly between the methods. According to both REPLI-g and RAMP datasets, the Chloroflexi, Crenarchaeota, and Euryarchaeota all show a notable increase with depth (6.9 and 5.7% higher at depth, respectively), while the Proteobacteria show a notable decrease with depth (15.3 and 13.5% lower at depth, respectively). Most other taxa also decrease with depth, perhaps only as a result of the increased dominance of the Chloroflexi and archaeal sequences. The exceptions are the Firmicutes, which increase slightly in the REPLI-g dataset only, and the minor taxa Thermotogae, which increases slightly in both datasets. These conclusions are consistent with those in the Biddle et al. (2008) study.
As another perspective of community composition, the metagenomes were also compared against the Silva 16S rRNA database. Using classification of organisms based on the most significant hit in these BLAST results, the community composition between metagenomes is not as consistent for either sample as it was when viewed via the nr results. In the 1-mbsf sample, the REPLI-g metagenome has several stark differences from the unamplified metagenome, including the complete absence of the Crenarchaeota and Proteobacteria, which make up 16.7 and 24.1% of the unamplified metagenome, respectively. Instead the REPLI-g sample is almost entirely dominated by Chloroflexi, at 71.4% of the 16S hits compared with 22.2% in the unamplified metagenome. The REPLI-g metagenome, however, had only 21 16S hits, compared with 54 in the unamplified metagenome, so the absence of some groups may be a result of under-sampling.
In contrast to the REPLI-g metagenome for the 1-mbsf sample, the RAMP metagenome for this sample had sequences from all of the major taxonomic groups represented in the unamplified metagenome, differing only in their relative proportions. In this case, the most apparent differences are the over-representation of the Firmicutes (8.6% higher) and the under-representation of the Euryarchaeota (9.5% lower) in the RAMP 1 mbsf metagenome as compared with the unamplified.
For the 32-mbsf sample, the RAMP metagenome shows a much larger proportion of Proteobacteria and Firmicutes than the REPLI-g sample (28.7 and 21.1% higher), which is consistent with the comparisons in the 1-mbsf sample. However, there is a discrepancy between the 1- and 32-mbsf samples concerning the Crenarchaeota. In the 1-mbsf sample, the RAMP metagenome contained 13.1% Crenarchaeota sequences while the REPLI-g metagenome contained less than 1%. However, for the 32-mbsf sample, the REPLI-g metagenome contains 41.7% Crenarchaeota sequences while the RAMP metagenome contains less than 1%, essentially opposite trends.
Due to the inconsistent finding of the nr and 16S methods, the metagenomes were also compared against a dataset of rpoB gene sequences. In the rpoB classification, Proteobacteria heavily dominated all metagenomes, followed by Chloroflexi. In contrast to the nr classifications, and consistent with results of the 16S classification, the Proteobacteria increase with depth for both the REPLI-g and RAMP metagenomes (7.8 and 19.6% higher), while Chloroflexi decrease (8.7 and 3.0% lower). Also in contrast to the nr classification results, the Euryarchaeota decrease with depth in both the REPLI-g and RAMP metagenomes (1.9 and 2.4% lower). This is consistent with the 16S classification results for the REPLI-g metagenomes only. Finally, in this rpoB classification, the Crenarchaeota make up a significant percent of the classified sequences of the unamplified sample (9.1%), and while present to a lesser degree in the REPLI-g dataset, still reveal an increase with depth (1.7–4.8%), consistent with the nr classification data and the 16S data for this amplification method. In the RAMP metagenomes however, the Crenarchaeota make up less than 1% of the rpoB-classified sequences.
In an attempt to clear up the trends associated with the archaea at the two depths in the different metagenomes, a classification of sequences at the domain level was performed, again using comparisons to the nr, 16S, and rpoB databases (Figure 4). In this interpretation, archaea increase in relative proportion with depth while bacteria decrease in relative proportion with depth in all cases except in the 16S classification of the RAMP metagenomes, where the opposite trend exists. Specifically, in the REPLI-g metagenomes, the proportion of archaeal sequences increases with depth by 11.9, 29.8, and 8.9% in the nr, 16S, and rpoB analyses, respectively. In the RAMP metagenomes, the proportion of archaeal sequences increases with depth by 12.2 and 12.3% in the nr and rpoB analyses, respectively, but decreases by 10.3% in the 16S analysis.
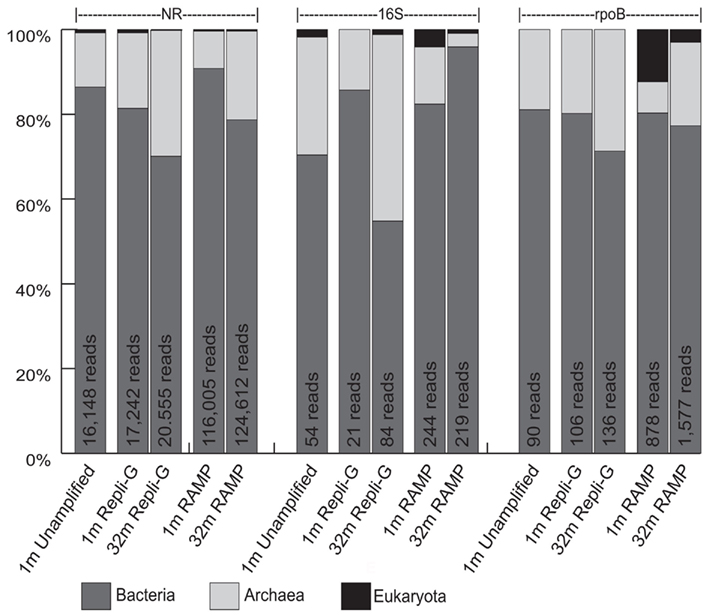
Figure 4. Domain classification of sequences in all five metagenomes, as revealed through comparison to the nr, 16S, and rpoB databases. Shown are percentages of total identifiable hits. Total number of identifiable hits for each condition is listed at the top of the bars.
Discussion
Method Assessment
General amplification of environmental DNA via the developed RAMP protocol is a means of producing DNA fragments suitable for immediate use in 454 metagenomic sequencing. The utilization of the PCR reaction with primers possessing a degenerate 3′ end results in general amplification of the genetic material without the creation of products in amplification reaction negative controls, allowing for confident amplification of low-biomass samples compared to MDA methods. Bias in coverage of starting genetic material is suggested by the estimated levels of coverage of the E. coli genome after sequencing of amplified products (Table 2). An increase in coverage levels with relation to sequencing effort (Figure 2), however, suggests that even with no improvements to the method, full coverage could be achieved with high enough levels of sequencing (estimated at 813 million base pairs or about two full plates of 454 sequencing). We had several hypotheses about the cause of bias in coverage. The first was that the bias was occurring due to the initial locations of primer annealing in the first cycle of PCR. After the first round of PCR, those regions amplified would be present in higher number than other regions and hence, more likely to amplify in subsequent rounds of PCR. This hypothesis was tested by combining the amplification products of 15 separate reactions, in theory allowing for 15 different sets of initial primer annealing locations due to random chance (test 2, Table 2). Analysis of sequenced products, however, revealed no significant reduction in bias. A second hypothesis was that the annealing temperature of the PCR reaction was favoring amplification of GC-rich regions of the genome, and that a lower annealing temperature may permit annealing at less GC-rich regions to be competitive. Sequencing after the use of a lower annealing temperature revealed a small reduction in bias, however, analysis of the GC content of sequenced products suggested no change in the proportion of GC-rich to GC-poor regions (test 3, Table 2). One observed problem with lowering the annealing temperature was a decrease in the yield of amplified DNA by a couple orders of magnitude.
Our current hypothesis on the cause of the bias is that it is related to the specific DNA sequence of the primer, particularly, the region closest to the degenerate 3′ end of the primer. Portions of the genome that match not only the five base pairs at the end of a primer, but also the next base or bases, would have a higher likelihood of primer annealing. This could explain the slight improvement in coverage resulting from a reduced annealing temperature, as less stringent PCR conditions allow for more non-specific annealing. One of the goals of primer development had been to maximize degeneracy while limiting the creation of DNA artifacts such as primer-dimers. In this effort, we attempted to increase the number of degenerate base pairs at the 3′ end of the primer. With six degenerate bases at the 3′ end of the primer, however, amplification failed for all PCR conditions attempted. This was more likely a result of hairpin formation of the primers than rampant primer-dimer formation, as primer-dimers viewed on the gel were less bright than those for the five degenerate base primer, ran as a control. One possible way to increase degeneracy of the primers without causing an increase in hairpin formation or primer-dimers, might be to create multiple primers, each with a different base preceding the 5-nucleotide degenerate 3′ end, carry out separate reactions with each primer, and combine the products at the end. In this way, the same effect of adding a sixth degenerate base might be achieved, without the problems of hairpin formation resulting from too many degenerate bases at the end of any one primer. However, this idea has not yet been tested.
Another possible limitation of the method is the sensitivity of the reaction to the concentration of starting DNA template. The E. coli dilution series test indicated that below a certain starting concentration of DNA, no significant amplification would occur (Figure 1C). In this test, amplification occurred with a starting concentration of 0.125 ng/μL but not for a starting concentration of 0.0625 ng/μL. As experimentation with environmental samples showed, increasing the number of cycles is one way to increase the sensitivity of the method, but this has not been quantified. Also, testing RAMP at 60 cycles revealed that there is a limit to how many cycles can be run before DNA artifacts appear in negative controls. Even so, there is likely much to be gained in terms of sensitivity between 25 and 50 cycles, and this is something we would like to test further in the future.
Application to Environment
When applied to environmental samples from the Peru Margin subsurface, at 1 and 32 mbsf, RAMP again produced amplified DNA product suitable for use with 454 metagenomic sequencing. In order to make a qualitative assessment of coverage bias, the community composition of these environmental samples was analyzed, and compared to that obtained using REPLI-g amplification for both samples, as well as to that obtained by sequencing the unamplified 1 mbsf sample, using data from Biddle et al. (2008).
In terms of the quality of data obtained (length of sequence, genuine sequence versus amplification artifact, etc.), it is difficult to compare the sequenced RAMP products to the unamplified and REPLI-g amplified samples, as sequencing technology was greatly improved in the length of time between studies. However, we can say that both methods resulted in sequenced products of a length which was typical for the sequencing technology of the time [∼100 bp (2008) and ∼400 bp (2010)]. When the sequenced products were compared to the nr, rpoB, and 16S rRNA databases, RAMP products had much higher percentages of sequences with identifiable homologs in all cases except the 16S database, where they were still higher, but to a lesser degree (Table 3). This might be contributable entirely to the increase in read length capable with the newer sequencing technology, as longer reads would increase the likelihood of a match in the protein databases, but may make less of a difference in the nucleic acid-based 16S rRNA database, where shorter reads lead to identification almost as often as longer ones (Biddle et al., 2008). The fact that both of the 2008 amplified samples have similar values to the unamplified sample for percent of the metagenome with identifiable homologs in the three databases, suggests that the cause of the low identification rates is likely not due to the production of amplification artifacts by MDA, a question we had wanted to explore. These results are slightly different than those obtained by analysis of the data in 2008, where the amplified 32 mbsf sample did have a significantly lower percentage of identified sequences than the 1-mbsf unamplified sample (5.83% to nr as compared with 13.39%). The current analysis was done using more recent databases, and the identifiable percentage of all three 2008 metagenomes was higher than in the 2008 analysis, but it may be that the 32-mbsf sample benefited most from the addition of new sequences to the available databases.
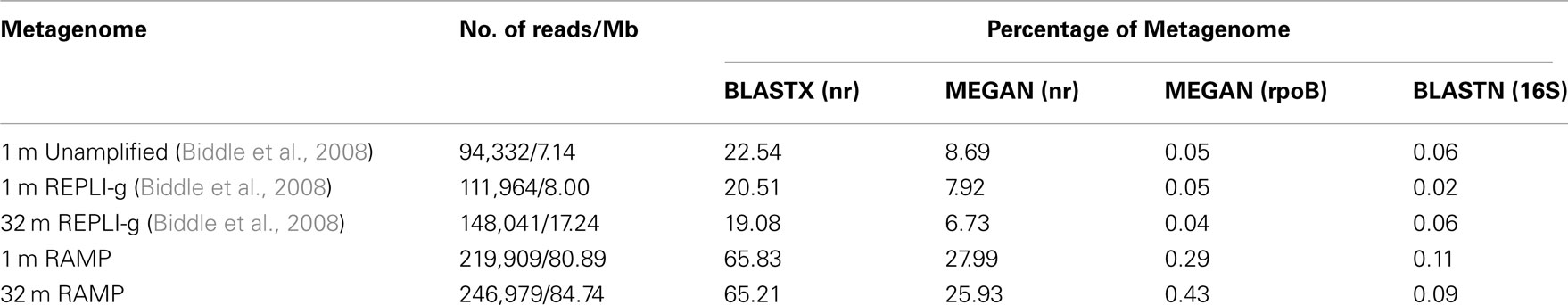
Table 3. Analysis of environmental sequence data.
Comparing the amplification methods using assessments of community composition via matches to the nr, rpoB, and 16S rRNA databases proved problematic, largely as a result of conflicting data between databases. When examining the community composition of all five metagenomes using results from the nr database, both methods appear successful in paralleling the unamplified 1 mbsf sample in terms of major taxonomic groups. In addition, RAMP and REPLI-g appear to give similar results to each other for the deeper 32 mbsf, which had no unamplified control. Further, patterns of increasing and decreasing of the proportions of taxa are consistent between RAMP and REPLI-g. In this analysis, both methods imply that the Chloroflexi and the archaeal groups increase with depth, while the Proteobacteria decrease with depth. These trends are consistent with results from the Biddle et al. (2008) study.
The nr database, however, suffers from bias toward groups of organisms with many sequenced representatives; hence, the relative proportions of taxa may be misleading. The Silva 16S rRNA database, on the other hand, has a far greater diversity of organisms represented, including many environmental sequences from organisms not yet cultivated or sequenced. We chose to compare our metagenomes to this database, as another perspective of community composition. The results of this method of community analysis were much less consistent between amplification techniques and between the amplified and unamplified samples. Several factors may be contributing to these results. First, unlike the analysis using the nr database, the 16S rRNA database is only of a single gene. Therefore, there is the possibility of missing that one gene in any given organism due to under-sampling, whereas, one would have a higher chance of hitting at least one gene in the organism’s genome that would provide a match to the nr database. This may be the issue with the 1-mbsf REPLI-g sample, as only 21 16S rRNA genes were identified (as contrasted with 84 in the 32-mbsf sample, of a similar dataset size). In the 2008 study, where more depths were examined, this 1 mbsf sample was a sort of outlier in the 16S analysis, where trends across the other depths were much more consistent (Biddle et al., 2008).
Other possibilities for the inconsistencies in this analysis could be related to WGA-induced bias as a result of DNA quantity or quality. It has been noted before for MDA that bias increases when less DNA template is used (Binga et al., 2008). In addition, the quality of the extracted DNA may result in differences in the success of amplification, favoring some DNA sequences over others. Perhaps such a bias in amplification could explain why the Crenarchaeota 16S rRNA genes are nearly completely absent from the 32-mbsf RAMP metagenome (matches were found, but constituted less than 1% of the identified reads). Alternatively, it is possible that REPLI-g has a strong bias toward the 16S rRNA of Crenarchaeota when starting DNA template is low or poor. As we do not have an unamplified metagenome for this deeper sample, it is very difficult to determine whether or not the trends we see are a result of WGA-induced bias of either or both of the amplification methods employed.
Attempting to use another marker gene, rpoB, did little to answer any questions about bias induced by WGA at depth, and added more confusion to the issue of what happens to the archaeal groups with depth, in some cases agreeing with the nr analysis, and in some cases with the 16S analysis. Hypothesizing that many of the archaeal sequences at depth were not being classified at the phylum level in the nr or rpoB analyses, we decided to carry out an analysis at the domain level, to get a better idea of archaeal trends in the subsurface, an ongoing question in deep biosphere research. This analysis, at least, revealed more consistency among the nr, 16S, and rpoB analysis, and showed that in nearly all cases, the proportion of archaea increased with depth (Figure 4). This is most consistent with the phylum level nr analysis, where both Crenarchaeota and Euryarchaeota increased with depth.
This study has introduced a new method of WGA, termed RAMP, which is a viable option for increasing DNA concentration for use with metagenomic sequencing. The method was designed specifically for the pyrosequencing platform of Roche 454, and helps to reduce preparation steps, as the 454 sequencing primers are attached to each fragment of DNA as it is amplified. With modifications to the primers and further optimization, this method may also be an option for use with other sequencing technologies, such as the Illumina or Ion Torrent sequencing systems, which utilize similar preparations as the 454 system. The method may have particular utility in very low-biomass samples, where the production of DNA artifacts produced by other methods of WGA may inhibit the amplification of sample DNA altogether. The method has room and flexibility for improvement, such as through further optimization of the primer design and cycling conditions, which may reduce some of the problems with potential biases. Based on the results of this study, however, it is clear that using any method of WGA may have a significant impact in community composition and diversity analyses. In addition, the results displayed herein should reinforce the caution that must be employed when analyzing community composition using any one method of analysis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the ODP shipboard scientific party, and D. Jones and Z. Zhang for technical assistance. Samples for this research were provided by the Integrated Ocean Drilling Program [sponsored by the National Science Foundation (NSF) and participating countries]. This work was supported by NSF Grant OCE 05-50601, by the National Aeronautics and Space Administration (NASA) Astrobiology Institute (NAI) under NASA–Ames Cooperative Agreement NNA09DA76A, and through support from the Gordon and Betty Moore Foundation. The Roche 454 facility at the Pennsylvania State University Center for Comparative Genomics and Bioinformatics was funded, in part, by a grant from the Pennsylvania Department of Health using Tobacco Settlement Funds appropriated by the legislature. Jennifer F. Biddle was also supported by a NASA Postdoctoral Program Fellowship administered by Oak Ridge Associated Universities (ORAU).
References
Bale, S. J., Goodman, K., Rochelle, P. A., Marchesi, J. R., Fry, J. C., Weightman, A. J., and Parkes, R. J. (1997). Desulfovibrio profundus sp. nov., a novel barophilic sulfate-reducing bacterium from deep sediment layers in the Japan Sea. Int. J. Syst. Bacteriol. 47, 515–521.
Biddle, J. F. (2006). Microbial Populations and Processes in Subseafloor Marine Environments. ProQuest Dissertations and Theses, The Pennsylvania State University, University Park, PA, 179.
Biddle, J. F., Fitz-Gibbon, S., Schuster, S. C., Brenchley, J. E., and House, C. H. (2008). Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. U.S.A. 105, 10583–10588.
Biddle, J. F., White, J. R., Teske, A. P., and House, C. H. (2011). Metagenomics of the subsurface Brazos-Trinity Basin (IODP site 1320): comparison with other sediment and pyrosequenced metagenomes. ISME J. 5, 1038–1047.
Binga, E. K., Lasken, R. S., and Neufeld, J. D. (2008). Something from (almost) nothing: the impact of multiple displacement amplification on microbial ecology. ISME J. 2, 233–241.
Blankenberg, D., Von Kuster, G., Coraor, N., Ananda, G., Lazarus, R., Mangan, M., Nekrutenko, A., and Taylor, J. (2010). Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 19, 1–21.
Dean, F. B., Hosono, S., Fang, L., Wu, X., Faruqi, A. F., Bray-Ward, P., Sun, Z., Zong, Q., Du, Y., Du, J., Driscoll, M., Song, W., Kingsmore, S. F., Egholm, M., and Lasken, R. S. (2002). Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. U.S.A. 99, 5261–5266.
Dietmaier, W., Hartmann, A., Wallinger, S., Heinmöller, E., Kerner, T., Endl, E., Jauch, K. W., Hofstädter, F., and Rüshoff, J. (1999). Multiple mutation analysis in single tumor cells with improved whole genome amplification. Am. J. Pathol. 154, 83–95.
Giardine, B., Riemer, C., Hardison, R. C., Burhans, R., Elnitski, L., Shah, P., Zhang, Y., Blankenberg, D., Albert, I., Taylor, J., Miller, W., Kent, W. J., and Nekrutenko, A. (2005). Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15, 1451–1455.
Goecks, J., Nekrutenko, A., Taylor, J., The Galaxy Team. (2010). Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11, R86.
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386.
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., Suzuki, M., Takai, K., Delwiche, M., Colwell, F. S., Nealson, K. H., Horikoshi, K., D’Hondt, S., and Jørgensen, B. B. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc. Natl. Acad. Sci. U.S.A. 103, 2815–2820.
Jørgensen, B. B., and Boetius, A. (2007). Feast and famine – microbial life in the deep-sea bed. Nature 5, 770–781.
Kittler, R., Stoneking, M., and Kayser, M. (2002). A whole genome amplification method to generate long fragments from low quantities of genomic DNA. Anal. Biochem. 300, 237–244.
Lipp, J. S., Morono, Y., Inagaki, F., and Hinrichs, K.-U. (2008). Significant contribution of Archaea to extant biomass in marine subsurface sediments. Nature 454, 991–994.
Mikucki, J. A., Liu, Y., Delwiche, M., Colwell, F. S., and Boone, D. R. (2003). Isolation of a methanogen from deep marine sediments that contain methane hydrates, and description of Methanoculleus submarinus sp. nov. Appl. Environ. Microbiol. 69, 3311–3316.
Nelson, D. L., Ledbetter, S. A., Corbo, L., Victoria, M. F., Ramirez-Solis, R., Webster, T. D., Ledbetter, D. H., and Caskey, C. T. (1989). Alu polymerase chain reaction: a method for rapid isolation of human-specific sequences from complex DNA sources. Proc. Natl. Acad. Sci. U.S.A. 86, 6686–6690.
Orcutt, B. N., Sylvan, J. B., Knab, N. J., and Edwards, K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 75, 361–422.
Pinard, R., de Winter, A., Sarkis, G. J., Gerstein, M. B., Tartaro, K. R., Plant, R. N., Egholm, M., Rothberg, J. M., and Leamon, J. H. (2006). Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics 7, 216–236.
Poinar, H. N., Schwarz, C., Qi, J., Shapiro, B., Macphee, R. D., Buigues, B., Tikhonov, A., Huson, D. H., Tomsho, L. P., Auch, A., Rampp, M., Miller, W., and Schuster, S. C. (2006). Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA. Science 311, 392–394.
Raghunathan, A., Ferguson, H. R. Jr., Bornarth, C. J., Song, W., Driscoll, M., and Lasken, R. S. (2005). Genomic DNA amplification from a single bacterium. Appl. Environ. Microbiol. 71, 3342–3347.
Sørensen, K. B., Lauer, A., and Teske, A. (2004). Archaeal phylotypes in a metal-rich and low-activity deep subsurface sediment of the Peru Basin, ODP Leg 202, Site 1231. Geobiology 2, 151–161.
Telenius, H., Carter, N. P., Bebb, C. E., Nordenskjöld, M., Ponder, B. A. J., and Tunnacliffe, A. (1992). Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 13, 718–725.
Toffin, L., Webster, G., Weightman, A. J., Fry, J. C., and Prieur, D. (2004). Molecular monitoring of culturable bacteria from deep-sea sediment of the Nankai Trough, Leg 190 Ocean Drilling Program. FEMS Microbiol. Ecol. 48, 357–367.
Webster, G., Newberry, C. J., Fry, J. C., and Weightman, A. J. (2003). Assessment of bacterial community structure in the deep sub-seafloor biosphere by 16S rDNA-based techniques: a cautionary tale. J. Microbiol. Methods 55, 155–164.
Keywords: whole-genome amplification, metagenomics, deep biosphere, low biomass, bacteria, archaea, next-gen sequencing
Citation: Martino AJ, Rhodes ME, Biddle JF, Brandt LD, Tomsho LP and House CH (2012) Novel degenerate PCR method for whole-genome amplification applied to Peru Margin (ODP Leg 201) subsurface samples. Front. Microbio. 3:17. doi: 10.3389/fmicb.2012.00017
Received: 14 October 2011; Paper pending published: 03 November 2011;
Accepted: 09 January 2012; Published online: 23 January 2012.
Edited by:
Andreas Teske, University of North Carolina at Chapel Hill, USAReviewed by:
Julie A. Huber, Marine Biological Laboratory, USAJohn R. Spear, Colorado School of Mines, USA
Copyright: © 2012 Martino, Rhodes, Biddle, Brandt, Tomsho and House. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Christopher H. House, Department of Geosciences, Penn State Astrobiology Research Center, Pennsylvania State University, 220 Deike Building, University Park, PA 16802, USA. e-mail: chrishouse@psu.edu