 Akira Ashizawa1
Akira Ashizawa1- 1 Department of Molecular Cell Biology, Interdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, Yamanashi, Japan
- 2 Department of Cell Biology, Kyoto Pharmaceutical University, Kyoto, Japan
Ubiquitination is a post-translational modification in which one or more ubiquitin molecules are covalently linked to lysine residues of target proteins. The ubiquitin system plays a key role in the regulation of protein degradation, which contributes to cell signaling, vesicular trafficking, apoptosis, and immune regulation. Bacterial and viral pathogens exploit the cellular ubiquitin system by encoding their own proteins to serve their survival and replication in infected cells. Recent studies have revealed that Kaposi’s sarcoma-associated herpesvirus (KSHV) manipulates the ubiquitin system of infected cells to facilitate cell proliferation, anti-apoptosis, and evasion from immunity. This review summarizes recent developments in our understanding of the molecular mechanisms used by KSHV to interact with the cellular ubiquitin machinery.
Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8, is classified as a rhadinovirus in the γ-herpesvirus subfamily and was the eighth human herpesvirus to be discovered from Kaposi’s sarcoma (KS) specimens (Chang et al., 1994). KSHV is closely associated with KS and is also linked to AIDS-related lymphoproliferative disorders, such as primary effusion lymphoma (PEL, also known as body cavity-based lymphoma) and plasmablastic variant multicentric Castlemans disease (Russo et al., 1996; Ganem, 2007). The neoplastic potential of KSHV is well-established, especially in the context of immunosuppressed patients who are undergoing organ transplants, or who are co-infected with human immunodeficiency virus (HIV).
Like other herpesviruses, KSHV has two life cycles (i.e., latency and lytic replication). In latency, the KSHV genome circularizes and forms a double-stranded DNA, the episome, in the nucleus of a host cell. The KSHV genome contains several homologs of human genes such as viral cyclin D (v-cyclin), viral FLICE inhibitory protein (v-FLIP), viral G-protein coupled receptor (vGPCR), vIRF1, vBcl2, and vIL6 (Jenner and Boshoff, 2002), which contribute directly to KSHV-induced pathogenesis. KSHV establishes a latent infection in KS and PEL expressing several viral genes, including latency associated nuclear antigen (LANA), v-FLIP, v-cyclin, kaposin, and microRNAs. LANA (also called LANA1) encoded by ORF73 is required for replication and maintenance of the KSHV episomal DNA (Ballestas et al., 1999) and is expressed in all KSHV-associated malignancies (Järviluoma and Ojala, 2006). LANA also contributes to KSHV-associated oncogenesis through interaction with cellular molecules. KSHV alternates between lytic replication (productive infection) and latency (latent infection) by way of replication and transcription activator (RTA) expression. RTA, encoded by the immediate-early gene ORF50, is a critical switch molecule for initiating lytic replication (Deng et al., 2007). During lytic infection, progeny viruses are generated and released from host cells.
Kaposi’s sarcoma-associated herpesvirus and other herpesviruses have a unique feature: KSHV can manipulate cellular machineries, such as cell proliferation, anti-apoptosis, and immune surveillance, by hijacking the cellular ubiquitin system, including polyubiquitin-dependent degradation by the 26S proteasome. The ubiquitin system plays fundamental roles in the regulation of cellular events, such as protein degradation, signal transduction, endocytosis, protein trafficking, and immune responses (Pickart, 2001; Ciechanover, 2005). It is well known that many viruses exploit the ubiquitin system in various ways for entry or release of progeny virus (Bieniasz, 2006) and also manipulate the system to overcome the cellular apoptosis machinery and immune responses. Additionally, viruses target deubiquitinating enzymes (DUBs) to stabilize cellular and viral proteins needed for viral processes (Sulea et al., 2006). In this review, we discuss how KSHV can hijack the cellular ubiquitin system to promote successful viral infection.
The Ubiquitin System and 26S Proteasome as Targets of KSHV’s Strategy
The Ubiquitin System and KSHV
In the ubiquitin system, a substrate protein is tagged with a polyubiquitin chain or a single ubiquitin molecule via isopeptide bonds that are formed between the carboxyl termini of ubiquitin molecules and either the ε-amino groups of the lysine residues in the target protein or the ubiquitin molecules themselves. Ubiquitin conjugation is catalyzed by the sequential actions of ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3). In this system, ubiquitin, a polypeptide of 76 amino acid residues, is first transferred to the E1 in an ATP-dependent manner (Figure 1A). This activated ubiquitin is then transferred to the E2. Finally, the ubiquitin is covalently attached to the target protein by E3 ubiquitin ligase, leading to the formation of a polyubiquitin chain (Pickart, 2001; Ciechanover, 2005; Weissman et al., 2011).
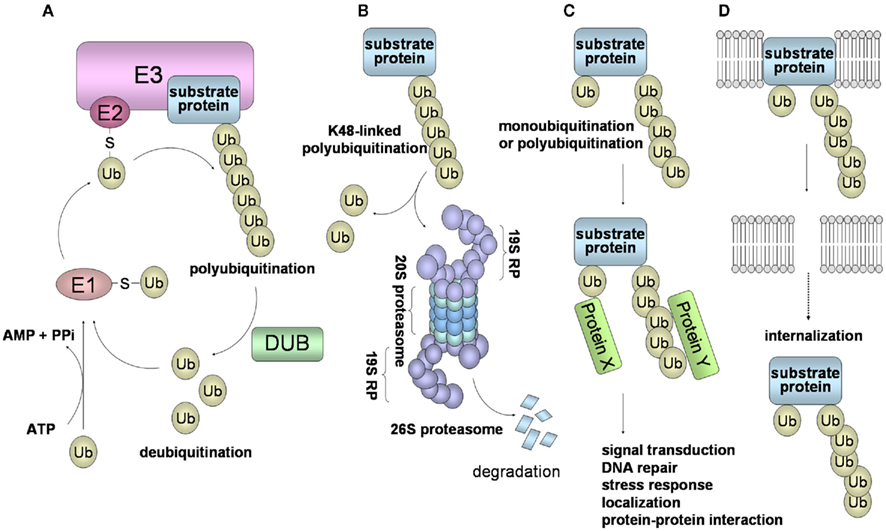
Figure 1. Schematic of the ubiquitin system and 26S proteasome-mediated degradation. (A) Ubiquitin is first transferred to E1 in an ATP-dependent manner. This process requires the initial ATP-dependent activation of ubiquitin, with the release of PPi, followed by binding of ubiquitin to a cysteine residue of E1 through a thioester linkage. Then, the activated ubiquitin is transferred to E2. Some selectivity for substrate occurs in this step, but the exquisite selectivity of intracellular proteolysis comes from the E3 ubiquitin ligases, which couple with E2 to bind substrates and catalyze the transfer of the activated ubiquitin from the E2 to the substrate, processively forming a chain of ubiquitins. (B) In the case of conjugation with K48-linked polyubiquitin, the polyubiquitinated protein is recognized by the 26S proteasome and is destroyed in an ATP-dependent manner. The 26S proteasome consists of the 19S regulatory complex (19RP) and a proteolytic core particle, the 20S proteasome. The 20S proteasome is arranged as a stack of four rings, composed of two outer α rings and two inner β rings. Three β subunits (β2, β5, β1) of a β ring are responsible for the distinct trypsin-like, chymotrypsin-like, and caspase-like proteolytic activities, respectively. (C) Modifications of a monoubiquitin and a polyubiquitin chain linked through Lys other than Lys48 (for example, Lys63, Lys11, and Lys29) play a role in many cellular processes, such as signal transduction, DNA repair, and localization through protein–protein interactions. (D) Monoubiquitination and K63-linked polyubiquitination are also involved in the internalization and endocytosis of membrane proteins.
Ubiquitin has seven internal Lys residues, at positions 6, 11, 27, 29, 33, 48, and 63; Lys48 is most commonly used for the formation of a polyubiquitin chain. Ubiquitination consists of monoubiquitination and polyubiquitination, and ubiquitination can be classified into two types: a degradation signal for the 26S proteasome (Figure 1B), and a non-degradation signal (Figures 1C,D). Polyubiquitination can be reversed by the isopeptidase activities of DUBs, also called deubiquitinase and isopeptidase, and of the POH1/Rpn11 subunit of the 26S proteasome. DUBs disassemble polyubiquitin chains from polyubiquitinated substrates, and also replenish the pool of free ubiquitin. Monoubiquitination, the conjugation of a single ubiquitin molecule, is involved in DNA repair, protein–protein interactions, and endocytosis of membrane proteins (Hicke, 2001). The polyubiquitin chain, linked through Lys48 (K48-linked polyubiquitin), functions as a signal for degradation by the 26S proteasome. On the other hand, it has been thought that the polyubiquitin chain linked through Lys other than Lys48 (i.e., Lys6, 11, 27, 29, 33, and 63), plays a role in numerous cellular events. In particular, K63-linked polyubiquitin is involved in protein–protein interactions, cell signaling pathways, DNA repair, the stress response, and ribosomal function (Weissman et al., 2011; Figure 1C). Moreover, K63-linked polyubiquitination functions as a signal that induces internalization and lysosome-mediated degradation through endocytosis toward cell surface proteins (Hicke, 1999; Figure 1D).
Recent studies have shown that KSHV targets the ubiquitin system to manipulate cell signaling pathways, apoptosis, cell cycle regulation, IFN response, and antigen presentation. KSHV dysregulates the stabilities of regulator molecules p53, p27, IκB, Notch, and β-catenin. By hijacking regulator molecules, KSHV is able to alter viral and cellular gene expression. Additionally, KSHV can utilize the ubiquitin system by providing its own viral protein as an E3 enzyme or as a component protein of the E3 complex. It was reported that KSHV-encoded RTA acted as the E3 ubiquitin ligase, and mediates polyubiquitination of IFN regulatory factor 7 (IRF7) for proteasomal degradation. KSHV-encoded LANA functions as a potential component of the E3 ubiquitin ligase complex for polyubiquitination of p53 and VHL (Figure 1B). KSHV also encodes ubiquitin ligases (K3 and K5) that induce the K63-linked polyubiquitination and internalization of major histocompatibility complex class I (MHC-I) molecules (Figure 1D). Furthermore, KSHV encodes a viral DUB which can disassemble polyubiquitin chains to stabilize the cellular and viral proteins. Thus, KSHV can create a favorable environment for the proliferation and survival of KSHV-infected host cells using these encoded proteins.
KSHV and the 26S Proteasome
Proteins conjugated with a K48-linked polyubiquitin chain are rapidly degraded by the 26S proteasome in an ATP-dependent manner (Figure 1B). Attachment of a chain composed of four or more ubiquitins is required for 26S proteasome-mediated protein degradation. The 26S proteasome consists of the 19S regulatory complex (19RP) and the 20S proteasome (Ciechanover, 2005; Weissman et al., 2011). The 20S proteasome is arranged as a stack of four rings, two outer α rings and two inner β rings, that have trypsin-like, chymotrypsin-like, and caspase-like proteolytic activities. The 19S RP can bind to a polyubiquitinated protein through the S5a/Rpn10 subunit, detach the polyubiquitin chain through the action of the Rpn11 subunit, and unfold the substrate and move it into the 20S proteasome for degradation. In the case of the globular proteins, the substrate undergoes ATP-dependent unfolding by the association with the 19S ATPases, followed by translocation into the entrance opening of the 20S proteasome. Finally, peptides ranging in length from 10 to 25 amino acid residues emerge from the 20S proteasome.
Kaposi’s sarcoma-associated herpesvirus dysregulates cellular antiviral responses in various ways: (1) downregulation of MHC-I molecules, (2) suppression of the IFN response by IRF7 degradation, and (3) inhibition of the proteasomal processing process needed for antigen production. Virus-infected cells are recognized by CD8+ cytotoxic T lymphocytes (CTL) upon display of virus-derived peptides by MHC-I. These virus-derived peptides are generated by the 26S proteasome, and then the viral peptides are translocated into the endoplasmic reticulum where they form a complex with MHC-I, with the assistance of peptide transporters (TAP) and chaperones. In particular, it is known that EBV prevents antigen presentation by inhibiting the 26S proteasome. EBV-encoded EBNA1 is required for the maintenance of EBV genomes. Furthermore, EBNA1 blocks the ability of the 26S proteasome to produce antigenic peptides (Levitskaya et al., 1995). EBNA1 is separated into N- and C-terminal domains by a Gly-Ala repeat domain, which disturbs the presentation of EBNA1 epitopes to MHC-I and also inhibits EBNA1 mRNA translation, thus minimizing protein levels in infected cells (Yin et al., 2003). EBNA1 is thus protected from endogenous presentation through the MHC-I pathway and this is also likely to be responsible for the long half-life of the EBNA1 protein. KSHV-encoded LANA, like EBNA1, may contribute to disturbing the antigen presentation by inhibition of the 26S proteasome activity. In fact, LANA also functions as a cis-acting inhibitor of antigen presentation (Zaldumbide et al., 2007). LANA has no sequence homology to EBNA1 but is apparently functionally analogous. LANA is a 220 kDa protein with unique N- and C-terminal regions separated by an acidic central repeat (CR) region, which contains three sets of repeats that are rich in Asp and Glu, Pro and Gln, and Pro and Glu, respectively. The CR region does not contain any proteolytic site for the proteasome. The LANA CR region can be further subdivided into three subdomains, based on differences in amino acid (aa) repeat sequences: CR1 (aa 321–428 in ORF73 of the BC-1 sequence, CR2 (aa 430–768), and CR3 (aa 769–937). It is known that the subdomain containing CR2CR3 contributes to LANA stability in vitro and in vivo and also retards LANA protein synthesis, minimizing LANA protein levels (Kwun et al., 2007). A recent study showed that the LANA CR1 subdomain inhibited MHC-I peptide antigen presentation in cis (Kwun et al., 2011). The CR2 subdomains retard LANA proteasomal processing but do not inhibit LANA peptide processing by MHC-I. These findings suggest that LANA physically inhibits the proteasome by insertion of the repeats into the proteolytic core particle of the 26S proteasome; however, KSHV and EBV use different mechanisms to evade host CTL-dependent surveillance.
KSHV and E3 Ubiquitin Ligases
Viral Proteins as Components of E3 Ubiquitin Ligases
Various combinations of E2 and E3 ubiquitin ligases determine the specificity toward target proteins and the types of ubiquitin linkages of the polyubiquitin chains. It is believed that there are in excess of 1000 different ubiquitin ligases in our genomes. E3 ubiquitin ligases are a large family of proteins that can be classified into three major and structurally distinct types: (1) HECT-type, (2) RING-finger-type, and (3) U-box-type E3 ubiquitin ligases (Pickart, 2001; Weissman et al., 2011). HECT-type E3s directly bind the substrate and contain a HECT domain that binds E2 and catalyzes the ubiquitin ligation (Figure 2A). Ubiquitin is transferred from the E2 to an active-site cysteine residue within the HECT domain, forming an E3 ubiquitin thioester complex. Ubiquitin is then transferred to the target protein. In humans, there are more than 50 kinds of HECT-type E3 enzymes. E6AP was the first HECT-type E3 identified, and this protein mediates K48-linked polyubiquitination of p53 in cells that express the E6 of human papillomavirus (HPV), types 16 and 18. The HPV E6 protein forms a complex with cellular E6AP, and increases the affinity for p53. RING-finger-type E3s contain RING-finger domains that interact with E2. RING-finger-type E3s can be sub-classified into monomeric RING-finger E3s (Figure 2B) and multimeric complex RING-finger E3s (Figure 2C), such as SCF and ECS (Lipkowitz and Weissman, 2011). RING-finger-type E3s function primarily as scaffolds, orienting the E2-ubiquitin thioester complex and the target protein for ubiquitin transfer. Mdm2 (Figure 2B-2) is monomeric RING-finger E3 for K48-linked polyubiquitination of p53 in normal cells. Multimeric RING-finger E3s [also called Cullin (Cul)-based E3s] constitute the largest subfamily and include the SKP1-Cul1-F-box protein (SCF) complex, the elongin C-elongin B-Cul5-SOCS-box (ECS) complex, and the anaphase-promoting complex/cyclosome (APC/C). The SCF complex (Figure 2C-4) is composed of Skp1 (adaptor protein), Cul1 (scaffold protein), Rbx1/Roc1 (E2-binding protein), and F-box protein. F-box protein, classified into Fbw (β-TrCP and Fbw7), Fbl (Skp2), and Fbx types, binds through its F-box motif to Skp1 and recognizes the substrate. The F-box protein also binds to Skp1, which can link the F-box protein to Cul1. Cul1 functions as a scaffold, binding the RING-finger protein, Rbx1, and an E2. The ECS complex (Figure 2C-5) is composed of elongin B, elongin C, Cul5, Rbx2, and SOCS-box protein. SOCS-box binds to the Skp1 homologs elongin C and elongin B. ECV consists of Cul2 in place of Cul5 in ECS, Rbx1 in place of Rbx2 in ECS, and also the VHL-box protein that binds HIF-1α.
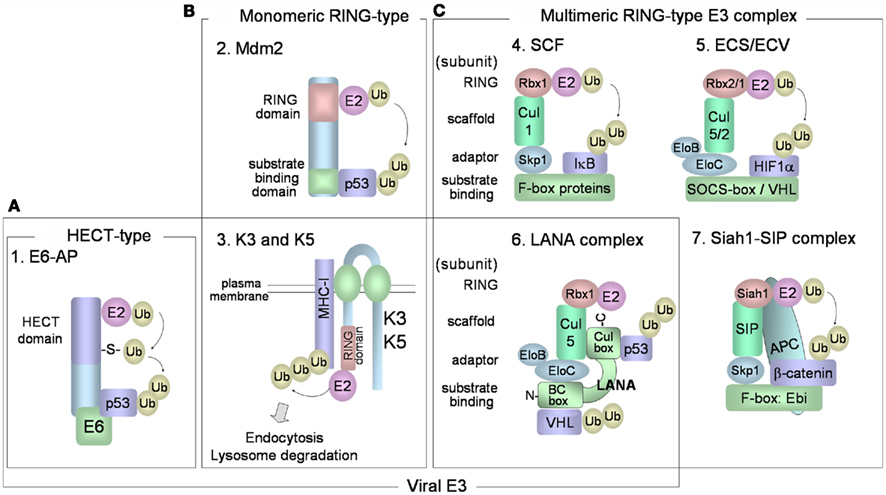
Figure 2. E3 ubiquitin ligases. (A) HECT-type E3 has a HECT domain that binds to E2. E6-E6AP complex mediates polyubiquitination of p53. (B,C) RING-finger-type E3 enzymes have RING-finger domains that bind to E2 enzymes and can be categorized into monomeric (B) and multimeric RING-finger E3 enzymes (C). (B-2) Mdm2 is monomeric RING-finger E3 for the K48-linked polyubiquitination of p53 in normal cells. (B-3) K3 and K5 bind to target substrates (MHC class I) via their transmembrane regions, and contain a RING-CH domain that interacts with E2. The cytoplasmic tails of the target substrates are mono- and poly-ubiquitinated, and the substrates are then endocytosed from the plasma membrane for degradation in lysosomes. (C-4) The SCF complex consists of Skp1 (adaptor protein), Cul1 (scaffold protein), Rbx1/Roc1 (E2-binding protein), and F-box protein that binds to Skp1 and recognizes the substrate. (C-5) The ECS complex is composed of elongin B, elongin C, Cul5, Rbx2, and substrate receptor SOCS-box protein. ECV is composed of elongin B, elongin C, Cul2, Rbx1, and VHL-box protein for binding HIF-1α. (C-6) KSHV LANA functions as a SOCS-box-like protein in the ECS/ECV complex, consisting of elongin B, elongin C, Cul5, and Rbx1. LANA SOCS-box-like motif consists of an elongin B/C box (N-terminus) and a Cul box (C-terminus). LANA binds to p53 protein through its C-terminus and binds to VHL protein through its N-terminus. (C-7) The human homolog of Drosophila Siah-1 has a RING-finger domain to bind E2 and forms an E3 complex composed of SIP, Skp1, and Ebi that recognizes β-catenin. Siah-1 binds to APC and this binding promotes degradation of β-catenin.
Kaposi’s sarcoma-associated herpesvirus proteins may function as potential components of E3 ubiquitin ligase complexes. Because E3 ubiquitin ligase can directly destabilize target proteins, the cellular polyubiquitin machinery is dysregulated by a spurious E3 ubiquitin ligase containing viral components. KSHV-encoded LANA plays an important role in the maintenance of the KSHV genome, and also contributes to KSHV-associated oncogenesis through interaction with pRb (Radkov et al., 2000), p53 (Friborg et al., 1999), and GSK-3 (Fujimuro et al., 2003). LANA also functions as a potential component within the ECV/ECS complex, and targets the tumor suppressor, VHL, and p53 protein for polyubiquitination (Cai et al., 2006). LANA contains a putative SOCS-box motif, which is necessary for its assembly into the Rbx1-Cul5-elongin B-elongin C complex (Figure 2C-6). LANA binds to p53 protein through its C-terminus and binds to VHL protein through its N-terminus. VHL protein, a component of ECV, functions as a receptor for hydroxylated HIF-1α and induces K48-linked polyubiquitination of HIF-1α (Figure 2C-5). The oxygen-sensitive transcription factor, HIF-1α (or HIF-2α), binds to hypoxia-responsive DNA promoters involved in angiogenesis and cell proliferation. During normoxia, hydroxylated HIF-1α and 2α are recognized by VHL protein, resulting in their polyubiquitination and degradation. LANA stabilizes HIF-1α and increases HIF-1α-dependent transactivation by down-regulating VHL protein. Thus, LANA directly serves as a substrate receptor in ECS to promote degradation of the VHL and p53. It is known that multiple KSHV proteins, apart from LANA, are involved in p53 polyubiquitination for proteasomal degradation. KSHV encodes viral IFN regulatory factor 4 (vIRF4) that binds Mdm2, leading to the proteasomal degradation of p53 in KSHV-infected cells (Lee et al., 2009). The central region of vIRF4 interacts with MDM2, leading to inhibition of MDM2 autoubiquitination and, thereby, increased MDM2 stability. Moreover, KSHV-encoded vIRF1 also contributes to destabilizing the protein level of p53 by manipulating ATM kinase, which is activated by DNA damage. ATM can phosphorylate the Ser15 residues of p53, and this phosphorylation inhibits the interaction with Mdm2, resulting in p53 stabilization. However, vIRF1 suppresses Ser15 phosphorylation of p53 by ATM, resulting in an increase of p53 ubiquitination and degradation by the proteasome (Shin et al., 2006).
Replication and transcription activator, a key regulator for KSHV reactivation from latent to lytic infection, may function as a E3 ubiquitin ligase for IRF7. In concert with UbcH5α as an E2 enzyme, RTA mediates the polyubiquitination of IRF7 depending on its Cys/His-rich domain, resulting in proteasomal degradation of IRF7 (Yu et al., 2005). RTA also autoregulates its own polyubiquitination and stability. The Cys/His-rich domain in the RTA N-terminal region is not categorized into a RING-finger domain and is composed of six Cys and one His residues (Cys3HisCys3). RAUL, a HECT-type E3, mediates polyubiquitination of IRF7 and IRF3. It is known that RTA induces the stabilization of RAUL and enhances the ability of RAUL to degrade IRF7 and IRF3 (Yu and Hayward, 2010). Furthermore, RTA induces the downregulation of “Toll-interleukin-1 receptor domain-containing adaptor-inducing beta-interferon” (TRIF; Ahmad et al., 2011), the cellular transcriptional repressor, Hey1 (Gould et al., 2009), and K-RBP (KSHV RTA binding protein)/human hypothetical protein MGC2663 (Yang et al., 2008). RTA interacts with Hey1 and K-RBP, and RTA mediates polyubiquitination, dependent on the Cys/His-rich domain of RTA, resulting in proteasomal degradation, while the Cys/His-rich domain of RTA is partially necessary for TRIF degradation, and RTA-mediated degradation of TRIF is partially mediated through the ubiquitin system. Thus, RTA manipulates the stabilities of various cellular proteins to dysregulate innate immunity and transcriptional regulation. However, it remains to be determined whether RTA Cys/His-rich domain directly binds E2. It is not known what kind of signal, such as protein–protein interaction, conformational switch or post-transcriptional modification of the substrate, is necessary to trigger interaction between RTA and it’s substrates. These issues are of great importance to many researchers and are expected to be resolved in future studies.
K3 and K5
Kaposi’s sarcoma-associated herpesvirus has elaborate machineries to escape innate and adaptive antiviral immune responses. KSHV establishes a life-long infection in healthy human hosts, and it is fundamentally important for KSHV to evade the host immune response. KSHV thus exploits the cellular ubiquitin system by providing its own viral E3 enzymes. KSHV-encoded K3 and K5 proteins (Figure 2B-3), a class of viral RING-CH-finger E3 ubiquitin ligases, also called the “membrane-associated RING-CH” (MARCH) family E3 ubiquitin ligases, can downregulate the cellular surface display of MHC-I proteins, by inducing endocytosis (Brander et al., 2000; Coscoy and Ganem, 2000; Ishido et al., 2000a). Furthermore, K3 and K5 both target γ-interferon receptor 1 (IFN-γR1) and induce its ubiquitination, endocytosis, and degradation, resulting in downregulation of IFN-γR1 surface expression (Li et al., 2007). K3 and K5 proteins exhibit 40% amino acid homology to each other and localize to the plasma membrane. The RING-CH domains in their cytosolic N-termini are responsible for polyubiquitination of the cytoplasmic tail region of the MHC-I protein, and the two transmembrane regions are required for recognizing MHC-I. K3 and K5-mediated polyubiquitination of MHC-I leads to rapid internalization of MHC-I from the cell surface and subsequent endolysosomal degradation.
The RING-CH domain of K5 interacts with UbcH5a as an E2 enzyme (Coscoy et al., 2001), which is involved in K63-, K11-, and K48-linked polyubiquitination (Bosanac et al., 2011). K5 also downregulates other cell surface molecules, such as natural killer cell activators ICAM-1, B7-2 (Ishido et al., 2000b; Coscoy and Ganem, 2001), CD1d (Sanchez et al., 2005), MHC class I-related chain A/MICA (Thomas et al., 2008), HFE (Rhodes et al., 2010), and CD31/PECAM (Mansouri et al., 2006) by enhancing endocytosis and subsequent endolysosomal degradation, leading to the escape of infected cells from natural killer cells. K5 also targets the cytoplasmic tail of tetherin (also known as BST2 and CD317) for ubiquitination, leading to relocalization of tetherin to endosomal compartments (Mansouri et al., 2009; Pardieu et al., 2010). Tetherin is an interferon-induced antiviral membrane protein that prevents the release of many enveloped viral particles, including KSHV and HIV. Thus, K5 contributes to efficient KSHV particle release by down-regulating tetherin.
K3 mediates the K63-linked polyubiquitination of MHC-I, which is necessary for the efficient endocytosis and lysosomal degradation of MHC-I. K3 also mediates MHC-I monoubiquitination, and K3-mediated polyubiquitination through the K63-linkage requires both UbcH5 and Ubc13 as E2 enzymes for initiating monoubiquitination and subsequent K63-linked polyubiquitination, respectively, of MHC-I (Duncan et al., 2006). Ubc13 has been implicated in a variety of cellular processes, such as cell signaling and DNA repair, and has the ability to catalyze formation of K63-linked polyubiquitin chains on various substrates.
Cellular Events Modulated by KSHV Through Ubiquitination
Substrate Recognition by the SCF Complex
Many substrates of the 26S proteasome are modified by the post-translational modification before their K48-linked polyubiquitination. E3 ubiquitin ligases strictly recognize the post-translational modified forms of the target substrates: several different types of triggers for polyubiquitination – phosphorylation, proline hydroxylation and glycosylation – function as recognition signals for E3s. However, it is not clear how KSHV E3s (i.e., K3, K5, LANA, and RTA) recognize their substrates. There is very limited information available concerning LANA and RTA substrate recognition. It is not known what type of post-transcriptional modification of the substrate triggers interaction with LANA or RTA. Prospective studies are expected to resolve these important issues. Furthermore, it remains to be determined which amino acid residue of the substrate is modified and what type of polyubiquitin chain is conjugated with their substrate. These issues are also expected to be cleared in future studies.
In contrast to KSHV E3s, the substrate recognitions of cellular SCF-type E3s are well known. Many F-box proteins of SCF-type E3s can recognize phosphorylated forms of target substrates (Lipkowitz and Weissman, 2011). The SCF complex consists of Skp1, Cul1, Rbx1/Roc1, and F-box protein that binds the substrate (Figure 2C-4). More than 70 F-box proteins in humans can be categorized into three types (Figure 3): F-box proteins containing WD40 repeats domain (Fbw/FBXW), F-box proteins containing leucine-rich repeats domain (Fbl/FBXL), and F-box proteins containing other domains (Fbx/FBXO; Nakayama and Nakayama, 2006). The SCFβTrCP complex, containing β-TrCP/Fbw1 as the Fbw-type F-box protein, binds to cellular signaling regulators, such as β-catenin, NF-κB inhibitors (IκBα, β, ε), ATF4, Snail, DIG1, Cdc25, and Wee1A. β-TrCP recognizes the conserved DpSGXXpS motif (with phosphorylated serine residues) in the substrate. The phosphorylation of Ser32 and Ser36 of IκBα is necessary for binding of β-TrCP, and the phosphorylation of Ser33 and Ser37 of β-catenin is necessary for binding of β-TrCP. As described in the following sections, KSHV can promote these phosphorylations of IκBα and β-catenin in order to up-regulate NF-κB and Wnt signaling. The SCFFbw7 complex, containing Fbw7 as the Fbw-type F-box protein, recognizes c-Myc, c-Jun, Notch-1, Notch-4, cyclin E, and presenilin. The SCFSkp2 complex, containing Skp2/Fbl1 as the Fbl-type F-box protein, recognizes p21, p27, p57, p130, cyclin A, -D, -E, E2F, Foxo1, Orc1, and Cdk9. SCFFbw7 complex is involved in downregulation of cell cycle accelerators, such as c-Myc, c-Jun, and Notch, whereas SCFSkp2 is involved in downregulation of cell cycle break molecules, such as p21, p27, p57, and p130. As described in the next section, KSHV can also promote the phosphorylation of Thr187 of p27, leading to polyubiquitination of p27. For the many substrates recognized by the SCF complex, phosphorylation is required for binding with the respective F-box proteins and for initiating K48-linked polyubiquitination.
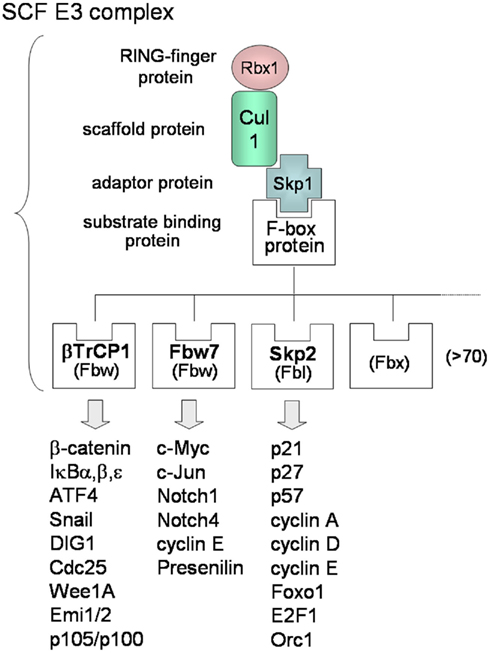
Figure 3. Molecular diversity of SCF E3 complexes. The SCF complex consists of Skp1, Cul1, Rbx1 (E2-binding protein), and F-box protein that binds Skp1 and recognizes the substrate. There are more than 70 F-box proteins in humans and the F-box protein decides the substrate specificity of SCF. F-box proteins can be categorized into three types: F-box proteins containing a WD40-repeat domain (Fbw), F-box proteins containing a leucine-rich-repeat domain (Fbl), and F-box proteins containing other domains “X” (Fbx). Each F-box protein binds the phosphorylated consensus sequence of the specific substrate, resulting in K48-linked polyubiquitination.
As described previously, the ubiquitin system controls the stabilization of regulatory factors involved in cell signaling pathways, which serve in the progression of cell proliferation, anti-apoptosis, and control of the viral life-cycle. It is known that KSHV dysregulates recognition signals for cellular SCF complex E3s and exploits polyubiquitinating activities of SCF E3s. KSHV manipulates the upstream regulators of these cell signals in order to utilize cellular SCF E3s and control cell signalings. By targeting the specific recognition signals for the SCF complex – the phosphorylation of substrates such as IκB, β-catenin, and p27 – KSHV can affect cell cycle regulation, NF-κB signaling, and Wnt signaling (described in the following sections).
Cell Cycle Regulation
G1-S phase progression is strictly regulated by cyclin-dependent kinase (Cdk)-cyclin complexes. The cyclin D-Cdk4 (or Cdk6) and cyclin E-Cdk2 complexes phosphorylate pRb from mid-G1 to the late-G1 stage. These pRb phosphorylations lead to release, and activate E2F for the S phase progression. Cdk inhibitors (CKIs), negative-regulators of cyclin-Cdk complexes, can be classified into the Cip/Kip family (p21Cip1, p27Kip1, and p57Kip2) and the INK4 family (p15INK4b, p16INK4a, p18INK4c, and p19INK4d). Cip/Kip family proteins bind cyclin-Cdk complexes, leading to inhibition of Cdk activity, and are targeted for K48-linked polyubiquitination by SCFSkp2 complex E3 (Nakayama and Nakayama, 2006).
Kaposi’s sarcoma-associated herpesvirus mimics and uses these regulatory factors. KSHV v-cyclin/ORF72, a human cyclin D2 homolog, interacts with and activates Cdk4 and Cdk6 to achieve phosphorylation of pRb and cellular cyclin D, leading to G1-S phase progression (Chang et al., 1996; Mittnacht and Boshoff, 2000; Järviluoma and Ojala, 2006). Interestingly, unlike the cellular cyclins, v-cyclin-Cdk complex is resistant to the CKIs, p21, p27, and p16 (Swanton et al., 1997), and this complex thus functions as a constitutively active kinase. The v-cyclin-Cdk6 complex phosphorylates p21 on Ser130, and this phosphorylated p21 cannot inhibit Cdk2 (Järviluoma et al., 2006). Furthermore, v-cyclin-Cdk6 complex interacts with p27 and phosphorylates p27 at Thr187, leading to polyubiquitination of p27 for degradation (Ellis et al., 1999; Mann et al., 1999; Järviluoma et al., 2004). This regulation of p27 is normally performed by cellular cyclin E-Cdk2. Skp2 F-box protein of SCFSkp2 complex can bind Thr187-phosphorylated p27 and mediates its modification with a K48-linked polyubiquitin chain (Figure 3), leading to its degradation.
Notch Signaling
Kaposi’s sarcoma-associated herpesvirus-encoded proteins manipulate upstream regulators in cell signaling and exploit the ubiquitin system to alter the stabilization of target proteins. Furthermore, KSHV proteins can directly bind a component of E3 ubiquitin ligases to inhibit the E3 activity for polyubiquitination, leading to stabilization of target substrates. The SCFFbw7 complex (Figure 3) is composed of Fbw7 as an F-box protein, which recognizes and binds the intracellular regions of Notch-1, -3, and -4 (Nakayama and Nakayama, 2006; Matsumoto et al., 2011). The F-box protein Fbw7, also known as Sel-10 in C. elegans and hCDC4 in humans, is thus a critical component of SCFFbw7, while it is a negative regulator of Notch signaling. Notch signaling plays roles in cell fate decisions, proliferation, and differentiation. When a Notch ligand, such as Jagged or Delta, binds the Notch receptor, a γ-secretase complex, including presenilin, cleaves the intramembrane region of the Notch receptor, Notch-1, 2, 3, and 4, resulting in release of intracellular Notch (ICN, also called the Notch intracellular domain, NICD). ICN, which functions as a transcriptional activator, enters the nucleus and binds “CBF1/Suppressor of Hairless/Lag-1” (CSL), upregulating transcription of its targets (Hayward et al., 2006).
It is known that, in addition to ICN, KSHV RTA interacts with CSL and activates target gene expression in cooperation with co-activators (Liang and Ganem, 2003). KSHV also has another way to activate Wnt signaling. KSHV LANA binds Fbw7/Sel-10, a component of SCFFbw7, to inhibit polyubiquitination of intracellular Notch-1 (ICN), which leads to stabilization of ICN. The carboxyl-terminus of LANA binds the F-box domain and the WD40 domain of Fbw7/Sel-10, whereas it competes with ICN for interaction with Fbw7/Sel-10, resulting in inhibition of ICN polyubiquitination and degradation (Lan et al., 2007). Stabilization of ICN by LANA is also related to the proliferation of KSHV-infected cells.
NF-κB Signaling
NF-κB signaling, regulated by the ubiquitin system, influences cell proliferation, anti-apoptosis, and inflammation. KSHV activates NF-κB pathways to promote host cell proliferation, survival, and angiogenesis. Additionally, constitutive activation of NF-κB signaling is essential for latent infection of KSHV and survival in KSHV-infected PEL cells. In normal resting cells, the canonical NF-κB signaling pathway is suppressed by IκBα protein. The NF-κB pathway is further regulated by IKK complex, consisting of IKKα, β, and a scaffold subunit (IKKγ/NEMO). A stimulus, such as TNF-α or IL-1β, induces activation of the IKK complex via TRAF6 and TRAF2. TRAF6 can modify NEMO by K63-linked polyubiquitination, in collaboration with Ubc13. K63-linked polyubiquitinated NEMO then activates the IKK complex, which phosphorylates IκBα. Phosphorylation of Ser32 and Ser36 IκBα can be a trigger for K48-linked polyubiquitination by SCFβTrCP (Figure 3). Phosphorylated IκBα is then subjected to SCFβTrCP-mediated K48-linked polyubiquitination and subsequent proteasomal degradation. This releases active NF-κB (p50–p65/RelA heterodimer). There is another upregulation system for IKK complex: TGF-β-activated kinase (TAK1) also phosphorylates and activates IKK. TAK1 forms a complex with TAK1-binding protein 1 (TAB1) and TAB2 (or TAB3). TAB2 stimulates TAK1 kinase activity and binds to K63-linked polyubiquitinated NEMO (or TRAF6) through the Zn-finger domain of TAB2. In response to TNF-α stimuli, receptor-interacting protein (RIP) is modified with K63-linked polyubiquitin chain, and polyubiquitinated RIP recruits NEMO into the IKK complex.
Kaposi’s sarcoma-associated herpesvirus-encoded v-FLIP, K7, and microRNA can induce NF-κB transcriptional activation by targeting the protein level of IκB. v-FLIP (ORF 71) achieves anti-apoptosis by activating NF-κB signaling (Chaudhary et al., 1999; Keller et al., 2000; Chugh et al., 2005). v-FLIP can activate the IKK complex through interaction with NEMO of the IKK complex, including Hsp90 (Field et al., 2003) and RIP (Chaudhary et al., 1999; Liu et al., 2002). Additionally, TRAF2 leads to the association of v-FLIP with the NEMO of the IKK complex (Guasparri et al., 2006). Activated IKK complex phosphorylates IκBα, leading to K48-linked polyubiquitination and proteasomal degradation. miR-K1 of the miRNA cluster, encoded by the KSHV genome, can decrease IκBα in a ubiquitin system-independent manner. miR-K1 reduces expression of IκBα protein by targeting the 3′-UTR of its transcript (Lei et al., 2010). Thus, miR-K1 upregulates NF-κB activity and inhibits viral lytic replication in PEL cells. KSHV small membrane protein, K7, inhibits caspase-3-induced apoptosis and interacts with ubiquitin/PLIC1 (Feng et al., 2004). Ubiquilin has an N-terminal ubiquitin-like (UBL) domain and a C-terminal ubiquitin-associated (UBA) domain. The UBA domain binds to the K7 protein and the polyubiquitin chain. The interaction between K7 and ubiquilin induces the degradation of IκB and concomitant activation of NF-κB signaling. Thus, to activate the NF-κB pathway, KSHV may induce the phosphorylation of IκBα for SCFβTrCP-mediated K48-linked polyubiquitination, resulting in reduced IκBα protein. Interestingly, K7 induces the degradation of KSHV-encoded ORF74/vGPCR, which is a pirated version of the human IL-8 receptor gene. vGPCR is a ligand-independent, constitutively active signaling molecule that activates NF-κB signaling and promotes cell proliferation. K7 causes vGPCR to be retained in the endoplasmic reticulum and induces the proteasomal degradation of vGPCR. Furthermore, K7 expression reduces vGPCR tumorigenicity in nude mice (Feng et al., 2008).
Wnt Signaling
The Wnt signaling pathway, also regulated by ubiquitin and the proteasome, is involved in several key developmental processes and in tumorigenesis. The Wnt signaling cascade is triggered when Wnt ligands bind to Frizzled receptors and the signal is transmitted through Disheveled to the axin-APC-GSK-3β complex, resulting in the disruption of this complex. The β-catenin level is regulated by GSK-3β (GSK-3), Ser/Thr kinase, which phosphorylates β-catenin and targets it for K48-linked polyubiquitination and, subsequently, proteasomal degradation. Displacement of GSK-3 from the complex leads to stabilization of β-catenin, which translocates to the nucleus and forms a complex with Lef (or Tcf4) transcription factors, stimulating the expression of c-Myc, c-Jun, and cyclin D1. Phosphorylation of β-catenin is essential for binding SCFβTrCP complex, containing the β-TrCP that recognizes the DpSGXXpS motif in β-catenin. SCFβTrCP complex can mediate K48-linked polyubiquitination for β-catenin (Figure 3). GSK-3 kinase recognizes and phosphorylates serine and/or threonine residues in the context of the S/Txxx pS/pT motif. The downstream Ser or Thr residue must first be phosphorylated by a priming kinase. In β-catenin, phosphorylation of Ser45 by casein kinase I serves as the priming event for GSK-3 kinase, which then phosphorylates the Thr41, Ser37, and Ser33 residues. Phosphorylation of Ser33 and Ser37 of β-catenin is necessary for binding of the SCFβTrCP complex and K48-linked polyubiquitination.
Kaposi’s sarcoma-associated herpesvirus dysregulates Wnt signaling by LANA (Fujimuro and Hayward, 2003; Fujimuro et al., 2003, 2005). The β-catenin protein accumulates in latently KSHV-infected PEL cells and KS cells. The mechanism of β-catenin dysregulation is related to the interaction of LANA and GSK-3 kinase. LANA associates with GSK-3, leading to nuclear translocation of GSK-3. Although GSK-3 is localized primarily in the cytoplasm, a small proportion of GSK-3 is known to enter the nucleus during S phase (Diehl et al., 1998), and LANA increases the number of cells in S phase (Fujimuro et al., 2003). Because LANA binds to nuclear GSK-3, LANA induces depletion of cytoplasmic GSK-3. In the absence of cytoplasmic GSK-3, β-catenin accumulates in the cytoplasm and enters the nucleus to stimulate transcriptional activation of downstream target genes. Additionally, LANA binds ERK1/2, which participates in the phosphorylation of Ser 9 of GSK-3, resulting in inactivation of GSK-3 (Liu et al., 2007). The stability of c-Myc is regulated by the c-Myc phosphorylation at Ser62, mediated by Erk, and at Thr58, mediated by GSK-3. This phosphorylation of c-Myc is necessary for the interaction with Fbw7 protein of SCFFbw7 and K48-linked polyubiquitination (Yada et al., 2004). Inactivation of nuclear GSK-3 by LANA may increase the stability and activity of c-Myc and further contribute to LANA-mediated growth dysregulation. In fact, abnormally stable c-Myc protein can be observed, and LANA is responsible for this stability in PEL cells (Bubman et al., 2007).
EBV upregulates Wnt signaling through a different mechanism than that in KSHV (Hayward et al., 2006). EBV LMP1 represses expression of the transcripts for Siah-1 (Jang et al., 2005), the human homolog of Drosophila seven in absentia. Siah-1 binds E2 enzyme with its N-terminal RING-finger domain and forms an E3 ubiquitin ligase complex, consisting of Siah-1, SIP, Skp1, and Ebi that binds the β-catenin protein (Figure 2C-7). This E3 ubiquitin ligase promotes the degradation of β-catenin through a mechanism independent of GSK-3β-mediated phosphorylation; Siah-1 binds to APC and this binding directly promotes degradation of β-catenin. On the other hand, an E3 complex including Siah-1 targets KSHV protein for polyubiquitination. The KSHV-encoded ORF45 protein is capable of inhibiting virus-dependent interferon induction and appears to be essential for both early and late stages of infection. Siah-1 complex induces the destabilization of ORF45 protein through polyubiquitination (Abada et al., 2008).
KSHV and Deubiquitinating Enzyme
Polyubiquitination is reversed by the isopeptidase activities of DUBs. DUBs disassemble polyubiquitin chains from substrates, resulting in inhibition of the recognition by the 26S proteasome, and induce stabilization of substrate proteins. It is well known that human herpesviruses utilize activities of DUBs for stabilization of their viral proteins. DUBs disassemble polyubiquitin chains from polyubiquitinated proteins, resulting in stabilization of substrate proteins.
USP7 (also called HAUSP) is an evolutionary-conserved mammalian DUB, which was originally identified as a protein binding to the EBNA1 of EBV and ICP0 of HSV-1 (Canning et al., 2004; Holowaty and Frappier, 2004). USP7 can remove the K48-linked polyubiquitin from the EBNA1 and ICP0, and stabilize them. Interestingly, recent studies show that KSHV also targets DUBs to stabilize the cellular and viral proteins needed for viral processes. KSHV encodes a viral DUB (ORF64) that can disassemble both K48- and K63-linked polyubiquitin chains (González et al., 2009), while ORF64 protein suppresses RIG-I-mediated IFN signaling by reducing the polyubiquitination of RIG-I (Inn et al., 2011). Additionally, RTA targets the USP7 to stabilize the HECT-type E3, RAUL (Yu and Hayward, 2010). KSHV, directly or indirectly, may exploit DUBs to stabilize its own proteins and other cellular proteins involved in its survival and replication in infected cells.
Conclusion
Ubiquitination plays a key role in controlling the function and stability of target proteins. KSHV hijacks the ubiquitin system and the degradation-by-proteasome system for the purpose of cell proliferation, anti-apoptosis, and evasion from immune surveillance in infected cells. KSHV-encoded proteins and E3 enzymes take advantage of cellular ubiquitins and induce the degradation of host proteins that are antagonistic to infection. KSHV also utilizes the activities of DUBs for stabilization of their viral proteins. However, there are many questions about these KSHV dysregulating mechanisms. It remains to be determined how K3, K5, LANA, and RTA recognize and dock their substrates. Particularly, it is not known what type of signal (such as post-transcriptional modification of the substrate, conformation-changing of substrate, or other protein interaction with substrate) can trigger interactions with KSHV E3s. These questions need to be resolved in a future study. Additionally, a very interesting discovery has been made with RTA Cys/His-rich domain, which is not categorized into RING-finger domain, and which may be a new type of RING-finger. However, it is unclear whether or not RTA Cys/His-rich domain binds to E2. This important question also needs to be answered in a future study.
With the exception of ubiquitination, several post-translational modifications of cellular and viral proteins, such as glycosylation, methylation, acetylation, sumoylation, and S-nitrosylation, have been described by numerous studies. Because these modifications play critical roles in regulating functions of substrate proteins, we need to consider these modification as well as ubiquitination. Further study is expected to reveal how KSHV associates with these post-translational modifications.
To further elucidate KSHV pathogenesis, it should be resolved how KSHV dysregulates and exploits ubiquitin pathways through advanced proteomic approaches. A better understanding of the interaction between KSHV infection and the ubiquitin system will provide new insights into the viral evasion of host immunity and the process of carcinogenesis triggered by KSHV, and may provide the basis for theories for the development of novel therapeutic interventions against KSHV-related cancers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was partly supported by Health and Labor Sciences Research Grants (grant no. H23-AIDS-Ippan-002) from the Ministry of Health, Labor and Welfare of Japan, Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Takeda Science Foundation, the Japan Health Sciences Foundation, and New Energy and Industrial Technology Development Organization (NEDO) of Japan.
References
Abada, R., Dreyfuss-Grossman, T., Herman-Bachinsky, Y., Geva, H., Masa, S. R., and Sarid, R. (2008). SIAH-1 interacts with the Kaposi’s sarcoma-associated herpesvirus-encoded ORF45 protein and promotes its ubiquitylation and proteasomal degradation. J. Virol. 82, 2230–2240.
Ahmad, H., Gubbels, R., Ehlers, E., Meyer, F., Waterbury, T., Lin, R., and Zhang, L. (2011). Kaposi sarcoma-associated herpesvirus degrades cellular Toll-interleukin-1 receptor domain-containing adaptor-inducing beta-interferon (TRIF). J. Biol. Chem. 286, 7865–7872.
Ballestas, M. E., Chatis, P. A., and Kaye, K. M. (1999). Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284, 641–644.
Bieniasz, P. D. (2006). Late budding domains and host proteins in enveloped virus release. Virology 344, 55–63.
Bosanac, I., Phu, L., Pan, B., Zilberleyb, I., Maurer, B., Dixit, V. M., Hymowitz, S. G., and Kirkpatrick, D. S. (2011). Modulation of K11-linkage formation by variable loop residues within UbcH5A. J. Mol. Biol. 408, 420–431.
Brander, C., Suscovich, T., Lee, Y., Nguyen, P. T., O’Connor, P., Seebach, J., Jones, N. G., van Gorder, M., Walker, B. D., and Scadden, D. T. (2000). Impaired CTL recognition of cells latently infected with Kaposi’s sarcoma-associated herpes virus. J. Immunol. 165, 2077–2083.
Bubman, D., Guasparri, I., and Cesarman, E. (2007). Deregulation of c-Myc in primary effusion lymphoma by Kaposi’s sarcoma herpesvirus latency-associated nuclear antigen. Oncogene 26, 4979–4986.
Cai, Q. L., Knight, J. S., Verma, S. C., Zald, P., and Robertson, E. S. (2006). EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2, e116. doi: 10.1371/journal.ppat.0020116
Canning, M., Boutell, C., Parkinson, J., and Everett, R. D. (2004). A RING finger ubiquitin ligase is protected from autocatalyzed ubiquitination and degradation by binding to ubiquitin-specific protease USP7. J. Biol. Chem. 279, 38160–38168.
Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M., and Moore, P. S. (1994). Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposis sarcoma. Science 266, 1865–1869.
Chang, Y., Moore, P. S., Talbot, S. J., Boshoff, C. H., Zarkowska, T., Godden-Kent, Paterson, H., Weiss, R. A., and Mittnacht, S. (1996). Cyclin encoded by KS herpesvirus. Nature 382, 410.
Chaudhary, P. M., Jasmin, A., Eby, M. T., and Hood, L. (1999). Modulation of the NF-(κ B pathway by virally encoded death effector domains-containing proteins. Oncogene 18, 5738–5746.
Chugh, P., Matta, H., Schamus, S., Zachariah, S., Kumar, A., Richardson, J. A., Smith, A. L., and Chaudhary, P. M. (2005). Constitutive NF-κB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 102, 12885–12890.
Ciechanover, A. (2005). Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 6, 79–87.
Coscoy, L., and Ganem, D. (2000). Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. U.S.A. 97, 8051–8056.
Coscoy, L., and Ganem, D. (2001). A viral protein that selectively downregulates ICAM-1 and B7–2 and modulates T cell costimulation. J. Clin. Invest. 107, 1599–1606.
Coscoy, L., Sanchez, D. J., and Ganem, D. (2001). A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J. Cell Biol. 155, 1265–1273.
Deng, H., Liang, Y., and Sun, R. (2007). Regulation of KSHV lytic gene expression, Curr. Top. Microbiol. Immunol. 312, 157–183.
Diehl, J. A., Cheng, M., Roussel, M. F., and ana Sherr, C. J. (1998). Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 12, 3499–3511.
Duncan, L. M., Piper, S., Dodd, R. B., Saville, M. K., Sanderson, C. M., Luzio, J. P., and Lehner, P. J. (2006). Lysine-63-linked ubiquitination is required for endolysosomal degradation of class I molecules. EMBO J. 25, 1635–1645.
Ellis, M., Chew, Y. P., Fallis, L., Freddersdorf, S., Boshoff, C., Weiss, R. A., Lu, X., and Mittnacht, S. (1999). Degradation of p27 (Kip) cdk inhibitor triggered by Kaposi’s sarcoma virus cyclin-cdk6 complex. EMBO J. 18, 644–653.
Feng, H., Dong, X., Negaard, A., and Feng, P. (2008). Kaposi’s sarcoma-associated herpesvirus K7 induces viral G protein-coupled receptor degradation and reduces its tumorigenicity. PLoS Pathog. 4, e1000157. doi: 10.1371/journal.ppat.1000157
Feng, P., Scott, C. W., Cho, N. H., Nakamura, H., Chung, Y. H., Monteiro, M. J., and Jung, J. U. (2004). Kaposi’s sarcoma-associated herpesvirus K7 protein targets a ubiquitin-like/ubiquitin-associated domain-containing protein to promote protein degradation. Mol. Cell. Biol. 24, 3938–3948.
Field, N., Low, W., Daniels, M., Howell, S., Daviet, L., Boshoff, C., and Collins, M. (2003). KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 116, 3721–3728.
Friborg, J. J., Kong, W., Hottiger, M. O., and Nabel, G. J. (1999). p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402, 889–894.
Fujimuro, M., and Hayward, S. D. (2003). The latency-associated nuclear antigen of Kaposis sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J. Virol. 77, 8019–8030
Fujimuro, M., Liu, J., Zhu, J., Yokosawa, H., and Hayward, S. D. (2005). Regulation of the interaction between glycogen synthase kinase 3 and the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen. J. Virol. 79, 10429–10441.
Fujimuro, M., Wu, F. Y., Aprhys, C., Kajumbula, H., Young, D. B., Hayward, G. S., and Hayward, S. D. (2003). A novel viral mechanism for dysregulation of beta-catenin in Kaposis sarcoma-associated herpesvirus latency. Nat. Med. 9, 300–306.
Ganem, D. (2007). “Kaposi’s sarcoma-associated herpesvirus,” in Fields’ Virology, 5th Edn, eds D. M. Knipe, and P. M. Howley (Philadelphia, PA: Lippincott Williams and Wilkins), 2847–2888.
González, C. M., Wang, L., and Damania, B. (2009). Kaposi’s sarcoma-associated herpesvirus encodes a viral deubiquitinase. J. Virol. 83, 10224–11033.
Gould, F., Harrison, S. M., Hewitt, E. W., and Whitehouse, A. (2009). Kaposi’s sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 83, 6727–6738.
Guasparri, I., Wu, H., and Cesarman, E. (2006). The KSHV oncoprotein vFLIP contains a TRAF-interacting motif and requires TRAF2 and TRAF3 for signalling, EMBO Rep. 7, 114–119.
Hayward, S. D., Liu, J., and Fujimuro, M. (2006). Notch and Wnt Signaling: Mimicry and manipulation by γ herpesviruses. Sci. STKE 335, re4.
Hicke, L. (1999). Gettin’ down with ubiquitin: turning off cell-surface receptors, transporters and channels. Trends Cell Biol. 9, 107–112.
Holowaty, M. N., and Frappier, L. (2004). HAUSP/USP7 as an Epstein–Barr virus target. Biochem. Soc. Trans. 32, 731–732.
Inn, K. S., Lee, S. H., Rathbun, J. Y., Wong, L. Y., Toth, Z., Machida, K., Ou, J. H., and Jung, J. U. (2011). Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 85, 10899–10904.
Ishido, S., Wang, C., Lee, B. S., Cohen, G. B., and Jung, J. U. (2000a). Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74, 5300–5309.
Ishido, S., Choi, J. K., Lee, B. S., Wang, C., DeMaria, M., Johnson, R. P., Cohen, G. B., and Jung, J. U. (2000b). Inhibition of natural killer cell-mediated cytotoxicity by Kaposi’s sarcoma-associated herpesvirus K5 protein. Immunity 13, 365–374.
Jang, K. L., Shackelford, J., Seo, S. Y., and Pagano, J. S. (2005). Up-regulation of beta-catenin by a viral oncogene correlates with inhibition of the seven in absentia homolog 1 in B lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 102, 18431–18436.
Järviluoma, A., Child, E. S., Sarek, G., Sirimongkolkasem, P., Peters, G., Ojala, P. M., and Mann, D. J. (2006). Phosphorylation of the cyclin-dependent kinase inhibitor p21Cip1 on serine 130 is essential for viral cyclin-mediated bypass of a p21Cip1-imposed G1 arrest. Mol. Cell. Biol. 26, 2430–2440.
Järviluoma, A., Koopal, S., Rasanen, S., Makela, T. P., and Ojala, P. M. (2004). KSHV viral cyclin binds to p27KIP1 in primary effusion lymphomas. Blood 104, 3349–3354.
Järviluoma, A., and Ojala, P. M. (2006). Cell signaling pathways engaged by KSHV. Biochim. Biophys. Acta 1766, 140–158.
Jenner, R. G., and Boshoff, C. (2002). The molecular pathology of Kaposis sarcoma-associated herpesvirus. Biochim. Biophys. Acta 1602, 1–22.
Keller, S. A., Schattner, E. J., and Cesarman, E. (2000). Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood 96, 2537–2542.
Kwun, H. J., da Silva, S. R., Qin, H., Ferris, R. L., Tan, R., Chang, Y., and Moore, P. S. (2011). The central repeat domain 1 of Kaposi’s sarcoma-associated herpesvirus (KSHV) latency associated-nuclear antigen 1 (LANA1) prevents cis MHC class I peptide presentation. Virology 412, 357–365.
Kwun, H. J., da Silva, S. R., Shah, I. M., Blake, N., Moore, P. S., and Chang, Y. (2007). Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J. Virol. 81, 8225–8235.
Lan, K., Verma, S. C., Murakami, M., Bajaj, B., Kaul, R., and Robertson, E. S. (2007). Kaposi’s sarcoma herpesvirus-encoded latency-associated nuclear antigen stabilizes intracellular activated Notch by targeting the Sel10 protein. Proc. Natl. Acad. Sci. U.S.A. 104, 16287–16292.
Lee, H. R., Toth, Z., Shin, Y. C., Lee, J. S., Chang, H., Gu, W., Oh, T. K., Kim, M. H., and Jung, J. U. (2009). Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to deregulate the p53 tumor suppressor pathway. J. Virol. 83, 6739–6747.
Lei, X., Bai, Z., Ye, F., Xie, J., Kim, C. G., Huang, Y., and Gao, S. J. (2010). Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 12, 193–199.
Levitskaya, J., Coram, M., Levitsky, V., Imreh, S., Steigerwald-Mullen, P. M., Klein, G., Kurilla, M. G., and Masucci, M. G. (1995). Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 375, 685–688.
Li, Q., Means, R., Lang, S., and Jung, J. U. (2007). Downregulation of gamma interferon receptor 1 by Kaposi’s sarcoma-associated herpesvirus K3 and K5. J. Virol. 81, 2117–2127.
Liang, Y., and Ganem, D. (2003). Lytic but not latent infection by Kaposi’s sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 8490–8495.
Lipkowitz, S., and Weissman, A. M. (2011). RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat. Rev. Cancer 11, 629–643.
Liu, J., Martin, H., Shamay, M., Woodard, C., Tang, Q. Q., and Hayward, S. D. (2007). Kaposi’s sarcoma-associated herpesvirus LANA protein downregulates nuclear glycogen synthase kinase 3 activity and consequently blocks differentiation. J. Virol. 81, 4722–4731.
Liu, L., Eby, M. T., Rathore, N., Sinha, S. K., Kumar, A., and Chaudhary, P. M. (2002). The human herpesvirus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the ikappa B kinase complex. J. Biol. Chem. 277, 13745–13751.
Mann, D. J., Child, E. S., Swanton, C., Laman, H., and Jones, N. (1999). Modulation of p27 (Kip1) levels by the cyclin encoded by Kaposi’s sarcoma-associated herpesvirus. EMBO J. 18, 654–663.
Mansouri, M., Douglas, J., Rose, P. P., Gouveia, K., Thomas, G., Means, R. E., Moses, A. V., and Früh, K. (2006). Kaposi sarcoma herpesvirus K5 removes CD31/PECAM from endothelial cells. Blood 108, 1932–1940.
Mansouri, M., Viswanathan, K., Douglas, J. L., Hines, J., Gustin, J., Moses, A. V., and Früh, K. (2009). Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 83, 9672–9681.
Matsumoto, A., Onoyama, I., Sunabori, T., Kageyama, R., Okano, H., and Nakayama, K. I. (2011). Fbxw7-dependent degradation of Notch is required for control of “stemness” and neuronal-glial differentiation in neural stem cells. J. Biol. Chem. 286, 13754–13764.
Nakayama, K. I., and Nakayama, K. (2006). Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381.
Pardieu, C., Vigan, R., Wilson, S. J., Calvi, A., Zang, T., Bieniasz, P., Kellam, P., Towers, G. J., and Neil, S. J. (2010). The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PLoS Pathog. 6, e1000843. doi: 10.1371/journal.ppat.1000843
Radkov, S. A., Kellam, P., and Boshoff, C. (2000). The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene H-ras transforms primary rat cells. Nat. Med. 6, 1121–1127.
Rhodes, D. A., Boyle, L. H., Boname, J. M., Lehner, P. J., and Trowsdale, J. (2010). Ubiquitination of lysine-331 by Kaposi’s sarcoma-associated herpesvirus protein K5 targets HFE for lysosomal degradation. Proc. Natl. Acad. Sci. U.S.A. 107, 16240–16245.
Russo, J. J., Bohenzky, R. A., Chein, M. C., Chen, J., Yan, M., Maddalena, D., Parry, J. P., Peruzzi, D., Edelman, I. S., Chang, Y., and Moore, P. S. (1996). Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U.S.A. 93, 14862–14867.
Sanchez, D. J., Gumperz, J. E., and Ganem, D. (2005). Regulation of CD1d expression and function by a herpesvirus infection. J. Clin. Invest. 115, 1369–1378.
Shin, Y. C., Nakamura, H., Liang, X., Feng, P., Chang, H., Kowalik, T. F., and Jung, J. U. (2006). Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 80, 2257–2266.
Sulea, T., Lindner, H. A., and Menard, R. (2006). Structural aspects of recently discovered viral deubiquitinating activities. Biol. Chem. 387, 853–862.
Swanton, C., Mann, D. J., Fleckenstein, B., Neipel, F., Peters, G., and Jones, N. (1997). Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature 390, 184–187.
Thomas, M., Boname, J. M., Field, S., Nejentsev, S., Salio, M., Cerundolo, V., Wills, M., and Lehner, P. J. (2008). Down-regulation of NKG2D and NKp80 ligands by Kaposi’s sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 1656–1661.
Weissman, A. M., Shabek, N., and Ciechanover, A. (2011). The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell Biol. 12, 605–620.
Yada, M., Hatakeyama, S., Kamura, T., Nishiyama, M., Tsunematsu, R., Imaki, H., Ishida, N., Okumura, F., Nakayama, K., and Nakayama, K. I. (2004). Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23, 2116–2125.
Yang, Z., Yan, Z., and Wood, C. (2008). Kaposi’s sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 82, 3590–35603.
Yin, Y., Manoury, B., and Fahraeus, R. (2003). Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science 301, 1371–1374.
Yu, Y., and Hayward, G. S. (2010). The ubiquitin E3 ligase RAUL negatively regulates type-I interferon through ubiquitination of IRF7 and IRF3. Immunity 33, 863–877.
Yu, Y., Wang, S. E., and Hayward, G. S. (2005). The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22, 59–70.
Keywords: Kaposi’s sarcoma-associated herpesvirus, ubiquitin, proteasome, polyubiquitin, RING
Citation: Ashizawa A, Higashi C, Masuda K, Ohga R, Taira T and Fujimuro M (2012) The ubiquitin system and Kaposi’s sarcoma-associated herpesvirus. Front. Microbio. 3:66. doi: 10.3389/fmicb.2012.00066
Received: 15 November 2011; Accepted: 07 February 2012;
Published online: 23 February 2012.
Edited by:
Keiji Ueda, Osaka University Graduate School of Medicine, JapanReviewed by:
Keiji Ueda, Osaka University Graduate School of Medicine, JapanKohji Noguchi, Keio University, Japan
Satoshi Ishido, RIKEN Research Center for Allergy and Immunology, Japan
Copyright: © 2012 Ashizawa, Higashi, Masuda, Ohga, Taira and Fujimuro. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Masahiro Fujimuro, Department of Cell Biology, Kyoto Pharmaceutical University, Misasagi-Shichonocho 1, Yamashinaku, Kyoto-shi, Kyoto 607-8412, Japan. e-mail: fuji2@mb.kyoto-phu.ac.jp