 Fabian Horn1
Fabian Horn1 Thorsten Heinekamp2
Thorsten Heinekamp2 Olaf Kniemeyer2
Olaf Kniemeyer2 Johannes Pollmächer3
Johannes Pollmächer3 Vito Valiante2
Vito Valiante2 Axel A. Brakhage2,4*
Axel A. Brakhage2,4*- 1 Systems Biology/Bioinformatics, Leibniz Institute for Natural Product Research and Infection Biology – Hans Knöll Institute, Jena, Germany
- 2 Molecular and Applied Microbiology, Leibniz Institute for Natural Product Research and Infection Biology – Hans Knöll Institute, Jena, Germany
- 3 Applied Systems Biology, Leibniz Institute for Natural Product Research and Infection Biology – Hans Knöll Institute, Jena, Germany
- 4 Department of Microbiology and Molecular Biology, Institute of Microbiology, Friedrich Schiller University, Jena, Germany
Elucidation of pathogenicity mechanisms of the most important human-pathogenic fungi, Aspergillus fumigatus and Candida albicans, has gained great interest in the light of the steadily increasing number of cases of invasive fungal infections. A key feature of these infections is the interaction of the different fungal morphotypes with epithelial and immune effector cells in the human host. Because of the high level of complexity, it is necessary to describe and understand invasive fungal infection by taking a systems biological approach, i.e., by a comprehensive quantitative analysis of the non-linear and selective interactions of a large number of functionally diverse, and frequently multifunctional, sets of elements, e.g., genes, proteins, metabolites, which produce coherent and emergent behaviors in time and space. The recent advances in systems biology will now make it possible to uncover the structure and dynamics of molecular and cellular cause-effect relationships within these pathogenic interactions. We review current efforts to integrate omics and image-based data of host-pathogen interactions into network and spatio-temporal models. The modeling will help to elucidate pathogenicity mechanisms and to identify diagnostic biomarkers and potential drug targets for therapy and could thus pave the way for novel intervention strategies based on novel antifungal drugs and cell therapy.
1. Background
1.1. Human-Pathogenic Fungi
It is estimated that the total number of fungal species exceeds 1.5 million (Hawksworth, 2001). However, only a small minority of approximately 100 species of fungi are associated with human diseases. Nevertheless, infections caused by fungal pathogens lead to a wide range of diseases including allergies, superficial infections, and invasive mycoses. The outcome of an infection with a human-pathogenic fungus often depends on the immune status of the host organism. Patients suffering from a weakened immune system are at high risk of developing a serious fungal infection. Continuous progress in medicine, e.g., in chemotherapy and organ or bone marrow transplantation, has led to an increasing number of patients with impaired immune status. In recent decades, the frequency of invasive fungal infections has increased steadily, resulting in considerable morbidity and mortality. In the USA, the incidence of sepsis caused by fungi has increased by more than 200% since 1991, whereas cases of bacterial sepsis have only increased moderate (Martin et al., 2003). Invasive mycoses are characterized by a high mortality rate. The increasing number of fungal infections has significantly contributed to health-related costs (Pfaller and Diekema, 2007).
The yeast Candida albicans and the filamentous fungus Aspergillus fumigatus are by far the most important causes of life-threatening invasive mycoses. Apart from A. fumigatus, around 10% of the more than 200 species of the genus Aspergillus are regarded as human pathogens or as having other adverse effects, e.g., A. terreus, A. flavus, and A. niger (Brakhage, 2005). The prevalence of C. albicans in clinical Candida samples is 50–70%, followed by infections with Candida glabrata, which is found in 20–25% of clinical Candida samples. Other pathogenic Candida species include C. tropicalis, C. dubliniensis, C. krusei, and C. parapsilosis (overview in Pfaller and Diekema, 2007). Another important human-pathogenic fungus of clinical relevance is the fungus Cryptococcus neoformans. The most common fungal infection among AIDS patients, cryptococcal meningitis, is caused by this basidiomycete. Furthermore, other fungal species, such as Pneumocystis jiroveci, Zygomycetes, Fusarium species, and Scedosporium species, have emerged as causal agents of invasive mycoses (Pfaller and Diekema, 2007).
Despite the different pathogenesis of infections caused by C. albicans and A. fumigatus, there are several common traits, particularly when the host response is considered: (i) the pathogens must be able to overcome epithelial barriers, (ii) innate immunity represents the major defense system, (iii) pathogenic fungi possess physiological characteristics, virulence determinants, and capabilities for immune evasion that make them aggressive pathogens, and (iv) invasive candidiasis and invasive aspergillosis are mainly found in patients with a weakened immune system either due to reduced activity of immune effector cells or defects in epithelial barriers.
Consequently, the aims of research on human-pathogenic fungi are (i) to unravel the pathogenic determinants specific to each fungus, (ii) to investigate the distinct roles of epithelial barriers, the mechanisms of the innate immunity, and potential contributions of the adaptive immune system to the pathogenesis of fungal infections, and (iii) to elucidate the complex mechanisms of fungal infections and identify common principles of fungal pathogenesis.
1.2. Pathobiology of Aspergillus fumigatus: From Environmental Microorganism to Pathogen
Within the last two decades, the filamentous fungus A. fumigatus has become one of the most important fungal pathogens. Conidia of this saprophytic fungus can be found almost everywhere, from the winds of the Sahara to the snow of the Antarctic. The most severe disease caused by A. fumigatus is invasive aspergillosis (IA), which occurs almost exclusively in immunocompromised patients (Brakhage, 2005). There is currently a lack of reliable diagnostic tools and effective treatment options for this condition, resulting in a high mortality rate despite therapy. Remarkably, A. fumigatus causes 90% of all systemic Aspergillus infections. This indicates that A. fumigatus possesses certain virulence determinants that favor this species becoming an opportunistic human pathogen. Because of their ubiquitous presence in the air, each person inhales several hundred A. fumigatus conidia daily. In immunosuppressed patients, the lung is the primary site of infection. In immunocompetent individuals, mucociliary clearance and phagocytic cells normally prevent the disease (Brakhage et al., 2010). However, there is a correlation between the degree of immunosuppression and the risk of contracting IA. Consequently, important risk factors include neutropenia, T cell depletion, CD34-selected stem cell products, corticosteroid therapy, and cytomegalovirus infections (Marr et al., 2002).
Since 2005, considerable progress has been made in the analysis of A. fumigatus. The genome sequence of A. fumigatus is available, and the transformation efficiency of the fungus was drastically increased by generation of ku70 and ku80 mutants of A. fumigatus (Nierman et al., 2005; da Silva Ferreira et al., 2006; Krappmann et al., 2006) making the generation of mutants by targeted gene deletion much easier. As a result of this improvement, the number of deletion mutants has increased from a handful, in the year 2000, to more than 400, today.
However, only a few virulence determinants of A. fumigatus have been characterized to date. These determinants include the siderophore-mediated iron uptake system (Schrettl et al., 2004) or the pksP gene, which is involved in the biosynthesis of the gray-green spore pigment (Langfelder et al., 1998; Thywißen et al., 2011; Volling et al., 2011). How these virulence determinants influence the infection is currently under investigation. DHN melanin was shown to inhibit both apoptosis and the acidification of conidia-containing phagolysosomes of macrophages (Thywißen et al., 2011; Volling et al., 2011). Because virulence is a multifactorial process, it can safely be expected that many more virulence-associated traits will be discovered, e.g., A. fumigatus is able to grow under hypoxic conditions. This ability is essential for pathogenicity (Willger et al., 2008). A. fumigatus also possesses immune-evasion mechanisms which reduce recognition, both by immune effector cells and the complement system (Behnsen et al., 2008, 2010; Aimanianda et al., 2009).
Innate immunity is of great importance in defense against A. fumigatus. Alveolar macrophages are the major resident cells of the lung alveoli and they phagocytose conidia. However, conidia have the ability to interfere with functions of the macrophages such as the maturation of phagolysosomes (Jahn et al., 2002; Ibrahim-Granet et al., 2003; Thywißen et al., 2011). Unphagocytosed conidia and outgrowing hyphae are killed by neutrophilic granulocytes, whose activity is essential for preventing IA (Feldmesser, 2006).
Also, the complement system appears to contribute to the defense mechanism (Moalli et al., 2010). Human pattern recognition receptors sensing fungal cell wall components include TLRs (Toll-like receptors), Galactin 3, DC-Sign (C-type lectin receptors), dendritic cell-specific intracellular adhesion molecule 3 (ICAM-3)-grabbing non-integrin, Dectin-1, SCARF1, and CD36. In line with the importance of these receptors for fungal recognition, a growing number of defined single nucleotide polymorphisms in the respective genes that appear to determine host susceptibility to A. fumigatus were identified (overview in Romani, 2011).
Furthermore, neutrophils possess recently discovered extracellular killing mechanisms: they degranulate, release DNA, and form neutrophil extracellular traps (NETs) both in vitro and in vivo against A. fumigatus (Bruns et al., 2010; McCormick et al., 2010). However, whether NET formation is detrimental for A. fumigatus overall is currently under investigation, and it remains unclear how neutrophils ultimately kill A. fumigatus. Reactive oxygen intermediates (ROI) most probably do not play a role as primary killing agents, but are required as signaling molecules (Lessing et al., 2007). Incubation of dendritic cells (DCs) with A. fumigatus in vitro resulted in the release of chemokine CXCL8, which attracts neutrophils (Gafa et al., 2007). Secretion of additional factors increased surface expression of CD11b and CD18 on neutrophils. Dectin-1, which is an important receptor on macrophages and neutrophils, is also expressed on the surface of immature DCs and is involved in the induction of a proinflammatory cytokine response (Mezger et al., 2008). DCs thus play an important role in defense against A. fumigatus. The pathogen recognition receptor Dectin-1 acts upstream of the Syk tyrosine kinase in response to an infection with A. fumigatus. Signaling via the Syk tyrosine kinase was recently found to be essential for the activation of NLRP3 inflammasome, another component of the innate immune system (Saïd-Sadier et al., 2010). Despite these findings, we are still in the early stages of understanding their role in organizing the immune defense mechanism.
Epithelial and endothelial cells in the lung can internalize conidia. It cannot, therefore, be excluded that these cells form sites of persistence and foci of infection (Latgé, 1999). The role of T cells has not yet been clarified. It appears most likely that they initiate the adaptive immune responses to Aspergillus species and directly influence the outcome of an infection (Dagenais and Keller, 2009). Phagocytosis of conidia by DCs leads to a protective Th1 response, whereas hyphal phagocytosis results in non-favorable Th2 responses and the generation of IL-10-producing CD4 cells (Romani, 2011).
1.3. Pathobiology of Candida albicans: From Commensal to Pathogen
Candida albicans normally exists as harmless commensal yeast on mucosal surfaces of the majority of the human population. Only under certain circumstances (imbalance of the normal microbial flora, immunosuppression, damage of tissue barriers), can C. albicans cause superficial (oral thrush in 90% of all untreated HIV patients, vaginal thrush in 75% of all women once in their lifetime) or life-threatening systemic infections (nosocomial candidiasis, candidemia; reviewed in Pfaller and Diekema, 2007; Martin et al., 2011). C. albicans is currently identified as the fourth most common blood isolate in US hospitals, accounting for around 10% of hospital-acquired bloodstream infections (Wisplinghoff et al., 2004). Cases of sepsis caused by this fungus lead to mortality rates of about 40% and are thus higher than observed for any bacterial sepsis (Gudlaugsson et al., 2003; Wisplinghoff et al., 2004; Picazo et al., 2008). Although C. albicans is an opportunistic pathogen, only a minority of cases (20%) of disseminated candidiasis occurs in patients with severe immunosuppression such as individuals with neutropenia, corticosteroid therapy, or HIV infection. Patients with severe illnesses who have prolonged periods of hospitalizations, a central venous catheter, gastrointestinal/cardiac surgery, or burns are at especially high risk of developing an invasive Candida infection (reviewed in Perlroth et al., 2007).
Almost all C. albicans infections are endogenous infections, caused by commensal strains of patients’ own microflora. Despite numerous studies, it remains unclear how the transition from a harmless commensal to an aggressive pathogen is triggered. It seems certain that it is not only modifications of the microbial flora and host factors, but also specific attributes of the fungus that play an important role in this transition.
The ability to switch from yeast to pseudohyphal or hyphal growth is an important virulence trait for C. albicans. Several environmental conditions such as temperatures above 37°C, pH values of 7.0 or higher, high exogenous CO2 concentrations of more than 5%, or the presence of serum (Liu, 2002) trigger this transition. Numerous genes are involved in the regulation of the morphological switch, but molecular details are still poorly understood. The transcription factor Efg1p plays a central role in the control of morphogenesis (Stoldt et al., 1997; Doedt et al., 2004) and the loss of the EFG1 gene led to mutants which were locked in the yeast form and showed reduced virulence in a murine model of candidiasis (Lo et al., 1997; Stoldt et al., 1997). In the regulation of the Efg1p pathway, cAMP-mediated signal transduction plays an essential role and mutants lacking the adenylyl cyclase Cdc35p do not form hyphae. As well as Efg1p, a large number of other transcription factors have also been reported to inhibit or trigger the yeast to hyphal transition, e.g., Efh1p, Mcm1p, Cph2p, and Tec1p (reviewed in Whiteway and Bachewich, 2007).
Besides the morphological plasticity, the ability of C. albicans to adhere to host cells and tissue and form biofilms is another important virulence factor. Amongst others, the ALS (agglutinin-like sequence) proteins are a well-studied group of proteins that form a family of peptide-binding proteins and which mediate adhesion (Salgado et al., 2011). They bind to extracellular matrix proteins, such as collagen, fibronectin and laminin (Als1p, Als3p, Als5p, Als6p, Als9p), endothelial and epithelial cells (Als1p, Als3p, Als5p), and also mediate cell-to-cell aggregation (Als5p) and iron acquisition (Als3p; Filler, 2006; Almeida et al., 2009). Hwp1p is another important adhesin, expressed only on hyphae, which binds tightly to oral epithelial cells and that is involved in biofilm formation (Nobile et al., 2006).
Secreted enzymes with proteolytic or lipolytic activity represent another group of proteins which contributes significantly to C. albicans’ pathogenicity. A large proportion of the proteolytic activity is attributed to a multigene family of secreted aspartic proteinases (SAPs). Ten different SAPs have been described in C. albicans, eight of which are secreted extracellularly and two of which are anchored to the membrane via GPI linkage. Their contribution to the pathogenesis of C. albicans infections has been extensively investigated. SAP1–SAP3 genes were considered to play a role in localized C. albicans infections and complement evasion, whereas SAP4–SAP6 were postulated to play an important role in the pathogenesis of invasive candidiasis (Schaller et al., 2005; Gropp et al., 2009). However, in a recent study by Correia et al. (2010) the importance of SAP1 to SAP6 for virulence was reassessed in a murine model of candidemia. In contrast to previous findings, SAP1 to SAP6 were found to play no significant role in disseminated C. albicans infections. In addition to the virulence determinants described above, physiological fitness, in other words, high stress tolerance and metabolic flexibility, is another important factor that contributes to the pathogenicity of C. albicans (Brown et al., 2007; Fleck et al., 2011).
The fact that C. albicans is a diploid fungus and was long thought to be an obligate asexual organism (Alby et al., 2009), has long hindered the production of genetically defined mutants. In contrast to the well-studied yeast Saccharomyces cerevisiae, C. albicans does not have any natural DNA plasmids that could be used for transformation. In addition to this, C. albicans shows a non-standard codon usage and translates the CUG codon as serine instead of leucine (Lloyd and Sharp, 1992). Only after establishing protocols for targeted gene disruptions of both alleles, creating conditional null mutants based on tetracycline-regulatable systems, sequencing of the entire genome, establishment of a genome database, production of genome-wide microarrays, and production of reporter strains and other molecular tools as well as infection models, has C. albicans reached the status of a model organism for yeast infections (Theiss et al., 2002; Fradin et al., 2003; Jones et al., 2004; Braun et al., 2005; Samaranayake and Hanes, 2011; Szabo and MacCallum, 2011).
Due to this technical progress, interactions of C. albicans with host cells and the immune system have been the focus of many studies in recent years. Fungal recognition is the first step in the antifungal immune response and is mediated by pattern recognition receptors (PRR). The mannan cell wall component is recognized by the mannose receptor, the C-type lectin-like receptor Dectin-2, and the TLR4. Furthermore, TLR2 triggers an immune response by binding to phospholipomannans, as the Dectin-1 receptor does by binding to β-glucan (Netea et al., 2008). TLR2 and Dectin-1 regulate also the gene transcription of proinflammatory cytokines such as the pro-IL-1β. This interleukin is further processed into its active mature form via the NLRP3 inflammasome, a multiprotein complex. It has recently been shown that it is crucial for antifungal host defense (Gross et al., 2009; Hise et al., 2009). After recognition, phagocytes, like macrophages or neutrophilic granulocytes, kill C. albicans cells by phagocytosis or secretion of antimicrobials. One mechanism discovered recently is the formation of neutrophil extracellular traps as mentioned above in the section on A. fumigatus (Urban et al., 2006). To link these interactions to the highly complex setting of clinical infections, complex in vitro, ex vivo, and in vivo infection models have been established and genome-wide transcriptional profiles, including direct C. albicans transcriptomes from patient samples, have been produced (Wilson et al., 2009; Cairns et al., 2010).
1.4. Systems Biology of Infection
Due to their high complexity, it is conceivable that invasive infections caused by human-pathogenic fungi can be described and understood in a comprehensive manner by taking a systems biological approach. There are two complementary strategies in systems biology: (i) Starting from smaller, even minimal models capturing the essential and abstract interactions in the system under study and (ii) using experimental measurements such as large omics datasets in combination with large-scale models. Hybrid approaches, which integrate these top-down and bottom-up perspectives, contribute to our understanding of the multiple interdependencies of different hierarchical levels in biological systems (Forst, 2006). The global dynamics of a system can only be understood and quantified if the functionality of modular subsystems is elucidated, while considering the most important interactions on different levels in the biological system. With the aim of revealing molecular and cellular cause-effect relationships within the host-pathogen interaction in a non-ambiguous and efficient way, the setup of experiments and the design of experimental series can be optimized on the basis of established mathematical and computational models. Data exchange proceeds in an iterative cycle between model and experiment, with a constant refinement and validation of the models and the model-based planning of experiments (Ideker et al., 2001).
Current experimental and modeling techniques focus on specific perspectives at different scales. At present, experimental data from high-throughput experiments are increasingly and routinely used as the basis for mathematical modeling. This wealth of information has become available recently. Nevertheless, the history of the mathematical modeling of infectious diseases can be traced back to the eighteenth century, when today’s basic concepts of evolution, genetics, and molecular biology were still unknown. Back in 1760, Daniel Bernoulli predicted the life expectancy of a population which has been immunized with cowpox (Bernoulli, 1760). In the early twentieth century, mathematical models were developed that mainly focused on the spread of diseases such as measles and malaria (Ross, 1911; Bailey, 1975). Most models would be related to today’s research field of population biology, meaning that they dealt with fluctuations in population size under different modes of disease transmission. Since the second half of the twentieth century, these models have become more sophisticated. Amongst other concepts, they incorporated new aspects (e.g., population variables, May and Anderson, 1979; transmission rates, Real and Biek, 2007; pathogen life cycles, and host specificities, Woolhouse et al., 2001; Barrett et al., 2008) and extended the model to allow multi-level modeling (Roux and Aiello, 2005) or concepts of evolution (reviewed in Tong and Ng, 2011).
Naturally, each infection process is unique. Nevertheless, the modeling makes it possible to reveal fundamental similarities and differences in the underlying processes. The influence of single model parameters and their interdependency can thus be deduced. These parameters are also assessed if they serve as effective control options for the implementation of governmental public health risk management programs (Tong and Ng, 2011).
With the progress in molecular biology, infection biology, and biotechnology, it is now possible to study species-specific host-pathogen interactions at the molecular level in order to search directly for biomarkers with diagnostic potential and drug targets for novel therapeutic treatment strategies. The focus of research has diversified, resulting in specialized databases and research groups. Only early steps toward the computational systems biology of A. fumigatus and C. albicans have been made, including genome-scale data mining and mathematical modeling of infection processes by these fungi (reviewed for human-pathogenic fungi in Albrecht et al., 2008, 2011 and Rizzetto and Cavalieri, 2011).
2. Data Basis and Data Analysis
The aim of understanding the complexity of host-pathogen interactions can be achieved by exploiting the increasing amount of experimental data, including high-throughput data and information available from public repositories as well as from biomolecular databases (see Table 1). Typically, experimental series comprise knock-down experiments along with global and specific screening using knockout mutants of pathogenic fungi. Procedures can be designed to analyze the complex structured data obtained from these experimental series. Standardized pre-processing of the raw data and integrative analyses allow the identification of key regulators involved in pathogenicity. The management and integrative analysis of experimental data is challenging and constitutes a research field itself within bioinformatics (Albrecht et al., 2008). It was presented exemplary for genome, transcriptome, and proteome analysis of the heat shock response of A. fumigatus (Albrecht et al., 2010). Each technique applied generates different types of data, which have been acquired at the various levels of information. For each level, all of the available data types and their accompanying computational methods are usually referred to as “omics.” New technologies, such as high-throughput sequencing, impose new challenges on efficient data storage, data retrieval, and statistical analysis. In order to handle the tremendous amount of data required for systems biology, the heterologous data has to be linked between existing databases. Automatic computer access has to be provided, and different user perspectives have to be taken into account. The standardization of biological terms (GO, FunCat, SBO, Ruepp et al., 2004; Arnaud et al., 2009; Courtot et al., 2011), data formats (SBML, Hucka et al., 2003), experimental metadata (MIBBI, Taylor et al., 2008), and operating procedures (Taverna, Hull et al., 2006) contribute to this objective.

Table 1. Bioinformatic resources of special interest for fungal systems biology.
Despite the wealth of information gained from analyzing high-throughput data, research into biological systems is mostly focused on a single data level. To overcome these limitations, data warehousing approaches (Omnifung, IntegromeDB, Albrecht et al., 2007; Kozhenkov et al., 2011) aim to integrate the different data layers. At the same time, most integration tools offer different visualization techniques as an additional key method for querying and understanding large datasets (Köhler et al., 2006; Smoot et al., 2011). Even though efforts have been made in this respect, an integrated view of different omics data levels is still a major challenge. Regulatory processes, different time scales, and non-linear processes in the biological system as well as technical limitations of omics technologies hamper the detection of causality or even correlation between different data layers (Albrecht et al., 2011).
2.1. “Omics” Based Data
2.1.1. Genome
The importance of fungal-derived infections is reflected impressively by the number of fungal genomes that have been sequenced in recent years. The genome of the diploid C. albicans was published in 2004 (Jones et al., 2004; Braun et al., 2005), followed by the C. neoformans genome (Loftus et al., 2005) and by the A. fumigatus genome (Nierman et al., 2005). Since then, other Candida and Aspergilli genomes have been sequenced (Arnaud et al., 2007; Fedorova et al., 2008). Also, the first genomes of dermatophytes have recently been published (Burmester et al., 2011). Table 2 lists all human-pathogenic fungi for which the full genome sequence is available. This list does not include genome projects currently in progress such as the sequencing of several C. albicans strains to evaluate Candida genome plasticity. New sequencing technologies now allow even greater numbers of genomes and transcriptomes to be deciphered, which will contribute to a better understanding of fungal pathogenesis. This information has been collected and released in suitable and easy to use web tools (see Table 1). For Candida sp., the main websites used are the CandidaDB (d’Enfert et al., 2005) and the Candida Genome Database (CGD; Arnaud et al., 2007). For A. fumigatus, the community normally refers to the Central Aspergillus Data REpositery (CADRE, Gilsenan et al., 2012) and the new Aspergillus Genome Database (AspGD, Arnaud et al., 2012).
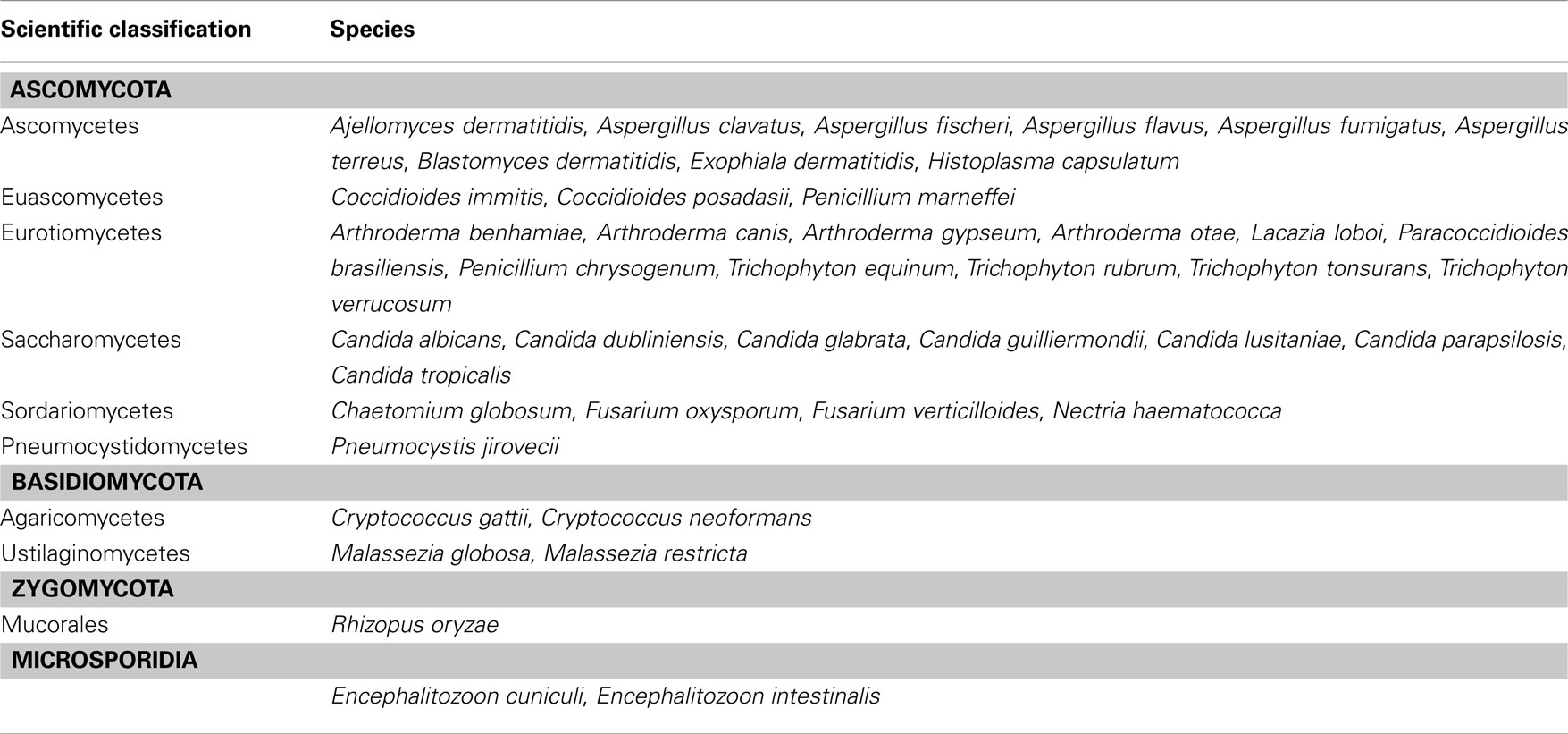
Table 2. List of human-pathogenic fungi of which the genomes have been sequenced.
The information available in the different genome databases allowed us to apply studies of comparative genomics to human-pathogenic fungi focusing on evolutionary aspects of virulence genes. Studies of different Candida sp. highlighted that cell wall-associated genes, important for host recognition and virulence of Candida sp., have been subject to gene duplications (Butler et al., 2009). Furthermore, the high number of lipases or GPI anchored proteins (normally clustered) is important for the virulence of C. albicans and C. glabrata (van het Hoog et al., 2007; Dujon, 2010). This phenomenon is known as gene family expansion and results in an increase of enzymatic power during infection processes or, where possible, easier rearrangement of the genome. In both cases, there is an increase in the competitiveness of the pathogens during infection processes (Moran et al., 2011).
Many clustered genes present in Aspergillus sp. encode proteins for secondary metabolite production, such as mycotoxins and antibiotics, and do not normally represent repetition of genes with similar enzymatic activity. A higher degree of genome rearrangement was observed in telomeric regions, where many of these clusters are located. The origins of these gene clusters have always been associated with the possibility of vertical gene transfer from other microbes. However, extensive comparative studies on Aspergilli genomes suggest that the presence of paralogs in the different species could be ascribed to gene duplication within the genus and subsequent translocation to telomere-proximal locations (Fedorova et al., 2008).
In addition to the importance for taxonomic studies, comparative genomics also highlighted peculiarities and similarities between different fungal species. One example was given by comparing signaling pathways in different pathogenic fungi. The mitogen activated protein kinase (MAPK) signaling pathways and the calcineurin pathway have been extensively studied in pathogenic fungi because of their involvement in pathogenesis. Global sequence similarity analysis indicated that, on the one hand, core structures involved in signaling are highly conserved while, on the other hand, upstream (e.g., receptors) and downstream factors (e.g., transcription factors) are far more species-specific (Rispail et al., 2009). Knowledge obtained in these studies can be used to identify suitable intraspecific or interspecific targets for therapeutic intervention.
2.1.2. Transcriptome
A genome, apart from its arrangement and complexity, can always be regarded as a static feature. In contrast to this, transcriptome analysis provides information about the dynamics of a genome’s expression. Due to the availability of fungal genome data, it is now possible to design microarrays for genome-wide expression analysis.
For more than 10 years, scientists have had access to various C. albicans transcriptome studies. Many experiments have focused on studying gene expression in response to antifungal agents (e.g., azole derivatives, amphotericin B, echinocandins; Backer et al., 2001; Barker et al., 2004; Liu et al., 2005) but also at different stages of development and during biofilm formation (Doedt et al., 2004; Murillo et al., 2005). For C. albicans, already in 2005 a comparative gene expression analysis was published (Ihmels et al., 2005).
Aspergillus fumigatus’ transcriptome history is comparatively recent. The first entire global transcriptome analysis was published together with the release of the first genome sequence (Nierman et al., 2005). After that, scientists had access to various transcriptome studies of developmental stages, during iron starvation, of biofilm formation, and response to antifungals (e.g., da Silva Ferreira et al., 2006; Schrettl et al., 2008; Bruns et al., 2010; Cagas et al., 2011; Jain et al., 2011). Fewer transcriptome studies have been carried out for A. fumigatus than for C. albicans. Furthermore, the comparability of transcriptome data is low. One reason for this is the development of different microarray platforms that made it harder to compare different gene expression data. In addition to this, the experimental design of transcriptome studies has been quite heterogenous with respect to media, strains, and general growth conditions. At present, we can only reliably compare different transcriptome data on the basis of genes that have high fold-changes in expression levels.
The main challenge in infection biology is to understand gene responses during infection. Such studies have been performed in several ways (co-culture of fungi and immune cells or direct tissue infection). The main problem remains the enrichment of the RNA from the pathogen prior to the hybridization step in order to avoid a decrease in data quality caused by cross-hybridization. This step is normally performed by separation of the different RNA species that can eventually be amplified to increase the nucleic acid quantity (Nygaard and Hovig, 2006). Previous studies identified genes involved in nutrient acquisition, oxidative stress response, and metal homeostasis, which are differentially regulated in C. albicans when co-cultured with macrophages and neutrophils (Lorenz and Fink, 2001; Lorenz et al., 2004; Wilson et al., 2009). Similar results have also been found in A. fumigatus when co-cultured with neutrophils and dendritic cells (Lessing et al., 2007; Sugui et al., 2008; Morton et al., 2011). A recent review described a comparison of transcriptome data obtained from pathogenic fungi during organ or tissue infections (Cairns et al., 2010). Many genes involved in primary metabolism appeared to be differentially expressed during infection in both human and plant pathogens. This data suggested that physiological reprogramming during infection remains relatively well conserved among various pathogens. On the other hand, these types of studies highlighted the limitations of hybridization-based techniques such as microarrays.
Recently, new techniques based on deep RNA sequencing have been introduced, i.e., the RNA-seq technique to analyze transcriptomes (Wang et al., 2009). Recent work on C. albicans and A. fumigatus indicated that with this technology it is possible to identify misannotated genes and differences in the level of low expressed genes which, for example, is relevant for many secondary metabolite gene clusters (Bruno et al., 2010; Gibbons et al., 2012). Furthermore, in infection biology it is important to study the gene expression profiles during infection, not only from the pathogen side, but also from the host side. To date, microarray analysis was limited in this respect, because separation of organism-specific RNA prior the hybridization is hard to achieve. Theoretically, RNA-seq analysis is capable of handling this problem. Technically, RNA from different species can be pooled, and then the data obtained can be separated during the analysis by aligning raw sequence data to different genomes. This approach seems possible because the sequences not matching a genome are normally discarded. The RNA-seq technique could potentially give us a way of monitoring gene expression profiles from the pathogen and the host simultaneously.
2.1.3. Proteome
To gain global insights into the biology of fungal pathogens and their interactions, genome-wide studies should focus not only on the transcript level, but also on the protein level. Proteins are the molecules that are catalytically active, build up the cellular structure, and mediate signal transduction and gene regulation. The release of the genome sequences of C. albicans and A. fumigatus paved the way for studies on the fungal proteome, i.e., the entire set of proteins which is synthesized and modified at a given time under defined conditions. Two dimensional-gel electrophoresis, a method invented in the mid-1970s, was the first technique used to study the presence of proteins on a global scale (O’Farrell, 1975; Klose and Kobalz, 1995). Meanwhile, mass spectrometry (MS)-based methods have become more and more popular. Here, the separation of tryptically digested peptides by liquid chromatography is coupled to mass spectrometry (reviewed in Aebersold and Mann, 2003). Despite the significant technical progress made in recent years in the field of MS-based proteomics, no technique currently available allows us to entirely profile the highly dynamic range and complexity of the protein set of a eukaryotic organism. However, a lot of knowledge about the proteome of C. albicans and A. fumigatus has already been obtained, which is summarized in several recent reviews (Rupp, 2004; Thomas et al., 2006; Kim et al., 2008; Kniemeyer and Brakhage, 2008; Andersen and Nielsen, 2009; Kniemeyer, 2011; Kniemeyer et al., 2011). Here, a brief overview will be given and current trends will be highlighted.
For C. albicans, the first studies on the extent of changes in cytoplasmic proteins during yeast-mycelial transition were conducted in the early 1980s, 25 years before the start of the post-genomic era in the field of fungal pathogenicity (Manning and Mitchell, 1980). More 2D-PAGE studies followed (Niimi et al., 1996), and differences in protein expression between the two morphotypes were also analyzed using MS-based techniques (Melanson et al., 2006). Several proteins that also play a role in the virulence of C. albicans were found to have increased expression levels in hyphal cells (reviewed in Kniemeyer and Brakhage, 2008). Recently, a study by Monteoliva et al. (2011) indicated that the primary metabolism undergoes a reorganization during morphotype-switching. Also, the composition of the C. albicans cell surface proteome undergoes changes during the yeast-mycelial transition. Several extraction techniques have been established in recent years to study this phenomenon (Pitarch et al., 2002; de Groot et al., 2004; Castillo et al., 2008; Hernáez et al., 2010). Heilmann et al. (2011) gave a first quantitative proteomic snapshot of the changes occurring in the cell wall proteome of C. albicans during the transition from yeast to hyphal cells. Several proteins were identified as indicators of hyphal growth, including the adhesin Als3p.
Due to the fact that C. albicans faces a multitude of diverse stresses, e.g., oxidative stress, higher temperatures, hypoxia, and low pH during infection, the proteomic response to these and other adverse conditions have been investigated by many groups. Kusch et al. (2007) and Yin et al. (2009) showed that the levels of many proteins with antioxidative functions were significantly increased during the oxidative stress response. Sosinska et al. (2008) and Sosinska et al. (2011) investigated the variability of the cell wall proteome during iron depletion, hypoxia, and at different pH values (pH 4 and 7).
Proteomics is also a suitable approach for gaining a deeper insight into the response of C. albicans toward antifungal compounds. Bruneau et al. (2003) characterized the proteome changes in C. albicans induced by triazoles (fluconazole and itraconazole) and an echinocandin-like lipopeptide (mulundocandin). The different modes of action of triazoles and echinocandins, two different classes of antifungal agents, were also reflected at the protein level. Similar results were obtained by Hoehamer et al. (2010), who additionally included the polyene amphotericin B in their study. Results of a recent study using a liquid chromatography–mass spectrometry (LC-MS) based approach suggested a cell wall destabilizing effect of the triazole fluconazole (Sorgo et al., 2011). Other studies addressed the mechanism of drug resistance by comparing the proteome of a drug-resistant mutant strain with a drug-susceptible wild-type strain (Hooshdaran et al., 2004; Yan et al., 2007).
A lot of progress has also been made in the field of A. fumigatus proteomics. In contrast to C. albicans, this pathogenic mold shows not only a mycelial growth form, but also produces spores for dispersal. Proteome maps of both morphotypes and the composition of the secretome have been established (Vödisch et al., 2009; Teutschbein et al., 2010; Cagas et al., 2011; Wartenberg et al., 2011). Also, the stress response to oxidative and heat stress was characterized (Lessing et al., 2007; Albrecht et al., 2010). Chaperones and antioxidative enzymes were produced under both conditions. Additionally, the results showed that the thioredoxin system seems to play an important role in maintaining the cellular redox balance for A. fumigatus. Recent findings revealed that A. fumigatus is exposed to oxygen-depleted microenvironments during infection (Grahl et al., 2011). The response to hypoxia was also studied at the protein level using an oxygen-controlled chemostat (Vödisch et al., 2011). Under hypoxic conditions, A. fumigatus cells developed a higher respiratory capacity, induced the synthesis of enzymes of the nitrosative stress response, and activated a secondary metabolite gene cluster (pseurotin A). Also, other growth conditions triggered the production of secondary metabolites with biological activity. When A. fumigatus attaches to surfaces, it can grow into a biofilm-like structure including the formation of an extracellular matrix. Under these conditions, higher levels of the immunosuppressive secondary metabolite gliotoxin were produced (Bruns et al., 2010).
Several groups profiled the A. fumigatus proteome in response to the antifungal compounds caspofungin (Cagas et al., 2011) and amphotericin B (Gautam et al., 2008). Amphotericin B influenced various metabolic processes including the ergosterol pathway, whereas caspofungin induced a strong increase in levels of ribosomal proteins.
The study of the interplay between fungal pathogens and human cells at the level of the proteome remains a challenging task due to the complexity and the limited number of methods available for the separation of fungal cells from the human effector cells. Several studies characterized the proteome of murine macrophages, which had been exposed to living or heat-inactivated C. albicans yeast cells (Shin et al., 2005; Martínez-Solano et al., 2006, 2009). To complement these studies, proteomic changes of C. albicans yeast cells due to macrophage confrontation were investigated and revealed an increase of the level of chaperones and other stress-related proteins. No proteomic data is available on the interaction with other cells of the immune system or on the interaction of A. fumigatus with cells of the immune system. Nevertheless, proteomic technologies, such as targeted MS methods with high-performance instruments, have the potential to determine the mode of interaction between pathogenic fungi and their host. Targeted proteomic approaches are based on the selection of specific peptides of a protein for mass analysis. This process is termed multiple reaction monitoring (MRM). It allows the identification of very low-abundance proteins (Domon and Aebersold, 2010). However, this technique is still limited to the analysis of several hundred proteins in a single LC-MS/MS run, but technical progress in this field can be expected and may help to make a leap forward to the systematic investigation of host-pathogen interplay.
2.2. Image-Based Data
Investigation and elucidation of the pathobiology of fungal infections strictly requires analysis of the interaction of the pathogen with the host immune effector cells (Brakhage et al., 2010). Image analysis is an indispensable tool for doing this. In combination with advances in computer performance and computing resources, the use of several imaging technologies led to the generation of large amounts of data (Behnsen et al., 2007; Hickey and Read, 2009; Brock, 2012) awaiting integration via a systems biological approach for analysis and interpretation. In general, image data can be obtained by different experimental approaches, e.g., microscopy, positron emission tomography (PET)/computer tomography (CT), or bioluminescence imaging. Due to the fact that the latter two techniques for monitoring fungal infections in living hosts are still in the early stages of development (Avet et al., 2009; Ibrahim-Granet et al., 2010), we focus here on recent studies using microscopy-generated image data. A good example of such data is represented by monitoring the interaction of labeled fungal cells, i.e., conidia, germlings, or hyphae, with immune effector cells using microscopy, especially fluorescence microscopy or confocal laser scanning microscopy (CLSM). Interaction of A. fumigatus and phagocytes based on fluorescence microscopy and manual image analysis was described, for example, by Ibrahim-Granet et al. (2003) and Jahn et al. (2002). In theses studies, interaction of A. fumigatus with phagocytes was monitored in detail, which revealed that A. fumigatus is able to inhibit acidification of phagolysosomes. More recently, Thywißen et al. (2011) were able to assign this ability to the presence of an active PksP, the polyketide synthase involved in dihydroxynaphthalene melanin biosynthesis. All of these studies were based on differentially labeled conidia, germlings, phagocytes, and their structures and compartments. It is even possible to monitor different pH values within distinct compartments of the phagocytes, allowing us to monitor phagocytosis rates in general and the fate of conidia after confrontation with immune cells in detail (Thywißen et al., 2011). In the future, a large number of mutants can be screened using this assay to identify further pathogen-derived components interfering with phagocytosis. Although phagocytosis of A. fumigatus by different phagocytes from both human and mouse has been analyzed in detail in several studies (Jahn et al., 2002; Ibrahim-Granet et al., 2003), only a limited amount of data is available with regard to direct observation of the phagocytosis process itself by live cell imaging. This applies to live cell imaging of phagocytosis of Candida species. Cell motility, however, is an essential requirement for the function of phagocytes. A detailed spatio-temporal analysis of the dynamics of the interaction of phagocytes with A. fumigatus and C. albicans using time-lapse microscopy and single-cell tracking was performed by Behnsen et al. (2007). In this study, the natural environments of different phagocytes were simulated by 2D liquid cultures and by generation of a 3D collagen environment. Live imaging showed that the interaction of phagocytes with A. fumigatus conidia or C. albicans cells in both 2D and 3D environments is a highly dynamic process that includes touching, dragging, and phagocytosis of fungal structures. Interestingly, the different immune cells, i.e., neutrophils, macrophages, and dendritic cells, exhibited different behavior with regard to the dependence on environmental dimensionality and as well as to the processing of A. fumigatus and C. albicans. Whereas neutrophils and alveolar macrophages efficiently phagocytosed or dragged A. fumigatus conidia in a 2D environment, their function was severely impaired in a 3D matrix. The opposite was found for processing of C. albicans cells. Phagocytosis was reduced in 2D environments, while in 3D environments most neutrophils internalized multiple yeast cells. These differences were also found in competitive assays, when both C. albicans and A. fumigatus were confronted with immune cells in the respective environment. Despite frequent touching of the other pathogen, neutrophils primarily incorporated A. fumigatus conidia in 2D and C. albicans yeast cells in a 3D environment. It is therefore conceivable that the activity and efficacy of the different phagocytes is best in the environment where a pathogen is naturally encountered.
Analysis of image data with regard to host-pathogen interaction was performed almost exclusively manually, with all of the inherent drawbacks and disadvantages. Manual data analysis is very time-consuming, error-prone, and last but not least, dependent on subjective criteria of the person performing the analysis. A first approach to automatize image analysis was performed by Mech et al. (2011). As a proof of principle, the interaction of A. fumigatus conidia with macrophages was monitored (see Figure 1). Data was collected by CLSM using cells labeled with different fluorescent dyes. The ruleset developed for processing microscopic raw data allows fully automated and context-based analysis of image data. By applying this method, discrimination between different cell types, i.e., phagocytes and conidia, was facilitated. Furthermore, cell counting based on discrimination between phagocytosed, adherent and non-adherent exterior conidia was performed. This is of particular importance since the different steps in conidia-macrophage interaction, i.e., recognition, adherence, ingestion, and intracellular processing of inhaled spores, define important pathogenesis-related processes. A prerequisite for automated image analysis, e.g., to determine phagocytosis rates, is the digitization of images. However, due to the fact that phagocytosis of A. fumigatus conidia by macrophages is a complex process, current image analysis tools that require precisely defined and homogeneous objects (overview in Shamir et al., 2010; Sysko and Davis, 2010) cannot be applied here. During co-incubation, conidia and macrophages tend to attach and form clusters. In addition to this, the relevant structures vary in their intensities as a result of different labeling efficacies and, last but not least, the background differs considerably. These parameters were taken into consideration by the ruleset developed by Mech et al. (2011) and thereby allowed fully automated and context-based analysis of spatially resolved biological data, i.e., phagocytosis rates of A. fumigatus conidia. As this ruleset can be easily transferred to high-throughput microscopy measurements of other pathogens, e.g., Candida sp. or Cryptococcus neoformans, it will contribute to the further elucidation of host-pathogen interactions.
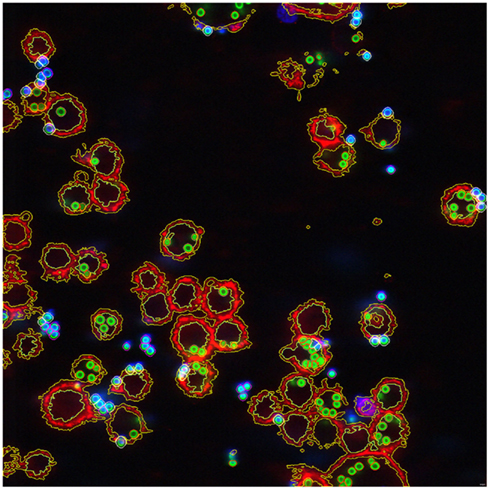
Figure 1. Image of the phagocytosis assay showing all conidia and macrophages after segmentation and classification. Exterior non-adherent conidia outlined in magenta, adherent conidia outlined in white, interior conidia outlined in orange, and macrophages outlined in yellow. ( Taken from Mech et al., 2011.)
Taken from Mech et al., 2011.)
3. Modeling
The identification of pathogenic traits will increasingly be supported by mathematical and computational modeling and by the integration of experimental data and prior knowledge into complex models that describe the underlying virulence mechanisms. The generation of models and their subsequent application aim to support the optimal and standardized design of experiments and to generate and validate hypotheses. This allows that new knowledge is gained and predictions for novel strategies for diagnostics (biomarker design) and therapy (drug discovery, drug administration, therapy decision support) are made.
Systems biology of fungal infections describes and analyses various aspects of the confrontation of the host and its pathogen under defined conditions. The interactions and co-evolution of host and pathogen can be described with the help of evolutionary models (May and Anderson, 1979). Currently, molecular modeling of the host-pathogen interaction generally takes a reductionistic approach.
In general, models either describe a biological perspective on a single scale, or they span several different orders of magnitude (see Figure 2). Molecular mechanisms of host-pathogen interaction were identified with the help of statistical and integrated analysis of experimental data (see section 2). Single interactions represent only a reduced level of complexity, and a combination of several different mechanisms is capable of reflecting the global behavior at a cellular level. Such multi-systems interactions will become more complex when several species are considered. More recent approaches incorporate different biological levels to get an interrelated view of fungal infections. These multi-scale approaches need to integrate data from experiments on two or more scales (Walker and Southgate, 2009). The greatest challenge arises from the fact that an effective computational framework has to deal with the complexity of different length and time scales spanning several orders of magnitude. This leads to quantifiable differences of crucial constituents (e.g., one cell and its sizeable number of molecules) and to newly considered events (e.g., movement of the cell and the corresponding events at the molecular scale).
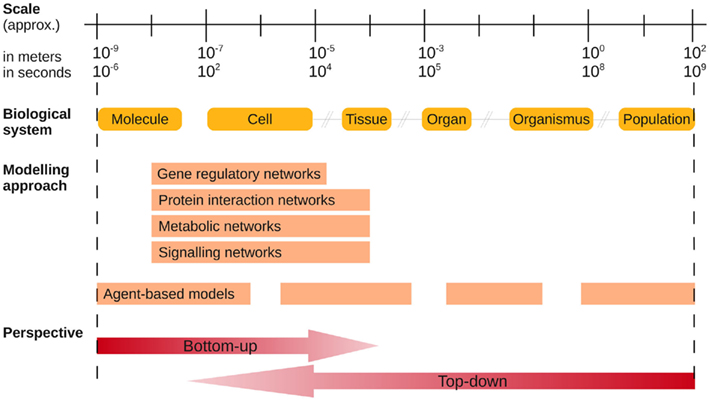
Figure 2. Schematic diagram of different biological and modeling levels of systems biology of infectious diseases. Biological systems span several orders of magnitude. Mathematical methods, which are presented in this paper, focus on different biological levels of a fungal or similar infection. Modeling approaches can be applied to host and pathogen systems and their interplay. The flexibility and adaptability of agent-based modeling allows analysis at multiple levels without any restrictions with respect to the biological system. In order to generate new hypotheses, the advantages of bottom-up and top-down models are usually incorporated into the analysis. (Figure adapted from Forst, 2006.)
To date, only a few models of fungal infections of humans have been studied, which is mainly due to the complex modeling challenges and the previous lack of measurements of model parameters. In perspective, the host-pathogen interaction should be described by a combination of spatio-temporal models with interacting molecular network models.
3.1. Network Modeling
Nodes in networks stand for interacting molecular entities (e.g., genes, proteins, metabolites) whose concentration or activity can be quantified by discrete or continuous variables. Edges, which stand for the relationships between the nodes, can be modeled in different ways, e.g., by directed or undirected edges and labeled by linear or non-linear functionality (Hecker et al., 2009). The cellular behavior of a system is usually represented by gene-regulatory networks, signaling networks, protein-protein interaction networks (PPI), and metabolic networks (see Figure 3). In addition to this, a confrontation between pathogen and host can be viewed as two interacting molecular networks, for example one within the host epithelial or immune cells and the other in the colonizing, persisting, or invading pathogens. Network models are capable of reflecting the non-linear dynamic behavior of the systems. Despite the fact that network visualization and handling is not scalable, its representation is intuitive and, as an example, there are ongoing projects which aim to standardize the graphical representation (BioPax, PSI-MI, SBGN, Hermjakob et al., 2004; Strömbäck and Lambrix, 2005; Le Novère et al., 2009). Basic networks only model the coordinated behavior of biological entities, and experimental data can be mapped to the network in order to confirm and annotate experimental results. The integration of general and specific knowledge transforms those “influence networks” into “mechanistic networks” of higher quality (Hecker et al., 2009; Santamaría et al., 2011) which themselves already represent a molecular interaction model that can be tested with the help of perturbation experiments. The dynamic nature of a system is partly reflected in the network topology. The investigation of the interactivity, the distribution and regulation of hubs, network motifs, and cross-talk of functional modules and reaction sets (e.g., coupled reaction sets and elementary flux patterns in metabolic networks) contribute to our understanding of the robustness and flexibility of a system (Barabási and Oltvai, 2004).
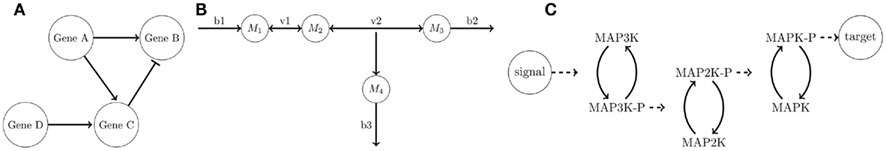
Figure 3. Graphical representation of (A) gene-regulatory network, (B) metabolic network, and (C) signaling network. (A) The activation of genes is shown by arrows whereas repression is marked by bars. (B) Metabolites (Mi) are connected with internal (vi) or external (bi) metabolic fluxes. (C) A signal can lead to a phosphorylation (“-P”) cascade of MAP kinases, and eventually the target is activated or repressed.
The major challenge of a systems biology-based approach to understanding the host-pathogen interaction arises from the robustness of the pathogenic system. The robustness originates from the network structure of the biological system that makes it unlikely that it will be possible to develop a single biomarker or drug against fungal infection. In the interests of clinical success, a system-oriented drug design with multiple antifungal strategies needs to affect sufficient points in the infection process (Kitano, 2007).
3.1.1. Gene-regulatory networks
Gene expression is mainly regulated by transcription factors and co-factors and additionally by post-transcriptional modification as well as mRNA and protein degradation. The reverse engineering of genome-wide interdependencies between these molecular entities relies on comprehensive datasets. Since there are only a few datasets on infectious processes available, one of the major tasks is to collect and process data and prior knowledge required for the development of novel parsimonious network models, describing essential fungus-host interactions. The inference process is mathematically challenging because the search space (number of possible gene regulations) increases exponentially with the number of nodes (genes). The modeling, on the other hand, relies on a small amount of data, which is usually obtained from microarray time series experiments. The experimental design is a trade-off that minimizes the cost and effort of the experiment, while ensuring that the data reliably reflects the underlying processes of the perturbation experiment. Many different network model architectures and inference methods have been proposed (reviewed in Hecker et al., 2009; Marbach et al., 2010). As part of the DREAM5-initiative1 (Dialog for Reverse Engineering Assessments and Methods), gene expression data of the bacterial pathogen Staphylococcus aureus was presented to infer large-scale gene-regulatory networks. The inferred networks have been used to create a predictive community network of 1054 genes and 1688 edges, which need to be validated experimentally (Stolovitzky, 2011). The infection process of the fungi C. albicans and A. fumigatus were modeled using tools such as NetGenerator (Guthke et al., 2005; Toepfer et al., 2007), which are based on ordinary differential equation systems and linear regression methods that are capable of describing the dynamic behavior of systems, even on a global scale (Altwasser et al., 2012). The high dimensionality of the mathematical problem can be reduced at several stages in the network reconstruction process (Hecker et al., 2009). For example, Guthke et al. (2007) reconstructed the underlying gene-regulatory events when A. fumigatus was exposed to a temperature shift. In order to select features for the network modeling, expression profiles with similar significantly regulated time courses were clustered to functional entities. Cluster representatives, indicating main biological functions, were assigned to these entities using gene annotation (Priebe et al., 2011).
The dimensionality of the problem can be further reduced when known global characteristics of a gene-regulatory network are taken into account. The observed high modularity, hierarchical structure, and over-representation of network motifs can guide the optimization of the structure of the model. A widely used criterion is the network sparseness, meaning that each gene is regulated only by a small number of regulatory genes. This feature of gene networks was incorporated in studies by Linde et al. (2010, 2012), who analyzed transcriptomic data focused on the iron homeostasis of C. albicans during infection and of A. fumigatus after a change in available iron concentration. The authors also used prior knowledge in order to reduce the search space. Interactions between genes and transcription factors found in literature were used to build a network template, and this prior knowledge was integrated into the network reconstruction procedures with the help of a weighting function. Currently, prior knowledge about fungi is scarcely available in databases and needs to be extracted from literature or genome sequences, e.g., by predicting transcription factor binding sites (Fazius et al., 2011). The prior knowledge also forms the basis for the analysis of the validity of the inferred network and the performance of the applied reconstruction method.
Recently, the inference of gene-regulatory network was applied to predict host-pathogen interactions (Tierney et al., 2012). RNA-seq data from a C. albicans infection of Mus musculus were used to predict subnetworks which were subsequently combined into an interspecies network. This model contained predicted regulations between the two species during the infection process. The results were supported by experimental findings which, overall, demonstrated that network inference can be used to support the deciphering of complex infectious processes.
3.1.2. Protein-protein interaction network
The biological function of genes originates from encoded proteins which form molecular structures, catalyze metabolic reactions, and are involved in signaling and regulation. With the help of technologies such as yeast two-hybrid or tandem affinity purification combined with mass spectrometry, it is possible to identify interacting protein partners which form an “interactome” network. The interactome shifts the focus from the detection of single protein interactions to the decoding of the global organization of proteomes (Barabási and Oltvai, 2004). Protein networks are increasingly used to identify host immune molecules and pathogenic effector proteins associated with host infection and drug targets (reviewed in Ideker and Sharan, 2008). For fungal infections, the main challenge is the lack of experimental protein interaction data, which is required to (semi)automatically construct such a network. The bioinformatic approaches either rely on text-mining (Zhou and He, 2008; Rao et al., 2010), or the interaction is predicted on the basis of sequence analyses (Dyer et al., 2007; Skrabanek et al., 2008). For the modeling of infectious diseases, Dyer et al. (2008) mapped all available host-pathogen protein interactions to a single protein network. Many pathogens were found to target the same process and therefore supported the hypothesis that topological properties of the protein-protein interaction network can be used to identify pathogenetic traits (Mukhtar et al., 2011) and drug targets (Hase et al., 2009; Zhu et al., 2009). In addition to this, the functional annotation of single gene products is supported by the interaction context of proteins within the global protein-protein interaction network (Xu and Li, 2006; Sharan et al., 2007).
3.1.3. Signaling Networks
The cell’s response to an internal and external stimulus is triggered by a signaling network whose regulation is the key control for cellular behavior. The understanding and modeling of this network holds great promise for the development of new therapeutic strategies and, consequently, many different modeling techniques have been developed (reviewed in Aldridge et al., 2006). The elements of the signal transduction networks help to identify the effect of positive or negative feedback loops (Blüthgen et al., 2009) and also shed light on the cross-talk between different signaling pathways (Borisov et al., 2009).
Successful application of signaling models depends on careful validation of the underlying data, especially since molecular signals are hard to measure experimentally. This is one reason why many applications in this research area concentrate on the perspective of the human host where, for example, the JAK/Stat pathway (Vera et al., 2011) and macrophage activation (Raza et al., 2008) have been modeled. Currently, the yeast Saccharomyces cerevisiae is a model organism for the analysis of fungal signaling networks (Waltermann and Klipp, 2010). The modeling of fungal pathogen signaling is still in its early stages. Efforts are being made to reconstruct the underlying signaling network, e.g., for C. albicans, with the help of sequence analysis and molecular biological experiments (Rispail et al., 2009). Additionally, the effects of host-pathogen interaction on the signaling pathways of the host are the subject of several studies (reviewed in Brodsky and Medzhitov, 2009; Hajishengallis and Lambris, 2011). For example, Franke et al. (2008) modeled the c-Met signaling network of hepatocytes after infection with the pathogenic bacterium Helicobacter pylori and predicted the effects of gene knock-outs, which were subsequently confirmed experimentally.
3.1.4. Metabolic networks
Supported by the increasing number of sequenced fungal genomes, the modeling of host-pathogen interactions with the help of genome-scale metabolic networks is feasible. The functionality of thousands of genes can be associated with a set of metabolic reactions, because they either encode enzymes or they regulate related reactions (Cavalieri and Filippo, 2005). The steady state of metabolic fluxes through a metabolic network structure can be understood as a phenotypic state of an organism. For example, the metabolic capacity of the pathogen needs to be able to produce a variety of secretory metabolites and proteins which correspond to the pathogenic traits of the species and which are interesting drug targets (Fang et al., 2009). On the other hand, fungi also rely on the uptake of essential minerals which are bound to storage proteins in the host, e.g., iron bound to hemoglobin or ferritin during C. albicans infection (Almeida et al., 2009). The intertwined nature of the host-pathogen interaction can thus be modeled by two interacting metabolic networks (Raghunathan et al., 2009).
Despite the increasing number of sequencing projects for pathogenic fungi (see Table 2), the number of reconstructed networks is limited to species which are in the focus of metabolic engineering. To the best of our knowledge, only two large-scale, manually curated metabolic network reconstructions of fungal pathogens exist, namely for A. fumigatus (Tuckwell et al., 2011) and C. albicans2. Despite the recent efforts made at automation, a high quality network reconstruction process needs manual curation, a step which acts as the bottleneck in this modeling technique (Pitkänen et al., 2010). During the curation of specific metabolic pathways, genes that encode missing enzymes are annotated with the help of bioinformatic tools or experimental validation (Pitkänen et al., 2010). The reconstruction process itself thus already contributes to the elucidation of the underlying molecular processes.
The mathematical modeling and simulation of metabolic networks allow us to address questions such as (i) the influence of single enzymes (and corresponding genes) within the network, (ii) the search for invariant steady states, (iii) the prediction of elementary flux modes, and even (iv) the consideration of different optimization strategies for the organism (Ruppin et al., 2010). With these objectives in mind, specific methods and concepts of metabolic modeling, which do not rely on mostly unknown specific kinetic parameters, can be applied to fungi.
Nevertheless, it is promising to compare metabolic networks of closely related pathogenic and non-pathogenic species in order to understand the physical features of pathogenicity (Lee et al., 2009). Recently, Oberhardt et al. (2011) examined differences and similarities between a pathogenic and non-pathogenic Pseudomonas species. Prior to analysis, the authors reconciled both metabolic networks in order to minimize the influence of different reconstruction approaches on the results. The subsequent analysis confirmed the multifactorial aspect of pathogenicity, but differences in the flexibility of sulfur related pathways were also found.
3.2. Spatio-Temporal Modeling
In contrast to the network models discussed in section 3.1, which are based on “omics”-data that does not contain any spatial information, spatio-temporal models are developed to include this type of information. For example, modeling the affinity maturation of antibodies in germinal centers in response to the recognition of antigens has been successfully performed in recent years in joint experimental and theoretical studies (Figge, 2005; Figge et al., 2008; Garin et al., 2010). Similarly, this modeling approach can be applied to the field of fungal infections to model the innate immune response (see Figure 4). With the application of special imaging techniques to generate spatially and spatio-temporally resolved data and their automated analysis (see section 2.2), the way has been paved to model, simulate, and study interactions between the host and pathogenic fungi from this point of view.

Figure 4. Schematic illustration of a continuous 3D spatial environment in silico within an agent-based model. Spherical objects represent cells, the different colors depict different cell types.
Two common approaches to model spatio-temporally resolved systems are partial differential equations (PDEs) and agent-based models (ABMs). The former treats systemic constituents (e.g., molecules or cells) as concentrations (or populations), whereas the latter deals with them as discrete objects. When it comes to the number of constituents and their quantities, scalability is one of the computational strengths of PDEs. Apart from this advantage, crucial limitations such as the identification of single objects, their specific local interactions and their particular internal states exist (Van Dyke Parunak et al., 1998). ABMs can cope with all of the constraints mentioned above, but come at the price of more expensive computer resources in terms of computer memory and computing time (Chavali et al., 2008).
Each modeling approach has its own right to exist. ABM systems proved to have the necessary properties when higher levels of granularity for single constituents are considered. PDEs are adequate when the general behavior, such as Brownian motion, for a whole population of constituents is considered (Guo and Tay, 2005). Another criterion for making the right choice is the amount of data and knowledge available for the scale under consideration. Cells can be differentiated in various states. They may either move in a straight line or show random migration behavior and interact individually, depending on the interaction partners involved. These characteristics favor the choice of an ABM. However, modeling molecules at the level of individual molecules also has several advantages. Nevertheless, describing innate immune responses against fungal infections at all levels within ABMs renders the multi-scale system intractable because of the overwhelmingly large number of cells and molecules. A promising approach toward modeling multi-scale systems was proposed by Guo et al. (2008), where, within a hybrid model, cells are described in continuous space as individual agents by an ABM approach, while molecular interaction is described by PDEs via reaction-diffusion equations for molecular populations. Such a hybrid multi-scale approach has the potential to model fungal infections since it is adaptable and extensible, thus providing the required flexibility.
At the beginning of the modeling process of this multi-scale system, one major issue is the definition of the necessary features of cells at the cellular scale. Examples of macroscopic characteristics are the cellular migration type as well as their morphology and interaction with other cells. A major source of these essential parameters are images and time-lapse videos, which are recorded during microscopy experiments. With the analysis of single frames, cellular sizes, morphological properties, and population quantities can be computationally extracted in an automated fashion (see Figure 1). An automated analysis of the interaction between macrophages and A. fumigatus was recently presented by Mech et al. (2011). Furthermore, the analysis of cell tracks extracted from videos plays an important role in the calculation of characteristic properties of the cellular movement such as speed, motility coefficient, and diffusion constant. The intercellular interactions can be defined at both the cellular and molecular scale. The initial setup of the interaction model can be arranged by implementing simple interaction rules of cellular behavior based on phenomenological knowledge. An extension toward a complex rule-system is also conceivable as well as the implementation of specific strategic behavior of the pathogenic fungi, as recently unraveled by Hummert et al. (2010), by applying methods of game theory. It would also be conceivable to include cell-cell interactions by considering interacting molecules at the cellular surface and their subsequent impact on internal and external signaling. These processes, taking place at the molecular level, require the data basis that underlies the network models (see section 3.1) and can be integrated together with the microscopy data basis into one and the same multi-scale model.
In addition to the integration of data, there are also other challenging tasks, such as the estimation of unknown parameters and the establishment of efficient algorithms and adequate data structures, in order to make simulations computational feasible. The former issue is strongly related to the generation of hypotheses. The efficiency of the applied algorithms needs to be verified experimentally in further studies. We are still at the beginning of the systems biology cycle regarding the spatio-temporal modeling of fungal infections, but the first promising steps have successfully been made.
4. Outlook
Although great advances have been made in the understanding of molecular and cellular mechanisms of fungal infections, they do not currently provide a genome-wide view on the pathogenic processes of both fungal pathogens and hosts. New technologies such as RNA-seq, single-cell measurements, and PET/CT imaging open up new opportunities to unravel molecular and cellular mechanisms in greater detail and complexity. However, integrative analysis of high-throughput and spatio-temporal data on several molecular and cellular levels is increasingly becoming the limiting step when identifying molecular key regulators and mechanisms involved in fungal pathogenicity. Efforts to standardize data management and annotation promise advances for computational approaches to the mathematical modeling of host-pathogen interactions. Bioinformatic tools also need to be further adapted to the specific biological context of fungi. The current focus of systems biology is on the application of network and agent-based modeling techniques to genome-wide and spatio-temporally resolved dynamic systems thus exploiting the full range of high-throughput and image data available. In order to fully facilitate existing computational methods, it is necessary to deepen our understanding of molecular mechanisms thus improving gene annotation and establishing wider knowledge bases.
Genomic knowledge, coupled with high-throughput gene knock-out methodologies, are already advancing. The availability of mutant strains has grown rapidly over the last 10 years (McCluskey et al., 2010). We now have the possibility to screen more than 3,000 C. albicans single deletion mutants, but we still lack complete A. fumigatus or C. albicans knock-out libraries. At present, many high-throughput investigations are performed using complete gene knock-out libraries from S. cerevisiae or Neurospora crassa. This approach can be exploited to discover wide spectra of antimycotic drugs, but is not helpful for virulence studies. However, complete sets of viable genome-wide mutants for many pathogenic fungi are expected to become available in the next few years. In parallel, many efforts are being made to create high-throughput screening infection model systems. Recently, alternative systems using insects, nematodes, or embryonated eggs have been used (Ferrandon et al., 2007; Mylonakis, 2008; Moy et al., 2009; Jacobsen et al., 2010).
In the meantime, the scientific community continues to investigate the role of innate immunity during fungal infections. It has yet to be explored how neutrophil granulocytes actually kill outgrowing hyphae of A. fumigatus. The cellular mechanisms can be explored with the help of images which are a promising data source for driving the systems biology cycle. Image-based Systems Biology focuses on spatial properties such as cellular morphology or the mechanical features of interactions, and allows further insights which contribute to a better understanding of biological systems.
In clinical practice, differential diagnosis of infectious diseases and sepsis is based primarily on clinical criteria. These criteria lack the required sensitivity and specificity (Alberti et al., 2003) making the identification of novel biomarkers essential to identify the pathogen causing the infection on time. In clinical decision-making, fast and reliable diagnosis of specific pathogens is required. Theranostics (Chen, 2011) is a novel concept of combining novel platforms and technologies in clinical diagnostics and therapy such as image-guided therapy by, e.g., PET (Walther et al., 2011) or Raman spectroscopy (Neugebauer et al., 2006). There is currently a great deal of effort being made to push commercialization and collaboration strategies to establish the so-called companion diagnostics (CDx) marketplace. So-called Translational Systems Biology describes the clinical application of mathematical or computational models by enhancing the understanding of the complex dynamics of biomedical processes in an integrative, genome-wide way (Vodovotz et al., 2008). It integrates the scientific background of these trends and is based on current experimental findings. Apart from integrating different “omics” data levels, models should not exclusively focus on the host’s and the pathogen’s aspects of the infection process, but also on the interaction between both biological systems. This interconnected perspective supports the elucidation of mechanisms underlying the complex process of fungal pathogenicity. Translational Systems Biology is primarily directed at drug target identification and validation as well as rational drug design, supported by analysis of the inferred molecular network models (Klipp et al., 2010). Furthermore, Translational Systems Biology of infection aims (i) to recognize pathogens by their molecular signatures, (ii) to make the outcome and responsiveness of therapeutic interventions more predictable, and (iii) to identify more effective therapies using mathematical and computational models. The first steps in Translational Systems Biology were taken in the area of sepsis control (Vodovotz et al., 2008) and tuberculosis research (Day et al., 2010). Translational Systems Biology of fungal infections with applications in personalized medicine (Willard and Ginsburg, 2009) can be expected to be developed in the near future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Part of this work was supported by the excellence graduate school Jena School for Microbial Communication (JSMC), funded by the Deutsche Forschungsgemeinschaft, as part o fit, by the International Leibniz Research School for Microbial and Biomolecular Interactions (ILRS Jena) and by a grant from the Federal Ministry of Education and Research (BMBF) in the frame of ERA-NetPathoGenoMics.
Footnotes
- ^http://wiki.c2b2.columbia.edu/dream/
- ^http://www.candidagenome.org and commercially available at Insilico Biotechnology AG and ERGO
References
Aimanianda, V., Bayry, J., Bozza, S., Kniemeyer, O., Perruccio, K., Elluru, S. R., Clavaud, C., Paris, S., Brakhage, A. A., Kaveri, S. V., Romani, L., and Latgé, J.-P. (2009). Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature 460, 1117–1121.
Alberti, C., Brun-Buisson, C., Goodman, S. V., Guidici, D., Granton, J., Moreno, R., Smithies, M., Thomas, O., Artigas, A., Gall, J. R. L., and Group, E. S. (2003). Influence of systemic inflammatory response syndrome and sepsis on outcome of critically ill infected patients. Am. J. Respir. Crit. Care Med. 168, 77–84.
Albrecht, D., Guthke, R., Brakhage, A. A., and Kniemeyer, O. (2010). Integrative analysis of the heat shock response in Aspergillus fumigatus. BMC Genomics 11, 32. doi:10.1186/1471-2164-11-32
Albrecht, D., Guthke, R., Kniemeyer, O., and Brakhage, A. A. (2008). “Systems biology of human-pathogenic fungi,” in Handbook of Research on Systems Biology Applications in Medicine, Vol. 1, ed. A. Daskalaki (Hershey: IGI Global), 400–:418.
Albrecht, D., Kniemeyer, O., Brakhage, A. A., Berth, M., and Guthke, R. (2007). Integration of transcriptome and proteome data from human-pathogenic fungi by using a data warehouse. J. Integr. Bioinform. 4, 52.
Albrecht, D., Kniemeyer, O., Mech, F., Gunzer, M., Brakhage, A. A., and Guthke, R. (2011). On the way toward systems biology of Aspergillus fumigatus infection. Int. J. Med. Microbiol. 301, 453–459.
Alby, K., Schaefer, D., and Bennett, R. J. (2009). Homothallic and heterothallic mating in the opportunistic pathogen Candida albicans. Nature 460, 890–893.
Aldridge, B. B., Burke, J. M., Lauffenburger, D. A., and Sorger, P. K. (2006). Physicochemical modelling of cell signalling pathways. Nat. Cell Biol. 8, 1195–1203.
Almeida, R. S., Wilson, D., and Hube, B. (2009). Candida albicans iron acquisition within the host. FEMS Yeast Res. 9, 1000–1012.
Altwasser, R., Linde, J., Buyko, E., Hahn, U., and Guthke, R. (2012). Genome-wide scale-free network inference for Candida albicans. Front. Microbiol. 3:51. doi:10.3389/fmicb.2012.00051
Andersen, M. R., and Nielsen, J. (2009). Current status of systems biology in aspergilli. Fungal Genet. Biol. 46(Suppl. 1), S180–S190.
Arnaud, M. B., Cerqueira, G. C., Inglis, D. O., Skrzypek, M. S., Binkley, J., Chibucos, M. C., Crabtree, J., Howarth, C., Orvis, J., Shah, P., Wymore, F., Binkley, G., Miyasato, S. R., Simison, M., Sherlock, G., and Wortman, J. R. (2012). The Aspergillus Genome Database (AspGD): recent developments in comprehensive multispecies curation, comparative genomics and community resources. Nucleic Acids Res. 40, D653–D659.
Arnaud, M. B., Costanzo, M. C., Shah, P., Skrzypek, M. S., and Sherlock, G. (2009). Gene ontology and the annotation of pathogen genomes: the case of Candida albicans. Trends Microbiol. 17, 295–303.
Arnaud, M. B., Costanzo, M. C., Skrzypek, M. S., Shah, P., Binkley, G., Lane, C., Miyasato, S. R., and Sherlock, G. (2007). Sequence resources at the Candida Genome Database. Nucleic Acids Res. 35, D452–D456. [Database issue].
Avet, J., Granjon, D., Prevot-Bitot, N., Isnardi, V., Berger, C., Stephan, J. L., and Dubois, F. (2009). Monitoring of systemic candidiasis by 18F-FDG PET/CT. Eur. J. Nucl. Med. Mol. Imaging 36, 1900.
Backer, M. D. D., Ilyina, T., Ma, X. J., Vandoninck, S., Luyten, W. H., and Bossche, H. V. (2001). Genomic profiling of the response of Candida albicans to itraconazole treatment using a DNA microarray. Antimicrob. Agents Chemother. 45, 1660–1670.
Bailey, N. J. T. (1975). The Mathematical Theory of Infectious Diseases and its Application. London: Griffin.
Barabási, A.-L., and Oltvai, Z. N. (2004). Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113.
Barker, K. S., Crisp, S., Wiederhold, N., Lewis, R. E., Bareither, B., Eckstein, J., Barbuch, R., Bard, M., and Rogers, P. D. (2004). Genome-wide expression profiling reveals genes associated with amphotericin B and fluconazole resistance in experimentally induced antifungal resistant isolates of Candida albicans. J. Antimicrob. Chemother. 54, 376–385.
Barrett, L. G., Thrall, P. H., Burdon, J. J., and Linde, C. C. (2008). Life history determines genetic structure and evolutionary potential of host-parasite interactions. Trends Ecol. Evol. (Amst.) 23, 678–685.
Behnsen, J., Hartmann, A., Schmaler, J., Gehrke, A., Brakhage, A. A., and Zipfel, P. F. (2008). The opportunistic human pathogenic fungus Aspergillus fumigatus evades the host complement system. Infect. Immun. 76, 820–827.
Behnsen, J., Lessing, F., Schindler, S., Wartenberg, D., Jacobsen, I. D., Thoen, M., Zipfel, P. F., and Brakhage, A. A. (2010). Secreted Aspergillus fumigatus protease Alp1 degrades human complement proteins C3, C4, and C5. Infect. Immun. 78, 3585–3594.
Behnsen, J., Narang, P., Hasenberg, M., Gunzer, F., Bilitewski, U., Klippel, N., Rohde, M., Brock, M., Brakhage, A. A., and Gunzer, M. (2007). Environmental dimensionality controls the interaction of phagocytes with the pathogenic fungi Aspergillus fumigatus and Candida albicans. PLoS Pathog. 3, e13. doi:10.1371/journal.ppat.0030013
Bernoulli, D. (1760). Essai d’une nouvelle analyse de la mortalité causée par la petite vérole et des avantages de l’inoculation pour la prévenir. Mémoires de Mathématiques et de Physique Paris Académie Royale des Sciences 1, 1–45.
Blüthgen, N., Legewie, S., Kielbasa, S. M., Schramme, A., Tchernitsa, O., Keil, J., Solf, A., Vingron, M., Schäfer, R., Herzel, H., and Sers, C. (2009). A systems biological approach suggests that transcriptional feedback regulation by dual-specificity phosphatase 6 shapes extracellular signal-related kinase activity in RAS-transformed fibroblasts. FEBS J. 276, 1024–1035.
Borisov, N., Aksamitiene, E., Kiyatkin, A., Legewie, S., Berkhout, J., Maiwald, T., Kaimachnikov, N. P., Timmer, J., Hoek, J. B., and Kholodenko, B. N. (2009). Systems-level interactions between insulin-EGF networks amplify mitogenic signaling. Mol. Syst. Biol. 5, 256.
Brakhage, A. A. (2005). Systemic fungal infections caused by Aspergillus species: epidemiology, infection process and virulence determinants. Curr. Drug Targets 6, 875–886.
Brakhage, A. A., Bruns, S., Thywissen, A., Zipfel, P. F., and Behnsen, J. (2010). Interaction of phagocytes with filamentous fungi. Curr. Opin. Microbiol. 13, 409–415.
Braun, B. R., van Het Hoog, M., d’Enfert, C., Martchenko, M., Dungan, J., Kuo, A., Inglis, D. O., Uhl, M. A., Hogues, H., Berriman, M., Lorenz, M., Levitin, A., Oberholzer, U., Bachewich, C., Harcus, D., Marcil, A., Dignard, D., Iouk, T., Zito, R., Frangeul, L., Tekaia, F., Rutherford, K., Wang, E., Munro, C. A., Bates, S., Gow, N. A., Hoyer, L. L., Köhler, G., Morschhäuser, J., Newport, G., Znaidi, S., Raymond, M., Turcotte, B., Sherlock, G., Costanzo, M., Ihmels, J., Berman, J., Sanglard, D., Agabian, N., Mitchell, A. P., Johnson, A. D., Whiteway, M., and Nantel, A. (2005). A human-curated annotation of the Candida albicans genome. PLoS Genet. 1, 36–57.
Brock, M. (2012). Application of bioluminescence imaging for in vivo monitoring of fungal infections. Int. J. Microbiol. 2012, 956794.
Brodsky, I. E., and Medzhitov, R. (2009). Targeting of immune signalling networks by bacterial pathogens. Nat. Cell Biol. 11, 521–526.
Brown, A. J. P., Odds, F. C., and Gow, N. A. R. (2007). Infection-related gene expression in Candida albicans. Curr. Opin. Microbiol. 10, 307–313.
Bruneau, J. -M., Maillet, I., Tagat, E., Legrand, R., Supatto, F., Fudali, C., Caer, J.-P. L., Labas, V., Lecaque, D., and Hodgson, J. (2003). Drug induced proteome changes in Candida albicans: comparison of the effect of beta(1,3) glucan synthase inhibitors and two triazoles, fluconazole and itraconazole. Proteomics 3, 325–336.
Bruno, V. M., Wang, Z., Marjani, S. L., Euskirchen, G. M., Martin, J., Sherlock, G., and Snyder, M. (2010). Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res. 20, 1451–1458.
Bruns, S., Seidler, M., Albrecht, D., Salvenmoser, S., Remme, N., Hertweck, C., Brakhage, A. A., Kniemeyer, O., and Müller, F.-M. C. (2010). Functional genomic profiling of Aspergillus fumigatus biofilm reveals enhanced production of the mycotoxin gliotoxin. Proteomics 10, 3097–3107.
Burmester, A., Shelest, E., Glöckner, G., Heddergott, C., Schindler, S., Staib, P., Heidel, A., Felder, M., Petzold, A., Szafranski, K., Feuermann, M., Pedruzzi, I., Priebe, S., Groth, M., Winkler, R., Li, W., Kniemeyer, O., Schroeckh, V., Hertweck, C., Hube, B., White, T. C., Platzer, M., Guthke, R., Heitman, J., Wöstemeyer, J., Zipfel, P. F., Monod, M., and Brakhage, A. A. (2011). Comparative and functional genomics provide insights into the pathogenicity of dermatophytic fungi. Genome Biol. 12, R7.
Butler, G., Rasmussen, M. D., Lin, M. F., Santos, M. A. S., Sakthikumar, S., Munro, C. A., Rheinbay, E., Grabherr, M., Forche, A., Reedy, J. L., Agrafioti, I., Arnaud, M. B., Bates, S., Brown, A. J. P., Brunke, S., Costanzo, M. C., Fitzpatrick, D. A., de Groot, P. W. J., Harris, D., Hoyer, L. L., Hube, B., Klis, F. M., Kodira, C., Lennard, N., Logue, M. E., Martin, R., Neiman, A. M., Nikolaou, E., Quail, M. A., Quinn, J., Santos, M. C., Schmitzberger, F. F., Sherlock, G., Shah, P., Silverstein, K. A. T., Skrzypek, M. S., Soll, D., Staggs, R., Stansfield, I., Stumpf, M. P. H., Sudbery, P. E., Srikantha, T., Zeng, Q., Berman, J., Berriman, M., Heitman, J., Gow, N. A. R., Lorenz, M. C., Birren, B. W., Kellis, M., and Cuomo, C. A. (2009). Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459, 657–662.
Cagas, S. E., Jain, M. R., Li, H., and Perlin, D. S. (2011). Profiling the Aspergillus fumigatus proteome in response to caspofungin. Antimicrob. Agents Chemother. 55, 146–154.
Cairns, T., Minuzzi, F., and Bignell, E. (2010). The host-infecting fungal transcriptome. FEMS Microbiol. Lett. 307, 1–11.
Castillo, L., Calvo, E., Martínez, A. I., Ruiz-Herrera, J., Valentín, E., Lopez, J. A., and Sentandreu, R. (2008). A study of the Candida albicans cell wall proteome. Proteomics 8, 3871–3881.
Cavalieri, D., and Filippo, C. D. (2005). Bioinformatic methods for integrating whole-genome expression results into cellular networks. Drug Discov. Today 10, 727–734.
Chavali, A. K., Gianchandani, E. P., Tung, K. S., Lawrence, M. B., Peirce, S. M., and Papin, J. A. (2008). Characterizing emergent properties of immunological systems with multi-cellular rule-based computational modeling. Trends Immunol. 29, 589–599.
Chen, X. S. (2011). Introducing Theranostics journal – from the editor-in-chief. Theranostics 1, 1–2.
Correia, A., Lermann, U., Teixeira, L., Cerca, F., Botelho, S., da Costa, R. M. G., Sampaio, P., Gärtner, F., Morschhäuser, J., Vilanova, M., and Pais, C. (2010). Limited role of secreted aspartyl proteinases Sap1 to Sap6 in Candida albicans virulence and host immune response in murine hematogenously disseminated candidiasis. Infect. Immun. 78, 4839–4849.
Courtot, M., Juty, N., Knüpfer, C., Waltemath, D., Zhukova, A., Dräger, A., Dumontier, M., Finney, A., Golebiewski, M., Hastings, J., Hoops, S., Keating, S., Kell, D. B., Kerrien, S., Lawson, J., Lister, A., Lu, J., Machne, R., Mendes, P., Pocock, M., Rodriguez, N., Villeger, A., Wilkinson, D. J., Wimalaratne, S., Laibe, C., Hucka, M., and Novère, N. L. (2011). Controlled vocabularies and semantics in systems biology. Mol. Syst. Biol. 7, 543.
da Silva Ferreira, M. E., Malavazi, I., Savoldi, M., Brakhage, A. A., Goldman, M. H. S., Kim, H. S., Nierman, W. C., and Goldman, G. H. (2006). Transcriptome analysis of Aspergillus fumigatus exposed to voriconazole. Curr. Genet. 50, 32–44.
Dagenais, T. R. T., and Keller, N. P. (2009). Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin. Microbiol. Rev. 22, 447–465.
Day, J., Schlesinger, L. S., and Friedman, A. (2010). Tuberculosis research: going forward with a powerful “translational systems biology” approach. Tuberculosis (Edinb.) 90, 7–8.
de Groot, P. W. J., de Boer, A. D., Cunningham, J., Dekker, H. L., de Jong, L., Hellingwerf, K. J., de Koster, C., and Klis, F. M. (2004). Proteomic analysis of Candida albicans cell walls reveals covalently bound carbohydrate-active enzymes and adhesins. Eukaryotic Cell 3, 955–965.
d’Enfert, C., Goyard, S., Rodriguez-Arnaveilhe, S., Frangeul, L., Jones, L., Tekaia, F., Bader, O., Albrecht, A., Castillo, L., Dominguez, A., Ernst, J. F., Fradin, C., Gaillardin, C., Garcia-Sanchez, S., de Groot, P., Hube, B., Klis, F. M., Krishnamurthy, S., Kunze, D., Lopez, M.-C., Mavor, A., Martin, N., Moszer, I., Onésime, D., Martin, J. P., Sentandreu, R., Valentin, E., and Brown, A. J. P. (2005). CandidaDB: a genome database for Candida albicans pathogenomics. Nucleic Acids Res. 33, D353–D357. [Database issue].
Doedt, T., Krishnamurthy, S., Bockmühl, D. P., Tebarth, B., Stempel, C., Russell, C. L., Brown, A. J. P., and Ernst, J. F. (2004). APSES proteins regulate morphogenesis and metabolism in Candida albicans. Mol. Biol. Cell 15, 3167–3180.
Domon, B., and Aebersold, R. (2010). Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721.
Dyer, M. D., Murali, T. M., and Sobral, B. W. (2007). Computational prediction of host-pathogen protein-protein interactions. Bioinformatics 23, i159–i166.
Dyer, M. D., Murali, T. M., and Sobral, B. W. (2008). The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 4, e32. doi:10.1371/journal.ppat.0040032
Fang, X., Wallqvist, A., and Reifman, J. (2009). A systems biology framework for modeling metabolic enzyme inhibition of Mycobacterium tuberculosis. BMC Syst. Biol. 3, 92.
Fazius, E., Shelest, V., and Shelest, E. (2011). SiTaR: a novel tool for transcription factor binding site prediction. Bioinformatics 27, 2806–2811.
Fedorova, N. D., Khaldi, N., Joardar, V. S., Maiti, R., Amedeo, P., Anderson, M. J., Crabtree, J., Silva, J. C., Badger, J. H., Albarraq, A., Angiuoli, S., Bussey, H., Bowyer, P., Cotty, P. J., Dyer, P. S., Egan, A., Galens, K., Fraser-Liggett, C. M., Haas, B. J., Inman, J. M., Kent, R., Lemieux, S., Malavazi, I., Orvis, J., Roemer, T., Ronning, C. M., Sundaram, J. P., Sutton, G., Turner, G., Venter, J. C., White, O. R., Whitty, B. R., Youngman, P., Wolfe, K. H., Goldman, G. H., Wortman, J. R., Jiang, B., Denning, D. W., and Nierman, W. C. (2008). Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 4, e1000046. doi:10.1371/journal.pgen.1000046
Ferrandon, D., Imler, J.-L., Hetru, C., and Hoffmann, J. A. (2007). The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nat. Rev. Immunol. 7, 862–874.
Figge, M. T. (2005). Stochastic discrete event simulation of germinal center reactions. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 71, 051907.
Figge, M. T., Garin, A., Gunzer, M., Kosco-Vilbois, M., Toellner, K.-M., and Meyer-Hermann, M. (2008). Deriving a germinal center lymphocyte migration model from two-photon data. J. Exp. Med. 205, 3019–3029.
Filler, S. G. (2006). Candida-host cell receptor-ligand interactions. Curr. Opin. Microbiol. 9, 333–339.
Fleck, C. B., Schöbel, F., and Brock, M. (2011). Nutrient acquisition by pathogenic fungi: nutrient availability, pathway regulation, and differences in substrate utilization. Int. J. Med. Microbiol. 301, 400–407.
Fradin, C., Kretschmar, M., Nichterlein, T., Gaillardin, C., d’Enfert, C., and Hube, B. (2003). Stage-specific gene expression of Candida albicans in human blood. Mol. Microbiol. 47, 1523–1543.
Franke, R., Müller, M., Wundrack, N., Gilles, E.-D., Klamt, S., Kähne, T., and Naumann, M. (2008). Host-pathogen systems biology: logical modelling of hepatocyte growth factor and Helicobacter pylori induced c-Met signal transduction. BMC Syst. Biol. 2, 4. doi:10.1186/1752-0509-2-4
Gafa, V., Remoli, M. E., Giacomini, E., Gagliardi, M. C., Lande, R., Severa, M., Grillot, R., and Coccia, E. M. (2007). In vitro infection of human dendritic cells by Aspergillus fumigatus conidia triggers the secretion of chemokines for neutrophil and Th1 lymphocyte recruitment. Microbes Infect. 9, 971–980.
Garin, A., Meyer-Hermann, M., Contie, M., Figge, M. T., Buatois, V., Gunzer, M., Toellner, K. -M., Elson, G., and Kosco-Vilbois, M. H. (2010). Toll-like receptor 4 signaling by follicular dendritic cells is pivotal for germinal center onset and affinity maturation. Immunity 33, 84–95.
Gautam, P., Shankar, J., Madan, T., Sirdeshmukh, R., Sundaram, C. S., Gade, W. N., Basir, S. F., and Sarma, P. U. (2008). Proteomic and transcriptomic analysis of Aspergillus fumigatus on exposure to amphotericin B. Antimicrob. Agents Chemother. 52, 4220–4227.
Gibbons, J. G., Beauvais, A., Beau, R., McGary, K. L., Latge, J.-P., and Rokas, A. (2012). Global transcriptome changes underlying colony growth in the opportunistic human pathogen Aspergillus fumigatus. Eukaryotic Cell. 11, 68–78.
Gilsenan, J. M., Cooley, J., and Bowyer, P. (2012). CADRE: The Central Aspergillus Data Repository 2012. Nucleic Acids Res. 40, D660–D666.
Grahl, N., Puttikamonkul, S., MacDonald, J. M., Gamcsik, M. P., Ngo, L. Y., Hohl, T. M., and Cramer, R. A. (2011). In vivo hypoxia and a fungal alcohol dehydrogenase influence the pathogenesis of invasive pulmonary aspergillosis. PLoS Pathog. 7, e1002145. doi:10.1371/journal.ppat.1002145
Gropp, K., Schild, L., Schindler, S., Hube, B., Zipfel, P. F., and Skerka, C. (2009). The yeast Candida albicans evades human complement attack by secretion of aspartic proteases. Mol. Immunol. 47, 465–475.
Gross, O., Poeck, H., Bscheider, M., Dostert, C., Hannesschläger, N., Endres, S., Hartmann, G., Tardivel, A., Schweighoffer, E., Tybulewicz, V., Mocsai, A., Tschopp, J., and Ruland, J. (2009). Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459, 433–436.
Gudlaugsson, O., Gillespie, S., Lee, K., Berg, J. V., Hu, J., Messer, S., Herwaldt, L., Pfaller, M., and Diekema, D. (2003). Attributable mortality of nosocomial candidemia, revisited. Clin. Infect. Dis. 37, 1172–1177.
Guo, Z., Sloot, P. M., and Tay, J. C. (2008). A hybrid agent-based approach for modeling microbiological systems. J. Theor. Biol. 255, 163–175.
Guo, Z., and Tay, J. (2005). A comparative study on modeling strategies for immune system dynamics under HIV-1 infection. Artif. Immune Syst. 3627, 220–233.
Guthke, R., Kniemeyer, O., Albrecht, D., Brakhage, A. A., and Möller, U. (2007). Discovery of gene regulatory networks in Aspergillus fumigatus. Lect. Notes Bioinform. 4366, 22–41.
Guthke, R., Möller, U., Hoffmann, M., Thies, F., and Töpfer, S. (2005). Dynamic network reconstruction from gene expression data applied to immune response during bacterial infection. Bioinformatics 21, 1626–1634.
Hajishengallis, G., and Lambris, J. D. (2011). Microbial manipulation of receptor crosstalk in innate immunity. Nat. Rev. Immunol. 11, 187–200.
Hase, T., Tanaka, H., Suzuki, Y., Nakagawa, S., and Kitano, H. (2009). Structure of protein interaction networks and their implications on drug design. PLoS Comput. Biol. 5, e1000550. doi:10.1371/journal.pcbi.1000550
Hawksworth, D. (2001). The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol. Res. 105, 1422–1432.
Hecker, M., Lambeck, S., Toepfer, S., van Someren, E., and Guthke, R. (2009). Gene regulatory network inference: data integration in dynamic models – a review. BioSystems 96, 86–103.
Heilmann, C. J., Sorgo, A. G., Siliakus, A. R., Dekker, H. L., Brul, S., de Koster, C. G., de Koning, L. J., and Klis, F. M. (2011). Hyphal induction in the human fungal pathogen Candida albicans reveals a characteristic wall protein profile. Microbiology 157, 2297–2307.
Hermjakob, H., Montecchi-Palazzi, L., Bader, G., Wojcik, J., Salwinski, L., Ceol, A., Moore, S., Orchard, S., Sarkans, U., von Mering, C., Roechert, B., Poux, S., Jung, E., Mersch, H., Kersey, P., Lappe, M., Li, Y., Zeng, R., Rana, D., Nikolski, M., Husi, H., Brun, C., Shanker, K., Grant, S. G. N., Sander, C., Bork, P., Zhu, W., Pandey, A., Brazma, A., Jacq, B., Vidal, M., Sherman, D., Legrain, P., Cesareni, G., Xenarios, I., Eisenberg, D., Steipe, B., Hogue, C., and Apweiler, R. (2004). The HUPO PSI’s molecular interaction format – a community standard for the representation of protein interaction data. Nat. Biotechnol. 22, 177–183.
Hernáez, M. L., Ximénez-Embún, P., Martínez-Gomariz, M., Gutiérrez-Blázquez, M. D., Nombela, C., and Gil, C. (2010). Identification of Candida albicans exposed surface proteins in vivo by a rapid proteomic approach. J. Proteomics 73, 1404–1409.
Hickey, P. C., and Read, N. D. (2009). Imaging living cells of Aspergillus in vitro. Med. Mycol. 47(Suppl. 1), S110–S119.
Hise, A. G., Tomalka, J., Ganesan, S., Patel, K., Hall, B. A., Brown, G. D., and Fitzgerald, K. A. (2009). An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5, 487–497.
Hoehamer, C. F., Cummings, E. D., Hilliard, G. M., and Rogers, P. D. (2010). Changes in the proteome of Candida albicans in response to azole, polyene, and echinocandin antifungal agents. Antimicrob. Agents Chemother. 54, 1655–1664.
Hooshdaran, M. Z., Barker, K. S., Hilliard, G. M., Kusch, H., Morschhäuser, J., and Rogers, P. D. (2004). Proteomic analysis of azole resistance in Candida albicans clinical isolates. Antimicrob. Agents Chemother. 48, 2733–2735.
Hucka, M., Finney, A., Sauro, H. M., Bolouri, H., Doyle, J. C., Kitano, H., Arkin, A. P., Bornstein, B. J., Bray, D., Cornish-Bowden, A., Cuellar, A. A., Dronov, S., Gilles, E. D., Ginkel, M., Gor, V., Goryanin, I. I., Hedley, W. J., Hodgman, T. C., Hofmeyr, J.-H., Hunter, P. J., Juty, N. S., Kasberger, J. L., Kremling, A., Kummer, U., Novère, N. L., Loew, L. M., Lucio, D., Mendes, P., Minch, E., Mjolsness, E. D., Nakayama, Y., Nelson, M. R., Nielsen, P. F., Sakurada, T., Schaff, J. C., Shapiro, B. E., Shimizu, T. S., Spence, H. D., Stelling, J., Takahashi, K., Tomita, M., Wagner, J., Wang, J., and Forum, S. B. M. L. (2003). The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19, 524–531.
Hull, D., Wolstencroft, K., Stevens, R., Goble, C., Pocock, M. R., Li, P., and Oinn, T. (2006). Taverna: a tool for building and running workflows of services. Nucleic Acids Res. 34, W729–W732. [Web Server issue].
Hummert, S., Hummert, C., Schröter, A., Hube, B., and Schuster, S. (2010). Game theoretical modelling of survival strategies of Candida albicans inside macrophages. J. Theor. Biol. 264, 312–318.
Ibrahim-Granet, O., Jouvion, G., Hohl, T. M., Droin-Bergère, S., Philippart, F., Kim, O. Y., Adib-Conquy, M., Schwendener, R., Cavaillon, J.-M., and Brock, M. (2010). In vivo bioluminescence imaging and histopathopathologic analysis reveal distinct roles for resident and recruited immune effector cells in defense against invasive aspergillosis. BMC Microbiol. 10, 105. doi:10.1186/1471-2180-10-105
Ibrahim-Granet, O., Philippe, B., Boleti, H., Boisvieux-Ulrich, E., Grenet, D., Stern, M., and Latgé, J. P. (2003). Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infect. Immun. 71, 891–903.
Ideker, T., Galitski, T., and Hood, L. (2001). A new approach to decoding life: systems biology. Annu. Rev. Genomics Hum. Genet. 2, 343–372.
Ihmels, J., Bergmann, S., Berman, J., and Barkai, N. (2005). Comparative gene expression analysis by differential clustering approach: application to the Candida albicans transcription program. PLoS Genet. 1, e39. doi:10.1371/journal.pgen.0010039
Jacobsen, I. D., Grosse, K., Slesiona, S., Hube, B., Berndt, A., and Brock, M. (2010). Embryonated eggs as an alternative infection model to investigate Aspergillus fumigatus virulence. Infect. Immun. 78, 2995–3006.
Jahn, B., Langfelder, K., Schneider, U., Schindel, C., and Brakhage, A. A. (2002). PKSP-dependent reduction of phagolysosome fusion and intracellular kill of Aspergillus fumigatus conidia by human monocyte-derived macrophages. Cell. Microbiol. 4, 793–803.
Jain, R., Valiante, V., Remme, N., Docimo, T., Heinekamp, T., Hertweck, C., Gershenzon, J., Haas, H., and Brakhage, A. A. (2011). The MAP kinase MpkA controls cell wall integrity, oxidative stress response, gliotoxin production and iron adaptation in Aspergillus fumigatus. Mol. Microbiol. 82, 39–53.
Jones, T., Federspiel, N. A., Chibana, H., Dungan, J., Kalman, S., Magee, B. B., Newport, G., Thorstenson, Y. R., Agabian, N., Magee, P. T., Davis, R. W., and Scherer, S. (2004). The diploid genome sequence of Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 101, 7329–7334.
Kim, Y., Nandakumar, M. P., and Marten, M. R. (2008). The state of proteome profiling in the fungal genus Aspergillus. Brief. Funct. Genomic. Proteomic. 7, 87–94.
Kitano, H. (2007). A robustness-based approach to systems-oriented drug design. Nat. Rev. Drug Discov. 6, 202–210.
Klipp, E., Wade, R. C., and Kummer, U. (2010). Biochemical network-based drug-target prediction. Curr. Opin. Biotechnol. 21, 511–516.
Klose, J., and Kobalz, U. (1995). Two-dimensional electrophoresis of proteins: an updated protocol and implications for a functional analysis of the genome. Electrophoresis 16, 1034–1059.
Kniemeyer, O. (2011). Proteomics of eukaryotic microorganisms: the medically and biotechnologically important fungal genus Aspergillus. Proteomics 11, 3232–3243.
Kniemeyer, O., and Brakhage, A. A. (2008). “Proteomics and its application to the human-pathogenic fungi,” in Human and Animal Relationships: The Mycota VI, eds A. A. Brakhage, and P. F. Zipfel (Berlin: Springer), 154–186.
Kniemeyer, O., Schmidt, A. D., Vödisch, M., Wartenberg, D., and Brakhage, A. A. (2011). Identification of virulence determinants of the human pathogenic fungi Aspergillus fumigatus and Candida albicans by proteomics. Int. J. Med. Microbiol. 301, 368–377.
Köhler, J., Baumbach, J., Taubert, J., Specht, M., Skusa, A., Rüegg, A., Rawlings, C., Verrier, P., and Philippi, S. (2006). Graph-based analysis and visualization of experimental results with ONDEX. Bioinformatics 22, 1383–1390.
Kozhenkov, S., Sedova, M., Dubinina, Y., Gupta, A., Ray, A., Ponomarenko, J., and Baitaluk, M. (2011). Biological networks – tools enabling the integration of multi-scale data for the host-pathogen studies. BMC Syst. Biol. 5, 7. doi:10.1186/1752-0509-5-7
Krappmann, S., Sasse, C., and Braus, G. H. (2006). Gene targeting in Aspergillus fumigatus by homologous recombination is facilitated in a nonhomologous end-joining-deficient genetic background. Eukaryotic Cell 5, 212–215.
Kusch, H., Engelmann, S., Albrecht, D., Morschhäuser, J., and Hecker, M. (2007). Proteomic analysis of the oxidative stress response in Candida albicans. Proteomics 7, 686–697.
Langfelder, K., Jahn, B., Gehringer, H., Schmidt, A., Wanner, G., and Brakhage, A. A. (1998). Identification of a polyketide synthase gene (pksP) of Aspergillus fumigatus involved in conidial pigment biosynthesis and virulence. Med. Microbiol. Immunol. 187, 79–89.
Le Novère, N., Hucka, M., Mi, H., Moodie, S., Schreiber, F., Sorokin, A., Demir, E., Wegner, K., Aladjem, M. I., Wimalaratne, S. M., Bergman, F. T., Gauges, R., Ghazal, P., Kawaji, H., Li, L., Matsuoka, Y., Villéger, A., Boyd, S. E., Calzone, L., Courtot, M., Dogrusoz, U., Freeman, T. C., Funahashi, A., Ghosh, S., Jouraku, A., Kim, S., Kolpakov, F., Luna, A., Sahle, S., Schmidt, E., Watterson, S., Wu, G., Goryanin, I., Kell, D. B., Sander, C., Sauro, H., Snoep, J. L., Kohn, K., and Kitano, H. (2009). The systems biology graphical notation. Nat. Biotechnol. 27, 735–741.
Lee, D.-S., Burd, H., Liu, J., Almaas, E., Wiest, O., Barabási, A.-L., Oltvai, Z. N., and Kapatral, V. (2009). Comparative genome-scale metabolic reconstruction and flux balance analysis of multiple Staphylococcus aureus genomes identify novel antimicrobial drug targets. J. Bacteriol. 191, 4015–4024.
Lessing, F., Kniemeyer, O., Wozniok, I., Loeffler, J., Kurzai, O., Haertl, A., and Brakhage, A. A. (2007). The Aspergillus fumigatus transcriptional regulator AfYap1 represents the major regulator for defense against reactive oxygen intermediates but is dispensable for pathogenicity in an intranasal mouse infection model. Eukaryotic Cell 6, 2290–2302.
Linde, J., Hortschansky, P., Fazius, E., Brakhage, A. A., Guthke, R., and Haas, H. (2012). Regulatory interactions for iron homoeostatis in Aspergillus fumigatus inferred by a systems biology approach. BMC Syst. Biol. 6, 6. doi:10.1186/1752-0509-6-6
Linde, J., Wilson, D., Hube, B., and Guthke, R. (2010). Regulatory network modelling of iron acquisition by a fungal pathogen in contact with epithelial cells. BMC Syst. Biol. 4, 148. doi:10.1186/1752-0509-4-148
Liu, H. (2002). Co-regulation of pathogenesis with dimorphism and phenotypic switching in Candida albicans, a commensal and a pathogen. Int. J. Med. Microbiol. 292, 299–311.
Liu, T. T., Lee, R. E. B., Barker, K. S., Lee, R. E., Wei, L., Homayouni, R., and Rogers, P. D. (2005). Genome-wide expression profiling of the response to azole, polyene, echinocandin, and pyrimidine antifungal agents in Candida albicans. Antimicrob. Agents Chemother. 49, 2226–2236.
Lloyd, A. T., and Sharp, P. M. (1992). Evolution of codon usage patterns: the extent and nature of divergence between Candida albicans and Saccharomyces cerevisiae. Nucleic Acids Res. 20, 5289–5295.
Lo, H. J., Köhler, J. R., DiDomenico, B., Loebenberg, D., Cacciapuoti, A., and Fink, G. R. (1997). Nonfilamentous C. albicans mutants are avirulent. Cell 90, 939–949.
Loftus, B. J., Fung, E., Roncaglia, P., Rowley, D., Amedeo, P., Bruno, D., Vamathevan, J., Miranda, M., Anderson, I. J., Fraser, J. A., Allen, J. E., Bosdet, I. E., Brent, M. R., Chiu, R., Doering, T. L., Donlin, M. J., D’Souza, C. A., Fox, D. S., Grinberg, V., Fu, J., Fukushima, M., Haas, B. J., Huang, J. C., Janbon, G., Jones, S. J. M., Koo, H. L., Krzywinski, M. I., Kwon-Chung, J. K., Lengeler, K. B., Maiti, R., Marra, M. A., Marra, R. E., Mathewson, C. A., Mitchell, T. G., Pertea, M., Riggs, F. R., Salzberg, S. L., Schein, J. E., Shvartsbeyn, A., Shin, H., Shumway, M., Specht, C. A., Suh, B. B., Tenney, A., Utterback, T. R., Wickes, B. L., Wortman, J. R., Wye, N. H., Kronstad, J. W., Lodge, J. K., Heitman, J., Davis, R. W., Fraser, C. M., and Hyman, R. W. (2005). The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science 307, 1321–1324.
Lorenz, M. C., Bender, J. A., and Fink, G. R. (2004). Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryotic Cell 3, 1076–1087.
Lorenz, M. C., and Fink, G. R. (2001). The glyoxylate cycle is required for fungal virulence. Nature 412, 83–86.
Manning, M., and Mitchell, T. G. (1980). Morphogenesis of Candida albicans and cytoplasmic proteins associated with differences in morphology, strain, or temperature. J. Bacteriol. 144, 258–273.
Marbach, D., Prill, R. J., Schaffter, T., Mattiussi, C., Floreano, D., and Stolovitzky, G. (2010). Revealing strengths and weaknesses of methods for gene network inference. Proc. Natl. Acad. Sci. U.S.A. 107, 6286–6291.
Marr, K. A., Carter, R. A., Boeckh, M., Martin, P., and Corey, L. (2002). Invasive aspergillosis in allogeneic stem cell transplant recipients: changes in epidemiology and risk factors. Blood 100, 4358–4366.
Martin, G. S., Mannino, D. M., Eaton, S., and Moss, M. (2003). The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 348, 1546–1554.
Martin, R., Wächtler, B., Schaller, M., Wilson, D., and Hube, B. (2011). Host-pathogen interactions and virulence-associated genes during Candida albicans oral infections. Int. J. Med. Microbiol. 301, 417–422.
Martínez-Solano, L., Nombela, C., Molero, G., and Gil, C. (2006). Differential protein expression of murine macrophages upon interaction with Candida albicans. Proteomics 6(Suppl. 1), S133–S144.
Martínez-Solano, L., Reales-Calderón, J. A., Nombela, C., Molero, G., and Gil, C. (2009). Proteomics of RAW 264.7 macrophages upon interaction with heat-inactivated Candida albicans cells unravel an anti-inflammatory response. Proteomics 9, 2995–3010.
May, R. M., and Anderson, R. M. (1979). Population biology of infectious diseases. Part II. Nature 280, 455–461.
McCluskey, K., Wiest, A., and Plamann, M. (2010). The Fungal Genetics Stock Center: a repository for 50 years of fungal genetics research. J. Biosci. 35, 119–126.
McCormick, A., Heesemann, L., Wagener, J., Marcos, V., Hartl, D., Loeffler, J., Heesemann, J., and Ebel, F. (2010). NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect. 12, 928–936.
Mech, F., Thywissen, A., Guthke, R., Brakhage, A. A., and Figge, M. T. (2011). Automated image analysis of the host-pathogen interaction between phagocytes and Aspergillus fumigatus. PLoS ONE 6, e19591. doi:10.1371/journal.pone.0019591
Melanson, J. E., Chisholm, K. A., and Pinto, D. M. (2006). Targeted comparative proteomics by liquid chromatography/matrix-assisted laser desorption/ionization triple-quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 20, 904–910.
Mezger, M., Kneitz, S., Wozniok, I., Kurzai, O., Einsele, H., and Loeffler, J. (2008). Proinflammatory response of immature human dendritic cells is mediated by dectin-1 after exposure to Aspergillus fumigatus germ tubes. J. Infect. Dis. 197, 924–931.
Moalli, F., Doni, A., Deban, L., Zelante, T., Zagarella, S., Bottazzi, B., Romani, L., Mantovani, A., and Garlanda, C. (2010). Role of complement and fcgamma receptors in the protective activity of the long pentraxin PTX3 against Aspergillus fumigatus. Blood 116, 5170–5180.
Monteoliva, L., Martinez-Lopez, R., Pitarch, A., Hernaez, M. L., Serna, A., Nombela, C., Albar, J. P., and Gil, C. (2011). Quantitative proteome and acidic subproteome profiling of Candida albicans yeast-to-hypha transition. J. Proteome Res. 10, 502–517.
Moran, G. P., Coleman, D. C., and Sullivan, D. J. (2011). Comparative genomics and the evolution of pathogenicity in human pathogenic fungi. Eukaryotic Cell 10, 34–42.
Morton, C. O., Varga, J. J., Hornbach, A., Mezger, M., Sennefelder, H., Kneitz, S., Kurzai, O., Krappmann, S., Einsele, H., Nierman, W. C., Rogers, T. R., and Loeffler, J. (2011). The temporal dynamics of differential gene expression in Aspergillus fumigatus interacting with human immature dendritic cells in vitro. PLoS ONE 6, e16016. doi:10.1371/journal.pone.0016016
Moy, T. I., Conery, A. L., Larkins-Ford, J., Wu, G., Mazitschek, R., Casadei, G., Lewis, K., Carpenter, A. E., and Ausubel, F. M. (2009). High-throughput screen for novel antimicrobials using a whole animal infection model. ACS Chem. Biol. 4, 527–533.
Mukhtar, M. S., Carvunis, A.-R., Dreze, M., Epple, P., Steinbrenner, J., Moore, J., Tasan, M., Galli, M., Hao, T., Nishimura, M. T., Pevzner, S. J., Donovan, S. E., Ghamsari, L., Santhanam, B., Romero, V., Poulin, M. M., Gebreab, F., Gutierrez, B. J., Tam, S., Monachello, D., Boxem, M., Harbort, C. J., McDonald, N., Gai, L., Chen, H., He, Y., E. U. E. C., Vandenhaute, J., Roth, F. P., Hill, D. E., Ecker, J. R., Vidal, M., Beynon, J., Braun, P., and Dangl, J. L. (2011). Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 333, 596–601.
Murillo, L. A., Newport, G., Lan, C.-Y., Habelitz, S., Dungan, J., and Agabian, N. M. (2005). Genome-wide transcription profiling of the early phase of biofilm formation by Candida albicans. Eukaryotic Cell 4, 1562–1573.
Mylonakis, E. (2008). Galleria mellonella and the study of fungal pathogenesis: making the case for another genetically tractable model host. Mycopathologia 165, 1–3.
Netea, M. G., Brown, G. D., Kullberg, B. J., and Gow, N. A. R. (2008). An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 6, 67–78.
Neugebauer, U., Schmid, U., Baumann, K., Holzgrabe, U., Ziebuhr, W., Kozitskaya, S., Kiefer, W., Schmitt, M., and Popp, J. (2006). Characterization of bacterial growth and the influence of antibiotics by means of UV resonance Raman spectroscopy. Biopolymers 82, 306–311.
Nierman, W. C., Pain, A., Anderson, M. J., Wortman, J. R., Kim, H. S., Arroyo, J., Berriman, M., Abe, K., Archer, D. B., Bermejo, C., Bennett, J., Bowyer, P., Chen, D., Collins, M., Coulsen, R., Davies, R., Dyer, P. S., Farman, M., Fedorova, N., Fedorova, N., Feldblyum, T. V., Fischer, R., Fosker, N., Fraser, A., García, J. L., García, M. J., Goble, A., Goldman, G. H., Gomi, K., Griffith-Jones, S., Gwilliam, R., Haas, B., Haas, H., Harris, D., Horiuchi, H., Huang, J., Humphray, S., Jiménez, J., Keller, N., Khouri, H., Kitamoto, K., Kobayashi, T., Konzack, S., Kulkarni, R., Kumagai, T., Lafon, A., Lafton, A., Latgé, J.-P., Li, W., Lord, A., Lu, C., Majoros, W. H., May, G. S., Miller, B. L., Mohamoud, Y., Molina, M., Monod, M., Mouyna, I., Mulligan, S., Murphy, L., O’Neil, S., Paulsen, I., Peñalva, M. A., Pertea, M., Price, C., Pritchard, B. L., Quail, M. A., Rabbinowitsch, E., Rawlins, N., Rajandream, M.-A., Reichard, U., Renauld, H., Robson, G. D., de Córdoba, S. R., Rodríguez-Peña, J. M., Ronning, C. M., Rutter, S., Salzberg, S. L., Sanchez, M., Sánchez-Ferrero, J. C., Saunders, D., Seeger, K., Squares, R., Squares, S., Takeuchi, M., Tekaia, F., Turner, G., de Aldana, C. R. V., Weidman, J., White, O., Woodward, J., Yu, J. H., Fraser, C., Galagan, J. E., Asai, K., Machida, M., Hall, N., Barrell, B., and Denning, D. W. (2005). Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438, 1151–1156.
Niimi, M., Shepherd, M. G., and Monk, B. C. (1996). Differential profiles of soluble proteins during the initiation of morphogenesis in Candida albicans. Arch. Microbiol. 166, 260–268.
Nobile, C. J., Nett, J. E., Andes, D. R., and Mitchell, A. P. (2006). Function of Candida albicans adhesin Hwp1 in biofilm formation. Eukaryotic Cell 5, 1604–1610.
Nygaard, V., and Hovig, E. (2006). Options available for profiling small samples: a review of sample amplification technology when combined with microarray profiling. Nucleic Acids Res. 34, 996–1014.
Oberhardt, M. A., Puchalka, J., Martins dos Santos, V. A. P., and Papin, J. A. (2011). Reconciliation of genome-scale metabolic reconstructions for comparative systems analysis. PLoS Comput. Biol. 7, e1001116. doi:10.1371/journal.pcbi.1001116
O’Farrell, P. H. (1975). High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 250, 4007–4021.
Perlroth, J., Choi, B., and Spellberg, B. (2007). Nosocomial fungal infections: epidemiology, diagnosis, and treatment. Med. Mycol. 45, 321–346.
Pfaller, M. A., and Diekema, D. J. (2007). Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 20, 133–163.
Picazo, J. J., González-Romo, F., and Candel, F. J. (2008). Candidemia in the critically ill patient. Int. J. Antimicrob. Agents 32(Suppl. 2), S83–S85.
Pitarch, A., Sánchez, M., Nombela, C., and Gil, C. (2002). Sequential fractionation and two-dimensional gel analysis unravels the complexity of the dimorphic fungus Candida albicans cell wall proteome. Mol. Cell Proteomics 1, 967–982.
Pitkänen, E., Rousu, J., and Ukkonen, E. (2010). Computational methods for metabolic reconstruction. Curr. Opin. Biotechnol. 21, 70–77.
Priebe, S., Linde, J., Albrecht, D., Guthke, R., and Brakhage, A. A. (2011). FungiFun: a web-based application for functional categorization of fungal genes and proteins. Fungal Genet. Biol. 48, 353–358.
Raghunathan, A., Reed, J., Shin, S., Palsson, B., and Daefler, S. (2009). Constraint-based analysis of metabolic capacity of Salmonella typhimurium during host-pathogen interaction. BMC Syst. Biol. 3, 38. doi:10.1186/1752-0509-3-38
Rao, A., Kumar, M. K., Joseph, T., and Bulusu, G. (2010). Cerebral malaria: insights from host-parasite protein-protein interactions. Malar. J. 9, 155.
Raza, S., Robertson, K. A., Lacaze, P. A., Page, D., Enright, A. J., Ghazal, P., and Freeman, T. C. (2008). A logic-based diagram of signalling pathways central to macrophage activation. BMC Syst. Biol. 2, 36. doi:10.1186/1752-0509-2-36
Real, L. A., and Biek, R. (2007). Infectious disease modeling and the dynamics of transmission. Curr. Top. Microbiol. Immunol. 315, 33–49.
Rispail, N., Soanes, D. M., Ant, C., Czajkowski, R., Grünler, A., Huguet, R., Perez-Nadales, E., Poli, A., Sartorel, E., Valiante, V., Yang, M., Beffa, R., Brakhage, A. A., Gow, N. A. R., Kahmann, R., Lebrun, M.-H., Lenasi, H., Perez-Martin, J., Talbot, N. J., Wendland, J., and Di Pietro, A. (2009). Comparative genomics of MAP kinase and calcium-calcineurin signalling components in plant and human pathogenic fungi. Fungal Genet. Biol. 46, 287–298.
Rizzetto, L., and Cavalieri, D. (2011). Friend or foe: using systems biology to elucidate interactions between fungi and their hosts. Trends Microbiol. 19, 509–515.
Ross, R. (1911). The Prevention of Malaria, with Addendum on the Theory of Happenings. London: Murray.
Roux, A. V. D., and Aiello, A. E. (2005). Multilevel analysis of infectious diseases. J. Infect. Dis. 191(Suppl. 1), S25–S33.
Ruepp, A., Zollner, A., Maier, D., Albermann, K., Hani, J., Mokrejs, M., Tetko, I., Güldener, U., Mannhaupt, G., Münsterkötter, M., and Mewes, H. W. (2004). The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 32, 5539–5545.
Rupp, S. (2004). Proteomics on its way to study host-pathogen interaction in Candida albicans. Curr. Opin. Microbiol. 7, 330–335.
Ruppin, E., Papin, J. A., de Figueiredo, L. F., and Schuster, S. (2010). Metabolic reconstruction, constraint-based analysis and game theory to probe genome-scale metabolic networks. Curr. Opin. Biotechnol. 21, 502–510.
Saïd-Sadier, N., Padilla, E., Langsley, G., and Ojcius, D. M. (2010). Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS ONE 5, e10008. doi:10.1371/journal.pone.0010008
Salgado, P. S., Yan, R., Taylor, J. D., Burchell, L., Jones, R., Hoyer, L. L., Matthews, S. J., Simpson, P. J., and Cota, E. (2011). Structural basis for the broad specificity to host-cell ligands by the pathogenic fungus Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 108, 15775–15779.
Samaranayake, D. P., and Hanes, S. D. (2011). Milestones in Candida albicans gene manipulation. Fungal Genet. Biol. 48, 858–865.
Santamaría, R., Rizzetto, L., Bromley, M., Zelante, T., Lee, W., Cavalieri, D., Romani, L., Miller, B., Gut, I., Santos, M., Pierre, P., Bowyer, P., and Kapushesky, M. (2011). Systems biology of infectious diseases: a focus on fungal infections. Immunobiology 216, 1212–1227.
Schaller, M., Korting, H. C., Borelli, C., Hamm, G., and Hube, B. (2005). Candida albicans-secreted aspartic proteinases modify the epithelial cytokine response in an in vitro model of vaginal candidiasis. Infect. Immun. 73, 2758–2765.
Schrettl, M., Bignell, E., Kragl, C., Joechl, C., Rogers, T., Arst, H. N., Haynes, K., and Haas, H. (2004). Siderophore biosynthesis but not reductive iron assimilation is essential for Aspergillus fumigatus virulence. J. Exp. Med. 200, 1213–1219.
Schrettl, M., Kim, H. S., Eisendle, M., Kragl, C., Nierman, W. C., Heinekamp, T., Werner, E. R., Jacobsen, I., Illmer, P., Yi, H., Brakhage, A. A., and Haas, H. (2008). SreA-mediated iron regulation in Aspergillus fumigatus. Mol. Microbiol. 70, 27–43.
Shamir, L., Delaney, J. D., Orlov, N., Eckley, D. M., and Goldberg, I. G. (2010). Pattern recognition software and techniques for biological image analysis. PLoS Comput. Biol. 6, e1000974. doi:10.1371/journal.pcbi.1000974
Sharan, R., Ulitsky, I., and Shamir, R. (2007). Network-based prediction of protein function. Mol. Syst. Biol. 3, 88.
Shin, Y.-K., Kim, K.-Y., and Paik, Y.-K. (2005). Alterations of protein expression in macrophages in response to Candida albicans infection. Mol. Cells 20, 271–279.
Skrabanek, L., Saini, H. K., Bader, G. D., and Enright, A. J. (2008). Computational prediction of protein-protein interactions. Mol. Biotechnol. 38, 1–17.
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P.-L., and Ideker, T. (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432.
Sorgo, A. G., Heilmann, C. J., Dekker, H. L., Bekker, M., Brul, S., de Koster, C. G., de Koning, L. J., and Klis, F. M. (2011). Effects of fluconazole on the secretome, the wall proteome, and wall integrity of the clinical fungus Candida albicans. Eukaryotic Cell 10, 1071–1081.
Sosinska, G. J., de Groot, P. W. J., de Mattos, M. J. T., Dekker, H. L., de Koster, C. G., Hellingwerf, K. J., and Klis, F. M. (2008). Hypoxic conditions and iron restriction affect the cell-wall proteome of Candida albicans grown under vagina-simulative conditions. Microbiology 154(Pt 2), 510–520.
Sosinska, G. J., de Koning, L. J., de Groot, P. W. J., Manders, E. M. M., Dekker, H. L., Hellingwerf, K. J., de Koster, C. G., and Klis, F. M. (2011). Mass spectrometric quantification of the adaptations in the wall proteome of Candida albicans in response to ambient pH. Microbiology 157(Pt 1), 136–146.
Stoldt, V. R., Sonneborn, A., Leuker, C. E., and Ernst, J. F. (1997). Efg1p, an essential regulator of morphogenesis of the human pathogen Candida albicans, is a member of a conserved class of bHLH proteins regulating morphogenetic processes in fungi. EMBO J. 16, 1982–1991.
Stolovitzky, G. (2011). “Wisdom of crowds for gene network inference,” in 12th International Conference on Systems Biology, August 28 to September 1st, 2011, Heidelberg.
Strömbäck, L., and Lambrix, P. (2005). Representations of molecular pathways: an evaluation of SBML, PSI MI and BioPAX. Bioinformatics 21, 4401–4407.
Sugui, J. A., Kim, H. S., Zarember, K. A., Chang, Y. C., Gallin, J. I., Nierman, W. C., and Kwon-Chung, K. J. (2008). Genes differentially expressed in conidia and hyphae of Aspergillus fumigatus upon exposure to human neutrophils. PLoS ONE 3, e2655. doi:10.1371/journal.pone.0002655
Sysko, L. R., and Davis, M. A. (2010). From image to data using common image-processing techniques. Curr. Protoc. Cytom. Chapter 12, Unit 12.21.
Szabo, E. K., and MacCallum, D. M. (2011). The contribution of mouse models to our understanding of systemic candidiasis. FEMS Microbiol. Lett. 320, 1–8.
Taylor, C. F., Field, D., Sansone, S.-A., Aerts, J., Apweiler, R., Ashburner, M., Ball, C. A., Binz, P.-A., Bogue, M., Booth, T., Brazma, A., Brinkman, R. R., Clark, A. M., Deutsch, E. W., Fiehn, O., Fostel, J., Ghazal, P., Gibson, F., Gray, T., Grimes, G., Hancock, J. M., Hardy, N. W., Hermjakob, H., Julian, R. K., Kane, M., Kettner, C., Kinsinger, C., Kolker, E., Kuiper, M., Novère, N. L., Leebens-Mack, J., Lewis, S. E., Lord, P., Mallon, A.-M., Marthandan, N., Masuya, H., McNally, R., Mehrle, A., Morrison, N., Orchard, S., Quackenbush, J., Reecy, J. M., Robertson, D. G., Rocca-Serra, P., Rodriguez, H., Rosenfelder, H., Santoyo-Lopez, J., Scheuermann, R. H., Schober, D., Smith, B., Snape, J., Stoeckert, C. J., Tipton, K., Sterk, P., Untergasser, A., Vandesompele, J., and Wiemann, S. (2008). Promoting coherent minimum reporting guidelines for biological and biomedical investigations: the MIBBI project. Nat. Biotechnol. 26, 889–896.
Teutschbein, J., Albrecht, D., Pötsch, M., Guthke, R., Aimanianda, V., Clavaud, C., Latgé, J.-P., Brakhage, A. A., and Kniemeyer, O. (2010). Proteome profiling and functional classification of intracellular proteins from conidia of the human-pathogenic mold Aspergillus fumigatus. J. Proteome Res. 9, 3427–3442.
Theiss, S., Köhler, G. A., Kretschmar, M., Nichterlein, T., and Hacker, J. (2002). New molecular methods to study gene functions in Candida infections. Mycoses 45, 345–350.
Thomas, D. P., Bachmann, S. P., and Lopez-Ribot, J. L. (2006). Proteomics for the analysis of the Candida albicans biofilm lifestyle. Proteomics 6, 5795–5804.
Thywißen, A., Heinekamp, T., Dahse, H.-M., Schmaler-Ripcke, J., Nietzsche, S., Zipfel, P. F., and Brakhage, A. A. (2011). Conidial dihydroxynaphthalene melanin of the human pathogenic fungus Aspergillus fumigatus interferes with the host endocytosis pathway. Front. Microbiol. 2, 96. doi:10.3389/fmicb.2011.00096
Tierney, L., Linde, J., Müller, S., Brunke, S., Molina, J., Hube, B., Schöck, U., Guthke, R., and Kuchler, K. (2012). An interspecies regulatory network inferred from simultaneous RNA-seq of Candida albicans invading innate immune cells. Front. Microbiol. 3, 85. doi:10.3389/fmicb.2012.00085
Toepfer, S., Guthke, R., Driesch, D., Woetzel, D., and Pfaff, M. (2007). The NetGenerator algorithm: reconstruction of gene regulatory networks. Lect. Notes Bioinform. 4366, 119–130.
Tong, J. C., and Ng, L. F. P. (2011). Understanding infectious agents from an in silico perspective. Drug Discov. Today 16, 42–49.
Tuckwell, D., Denning, D. W., and Bowyer, P. (2011). A public resource for metabolic pathway mapping of Aspergillus fumigatus Af293. Med. Mycol. 49(Suppl 1), S114–S119.
Urban, C. F., Reichard, U., Brinkmann, V., and Zychlinsky, A. (2006). Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 8, 668–676.
Van Dyke Parunak, H., Savit, R., and Riolo, R. (1998). “Agent-based modeling vs. equation-based modeling: a case study and users’ guide,” in Multi-Agent Systems and Agent-Based Simulation, eds J. Sichman, R. Conte, and N. Gilbert Lecture Notes in Computer Science, Vol. 1534 (Berlin: Springer), 277–283.
van het Hoog, M., Rast, T. J., Martchenko, M., Grindle, S., Dignard, D., Hogues, H., Cuomo, C., Berriman, M., Scherer, S., Magee, B. B., Whiteway, M., Chibana, H., Nantel, A., and Magee, P. T. (2007). Assembly of the Candida albicans genome into sixteen supercontigs aligned on the eight chromosomes. Genome Biol. 8, R52.
Vera, J., Rateitschak, K., Lange, F., Kossow, C., Wolkenhauer, O., and Jaster, R. (2011). Systems biology of JAK-STAT signalling in human malignancies. Prog. Biophys. Mol. Biol. 106, 426–434.
Vödisch, M., Albrecht, D., Lessing, F., Schmidt, A. D., Winkler, R., Guthke, R., Brakhage, A. A., and Kniemeyer, O. (2009). Two-dimensional proteome reference maps for the human pathogenic filamentous fungus Aspergillus fumigatus. Proteomics 9, 1407–1415.
Vödisch, M., Scherlach, K., Winkler, R., Hertweck, C., Braun, H.-P., Roth, M., Haas, H., Werner, E. R., Brakhage, A. A., and Kniemeyer, O. (2011). Analysis of the Aspergillus fumigatus proteome reveals metabolic changes and the activation of the pseurotin A biosynthesis gene cluster in response to hypoxia. J. Proteome Res. 10, 2508–2524.
Vodovotz, Y., Csete, M., Bartels, J., Chang, S., and An, G. (2008). Translational systems biology of inflammation. PLoS Comput. Biol. 4, e1000014. doi:10.1371/journal.pcbi.1000014
Volling, K., Thywissen, A., Brakhage, A. A., and Saluz, H. P. (2011). Phagocytosis of melanized Aspergillus conidia by macrophages exerts cytoprotective effects by sustained PI3K/Akt signalling. Cell. Microbiol. 13, 1130–1148.
Walker, D. C., and Southgate, J. (2009). The virtual cell – a candidate co-ordinator for ‘middle-out’ modelling of biological systems. Brief. Bioinformatics 10, 450–461.
Waltermann, C., and Klipp, E. (2010). Signal integration in budding yeast. Biochem. Soc. Trans. 38, 1257–1264.
Walther, M., Gebhardt, P., Grosse-Gehling, P., Würbach, L., Irmler, I., Preusche, S., Khalid, M., Opfermann, T., Kamradt, T., Steinbach, J., and Saluz, H.-P. (2011). Implementation of 89Zr production and in vivo imaging of B-cells in mice with 89Zr-labeled anti-B-cell antibodies by small animal PET/CT. Appl. Radiat. Isot. 69, 852–857.
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63.
Wartenberg, D., Lapp, K., Jacobsen, I. D., Dahse, H.-M., Kniemeyer, O., Heinekamp, T., and Brakhage, A. A. (2011). Secretome analysis of Aspergillus fumigatus reveals Asp-hemolysin as a major secreted protein. Int. J. Med. Microbiol. 301, 602–611.
Whiteway, M., and Bachewich, C. (2007). Morphogenesis in Candida albicans. Annu. Rev. Microbiol. 61, 529–553.
Willard, H., and Ginsburg, G. (2009). Genomic and Personalized Medicine. Number Vol. 2 in Genomic and Personalized Medicine. Amsterdam: Academic Press.
Willger, S. D., Puttikamonkul, S., Kim, K.-H., Burritt, J. B., Grahl, N., Metzler, L. J., Barbuch, R., Bard, M., Lawrence, C. B., and Cramer, R. A. (2008). A sterol-regulatory element binding protein is required for cell polarity, hypoxia adaptation, azole drug resistance, and virulence in Aspergillus fumigatus. PLoS Pathog. 4, e1000200. doi:10.1371/journal.ppat.1000200
Wilson, D., Thewes, S., Zakikhany, K., Fradin, C., Albrecht, A., Almeida, R., Brunke, S., Grosse, K., Martin, R., Mayer, F., Leonhardt, I., Schild, L., Seider, K., Skibbe, M., Slesiona, S., Waechtler, B., Jacobsen, I., and Hube, B. (2009). Identifying infection-associated genes of Candida albicans in the postgenomic era. FEMS Yeast Res. 9, 688–700.
Wisplinghoff, H., Bischoff, T., Tallent, S. M., Seifert, H., Wenzel, R. P., and Edmond, M. B. (2004). Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 39, 309–317.
Woolhouse, M. E., Taylor, L. H., and Haydon, D. T. (2001). Population biology of multihost pathogens. Science 292, 1109–1112.
Xu, J., and Li, Y. (2006). Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics 22, 2800–2805.
Yan, L., Zhang, J.-D., Cao, Y.-B., Gao, P.-H., and Jiang, Y.-Y. (2007). Proteomic analysis reveals a metabolism shift in a laboratory fluconazole-resistant Candida albicans strain. J. Proteome Res. 6, 2248–2256.
Yin, Z., Stead, D., Walker, J., Selway, L., Smith, D. A., Brown, A. J. P., and Quinn, J. (2009). A proteomic analysis of the salt, cadmium and peroxide stress responses in Candida albicans and the role of the Hog1 stress-activated MAPK in regulating the stress-induced proteome. Proteomics 9, 4686–4703.
Zhou, D., and He, Y. (2008). Extracting interactions between proteins from the literature. J. Biomed. Inform. 41, 393–407.
Keywords: Aspergillus fumigatus, Candida albicans, gene-regulatory network, network modeling, pathogenicity, pathogen-host interaction, spatio-temporal modeling, systems biology
Citation: Horn F, Heinekamp T, Kniemeyer O, Pollmächer J, Valiante V and Brakhage AA (2012) Systems biology of fungal infection. Front. Microbio. 3:108. doi: 10.3389/fmicb.2012.00108
Received: 12 December 2011; Accepted: 05 March 2012;
Published online: 02 April 2012.
Edited by:
Jörg Linde, Leibniz Institute for Natural Product Research and Infection Biology – Hans Knöll Institute, GermanyReviewed by:
Rene Alvarez, Naval Medical Research Unit-San Antonio, USAGuoku Hu, Creighton University, USA
Scott G. Filler, Los Angeles Biomedical Research Institute, USA
Copyright: © 2012 Horn, Heinekamp, Kniemeyer, Pollmächer, Valiante and Brakhage. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Axel A. Brakhage, Molecular and Applied Microbiology, Leibniz Institute for Natural Product Research and Infection Biology – Hans Knöll Institute, Beutenbergstrasse 11a, 07745 Jena, Germany. e-mail:axel.brakhage@hki-jena.de