 Heath J. Mills1* Brandi Kiel Reese1,2 Alicia K. Shepard3 Natascha Riedinger4 Scot E. Dowd5 Yuki Morono6 Fumio Inagaki6
Heath J. Mills1* Brandi Kiel Reese1,2 Alicia K. Shepard3 Natascha Riedinger4 Scot E. Dowd5 Yuki Morono6 Fumio Inagaki6
- 1 Department of Oceanography, Texas A&M University, College Station, TX, USA
- 2 Department of Biological Sciences, University of Southern California, Los Angeles, CA, USA
- 3 Department of Marine Biology, Texas A&M University at Galveston, Galveston, TX, USA
- 4 Department of Earth Sciences, University of California, Riverside, CA, USA
- 5 Research and Testing Laboratory, Medical Biofilm Research Institute, Lubbock, TX, USA
- 6 Geomicrobiology Group, Kochi Institute for Core Sample Research, Japan Agency for Marine-Earth Science and Technology, Kochi, Japan
A remarkable number of microbial cells have been enumerated within subseafloor sediments, suggesting a biological impact on geochemical processes in the subseafloor habitat. However, the metabolically active fraction of these populations is largely uncharacterized. In this study, an RNA-based molecular approach was used to determine the diversity and community structure of metabolically active bacterial populations in the upper sedimentary formation of the Nankai Trough seismogenic zone. Samples used in this study were collected from the slope apron sediment overlying the accretionary prism at Site C0004 during the Integrated Ocean Drilling Program Expedition 316. The sediments represented microbial habitats above, within, and below the sulfate–methane transition zone (SMTZ), which was observed approximately 20 m below the seafloor (mbsf). Small subunit ribosomal RNA were extracted, quantified, amplified, and sequenced using high-throughput 454 pyrosequencing, indicating the occurrence of metabolically active bacterial populations to a depth of 57 mbsf. Transcript abundance and bacterial diversity decreased with increasing depth. The two communities below the SMTZ were similar at the phylum level, however only a 24% overlap was observed at the genus level. Active bacterial community composition was not confined to geochemically predicted redox stratification despite the deepest sample being more than 50 m below the oxic/anoxic interface. Genus-level classification suggested that the metabolically active subseafloor bacterial populations had similarities to previously cultured organisms. This allowed predictions of physiological potential, expanding understanding of the subseafloor microbial ecosystem. Unique community structures suggest very diverse active populations compared to previous DNA-based diversity estimates, providing more support for enhancing community characterizations using more advanced sequencing techniques.
Introduction
Microbial populations in subseafloor sediments on the global continental margins account for one tenth to one third of the total biomass on Earth (Whitman et al., 1998; Parkes et al., 2000; Lipp et al., 2008). Although microbial populations have been detected ubiquitously in the marine subseafloor environment, the diversity, activity, metabolic processes, and interactions with geochemistry are still largely unknown (D’Hondt et al., 2007; Bach et al., 2010). Microbial processes in the marine subsurface are potentially significant to global carbon and nutrient cycles (Whiticar, 1999; D’Hondt et al., 2002) and provide relevant analogs to the emerging astrobiology field (Gold, 1992; Chapelle et al., 2002). To better understand the subseafloor biosphere, the Integrated Ocean Drilling Program (IODP) has made microbiology and biogeochemistry a prominent initiative on recent drilling expeditions recognizing the broad scientific benefit (Bickle et al., 2011). Microbiological samples were collected during the IODP Nankai Trough Seismogenic Zone Experiment (NanTroSEIZE) Expedition 316. The expedition provided an opportunity to advance the understanding of the subseafloor biosphere in an active crustal seismogenic zone characterized by large-scale earthquake- and tsunami-genesis (Kimura et al., 2008).
The sulfate–methane transition zone (SMTZ) represents a distinct geochemical demarcation within the marine subseafloor (Iversen and Jørgensen, 1985; D’Hondt et al., 2002). Geochemistry predicts a thermodynamic driven stratification of the associated microbial community structure within this zone (D’Hondt et al., 2004; Parkes et al., 2005; Inagaki et al., 2006). While some microbial processes associated with the SMTZ have been previously examined (Parkes et al., 2005; Biddle et al., 2006), recent findings suggest that microbial activity may not be as restricted as predicted by redox potential (Orcutt et al., 2011). Geochemical descriptions of microbial activity rely on the consumption or production of measurable compounds. Transformations of such compounds may be undetectable using standard geochemical analysis if the end products of the reactions are consumed by a secondary reaction. For example, sulfur-related cryptic cycles have been previously noted in the oxygen minimum zone off the Chilean coast where sulfur is being reduced but rapidly re-oxidized leaving no geochemical marker of the reduction pathway (Canfield et al., 2010). The active reduction pathway was determined by molecular analysis active sulfate reducing bacteria. Therefore, molecular characterizations of metabolically active microbial populations is necessary to determine these geochemically cryptic cycles and provide a better understanding of the subsurface biosphere.
Previous analyses of subseafloor microbial populations have targeted ribosomal RNA genes (DNA-based) to describe the total microbial community, which includes metabolically active populations as well as potentially dormant and/or dead cells (e.g., Newberry et al., 2004; Inagaki et al., 2006; Polymenakou et al., 2009). DNA-based studies reported few shifts in microbial population structure associated with varying geochemical conditions (Parkes et al., 2005). Metagenomic analyses have observed shifts with depth in gene abundance associated with sulfur metabolism but lacked variability in genes associated with methanogenesis despite a pronounced SMTZ (Biddle et al., 2008). DNA-based analysis of microbial communities within the subseafloor, as in other environments, may detect dormant or extremely low metabolically active populations in the natural environment and thus may not correspond with observed geochemistry (Davis et al., 1986).
In contrast, the ribosomal RNA transcripts (RNA-based) can be used to describe the metabolically active community, providing a better link to the geochemistry. Detection of rRNA transcripts have been correlated to reproducing cells (Neidhardt and Magasanik, 1960) as they rapidly degrade upon cell dormancy or death (Davis et al., 1986). Two previous molecular studies analyzed metabolically active Archaea using cloning and Sanger sequencing of RNA targets (Biddle et al., 2006; Sørensen and Teske, 2006), however a detailed description of the active bacterial population using more robust techniques is lacking. Recent advances in pyrosequencing technologies have increased the accuracy and decreased the associated costs, providing access to the depth of sequencing required to more adequately sample environmental microbial diversity compared to previous methods, such as cloning (Edwards et al., 2006; Sogin et al., 2006; Liu et al., 2008). The authors recognize that this method does not eliminate PCR amplification biases common with cloning and Sanger based sequencing. This study is the first report to utilize high-throughput sequencing of RNA targets from the marine subseafloor providing a novel analysis of the metabolically active and ecologically relevant bacterial community structure and function.
This study advances current understanding of subseafloor microbial communities by characterizing the metabolically active bacterial populations surrounding and within the SMTZ in the Nankai Trough subseafloor sediments. Structural diversity of the active community was used to interpret potential metabolic function and was compared to measured geochemical concentrations. We hypothesized that the functional diversity of subseafloor microbial populations will exceed the observed geochemically predicted zones as some carbon and nutrient measured below detection limits. Therefore, RNA characterizations of microbial communities will provide a more informative description of environmental microbial ecology than DNA or geochemical-based methods alone.
Materials and Methods
Site and Sample Description
Sediment samples were obtained during IODP Expedition 316 from Site C0004 in the Nankai Trough, located approximately 63 nautical miles southeast from the city of Shingu off the Kii Peninsula of Japan (see Kinoshita et al., 2009 for IODP Expedition 316 Site Map). A detailed geological description of this area is available as part of the IODP site description report (Kimura et al., 2008). Site C0004 Hole C (33°13.0′N, 136°43.0′E) was drilled on December 20, 2008 using the hydraulic piston coring system. Core sediments were processed less than 1 h after core recovery (Kimura et al., 2008; Strasser et al., 2009, 2011). Whole round cores were sectioned, immediately stored at −80°C for preservation of both RNA and DNA, and were shipped to the Mills laboratory on dry ice. Samples from three different depths of 1, 19, and 57 m below sea floor (mbsf) were selected for RNA and geochemical analysis surrounding and within the SMTZ.
Geochemical Analyses
Standard shipboard operations obtained concentrations of methane, sulfate, and iron (Fe2+; Kimura et al., 2008). To assess the amount of iron sulfide minerals in these sediments, chromium reducible sulfur (CRS = FeS2, S0 and remaining part of Fe3S4) concentrations were determined by treating samples of approximately 0.5 g with the two-step acid Cr (II) method (Fossing and Jørgensen, 1989). Trapped sulfide was analyzed by the methylene blue method (Cline, 1969).
Cell Counts
Cells were stained with SYBR Green I and then enumerated by fluorescent image-based cell count technique (Morono et al., 2009) using an automated slide-loader system (Morono et al., 2009; Morono and Inagaki, 2010).
Nucleic Acid Extraction and Reverse Transcription-PCR
Microbial SSU ribosomal RNA (rRNA) was extracted from approximately 0.5 g of sediment from the center of a whole round core using the Mills Extraction Method as described in Mills et al. (2008). Extraction controls containing no sediment were performed simultaneously with environmental samples to confirm lack of contamination during the extraction process.
All incubation steps during the reverse transcription (RT)-PCR were conducted on a Veriti 96-well Fast Thermal Cycling System (Applied Biosystems, Foster City, CA, USA). Ribosomal RNA was reverse transcribed to cDNA using the Moloney murine leukemia virus (M-MLV) reverse transcriptase and Bacteria domain specific Small subunit ribosomal RNA (SSU rRNA) reverse primer 518R (Nogales et al., 1999). RNA was incubated at 65°C for 5 min followed by an incubation of both RNA extract and primer at 70°C for 5 min. The total RT-PCR reaction mixture [1X M-MLV buffer containing 50 mM Tris–HCl, 3 mM MgCl2, 75 mM KCl, and 10 mM DTT (Promega, USA), 10 mM deoxynucleotide triphosphate solution mix (New England Biolabs, USA), 1 U of M-MLV enzyme (Promega, USA), and molecular grade water] was incubated at 37°C for 60 min. PCR amplification of cDNA used Bacteria domain specific SSU rRNA forward primer 27F (Giovannoni, 1991) and reverse primer 518R (Nogales et al., 1999). This fragment of the 16S rRNA gene includes the hypervariable V3 region. The reaction mixture was as follows: 1× buffer [10 mM KCl, 10 mM (NH4)2 SO4, 20 mM Tris–HCl, 2 mM Mg SO4, 0.1% Triton X-100; New England Biolabs, USA], 10 mM deoxynucleotide triphosphate solution mix (New England Biolabs, USA), 1 U of Taq DNA Polymerase (New England Biolabs, USA), and molecular grade water. Thermal cycling conditions were 95°C for 5 min, 40 cycles of 95°C for 30 s, 50°C for 30 s, and 72°C for 30 s with a final extension of 72°C for 10 min. Amplicons were visualized by gel electrophoresis on 0.7% agarose gels, stained with ethidium bromide, and illuminated by UV exposure. DNA contamination of RNA extracts was determined by omitting the RT step. No amplifiable DNA remained in RNA extracts. Standard negative controls for the RT and PCR steps were also incorporated, both indicated no contamination of the reactions.
Quantitative RT-PCR
To quantify the bacterial SSU rRNA transcripts, a series of standards were amplified from purified pure culture bacterial SSU rRNA gene amplicons. The PCR product was obtained using primers 331F and 797R (Nadkarni et al., 2002). Amplicon size was confirmed by gel electrophoresis. PCR purification was completed using the QIAquick PCR purification kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. The concentration of standards was determined on a NanoDrop 1000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA). Copy number was calculated assuming a molecular mass of 660 Da for a base pair of DNA and using the following formula: Copy Number = [6.023 × 1023 (bp mol−1 bp−1) × concentration of standard (ng μL−1)]/[PCR Product Size (bp gene copy−1) × 1 × 109 (ng g−1) × 660 (g mol−1 bp−1)] (Mattes and Jin, 2010). Standard concentrations ranged over five orders of magnitude from 2.6 to 2.6 × 104 copies μL−1 and were amplified in triplicate during Q-PCR. Environmental samples and extract controls were reverse transcribed as described above using the 797R primer and was Q-PCR amplified from two dilutions, 1× and 1/100× of the reverse transcribed product, in duplicate. All samples were amplified using the primers 331F and 797R and TaqMan probe BacTaq (Nadkarni et al., 2002). Manufacturers suggested Q-PCR reaction mixes were used with thermal cycling conditions as follows: initial warming step at 50°C for 2 min, a denaturation step at 95°C for 10 min followed by 40 cycles of 95°C for 30 s, 52°C for 1 min, 65°C for 30 s, and a final extension step at 65°C for 6 min. The quantification and data analysis was conducted using a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA1) and the StepOne Software 2.0 (Applied Biosystems, Foster City, CA, USA). Baseline and Ct values were automatically selected by the provided software and visually verified by the user.
Pyrosequencing and Sequence Analysis
Sequences were obtained from two dilutions of cDNA reverse transcribed from a single RNA extract. The dilutions were sequenced as described below with the libraries combined after the sequences were quality checked. Pyrosequencing, including initial amplification of the cDNA with primers containing unique sequence identifiers, was conducted at the Medical Biofilm Research Institute (Research and Testing Laboratory, Lubbock, TX, USA) according to standard laboratory procedures using a 454 FLX Sequencer (454 Life Sciences, Branford, CT, USA). Pyrosequencing conditions are routinely optimized by the Research and Testing Laboratory to limit multiple strand attachment. Long homopolymer regions can be misread by 454 (Jones, 2010), however such misreads are less problematic for SSU rRNA analysis. Resulting sequences were first de-noised and then individual sequences were parsed the into sample specific libraries. Libraries were screened for reads less than 200 bases, reads lacking a Roche-designed four base key sequence, and non-bacterial reads lacking specific 28F primer recognition site. Chimera detection was completed using the Chimera Slayer system adapted for the Mothur Program (Schloss et al., 2009; Haas et al., 2011). Sequences were uploaded to the National Center for Biotechnology Information Sequence Read Archive (NCBI-SRA) under accession number SRA049352.
Phylogenetic analyses were conducted on the combined sequence data sets to reduce the potential for RT-PCR and pyrosequencing biases. Sequences were aligned using the Ribosomal Database Project (RDP) Pyrosequencing Aligner tool, which is based on Infernal aligner (Nawrocki and Eddy, 2007). Sequences were clustered at a 95% sequence similarity using farthest-neighbor method on the RDP Complete Linkage Clustering program available on the RDP pyrosequencing pipeline2 (Cole et al., 2009) and verified using the Mothur Program3 (Schloss et al., 2009). Taxonomic classification of the sequences was completed using the NET Network Distributed Basic Local Alignment Search Toolkit (W.ND-BLAST) program (Dowd et al., 2005) and randomly checked against the Basic Local Alignment Search Tool (BLAST) pipeline through National Center for Biotechnology Information (NCBI) database. The highest classification level with a confidence interval greater than 80% was used in downstream analysis. Genera identified by more then 1% of the sequence data set at a given depth were used in community functional group characterization.
Diversity indices including Shannon, Chao1, and rarefaction were calculated from the 95% similarity operational taxonomic units (OTU) defined by the RDP clustering at. Comparison of OTUs that were absent or present between samples was completed using the shared OTU and Venn diagram systems in the mothur program (Schloss et al., 2009).
The authors acknowledge and have taken steps to limit potential for bias associated with extraction, amplification, and sequencing of SSU rRNA from marine sediments. The authors recognize that the use of one primer set may reduce the detection of overall diversity. Multiple sediment samples at each depth were combined to limit effects of community and sediment chemistry heterogeneity. PCR and Q-PCR cycling conditions were optimized for the specific reagents to reduce effects of mis-priming, primer–dimer formation, unequal amplification efficiency of template, and the potential formation of chimeric sequence formation. In addition, amplicons were obtained from two dilutions of cDNA and combined according to depth reduce the potential for RT-PCR biases. Despite the biases inherent with molecular studies, the steps taken to reduce the effect of these biases have produced a data set that is comparable to previous studies and is of sufficient quality for future comparisons.
Statistical Analysis
Geochemical data was correlated to phylogenetic profiles through Singular value decomposition (SVD) and principal component analysis (PCA) using Microsoft Excel Pop Tools. Geochemical and phylogenetic data was first transformed using either chi-square or arcsine statistical tests, where appropriate, and normalized prior to SVD and PCA analyses. Statistical significance (p < 0.01) was determined using the Monte Carlo significance test with 10,000 iterations.
Results
Site Description
Site C0004 Hole C was located on the slope of the accretionary prism in the Nankai Trough, over an active megasplay fault that has repeatedly caused episodic earthquakes and tsunamis (Strasser et al., 2009). Water depth at Site C0004 Hole C was 2,627 m. A total of 403 m of sediment core was retrieved during IODP Expedition 316. In this study, sediment samples were selected from above, within, and below the SMTZ (1, 19, and 57 mbsf, respectively) in the upper sedimentary unit of the slope apron overlying the accretionary prism (i.e., Lithologic Unit I, 0–78.08 mbsf). The sediment samples are mainly composed of nannofossil-rich hemi-pelagic mud (Strasser et al., 2009).
Geochemical Profiles
Pore water geochemical characteristics for sediments at Site C0004 were determined onboard during IODP Expedition 316 (Kimura et al., 2008). Sulfate and methane concentration profiles showed that the SMTZ was located between 18.5 and 22.9 mbsf (Figure 1). The sulfate concentration was 25.3 mM at 2.7 mbsf and decreased with increasing depth down to below detection limits at 22.9 mbsf (Figure 1). Methane concentrations increased three orders of magnitude from an initial 1.8 × 10−3 mM at 1.4 mbsf to 1.4 mM at 18.5 mbsf and continued to increase to a maximum concentration of 9.7 mM at 35.3 mbsf (Figure 1). The Fe (II) concentration was 2.5 μM at 1 mbsf but then remained less than 1.0 μM until approximately 48.6 mbsf where it began to increase to a concentration of 4.7 μM at 59.5 mbsf (Figure 1). The amount of iron sulfide phases (determined by CRS) increased from 78.1 μmol g−1 at 2.8 mbsf and to a maximum of 428.4 μmol g−1 at 29.5 mbsf. Below this depth the concentration of iron sulfides decreases consistently down to 99.4 μmol g−1 at approximately 59.5 mbsf (Figure 1).
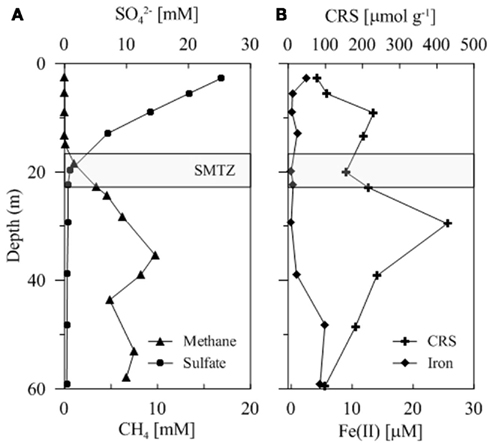
Figure 1. (A) Concentration of  in millimolar and CH4 in millimolar with depth to 60 mbsf. (B) Concentration of
in millimolar and CH4 in millimolar with depth to 60 mbsf. (B) Concentration of  (μM) and iron sulfides, determined as chromium reducible sulfur (CRS; μmol g−1), with depth to 60 mbsf.
(μM) and iron sulfides, determined as chromium reducible sulfur (CRS; μmol g−1), with depth to 60 mbsf.
Cell Abundance and Quantification of Bacterial SSU RRNA
Cell abundance in sediment at 1 mbsf was 3.4 × 107 cells cm−3 and decreased approximately two orders of magnitude to 1.3 × 105 cells cm−3 at both the 19- and 57-mbsf sampling depths. The trend of cell concentration matched the yield of SSU rRNA gene transcript. Quantitative real-time PCR analyses showed that the bacterial SSU cDNA reverse transcribed from Hole C0004C sediment RNA extracts were estimated to be 6.5 × 106 copies g−1 in the 1.0-mbsf sample and 3.3 × 103 copies g−1 at 19 mbsf and 9.0 × 104 copies g−1 at 57 mbsf. The ratio of transcripts per cell increases at the lowest depth from 0.19 at 1 mbsf to 0.03 at 19 mbsf to 0.69 at 57 mbsf.
Bacterial Community Structure
Pyrosequencing of reverse transcribed bacterial SSU rRNA gene transcripts from the metabolically active fraction of the total microbial community resulted in a total of 12,020 sequences with an average length of 426 base pairs (Table 1). Two different species diversity indices were calculated from the pyrosequencing data. For sample depths 1, 19, and 57 mbsf, Shannon diversity index calculated values of 6.32, 3.02, and 3.13 and Chao1 index calculated values of 2,638, 67, 90, respectively (Table 1). The rarefaction curve generated for the 1-mbsf sample did not indicate exhaustive sampling of the sequence data set whereas the rarefaction curves generated from the 19- and 57-mbsf libraries indicated that sequencing efforts sufficiently represented the detectable diversity of the community based on the methods used in this study (Figure 2). Comparison of OTUs observed between those depths showed that 1.3% of OTUs were shared between 1 and 19 mbsf, 1.2% of OTUs were shared between 1 and 57 mbsf, and 23.6% of OTUs were shared between 19 and 57 mbsf (Figure 3).
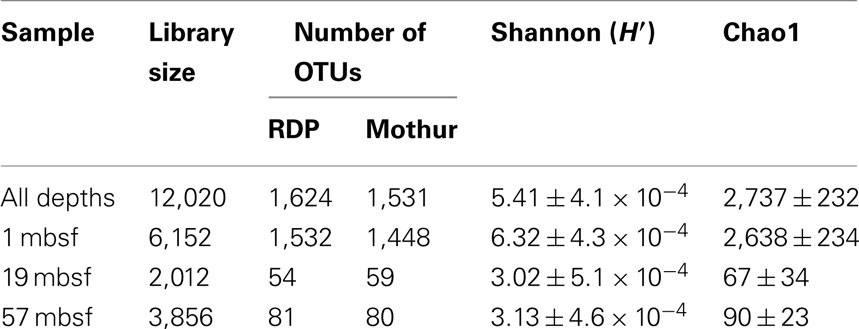
Table 1. Summary of sequence analysis data Nankai Site C0004 Hole C.
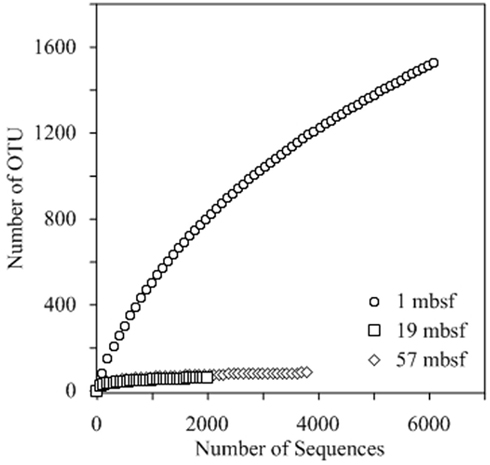
Figure 2. Rarefaction analysis based on pyrosequencing of community bacterial SSU rRNA from 1, 20, and 57mbsf. Sequences with 95% sequence similarity were combined as a single OTU. These data indicate that the sampling effort at 19 and 57 mbsf adequately represents the phylogenetic diversity of the population. Although additional sequencing would be required to fully annotate the entire diversity of the bacterial population at 1 mbsf, sufficient data have been collected to make conclusions regarding the more frequently detected lineages.
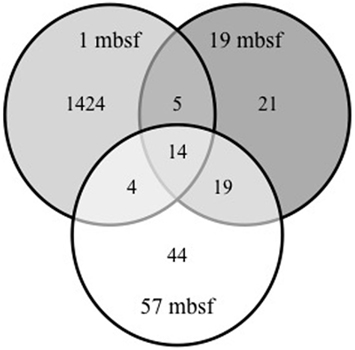
Figure 3. Venn diagram comparing OTUs shared by and unique to the 1, 19, and 57mbsf sampling depths. Sequences with 95% sequence similarity were combined as a single OTU. These data indicate that the bacterial diversity at 1 mbsf is greater than at 19 and 57 msbf. Although the abundance of OTUs at 19 and 57 mbsf is similar only 24% are shared between depths.
Phylogenetic analysis detected 18 unique phyla (Figure 4). At all depths sampled, Proteobacteria had the highest sequence abundance (68.4, 49.8, and 58.6% of total sequences obtained from 1, 19, and 57 mbsf, respectively). Additional lineages detected and their associated sequence detection frequency at 1 mbsf included Chloroflexi (10.3%), Cyanobacteria (5.3%), Bacteroidetes (3.8%), Deferribacteres (3.0%), Actinobacteria (2.0%), and Firmicutes (1.7%; Figure 4). A shift in community composition occurred between the 1 mbsf and the 19 and 57 mbsf depth ranges.
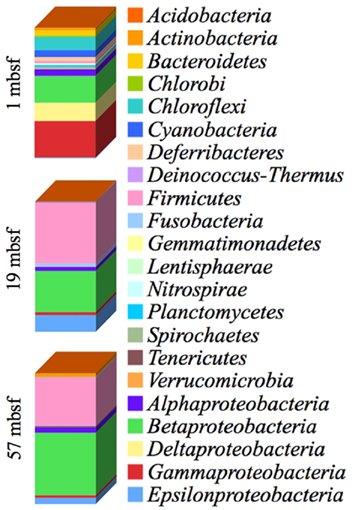
Figure 4. Combined bacterial diversity at the phylum level based on small subunit ribosomal (SSU rRNA) at 1, 19, and 57 m below the seafloor (mbsf) from PCR amplicons at 1× and 1/10× dilutions. The phylum Proteobacteria is separated into the classes Alphaproteobacteria, Betaproteobacteria, Deltaproteobacteria, Epsilonproteobacteria, and Gammaproteobacteria. The presence and abundance of different phyla varied between depths and treatments. The majority of the sequences were contained within the Proteobacteria, Bacteroidetes, and Firmicutes phyla.
In the 19- and 57-mbsf depths the majority of non-Proteobacteria sequences were related to the Firmicutes (46.8, 37.0%) phyla while the phyla Chloroflexi, Cyanobacteria, Bacteroidetes, and Deferribacteres individually represented less than 0.5% of the sequences obtained in both samples or were not observed (Figure 4). However, the Fusobacteria, which were identified as 0.1% of the sequences at 1 mbsf and 0.0% at 57 mbsf, comprised 2.4% of the sequences at 19 mbsf (Figure 4). The Actinobacteria accounted for 1.0% at 1 mbsf and only 0.2% of the sequences at 19 mbsf but represented 2.9% of the sequences at 57 mbsf (Figure 4). The Tenericutes composed 1.0% of the sequences at 57 mbsf whereas they were not observed at the other depths.
Potential Metabolic Function
The authors acknowledge the limitations of using SSU rRNA for functional characterization of microbial communities. The trends in potential metabolic function are presented recognizing that pyrosequencing technology is semi-quantitative. Lineages with known metabolic functions were described and the abundance of each genus was noted as a percentage of the total sequences obtained at 1, 19, and 57 mbsf. Forty-two genera were observed to compose greater than 1.0% of the sequences obtained within at least one of the sampling depths (Table S1 in Supplementary Material). In general, sequences representing lineages capable of sulfur cycling accounted for 15.5, 12.83, and 4.66% of the total sequences at 1, 19, and 57 mbsf, respectively. Genera previously linked with iron cycling composed 2.7, 2.7, and 1.4% of the total sequences from 1, 19, and 57 mbsf, respectively. Genera that are potentially associated with nitrogen cycling were associated with 29.5, 29.1, and 49.5% of the total sequences at 1, 19, and 57 mbsf respectively. Finally, 14.7, 43.3, and 27.9% of the sequences were observed to be associated with lineages capable of fermentative-based metabolism at 1, 19, and 57 mbsf, respectively.
Sulfur cycling
Metabolically active genera were identified having the potential to contribute to sulfur cycling in the marine subseafloor. A majority of these sequences were detected in the 1-mbsf sample (Table S1 in Supplementary Material). The genera Desulfonema (Kuever and Meyer, 2007), Desulfobacterium (Brysch et al., 1987), Desulfosarcina (Leloup et al., 2009), and Desulfuromonas have the capacity to use sulfate as a terminal electron acceptor, while a Helicobacter-related lineage within the Epsilonproteobacteria has been shown to reduce sulfur to hydrogen sulfide (Campbell et al., 2006). Previous studies have shown that the genera Achromatium (Head et al., 2000), Thioalkalispira (Sorokin et al., 2002), and Thiothrix (Macalady et al., 2008) have the capacity for full or partial oxidation of sulfur species. The number of sequences related to sulfate reducing and sulfur-oxidizing lineages detected in the database were negatively correlated with the amount of iron sulfide and depth (p < 0.01; i.e., as depth from surface increases, the number of sequences related to sulfate reducing and sulfur-oxidizing lineages detected decreases) but positively correlated (p < 0.01) with a loss of sulfate. The percentage of sequences associated with each lineage is presented in Table S1 in Supplementary Material.
Iron cycling
The potential for metabolically active iron cycling by bacteria was observed at all depths (Table S1 in Supplementary Material). In the 1-mbsf sample, the genera Acidimicrobium has the potential to oxidize iron (Clark and Norris, 1996). Genera capable of reducing iron for energy conservation detected in this study included Anaeromyxobacter (Sanford et al., 2002), Dyella (Lu et al., 2010), and Ralstonia (Roling et al., 2007). These lineages were detected at all depths with the highest percentage of sequences at the 19-mbsf depth. The frequency of detecting iron reducing lineage sequences were positively correlated (p < 0.01) with depth, whereas Fe(II) accumulation was negatively correlated (p < 0.01) with iron oxidizing lineage sequences, as observed through SVD.
Nitrogen cycling
Multiple genera capable of using nitrogen species as a terminal electron acceptor were throughout the sediment samples (Table S1 in Supplementary Material). The most frequently detected lineage with the sequence database was Cupriavidus (described in Van Damme and Coenye, 2004), representing 14.7, 21.9, and 36.3% of the total sequences at 1, 19, and 57 mbsf (Table S1 in Supplementary Material). Additional nitrogen-reducing lineages detected as being metabolically active included Alcanivorax (Nakano et al., 2008), Burkholderia (Igual et al., 2006), Caldithrix (Miroshnichenko et al., 2003), Corynebacterium (Renner and Becker, 1970), and Stenotrophomonas. The number of nitrate reducing lineage sequences detected did not significantly change with depth. Genera detected as metabolically active with the capacity for nitrogen fixation included Herbaspirillum (Rigo et al., 2007), Alkalilimnicola (Tourova et al., 2007), Spirochaeta (Rainey et al., 1991), and Cyanothece (Pakrasi et al., 2008) were identified in all samples (Table S1 in Supplementary Material), despite ammonia concentrations being positively correlated (p < 0.01) with depth.
Fermentation and methylotrophy metabolism
Genera with the capacity for fermentative metabolism were frequently detected at each depth, accounting for 14.7, 43.3, and 27.9% of the total sequences (Table S1 in Supplementary Material). These genera included Anaerophaga (Schink et al., 2002), Byssovorax (Kunze et al., 2006), Caldilinea (Ollivier et al., 2011), Faecalibacterium (Fidopiastis et al., 2006), Lactobacillus (Kandler et al., 1983), Leptolinea (Yamada et al., 2006), Levillnea (Yamada et al., 2006), Staphylococcus (Gregory et al., 2003), and Turicibacter (Bosshard et al., 2002). Group 1 methylotrophs, Methylobacter and Methylomicrobium (Bowman et al., 1993), were detected in the 1-mbsf sequence database but were absent from the deeper depths (Table S1 in Supplementary Material).
Discussion
The community characterization data presented in this study represent one of the first RNA-based pyrosequencing and quantitative PCR analyses of microbial communities associated with the subseafloor sedimentary habitat. The highly resolved sequence dataset produced a novel structural view of the metabolically active fraction of subseafloor bacterial populations. Sequence data also revealed numerous genera that individually represented less than 1% of the sequence dataset highlighting the overall diversity of the community. Active microbial populations and their environmental factors were compared by combining geochemical analysis to robust molecular data to predict the local microbial ecology and metabolic processes. In addition, this study enhances sequence databases produced by previous RNA-based community pyrosequencing efforts from marine water samples (Frias-Lopez et al., 2008; Gilbert et al., 2008) and from soil samples (Leininger et al., 2006; Urich et al., 2008) to improve understanding of the metabolically active fraction of the microbial communities.
Metabolically Active Community Structure
Quantification of cell and SSU rRNA transcript abundance indicated a decreasing population density and reduced overall metabolic activity from the top depth horizon to within and below the SMTZ. The reported cell abundance was in agreement with previously observed ranges within the subsurface and follows a similar decreasing trend with depth (Parkes et al., 1994, 2000). Similar trends were detected in Peru margin sediments where bacterial SSU rRNA gene abundance (DNA) decreased rapidly over the top 20 mbsf but remained unchanged to 50 mbsf (Biddle et al., 2008). A similar trend was observed with transcript abundance. It was interesting to note that the ratio of transcripts detected to cells counted increased over 3.5 fold from the 1-mbsf sample to the 57-mbsf sample. This does not imply higher activity at depth, as there are both more cells and more transcripts in the surfaces communities. However, it does suggest a higher proportion of the total population may be metabolically active at depth compared to the shallow communities. The concentration of ribosomes, and thus copies of SSU rRNA transcripts within a cell, is linearly correlated to cellular metabolic activity (DeLong et al., 1989; Kemp et al., 1993; Kerkhof and Ward, 1993; Lee and Kemp, 1994), with dormant and dead cells having few to no ribosomes present (Davis et al., 1986; Fegatella et al., 1998). The shallow sediments may have more dormant or dead cells, elevating the cell counts while not increasing the transcript abundance. The potential for geochemical flux in the shallow sediments may be the driver for this community composition, while geochemical conditions in the deeper sediments would remain more stable. These data suggest the deeper subsurface environments may select for fewer dormant populations and may effectively recycle dead cells. Previous results have made similar observations reporting predominantly metabolically active populations (Morono et al., 2011) and a limited of number of dead cells within biomass calculations (Takano et al., 2010). Further analysis specifically targeting dormant populations should be completed to test the role these communities play in the environment and diversity of the community.
Cell and transcript abundances in the SMTZ (19 mbsf) and the deeper horizon (57 mbsf) were similar, suggesting that abundance of active bacterial populations below the SMTZ decreases less rapidly than above the SMTZ in these sediments. The observation of a similar trend in transcript abundance indicates that the fraction of active microbial population is positively related to the total cell abundance. Therefore, the decrease of SSU rRNA gene abundance in subseafloor sediments may be attributed to decreasing cell abundance. The bacterial SSU rRNA transcript abundance in this study were four to seven orders of magnitude lower than that of marine estuarine sedimentary environments (Smith et al., 2006), which was expected given lower organic carbon concentrations. While the results of this study indicate that the metabolic activity levels within the subseafloor sedimentary habitats were substantially less than other shallow, higher organic carbon environments, it is important to note that a metabolically active population was detected at all depths selected.
Sequences were grouped into OTUs based on 95% sequence similarity to obtain a genus-level clustering of the metabolically active bacterial community. For the purpose of this study, the designation of genus will be used as a descriptor of relative taxonomic level but may not reflect a true genus-level classification despite the use of 95% sequence similarity in other studies (Zhang et al., 2010). The sequencing capacity required to reach the depth of phylogenetic analyses presented here is unique for the characterization of subseafloor microbial communities. In addition, the V1–V3 regions were selected based on the overall volume of sequences available for comparison and the widely accepted and used primer sets available. As deep subsurface sediments have not been well characterized for primer optimization (Teske and Sørensen, 2008), this initial study provides a resource for future primer development efforts.
The bacterial diversity observed in sediments at 1 mbsf was similar to that found in other marine sedimentary environments considered to host diverse active microbial communities, such as near-shore sediments (Reese et al., 2012). The level of diversity determined in this study indicated that this system was more complex than anticipated. It is important to note that a majority of the genera observed comprised less than 1% of the total population. This is supported by high Shannon index values indicating species richness. The identification of these populations was possible as a result of the high-throughput sequencing efforts and may still play important roles in the overall community function. However, many of these genera have not been physiologically characterized in marine systems. To enhance the descriptive nature of molecular datasets, additional effort is required in cultivation-based molecular ecological and biogeochemical studies.
A limited number of shared OTUs at each depth indicated that the observed metabolically active populations were unique from each other. Although the samples had similar phyla characterization and similar abundance of genera at both 19 and 57 mbsf, they only shared 24% of their OTUs. Statistical data including rarefaction and Chao1 estimates suggested that a majority of the active microbial community was likely characterized at 19 and 57 mbsf. While these communities are both deep in the subsurface, the different community structure can be in part attributed to local variations in geochemistry, such as proximity to the SMTZ. Previous studies have shown that geological variability can support differences in microbial population diversity in subseafloor sediments (Inagaki et al., 2006; Hoshino et al., 2011). A broad study describing the horizontal spatial variability of microbial populations in marine subseafloor has yet to be compiled.
The OTU abundance was significantly at 19 and 57 mbsf compared to the community at 1 mbsf. These data supported a reduction in diversity with increasing sediment depth in subseafloor microbial populations. A similar loss of diversity with increasing depth below surface was previously shown in subseafloor microbial communities using bacterial SSU rRNA gene targets (Parkes et al., 2005; Biddle et al., 2008). Increased sampling resolution should be performed in future studies to determine the level of heterogeneity within the subseafloor biosphere. It is interesting to note that the diversity at the phylum level observed in this study was greater than previous subseafloor microbial characterizations despite using RNA to target the metabolically active fraction of the total population. Furthermore, the total diversity in these sediments are predicted to be higher than indicated by an RNA-based analysis, as only a fraction of the total diversity would be active. These results suggest that previous studies have underestimated the total diversity of the subseafloor microbial community and that more robust techniques should be routinely applied. The combination of extraction method and pyrosequencing techniques may have produced a higher nucleic acid yield and deeper sequencing effort, yielding the increased diversity observations. Therefore, the Nankai sediments may support increased diversity compared to other subsurface locations or these results represent an advancement of the procedures used to characterize microbial populations. Such uncertainty stresses the need for standardizing molecular methods between laboratories working on the subseafloor biosphere as well as expanding characterizations using RNA targets.
Predicted Biogeochemical Interactions
Combined biologic and geochemical comparisons provide a more complete understanding of the subseafloor biosphere. Geochemical profiles indicated that the SMTZ was at approximately 20 mbsf. Multiple microbial processes are predicted as being metabolically active within this zone including: sulfate reduction, methane oxidation, methanogenesis, and potentially metal reduction. These processes, along with fermentation are considered dominant metabolic processes throughout anaerobic subsurface sediments (D’Hondt et al., 2002, 2004), however geochemical analysis is used to predict stratification of the microbial process and thus populations. Presence and abundance of active genera were examined to determine potential changes in the overall metabolic function of the community corresponding to depth and local geochemical conditions. As previously noted, the authors acknowledge the limitation of using SSU rRNA for functional characterization of microbial communities.
Thermodynamically predicted processes
Thermodynamics predicts that microbial populations in the marine subseafloor will be stratified based on the potential energy gain available from the oxidation and reduction reactions of electron donors and acceptors (Froelich et al., 1979; Orcutt et al., 2011). Molecular data from this study support metabolically active subseafloor microbial populations can be linked to the predicted geochemical conditions, though anomalies do occur as discussed below. Previous studies have predicted that shallow subsurface sediments with high sulfate concentrations and low electron donor concentrations would support a wide zone of sulfate reduction and with a majority of the microbial population using sulfur metabolism (D’Hondt et al., 2002). Ship-based geochemical analysis indicated a gradual loss of sulfate from the surface to the top of the SMTZ followed by a more rapidly decrease in concentration through the SMTZ. These data support microbial sulfate reduction occurring above and within the SMTZ. Correspondingly, RNA-based molecular analysis identified metabolically active organisms capable of sulfate reduction in the 1- and 19-mbsf samples. Additionally, there were organisms capable of oxidizing sulfides generated in the 1-mbsf sample to completely cycle sulfur species at the surface. A shift to sulfur reduction was observed at 19 mbsf with over 12% of the sequences related to Helicobacteraceae. This lineage, also detected at 57 mbsf but not detected in the active fraction at 1 mbsf, has the potential to utilize the products of reduced sulfate to produce the hydrogen sulfide detected in geochemical analysis. The localization of this lineage within and below the SMTZ is supported by the inferred physiology and may provide a critical linkage to the complete reduction of sulfate to hydrogen sulfide. Members of this family have been noted as being understudied while frequently detected in marine systems (Campbell et al., 2006). Additional sequence analysis of the V1–V3 region will be completed to determine if detection of this lineage is biased in NCBI Blast results, and does not recognize the sequence similarity to other, sediment-associated Helicobacteraceae.
Processes other than sulfate reduction may contribute to the overall community metabolic function at 1 mbsf despite low organic carbon concentrations donors. Approximately 2.7% of the total population observed at the 1-mbsf was capable of iron oxidation and reduction, indicating that complete cycling of iron species is also possible in shallow subseafloor sediments. Additionally 30.9% of the total population observed in the sample at 1 mbsf is potentially associated with nitrogen cycling. Nitrogen cycling in these shallow sediments may play a substantial role in the overall microbial ecology despite previous predictions of a sulfate dominated environment (D’Hondt et al., 2002). However, recent reports suggest that sulfate reduction can be coupled to ammonium oxidation, providing a link between the two pathways in the shallow subsurface (Schrum et al., 2009). These results stress the need to couple geochemical analysis with molecular characterization of microbial populations. Studies limited to one of these procedures would result in inaccurate interpretations of the microbial ecology.
Lineages detected outside of thermodynamically predicted zones
Multiple lineages capable of nitrogen and iron reduction were detected as metabolically active outside of thermodynamically predicted zones, i.e., above the SMTZ (Froelich et al., 1979). Sequence data indicated lineages capable of iron reduction comprised 2.7 and 1.4% of the total sequences at 19 and 57 mbsf, respectively. A slight increase of dissolved iron [Fe(II)] concentration corresponding to a decrease of iron sulfides (CRS) potentially explains the observation of iron reducers below the SMTZ (Figure 1). It is important to note that since the increase of Fe(II) is small, it might be attributed to procedural biases associated with the determination of Fe(II) concentration. However, corresponding molecular evidence supports the geochemical result corroborating this as natural variability. As a result of the dissolution of the metal sulfide compounds, Fe(III) may be produced at depth through chemical reactions with fermentation products including hydrogen peroxide (Brooijmans et al., 2009). Iron reducers active at depth, as suggested by molecular results at 57 mbsf, would reduce the Fe(III) upon formation. Future research in this area would provide a more defined link between the products of fermentation and the oxidation of reduced substrates at depth.
Active iron and nitrogen cycling below the SMTZ may indicate the presence of geochemically cryptic cycles at depth. In addition to iron reduction, nitrogen-reducing lineages were detected within (23.5% of the total sequences) and below (43.7% of the total sequences) the SMTZ. Oxidizing populations were metabolically active at depth although they were less than 1% of the total sequences obtained. For example, the genus Dyella was detected at 19 and 57 mbsf and has species capable of both iron oxidation and iron reduction (Lu et al., 2010). Lineages at 57 mbsf included sulfur-oxidizing Thioalkalispira, and the Mn(II) oxidation genus Leptothrix (Boogerd and Devrind, 1987). Detection of these lineages being metabolically active supports the presence of complex interactions between microbial communities within the marine subseafloor that may not be solely directed geochemically. The co-occurrence of populations with the capacity to conduct both oxidative and reductive processes recently described as cryptic biogeochemical cycling (Holmkvist et al., 2011; Orcutt et al., 2011). The nature of the redox pairs would result in a lack of detectable geochemical end products at the standard resolution of analysis, keeping one half of the process geochemically undetectable. However the low abundance of oxidizers and the presence of reduced compounds suggest oxidizing processes may be the limiting component.
Further investigations into the active metabolisms of the populations found at various depths using culture assays is essential to gain a better understanding of how these organisms survive in marine subseafloor environments. Cultivation-based characterizations of lineages isolated from the subsurface are currently ongoing in the Mills laboratory and will be used to support community characterizations. Future research will focus on combining molecular techniques including the isolation of functional gene targets, metatranscriptomic, and proteomic analyses. In addition, increasing sequence resolution in these environments will provide a better characterization of genera detected at low frequency. Description of the rare biosphere is important to understand the extent of total diversity in the subseafloor biosphere.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the Integrated Ocean Drilling Program, the Science Party and Crew of the Chikyu during IODP Expedition 316, Tim Ferdelman, Raymond Cruz St. Peter, and Emily Hollister for their contributions to this research. Additionally, we would like to thank the Ocean Drilling and Sustainable Earth Sciences faculty and the Department of Oceanography at Texas A&M University for their contributions toward funding this research. This paper is contribution number 128 to the Center for Dark Energy Biosphere Investigations (CDEBI).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/extreme_microbiology/10.3389/fmicb.2012.00113/abstract
Table S1. Taxonomic identification of the Nankai subsurface sequence data set.
Footnotes
References
Bach, W., Ravelo, C., Behrmann, J., Camoin, G., Duncan, R., Edwards, K., Gulick, S., Inagaki, F., Pälike, H., and Tada, R. (2010). IODP New Ventures in Exploring Scientific Targets (INVEST): defining the new goals of an international drilling program. Sci. Drill. 9, 54–64.
Bickle, M., Arculus, R., Barrett, P., DeConto, R., Camoin, G., Edwards, K., Fisher, A., Inagaki, F., Kodaira, S., Ohkouchi, N., Pälike, H., Ravelo, C., Saffer, D., and Teagle, D. (2011). Illuminating Earth’s Past, Present and Future: Science Plan for 2013–2023. Washington: IODP Press, 92.
Biddle, J. F., Fitz-Gibbon, S., Schuster, S. C., Brenchley, J. E., and House, C. H. (2008). Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. U.S.A. 105, 10583–10588.
Biddle, J. F., Lipp, J. S., Lever, M. A., Lloyd, K. G., Sorensen, K. B., Anderson, R., Fredricks, H. F., Elvert, M., Kelly, T. J., Schrag, D. P., Sogin, M. L., Brenchley, J. E., Teske, A., House, C. H., and Hinrichs, K. U. (2006). Heterotrophic Archaea dominate sedimentary subsurface ecosystems off Peru. Proc. Natl. Acad. Sci. U.S.A. 103, 3846–3851.
Boogerd, F. C., and Devrind, J. P. M. (1987). Manganese oxidation by Leptothrix discophora. J. Bacteriol. 169, 489–494.
Bosshard, P. P., Zbinden, R., and Altwegg, M. (2002). Turicibacter sanguinis gen. nov., sp nov., a novel anaerobic, Gram-positive bacterium. Int. J. Syst. Evol. Microbiol. 52, 1263–1266.
Bowman, J. P., Sly, L. I., Nichols, P. D., and Hayward, A. C. (1993). Revised taxonomy of the methanotrophs – description of Methylobacter gen. nov, emendation of Methylococcus, validation of Methylosinus and Methylocystis species, and a proposal that the family Methylococcaceae includes only the Group I methanotrophs. Int. J. Syst. Evol. Microbiol. 43, 735–753.
Brooijmans, R. J. W., De Vos, W. M., and Hugenholtz, J. (2009). Lactobacillus plantarum WCFS1 electron transport chains. Appl. Environ. Microbiol. 75, 3580–3585.
Brysch, K., Schneider, C., Fuchs, G., and Widdel, F. (1987). Lithoautotrophic growth of sulfate-reducing bacteria, and description of Desulfobacterium autotrophicum gen nov, sp. nov. Arch. Microbiol. 148, 264–274.
Campbell, B. J., Engel, A. S., Porter, M. L., and Takai, K. (2006). The versatile ε-proteobacteria: key players in sulphidic habitats. Nat. Rev. Microbiol. 4, 458–468.
Canfield, D. E., Stewart, F. J., Thamdrup, B., De Brabandere, L., Dalsgaard, T., DeLong, E. F., Revsbech, N. P., and Ulloa, O. (2010). A cryptic sulfur cycle in oxygen-minimum-zone waters off the Chilean Coast. Science 330, 1375–1378.
Chapelle, F. H., O’Neil, K., Bradley, P. M., Methe, B. A., Cluto, S. A., Knobel, L. L., and Lovely, D. R. (2002). A hydrogen-based subsurface microbial community dominated by methanogens. Nature 415, 312–314.
Clark, D. A., and Norris, P. R. (1996). Acidimicrobium ferrooxidans gen nov, sp nov: mixed-culture ferrous iron oxidation with Sulfobacillus species. Microbiology 142, 785–790.
Cline, J. D. (1969). Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14, 454–458.
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., Kulam-Syed-Mohideen, A. S., McGarrell, D. M., Marsh, T., Garrity, G. M., and Tiedje, J. M. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145.
Davis, B. D., Luger, S. M., and Tai, P. C. (1986). Role of ribosome degradation in the death of starved Escherichia coli cells. J. Bacteriol. 166, 439–445.
DeLong, E. F., Wickham, G. S., and Pace, N. R. (1989). Phylogenetic stains – ribosomal RNA-based probes for the identification of single cells. Science 243, 1360–1363.
D’Hondt, S., Inagaki, F., Ferdelman, T., Jørgensen, B. B., Kato, K., Kemp, P., Sobecky, P., Sogin, M., and Takai, K. (2007). Exploring subseafloor life with the Integrated Ocean Drilling Program. Sci. Drill. 5, 26–37.
D’Hondt, S., Jørgensen, B. B., Miller, D. J., Batzke, A., Blake, R., Cragg, B. A., Cypionka, H., Dickens, G. R., Ferdelman, T., Hinrichs, K. U., Holm, N. G., Mitterer, R., Spivack, A., Wang, G. Z., Bekins, B., Engelen, B., Ford, K., Gettemy, G., Rutherford, S. D., Sass, H., Skilbeck, C. G., Aiello, I. W., Guerin, G., House, C. H., Inagaki, F., Meister, P., Naehr, T., Niitsuma, S., Parkes, R. J., Schippers, A., Smith, D. C., Teske, A., Wiegel, J., Padilla, C. N., and Acosta, J. L. S. (2004). Distributions of microbial activities in deep subseafloor sediments. Science 306, 2216–2221.
D’Hondt, S., Rutherford, S., and Spivack, A. J. (2002). Metabolic activity of subsurface life in deep-sea sediments. Science 295, 2067–2070.
Dowd, S. E., Zaragoza, J., Rodriguez, J. R., Oliver, M. J., and Payton, P. R. (2005). Windows.NET network distributed basic local alignment search toolkit (W.ND-BLAST). BMC Bioinformatics 6, 93. doi:10.1186/1471-2105-6-93
Edwards, R. A., Rodriguez-Brito, B., Wegley, L., Haynes, M., Breitbart, M., Peterson, D. M., Saar, M. O., Alexander, S., Alexander, E. C. Jr., and Rohwer, F. (2006). Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7, 57. doi:10.1186/1471-2164-7-57
Fegatella, F., Lim, J., Kjelleberg, S., and Cavicchioli, R. (1998). Implications of rRNA operon copy number and ribosome content in the marine oligotrophic ultramicrobacterium Sphingomonas sp. strain RB2256. Appl. Environ. Microbiol. 64, 4433–4438.
Fidopiastis, P. M., Bezdek, D. J., Horn, M. H., and Kandel, J. S. (2006). Characterizing the resident, fermentative microbial consortium in the hindgut of the temperate-zone herbivorous fish, Hermosilla azurea (Teleostei: Kyphosidae). Mar. Biol. 148, 631–642.
Fossing, H., and Jørgensen, B. B. (1989). Measurement of bacterial sulfate reduction in sediments. Evaluation of a single-step chromium reduction method. Biogeochemistry 8, 205–222.
Frias-Lopez, J., Shi, Y., Tyson, G. W., Coleman, M. L., Schuster, S. C., Chisholm, S. W., and DeLong, E. F. (2008). Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U.S.A. 105, 3805–3810.
Froelich, P. N., Klinkhammer, G. P., Bender, M. L., Luedtke, N. A., Heath, G. R., Cullen, D., Dauphin, P., Hammond, D., Hartman, B., and Maynard, V. (1979). Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic suboxic diagenesis. Geochim. Cosmochim. Acta 43, 1075–1090.
Gilbert, J., Field, D., Huang, Y., Edwards, R., Li, W., Gilna, P., and Joint, I. (2008). Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS ONE 3, e3042. doi:10.1371/journal.pone.0003042
Giovannoni, S. J. (1991). “The polymerase chain reaction,” in Nucleic Acid Techniques in Bacterial Systematics, eds. E. Stackenbrandt and M. Goodfellow (Chichester: Wiley), 177–203.
Gregory, L. G., Bond, P. L., Richardson, D. J., and Spiro, S. (2003). Characterization of a nitrate-respiring bacterial community using the nitrate reductase gene (narG) as a functional marker. Microbiology 149, 229–237.
Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., Ciulla, D., Tabbaa, D., Highlander, S. K., Sodergren, E., Methe, B., Desantis, T. Z., Petrosino, J. F., Knight, R., Birren, B. W., and Consortium, H. M. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504.
Head, I. M., Gray, N. D., Babenzien, H. D., and Glockner, F. O. (2000). Uncultured giant sulfur bacteria of the genus Achromatium. FEMS Microbiol. Ecol. 33, 171–180.
Holmkvist, L., Ferdelman, T. G., and Jørgensen, B. B. (2011). A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark). Geochim. Cosmochim. Acta 75, 3581–3599.
Hoshino, T., Morono, Y., Terada, T., Imachi, H., Ferdelman, T. F., and Inagaki, F. (2011). Comparative study of subseafloor microbial community structures in deeply buried coral fossils and sediment matrices from the Challenger Mound in the Porcupine Seabight. Front. Microbiol. 2:231. doi:10.3389/fmicb.2011.00231
Igual, J. M., Valverde, A., Delvasto, P., Peix, A., Velazquez, E., Santa-Regina, I., Ballester, A., Rodriguez-Barrueco, C., and Garcia-Balboa, C. (2006). Burkholderia ferrariae sp nov., isolated from an iron ore in Brazil. Int. J. Syst. Evol. Microbiol. 56, 2421–2425.
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., Suzuki, M., Takai, K., Delwiche, M., Colwell, F. S., Nealson, K. H., Horikoshi, K., D’Hondt, S., and Jørgensen, B. B. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments, on the Pacific ocean margin. Proc. Natl. Acad. Sci. U.S.A. 103, 2815–2820.
Iversen, N., and Jørgensen, B. B. (1985). Anaerobic methane oxidation rates at the sulfate methane transition in marine sediments from Kattegat and Skagerrak (Denmark). Limnol. Oceanogr. 30, 944–955.
Kandler, O., Schillinger, U., and Weiss, N. (1983). Lactobacillus bifermentans sp. nov, nom rev, an organism forming CO2 and H2 from lactic acid. Syst. Appl. Microbiol. 4, 408–412.
Kemp, P. F., Lee, S., and Laroche, J. (1993). Estimating the growth rate of slowly growing marine bacteria from RNA content. Appl. Environ. Microbiol. 59, 2594–2601.
Kerkhof, L. J., and Ward, B. B. (1993). Comparison of nucleic acid hybridization and fluorometry for measurement of RNA/DNA relationship with growth rate in a marine bacterium. Appl. Environ. Microbiol. 59, 1303–1307.
Kimura, G., Screaton, E. J., Curewitz, D., and Expedition 316 Scientists. (2008). NanTroSEIZE Stage 1A: NanTroSEIZE Shallow Megasplay and Frontal Thrusts. IODP Preliminary Report, College Station, 316.
Kinoshita, M., Tobin, H., Ashi, J., Kimura, G., Lallemant, S., Screaton, E. J., Curewitz, D., Masago, H., Moe, K. T., and the Expedition 314/315/316 Scientists. (2009). “NanTroSEIZE Stage 1: Investigations of seismogenesis Nankai Trough, Japan,” in Proceeding of the Integrated Ocean Drilling Program, 314/315/316 Expedition Reports, College Station.
Kuever, J., and Meyer, B. (2007). Phylogeny of the alpha and beta subunits of the dissimilatory adenosine-5′-phosphosulfate (APS) reductase from sulfate-reducing prokaryotes – origin and evolution of the dissimilatory sulfate-reduction pathway. Microbiology 153, 2026–2044.
Kunze, B., Steinmetz, H., Hofle, G., Huss, M., Wieczorek, H., and Reichenbach, H. (2006). Cruentaren, a new antifungal salicylate-type macrolide from Byssovorax cruenta (Myxobacteria) with inhibitory effect on mitochondrial ATPase activity – fermentation and biological properties. J. Antibiot. 59, 664–668.
Lee, S. H., and Kemp, P. F. (1994). Single cell RNA content of natural marine planktonic bacteria measured by hybridization with multiple 16S ribosomal RNA targeted fluorescent probes. Limnol. Oceanogr. 39, 869–879.
Leininger, S., Urich, T., Schloter, M., Schwark, L., Qi, J., Nicol, G. W., Prosser, J. I., Schuster, S. C., and Schleper, C. (2006). Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442, 806–809.
Leloup, J., Fossing, H., Kohls, K., Holmkvist, L., Borowski, C., and Jørgensen, B. B. (2009). Sulfate-reducing bacteria in marine sediment (Aarhus Bay, Denmark): abundance and diversity related to geochemical zonation. Environ. Microbiol. 11, 1278–1291.
Lipp, J. S., Morono, Y., Inagaki, F., and Hinrichs, K. U. (2008). Significant contribution of Archea to extant biomass in marine subsurface sediments. Nature 454, 991–994.
Liu, Z., Desantis, T. Z., Andersen, G. L., and Knight, R. (2008). Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 36, e120.
Lu, S. P., Gischkat, S., Reiche, M., Akob, D. M., Hallberg, K. B., and Kusel, K. (2010). Ecophysiology of Fe-cycling bacteria in acidic sediments. Appl. Environ. Microbiol. 76, 8174–8183.
Macalady, J. L., Dattagupta, S., Schaperdoth, I., Jones, D. S., Druschel, G. K., and Eastman, D. (2008). Niche differentiation among sulfur-oxidizing bacterial populations in cave waters. ISME J. 2, 590–601.
Mattes, T. E., and Jin, Y. O. (2010). A quantitative PCR assay for aerobic, vinyl chloride- and ethene-assimilating microorganisms in groundwater. Environ. Sci. Technol. 44, 9036–9041.
Mills, H. J., Hunter, E., Humphrys, M., Kerkhof, L., McGuinness, L., Huettel, M., and Kostka, J. E. (2008). Determination of high resolution microbial community structure in permeable marine sediments of the northeastern Gulf of Mexico. Appl. Environ. Microbiol. 74, 4440–4453.
Miroshnichenko, M. L., Kostrikina, N. A., Chernyh, N. A., Pimenov, N. V., Tourova, T. P., Antipov, A. N., Spring, S., Stackebrandt, E., and Bonch-Osmolovskaya, E. A. (2003). Caldithrix abyssi gen. nov., sp nov., a nitrate-reducing, thermophilic, anaerobic bacterium isolated from a Mid-Atlantic Ridge hydrothermal vent, represents a novel bacterial lineage. Int. J. Syst. Evol. Microbiol. 53, 323–329.
Morono, Y., and Inagaki, F. (2010). Automatic slide-loader fluorescence microscope for discriminative enumeration of subseafloor life. Sci. Drill. 9, 32–36.
Morono, Y., Terada, T., Masui, N., and Inagaki, F. (2009). Discriminative detection and enumeration of microbial life in marine subsurface sediments. ISME J. 3, 503–511.
Morono, Y., Terada, T., Nishizawa, M., Ito, M., Hillion, F., Takahata, N., Sano, Y., and Inagaki, F. (2011). Carbon and nitrogen assimilation in deep subseafloor microbial cells. Proc. Natl. Acad. Sci. U.S.A. 108, 18295–18300.
Nadkarni, M. A., Martin, F. E., Jacques, N. A., and Hunter, N. (2002). Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148, 257–266.
Nakano, M., Shimizu, Y., Okumura, H., Sugahara, I., and Maeda, H. (2008). Construction of a consortium comprising ammonia-oxidizing bacteria and denitrifying bacteria isolated from marine sediment. Biocontrol Sci. 13, 73–89.
Nawrocki, A. P., and Eddy, S. R. (2007). Query-dependant banding (QDB) for faster RNA similarity searches. PLoS Comput. Biol. 3, E56.
Neidhardt, F. C., and Magasanik, B. (1960). Studies on the role of ribonucleic acid in the growth of bacteria. Biochim. Biophys. Acta 42, 99–116.
Newberry, C. J., Webster, G., Cragg, B. A., Parkes, R. J., Weightman, A. J., and Fry, J. C. (2004). Diversity of prokaryotes and methanogenesis in deep subsurface sediments from the Nankai Trough, Ocean Drilling Program Leg 190. Environ. Microbiol. 6, 274–287.
Nogales, B., Moore, E. R. B., Abraham, W. R., and Timmis, K. N. (1999). Identification of the metabolically active members of a bacterial community in a polychlorinated biphenyl-polluted moorland soil. Environ. Microbiol. 1, 199–212.
Ollivier, B., Gregoire, P., Bohli, M., Cayol, J. L., Joseph, M., Guasco, S., Dubourg, K., Cambar, J., Michotey, V., Bonin, P., and Fardeau, M. L. (2011). Caldilinea tarbellica sp nov., a filamentous, thermophilic, anaerobic bacterium isolated from a deep hot aquifer in the Aquitaine Basin. Int. J. Syst. Evol. Microbiol. 61, 1436–1441.
Orcutt, B. N., Sylvan, J. B., Knab, N. J., and Edwards, K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 75, 361–422.
Pakrasi, H. B., Welsh, E. A., Liberton, M., Stoeckel, J., Loh, T., Elvitigala, T., Wang, C., Wollam, A., Fulton, R. S., Clifton, S. W., Jacobs, J. M., Aurora, R., Ghosh, B. K., Sherman, L. A., Smith, R. D., and Wilson, R. K. (2008). The genome of Cyanothece 51142, a unicellular diazotrophic cyanobacterium important in the marine nitrogen cycle. Proc. Natl. Acad. Sci. U.S.A. 105, 15094–15099.
Parkes, R. J., Cragg, B. A., Bale, S. J., Getliff, J. M., Goodman, K., Rochelle, P. A., Fry, J. C., Weightman, A. J., and Harvey, S. M. (1994). Deep bacterial biosphere in Pacific ocean sediments. Nature 371, 410–413.
Parkes, R. J., Cragg, B. A., and Wellsbury, P. (2000). Recent studies on bacterial populations and processes in subseafloor sediments: a review. Hydrogeol. J. 8, 11–28.
Parkes, R. J., Webster, G., Cragg, B. A., Weightman, A. J., Newberry, C. J., Ferdelman, T. G., Kallmeyer, J., Jørgensen, B. B., Aiello, I. W., and Fry, J. C. (2005). Deep sub-seafloor prokaryotes stimulated at interfaces over geological time. Nature 436, 390–394.
Polymenakou, P. N., Lampadariou, N., Mandalakis, M., and Tselepides, A. (2009). Phylogenetic diversity of sediment bacteria from the southern Cretan margin, Eastern Mediterranean Sea. Syst. Appl. Microbiol. 32, 17–26.
Rainey, F. A., Janssen, P. H., Wild, D. J. C., and Morgan, H. W. (1991). Isolation and characterization of an obligately anaerobic, polysaccharolytic, extremely thermophilic member of the genus Spirochaeta. Arch. Microbiol. 155, 396–401.
Reese, B. K., Mills, H. J., Dowd, S., and Morse, J. W. (2012). Benthic biogeochemistry of microbial iron and sulfate reduction in the Gulf of Mexico hypoxic zone. Geomicrobiol. J. (in press).
Renner, E. D., and Becker, G. E. (1970). Production of nitric oxide and nitrous oxide during denitrification by Corynebacterium nephridii. J. Bacteriol. 101, 821–826.
Rigo, L. U., Schwab, S., Ramos, H. J., Souza, E. M., Pedrosa, F. O., Yates, M. G., and Chubatsu, L. S. (2007). Identification of NH4+-regulated genes of Herbaspirillum seropedicae by random insertional mutagenesis. Arch. Microbiol. 187, 379–386.
Roling, W. F. M., Lin, B., Hyacinthe, C., Bonneville, S., Braster, M., and Van Cappellen, P. (2007). Phylogenetic and physiological diversity of dissimilatory ferric iron reducers in sediments of the polluted Scheldt estuary, Northwest Europe. Environ. Microbiol. 9, 1956–1968.
Sanford, R. A., Cole, J. R., and Tiedje, J. M. (2002). Characterization and description of Anaeromyxobacter dehalogenans gen. nov., sp nov., an aryl-halorespiring facultative anaerobic myxobacterium. Appl. Environ. Microbiol. 68, 893–900.
Schink, B., Denger, K., Warthmann, R., and Ludwig, W. (2002). Anaerophaga thermohalophila gen. nov., sp nov., a moderately thermohalophilic, strictly anaerobic fermentative bacterium. Int. J. Syst. Evol. Microbiol. 52, 173–178.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., Robinson, C. J., Sahl, J. W., Stres, B., Thallinger, G. G., Van Horn, D. J., and Weber, C. F. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541.
Schrum, H., Spivack, A. J., Kastner, M., and D’Hondt, S. (2009). Sulfate-reducing ammonium oxidation: a thermodynamically feasible metabolic pathway in subseafloor sediment. Geology 37, 939–942.
Smith, C. J., Nedwell, D. B., Dong, L. F., and Osborn, A. M. (2006). Evaluation of quantitative polymerase chain reaction-based approaches for determining gene copy and gene transcript numbers in environmental samples. Environ. Microbiol. 8, 804–815.
Sogin, M. L., Morrison, H. G., Huber, J. A., Welch, D. M., Huse, S. M., Neal, P. R., Arrieta, J. M., and Herndi, G. J. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120.
Sørensen, K. B., and Teske, A. (2006). Stratified communities of active archaea in deep marine subsurface sediments. Appl. Environ. Microbiol. 72, 4596–4603.
Sorokin, D. Y., Tourova, T. P., Kolganova, T. V., Sjollema, K. A., and Kuenen, J. G. (2002). Thioalkalispira microaerophila gen. nov., sp nov., a novel lithoautotrophic, sulfur-oxidizing bacterium from a soda lake. Int. J. Syst. Evol. Microbiol. 52, 2175–2182.
Strasser, M., Moore, G. F., Kimura, G., Kitamura, Y., Kopf, A. J., Lallemant, S., Park, J. O., Screaton, E. J., Su, X., Underwood, M. B., and Zhao, X. X. (2009). Origin and evolution of a splay fault in the Nankai accretionary wedge. Nat. Geosci. 2, 648–652.
Strasser, M., Moore, G. F., Kimura, G., Kopf, A. J., Underwood, M. B., Guo, J., and Screaton, E. J. (2011). Slumping and mass transport deposition in the Nankai fore arec: evidence from IODP drilling and 3-D reflection seismic data. Geochem. Geophys. Geosyst. 12, Q0AD13.
Takano, Y., Chikaraishi, Y., Ogawa, N. O., Nomaki, H., Morono, Y., Inagaki, F., Kitazato, H., Hinrichs, K.-U., and Ohkouchi, N. (2010). Sedimentary membrane lipids recycled by deep-sea benthic archaea. Nat. Geosci. 3, 858–861.
Teske, A., and Sørensen, K. (2008). Uncultured archaea in deep marine subsurface sediments: have we caught them all? ISME J. 2, 3–18.
Tourova, T. P., Spiridonova, E. M., Berg, I. A., Slobodova, N. V., Boulygina, E. S., and Sorokin, D. Y. (2007). Phylogeny and evolution of the family Ectothiorhodospiraceae based on comparison of 16S rRNA, cbbL and nifH gene sequences. Int. J. Syst. Evol. Microbiol. 57, 2387–2398.
Urich, T., Lanzen, A., Huson, D. H., Schleper, C., and Schuster, S. C. (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 3, e2527. doi:10.1371/journal.pone.0002527
Van Damme, P., and Coenye, T. (2004). Taxonomy of the genus Cupriavidus: a tale of lost and found. Int. J. Syst. Evol. Microbiol. 54, 2285–2289.
Whiticar, M. J. (1999). Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem. Geol. 161, 291–314.
Whitman, W. B., Coleman, D. C., and Wiebe, W. J. (1998). Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U.S.A. 95, 6578–6583.
Yamada, T., Sekiguchi, Y., Hanada, S., Imachi, H., Ohashi, A., Harada, H., and Kamagata, Y. (2006). Anaerolinea thermolimosa sp nov., Levilinea saccharolytica gen. nov., sp nov and Leptolinea tardivitalis gen. nov., so. nov., novel filamentous anaerobes, and description of the new classes Anaerolineae classis nov and Caldilineae classis nov in the bacterial phylum Chloroflexi. Int. J. Syst. Evol. Microbiol. 56, 1331–1340.
Keywords: microbial ecology, sulfate–methane transition zone, Nankai Trough sediment, 454 pyrosequencing, SSU rRNA, metabolically active
Citation: Mills HJ, Reese BK, Shepard AK, Riedinger N, Dowd SE, Morono Y and Inagaki F (2012) Characterization of metabolically active bacterial populations in subseafloor Nankai Trough sediments above, within, and below the sulfate–methane transition zone. Front. Microbio. 3:113. doi: 10.3389/fmicb.2012.00113
Received: 02 October 2011; Accepted: 12 March 2012;
Published online: 03 April 2012.
Edited by:
Andreas Teske, University of North Carolina at Chapel Hill, USAReviewed by:
William D. Orsi, Woods Hole Oceanographic Institution, USAAharon Oren, The Hebrew University of Jerusalem, Israel
Martin Krüger, Federal Institute for Geosciences and Natural Resources, Germany
Copyright: © 2012 Mills, Reese, Shepard, Riedinger, Dowd, Morono and Inagaki. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Heath J. Mills, Department of Oceanography, Texas A&M University, Mail Stop 3146, College Station, TX 77843, USA. e-mail: hmills@ocean.tamu.edu