
- 1Department of Biomolecular Engineering, University of California, Santa Cruz, CA, USA
- 2Janelia Farm Research Campus, Howard Hughes Medical Institute, Ashburn, VA, USA
Within the domain Archaea, the CRISPR immune system appears to be nearly ubiquitous based on computational genome analyses. Initial studies in bacteria demonstrated that the CRISPR system targets invading plasmid and viral DNA. Recent experiments in the model archaeon Pyrococcus furiosus have uncovered a novel RNA-targeting variant of the CRISPR system. Because our understanding of CRISPR system evolution in other archaea is limited, we have taken a comparative genomic and transcriptomic view of the CRISPR arrays across six diverse species within the crenarchaeal genus Pyrobaculum. We present transcriptional data from each of four species in the genus (P. aerophilum, P. islandicum, P. calidifontis, P. arsenaticum), analyzing mature CRISPR-associated small RNA abundance from over 20 arrays. Within the genus, there is remarkable conservation of CRISPR array structure, as well as unique features that are have not been studied in other archaeal systems. These unique features include: a nearly invariant CRISPR promoter, conservation of direct repeat families, the 5′ polarity of CRISPR-associated small RNA abundance, and a novel CRISPR-specific association with homologues of nurA and herA. These analyses provide a genus-level evolutionary perspective on archaeal CRISPR systems, broadening our understanding beyond existing non-comparative model systems.
Introduction
CRISPR immunity systems, like the vertebrate adaptive immune system (Boehm, 2011), include mechanisms to adapt to new pathogens, surveillance methods for detecting previously encountered pathogens, and means to inactivate those pathogens. In the case of the CRISPR system, the targeted molecule is a nucleic acid sequence, and the sequence of events moves from adaptation, where the invading nucleic acid sequence is recognized and acquired, to expression, where the CRISPR-specific small RNA recognition molecules (crRNA) are transcribed, processed and loaded by the CAScade complex of CRISPR-specific proteins (Brouns et al., 2008; Jore et al., 2011). The third phase, interference, is initiated upon detection of a targeted nucleic acid sequence and results in specific inactivation of the recognized nucleic acid from the invading “pathogen.” DNA of viral or plasmid origin has been shown to be the target of CRISPR defense in bacteria (Barrangou et al., 2007; Marraffini and Sontheimer, 2008) and the archaeon Sulfolobus solfataricus (Manica et al., 2011). RNA sequences are targeted in the CRISPR system present in Pyrococcus furiosus (Hale et al., 2009, 2012), opening the possibility of endogenous targeting of messenger RNA sequence.
Most archaeal and many bacterial genomes contain one or more loci that encode the CRISPR system. Each CRISPR locus consists of an array of short DNA sequences, and frequently includes a cluster of CRISPR-associated (CAS) protein coding genes (Haft et al., 2005). The DNA arrays are composed of a leader sequence, followed by a set of 24–47 nucleotide (nt) direct repeats (DR) that form the delimiting punctuation of the array. The sequences between DR, termed spacers, are found to be 26–72 nt in length and encode small RNAs that are the stored immune memory for the system. The transcriptional promoter for the array is likely to be encoded within the leader sequence (Haft et al., 2005; Lillestol et al., 2009; Horvath and Barrangou, 2010). In Escherichia coli, the specific promoters for the array and associated CAS genes have been identified (Pul et al., 2010).
CRISPR arrays are dynamic structures, some containing only a single sequence while others may be quite large; for example, crispr4 in Metallosphaera sedula is over 10,000 nt in length and contains over 160 spacer sequences (Grissa et al., 2007). The genomes of most strains of Methanococcus maripaludis contain only one CRISPR array locus whereas the genome of strain S2 has no CRISPR array present. In contrast, the genomes of Methanocaldococcus strains encode between seven and 20 individual CRISPR arrays. In Sulfolobus, recent work has shown that selective pressure can be introduced in vivo, which results in deletion of genomic loci containing all or part of the CRISPR/CAS system (Gudbergsdottir et al., 2011).
Individual spacer elements in CRISPR arrays are acquired in the adaptation phase, during exposure to an invading genetic element. Evidence from surviving, phage-challenged cells shows an addition of one or more spacer sequences at the leader-proximal end of the array. These new spacer sequences are identical to phage sequence, can be from either phage genome strand, and confer immunity to survivor progeny (Barrangou et al., 2007). During this spacer acquisition phase, the target sequence is integrated into the array, likely through the action of CAS1, CAS2, and possibly other CAS proteins (for example, CSN2 in the Streptococcus thermophilus Type II system). This adaptation process only requires a single direct repeat in the array (Yosef et al., 2012). It is unclear if the acquired DNA spacer is derived directly from invading DNA, or if the DNA spacer is a copy produced during the adaptation process.
The mechanism of immunity is still incompletely understood, but immunity is dependent on CAS genes (Barrangou et al., 2007; Brouns et al., 2008), usually located near one or more CRISPR arrays. Early studies showed that four CAS genes (cas1–4) were frequently associated with CRISPR arrays (Jansen et al., 2002; Haft et al., 2005). A role in CRISPR adaptation (acquisition of new spacers) has been proposed for cas1 and cas2 (Wiedenheft et al., 2012). Potentially, CAS4 is also involved during the acquisition phase; this hypothesis is based on the frequent cas4 genomic proximity to cas1 (Makarova et al., 2011).
The CAS genes have recently been reclassified into three main families based on gene content and mode of action of the associated system (Makarova et al., 2011). In Type I, II, and III-A CRISPR systems (Makarova et al., 2011), the target of the CRISPR immunity system is invading DNA (Marraffini and Sontheimer, 2008). In contrast, Type III-B systems target RNA instead of DNA (Hale et al., 2009, 2012). Type I systems have been studied in both bacteria and archaea, and have recently yielded low-resolution structures of the multimeric CAScade complex in both E. coli (Jore et al., 2011; Wiedenheft et al., 2011) and in the archaeon Sulfolobus solfataricus (Lintner et al., 2011). In Type I systems, the CAScade complex is required for maturing of CRISPR RNA (crRNA) that guide protective immunity during subsequent invasion by foreign DNA elements. This crRNA-enabled complex is also responsible for surveillance and eventual interference by recruiting additional CAS proteins (Wiedenheft et al., 2011). The primary transcript of the CRISPR array, pre-crRNA, is cleaved within the DR to generate the individual crRNA segments. In the Sulfolobus variant of CAScade, CAS6 is responsible for cleavage of pre-crRNA, while in E. coli this role is carried out by CAS6e, also known as CasE (Brouns et al., 2008). The short RNA segments that are released from pre-crRNA processing retain an 8 nt 5′ “handle” sequence from the upstream DR as part of the mature crRNA (Brouns et al., 2008). Processing of pre-crRNA transcripts in Sulfolobus has been reported to proceed from the 3′ distal end toward the 5′ leader sequence (Lillestol et al., 2009). It is unclear how this 3–5′ directionality is established, given the site-specific endonucleolytic nature of CAS6 (Carte et al., 2008).
The Type III-B RNA-targeting CRISPR systems have been investigated in Pyrococcus furiosus (Hale et al., 2009, 2012) and in Sulfolobus solfataricus (Zhang et al., 2012). These systems include the cmr family of CAS genes along with the nearly ubiquitous cas1, cas2, and cas6. The cmr complex is composed of the protein products of cmr1, cas10, and cmr3–cmr6, plus the cas6-derived crRNA. In Sulfolobus, an additional cmr component, cmr7, joins the complex.
All CRISPR systems examined to date load crRNAs with 5′ OH ends, although the crRNA length and mature state of the 3′ end varies by CRISPR type and by species. We have therefore utilized a cloning strategy that is independent of 5′ end chemistry and partially independent of 3′ end chemistry.
In this study, we show linkage of CAS protein types with families of CRISPR arrays, conservation of CRISPR array elements across the genus, a novel nurA-csm6-herA gene cluster associated with Pyrobaculum CRISPR arrays, and provide transcriptional support for polarity in crRNA abundance.
Methods
Culture Conditions
P. aerophilum cells were grown anaerobically in media containing 0.5 g/L yeast extract, 1X DSM390 salts, 10 g/L NaCl, 1X DSM 141 trace elements, 0.5 mg/L Fe(SO4)2(NH4)2, pH 6.5, with 10 mM NaNO3. P. islandicum and P. arsenaticum cells were grown anaerobically in media containing 10 g/L tryptone, 2 g/L yeast extract, 1X DSM390 salts, 1X DSM88 trace elements, and 20 mM Na2S2O3. P. calidifontis cells were grown aerobically in 1L flasks using 500 ml media containing 10 g/L tryptone, 2 g/L yeast extract, 1X DSM88 trace metals, 15 mM Na2S2O3, pH 6.8, loosely capped with moderate shaking at 125 rpm. Anaerobic cultures were grown in 2L flasks with 1L media, prepared under nitrogen with resazurin as a redox indicator at 0.5 mg/L; 0.25 mM Na2S was added as a reductant. All cultures were grown at 95C to late log or stationary phase, monitored at OD600.
The 10X DSM390 salts are comprised of (per liter ddH2O) 1.3 g (NH4)2SO4, 2.8 g KH2PO4, 2.5 g MgSO4·7H2O. The 100X DSM88 trace metal solution is comprised (per liter 0.12N HCl), 0.9 mM MnCl2, 4.7 mM Na2B4O7, 76 μM ZnSO4, 25 μM CuCl2, 12.4 μM NaMoO4, 18 μM VOSO4, 6 μM CoSO4. The 100X DSM141 trace metal solution is comprised of 7.85 mM Nitrolotriacetic acid, 12.2 mM MgSO4, 2.96 mM MnSO4, 17.1 mM NaCl, 0.36 mM FeSO4, 0.63 mM CoSO4, 0.68 mM CaCl2, 0.63 mM ZnSO4, 40 μM CuSO4, 42 μM KAl(SO4)2, 0.16 mM H3BO3, 41 μM Na2MoO4, 0.1 mM NiCl2, 1.14 μM Na2SeO3.
cDNA Library Preparation
The cDNA libraries were prepared using small RNA fractions collected from cells grown to stationary and exponential phase, using methods previously described (Bernick et al., 2012), with brief details given in Results. These two preparations were constructed for each of P. aerophilum, P. islandicum, P. arsenaticum, and P. calidifontis cultures, yielding a total of eight cDNA libraries.
The 3′ end chemistries of crRNA have been reported as either 2–3′ cyclic phosphate (Hale et al., 2012; Jore et al., 2011), or as 3′ OH (Hatoum-Aslan et al., 2011; Zhang et al., 2012). Under the acidic conditions (pH 5) used in RNA preparation in this study, we expect an equilibrium population of 3′ OH terminated RNA to exist under either scenario, providing a cloning method that is semi-independent of 3′ end chemistry.
Sequencing and Read Mapping
Sequencing was performed using a Roche/454 GS FLX sequencer, and the GS emPCR Kit II (Roche). Sequencing reads in support of this work are provided online via the UCSC Archaeal Genome Browser (http://archaea.ucsc.edu) (Chan et al., 2012).
Reads that included barcodes and sequencing linkers were selected from the raw sequencing data and used to identify reads from each of the eight pooled cDNA libraries. Reads were further consolidated, combining identical sequences with associated counts for viewing with the Archaeal Genome Browser. Reads were mapped to the appropriate genome [P. aerophilum (NC_003364.1); P. arsenaticum (NC_009376.1); P. calidifontis (NC_009073.1); P. islandicum (NC_008701.1); P. oguniense (NC_016885.1); P. neutrophilum (T. neutrophilus: NC_010525.1)] using BLAT (Kent, 2002), requiring a minimum of 90% identity (-minIdentity), a maximal gap of 3 (-maxIntron) and a minimum score (matches minus mismatches) of 16 (-minScore) using alignment parameters for this size range (-tileSize = 8-stepSize = 4). Reads that mapped equally well to multiple positions in the genome were excluded from this study. The remaining, uniquely mapped reads were formatted and visualized as BED tracks within the UCSC Archaeal Genome Browser.
Computational Prediction of Orthologous Gene Clusters
Computational prediction of orthologous groups was established by computing reciprocal best BLASTP (Altschul et al., 1990) (RBB) protein coding gene-pairs among pairs of four Pyrobaculum species. When at least three RBB gene-pairs select the same inter-species gene set (for example A pairs with B, B pairs with C, and C pairs with A), the cluster is considered an orthologous gene cluster.
CRISPR Array Mapping
Arrays were predicted using CRISPRfinder (Grissa et al., 2007). Arrays were merged in some cases-based on sequencing data evidence.
Results
CRISPR/CAS Protein Families
Three distinct types of CAS gene clusters exist within the six Pyrobaculum species examined (Figure 1 and Table A1) (Makarova et al., 2011). In most Pyrobaculum species, the Type I system is present, organized in submodules. Typically we find a submodule that includes: cas1, cas2, cas4, and a cas4 variant herein referred to as cas4′, previously described as csa1 (Haft et al., 2005) (submodule abbreviation cas4′-1-2-4). A second submodule is found nearby, comprising cas6, cas7, cas5, cas3′, cas3″, and cas8a2 (abbreviated cas6-7-5-3′−3″-8a2) (Figure 1). With the exception of P. islandicum, each species in the genus has these submodules or close variants, and one or more submodules may be duplicated. In some cases, terminal members of the submodule may be relocated, such as cas6 in P. calidifontis or P. neutrophilum. Type I subtypes are defined by the presence of specific genes: cas8a1 or cas8a2 (subtype I-A); cas8b (subtype I-B); cas8c (subtype I-C); cas10d (subtype I-D); cse1 (subtype I-E); and csy1 (subtype I-F) (Makarova et al., 2011). P. aerophilum, P. oguniense, and P. neutrophilum contain cas8a2, so fall within the definitive Type I-A subtype. P. arsenaticum and P. calidifontis do not appear to contain any recognized signature genes, so the subtype remains indeterminate. Notably, the Type I system is completely absent from P. islandicum.

Figure 1. Genomic arrangement of CRISPR modules within Pyrobaculum species. Colors indicate Type I CAS modules (red), Type III CAS modules (blue) and CRISPR-associated nurA-csm6-herA clusters (green). CRISPR DNA arrays represented as vertical bars. Arrangements of multiple genes in the same CAS family are indicated using filled triangles (cas1-cas2 is indicated as cas1 2). Genomic distances greater than 10 Kb are indicated using diagonal slashes (“/”). Gene strand is indicated relative to the solid black line for positive (above) and negative (below) orientations. Most Pyrobaculum species encode both Type I and III CAS modules; P. islandicum encodes only a Type III CAS module.
2). Genomic distances greater than 10 Kb are indicated using diagonal slashes (“/”). Gene strand is indicated relative to the solid black line for positive (above) and negative (below) orientations. Most Pyrobaculum species encode both Type I and III CAS modules; P. islandicum encodes only a Type III CAS module.
A second CAS group, the Type III-B family of RNA-targeting CAS genes, is present in four Pyrobaculum species but not in P. aerophilum or P. islandicum. Again, this second family is present as submodules, with cmr4, cmr5, cmr1, and cmr6 (cmr4-5-1-6) adjacent but on the opposite strand of the cmr3-cas10 submodule. One or both of these submodules include csx1, and are currently classified as members of Type III-U (unclassified Type III). We find that csx1 also appears in the Type I modules, so this suggests a broader role for csx1 among Pyrobaculum CAS modules.
The third kind of module found in the genus, Type III-A (csm), appears to be complete in P. aerophilum, and is the only apparent CAS family found in P. islandicum. Previously, Makarova suggested that CRISPR adaptation for Type III families may require use of cas1 and cas2 in trans from a resident Type I family member (Makarova et al., 2011). However, this option is unavailable in P. islandicum, suggesting that adaptation for Pyrobaculum Type III systems may not require cas1-cas2. Possibly, an undescribed enzyme fulfills this role, or P. islandicum may have lost the ability to further adapt its CRISPR arrays.
Curiously, csm6 is absent from P. islandicum, but is present in every other species examined in this study. This is notable because csm6 would be expected to be part of the Type III-A system in P. islandicum, and would not be expected in species that do not encode a complete Type III-A module. Both P. oguniense and P. neutrophilum encode a portion of the Type III-A module (csm3-csm5-cas10-csx1) but both species are missing csm2 and csm4. Where csm6 is present, it is located next to a conserved paralog of nurA and herA; these genes are near a CRISPR array in species of Pyrobaculum, Thermoproteus, and Vulcanisaeta, suggesting that this arrangement is widespread among the Thermoproteales.
The nurA-herA protein complex is comprised of a 5–3′ DNA exonuclease (nurA) and a bidirectional helicase (herA) with probable involvement in homologous recombination (HR) (Constantinesco et al., 2004). HR processing requires a 3′ single stranded DNA (ssDNA) resection of chromosomal ends resulting from a double-strand break, and in thermophilic archaea, that resection is carried out by the helicase-nuclease complex of HerA-NurA (Blackwood et al., 2012). In most Pyrobaculum spp., there are three or more paralogs of this gene-pair, one of which is clustered with csm6 and near a CRISPR array (Figure 1). Computationally predicted orthologs of the CRISPR-associated nurA-herA genes (RBB) show that this pair has been retained throughout the Pyrobaculum genus and more broadly among the Thermoproteales (Figure 2 and Table A2). In P. islandicum, however, the CRISPR-associated nurA-herA pair and csm6 are absent. We propose that the nurA-csm6-herA complex may be associated with adaptation in Pyrobaculum species. Three possibilities arise from this proposal: (1) adaptation in P. islandicum may have been lost; (2) adaptation in P. islandicum may occur using an alternative mechanism, possibly one of the nurA-herA paralogs; or (3) the nurA-csm6-herA trio may only be required in Type I CRISPR systems (Yosef et al., 2012).
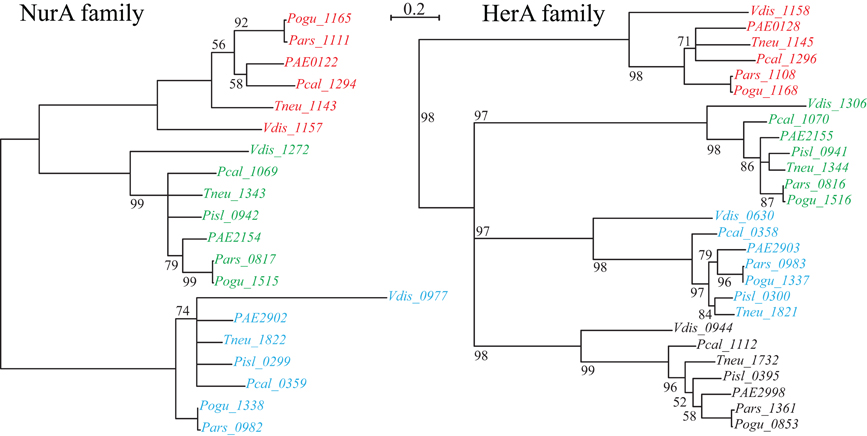
Figure 2. NurA and HerA gene families in Pyrobaculum species. Pyrobaculum species encode at least three nurA-herA gene-pairs, shown above to display parallel phylogenetic structure (left and right panels), with genomically adjacent genes matched by color. Within each gene clade (red, green or blue), the relationship of genes follows the expected species tree, indicating robust ortholog groups. The CRISPR-associated nurA-herA pairs (shown in red above) maintain the paired relationship throughout the Thermoproteales. An unmatched paralogous group of herA genes appears in black. The Vulcanisaeta distributa nurA-herA genes are provided as outgroups of the Pyrobaculum genus; note that only the CRISPR-associated nurA-herA pair (Vdis_1157-Vdis_1158) is collocated in V. distributa, further supporting the tight functional association. Bootstrap percentages are included in black (100% unless otherwise noted). Gene name prefixes: Vdis—Vulcanisaeta distributa; PAE—Pyrobaculum aerophilum; Pars—P. arsenaticum; Pisl—P. islandicum; Pcal—P. calidifontis; Pogu—P. oguniense; Tneu—P. neutrophilum. Multiple alignment of amino-acid sequences was performed using Clustal-Omega (v 1.04), maximum-likelihood trees were computed using Tree-puzzle (v 5.02), and the final tree was visualized using newicktops (v 1.0), available through the Pasteur Institute's Mobyle portal (http://mobyle.pasteur.fr/).
CRISPR Arrays
We have characterized three distinct families of CRISPR arrays present among six sequenced Pyrobaculum genomes (Table 1). These three families are defined by the sequences central to the DR and typically contain an A-rich core of 3–5 nt. These central motifs are flanked by short reverse complement (RC)-palindromes. The DR is terminated by an 8 nt-long sequence that becomes the 5′ handle of the mature crRNAs (Brouns et al., 2008). The various Pyrobaculum species encode between four and seven CRISPR arrays within their respective genomes. Except for P. islandicum, all species contain one or more representatives of family I and at least one additional representative from family III.
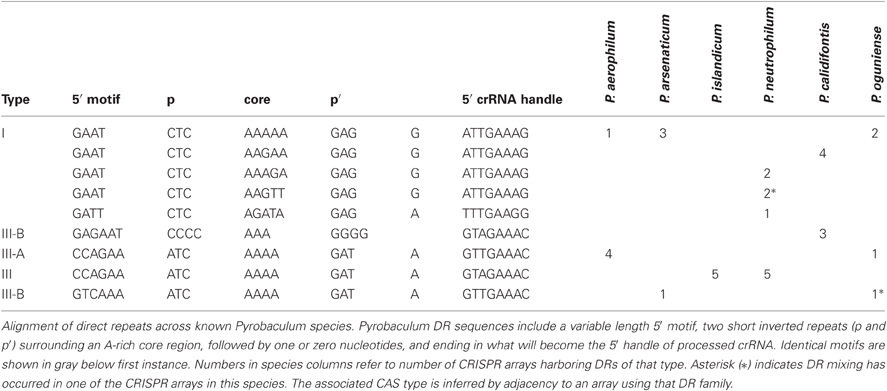
Table 1. Pyrobaculum direct repeat (DR) families.
A single array may include multiple families of DR sequences, as found in crispr1 of P. oguniense and crispr5 of P. neutrophilum. In these unusual cases, the DRs are clustered; for example in the P. neutrophilum case, the type I DR array begins with 11 repeats using the “AAGTT” core, followed by a set of four repeats mixing “AAAAA” with “AAAGA” cores, and terminating with three “AAAGA” core repeats. In P. oguniense, crispr1 has eight repeats with a 5′ motif of “GTCAAA” and five repeats with a 5′ motif of “CCAGAA.” In both cases where DR mixing was observed, the array type (based on CAS proteins) is maintained (Table 1). Previous studies in E. coli have shown that new DRs are added to an array during adaptation, by copying the first DR in the array (leader-proximal) (Yosef et al., 2012). We note that non-mixed arrays exist in P. neutrophilum whose leader-proximal repeats include the “AAAGA” and “AAGTT” cores. Potentially, DR mixing may come about through HR (duplication) events, or possibly by copying a leader-proximal DR from another array during adaptation.
A 5′ promoter-like sequence (AAAAACTTAAAAA) is ultra-conserved with only three single nt polymorphisms among all 37 CRISPR arrays in the six Pyrobaculum species studied. The same promoter-like element is also associated with some tRNA genes in these genomes. The sequence variation in the corresponding promoter elements for other genes is commonly much more diverse. This finding suggests that the invariant CRISPR promoter sequence is maintained either through strong purifying selection or through frequent gene-conversion (Liao, 2000).
CRISPR/CAS protein families appear to be associated with arrays of a given sequence family. This association is upheld to the CAS type, but does not extend to the subtype. For example, in P. islandicum, the only CAS family present is Type III-A (Figure 1) and the five encoded arrays in that species use a single DR type (Table 1). This same DR is also found in P. neutrophilum next to a Type III-B CAS cluster. In a second example, the mixed crispr1 in P. oguniense is made up of DRs associated with Type III-A CAS clusters as found in P. aerophilum, and Type III-B CAS clusters, as found in P. arsenaticum. Both of these examples demonstrate the association of CAS types (not subtypes) with CRISPR array families in the Pyrobaculum genus.
Pre-crRNA transcripts are subjected to endonucleolytic processing to yield individual crRNA sequences, which we detect within small-RNA libraries. Deep sequencing from four Pyrobaculum species yielded thousands of sequencing reads, representing between 3% (P. arsenaticum) and 20% (P. islandicum) of the total sequencing reads in the 20–70 nt size range (Table 2).
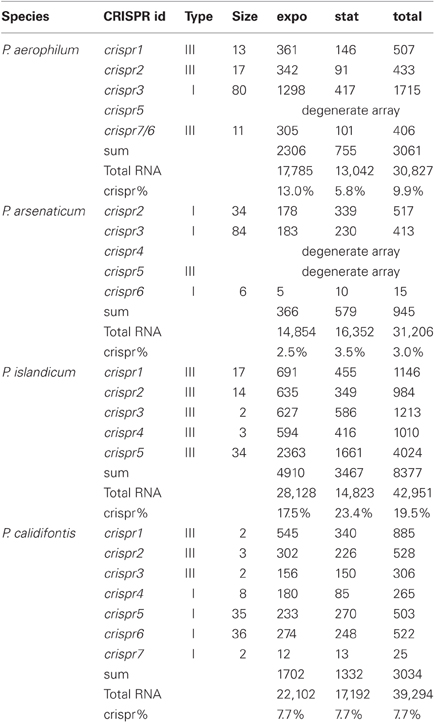
Table 2. CRISPR crRNA abundance (counts) in Pyrobaculum species from each CRISPR array, measured during exponential (expo) and stationary (stat) growth phases.
The abundance of individual crRNAs appears to be related to their position within the array (Figure 3). Abundance is generally highest when the spacer is located in the leader-proximal (5′) portion of the array, and decays distally (3′) (Figure 4), as seen in Pyrococcus (Hale et al., 2012). This pattern is evident in most Pyrobaculum arrays that contain more than five spacers. We also see significant variation in crRNA abundance against this decaying background pattern as described for Sulfolobus species (Zhang et al., 2012).
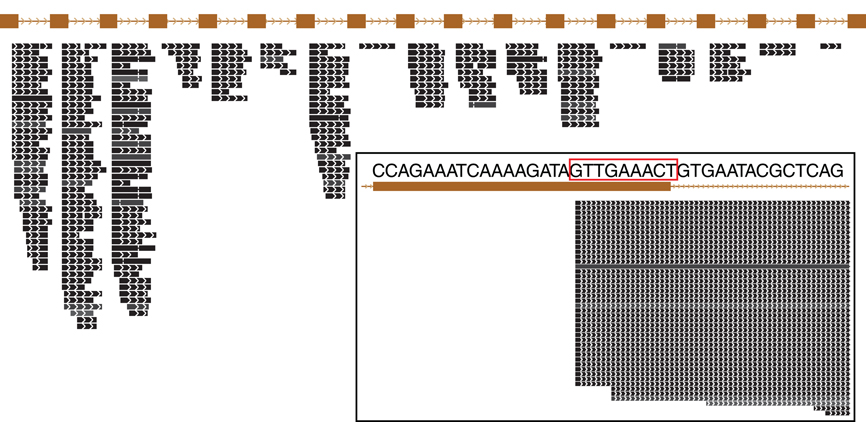
Figure 3. Small RNA expressed from a typical CRISPR array in Pyrobaculum aerophilum. The crispr2 array is shown, depicting the near-identical repeat regions (brown rectangles) with intervening spacer sequences. Mature crRNA reads (black bars) that map uniquely to individual spacers within CRISPR arrays are shown with strand indicated by interior arrowheads. The 5′ ends of crRNA are sharply terminated and includes an 8-base sequence (5′ handle boxed in red) derived from the upstream DR (inset panel). Images generated from the UCSC Archaeal browser (Chan et al., 2012). CRISPR annotation derived from CRISPRFinder (Grissa et al., 2007).
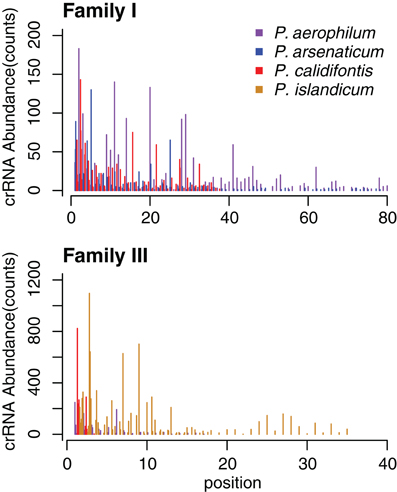
Figure 4. CRISPR small RNA abundance density in Pyrobaculum CRISPR arrays by family. Abundance is depicted on the vertical axes, in each species at every spacer position in the array. The horizontal axes plot crRNA genetic origin in terms of spacer number within CRISPR arrays, starting at the 5′ end of arrays. The abundance profile appears higher at the 5′ portion CRISPR arrays, with considerable variation deeper (3′) in the array. Multiple arrays of the same type within a species are plotted as adjacent bars by spacer position.
The majority of terminal positions of sequencing reads found in Pyrobaculum species include an 8-base portion of the upstream DR at the 5′ end (Figure A1); this corresponds to the 5′ handle (Brouns et al., 2008) (Figure 3). We also see a minority population of sequencing reads that include a 5-base portion of the upstream DR (Figure A1), though these are not present in P. islandicum.
We tested two models for 3′ maturation considering an upstream DR ruler-mechanism as seen in Staphylococcus species (Hatoum-Aslan et al., 2011), and a wrap-around model involving the downstream DR, as described for Pyrococcus furiosus (Wang et al., 2011). Because spacer sizes are not uniform in these species, we examined 3′ processing by testing distributions of 3′ end positions as measured from either the upstream DR or the downstream DR, under the assumption that spacer size variation would provide added noise to the incorrect model. Under the ruler-mechanism model, the 3′ distribution of end positions in P. aerophilum, P. arsenaticum, and P. calidifontis includes majority peaks at positions 40–41, and a minority peak at position 32 in P. aerophilum (Figure A2). Under the downstream DR based wrap-around model (Figure A3), P. aerophilum has a reduced peak at −25 (corresponding to position 40 in the ruler-mechanism model) and the minority peak is absent (seen previously at position 32). We consider this evidence as consistent with a ruler-mechanism for P. aerophilum CRISPR systems. In the remaining species, this analysis was inconclusive.
We find limited evidence for bidirectional CRISPR transcription as reported in Sulfolobus (Lillestol et al., 2009). Across all four of the Pyrobaculum species in the selected 16–70 nt size range, we see less than 1% of 15,417 CRISPR reads that map to the reverse strand of the array. Where those antisense reads are present, they appear to originate within the spacers and terminate at poly-T motifs within the DR regions. With the limited number of reverse reads seen in this size range, it appears that transcription from the opposite strand is either not processed down to the size range studied, or that reverse transcripts are much less abundant in Pyrobaculum. Potentially, this negative finding could be the result of the ubiquitous poly-A sequence present in every DR studied in this genus (Table 1). We anticipate that the poly-A sequence could mimic a poly-T terminator on the reverse strand, and thereby prevent significant reverse strand transcription.
Discussion
Within CRISPR arrays, we see an overabundance of reads emanating from the 5′ proximal portion in larger arrays, where transcription from these arrays is likely initiated from a single promoter. The polarity is not perfect given that the abundance of some distal spacers is greater in comparison to more proximal spacer positions. Clearly, there are a number of mechanisms or phenomena that could contribute to crRNA abundance across the array, including: (1) simple stochastic termination of the pre-crRNA transcript, (2) differential efficiency in the endonucleolytic processing of individual crRNAs, (3) transcriptional polarity within the array, (4) differential stability of individual crRNAs, (5) selective recovery and amplification of certain crRNA sequences during library preparation, and (6) recently evolved changes in spacer content (gain or loss or rearrangements) between the reference genome strain and the cultured strains used in our RNA-seq experiments.
It is unknown which or how many of the six possibilities are most relevant, although our data do not equally favor all. If we consider a model of passive, stochastic termination of the primary transcript, we could explain the 5′ polarity but fail to account for the intermediate crRNA variation. Alternatively, a model where individual spacers are matured (excised) from pre-crRNA with varying efficiency might explain the variation in spacer abundance, but the 5′ polarity would be more difficult to accommodate. Instead, we tend toward a model that relies on coupling of pre-crRNA transcription with processing of the transcript, which might explain both polarity and the intermediate variation; for example, if transcription is aborted under conditions of limiting processing capability. We note that some bacterial systems make use of rho-mediated termination, coupling transcription and translation in a manner that aborts transcription under conditions of limiting polysomes; this process yields an abundance polarity favoring genes that are near the 5′ end of an operon transcript. Recently, operon polarity has been described in the archaeon Thermococcus kodakaraensis (Santangelo et al., 2008). In a polarity model that couples CRISPR pre-crRNA transcription with crRNA processing, we hypothesize that given a limitation in processing by the CRISPR CAScade complex (or cmr-processing complex), the pre-crRNA transcript might be prematurely aborted, yielding an abundance of 5′ crRNA. Compelling evidence exists for incremental, endonucleolytic processing of the primary transcript in other species (Brouns et al., 2008; Hale et al., 2008). Under this 5′ polarity model, we would expect to see both polarity as well as a degree of variation in individual spacer abundance, which seems to match our data the closest. This model is necessarily incompatible with 3–5′ directional processing that has been suggested previously (Lillestol et al., 2006).
Within the Pyrobaculum genus, one of the conserved nurA-herA clusters of syntenic orthologs is always found next to a CRISPR array (Figure 2). This cluster includes csm6, a gene classified with the Type III-A CRISPR/CAS family. In every case observed, nurA-csm6 appear to be co-transcribed, in some cases with herA. The studied function of nurA-herA involves preparation of dsDNA ends as part of HR repair. If these genes participate in CRISPR processing, we suggest that they may be part of new spacer acquisition. That process requires the creation of a new DR and the integration of a novel spacer sequence into an existing array. Generally, this process yields an array with perfect copies, suggesting that the source of the novel DR sequence is an existing array element. In this model, a nurA-herA protein complex could provide the HR activity required to repair the array incision.
The phylogeny of the nurA-herA orthologous pairs suggests that they have been inherited vertically (Figure 2). Furthermore, a parsimonious interpretation of these gene trees indicates that the CRISPR-specific pair predates the divergence of Pyrobaculum species, and is well-represented across the Thermoproteaceae. The DR sequences that are in use throughout the Pyrobaculum are also remarkably conserved, with only three major sequence variants found, corresponding to the CAS proteins that make use of these structures. The structural conservation of the CAS operons is consistent across the Pyrobaculum clade, though not quite as invariant as seen in other archaeal or bacterial models. Finally, we find an ultra-conserved Pyrobaculum-specific promoter-like sequence across every CRISPR array examined. Taken together, we infer that the CRISPR system is endemic in the Pyrobaculum clade, and is unlikely to have been horizontally acquired through independent events for each of its members.
Cas6 is presumed to be responsible for cleavage of pre-crRNA, and through its association with the Cas complex is likely responsible for the association of Cas protein Types with CRISPR array families. Cas6 is believed to be responsible for recognition and cleavage of pre-crRNA (Hale et al., 2008). In Type I complexes, CAScade (Brouns et al., 2008) and aCAScade (Lintner et al., 2011), Cas6 is a co-purifying member of the complex. In Type III systems where Cas6 does not appear to be part of the Cas complex, specific proteins that are members of the complex are required for maturation of crRNA (Hatoum-Aslan et al., 2011). Furthermore, the binding of Cas6 in Pseudomonas aeruginosa has been shown to be quite specific (Sternberg et al., 2012), and in S. solfataricus, there are five distinct Cas6 proteins possibly specialized for specific repeats (Zhang et al., 2012). Taken together, we suggest that Cas6 mediates the association between Cas protein families and CRISPR array families in Pyrobaculum species. This mediation may be by direct participation in the Cas complex (Type I systems), or through an indirect association as suggested for Type III systems.
Our transcriptional data clearly show that the P. islandicum Type III-A system is capable of generating mature crRNA from each of its five arrays. This Type III-A system is operating without cas1, cas2, or csm6. In Pyrococcus abyssi, the Type I-A system generates crRNA (Phok et al., 2011) and is also missing cas1 and cas2. Possibly one or both of these systems has an alternative enzymatic method for incorporating novel spacers without CAS1, or one or both of these systems may be incapable of CRISPR adaptation. The missing csm6 in P. islandicum is equally surprising given that it has been considered essential in the Type III-A (csm) system, the only system present in this species. Establishing if P. islandicum is still capable of CRISPR adaptation could be a first step in identifying an alternative mechanism for spacer incorporation.
The classification system authored by Makarova (Makarova et al., 2011) has been instrumental in coordinating diverse efforts across the field of CRISPR research. As we examine new phylogenetic clades in detail, we have both a convenient mechanism for classifying our findings as well as adding variations brought into focus by new groups. In light of our new analyses, the consolidation of csa1 (described herein as cas4′) with cas4 may not be justified, as this would suggest many Pyrobaculum submodule examples with two copies of cas4 (cas4′-cas1-cas2-cas4). Alternatively, we suggest that the functions of cas4 and cas4′ (csa1) are distinct in Pyrobaculum and should be uniquely classified. Furthermore, we find csm6 (previously named APE2256) deeply associated with a CRISPR-associated nurA-herA pair, and not apparently part of the Type III-A module where it is currently classified. Finally, we observe that the csx1 classification (part of Type III-U) given to the numerous Pyrobaculum genes encoding a DXTHG domain (or MJ1666-like protein) may not be optimal; in Pyrobaculum, these genes appear to be found among Type I and III systems. Clearly, the unique comparative perspective afforded by Pyrobaculum provides numerous opportunities for future discovery.
Author Contributions
David L. Bernick designed and performed the experimental and computational analyses, and wrote the manuscript. Courtney L. Cox provided the analysis of the CRISPR promoter sequence conservation. Patrick P. Dennis provided assistance with the manuscript and collaborative review. Todd M. Lowe provided scientific advising, suggested analyses, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to members of the Joint Genome Institute for making 454 sequencing possible (P. Richardson and J. Bristow for providing resources, and E. Lindquist and N. Zvenigorodsky for sample preparation and analysis). This work was supported by National Science Foundation Grant EF-082277055 (Todd M. Lowe and David L. Bernick); the Graduate Research and Education in Adaptive Bio-Technology (GREAT) Training Program sponsored by the University of California Biotechnology Research and Education Program (David L. Bernick); and by the National Science Foundation while Patrick P. Dennis was working at the Foundation. The opinions, findings and conclusions expressed in this publication do not necessarily reflect the views of the National Science Foundation.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., Romero, D. A., and Horvath, P. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712.
Bernick, D. L., Dennis, P. P., Lui, L. M., and Lowe, T. M. (2012). Diversity of antisense and other non-coding RNAs in Archaea revealed by comparative small RNA sequencing in four Pyrobaculum species. Front. Microbio. 3:231. doi: 10.3389/fmicb.2012.00231
Blackwood, J. K., Rzechorzek, N. J., Abrams, A. S., Maman, J. D., Pellegrini, L., and Robinson, N. P. (2012). Structural and functional insights into DNA-end processing by the archaeal HerA helicase-NurA nuclease complex. Nucleic Acids Res. 40, 3183–3196.
Brouns, S. J., Jore, M. M., Lundgren, M., Westra, E. R., Slijkhuis, R. J., Snijders, A. P., Dickman, M. J., Makarova, K. S., Koonin, E. V., and Van Der Oost, J. (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964.
Carte, J., Wang, R., Li, H., Terns, R. M., and Terns, M. P. (2008). Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 22, 3489–3496.
Chan, P. P., Holmes, A. D., Smith, A. M., Tran, D., and Lowe, T. M. (2012). The UCSC archaeal genome browser: 2012 update. Nucleic Acids Res. 40, D646–D652.
Chenchik, A., Diachenko, L., Moqadam, F., Tarabykin, V., Lukyanov, S., and Siebert, P. D. (1996). Full-length cDNA cloning and determination of mRNA 5′ and 3′ ends by amplification of adaptor-ligated cDNA. Biotechniques 21, 526–534.
Constantinesco, F., Forterre, P., Koonin, E. V., Aravind, L., and Elie, C. (2004). A bipolar DNA helicase gene, herA, clusters with rad50, mre11 and nurA genes in thermophilic archaea. Nucleic Acids Res. 32, 1439–1447.
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8, 172.
Gudbergsdottir, S., Deng, L., Chen, Z., Jensen, J. V., Jensen, L. R., She, Q., and Garrett, R. A. (2011). Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol. Microbiol. 79, 35–49.
Haft, D. H., Selengut, J., Mongodin, E. F., and Nelson, K. E. (2005). A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 1:e60. doi: 10.1371/journal.pcbi.0010060
Hale, C., Kleppe, K., Terns, R. M., and Terns, M. P. (2008). Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA 14, 2572–2579.
Hale, C. R., Majumdar, S., Elmore, J., Pfister, N., Compton, M., Olson, S., Resch, A. M., Glover, C. V. 3rd, Graveley, B. R., Terns, R. M., and Terns, M. P. (2012). Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol. Cell 45, 292–302.
Hale, C. R., Zhao, P., Olson, S., Duff, M. O., Graveley, B. R., Wells, L., Terns, R. M., and Terns, M. P. (2009). RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139, 945–956.
Hatoum-Aslan, A., Maniv, I., and Marraffini, L. A. (2011). Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc. Natl. Acad. Sci. U.S.A. 108, 21218–21222.
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170.
Jansen, R., Embden, J. D., Gaastra, W., and Schouls, L. M. (2002). Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 43, 1565–1575.
Jore, M. M., Lundgren, M., Van Duijn, E., Bultema, J. B., Westra, E. R., Waghmare, S. P., Wiedenheft, B., Pul, U., Wurm, R., Wagner, R., Beijer, M. R., Barendregt, A., Zhou, K., Snijders, A. P., Dickman, M. J., Doudna, J. A., Boekema, E. J., Heck, A. J., Van Der Oost, J., and Brouns, S. J. (2011). Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat. Struct. Mol. Biol. 18, 529–536.
Liao, D. (2000). Gene conversion drives within genic sequences: concerted evolution of ribosomal RNA genes in bacteria and archaea. J. Mol. Evol. 51, 305–317.
Lillestol, R. K., Redder, P., Garrett, R. A., and Brugger, K. (2006). A putative viral defence mechanism in archaeal cells. Archaea 2, 59–72.
Lillestol, R. K., Shah, S. A., Brugger, K., Redder, P., Phan, H., Christiansen, J., and Garrett, R. A. (2009). CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol. Microbiol. 72, 259–272.
Lintner, N. G., Kerou, M., Brumfield, S. K., Graham, S., Liu, H., Naismith, J. H., Sdano, M., Peng, N., She, Q., Copie, V., Young, M. J., White, M. F., and Lawrence, C. M. (2011). Structural and functional characterization of an archaeal clustered regularly interspaced short palindromic repeat (CRISPR)-associated complex for antiviral defense (CASCADE). J. Biol. Chem. 286, 21643–21656.
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J., Charpentier, E., Horvath, P., Moineau, S., Mojica, F. J., Wolf, Y. I., Yakunin, A. F., Van Der Oost, J., and Koonin, E. V. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477.
Manica, A., Zebec, Z., Teichmann, D., and Schleper, C. (2011). In vivo activity of CRISPR-mediated virus defence in a hyperthermophilic archaeon. Mol. Microbiol. 80, 481–491.
Marraffini, L. A., and Sontheimer, E. J. (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845.
Phok, K., Moisan, A., Rinaldi, D., Brucato, N., Carpousis, A. J., Gaspin, C., and Clouet-D'orval, B. (2011). Identification of CRISPR and riboswitch related RNAs among novel noncoding RNAs of the euryarchaeon Pyrococcus abyssi. BMC Genomics 12, 312.
Pul, U., Wurm, R., Arslan, Z., Geissen, R., Hofmann, N., and Wagner, R. (2010). Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol. Microbiol. 75, 1495–1512.
Punta, M., Coggill, P. C., Eberhardt, R. Y., Mistry, J., Tate, J., Boursnell, C., Pang, N., Forslund, K., Ceric, G., Clements, J., Heger, A., Holm, L., Sonnhammer, E. L., Eddy, S. R., Bateman, A., and Finn, R. D. (2012). The Pfam protein families database. Nucleic Acids Res. 40, D290–D301.
Santangelo, T. J., Cubonova, L., Matsumi, R., Atomi, H., Imanaka, T., and Reeve, J. N. (2008). Polarity in archaeal operon transcription in Thermococcus kodakaraensis. J. Bacteriol. 190, 2244–2248.
Sternberg, S. H., Haurwitz, R. E., and Doudna, J. A. (2012). Mechanism of substrate selection by a highly specific CRISPR endoribonuclease. RNA 18, 661–672.
Wang, R., Preamplume, G., Terns, M. P., Terns, R. M., and Li, H. (2011). Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure 19, 257–264.
Wiedenheft, B., Lander, G. C., Zhou, K., Jore, M. M., Brouns, S. J., Van Der Oost, J., Doudna, J. A., and Nogales, E. (2011). Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 477, 486–489.
Wiedenheft, B., Sternberg, S. H., and Doudna, J. A. (2012). RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338.
Yosef, I., Goren, M. G., and Qimron, U. (2012). Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 40, 5569–5576.
Zhang, J., Rouillon, C., Kerou, M., Reeks, J., Brugger, K., Graham, S., Reimann, J., Cannone, G., Liu, H., Albers, S. V., Naismith, J. H., Spagnolo, L., and White, M. F. (2012). Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol. Cell 45, 303–313.
Appendix
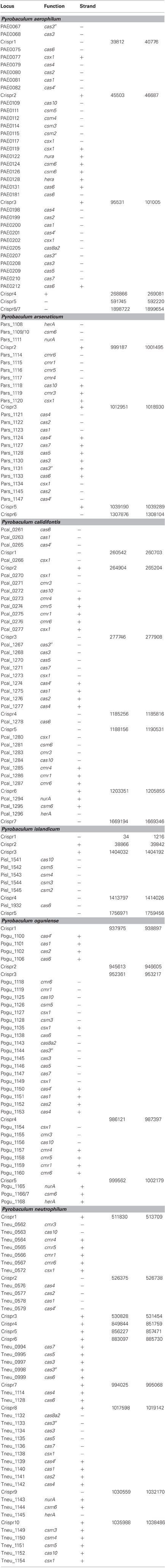
Table A1. Gene reannotations in Pyrobaculum species.
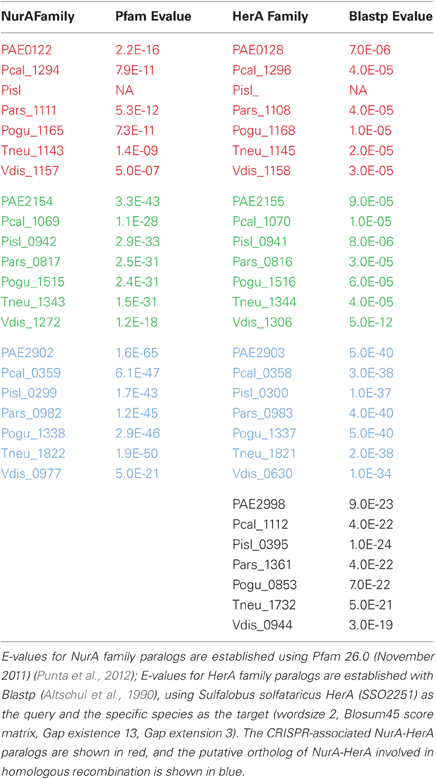
Table A2. NurA and HerA paralogs in Pyrobaculum species.
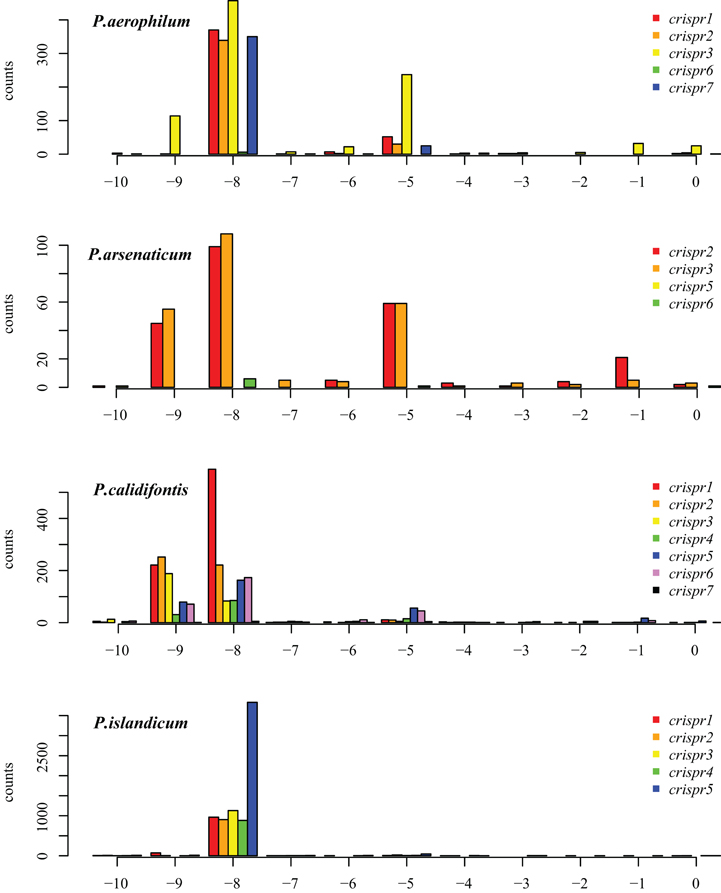
Figure A1. Distribution of mapped 5′ ends of crRNA associated reads within CRISPR arrays in P. aerophilum, P. arsenaticum, P. calidifontis, and P. islandicum. The majority of transcription sequencing reads begin at position −8 (relative to the beginning of the associated spacer (position 0). This finding implies that most crRNA associated sequencing reads include the 8 nucleotide 5′ handle sequence. A minority population of transcription reads begins at position −5. A third population of sequencing reads begin at position −9; these may be an artifact of the terminal transferase activity of MMLV derived reverse transcriptases. This activity most often yields a terminal cytosine residue to the 3′ end of the cDNA, yielding an implied “G” to the 5′ end of the sequencing read Chenchik et al. (1996).
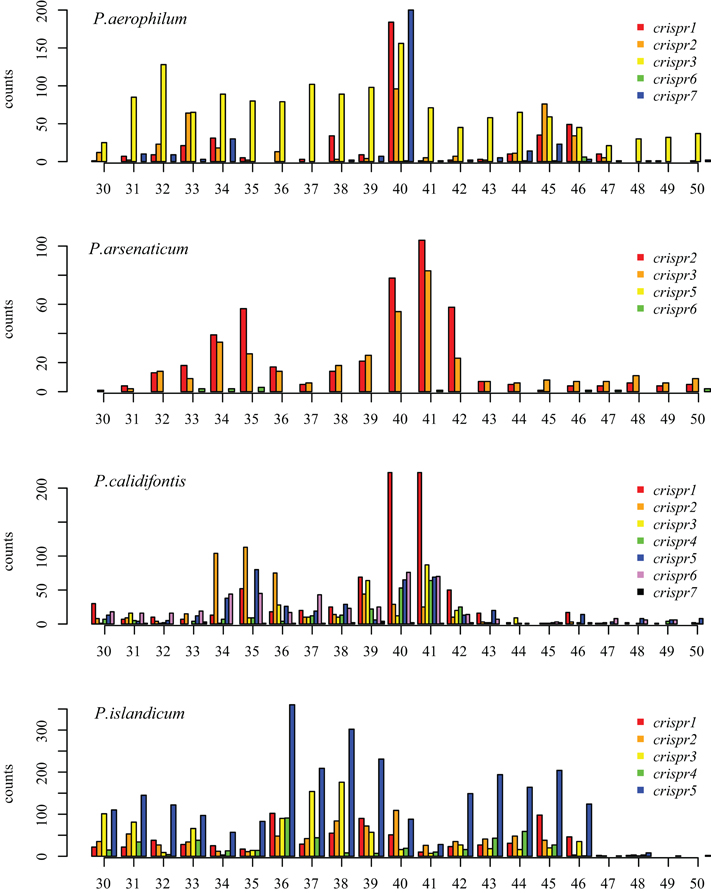
Figure A2. Distribution of mapped 3′ ends of crRNA associated sequencing reads, relative to the 5′ end of the associated spacer. This model proposes that cleavage of the 3′ end of crRNA associated reads utilizes a ruler-mechanism measured from the upstream DR. In P. aerophilum, P. arsenaticum, and P. calidifontis, the majority population of crRNA associated sequencing reads have a 3′ end centered around positions 40–41. A second minority population has a 3′ end centered around positions 32–35. In P. islandicum, the majority 3′ end is centered at positions 36–38, and a second minority 3′ end is centered around positions 42–46.
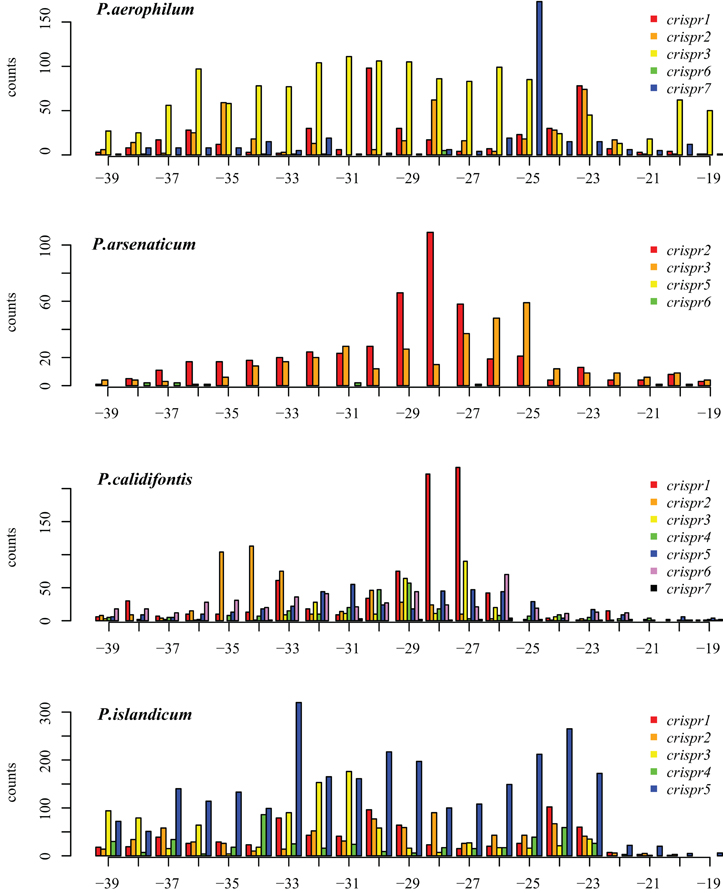
Figure A3. Distribution of mapped 3′ ends of crRNA associated sequencing reads, relative to the downstream Direct repeat. (position 0 is the start of the downstream spacer). This alternative model proposes that the downstream DR establishes the 3′ cut site. In P. aerophilum, the major population of 3′ ends shown for crispr 1–3 (Figure A2, position 40) is much more diffuse when measured in relation to the down stream direct repeat; this suggests that the 3′ cleavage of crRNA better modeled using the upstream DR as reference rather than the alternative, downstream DR reference. A single spacer region dominates abundance of crRNA in P. aerophilum.crispr7; this abundance provides the peak at −25 (corresponding to position 40 in Figure A2). In the remaining species, an attempt to distinguish between models of the underlying 3′ cleavage position was inconclusive.
Keywords: Pyrobaculum, CRISPR, sRNA, crRNA, repeat, RNAseq
Citation: Bernick DL, Cox CL, Dennis PP and Lowe TM (2012) Comparative genomic and transcriptional analyses of CRISPR systems across the genus Pyrobaculum. Front. Microbio. 3:251. doi: 10.3389/fmicb.2012.00251
Received: 30 April 2012; Paper pending published: 11 May 2012;
Accepted: 24 June 2012; Published online: 13 July 2012.
Edited by:
Zvi Kelman, University of Maryland, USACopyright © 2012 Bernick, Cox, Dennis and Lowe. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Todd M. Lowe, Department of Biomolecular Engineering, University of California, 1156 High St, Santa Cruz, CA 95064, USA. e-mail: lowe@soe.ucsc.edu