
- 1 Department of Bioinformatic Engineering, Graduate School of Information Science and Technology, Osaka University, Osaka, Japan
- 2 Division of Life Science and Engineering, School of Science and Engineering, Tokyo Denki University, Saitama, Japan
- 3 Computational Biology Research Center, Advanced Industrial Science and Technology, Tokyo, Japan
Chemokine receptors (CKRs) function in the inflammatory response and in vertebrate homeostasis. Decoy and viral receptors are two types of CKR homologs with modified functions from those of the typical CKRs. The decoy receptors are able to bind ligands without signaling. On the other hand, the viral receptors show constitutive signaling without ligands. We examined the sites related to the functional difference. At first, the decoy and viral receptors were each classified into five groups, based on the molecular phylogenetic analysis. A multiple amino acid sequence alignment between each group and the CKRs was then constructed. The difference in the amino acid composition between the group and the CKRs was evaluated as the Kullback–Leibler (KL) information value at each alignment site. The KL information value is considered to reflect the difference in the functional constraints at the site. The sites with the top 5% of KL information values were selected and mapped on the structure of a CKR. The comparisons with decoy receptor groups revealed that the detected sites were biased on the intracellular side. In contrast, the sites detected from the comparisons with viral receptor groups were found on both the extracellular and intracellular sides. More sites were found in the ligand binding pocket in the analyses of the viral receptor groups, as compared to the decoy receptor groups. Some of the detected sites were located in the GPCR motifs. For example, the DRY motif of the decoy receptors was often degraded, although the motif of the viral receptors was basically conserved. The observations for the viral receptor groups suggested that the constraints in the pocket region are loose and that the sites on the intracellular side are different from those for the decoy receptors, which may be related to the constitutive signaling activity of the viral receptors.
Introduction
The members of the chemokine (CK) family play important roles in regulating cell migration against inflammation, immune surveillance, and oncogenesis in vertebrates (Zlotnik and Yoshie, 2000). The CKs are classified into four subfamilies: CC, CXC, CX3C, and XC, based on the cysteine positions in their motifs (Zlotnik and Yoshie, 2000). CKs exert their activities through binding to their corresponding receptors. Presently, more than 40 CKs and 18 chemokine receptors (CKRs) have been identified in the human genome: 10 CCRs, six CXCRs, one XCR, and one CX3CR (Nomiyama et al., 2011). The CKR homologs are widely distributed among the vertebrate genomes. For example, homologs have even been identified from sea lampreys, which are one of the most primitive vertebrates (Nomiyama et al., 2011). The amino acid sequence identities among the CKRs and the homologs range from 25 to 80%, and the CKRs constitute a subfamily in the class A G protein-coupled receptors (GPCRs). The CKRs have broad ligand specificities (Nomiyama et al., 2011), and each receptor is able to interact with several CKs, and vice versa. This binding promiscuity makes it difficult to develop drugs to pinpoint the specific function of each CKR. Among these receptors, only the structure of CXCR4 has been solved by X-ray crystallography (Wu et al., 2010). Like the other GPCRs, this structure is characterized by the seven transmembrane (TM) helices, although T4 lysozyme was inserted within the intracellular loop (ICL) 3 between the TM helices 5 and 6, to stabilize the crystal. The extracellular cavity of CXCR4 is reportedly larger and wide open, as compared to those of other GPCR structures (Wu et al., 2010).
In addition to the traditional CKRs, five non-signaling CKR homologs have been identified in vertebrate genomes: CCRL1 (also known as CCX-CKR), CCRL2, CCBP2 (D6), CXCR7, and DARC (Duffy antigen receptor; Graham, 2009; Leick et al., 2010; Naumann et al., 2010). They are called “decoy” or “silent” receptors, because they are able to bind to several CKs without ligand-induced signaling. Most of them are constitutively internalized with or without ligands, and only the receptors are recycled to the cell membrane. Their functions are considered to regulate inflammatory responses by controlling the volume of free extracellular CKs, through internalization and degradation (Bonecchi et al., 2010). Like the traditional CKRs, these decoy receptors show a broad CK-binding spectrum. CCRL1 interacts with several homeostatic CC-type CKs (Comerford et al., 2006), whereas CCBP2 and DARC interact with inflammatory CKs (Graham, 2009). CXCR7 interacts with the dual-functional CXC-type CKs (Naumann et al., 2010) without activating G proteins (Thelen and Thelen, 2008). CCRL2 is known to be a multifunctional receptor (Yoshimura and Oppenheim, 2011). Like other decoy receptors, it regulates the amount of free CKs. At the same time, it functions as a receptor for an adipokine called chemerin, although the ligand binding does not induce signaling and the receptor is not internalized even after ligand engagement. DARC is the most distantly related to the CKRs among the five decoy receptors, and was originally identified as a malarial parasite receptor (Bonecchi et al., 2010). The receptor also binds to the CC- and CXC-type inflammatory CKs.
Chemokine receptors homologs have been detected in double stranded DNA viruses, such as herpesvirus and poxvirus. These viruses are considered to have gained these proteins by horizontal gene transfer during the course of evolution (Slinger et al., 2011). The viral receptors are constitutively active without ligands, although some of them can bind to CKs. We studied five groups of viral proteins as described below. E1 is derived only from equid herpesvirus 2 of γ-herpesvirinae, which interacts with CCL11 (Camarda et al., 1999). ORF74 is derived from several γ-herpesviruses, and interacts with a broad range of CXC-type CKs (Maussang et al., 2009). The β-herpesviruses also have several CKR homologs. Among them, UL33 is encoded by the genomes of various vertebrate viruses, although its ligands have not been identified (Gruijthuijsen et al., 2002). On the other hand, the US27, US28, and vGPCRs, which share high sequence similarity, have only been identified in the primate β-herpesviruses (Sahagun-Ruiz et al., 2004). US28 is characterized as a receptor for CC-type ligands (Maussang et al., 2009). Several poxviruses, such as capripox virus, deerpox virus, and yatapox virus, also encode CKR homologs in their genomes. The receptors of poxviruses not only share high amino acid sequence similarity to CCR8, but also the CCR8-like CK-binding profile; that is, high affinity to CCL1 (Najarro et al., 2006). These viral receptors are considered to contribute to the escape from and/or the perturbation of the host immune system, and are involved in inflammatory diseases and cancer (Slinger et al., 2011), although the mechanisms of these receptors in viral pathogenesis remain poorly understood.
The CKRs and their homologs have been classified into three functionally different types, from the viewpoints of ligand binding and signaling. The traditional CKRs bind their ligands, which induce signal transduction. The decoy receptors also bind ligands, although the process does not induce signal transduction. In contrast, the viral receptors exhibit signaling activity without ligand binding. The decoy receptors and the viral receptors are considered to have functionally differentiated after their divergence from the traditional CKRs, by gene duplication or horizontal gene transfer. Therefore, the functional differentiation of these three types is expected to have changed the functional constraints at the amino acid sites responsible for the ligand binding and/or signaling. If the sites involved in the functional differentiation can be identified, then the information about the sites would be helpful to understand the mechanisms for the signaling associated with ligand-induced conformational changes. Several different methods have been developed to detect the amino acid sites involved in the functional differentiation of homologous proteins from a multiple sequence alignment, and they are roughly classified into two types. One of them examines the difference in the evolutionary rate at each alignment site among the proteins with different functions (Gu, 1999; Simon et al., 2002), while the other compares the amino acid compositions at each alignment site among the proteins with different functions (Landgraf et al., 1999; Hannenhalli and Russell, 2000). We applied the latter method, the comparison of amino acid compositions, to identify the sites involved in the functional differentiation of CKR homologs. The difference in the amino acid composition at each alignment site between two groups (CKRs and decoy receptors, or CKRs and viral receptors) was calculated as the Kulback–Leibler (KL) information value (Hannenhalli and Russell, 2000; Ichihara et al., 2004). The sites with large KL information values were selected as the candidates for the functional differentiation. The amino acid residues corresponding to the selected sites were mapped on the tertiary structure of CXCR4. The comparison of the CKRs and decoy receptors revealed that the sites with large KL information values were concentrated on the cytosolic side of the CKR structure, with statistical significance. In contrast, there was no such bias in the distribution of the sites with large KL values between the CKRs and viral receptors. Based on the detected sites and the distribution of the corresponding residues on the tertiary structure, the underlying mechanisms for the functional divergence of the CKR homologs will be discussed.
Materials and Methods
Amino Acid Sequence Data
The amino acid sequences of the CKRs and their homologs, including decoy receptors and viral receptors, were collected by searching the non-redundant protein sequence database at the NCBI site1 with BLAST version 2.2.25 (Altschul et al., 1997). The amino acid sequence of human CXCR4 (GI number of NCBI: 1705894) was used as the query for the BLAST search. The sequence similarity search was also performed against the Ensembl2 and elephant shark genome project3 genome databases. When several amino acid sequences were almost identical, one of them was selected as the representative. The sequences used in this study are shown in Table 1.
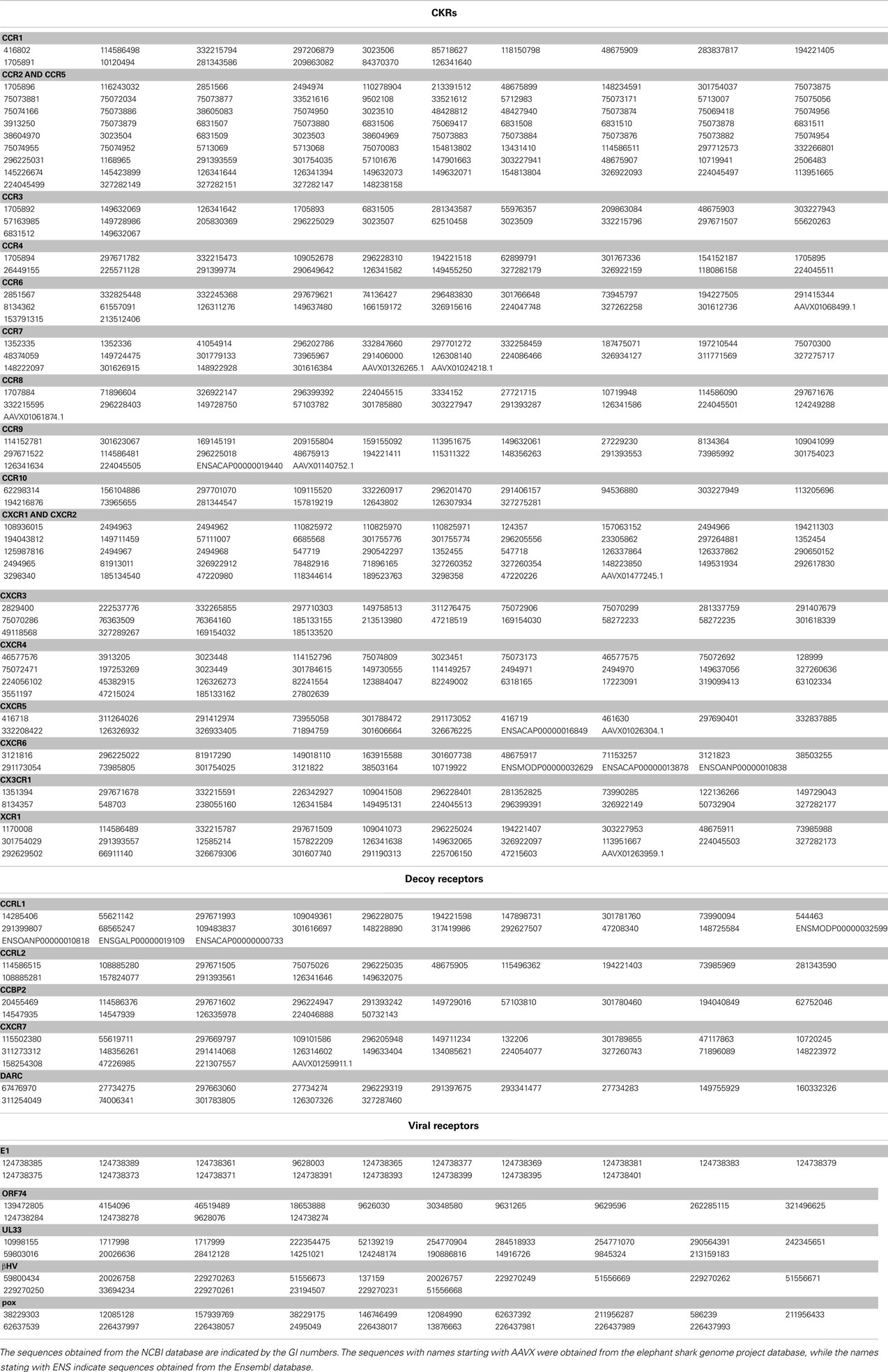
Table 1. The sequences of the CKRs, decoy receptors, and viral receptors.
Amino Acid Sequence Alignment and Phylogenetic Analysis
A multiple amino acid sequence alignment was produced with the alignment software MAFFT, version 6.857 (Katoh et al., 2002; Katoh and Toh, 2008). At first, 444 traditional CKRs were aligned. This result was manually refined, based on information about the secondary structures. Subsequently, 178 sequences consisting of decoy and viral receptors were added to the CKR alignment one by one, using the profile option of Clustal W (version 1.83; Thompson et al., 1994). Based on the alignment, an unrooted molecular phylogenetic tree was constructed by the neighbor-joining (NJ) method (Saitou and Nei, 1987). The genetic distance between every pair of aligned sequences was calculated as a maximum likelihood estimate (Felsenstein, 1996), under the JTT model (Jones et al., 1992) for the amino acid substitutions. The sites including gaps in the alignment were excluded from the calculation. The statistical significance of the NJ tree topology was evaluated by a bootstrap analysis (Felsenstein, 1985) with 1,000 iterative tree reconstructions. Two software packages, PHYLIP 3.5c (Felsenstein, 1993) and MOLPHY 2.3b3 (Adachi and Hasegawa, 1996), were used for the phylogenetic analyses. A cluster of decoy or viral receptors with a bootstrap probability greater than 80% was adopted as a group of receptors with different functions from the traditional CKRs.
Calculation of the Kullback–Leibler Information Value
The multiple alignment thus obtained was reconstructed into 10 alignments, each consisting of two groups, the traditional CKRs and one of the decoy receptor groups or viral receptor groups. We then calculated the amino acid compositions of the two groups at each alignment site, according to the multiple alignment. We used the method adopted in PSI-BLAST (Altschul et al., 1997) to estimate the site-specific amino acid composition. The weighting method of Henikoff and Henikoff (1994) was used for the residue count. The weight for the pseudocount β, was set to 0.1. For the calculation of the pseudocount, λu, a parameter for ungapped BLAST, was calculated for each alignment by the Newton–Raphson method (Ewens and Grant, 2001). When more than half of the sequences had gaps at an alignment site, the calculation of the site-specific amino acid composition and the following investigation were skipped. Next, the difference in the amino acid composition between two groups at each alignment site was calculated as the KL information value. The KL information value is defined as follows:
where p and q are the site-specific amino acid residue compositions for the two groups, which are estimated by the method described above. The parameter i indicates that the summation is obtained over 20 amino acid residues. KL information does not satisfy one of the distance axioms, symmetry. To satisfy this condition, the KL information was modified as follows:
Formula 2 was used to predict the sites subjected to different functional constraints between the two groups. In this study, the functional constraint at a site of a protein sequence is defined as the extent of intolerance to mutation at the site, due to a reduction of the protein function by the mutation. This is a special case of the cumulative relative entropy developed by Hannenhalli and Russell (2000), which is applicable to an alignment consisting of multiple groups. When the KL information value of an alignment site was located in the top 5% of the distribution of KL information values for all of the sites, the site was regarded as a site under different constraints between the groups (Ichihara et al., 2004). Among them, the sites that fell in the gap region of CXCR4 in the alignment were neglected, because the subsequent analyses were performed based on mapping onto the CXCR4 structure.
Statistical Evaluation for Bias in the Spatial Distribution of the Sites under Different Constraints
We examined the statistical significance for the bias in the positions of the selected sites by the KL information values on the reference CXCR4 structure (PDB ID: 3ODU), using the following procedure. At first, we calculated the geometric center of the three extracellular loops (ECLs) and the N-terminal region, and that of the three ICLs. The coordinates of the Cα atoms were used for the calculation. The C-terminal region (residues 303–328) was not used in the calculation of the geometric center of the intracellular side, since this region extended into the cytosolic region. The chimeric lysozyme region was also neglected from the calculation. A unit vector on the axis connecting the two geometric centers, which originated from the midpoint between the geometric centers toward the geometric center of the extracellular side, was calculated. The inner product between the unit vector and a vector from the midpoint to the Cα atom of every residue, except for those of the chimeric lysozyme region, was then calculated. The inner product score indicated the projected position of the residue on the axis (see Figure 1). The positive or negative score corresponded to the extracellular or cytosolic location of a residue, respectively, relative to the geometric center. The distribution of the inner product scores for the residues selected by the KL information values was compared with those of the remaining residues by the two-sided t-test. The null hypothesis was the same for all of the tests: the average of the residues corresponding to the sites with large KL information values is the same as that of the remaining residues. For the statistical test, the function in the statistical computing software R, “t-test,” was used for the evaluation.
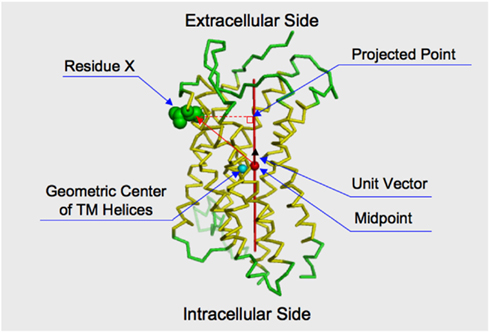
Figure 1. Projection of a residue on the axis connecting the intracellular and extracellular sides of the receptor. The structure of CXCR4 is shown by the ribbon model. The membrane spanning helices indicated by the structural element page for CXCR4 in GPCRDB (http://www.gpcr.org/7tm/) are colored yellow. The sphere colored cyan indicates the geometric center of the alpha carbons of the membrane spanning helices. The red axis connects the geometric center of extracellular loops and the N-terminal loop and that of the intracellular loops. The midpoint of the axis is indicated by a filled sphere colored red. The distance between the cyan and red spheres is close (3.26 Å). That is, the midpoint is considered to roughly reflect the geometric center of the transmembrane helices. How to take the orthogonal projection of an amino acid residue to the axis is shown by using Residue X. Consider a vector from the midpoint to the Cα atom of the residue. By taking an inner product between the vector and a unit vector, which runs along the axis and is originated from the midpoint. The projected point is obtained by taking the inner product.
Residue Indication
The sites of each group selected by the KL information values are indicated on the corresponding sites of CXCR4 in this study. When the site has the number based on Ballesteros–Weinstein nomenclature (Ballesteros and Weinstein, 1995), the figure is also shown in the superscript. In this notation, the first digit indicates the number of the TM helix, and the following digit is the position counted from the most conserved site in each TM, to which the number 50 is assigned.
Results
The Phylogenetic Analysis
The multiple alignment of 622 sequences were constructed, which is downloadable from the URL: http://seala.cbrc.jp/~toh/suppl.html. The alignment of the representative sequences is shown in Figure 2. Based on the alignment, the phylogenetic tree of the CKRs and the decoy and viral receptors was constructed (Figure 3). Several clusters with high bootstrap probability (>80%) were identified in the tree, which included five decoy receptor groups and five viral receptor groups. The decoy receptor groups are referred to as CCRL1, CCRL2, CCBP2, CXCR7, and DARC, according to the constituent receptors. The numbers in each group were 23, 15, 15, 24, and 15, respectively. On the other hand, the viral receptor groups are referred to as E1, ORF74, UL33, βHV, and pox. The first three groups were named according to the constituent receptors. The βHV group consists US28, US27, and vGPCRs. Pox is a group of receptors derived from poxviruses. The numbers in each viral group were 18, 14, 19, 16, and 19. The evolutionary relationships between the CKRs and the decoy and viral receptors shown in the figure were roughly similar to those reported previously (Rosenkilde et al., 2001; Zlotnik et al., 2006). Murphy et al. (2000) suggest that the evolutionary rates of the CKRs are faster than those of the other GPCRs, because of the immune functions of CKRs. The long branch lengths suggested that the evolutionary rates of the receptors belonging to CCRL2, DARC, ORF74, UL33, βHV, and pox may be higher than those of the traditional CKRs, although we refrained from further examination of evolutionary rate accelerations in this study. In the subsequent analyses, each group of the decoy and viral receptors thus obtained was compared with the group of the traditional CKRs.
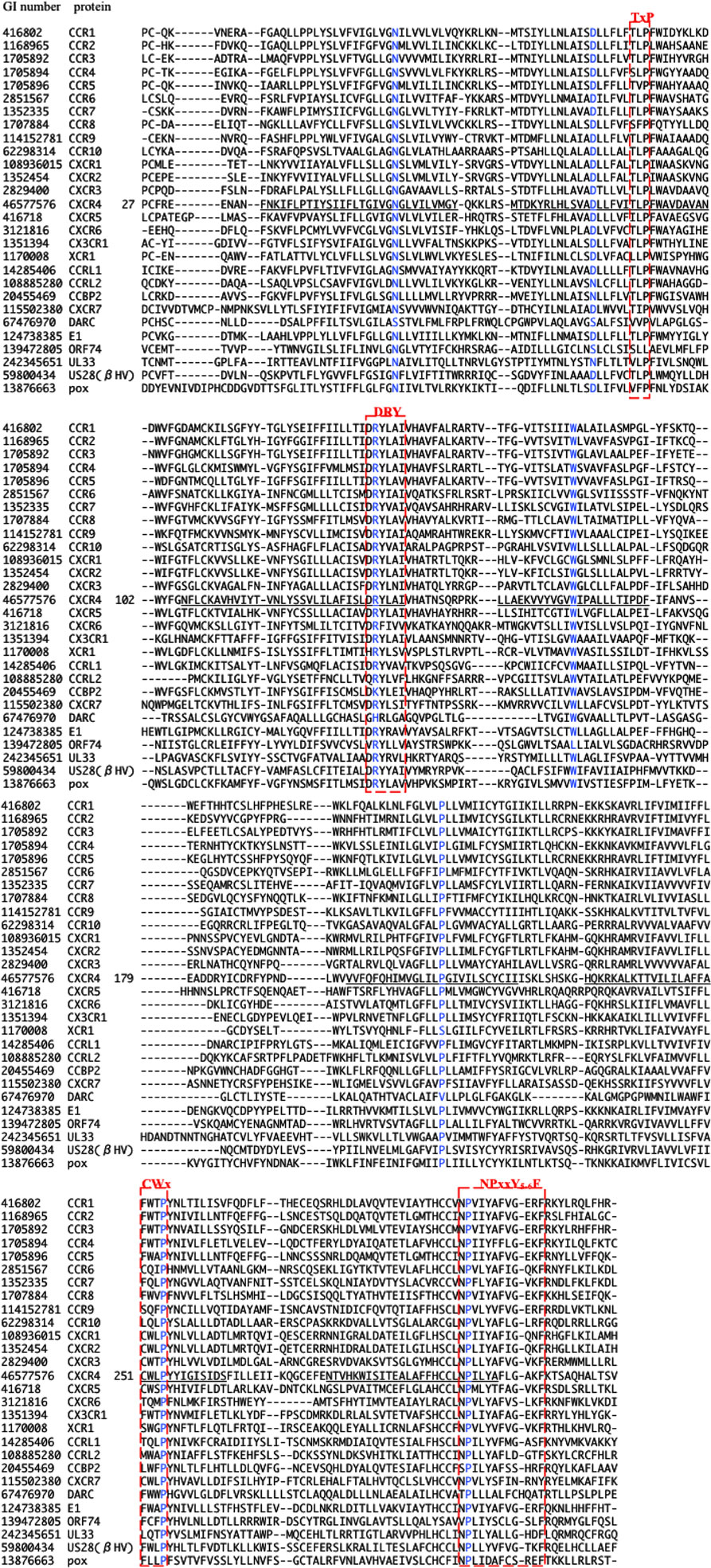
Figure 2. Multiple amino acid sequence alignment of CKRs, decoy, and viral receptors. The GI number and protein name of the representative protein from each group are shown at the left side of the aligned amino acid sequence. In the case of CXCR4, the residue number of the first residue of the aligned sequence is shown after the protein name, and the TM regions described in the GPCRDB (http://www.gpcr.org/7tm/) are indicated by underlines. The corresponding sites for x.50 of Ballesteros–Weinstein nomenclature are colored blue. Four motifs, TxP, DRY, CWxP, and NPxxY5-6F, are enclosed by red line.
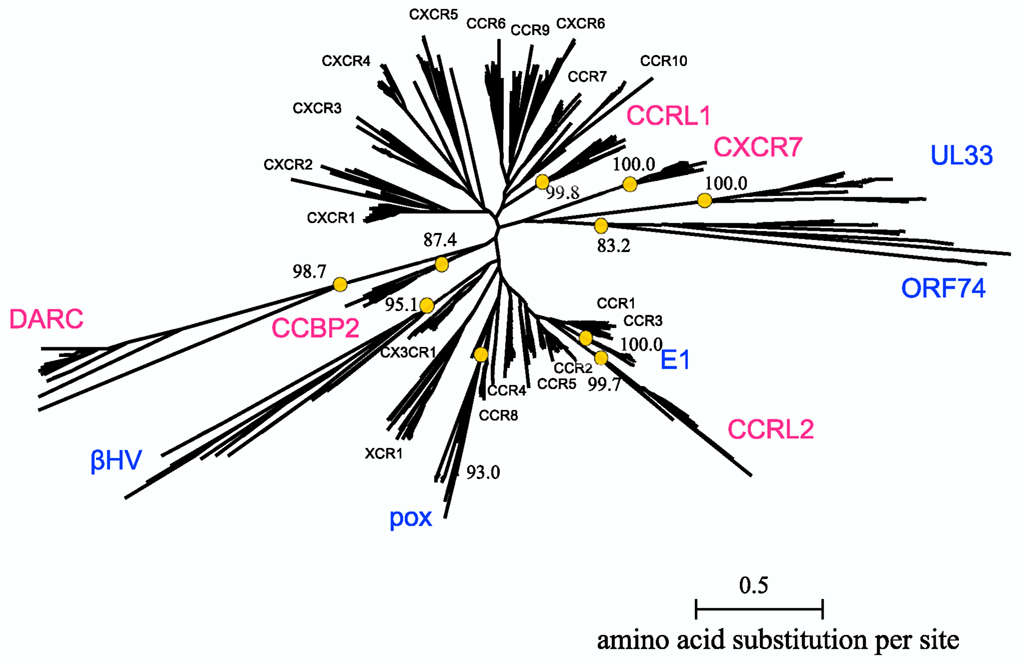
Figure 3. Phylogenetic tree of CKRs and their homologs. The tree for the 622 sequences listed in Table 1 is shown. The names of the CKRs (black), the decoy receptor groups (magenta), and the viral receptor groups (blue) are indicated near the receptor clusters. The bootstrap probabilities of the decoy and viral receptor groups are shown at the nodes corresponding to the common ancestors of the groups, which are indicated by circles.
Detection of Sites with Large KL Information Values
The differences in the amino acid composition at each alignment site were examined between the traditional CKRs and each group of decoy and viral receptors. The sites with large KL information values in the top 5% are summarized in Table 2. The residues corresponding to such sites were mapped on the structure of CXCR4 (Figure 4). As shown in Table 2, about 11 ~ 14 sites were selected from each group with the comparison of CKRs, and they included the sites in the sequences for GPCRs or the CKR-specific motif. Several sites that have been experimentally identified to be important for ligand binding or signaling were also selected. In addition, many uncharacterized sites were detected.
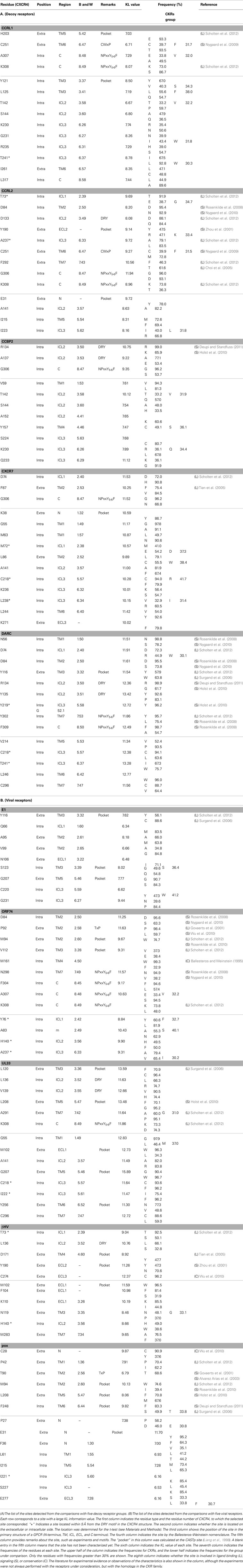
Table 2. Selected sites with large KL information values.
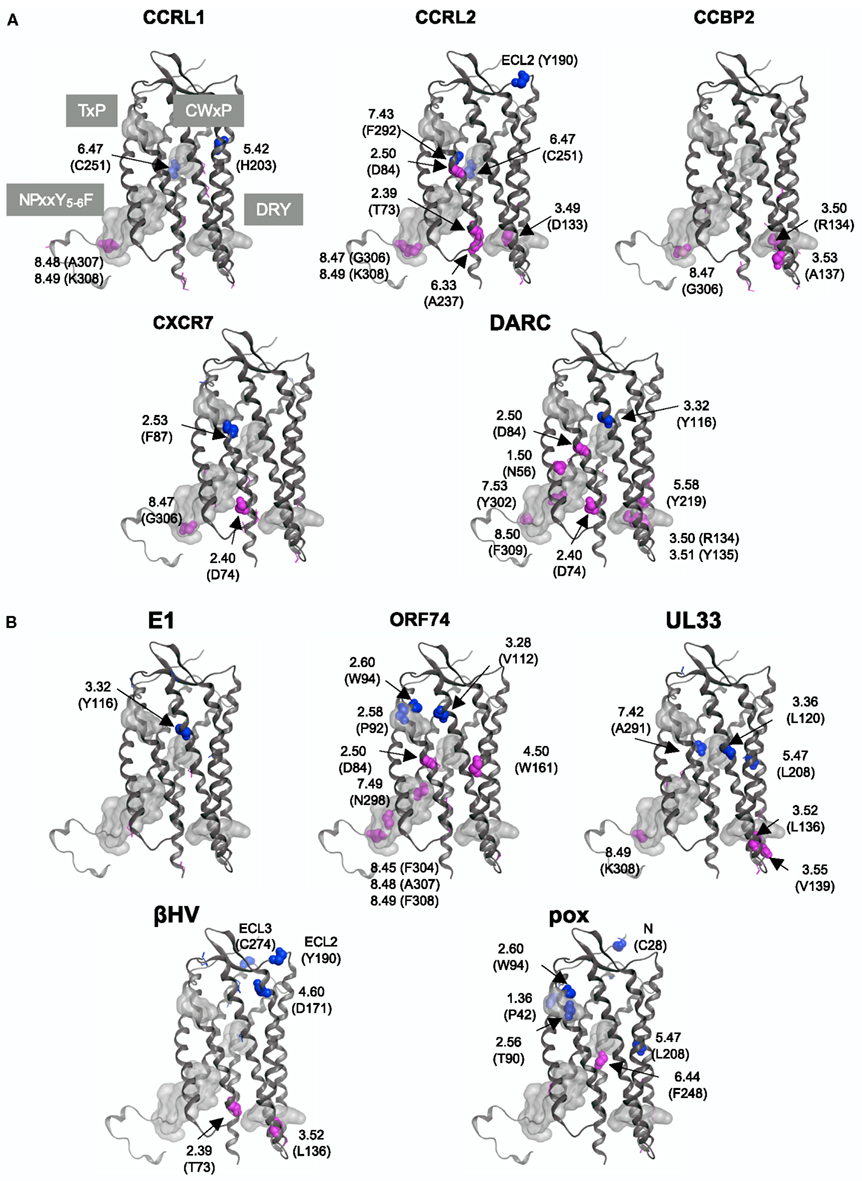
Figure 4. Mapping of the sites with large KL information values on the CXCR4 structure. The sites detected from the comparisons with (A) five decoy receptor groups and with (B) five viral receptor groups are mapped on the main chain structure of CXCR4. The residues corresponding to the detected sites with information about function and/or motif are depicted by space filling models, and are indicated according to the Ballesteros–Weinstein nomenclature. The corresponding amino acid residue types and numbers of CXCR4 are also shown in parentheses. On the other hand, the sites without any information are indicated by line models. The four motif regions are indicated by gray surface models. The residues that mapped on the extracellular side are colored blue, and those that mapped on the intracellular side are colored red.
The DRY (Asp-Arg-Tyr) motif of the GPCRs is conserved as the sequence DRYLAIV in the traditional CKRs, from the end of TM3 to ICL2 (Graham, 2009). The motif is related to signal transduction, through interactions with G proteins. The conserved R1343.50 is involved in the interchanges between the inactive and active conformations of GPCRs. In the inactive conformation, this Arg interacts with its neighboring residue, D1333.49, but in the active conformation, the residue interacts with Y2195.58 (Holst et al., 2010). The sites in the DRY region of the DRYLAIV sequence were only detected from the analyses with the decoy receptor groups, CCRL2, CCBP2, and DARC. In addition, Y2195.58 was also detected from the analysis with the DARC group. On the other hand, the sites in the LAIV3.52 ~ 3.55 region of the DRYLAIV sequence were detected from the examinations with the decoy and viral receptor groups, CCBP2, UL33, and βHV. The CWxP motif is located in the middle of TM6. This W2526.48 is believed to function as a micro-switch in the receptor activation mechanism, and P2546.50 creates a kink in this helix, around which TM7 performs its rigid body movements during activation (Nygaard et al., 2009). The corresponding sites of this motif were detected from the analyses of two decoy receptor groups, CCRL1 and CCRL2, but not from those of any viral receptor group. The fifth site of the NPxxY5-6F motif in TM7, Y3027.53 functions in the interchange of an inactive rotamer conformation (Nygaard et al., 2009). The sites of this motif were detected from the investigations with every decoy receptor group and two viral receptor groups, ORF74 and UL33. The TxP motif of TM2 is known as a specific motif of the traditional CKRs. It is known that the TxP motif in TM2 is specific for traditional CKRs (Govaerts et al., 2001). The third site of the TxP motif, P922.58, bends the helix, which determines the intra-helical location that is involved in the receptor activation. The sites of the motif were detected from the analyses of two viral receptor groups, the ORF74 and pox groups, but not from the assessment with any decoy receptor group. In addition, several sites corresponding to highly conserved positions in GPCRs, which are denoted as x.50 by the Ballesteros–Weinstein nomenclature, such as N561.50 and D842.50, were detected from analyses of several groups (see Table 2). Table 2 also shows the other important residues experimentally identified as having binding or signaling functions.
We examined which sites were commonly selected from the comparisons. No site was shared in all of the comparisons. Furthermore, there was no site commonly detected from the analyses with all of the decoy receptor groups or all of the viral receptor groups. However, several sites were detected from the different comparisons. For example, the sites corresponding to D742.40, D842.50, R1342.50, A1413.57, T1423.58, S1443.60, C2185.57, K2306.26, T2416.37, C2516.47, G3068.47, and K3088.49 were detected from at least two assessments with decoy receptor groups. Most of these sites are located in ICL2, 3, and the C-terminal region. Among these sites, D842.50, A1413.57, C2185.57, and K3088.49 were also detected from at least one analysis with the viral receptor group. W942.60, W102 (ECL1), L1363.52, H1403.56, G2075.46, L2085.47, and K3088.49 were detected from at least two analyses of the viral receptor groups. None of them, except for K3088.49, was detected from the analyses of any decoy receptor group.
Statistical Test for the Spatial Bias of the Sites with Large KL Information Values
As shown in Figure 4, the distribution of the sites selected from the analyses with the decoy receptor groups seemed to be biased toward the cytosolic side of the CKR structure. In contrast, there did not seem to be any trends in the distribution of the sites obtained from the analyses with the viral receptor groups. To quantitatively examine the observations, the residues corresponding to the selected sites and the remaining residues were projected on the axis connecting the center of gravity of the ECLs including the N-terminal region, and that of the ICLs (see Figure 5). Based on the projection on the axis, t-tests were performed as described in the Section “Materials and Methods.”

Figure 5. Projection of the residues on the axis connecting the intracellular and extracellular sides of the receptor. (A) Projections corresponding to the five comparisons with the decoy receptor groups. (B) Projections corresponding to the five comparisons with the viral receptor groups. The axis generation method and the projection method are described in the Section “Materials and Methods.” In each figure, the vertical line indicates the axis. Horizontal lines on the right side of the axis indicate the projected positions of the residues corresponding to the sites selected based on the KL information values. Horizontal lines on the left side indicate the projected positions of the remaining residues. The filled circles indicate the midpoint of the axis.
The results of the t-tests are summarized in Table 3. As shown in this table, the null hypothesis was rejected in three cases of the analyses with decoy receptor groups, CCRL1, CCBP2, and DARC, under the significance level of 5%. To examine the bias further, the one-sided t-test was applied to the observations about the decoy receptor groups. The null hypothesis was the same as that of the two-sided test, but the alternative hypothesis was that the average of the residue with the large KL value is smaller than that of the remaining residues. We found that the null hypothesis was rejected for four cases with decoy receptor groups, CCRL1, CCBP2, DARC, and CXCR7 (data not shown). That is, the distribution of the residues corresponding to the sites with large KL information values of the decoy receptor groups, except for CCRL2, was biased toward the intracellular side of the receptor. The two-sided t-test was also applied to the analyses of the viral receptor groups. In all cases, the null hypothesis was not rejected. This result suggested that the residues selected by the KL information values of the viral receptors were distributed on both the extracellular and intracellular sides.

Table 3. Results of t-tests.
Discussion
Decoy Receptors
The difference in the amino acid composition at an alignment site between two receptor groups, as evaluated by the KL information value, was considered to reflect the difference in the functional constraints at the site between the groups. As described above, decoy receptors are able to bind to CKs, but do not induce signaling. The sites detected by the KL information value would reflect the functional difference. Actually, the sites included in several motifs, such as DRY, CWxP, and NPxxY5-6F, which are involved in signaling, were detected. The bias in the locations of the detected sites on the intracellular side was statistically significant by the two-sided or one-sided t-test in four out of five decoy receptor groups. Especially, all of the sites detected from the analysis of CCBP2 were located on the intracellular side. The test with the CCRL2 group was the only one that did not suggest a statistically significant bias in the distribution of the detected sites. As described above, CCRL2 is also able to bind to chemerin (Yoshimura and Oppenheim, 2011). The adaptation to the new ligand may have introduced the change in the functional constraints on the extracellular side, which may be the reason why the null hypothesis was not rejected. This observation suggested that the functional divergence of CCRL2 was induced under different selective pressure, as compared to the other decoy receptors after gene duplication. CCRL2 forms a gene cluster together with the genes for CCR1, 2, 3, and 5 in several mammalian genomes (Nomiyama et al., 2011). The close relationship of CCRL2 to these CKRs and its distant relationship to the other decoy receptors in the phylogenetic tree (Figure 3) were consistent with the conservation of the gene orders in the genomes, although the bootstrap probabilities for the relationships were not always high. The evolutionary relationship and the conserved gene order, together with the acquisition of binding activity to a new ligand, suggested a unique evolutionary position of CCRL2 relative to the other decoy receptors.
The lack of signal transduction activity in the decoy receptors is attributed to the degeneration of the DRY motif (Comerford et al., 2007). Our study suggested that the degenerations of other motifs and functional residues may also be related to functional changes. For example, two decoy groups, CCRL1 and CXCR7, contained the typical DRY motif. However, the sites in other motifs that are related to the conformational change associated with the active-inactive switch had large KL information values in these decoy receptors. This observation suggested that the constraints for the residue conservation at the sites in the traditional CKRs are looser in the two decoy receptor groups (see Table 2). In addition to the motif sites, the highly conserved sites in the TM regions of GPCRs, including the traditional CKRs (x.50 in the Ballesteros–Weinstein nomenclature), had large KL information values in the analyses with several decoy receptor groups. The use of different amino acid residues at such sites may lead to functional and/or structural changes. Several sites with uncharacterized functional relationships also showed large KL information values. Most of them were found in ICLs 2 and 3. As these loops are considered to interact with G proteins, the sites detected on the loops may be involved in the loss of the signaling function of the decoy receptors.
Viral Receptors
We anticipated that the sites detected from the analyses with the viral receptor groups would be found on the extracellular side, since viral receptors exhibit signaling activity without ligand binding. As described above, however, the sites with the large KL information values were found not only on the extracellular side, but also on the intracellular side. We examined the detected sites from the different viewpoint. CASTp4 (Liang et al., 1998) is a program to identify pocket regions in a given tertiary structure. When we applied CASTp to the coordinates of CXCR4, the pocket region corresponding to the ligand binding cavities of GPCRs was reported with the highest score. The residues consisting of the pocket region were mainly projected on the extracellular side of the axis (see Figure 1), although some residues were projected on the intracellular side. The numbers of detected sites located in the pocket regions of the five decoy receptor groups were 2, 2, 0, 1, and 1, whereas 3, 2, 5, 3, and 4 sites were located in the pocket regions of the five viral receptor groups (see Table 2). The number of sites was transformed into the ratio to the total number of detected sites for each receptor group. The one-sided t-test showed that the difference in the ratios between the decoy and viral receptor groups was statistically significant (p-value = 0.003864). That is, more sites were detected in the pocket region in the viral receptor groups, as compared to the decoy receptor groups. As shown in Table 2, in addition, about half of the sites in the pocket region have been characterized as being involved in ligand recognition. These sites are often occupied by conserved, bulky amino acid residues in CKRs. The result suggested that the functional constraints at the ligand binding region are different between the viral receptors and the traditional CKRs, as we first expected.
The sites in the DRY motif were not detected in any of the viral receptor groups. This motif was basically conserved in the viral receptors, except for the ORF74 group. A previous study reported that ORF74 performs signal transduction, despite the fact that the DRY motif is changed to DTW (Rosenkilde et al., 2005). They also showed that the introduction of the DRY sequence into ORF74 induces functional reduction. In our study, the sequences collected as the ORF74 group showed variations in this region. For example, equid herpesvirus 2 has DTW, whereas the rodent and primate herpesviruses have xRC or xRY. Each variation includes the residues identical to those of the original DRY motif, which may have reduced the KL information value and led to the failure in the detection of the sites. Instead, the sites in the TxP and NPxxY5-6F motifs and the sites spatially surrounding the DRY motif were detected from the analysis of the ORF74 group (see Table 2). The amino acid replacements in the two motifs, which are considered to be involved in the conformational change, and those of the residues near the DRY motif may have contributed to the maintenance of the signaling activity of ORF74, despite the deviation from the typical DRY motif. In contrast, no sites in any motif were detected from the comparison with the E1 group. The E1 receptor reportedly lacks constitutive signaling activity (Rosenkilde et al., 2008). The conservation of the motifs suggested the difference in the signaling functions between the E1 group and other viral receptor groups.
We had not expected to detect the sites on the intracellular side from the comparisons with the viral receptor groups, since these receptors exhibit signaling activity without ligand binding. However, quite a few sites with large KL information values were also found on the intracellular side. As described above, the overlap of the selected sites between the decoy receptors and the viral receptors was small. The difference in the selected sites on the intracellular side between the viral receptor groups and the decoy receptor groups may be basically related to the difference in the activities of the receptor groups. That is, the sites of the viral receptor groups under the constraint to maintain the signaling without ligand binding may be different from the sites of the decoy receptor groups, where the functional constraints may have been weakened due to the loss of the signaling activity.
Conclusion
We have identified the alignment sites (and the corresponding amino acid residues) that may be responsible for the functional changes from CKRs to decoy receptors or viral receptors. The distributions of the identified residues on the tertiary structure seemed to reflect the functional differences. This prediction could be examined by an experimental study, such as amino acid replacement, or a computational study with molecular dynamic simulations. Such studies could provide deep insights into the mechanism of GPCR signaling through conformational changes. The experimental and computational confirmations of our prediction remain as future endeavors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Hiromi Daiyasu was supported by a Grant-in-Aid for Scientific Research and MEXT SPIRE Supercomputational Life Science. Hiroyuki Toh was supported by the Target Tanpaku program.
Footnotes
References
Adachi, J., and Hasegawa, M. (1996). MOLPHY (Programs for Molecular Phylogenetics), Version 2.3b3. Tokyo: Institute of Statistical Mathematics.
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Alvarez Arias, D., Navenot, J. M., Zhang, W. B., Broach, J., and Peiper, S. C. (2003). Constitutive activation of CCR5 and CCR2 induced by conformational changes in the conserved TXP motif in transmembrane helix 2. J. Biol. Chem. 278, 36513–36521.
Ballesteros, J. A., and Weinstein, H. (1995). Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428.
Bonecchi, R., Savino, B., Borroni, E. M., Mantovani, A., and Locati, M. (2010). Chemokine decoy receptors: structure-function and biological properties. Curr. Top. Microbiol. Immunol. 341, 15–36.
Camarda, G., Spinetti, G., Bernardini, G., Mair, C., Davis-Poynter, N., Capogrossi, M. C., and Napolitano, M. (1999). The equine herpesvirus 2 E1 open reading frame encodes a functional chemokine receptor. J. Virol. 73, 9843–9848.
Choi, W. T., Tian, S., Dong, C. Z., Kumar, S., Liu, D., Madani, N., An, J., Sodroski, J. G., and Huang, Z. (2005). Unique ligand binding sites on CXCR4 probed by a chemical biology approach: implications for the design of selective human immunodeficiency virus type 1 inhibitors. J. Virol. 79, 15398–15404.
Comerford, I., Litchfield, W., Harata-Lee, Y., Nibbs, R. J., and McColl, S. R. (2007). Regulation of chemotactic networks by “atypical” receptors. Bioessays 29, 237–247.
Comerford, I., Milasta, S., Morrow, V., Milligan, G., and Nibbs, R. (2006). The chemokine receptor CCX-CKR mediates effective scavenging of CCL19 in vitro. Eur. J. Immunol. 36, 1904–1916.
Deupi, X., and Standfuss, J. (2011). Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr. Opin. Struct. Biol. 21, 541–551.
Ewens, W. J., and Grant, G. R. (2001). Statistical Methods in Bioinformatics: An Introduction (Statistics for Biology and Health). New York: Springer.
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791.
Felsenstein, J. (1993). PHYLIP (Phylogeny Inference Package), Version 3.5c. Seattle: University of Washington.
Felsenstein, J. (1996). Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Meth. Enzymol. 266, 418–427.
Govaerts, C., Blanpain, C., Deupi, X., Ballet, S., Ballesteros, J. A., Wodak, S. J., Vassart, G., Pardo, L., and Parmentier, M. (2001). The TXP motif in the second transmembrane helix of CCR5. A structural determinant of chemokine-induced activation. J. Biol. Chem. 276, 13217–13225.
Graham, G. J. (2009). D6 and the atypical chemokine receptor family: novel regulators of immune and inflammatory processes. Eur. J. Immunol. 39, 342–351.
Gruijthuijsen, Y. K., Casarosa, P., Kaptein, S. J., Broers, J. L., Leurs, R., Bruggeman, C. A., Smit, M. J., and Vink, C. (2002). The rat cytomegalovirus R33-encoded G protein-coupled receptor signals in a constitutive fashion. J. Virol. 76, 1328–1338.
Gu, X. (1999). Statistical methods for testing functional divergence after gene duplication. Mol. Biol. Evol. 16, 1664–1674.
Hannenhalli, S. S., and Russell, R. B. (2000). Analysis and prediction of functional sub-types from protein sequence alignments. J. Mol. Biol. 303, 61–76.
Henikoff, S., and Henikoff, J. G. (1994). Position-based sequence weights. J. Mol. Biol. 243, 574–578.
Holst, B., Nygaard, R., Valentin-Hansen, L., Bach, A., Engelstoft, M. S., Petersen, P. S., Frimurer, T. M., and Schwartz, T. W. (2010). A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J. Biol. Chem. 285, 3973–3985.
Ichihara, H., Daiyasu, H., and Toh, H. (2004). How does a topological inversion change the evolutionary constraints on membrane proteins? Protein Eng. Des. Sel. 17, 235–244.
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282.
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066.
Katoh, K., and Toh, H. (2008). Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinformatics 9, 286–298.
Landgraf, R., Fisher, D., and Eisenberg, D. (1999). Analysis of heregulin symmetry by weighted evolutionary tracing. Protein Eng. 12, 943–951.
Leick, M., Catusse, J., Follo, M., Nibbs, R. J., Hartmann, T. N., Veelken, H., and Burger, M. (2010). CCL19 is a specific ligand of the constitutively recycling atypical human chemokine receptor CRAM-B. Immunology 129, 536–546.
Liang, J., Edelsbrunner, H., and Woodward, C. (1998). Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Science 7, 1884–1897.
Maussang, D., Vischer, H. F., Leurs, R., and Smit, M. J. (2009). Herpesvirus-encoded G protein-coupled receptors as modulators of cellular function. Mol. Pharmacol. 76, 692–701.
Murphy, P. M., Baggiolini, M., Charo, I. F., Hébert, C. A., Horuk, R., Matsushima, K., Miller, L. H., Oppenheim, J. J., and Power, C. A. (2000). International Union of Pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 52, 145–176.
Najarro, P., Gubser, C., Hollinshead, M., Fox, J., Pease, J., and Smith, G. L. (2006). Yaba-like disease virus chemokine receptor 7L, a CCR8 orthologue. J. Gen. Virol. 87(Pt 4), 809–816.
Naumann, U., Cameroni, E., Pruenster, M., Mahabaleshwar, H., Raz, E., Zerwes, H. G., Rot, A., and Thelen, M. (2010). CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 5, e9175. doi:10.1371/journal.pone.0009175
Nomiyama, H., Osada, N., and Yoshie, O. (2011). A family tree of vertebrate chemokine receptors for a unified nomenclature. Dev. Comp. Immunol. 35, 705–715.
Nygaard, R., Frimurer, T. M., Holst, B., Rosenkilde, M. M., and Schwartz, T. W. (2009). Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 30, 249–259.
Nygaard, R., Valentin-Hansen, L., Mokrosinski, J., Frimurer, T. M., and Schwartz, T. W. (2010). Conserved water-mediated hydrogen bond network between TM-I, -II, -VI, and -VII in 7TM receptor activation. J. Biol. Chem. 285, 19625–19636.
Rosenkilde, M. M., Benned-Jensen, T., Frimurer, T. M., and Schwartz, T. W. (2010). The minor binding pocket: a major player in 7TM receptor activation. Trends Pharmacol. Sci. 31, 567–574.
Rosenkilde, M. M., Kledal, T. N., and Schwartz, T. W. (2005). High constitutive activity of a virus-encoded seven transmembrane receptor in the absence of the conserved DRY motif (Asp-Arg-Tyr) in transmembrane helix 3. Mol. Pharmacol. 68, 11–19.
Rosenkilde, M. M., Smit, M. J., and Waldhoer, M. (2008). Structure, function and physiological consequences of virally encoded chemokine seven transmembrane receptors. Br. J. Pharmacol. 153(Suppl. 1), S154–S166.
Rosenkilde, M. M., Waldhoer, M., Lüttichau, H. R., and Schwartz, T. W. (2001). Virally encoded 7TM receptors. Oncogene 20, 1582–1593.
Sahagun-Ruiz, A., Sierra-Honigmann, A. M., Krause, P., and Murphy, P. M. (2004). Simian cytomegalovirus encodes five rapidly evolving chemokine receptor homologues. Virus Genes 28, 71–83.
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Scholten, D. J., Canals, M., Maussang, D., Roumen, L., Smit, M., Wijtmans, M., de Graaf, C., Vischer, H., and Leurs, R. (2012). Pharmacological modulation of chemokine receptor function. Br. J. Pharmacol. 165, 1617–1643.
Simon, A. L., Stone, E. A., and Sidow, A. (2002). Inference of functional regions in proteins by quantification of evolutionary constraints. Proc. Natl. Acad. Sci. U.S.A. 99, 2912–2917.
Slinger, E., Langemeijer, E., Siderius, M., Vischer, H. F., and Smit, M. J. (2011). Herpesvirus-encoded GPCRs rewire cellular signaling. Mol. Cell. Endocrinol. 331, 179–184.
Surgand, J. S., Rodrigo, J., Kellenberger, E., and Rognan, D. (2006). A chemogenomic analysis of the transmembrane binding cavity of human G-protein-coupled receptors. Proteins 62, 509–538.
Thelen, M., and Thelen, S. (2008). CXCR7, CXCR4 and CXCL12: an eccentric trio? J. Neuroimmunol. 198, 9–13.
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680.
Tian, S., Choi, W. T., Liu, D., Pesavento, J., Wang, Y., An, J., Sodroski, J. G., and Huang, Z. (2005). Distinct functional sites for human immunodeficiency virus type 1 and stromal cell-derived factor 1alpha on CXCR4 transmembrane helical domains. J. Virol. 79, 12667–12673.
Wu, B., Chien, E. Y., Mol, C. D., Fenalti, G., Liu, W., Katritch, V., Abagyan, R., Brooun, A., Wells, P., Bi, F. C., Hamel, D. J., Kuhn, P., Handel, T. M., Cherezov, V., and Stevens, R. C. (2010). Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071.
Yoshimura, T., and Oppenheim, J. J. (2011). Chemokine-like receptor 1 (CMKLR1) and chemokine (C-C motif) receptor-like 2 (CCRL2); two multifunctional receptors with unusual properties. Exp. Cell Res. 317, 674–684.
Zhou, N., Luo, Z., Luo, J., Liu, D., Hall, J. W., Pomerantz, R. J., and Huang, Z. (2001). Structural and functional characterization of human CXCR4 as a chemokine receptor and HIV-1 co-receptor by mutagenesis and molecular modeling studies. J. Biol. Chem. 276, 42826–42833.
Zlotnik, A., and Yoshie, O. (2000). Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127.
Keywords: chemokine receptors, decoy receptors, viral receptors, GPCR, molecular evolution
Citation: Daiyasu H, Nemoto W and Toh H (2012) Evolutionary analysis of functional divergence among chemokine receptors, decoy receptors, and viral receptors. Front. Microbio. 3:264. doi: 10.3389/fmicb.2012.00264
Received: 06 June 2012; Paper pending published: 17 June 2012;
Accepted: 05 July 2012; Published online: 26 July 2012.
Edited by:
Hironori Sato, National Institute of Infectious Diseases, JapanReviewed by:
Hironori Sato, National Institute of Infectious Diseases, JapanMikita Suyama, Kyushu University, Japan
Copyright: © 2012 Daiyasu, Nemoto and Toh. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Hiromi Daiyasu, Department of Bioinformatic Engineering, Graduate School of Information Science and Technology, Osaka University, 1-5, Yamadaoka, Suita, Osaka 565-0871, Japan. e-mail: daiyasu@ist.osaka-u.ac.jp