

- Pathogen Research Group, School of Science and Technology, Nottingham Trent University, Nottingham, UK
Cronobacter spp. (previously known as Enterobacter sakazakii) is a bacterial pathogen affecting all age groups, with particularly severe clinical complications in neonates and infants. One recognized route of infection being the consumption of contaminated infant formula. As a recently recognized bacterial pathogen of considerable importance and regulatory control, appropriate detection, and identification schemes are required. The application of multilocus sequence typing (MLST) and analysis (MLSA) of the seven alleles atpD, fusA, glnS, gltB, gyrB, infB, and ppsA (concatenated length 3036 base pairs) has led to considerable advances in our understanding of the genus. This approach is supported by both the reliability of DNA sequencing over subjective phenotyping and the establishment of a MLST database which has open access and is also curated; http://www.pubMLST.org/cronobacter. MLST has been used to describe the diversity of the newly recognized genus, instrumental in the formal recognition of new Cronobacter species (C. universalis and C. condimenti) and revealed the high clonality of strains and the association of clonal complex 4 with neonatal meningitis cases. Clearly the MLST approach has considerable benefits over the use of non-DNA sequence based methods of analysis for newly emergent bacterial pathogens. The application of MLST and MLSA has dramatically enabled us to better understand this opportunistic bacterium which can cause irreparable damage to a newborn baby’s brain, and has contributed to improved control measures to protect neonatal health.
Introduction
Cronobacter spp. (formerly known as Enterobacter sakazakii) is a genus consisting of Gram negative, motile, facultatively anaerobic opportunistic bacterial pathogens belonging to the Enterobacteriaceae family (Kucerova et al., 2011). The diverse genus accommodates seven species: C. sakazakii, C. malonaticus, C. turicensis, C. muytjensii, C. dublinensis, and the two newly defined species, C. universalis and C. condimenti (Joseph et al., 2012a). The primary niche of this organism is believed to be plant material (i.e., wheat, rice, herbs, and spices; Iversen and Forsythe, 2003). However, it is brought into contact with humans via food and environmental exposure. It has been isolated from a wide range of foods including cereals, rice, cheese, fruits, meat, milk, vegetables, grains, herbs, and spices as well as their by-products (Iversen and Forsythe, 2003; Friedemann, 2007). Cronobacter spp. have been isolated from other mammals and invertebrates such as rats and flies (Gakuya et al., 2001; Mramba et al., 2006).
Kucerova et al. (2010) were the first to publish a detailed genome description for Cronobacter. The sequenced strain, C. sakazakii BAA-894, had been isolated from formula powder. This was later followed by the announcement of two further genomes, C. sakazakii E899 and C. turicensis z3032, though without detailed descriptions (Stephan et al., 2010; Chen et al., 2011). Kucerova et al. (2010, 2011) in two detailed publications used whole genome sequence analysis of C. sakazakii strain BAA-894 and microarray based comparative genomic hybridization (CGH) to explore the genomes of strains across the Cronobacter genus. These strains had been chosen using MLST to ensure they represented the diverse genus. They identified several variable regions which are putative virulence factors, i.e., fimbriae and multidrug efflux systems, many of which are plasmid borne (Franco et al., 2011). Putative virulence traits of particular interest are iron-uptake mechanisms (Grim et al., 2012), superoxide dismutase (SodA) for macrophage survival (Townsend et al., 2008), hemolysin (Cruz et al., 2011a), flagella (Cruz et al., 2011b), pili, a metalloprotease (Kothary et al., 2007), an enterotoxin (Pagotto et al., 2003), and plasmid borne virulence factors such as Cronobacter plasminogen activator (Cpa) and type six secretion systems (T6SS; Franco et al., 2011).
The majority of reported Cronobacter cases are in adults (FAO/WHO, 2008); however neonates and infants are the major identified group at risk due to the associated high mortality rate following necrotizing enterocolitis (NEC), septicemia, and meningitis. In neonatal cases of Cronobacter meningitis, there is gross destruction of the brain, leading sadly to either death (40–80% of cases) or severe neurological damage. Due to the understandable sensitivity toward neonatal infections, such cases have attracted more attention than infections in other age groups. Cronobacter can attach to intestinal cells, invade, and survive in macrophages (Townsend et al., 2007, 2008). OmpA and OmpX possibly have a role in the organism penetrating the blood brain barrier. The reason for the destruction of the brain cells is unknown and could, in part, be a host response (Kim et al., 2010). Infections in older age groups are principally bacteremias as well as urosepsis and wound infections. To date, only strains from the three species C. sakazakii, C. malonaticus, and C. turicensis have been associated with neonatal infections (Joseph et al., 2012b). C. malonaticus appears to be more associated with adult than neonatal infections (Joseph and Forsythe, 2011). Therefore pathogenicity in humans may be an acquired trait in this genus.
A number of neonatal outbreaks and cases of Cronobacter spp. infections have been reported in intensive care units (van Acker et al., 2001; Block et al., 2002; Himelright et al., 2002; Caubilla-Barron et al., 2007). Many of these infections have been directly linked to reconstituted powdered infant formula (PIF) which may have been contaminated intrinsically or during preparation and administration (Himelright et al., 2002). A common feature in some of these outbreaks is the opportunity for temperature abuse of the prepared feed, which would permit bacterial growth (Caubilla-Barron et al., 2007). It is pertinent to note that there is also asymptomatic human carriage of the organism. The bacterium has been isolated from the tracheae and feces, and additionally has been recovered from the feeding tubes of neonates fed breast milk or ready-to-feed formula, and not infant formula (Hurrell et al., 2009). Therefore, it is important that robust and reliable typing schemes are readily available for Cronobacter spp., and that a wide range of possible sources of the organism during an outbreak need to be investigated.
The Cronobacter genus belongs to the bacterial class Gammaproteobacteria, and is within the family Enterobacteriaceae. The genus is closely related to the genera Enterobacter and Citrobacter, and some Enterobacter hormaechei and E. ludwiggii strains have been mis-identified as Cronobacter which has led to some confusion in the literature. 16S rDNA sequence diversity has been used to define genus (5%) and species (3%) boundaries in the Enterobacteriaceae. However the method has limitations with closely related species due to minimal sequence diversity. Additionally, the sequence diversity between multiple copies of the 16S rDNA operon within a bacterium can also introduce discrepancies (Acinas et al., 2004).
Initially the Cronobacter genus was composed of C. sakazakii, C. turicensis, C. muytjensii, and C. dublinensis (Iversen et al., 2007). This was quickly revised (Iversen et al., 2008) with the addition of C. malonaticus. This species had originally been described as a subspecies of C. sakazakii by Iversen et al. (2007) who could not distinguish C. sakazakii and C. malonaticus using 16S rDNA sequence analysis. The Cronobacter species were initially differentiated by Iversen et al. (2008) according to 16 E. sakazakii biotypes; C. sakazakii (biotypes 1–4, 7, 8, 11, and 13), C. malonaticus (biotypes 5, 9, and 14), C. turicensis (biotypes 16, 16a, and 16b), C. muytjensii (biotype 15), and C. dublinensis (biotypes 6, 10, and 12). In contrast, Joseph et al. (2012a) used strains selected by multilocus sequence analysis (MLSA; Baldwin et al., 2009) as representatives across the genus and therefore overcame the preconceived grouping of strains based on phenotyping. These recent studies, which will be described in more detail below, led to the naming of two new Cronobacter species, C. universalis and C. condimenti, by Joseph et al. (2012a). Due to numerous limitations such as subjectivity and reproducibility, the earlier phenotyping approach to Cronobacter, based on 10 biochemical and physiological tests, has been replaced by various DNA based techniques. Consequently, biotyping no longer has a role in designating the species of Cronobacter isolates.
Multilocus Sequence Typing Scheme for Cronobacter spp.
A number of DNA based methods for identification, speciation, and profiling have been proposed for Cronobacter spp. These include PCR probes for dnaG, rpsU, and rpoB genes (Seo and Brackett, 2005; Stoop et al., 2009). However they suffer in that they have either have not continued in use or have not been validated against a robust Cronobacter strain collection of the seven species. Some require different PCR primer pairs for each species. The most advanced non-MLST profiling method is serogrouping using PCR (Jarvis et al., 2011; Sun et al., 2011). However currently the method, using seven primer pairs, only covers four of the seven species and 48/231 (>20%) strains do not give a PCR product (Jarvis et al., 2011; Sun et al., 2011) which indicates that further unrecognized serogroups exist. Finally, there are contradictions in the literature with the same serogroup being across more than one species – C. sakazakii O3 and C. muytjensii O1 (Jarvis et al., 2011). In contrast to these methods, multilocus sequence typing (MLST) based on only seven primer pairs has been established for the whole Cronobacter genus which is robust and reliable. This article concerns the advances in our understanding of Cronobacter using the seven loci scheme, both for profiling and phylogeny. The MLST and MLSA approaches have:
1. Revealed the diversity of the Cronobacter genus.
2. Contributed to the recognition of two new species; C. universalis and C. condimenti.
3. Shown the close relatedness and groupings of the seven species.
4. Shown the evolutionary descent of the genus.
5. Revealed the majority of neonatal meningitis cases are being attributable to one clonal lineage, C. sakazakii ST4 clonal complex.
6. Formed the basis for future research regarding stain selection for investigating Cronobacter virulence and environmental fitness.
7. Established an open access curate database http://www.pubMLST.org/cronobacter. This is composed of ∼400 MLST profiled strains which are widely geographically, temporally, and sourced distributed, describing experimental protocols, contains DNA sequences of the seven alleles for offline analysis, and where users can determine the sequence type (ST) of their strains and undertake further advanced DNA sequence analysis. The database is curated (by S. Forsythe), and hosted at PubMLST by the University of Oxford, UK.
The Cronobacter MLST scheme requires the partial sequence analysis of seven housekeeping genes: atpD, fusA, glnS, gltB, gyrB, infB, and ppsA (Baldwin et al., 2009). These loci are distributed around the Cronobacter genome (Kucerova et al., 2010), and are under the least influence of any possible selective evolutionary pressures. Also, the primers for the seven genes have been designed to have similar annealing temperatures, enabling all reactions to be performed in a single run, thus reducing the time involved. Comparing the loci DNA sequences with the Cronobacter MLST reference database (http://www.pubMLST.org/cronobacter) generates the seven digit allele code, and the strain’s ST. When concatenated together the seven allele sequences form a 3036 nt length for MLSA and phylogenetic analysis.
The MLST scheme was announced first for C. sakazakii and C. malonaticus (Baldwin et al., 2009) to demonstrate its usefulness over 16S rDNA sequence analysis, which could not distinguish between them. Further publications have demonstrated the use of MLST for all seven formally recognized species of Cronobacter genus (Czerwicka et al., 2010; Kucerova et al., 2010, 2011; Hamby et al., 2011; Joseph and Forsythe, 2011; Hariri et al., 2012; Joseph et al., 2012a,b). The scheme has also shown the reliability of fusA as a single locus for speciation (Joseph et al., 2012b).
The significant contribution of MLSA and MLST to our understanding of the Cronobacter genus is covered later. Nevertheless, suffice it for now to point out that the initial use of biotypes (phenotyping) to support the description of Cronobacter species was unfortunately flawed as some biotype index strains were attributed to the wrong Cronobacter species, as shown by the DNA sequence based MLSA (Baldwin et al., 2009).
Phylogeny and Evolution of the Cronobacter Genus
By concatenating the seven loci sequences together (3036 base pair total length) the DNA sequences can be used to construct a phylogenetic tree of the genus. This enables the quantification of intraspecific and interspecific diversity of the genus, as well as potential characterization of the strains according to virulence groupings and source.
Using the Maximum Likelihood algorithm in MEGA5 (Tamura et al., 2011), one can, see the clear clustering of the various Cronobacter species within the genus (Figure 1). It is advisable when doing this analysis with newly profiled strains to include closely related organisms as outliers. We use the corresponding seven MLST loci sequences from the publicly available genomes of Citrobacter koseri (Accession number CP000822) and Enterobacter spp. 683 (Accession number CP000653). These two organisms are closely related to Cronobacter spp. yet none of their alleles are shared with any of the Cronobacter species (Figure 1). The reason for stressing the use of outliers is that we often receive queries from MLST users regarding problematic strains. These invariably are strains of Enterobacter spp. and less often Pantoea spp. which have been mis-identified as Cronobacter, or vice versa. These strains frequently have been provisionally identified by phenotyping and even MALDI-TOF. Unfortunately, both methods have inaccuracies in their databases.
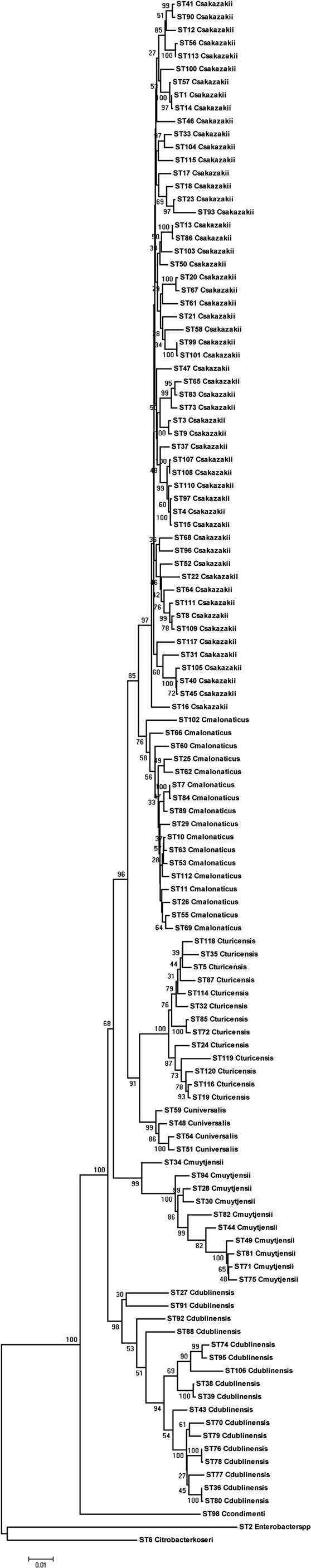
Figure 1. Maximum likelihood tree based on the concatenated sequences (3036 bp) of the seven LST loci for the genus Cronobacter. The STs and the corresponding species are indicated at the tip of each branch. The tree is drawn to scale using MEGA5, with 1000 bootstrap replicates.
Joseph et al. (2012b) undertook an evolutionary analysis of the genus by using the mean synonymous substitution values (Ds) of the Cronobacter MLST dataset, and previously published substitution rates for other Enterobacteriaceae members such as E. coli and Salmonella. This predicted that the Cronobacter genus split from its closest ancestor in the Enterobacteriaceae family approximately 45–68 million years ago (Figure 2). Accepting that such calculations are based on a number of assumptions, it is notable that this corresponds with the Paleogene period of the Cenozoic era when early flowering plants evolved. This is an interesting observation as it coincides with the suspected natural plant habitat of the organism and opens speculation for the evolution of the genus. The feeding of insect larvae on plants could have led to a host adaptation. It is notable that Cronobacter have been isolated from flies (Mramba et al., 2006; Pava-Ripoll et al., 2012). The evolution of other members of the Enterobacteriaceae family has been estimated by similar MLST studies and hence comparable. The evolution of the distinguishable Cronobacter species appears to have occurred over the same period as the divergence of the Salmonella species and subspecies, after its split from E. coli (McQuiston et al., 2008). Further analysis using whole genomes will be more informative.
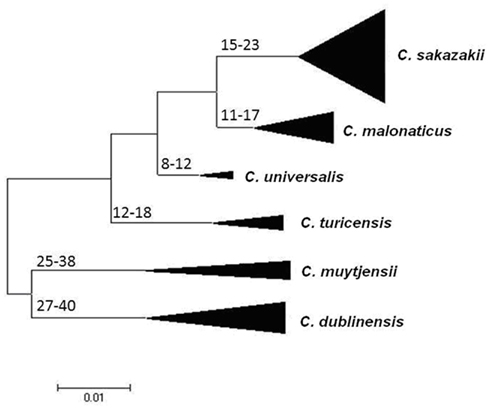
Figure 2. Maximum Likelihood tree of the Cronobacter MLST dataset indicating ranges of the hypothetical divergence dates of each species node measured in millions of years before the present. The tree has been drawn to scale using MEGA5. The bases of the triangles indicate the number of isolates used for the analysis, while the heights indicate the diversity of each branch. C. condimenti with the single isolate has been excluded from this analysis. Reproduced from Joseph et al. (2012b).
The estimates of the divergence dates of the individual species have been calculated. The earliest branches of the genus led to C. dublinensis and C. muytjensii (25–40 MYA), more recent divergences have led to C. malonaticus and C. universalis (8–17 MYA). C. condimenti has to date been excluded from this analysis because only one isolate has been identified for this species, and hence it was not possible to calculate the necessary Ds values for the species (Joseph et al., 2012b).
Splitstree Analysis for Homologous Recombination Visualization
There is evidence of homologous recombination (gene conversion) events having occurred in the evolution of the genus. This is covered in detail in Joseph et al. (2012b) and so only two loci, fusA and gltB, have been chosen to illustrate homologous recombination in Cronobacter spp. Figure 3 shows the neighbor-nets of these loci by Splitstree4 (Huson and Bryant, 2006) analysis to visualize recombination events and evolutionary relationships in the dataset. The formation of parallelograms in these figures denotes the possibility of recombination events. Among the seven loci, fusA is the most stable with the least number of shared alleles among species, and none of the profiles were shared between two or more species (Figure 3A). Thus fusA can be used for speciation of Cronobacter strains, and avoids the ambiguities of 16S rDNA sequence analysis. In contrast, there are some instances of allelic profiles being shared between two species. The gltB locus has the most (7) allelic profiles shared between species (Figure 3B). This sharing is mostly observed between C. sakazakii with C. muytjensii and C. sakazakii with C. dublinensis species. Despite these recombination events, the concatenation of the sequences of the seven loci for the MLSA helps to overcome any possible influence of this recombination and ensures a robust phylogeny for the genus.
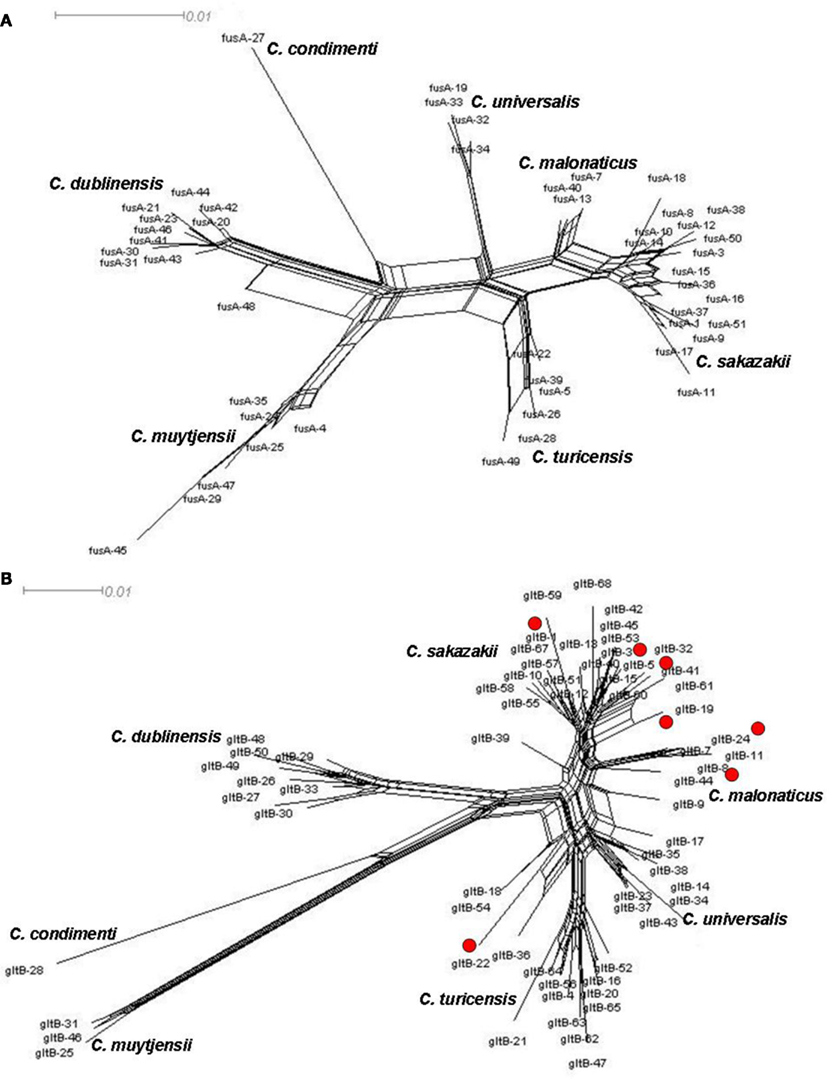
Figure 3. Neighbor-net of allele sequence alignment for (A) fusA and (B) gltB generated for the Cronobacter MLST dataset indicating diversity and recombination events. The figure has been drawn to scale using Splitstree4. The formation of parallelograms indicate possible recombination events.
Splitstree analysis also reveals a higher diversity in the C. muytjensii and C. dublinensis species than other Cronobacter species, with some individual branches being more genetically distant from the main cluster. This phenomenon has been described as “fuzzy” species in the MLST of Neisseria spp. (Hanage et al., 2005). Strain 1330 (ST 98) was one such candidate, seen as an lineage branching out from the C. dublinensis cluster which, following further phenotypic and DNA–DNA hybridization studies, was confirmed to be a previously unrecognized independent species, C. condimenti (Joseph et al., 2012a). Further recognition of new species in the Cronobacter genus is likely following MLST adoption, as the phylogenetic analysis is dynamic and not constrained to an analytical database.
Other divergent strains are evident in the C. muytjensii and C. dublinensis clusters and could indicate the presence of hitherto unrecognized Cronobacter species. The C. muytjensii species splits into two main clusters and a lone diverse branch (ST34). The C. dublinensis species shows greater diversity with two major clusters, and a few independent branches distant from each other. The diversity of these species has been investigated by Joseph et al. (2012b) using percent nucleotide divergence in the MLST sequence data, similar to the percent divergence used as cut-off thresholds for genus and species definitions using 16S rDNA analysis. Since the equivalent value for MLST has not been established, Joseph et al. (2012b) calculated the nucleotide divergence for the individual type strains of the Cronobacter species. The least divergence value was 2.8% (n = 3036 bp) which was between the type strains of C. sakazakii (NCTC 11467T; ST8) and C. malonaticus (CDC 1058-77T; ST7). This value was proposed as a minimum cut-off value for identifying potential candidates for new species. As a comparison these strains are only 0.3% different by full length 16S rDNA sequences (n = 1350 bp). The DNA divergence values between many strains in the C. muytjensii and C. dublinensis clusters were above this cut-off value. However, current taxonomic standards for recognizing new species by the International Journal for Systematic and Evolutionary Microbiology require a polyphasic analysis including DNA–DNA hybridization studies and phenotypic testing for biochemical traits. To date, our phenotypic profiling has not produced biochemical traits that could distinguish between the independent branches within C. muytjensii and C. dublinensis (Joseph, S., and Forsythe, S. J., unpublished data). The boundaries for defining a bacterial species have always been a topic of debate (Konstantinidis et al., 2006), and are ultimately a matter of taxonomic convenience. Some might argue about whether there is a real need for further delineation of already identified species. However, for an accurate understanding of an organism, it is very important to constantly evaluate the diversity of the organism at the genus and species level. From an evolutionary standpoint, this aids in an improved understanding of the relationships of the organisms with its possible closest ancestors. A whole genome level analysis should improve our understanding on this intra-species diversity, and certainly the need for subjective phenotypic tests to complement DNA sequence may mask further speciation.
Clonality
IA is a measure of the linkage of a population and when calculated using the MLST sequences the IA values for the genus Cronobacter were found to be significantly greater than zero (Joseph et al., 2012b). This indicates the presence of linkage disequilibrium or clonality. This analysis has been highly significant for our understanding of both the diversity of the genus and the specificity of clinically relevant strains.
In this review article, the goeBURST algorithm in PHYLOViZ (Francisco et al., 2012) has been used to visualize the relatedness of Cronobacter STs. Primarily the diagrams show where STs differ in a one, two, or three of the seven loci, i.e., single locus variant (SLV), double locus variant (DLV), and triple locus variant (TLV) respectively. For example, ST4 and ST107 are SLV as they differ in the fusA allele; 5-1-3-3-5-5-4 and 5-50-3-3-5-5-4. It should be noted that this is not the same analysis of DNA sequence variation which has already been considered above under phylogeny and MLSA.
The goeBURST analysis of 115 identified STs for the Cronobacter genus at the time of analysis in the pubMLST database, shows there were 13 SLV clonal complexes (CC). Nine of these belong to C. sakazakii, two to C. dublinensis and one each to C. turicensis and C. malonaticus. C. muytjensii, C. condimenti, and C. universalis did not show the formation of any such CC. Apart from these SLVs, a number of DLVs were also observed and these are the STs which shared five out of the seven definitive allelic profiles. Some of these large CC are especially significant with respect to strain clustering according to their isolation sources. The reader should note that new STs are frequently added to the database and therefore such analysis should be revised in any presentations. For the purposes of this review only CC 1, 2, and 4 have been chosen for further discussion. The reader should refer to Joseph et al. (2012b) for a more detailed account.
Clonal complex 1 currently comprises C. sakazakii STs 1 and 14. ST1 is a dominant ST consisting of strains isolated from across the world over a period of more than 25 years. These have been mainly isolated from PIF and clinical cases, and also more recently from milk powder processing factories in Germany and Australia (Craven et al., 2010; Jacobs et al., 2011), apart from a few food isolates. ST14, which is a SLV (ppsA allele) of ST1 was isolated from infant formula (Caubilla-Barron et al., 2007). Also linked to this clonal complex is ST57, which is a DLV (alleles atpD and glnS) to ST1. This is the profile for a PIF isolate from Denmark in 1988, as reported by Muytjens et al. (1988) in their milk powder survey.
Clonal complex 2 comprises C. malonaticus STs 7, 84, and 89. ST7 is the predominant ST in this complex, and is a mixture of clinical and PIF isolates from over 30 years. STs 84 and 89 comprise clinical isolates from the Czech Republic. These clinical isolates were recovered mainly from fecal, sputum, and blood samples. Where information is available, these clinical strains were primarily from non-infants (i.e., >1 year in age), especially adults there are also food and weaning food isolates in this complex.
Clonal complex 4 comprises C. sakazakii STs 4, 15, 97, 107, and 108. This is a key complex with respect to Cronobacter spp. epidemiology. ST4 is the most dominant ST in this MLST study with 78 isolates, and also the most frequent clinical ST (Baldwin et al., 2009). However, the description of “clinical” when referring to the origin of strains is ambiguous. Such strains are not necessarily from the site of infection, i.e., conjunctivae swabs from meningitis cases, or even isolates from colonized asymptomatic individuals. Fortunately sufficient detailed information was available for these clinical C. sakazakii strains to reveal what is possibly the most significant finding generated by MLST analysis regarding the epidemiology and trophism of neonatal Cronobacter infections. Clonal complex 4 has been identified as a genetic signature for C. sakazakii neonatal meningitis, with the majority of the isolates being linked to meningitis cases over a period of 50 years from six different countries (Joseph and Forsythe, 2011; Hariri et al., 2012). Out of the 480 strains and 133 STs in the pubMLST database (data collected September 2012), 18 isolates are from the cerebral spinal fluid of meningitis cases. Of these, 16 are C. sakazakii ST4 and loci variants (STs 15, 97, 107, 108, and 110) belonging in the clonal complex 4. These include isolates from the 2011 highly publicized cases in the USA (Hariri et al., 2012). Why C. sakazakii clonal complex 4 predominates neonatal meningitis cases is presently unclear and could be due to environmental fitness factors as well as virulence traits. It is plausible that adult meningitis cases are unreported to date due to the maturity of the blood brain barrier.
Within the 15 2011 US strains, there were two ST4 SLVs. The CSF strain 1565 (ST107) differed from the ST4 profile in the fusA loci by 6/438 nt. Strain 1572 (ST108) isolated from an opened tin of PIF differed in the fusA loci by 5/438 nt. These two strains only differ from each other by 1 nt out of 3036 (concatenated length) in the fusA loci position 378 (A: T). This level of discrimination is not possible using PFGE. It should be noted that PFGE and MLST analyze the bacterial DNA content differently, and there are no XbaI sites (the endonuclease most commonly used with PFGE of Enterobacteriaceae) within the seven MLST loci.
Two strains which are not from the clonal complex 4 have also been associated with neonatal meningitis, both are ST1. The most well known being C. sakazakii BAA-894 from the publicized Tennessee outbreak (Himelright et al., 2002). This indicates that non-clonal complex 4 strains may on occasion also cause severe brain damage. The additional importance of strain BAA-894 is that it was the first Cronobacter to be fully sequenced (Kucerova et al., 2010, 2011). Also within the 2011 US isolates that have been profiled is a strain of C. malonaticus ST112. The strain had been isolated from the blood of a <1-month-old infant who died from a fatal case of meningitis. This isolate is highly significant as previously it had been observed that C. malonaticus predominates adult infections and no previous neonatal meningitis cases have been attributed to this species (Joseph and Forsythe, 2011).
In contrast to the correlation of clonal complex 4 with meningitis, no correlation of ST4 or other STs has been found with other clinical presentations such as NEC. C. sakazakii ST4 strains have been isolated from PIF collected in 12 countries. They have also been isolated from milk powder processing factories in Australia and Germany, including roller dryers, tanker bays, and spray dryers. Other non-clinical sources included isolates from weaning food, chocolate, and a washing brush (van Acker et al., 2001; Turcovský et al., 2011).
In addition to the three dominant CC of C. sakazakii and C. malonaticus, described above, some of the other SLVs and DLVs also showed clustering patterns depending on source such as clonal complex 8 (ST13 and ST86) from food and feed.
Application of Multilocus Sequencing of Cronobacter spp. in Food Microbiology and Outbreak Investigations
A major impact of the multilocus sequencing of Cronobacter strains has been to support the recent definition of the Cronobacter genus. This has been essential for the evaluation of newly developed detection methods, and therefore of importance to the food industry and regulatory authorities for compliance. MLST has been used to profile Cronobacter strains from outbreaks in France, New Zealand, and the USA. This revealed the predominance of C. sakazakii ST4 in isolates from cerebral spinal fluid; Joseph and Forsythe (2011). More recently it was applied to Cronobacter strains submitted to CDC during 2011, and included the highly publicized cases last December. This confirmed the earlier study, by showing the CSF strains were C. sakazakii clonal complex 4 which includes ST4 (Hariri et al., 2012). The reason for this predominance is uncertain, but our studies of strains from the environmental sampling of milk powder processing factories, and also from PIF has also shown the predominance of the C. sakazakii species. Of particular interest is that C. sakazakii ST4 strains were isolated from powdered formulas as well as the roof, tanker bays, and roller driers of manufacturing plants. These dry environments may be selecting for desiccation resistant strains of Cronobacter spp. and in particular C. sakazakii ST4. In contrast, Cronobacter spp. isolates from a range of other foods, food ingredients, herbs, and spices are from a wide range of species and STs.
goeBURST Analysis of Cronobacter spp. Distribution
Analysis using the goeBURST algorithm has revealed an interesting link between clonal complex 4 and ST8, via ST108 and ST111 with respect to the pathogenicity of C. sakazakii; Figure 4. ST8 comprises 13 strains, 9 of which are clinical isolates. Unlike ST4, none of these are associated with meningitis, but are associated with diarrhea. ST108 and ST111 were isolated from an opened tin of PIF and formula reconstitution water, respectively. Based on the goeBURST analysis, it is difficult to conclude whether ST8 could be a clonal descendant of ST4, since these are only indicative evolutionary relationships. However, the connection, albeit distant, does correlate with the epidemiology of a majority of the clinical strains of these STs.
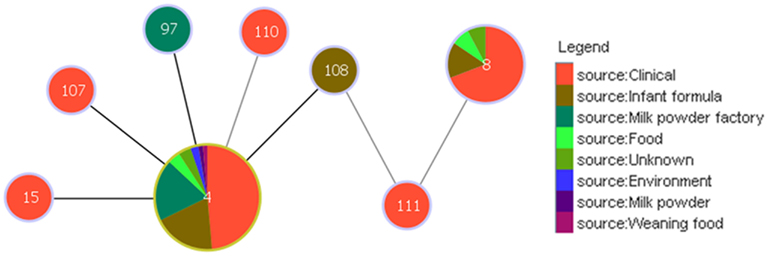
Figure 4. Relationship between the clinically significant C. sakazakii ST4 clonal complex and ST8. The threshold for the output was set to triple locus variation. The black lines denote the SLVs; while the gray lines indicate the TLVs. ST4 is the founder clone of the clonal complex.
Figure 5 shows the goeBURST analysis of Cronobacter STs according to species. The formation of 13 SLV CC among 115 STs are revealed. The minimum requirement for the CC formation was an SLV linkage with the founder ST. Apart from these, a number of DLVs and TLVs were observed which, though not defined as CCs, have been indicated in the figures. Figure 6 shows the goeBURST analysis of Cronobacter STs according to the diversity of the countries of isolation. It illustrates the stability of certain STs such as ST1, ST4, and ST7 isolated from a range of countries over >50 years. Figure 7 is the goeBURST analysis of Cronobacter STs according to the diversity of their sources of isolation. This figure shows the majority of the clinical isolates to be concentrated in ST4, ST8, ST1, and their related STs, while ST1 seemed to also predominate with the milk powder factory and infant formula isolates. See also Table 1. Thus, the Cronobacter MLST scheme has been successful in the clustering of the clinically associated Cronobacter strains into specific STs/CC to form stable virulent lineages. A similar phenomenon has also been seen in the MLST of the meningococcus, Neisseria meningitidis, with the formation of hyper-invasive lineages (Caugant and Maiden, 2009).
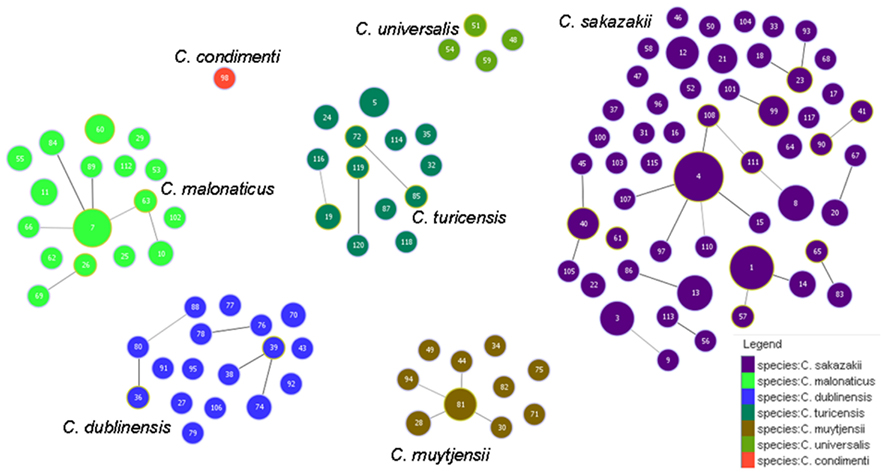
Figure 5. Population snapshot of the Cronobacter MLST database generated using the goeBURST algorithm, indicating the clonal complexes and the breakdown of the species of the strains. The threshold for the output was set to triple locus variation. The dominant STs are represented by the circles with larger diameters.

Figure 6. Population snapshot of the Cronobacter MLST database generated using goeBURST, indicating the clonal complexes and the diversity of the strains based on country of isolation. The threshold for the output was set to triple locus variation. The dominant STs are represented by the circles with larger diameters.
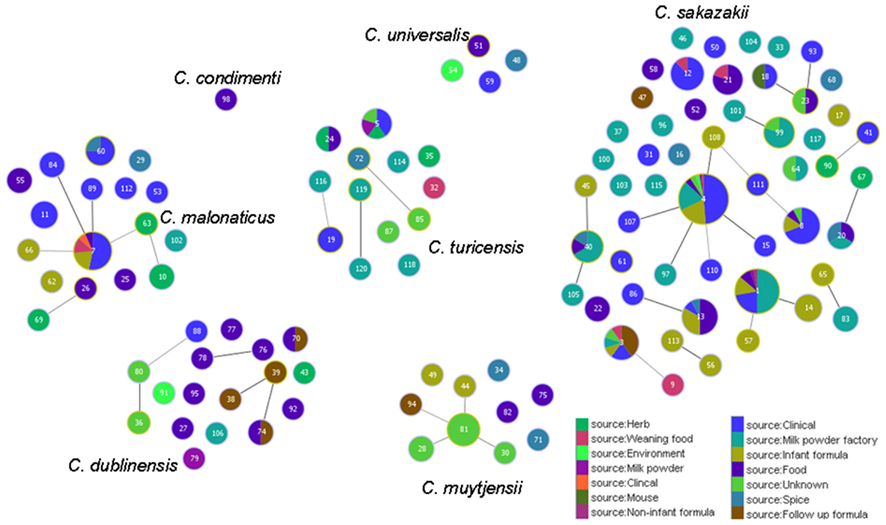
Figure 7. Population snapshot of the Cronobacter MLST database generated using the goeBURST algorithm, indicating the clonal complexes and the diversity of the sources of the strains. The threshold for the output was set to triple locus variation. The dominant STs are represented by the circles with larger diameters.
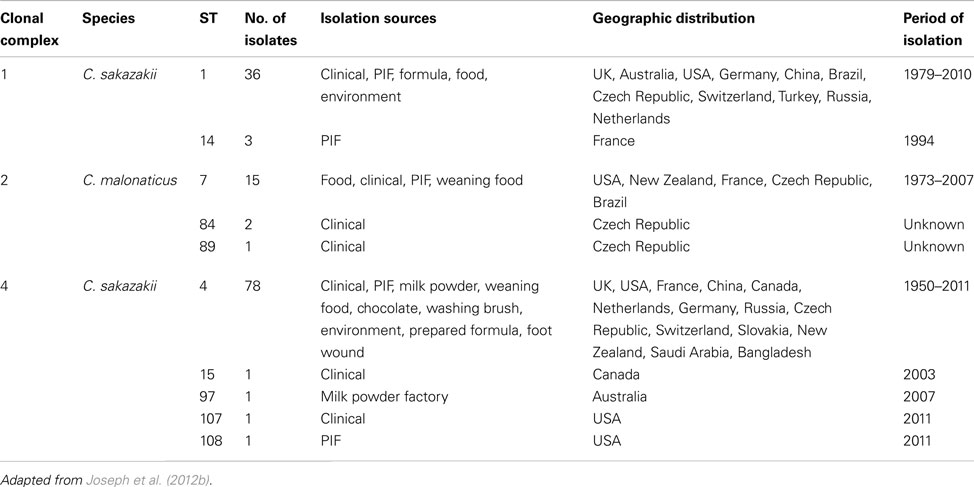
Table 1. Major clonal complexes in the Cronobacter MLST database.
Summary
The Cronobacter genus has come to the attention of the food industry, especially infant formula manufacturers, and regulators due to its association with life-threatening infections of neonates. Our current knowledge of the virulence and epidemiology of this organism is limited, and therefore an improved understanding of the diversity of the genus is warranted. Studying the organism through the application of MLST and MLSA has revealed a vast amount of information on the emergent bacterial pathogen, using just seven primer pairs with an costs equivalent to phenotyping and with all DNA sequence information being stored on an electronically portable open access database. This approach has established new Cronobacter species, the evolution of the genus and recognition of the clonal lineage attributed to the majority of neonatal meningitis cases. Supported by an open access curated database, the technique has become an established means of investigating an emergent bacterial pathogen which can cause a devastating infection in newborn babies. As an example of the benefits of rapid DNA sequencing, even without considering Next Generation Sequencing of genomes, the study of just seven loci is a model for modern microbial epidemiology and food microbiology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Nottingham Trent University for their financial support and the many contributors to the Cronobacter PubMLST database (http://www.pubMLST.org/cronobacter).
References
Acinas, S. G., Marcelino, L. A., Klepac-Ceraj, V., and Polz, M. F. (2004). Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 186, 2629–2635.
Baldwin, A., Loughlin, M., Caubilla-Barron, J., Kucerova, E., Manning, G., Dowson, C., et al. (2009). Multilocus sequence typing of Cronobacter sakazakii and Cronobacter malonaticus reveals stable clonal structures with clinical significance which do not correlate with biotypes. BMC Microbiol. 9, 223. doi:10.1186/1471-2180-9-223
Block, C., Peleg, O., Minster, N., Bar-Oz, B., Simhon, A., Arad, I., et al. (2002). Cluster of neonatal infections in Jerusalem due to unusual biochemical variant of Enterobacter sakazakii. Eur. J. Clin. Microbiol. Infect. Dis. 21, 613–616.
Caubilla-Barron, J., Hurrel, E., Townsend, S., Cheetham, P., Loc-Carrillo, C., Fayet, O., et al. (2007). Genotypic and phenotypic analysis of Enterobacter sakazakii strains from an outbreak resulting in fatalities in a neonatal intensive care unit in France. J. Clin. Microbiol. 45, 3979–3985.
Caugant, D. A., and Maiden, M. C. J. (2009). Meningococcal carriage and disease – population biology and evolution. Vaccine 27, B64–B70.
Chen, Y., Strain, E. A., Allard, M., and Brown, E. W. (2011). Genome sequence of Cronobacter sakazakii E899, a strain associated with human illness. J. Bacteriol. 193, 581.
Craven, H. M., McAuley, C. M., Duffy, L. L., and Fegan, N. (2010). Distribution, prevalence and persistence of Cronobacter (Enterobacter sakazakii) in the nonprocessing and processing environments of five milk powder factories. J. Appl. Microbiol. 109, 1044–1052.
Cruz, A., Xicohtencatl-Cortes, J., González-Pedrajo, B., Bobadilla, M., Eslava, C., and Rosas, I. (2011a). Virulence traits in Cronobacter species isolated from different sources. Can. J. Microbiol. 57, 735–744.
Cruz, A. L., Rocha-Ramirez, L. M., Gonzalez-Pedrajo, B., Ochoa, S. A., Eslava, C., and Rosas, I. (2011b). Flagella from Cronobacter sakazakii induced an inflammatory response in human monocytes. Cytokine 56, 95.
Czerwicka, M., Forsythe, S. J., Bychowska, A., Dziadziuszko, H., Kunikowska, D., Stepnowski, P., et al. (2010). Chemical structure of the O-polysaccharide isolated from Cronobacter sakazakii 767. Carbohydr. Res. 345, 908–913.
FAO/WHO. (2008). Enterobacter Sakazakii (Cronobacter spp.) in Powdered Follow-up Formula - Meeting Report (Microbiological Risk Assessment Series), Rome, 90. Available at: http://www.who.int/foodsafety/publications/micro/mra_followup/en/
Francisco, A. P., Vaz, C., Monteiro, P. T., Melo-Cristino, J., Ramirez, M., and Carrico, J. A. (2012). PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinformatics 13, 87. doi:10.1186/1471-2105-13-87
Franco, A. A., Hu, L., Grim, C. J., Gopinath, G., Sathyamoorthy, V., Jarvis, K. G., et al. (2011). Characterization of putative virulence genes on the related RepFIB plasmids harbored by Cronobacter spp. Appl. Environ. Microbiol. 77, 3255–3267.
Friedemann, M. (2007). Enterobacter sakazakii in food and beverages (other than infant formula and milk powder). Int. J. Food Microbiol. 116, 1–10.
Gakuya, F. M., Kyule, M. N., Gathura, P. B., and Kariuki, S. (2001). Antimicrobial resistance of bacterial organisms isolated from rats. East Afr. Med. J. 78, 646–649.
Grim, C. J., Kothary, M. H., Gopinath, G., Jarvis, K. G., Beaubrun, J. J., McClelland, M., et al. (2012). Identification and characterization of Cronobacter iron acquisition systems. Appl. Environ. Microbiol. 78, 6035–6050.
Hamby, S., Joseph, S., Forsythe, S. J., and Chuzhanova, N. (2011). In silico identification of pathogenic strains of Cronobacter from biochemical data reveals association of inositol fermentation with pathogenicity. BMC Microbiol. 11, 204. doi:10.1186/1471-2180-11-204
Hanage, W. P., Fraser, C., and Spratt, B. G. (2005). Fuzzy species among recombinogenic bacteria. BMC Biol. 7, 6. doi:10.1186/1741-7007-3-6
Hariri, S., Joseph, S., and Forsythe, S. J. (2012). Predominance of Cronobacter sakazakii ST4 clonal complex strains in Cronobacter neonatal meningitis infections in US 2011. Emerging Infect. Dis. (in press).
Himelright, I., Harris, E., Lorch, V., and Anderson, M. (2002). Enterobacter sakazakii infections associated with the use of powdered infant formula – Tennessee, 2001. J. Am. Med. Assoc. 287, 2204–2205.
Hurrell, E., Kucerova, E., Loughlin, M., Caubilla-Barron, J., Hilton, A., Armstrong, R., et al. (2009). Neonatal enteral feeding tubes as loci for colonisation by members of the Enterobacteriaceae. BMC Infect. Dis. 9, 146. doi:10.1186/1471-2334-9-146
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267.
Iversen, C., and Forsythe, S. (2003). Risk profile of Enterobacter sakazakii, an emergent pathogen associated with infant milk formula. Trends Food Sci. Technol. 14, 443–454.
Iversen, C., Lehner, A., Mullane, N., Bidlas, E., Cleenwerck, I., Marugg, J., et al. (2007). The taxonomy of Enterobacter sakazakii: proposal of a new genus Cronobacter gen. nov. and descriptions of Cronobacter sakazakii comb. nov. Cronobacter sakazakii subsp. sakazakii, comb. nov., Cronobacter sakazakii subsp. malonaticus subsp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov. and Cronobacter genomospecies 1. BMC Evol. Biol. 7, 64. doi:10.1186/1471-2148-7-64
Iversen, C., Mullane, N., McCardell, B., Tall, B. D., Lehner, A., Fanning, S., et al. (2008). Cronobacter gen. nov., a new genus to accommodate the biogroups of Enterobacter sakazakii, and proposal of Cronobacter sakazakii gen. nov., comb. nov., Cronobacter malonaticus sp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov., Cronobacter genomospecies 1, and of three subspecies, Cronobacter dublinensis subsp. dublinensis subsp. nov., Cronobacter dublinensis subsp. lausannensis subsp. nov. and Cronobacter dublinensis subsp. lactaridi subsp. nov. Int. J. Syst. Evol. Biol. 58, 1442–1447.
Jacobs, C., Braun, P., and Hammer, P. (2011). Reservoir and routes of transmission of Enterobacter sakazakii (Cronobacter spp.) in a milk powder-producing plant. J. Dairy Sci. 94, 3801–3810.
Jarvis, K. G., Grim, C. J., Franco, A. A., Gopinath, G., Sathyamoorthy, V., Hu, L., et al. (2011). Molecular characterization of Cronobacter lipopolysaccharide O-antigen gene clusters and development of serotype-specific PCR assays. Appl. Environ. Microbiol. 77, 4017–4026.
Joseph, S., Cetinkaya, E., Drahovska, H., Levican, A., Figueras, M. J., and Forsythe, S. J. (2012a). Cronobacter condimenti sp. nov., isolated from spiced meat and Cronobacter universalis sp. nov., a novel species designation for Cronobacter sp. genomospecies 1, recovered from a leg infection, water, and food ingredients. Int. J. Syst. Evol. Microbiol. 62, 1277–1283.
Joseph, S., Sonbol, H., Hariri, S., Desai, P., McClelland, M., and Forsythe, S. J. (2012b). Diversity of the Cronobacter genus as revealed by multi locus sequence typing. J. Clin. Microbiol. 50, 3031–3039.
Joseph, S., and Forsythe, S. J. (2011). Predominance of Cronobacter sakazakii ST4 with neonatal infections. Emerging Infect. Dis. 17, 1713–1715.
Kim, K., Kim, K.-P., Choi, J., Lim, J.-A., Lee, J., Hwang, S., et al. (2010). Outer membrane proteins A (OmpA) and X (OmpX) are essential for basolateral invasion of Cronobacter sakazakii. Appl. Environ. Microbiol. 76, 5188–5198.
Konstantinidis, K. T., Ramette, A., and Tiedje, J. M. (2006). The bacterial species definition in the genomic era. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1929–1940.
Kothary, M. H., McCardell, B. A., Frazar, C. D., Deer, D., and Tall, B. D. (2007). Characterization of the zinc-containing metalloprotease encoded by zpx and development of a species-specific detection method for Enterobacter sakazakii. Appl. Environ. Microbiol. 73, 4142–4151.
Kucerova, E., Clifton, S. W., Xia, X. Q., Long, F., Porwollik, S., Fulton, L., et al. (2010). Genome sequence of Cronobacter sakazakii BAA-894 and comparative genomic hybridization analysis with other Cronobacter species. PLoS ONE 5, e9556. doi:10.1371/journal.pone.0009556
Kucerova, E., Joseph, S., and Forsythe, S. (2011). The Cronobacter genus: ubiquity and diversity. Qual. Assur. Saf. Crop Food 3, 104–122.
McQuiston, J. R., Herrera-Leon, S., Wertheim, B. C., Doyle, J., Fields, P. I., Tauxe, R. V., et al. (2008). Molecular phylogeny of the Salmonellae: relationships among Salmonella species and subspecies determined from four housekeeping genes and evidence of lateral gene transfer events. J. Bacteriol. 190, 7060–7067.
Mramba, F., Broce, A., and Zurek, L. (2006). Isolation of Enterobacter sakazakii from stable flies, Stomoxys calcitrans L. (Diptera: Muscidae). J. Food Prot. 69, 671–673.
Muytjens, H. L., Roelofs-Willemse, H., and Jaspar, G. H. (1988). Quality of powdered substitutes for breast milk with regard to members of the family Enterobacteriaceae. J. Clin. Microbiol. 26, 743–746.
Pagotto, F. J., Nazarowec-White, M., Bidawid, S., and Farber, J. M. (2003). Enterobacter sakazakii: infectivity and enterotoxin production in vitro and in vivo. J. Food Prot. 66, 370–375.
Pava-Ripoll, M., Pearson, R. E., Miller, A. K., and Ziobro, G. C. (2012). Prevalence and relative risk of Cronobacter spp., Salmonella spp. and Listeria monocytogenes associated with the body surface and the guts of individual filth flies. Appl. Environ. Microbiol. [Epub ahead of print]. doi:10.1128/AEM.02195-12
Seo, K. H., and Brackett, R. E. (2005). Rapid, specific detection of Enterobacter sakazakii in infant formula using a real-time PCR assay. J. Food Prot. 68, 59–63.
Stephan, R., Lehner, A., Tischler, P., and Rattei, T. (2010). Complete genome sequence of Cronobacter turicensis LMG 23827, a food-borne pathogen causing deaths in neonates. J. Bacteriol. 193, 309–310.
Stoop, B., Lehner, A., Iversen, C., Fanning, S., and Stephan, R. (2009). Development and evaluation of rpoB based PCR systems to differentiate the six proposed species within the genus Cronobacter. Int. J. Food Microbiol. 136, 165–168.
Sun, Y., Wang, M., Liu, H., Wang, J., He, X., Zeng, J., et al. (2011). Development of an O-antigen serotyping scheme for Cronobacter sakazakii. Appl. Environ. Microbiol. 77, 2209–2214.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739.
Townsend, S. M., Hurrell, E., and Forsythe, S. (2008). Virulence studies of Enterobacter sakazakii isolates associated with a neonatal intensive care unit outbreak. BMC Microbiol. 8, 64. doi:10.1186/1471-2180-8-64
Townsend, S. M., Hurrell, E., Gonzalez-Gomez, I., Lowe, J., Frye, J. G., Forsythe, S., et al. (2007). Enterobacter sakazakii invades brain capillary endothelial cells, persists in human macrophages influencing cytokine secretion and induces severe brain pathology in the neonatal rat. Microbiology 153, 3538–3547.
Turcovský, I., Kuniková, K., Drahovská, H., and Kaclíková, E. (2011). Biochemical and molecular characterization of Cronobacter spp. (formerly Enterobacter sakazakii) isolated from foods. Antonie Van Leeuwenhoek 99, 257–269.
Keywords: Cronobacter, MLST, MLSA, sequence typing, ST4
Citation: Joseph S and Forsythe SJ (2012) Insights into the emergent bacterial pathogen Cronobacter spp., generated by multilocus sequence typing and analysis. Front. Microbio. 3:397. doi: 10.3389/fmicb.2012.00397
Received: 26 September 2012; Paper pending published: 17 October 2012;
Accepted: 30 October 2012; Published online: 22 November 2012.
Edited by:
Danilo Ercolini, Università degli Studi di Napoli Federico II, ItalyReviewed by:
Anderson D. Sant’Ana, University of São Paulo, BrazilGiorgio Giraffa, Agriculture Research Council, Fodder and Dairy Productions Research Centre, Italy
Copyright: © 2012 Joseph and Forsythe. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Stephen J. Forsythe, Pathogen Research Group, School of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK. e-mail:stephen.forsythe@ntu.ac.uk