
- Department of Biological Chemistry, University of Michigan Medical School, University of Michigan, Ann Arbor, MI, USA
Methyl-coenzyme M reductase (MCR) from methanogenic archaea catalyzes the rate-limiting and final step in methane biosynthesis. Using coenzyme B as the two-electron donor, MCR reduces methyl-coenzyme M (CH3-SCoM) to methane and the mixed disulfide, CoBS-SCoM. MCR contains an essential redox-active nickel tetrahydrocorphinoid cofactor, Coenzyme F430, at its active site. The active form of the enzyme (MCRred1) contains Ni(I)-F430. Rapid and efficient conversion of MCR to MCRred1 is important for elucidating the enzymatic mechanism, yet this reduction is difficult because the Ni(I) state is subject to oxidative inactivation. Furthermore, no in vitro methods have yet been described to convert Ni(II) forms into MCRred1. Since 1991, it has been known that MCRred1 from Methanothermobacter marburgensis can be generated in vivo when cells are purged with 100% H2. Here we show that purging cells or cell extracts with CO can also activate MCR. The rate of in vivo activation by CO is about 15 times faster than by H2 (130 and 8 min-1, respectively) and CO leads to twofold higher MCRred1 than H2. Unlike H2-dependent activation, which exhibits a 10-h lag time, there is no lag for CO-dependent activation. Based on cyanide inhibition experiments, carbon monoxide dehydrogenase is required for the CO-dependent activation. Formate, which also is a strong reductant, cannot activate MCR in M. marburgensis in vivo.
Introduction
Methanogens are responsible for all biological methane production on earth, generating 109 tons of methane annually. Being strict anaerobes, they are widely distributed in anoxic environments where electron acceptors such as , Fe3+, and are limiting, such as aquatic sediments, the digestive tract of animals, rice fields, sewage digesters, landfills, heart wood of living trees, and decomposing algal mats (Garcia et al., 2000). Methanogens play critical roles in the carbon cycle by converting products from anaerobic fermentation (such as hydrogen, carbon dioxide, methanol, formate, and acetate) into methane (Thauer, 1998; Ferry, 2010). Only by such means can methanogens obtain energy to grow (Thauer, 1998; Ferry, 2010). Methane, the principal component of natural gas, is an important source of clean renewable energy, with the highest heat production per mass unit (55.7 kJ g-1) among all hydrocarbons (Richard and Ball, 2008). However, methane also is a potent greenhouse gas (Hanson and Hanson, 1996) and, predominantly due to changing agricultural practices (e.g., increased development of rice production and livestock cultivation) over the past two centuries, the atmospheric concentration of methane has more than doubled, reaching a level (1770 ppb in 2005) that far exceeds the natural range (320–790 ppb) of the last 650,000 years (Wuebbles and Hayhoe, 2000). Therefore, because of the effect of greenhouse gases on climate change, understanding the basis of methane production is an important research goal, while controlling methane emissions is an important aim for governmental policy makers.
Methyl-coenzyme M reductase (MCR, EC 2.8.4.1), a nickel-containing enzyme, catalyzes the rate-limiting and final step of methane production and the first step in anaerobic methane oxidation (Dey et al., 2010b; Scheller et al., 2010). For methane formation, MCR converts methyl-coenzyme M (CH3-SCoM) and coenzyme B (CoBSH) to methane and the heterodisulfide of coenzyme M and coenzyme B (CoBS-SCoM) as shown in Eq. 1 (DiMarco et al., 1990; Thauer, 1998).
A number of high-resolution crystal structures of MCR are available (Ermler et al., 1997; Grabarse et al., 2000, 2001; Cedervall et al., 2010, 2011). These structures reveal MCR to be a dimer of heterotrimers (α2β2γ2) with a molecular mass of around 270 kDa (Ermler et al., 1997). The three subunits (αβγ) are tightly associated, especially between subunits α and α′ and between β and β′, forming two 50 Å hydrophobic channels (one in each heterotrimer) ending in a pocket that accommodates a deeply embedded redox-sensitive nickel tetrapyrrole cofactor called coenzyme F430, which plays an essential role in catalysis (Goubeaud et al., 1997; Becker and Ragsdale, 1998). The substrates and products thread through the channel toward F430. In each heterotrimer, there is one F430-containing active site, located 50 Å from the F430 in the other heterotrimer. Due to its extreme O2 sensitivity, MCR in all of these structures but one contains F430 in the inactive Ni(II) state (Ermler et al., 1997; Grabarse et al., 2000, 2001; Cedervall et al., 2010), the other being methyl-Ni(III) (Cedervall et al., 2011). More than 16 distinct oxidation and coordination states of MCR have been spectroscopically characterized (Cedervall et al., 2010). To initiate catalysis, the enzyme must be in the MCRred1 state, which contains the redox-active nickel as Ni(I) (Albracht et al., 1988; Goubeaud et al., 1997; Becker and Ragsdale, 1998). The binding site of HSCoM (and presumably CH3-SCoM) is more deeply buried within the enzyme, and so it must enter prior to CoBSH for productive chemistry to occur. Therefore, it was suggested that a conformational change upon CH3-SCoM binding might lower the Kd for CoBSH and promote an ordered substrate binding mechanism (Grabarse et al., 2001). Furthermore, electron paramagnetic resonance (EPR) and electron nuclear double resonance (ENDOR) studies indicate that CoBSH binding induces a conformational change that brings the methyl group of methyl-SCoM into closer proximity to the nickel, which could promote C–S bond cleavage (Ebner et al., 2010).
Two main catalytic mechanisms for MCR-catalyzed reaction have been under debate for a long time. These two mechanisms in principle differ in how they describe the initial cleavage of sulfur–carbon bond of the substrate CH3-SCoM (Ermler, 2005). Mechanism I involves a methyl-Ni(III) intermediate (Ermler et al., 1997), while mechanism II postulates formation of a methyl radical and a CoMS–Ni(II) complex (Pelmenschikov et al., 2002). Since none of the proposed intermediates have been trapped and spectroscopically or structurally characterized when using natural substrates, both mechanisms remain controversial. Based on spectroscopic and rapid kinetic studies, a hybrid mechanism involving both methyl-Ni and methyl radical has been proposed (Dey et al., 2010a,b; Li et al., 2010). In order to perform mechanistic studies, it is crucial to have a reliable protocol to generate the active state of MCR.
It is rather straightforward to purify native MCR from methanogenic cells; however, it has been extremely difficult to isolate and maintain MCR in its active state, due to the low midpoint redox potential of the Ni(II)/Ni(I) couple of bound F430 (between -700 and -600 mV compared to the normal hydrogen electrode, NHE; Holliger et al., 1993). Without special treatment of the cells and addition of reductants and HSCoM (or analogs) to the purification buffers, 99% of the MCR activity is lost upon cell lysis (Gunsalus and Wolfe, 1980; Brenner et al., 1993). In the late 1970s and early 1980s, several protein fractions (components A1, A2, A3a, A3b, and C) and compounds [component B and catalytic amounts of ATP, Mg2+, flavin adenine dinucleotide (FAD), and F420] were used to activate MCR from Methanothermobacter thermautotrophicus in vitro (Gunsalus and Wolfe, 1980; Nagle and Wolfe, 1983). However, only 1–5% of the MCR activity can be recovered by this activation.
In 1991, Thauer and colleagues found that the M. marburgensis MCR could be isolated in a much more active form (up to 50% of the in vivo activity) when cells are pre-incubated with 100% H2 and CH3-SCoM (or HSCoM) before lysis and purified under strict anaerobic conditions in the presence of Ti(III) citrate and HSCoM (Rospert et al., 1991a). CH3-SCoM and HSCoM were shown to stabilize both the activity and the EPR signal of MCRred1 (Rospert et al., 1991a). Since then, H2-dependent activation has been used to purify MCRred1 and 50–90% MCRred1 is routinely obtained (Li et al., 2010; Duin et al., 2011). Thus, although the steps involved in MCR activation are not known, it is clear that MCR activation involves a reductive activation to the Ni(I) state (Rospert et al., 1991b; Dey et al., 2006).
Here we report that carbon monoxide, which is a stronger reductant than H2, can activate MCR in vivo and in cell extracts of M. marburgensis. The rate of CO-dependent activation (130 min-1) is about 15-fold faster than H2-dependent activation (8 min-1). Furthermore, CO can activate MCR to a higher percentage. We observe no activation of MCR when cells are treated with formate, another strong reductant, using similar conditions as with CO or H2.
Materials and Methods
Biochemicals, Gas Mixes, and Cell Growth
All buffers, media ingredients, and other reagents were acquired from Sigma-Aldrich (St. Louis, MO, USA) and, unless otherwise stated, were of the highest purity available. Solutions were prepared using Nanopure deionized water. N2 (99.98%), CO (99.99%), argon (99.8%), H2/CO2 (80%/20%), and ultrahigh purity H2 (99.999%) were obtained from cryogenic gases (Grand Rapids, MI, USA). Ti(III) citrate solutions were prepared from a stock solution of 200 mM Ti(III) citrate, which was synthesized by adding sodium citrate to Ti(III) trichloride (30 wt% solution in 2 N hydrochloric acid; Acros Organics, Morris Plains, NJ, USA) under anaerobic conditions and adjusting the pH to 7.0 with sodium bicarbonate (Zehnder and Wuhrmann, 1976). The concentration of Ti(III) citrate was determined routinely by titrating a methyl viologen solution. M. marburgensis (formally M. thermoautotrophicum strain Marburg) was obtained from the Oregon Collection of Methanogens catalog as OCM82. M. marburgensis was cultured on H2/CO2 (80%/20%) at 65°C in a 14-L fermentor (New Brunswick Scientific Co. Inc., New Brunswick, NJ, USA). Culture media were prepared as previously described (Kunz et al., 2006) with a slight modification of the sulfur and reducing source, by adding 50 mM sodium sulfide (instead of H2S gas) at a flow rate of 1 ml min-1 during the entire growth period.
Activation of MCR in Whole Cells by H2, CO, or Formate
Solutions were prepared and all manipulations were performed under strictly anaerobic conditions in a Vacuum Atmospheres (Hawthorne, CA, USA) anaerobic chamber maintained under nitrogen gas at <1 ppm of oxygen (monitored continually by a Teledyne detector). Cells were grown under H2/CO2 as described (Dey et al., 2010a) and, before harvest, the cells were treated with H2 for 30 min in the fermenter (different gases were used in the experiments shown in Figure 1D). Then, the cells were harvested, transferred to the anaerobic chamber, and resuspended in 50 mM Tris, pH 7.6, 10 mM HSCoM, 0.1 mM Ti(III) citrate, and aliquoted into 150 ml serum-stoppered anaerobic bottles, as described (Dey et al., 2010a; Li et al., 2010). The headspaces of the vials containing the resuspended cells were then purged with CO or H2 for 10 min, or at timed intervals right before samples were taken for analysis. This second (and all subsequent) purge was also omitted for the formate-activation experiment as described in the next paragraph to ensure that the gas treatment did not interfere with the formate incubation. To facilitate equilibration of the gas with the solution, the vials were shaken every 2 min. In most experiments, the cell suspension was transferred into smaller vials, where the headspace was purged again for 10 min with the test gas. Purges with H2 were done inside of the chamber, while, for safety reasons, purges with CO were done in the fume hood outside of the chamber. To remove trace amounts of oxygen in the H2 or CO line for aerating the serum vials, the line was fitted with a high pressure Oxy-Trap oxygen scrub (Alltech, Deerfield, IL, USA) followed by an indicating Oxy-Trap (Alltech) to monitor the efficiency/remaining capacity of the oxygen scrub. Between the purges, the serum vials were inverted in a water bath in the anaerobic chamber to minimize any gas escape. For monitoring the conversion of MCR to the MCRred1 state, 200 μl samples were removed at different time points to be analyzed by EPR spectroscopy.
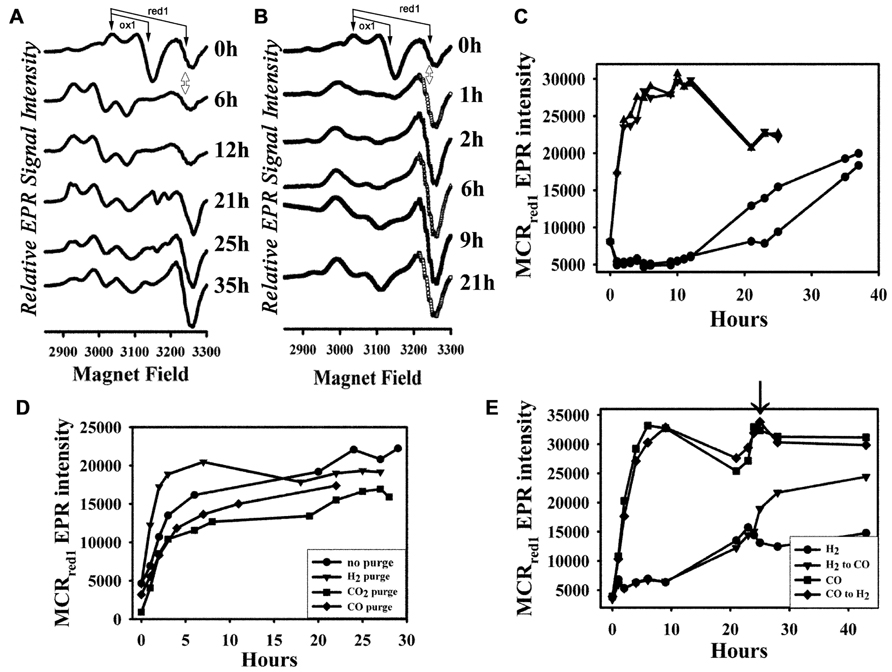
FIGURE 1. Activation of MCR by H2 and CO in vivo. (A) EPR spectra of the H2-activated cell samples at different time points; (B) EPR spectra of the CO-activated cell samples at different time points; (C) plot of intensity of the MCRred1 EPR signal (the “S”-shaped derivative signal at ~3250 G, double-sided arrow) over time with duplicate H2- (circles) and CO-(triangles) activated cell samples; (D) plot of MCRred1 EPR intensity over time with CO activation with no initial purge (circles) in the fermentor, or initially purged with H2 (triangle), CO2 (square), or CO (diamond) in the fermentor; (E) plot of MCRred1 EPR intensity over time of treatment (as in C) with H2 (circles) and CO (triangles), arrow indicates the switch of gases. The features of the MCRox1 and MCRred1 signals are depicted with arrows to demarcate their g values.
To determine whether formate activates MCR, the same protocol outlined above was followed (including the first 30 min H2 purge in the fermenter), except that sodium formate was added directly to the cell suspension to a final concentration of 100 or 500 mM (pH did not change in the Tris buffer that is described above for resuspending cells). Thus, there was only one gas purge in this experiment. To inhibit carbon monoxide dehydrogenase (CODH) activity, NaCN was added to the cell suspension to a final concentration of 0.2 or 0.4 mM before aerating with CO. Then, the cell suspension was incubated as described above.
Spectroscopy of MCR
UV-visible spectra of MCR were recorded in the anaerobic chamber using a diode array spectrophotometer (model DT 1000A, Analytical Instrument Systems, Inc., Flemington, NJ, USA). All samples for EPR spectroscopy were prepared in 50 mM Tris–HCl buffer, pH 7.6, in a Vacuum Atmospheres anaerobic chamber maintained under nitrogen gas at <1 ppm of oxygen. EPR spectra were recorded on a Bruker EMX spectrometer (Bruker Biospin Corp., Billerica, MA, USA), equipped with an Oxford ITC4 temperature controller, a Hewlett-Packard model 5340 automatic frequency counter, and Bruker gaussmeter. Unless otherwise noted, the EPR spectroscopic parameters were: temperature, 70 K; microwave power, 10 mW; microwave frequency, 9.43 GHz; receiver gain, 2 × 104; modulation amplitude, 10 G; and modulation frequency, 100 kHz. Double integrations of the EPR spectra were performed and referenced to a 1-mM copper perchlorate standard.
Protein Purification
The purification of MCRred1 from M. marburgensis was performed under strictly anaerobic conditions, as described previously (Kunz et al., 2006). The amount of MCRred1 was calculated according to the UV-visible and EPR spectra (Kunz et al., 2006; Li et al., 2010). Protein concentration was determined by Rose Bengal assay (Elliott and Brewer, 1978) with lysozyme as the standard.
Enzyme Activity Assays
Methyl-coenzyme M reductase assays for methane formation from methyl-CoM and CoBSH were performed at 60°C, basically as described by Kunz et al. (2006). The standard assay mixture contained 10 mM 14CH3-SCoM, 0.1 mM CoBSH, 1.8 mM aquacobalamin, 20 mM Ti(III) citrate, and 0.5 M Tris (pH 7.2) in a final volume of 0.2 ml. The reaction was started by adding MCR. MCR activity was measured by following the time-dependent loss of radioactivity from the methyl group of 14CH3-SCoM, which is converted to highly volatile and insoluble [14C] methane (Kunz et al., 2006). Rates of methane formation were calculated from the linear portion of the time course. One unit of MCR activity is equal to 1 μmol of methane produced per minute.
To determine formate dehydrogenase (FDH), hydrogenase, and CODH activities, M. marburgensis cell extracts were prepared under anaerobic conditions as described (Kunz et al., 2008). All assays were performed in a 1-ml sealed solution at 60°C. The anaerobic assay mixture contained 20 mM methyl viologen, 50 mM Tris, pH 7.5, and cell extract. For the hydrogenase and CODH assays, oxygen-free hydrogen gas or carbon monoxide (99.99%), respectively, was bubbled into the assay mix in a serum-stopped cuvette for 5 min (~1 mM of each gas in the solution) before enzyme was added to start the reaction. In the FDH assay, 20 mM formate was used and either 20 mM methyl viologen or 10 mM NADP+ was included as an electron acceptor. The activities were determined by following the reduction of methyl viologen at 578 nm with an extinction coefficient of 9.78 mM-1 cm-1 or the reduction of NADP+ at 340 nm with an extinction coefficient of 6.27 mM-1 cm-1.
Results
Generation of MCRred1 in vivo by Co
To date, the most effective way to activate MCR has been to incubate the cell suspension under a H2 atmosphere (Rospert et al., 1991a; Kunz et al., 2006). While this is effective for generating MCRred1 from organisms like M. marburgensis, for some methanogens, like Methanosarcina acetivorans, H2 activation is inefficient because the hydrogenases are weakly expressed (Guss et al., 2009; Wang et al., 2011). Therefore, there must be other pathways for in vivo activation of MCR. We hypothesized that the low-potential reductant, CO, with a standard reduction potential (vs. NHE for the CO2/CO couple) of -520 mV (in comparison to -420 mV for that of 2 H+/H2; Lehn and Ziessel, 1982) should be able to activate MCR.
To measure the effectiveness of CO in activating MCR, we purged the M. marburgensis culture for 30 min with H2 in the fermentor, harvested the cells, resuspended the cells in buffer and incubated the cell suspensions at 32°C in a water bath under a CO or H2 atmosphere. The whole-cell EPR spectra of the H2- and CO-treated cells at different time points after initiating H2 or CO gas purge are compared in Figures 1A,B, respectively. The activity of MCR is linearly related to the MCRred1 EPR signal, with g values at 2.24 and 2.065; thus EPR spectroscopy is a convenient method to assess the level of MCR activation achieved. It also reveals various other EPR-active forms of MCR that are present during the treatment – for example, MCRox1, which is an oxidized form of MCR that has a distinct EPR spectrum (g values at 2.23 and 2.16), can be converted into the MCRred1 state by the addition of low-potential reductants like Ti(III) citrate, and MCRred2, a Ni(I) state that is formed when the enzyme in the MCRred1 form is treated with HSCoM and CoBSH (Mahlert et al., 2002). Therefore, we feel that assessing the level of MCRred1 is the most accurate indicator of the level of activation achieved. The spectra of the cell suspension before the CO/H2 purge reveal about two to three times more MCRox1 than MCRred1, very little MCRred2, and the remainder in the EPR-silent Ni(II) form. For the H2-purged cells, after ~1 h of incubation, MCRox1 disappeared as MCRred2 increased; then, over the next ~20 h, MCRred1 appeared. Following a ~10 h lag time, the rate constant for H2-dependent activation was 8 min-1. The amount of MCRred1 formed at the end of activation, when compared with the amount of MCRox1 in the beginning, varied slightly among enzyme preparations.
In contrast, when cells were incubated with CO during the timed purge, within 1 h, MCR underwent rapid activation to the MCRred1 state without a noticeable lag time or intermediacy of MCRred2. The enhanced activation by CO was noticeable by eye – the cell suspension began turning green immediately after aerating with CO. However, for H2-dependent activation, this color development required overnight incubation. At the end of activation (when the whole-cell EPR signal reached maximum intensity), the CO-activated cell suspension was also greener than that of the H2-activated cells (data not shown). Quantitative analysis of the activation confirmed the visual observations. The rate constant for CO-dependent activation was 130 min-1, which is over 15-fold faster than that observed for activation by H2; furthermore, the amount of accumulated MCRred1 was ~twofold higher than with H2 (Figure 1C). Furthermore, with CO activation, the amount of MCRred1 formed at the end of activation was approximately two times more than the initial amount of MCRox1, indicating that CO activation converts the MCRsilent form to MCRred1.
As described above, the CO-dependent activation experiments shown in Figures 1B,C involved an initial 30 min H2 purge in the fermentor. To test whether the initial H2 treatment is required, we purged the cell suspension with pure H2, CO, or CO2 in the fermentor, or omitted the initial 30-min purge before harvesting, and then bubbled the cell suspension with CO after resuspending the cells in Tris–HCl buffer as above. The initial EPR spectra significantly differed with different purge conditions. MCRox1 was only observed when the cells were initially purged with H2 in the fermentor, while MCRred1 was only noticeable with the CO treatment or when the initial H2 purge was omitted. On the other hand, MCR appears to be completely in a Ni(II)-silent form when cells were initially purged with CO2, because no detectable MCR signal was present. However, MCR underwent activation in all these conditions (i.e., H2, CO, CO2, no initial purge) after treating the cell suspensions with CO, and the activation rates and the final levels of activation were similar (Figure 1D). Thus, an initial H2 purge is not required for CO-dependent activation of MCR. This experiment also clearly shows that the EPR-silent Ni(II)-MCR forms undergo conversion to MCRred1 by CO in vivo.
We also tested the effect of addition of CO to the H2-purged cells. Figure 1E shows that treatment with CO can further activate MCR in H2-purged cells to nearly reach the level of MCRred1 observed in CO-activated cells. On the other hand, treatment with H2 does not increase the MCRred1 level in CO-activated cells. These experiments indicate that H2- and CO-dependent activations pathways converge after the initial transfer of electrons to the acceptor of reducing equivalents from hydrogenase or CODH.
Effect of Temperature on Co-Dependent MCR Activation
H2-dependent activation of MCR is very sensitive to the incubation temperature; it is most effective at 30–33°C (Kunz et al., 2006). To test whether CO-dependent MCR activation is also temperature-dependent, we incubated the cell suspension with CO at different temperatures and followed the whole-cell EPR spectrum over 12 h. As shown in Figure 2, CO-dependent activation, like the H2-dependent process, is most effective at ~30°C. The complicating factor in these experiments is the inactivation of MCR, which occurs markedly faster as the temperature increases (Horng et al., 2001). At high temperatures (40–60°C), MCRred1 decays after 2 h of CO-dependent activation, while, at lower temperatures (10–20°C), activation by CO occurs more slowly, but, because MCRred1 decays more slowly, this form of the enzyme is more stable and accumulates to higher levels.
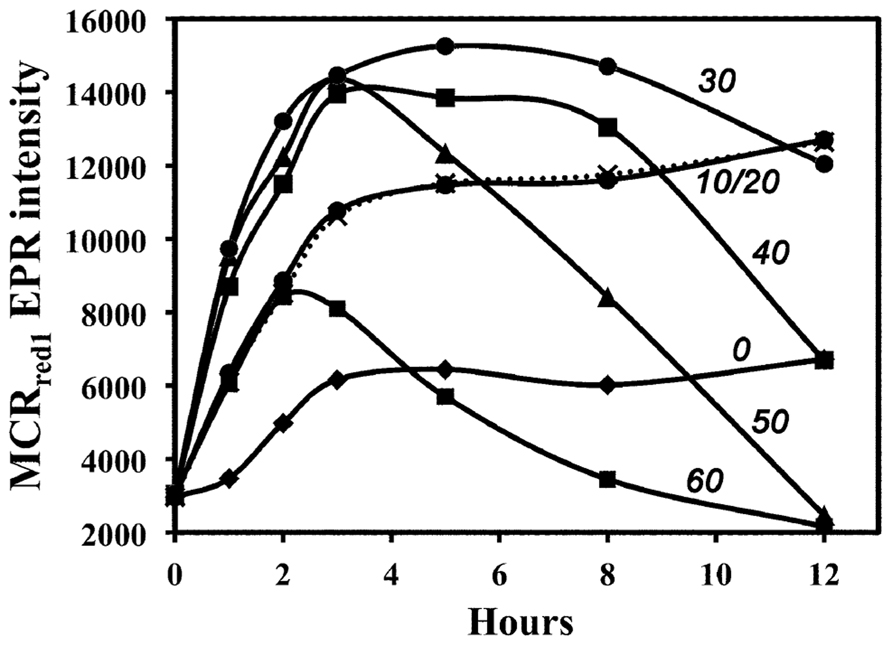
FIGURE 2. Temperature dependence of in vivo MCR activation in whole cells by CO. Activation temperatures (°C) are shown beside the relevant line.
Codh Activity Requirement for Co-Dependent Activation of MCR
To test whether CODH activity is involved in CO-dependent activation of MCR, CN-, which is a competitive slow-binding inhibitor of CODH with a Ki for CODH inhibition in the low micromolar range (Ragsdale et al., 1983; Ha et al., 2007), was added to the cell suspension. As shown in Figure 3, 0.2 mM CN- blocked the CO-dependent activation of MCR, indicating that CODH activity is required for CO-dependent activation of MCR. In contrast, 0.2–0.4 mM CN- does not block H2-dependent activation of MCR (data not shown). We measured a specific activity of 3–5 U mg-1 of CODH in cell extracts, which is significantly lower than that of hydrogenase (~80 U mg-1), but is sufficient to drive the activation of MCR.
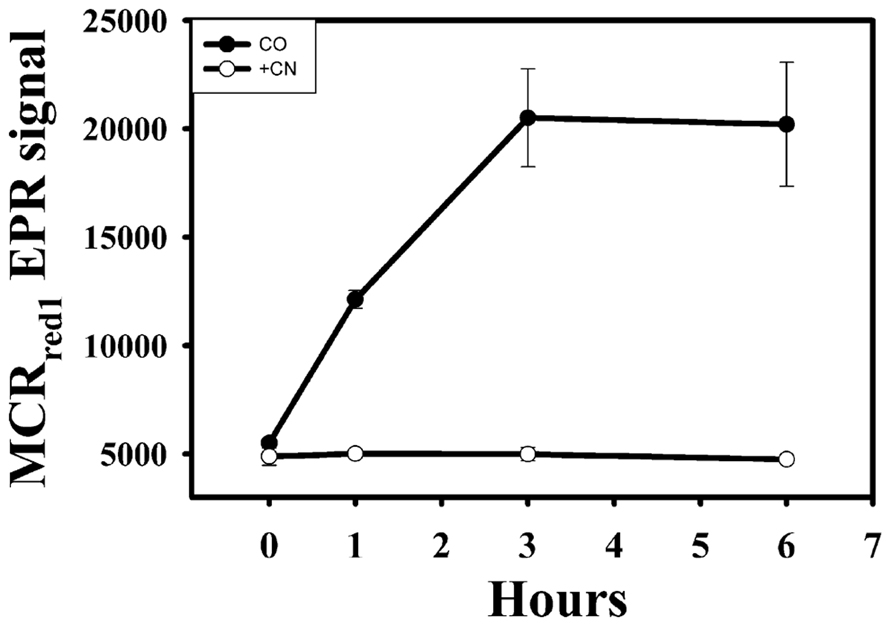
FIGURE 3. Activation of MCR by CO in the presence of CODH inhibitor, CN-. Filled circles, CO-purged samples; open circles, CO-purged samples in the presence of 0.2 mM CN-.
Properties of the Purified MCR from Co-Activated Cells
Methyl-coenzyme M reductase was purified from cells that had been undergone a 5-h incubation with CO. Based on the UV-visible (Figure 4A) and EPR spectra (Figure 4B) of the purified enzyme, MCR was about ~80% active. Based on the enzymatic assay for the conversion of methyl-SCoM to methane, the specific activity of the CO-activated enzyme was calculated to be 89 ±10 U mg-1, indicating 89% conversion to the active MCRred1 state. Thus, the CO-dependent activation is highly efficient in producing a stable Ni(I) form of the enzyme.
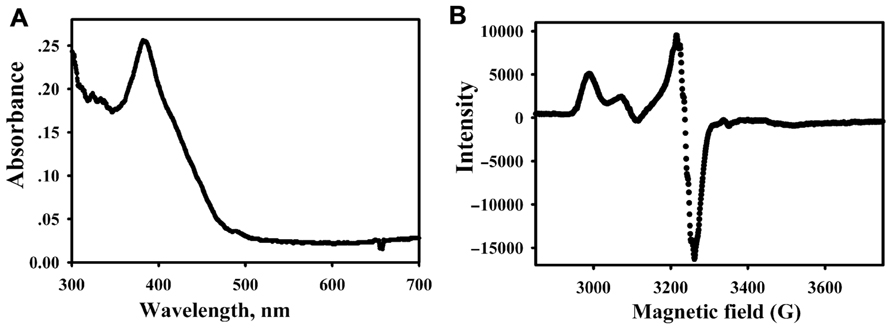
FIGURE 4. Spectra of the purified MCR (9.4 μM heterotrimer, 1.3 mg ml-1) from CO-activated cells. UV-visible (A) and EPR spectrum (B) of MCR purified from CO-activated cells.
Formate cannot Activate MCR
The standard redox potential of the CO2/HCOOH half-cell (-430 mV, Eq. 2) is equivalent to that of the 2H+/H2 half-cell (Eq. 3; Scherer and Thauer, 1978). Furthermore, the FDH gene (fdh) is present in the M. marburgensis genome and, based on sequence identity, is thought to use F420 as an electron donor (Liesegang et al., 2010; Kaster et al., 2011a). Thus, we expected that formate could also activate the M. marburgensis MCR. However, as shown in Figure 5, we did not observe any increase in the level of MCRred1 upon incubation of the cell suspension for over 20 h with formate. Correspondingly, we measured only a very low specific activity of FDH in cell extracts of M. marburgensis (3–4 mU mg-1, using NADP+ as electron acceptor and 2–3 mU mg-1, using MV), which is consistent with earlier reported values of 3–8 mU mg-1, depending on the electron acceptor and the pH of the assays (Tanner et al., 1989). We assayed using both NADP+ and MV, which is an electron acceptor for the F420-dependent enzyme, which the M. marburgensis enzyme is predicted to be (Kaster et al., 2011a). There are two forms of FDH: a tungsten- and a molybdenum-containing enzyme. The growth medium that we used has 1 μM molybdenum, but no tungsten (Kunz et al., 2006); however, addition of 1 μM tungsten and 1.1 μM selenium to the growth medium for M. marburgensis, also did not lead to activation of MCR in the presence of formate (data not shown). Apparently, the low level of FDH activity present in M. marburgensis is not sufficient to drive activation of MCR.
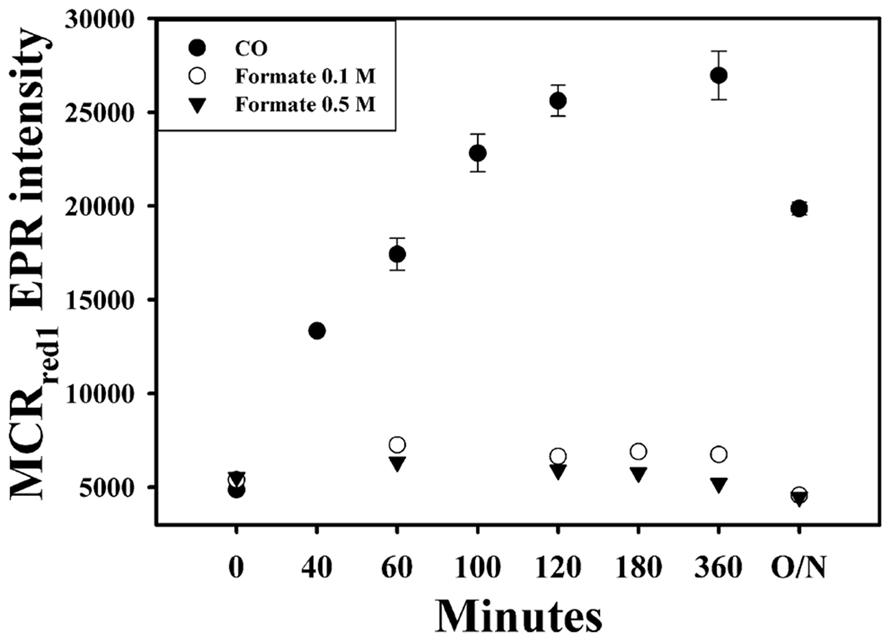
FIGURE 5. In vivo activation of MCR with CO (filled circles), 0.1 M formate (open circles), and 0.5 M formate (triangles). O/N, overnight.
Discussion
In order to study the mechanism of methane formation, it is crucial to develop an effective protocol for activation of MCR, the rate-limiting enzyme in this process. MCR has a number of different states, some of which have been detected in vivo (Albracht et al., 1988; Rospert et al., 1991a; Becker and Ragsdale, 1998) and others that have been observed in vitro (Mahlert et al., 2002; Finazzo et al., 2003; Duin et al., 2004; Kern et al., 2007; Harmer et al., 2008). Among these forms, only the Ni(I)-MCRred (red1 and red2) forms are active and there is evidence supporting (Kunz et al., 2006; Dey et al., 2007, 2010a) and questioning (Ghosh et al., 2001; Pelmenschikov et al., 2002; Pelmenschikov and Siegbahn, 2003) the catalytic relevance of the methyl-Ni(III) state.
Without treating cells before they are harvested, MCR is isolated in the inactive Ni(II) state. This is the stable state of the enzyme and, except for one structure of the methyl-Ni(III) state (Cedervall et al., 2011), it is the only form whose crystal structure is known. There exist no published methods for conversion of any of the Ni(II)-MCRsilent forms into MCRred1 in vitro. Thauer and colleagues discovered that MCRox1, an inactive EPR-active state that has been termed the “ready” state, can be generated by purging the cells with 80% N2/20% CO2 before harvesting; then MCRox1 can be isolated and reduced to active MCRred1 in vitro by addition of the reductant, Ti(III) citrate (Goubeaud et al., 1997). In lieu of the gas purging protocol, formation of MCRox1 can be optimized by adding sulfide to the culture prior to harvesting (Becker and Ragsdale, 1998). Then, as with the gas-switching method, the relatively stable MCRox1 state of the enzyme can be purified and quantitatively converted to MCRred1 by treating with Ti(III) citrate.
Here we show that CO, like H2, can activate the M. marburgensis MCR in vivo. When cells are purged with CO (Figure 1C), the whole-cell EPR signal of MCRred1 increases much more rapidly than that of H2-purged cells. The more rapidly MCR can be activated the better, because oxidants are present in the whole cells and in extracts that oxidize the Ni(I) state back to inactive Ni(II). However, once the enzyme is purified and maintained under anaerobic conditions, the MCRred1 state is rather stable. This CO-purging protocol yields purified MCR that is 80–90% MCRred1, as measured by the UV-visible and EPR spectral intensities and the specific activity.
Because growth of M. marburgensis on CO has not been reported, we were somewhat surprised that CO can activate MCR. However, there is a copy of the CODH gene (MTBMA c02870-02930) in the M. marburgensis genome (Liesegang et al., 2010; Kaster et al., 2011a) and relatively high CODH activities are measured in the cell extracts. This enzyme would be required for the autotrophic fixation of CO2 by M. marburgensis (Ferry, 2010). It is likely that it is this CODH activity that is required for CO-dependent activation of MCR, because we are unaware of other enzymes that can use CO as an electron donor and because CN-, a potent CODH inhibitor, blocks CO-dependent activation of MCR (Figure 3).
On the other hand, incubation of cells with formate under the same condition used for H2- and CO-dependent activation does not activate MCR. Although M. marburgensis can assimilate formate and has FDH activity, it is unlike the related strains, Methanothermobacter thermautotrophicus strain Z-245 and Methanothermobacter wolfeii (Nolling et al., 1993; Wasserfallen et al., 2000), in that it cannot grow on formate as an energy source (Kaster et al., 2011a). Mutants of M. marburgensis lacking FDH require formate for growth on H2 and CO2 (Tanner et al., 1989). Our experiments confirm earlier reports (Tanner et al., 1989) that the specific activity of FDH is very low – approximately 1000-fold lower than that of CODH, probably insufficient to support activation of MCR.
Thus, there are now two known electron sources that can reduce MCR to active MCRred1: H2 and CO. One possible activation mechanism involves H2 as an intermediate because CO-dependent H2 production has been reported in methanogens (Daniels et al., 1977; O’Brien et al., 1984), however, it is clear that this is not the pathway for CO-dependent activation of MCR because the rate of activation by CO is about 15 times faster than that with H2. Furthermore, the lag phase observed in H2-dependent activation is not present in the CO-dependent process. One might also argue that CO is an intermediate in the H2-dependent activation process and that the lag phase observed in H2-dependent activation is due to the time needed for formation of CO from H2 and CO2 in the suspensions. However, this possibility is ruled out by the fact that CN- blocks CO-, but not H2-dependent activation of MCR. Yet, that CO further activates the H2-activated enzyme to reach the level of MCRred1 observed in CO-activated cells (but not vice versa – H2 hardly activates MCR in CO-activated cells) indicates that H2- and CO-dependent activations pathways converge after the initial electron transfer from hydrogenase or CODH. The reason for the faster and higher activation level of MCR by CO may be related to the standard redox potential of the CO2/CO half-cell (-520 mV, Eq. 4), which is approximately 100 mV more negative than that of the 2H+/H2 half-cell (Lehn and Ziessel, 1982; Drake et al., 2006; Thauer et al., 2008; Kaster et al., 2011b). According to Marcus theory, for a one-electron redox reaction, a 100-mV greater driving force would be expected to increase the rate of electron transfer by ~30-fold at 30°C. Accordingly, we observe that CO-dependent activation is about 15 times faster that the H2-dependent process. This driving force argument is also supported by our results that CO activation is faster than H2 even though hydrogenase activity in M. marburgensis cells is 20-fold greater than that of CODH.
Activation of MCR involves reduction of the nickel center of F430 to the Ni(I) state (Rospert et al., 1991b; Dey et al., 2006). The electron carrier(s) needed for activation must interface with CODH (or hydrogenase, when cells are activated with H2) and with MCR. Although the methanogenic CODH is recognized to interface with ferredoxin (Terlesky and Ferry, 1988), our results indicate that there are additional redox steps involved in activation that couple CO-reduced ferredoxin to MCR. These components are probably also required in the H2-dependent activation. However, the activation by H2 is likely to be more complex than that for CO because the midpoint potential for the Ni(II)/(I) couple of bound F430 (less than -600 mV) is significantly lower those of ferredoxin (approximately -450 mV) and 2 H+/H2 (-414 mV), and only slight lower than that of CO2/CO (-520 mV). Thus, some type of bioenergetic coupling is required for activation of MCR (at least in the case of H2-dependent activation). We speculate that this mechanism of activation might involve electron bifurcation, which has been seen to drive various uphill enzymatic reactions (Lie et al., 2012; Thauer, 2012).
Cell extracts of M. thermoautotrophicus have been shown to slowly catalyze the reduction of CO2 to methane only upon addition of CH3-SCoMor CoBS-SCoM (Gunsalus and Wolfe, 1977; Rouviére and Wolfe, 1988). This phenomenon is referred to as the RPG effect, which couples the first step in methanogenesis from CO2 and H2 to the reduction of CoBS-SCoM to the free thiolate cofactors. Kaster et al. (2011a) demonstrated that the MvhADG/HdrABC complex from hydrogenotrophic methanogens couples the endergonic reduction of ferredoxin (midpoint potential of -450 to -500 mV) with H2 to the exergonic reduction of CoBS-SCoM with H2. Furthermore, reduction of ferredoxin by H2 (-414 mV at 100% gas phase under 1 bar pressure) at pH7 occurs only if CoBS-SCoM is present – thus, this is a coupled and energy driven reaction. We speculate that electron bifurcation involving CoBS-SCoM may be involved in the uphill reductive activation of MCR. The need for coupling of reduction of ferredoxin to the reduction of CoBS-SCoM by H2 may explain why there is a lag phase in the H2-dependent activation of MCR.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful for the support (DE-FG02-08ER15931) from the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy.
References
Albracht, S. P. J., Ankel-Fuchs, D., Böcher, R., Ellermann, J., Moll, J., Van Der Zwann, J. W., et al. (1988). Five new EPR signals assigned to nickel in methyl-coenzyme M reductase from Methanobacterium thermoautotrophicum, strain Marburg. Biochim. Biophys. Acta 955, 86–102.
Becker, D. F., and Ragsdale, S. W. (1998). Activation of methyl-SCoM reductase to high specific activity after treatment of whole cells with sodium sulfide. Biochemistry 37, 2639–2647.
Brenner, M. C., Zhang, H., and Scott, R. A. (1993). Nature of the low activity of S-methyl-coenzyme M reductase as determined by active site titrations. J. Biol. Chem. 268, 18491–18495.
Cedervall, P. E., Dey, M., Li, X., Sarangi, R., Hedman, B., Ragsdale, S. W., et al. (2011). Structural analysis of a Ni-methyl species in methyl-coenzyme M reductase from Methanothermobacter marburgensis. J. Am. Chem. Soc. 133, 5626–5628.
Cedervall, P. E., Dey, M., Pearson, A. R., Ragsdale, S. W., and Wilmot, C. M. (2010). Structural insight into methyl-coenzyme M reductase chemistry using coenzyme B analogues. Biochemistry 49, 7683–7693.
Daniels, L., Fuchs, G., Thauer, R. K., and Zeikus, J. G. (1977). Carbon monoxide oxidation by methanogenic bacteria. J. Bacteriol. 132, 118–126.
Dey, M., Kunz, R., Heuvelen, K. M. V., Craft, J. L., Horng, Y.-C., Tang, Q., et al. (2006). Spectroscopic and computational studies of reduction of the metal versus the tetrapyrrole ring of coenzyme F430 from methyl-coenzyme M reductase. Biochemistry 45, 11915–11933.
Dey, M., Kunz, R. C., Lyons, D. M., and Ragsdale, S. W. (2007). Characterization of alkyl-nickel adducts generated by reaction of methyl-coenzyme M reductase with brominated acids. Biochemistry 46, 11969–11978.
Dey, M., Li, X., Kunz, R. C., and Ragsdale, S. W. (2010a). Detection of organometallic and radical intermediates in the catalytic mechanism of methyl-coenzyme M reductase using the natural substrate methyl-coenzyme M and a coenzyme B substrate analogue. Biochemistry 49, 10902–10911.
Dey, M., Li, X., Zhou, Y., and Ragsdale, S. W. (2010b). Evidence for organometallic intermediates in bacterial methane formation involving the nickel coenzyme F. Met. Ions Life Sci. 7, 71–110.
DiMarco, A. A., Bobik, T. A., and Wolfe, R. S. (1990). Unusual coenzymes of methanogenesis. Annu. Rev. Biochem. 59, 355–394.
Drake, H. L., Kusel, K., and Matthies, C. (eds). (2006). Acetogenic Prokaryotes. New York: Springer-Verlag.
Duin, E. C., Prakash, D., and Brungess, C. (2011). Methyl-coenzyme M reductase from Methanothermobacter marburgensis. Methods Enzymol. 494, 159–187.
Duin, E. C., Signor, L., Piskorski, R., Mahlert, F., Clay, M. D., Goenrich, M., et al. (2004). Spectroscopic investigation of the nickel-containing porphinoid cofactor F(430). Comparison of the free cofactor in the (+)1, (+)2 and (+)3 oxidation states with the cofactor bound to methyl-coenzyme M reductase in the silent, red and ox forms. J. Biol. Inorg. Chem. 9, 563–576.
Ebner, S., Jaun, B., Goenrich, M., Thauer, R. K., and Harmer, J. (2010). Binding of coenzyme B induces a major conformational change in the active site of methyl-coenzyme M reductase. J. Am. Chem. Soc. 132, 567–575.
Elliott, J. I., and Brewer, J. M. (1978). The inactivation of yeast enolase by 2,3-butanedione. Arch. Biochem. Biophys. 190, 351–357.
Ermler, U., Grabarse, W., Shima, S., Goubeaud, M., and Thauer, R. K. (1997). Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological methane formation. Science 278, 1457–1462.
Finazzo, C., Harmer, J., Jaun, B., Duin, E. C., Mahlert, F., Thauer, R. K., et al. (2003). Characterization of the MCRred2 form of methyl-coenzyme M reductase: a pulse EPR and ENDOR study. J. Biol. Inorg. Chem. 8, 586–593.
Garcia, J. L., Patel, B. K., and Ollivier, B. (2000). Taxonomic, phylogenetic, and ecological diversity of methanogenic Archaea. Anaerobe 6, 205–226.
Ghosh, A., Wondimagegn, T., and Ryeng, H. (2001). Deconstructing F(430): quantum chemical perspectives of biological methanogenesis. Curr. Opin. Chem. Biol. 5, 744–750.
Goubeaud, M., Schreiner, G., and Thauer, R. K. (1997). Purified methyl-coenzyme-M reductase is activated when the enzyme-bound coenzyme F430 is reduced to the nickel(I) oxidation state by titanium(III) citrate. Eur. J. Biochem. 243, 110–114.
Grabarse, W. G., Mahlert, F., Duin, E. C., Goubeaud, M., Shima, S., Thauer, R. K., et al. (2001). On the mechanism of biological methane formation: structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. J. Mol. Biol. 309, 315–330.
Grabarse, W. G., Mahlert, F., Shima, S., Thauer, R. K., and Ermler, U. (2000). Comparison of three methyl-coenzyme M reductases from phylogenetically distant organisms: unusual amino acid modification, conservation and adaptation. J. Mol. Biol. 303, 329–344.
Gunsalus, R. P., and Wolfe, R. S. (1977). Stimulation of CO2 reduction to methane by methylcoenzyme M in extracts Methanobacterium. Biochem. Biophys. Res. Commun. 76, 790–795.
Gunsalus, R. P., and Wolfe, R. S. (1980). Methyl coenzyme M reductase from Methanobacterium thermoautotrophicum: resolution and properties of the components. J. Biol. Chem. 255, 1891–1895.
Guss, A. M., Kulkarni, G., and Metcalf, W. W. (2009). Differences in hydrogenase gene expression between Methanosarcina acetivorans and Methanosarcina barkeri. J. Bacteriol. 191, 2826–2833.
Ha, S. W., Korbas, M., Klepsch, M., Meyer-Klaucke, W., Meyer, O., and Svetlitchnyi, V. (2007). Interaction of potassium cyanide with the [Ni-4Fe-5S] active site cluster of CO dehydrogenase from carboxydothermus hydrogenoformans. J. Biol. Chem. 282, 10639–10646.
Harmer, J., Finazzo, C., Piskorski, R., Ebner, S., Duin, E. C., Goenrich, M., et al. (2008). A nickel hydride complex in the active site of methyl-coenzyme m reductase: implications for the catalytic cycle. J. Am. Chem. Soc. 130, 10907–10920.
Holliger, C., Pierik, A. J., Reijerse, E. J., and Hagen, W. R. (1993). A spectroelectrochemical study of Factor F430 Nickel(II/I) from methanogenic bacteria in aqueous solution. J. Am. Chem. Soc. 115, 5651–5656.
Horng, Y.-C., Becker, D. F., and Ragsdale, S. W. (2001). Mechanistic studies of methane biogenesis by methyl-coenzyme M reductase: evidence that coenzyme B participates in cleaving the C–S bond of methyl-coenzyme M. Biochemistry 40, 12875–12885.
Kaster, A. K., Goenrich, M., Seedorf, H., Liesegang, H., Wollherr, A., Gottschalk, G., et al. (2011a). More than 200 genes required for methane formation from H2 and CO2 and energy conservation are present in Methanothermobacter marburgensis and Methanothermobacter thermautotrophicus. Archaea 2011, 973848.
Kaster, A. K., Moll, J., Parey, K., and Thauer, R. K. (2011b). Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc. Natl. Acad. Sci. U.S.A. 108, 2981–2986.
Kern, D. I., Goenrich, M., Jaun, B., Thauer, R. K., Harmer, J., and Hinderberger, D. (2007). Two sub-states of the red2 state of methyl-coenzyme M reductase revealed by high-field EPR spectroscopy. J. Biol. Inorg. Chem. 12, 1097–1105.
Kunz, R. C., Dey, M., and Ragsdale, S. W. (2008). Characterization of the thioether product formed from the thiolytic cleavage of the alkyl-nickel bond in methyl-coenzyme M reductase. Biochemistry 47, 2661–2667.
Kunz, R. C., Horng, Y.-C., and Ragsdale, S. W. (2006). Spectroscopic and kinetic studies of the reaction of bromopropanesulfonate with methyl-coenzyme M reductase. J. Biol. Chem. 281, 34663–34676.
Lehn, J. M., and Ziessel, R. (1982). Photochemical generation of carbon-monoxide and hydrogen by reduction of carbon-dioxide and water under visible-light irradiation. Proc. Natl. Acad. Sci. U.S.A. 79, 701–704.
Li, X. H., Telser, J., Kunz, R. C., Hoffman, B. M., Gerfen, G., and Ragsdale, S. W. (2010). Observation of organometallic and radical intermediates formed during the reaction of methyl-coenzyme M reductase with bromoethanesulfonate. Biochemistry 49, 6866–6876.
Lie, T. J., Costa, K. C., Lupa, B., Korpole, S., Whitman, W. B., and Leigh, J. A. (2012). Essential anaplerotic role for the energy-converting hydrogenase Eha in hydrogenotrophic methanogenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 15473–15478.
Liesegang, H., Kaster, A. K., Wiezer, A., Goenrich, M., Wollherr, A., Seedorf, H., et al. (2010). Complete genome sequence of Methanothermobacter marburgensis, a methanoarchaeon model organism. J. Bacteriol. 192, 5850–5851.
Mahlert, F., Grabarse, W., Kahnt, J., Thauer, R. K., and Duin, E. C. (2002). The nickel enzyme methyl-coenzyme M reductase from methanogenic archaea: in vitro interconversions among the EPR detectable MCR-red1 and MCR-red2 states. J. Biol. Inorg. Chem. 7, 101–112.
Nagle, D. P. Jr., and Wolfe, R. S. (1983). Component A of the methyl coenzyme M methylreductase system of Methanobacterium: resolution into four components. Proc. Natl. Acad. Sci. U.S.A. 80, 2151–2155.
Nolling, J., Hahn, D., Ludwig, W., and Vos, W. M. D. (1993). Phylogenetic analysis of thermophilic Methanobacterium sp.: evidence for a formate-utilizing ancestor. Syst. Appl. Microbiol. 16, 208–215.
O’Brien, J. M., Wolkin, R. H., Moench, T. T., Morgan, J. B., and Zeikus, J. G. (1984). Association of hydrogen metabolism with unitrophic or mixotrophic growth of Methanosarcina barkeri on carbon monoxide. J. Bacteriol. 158, 373–375.
Pelmenschikov, V., Blomberg, M. R. A., Siegbahn, P. E. M., and Crabtree, R. H. (2002). A mechanism from quantum chemical studies for methane formation in methanogenesis. J. Am. Chem. Soc. 124, 4039–4049.
Pelmenschikov, V., and Siegbahn, P. E. (2003). Catalysis by methyl-coenzyme M reductase: a theoretical study for heterodisulfide product formation. J. Biol. Inorg. Chem. 8, 653–662.
Ragsdale, S. W., Clark, J. E., Ljungdahl, L. G., Lundie, L. L., and Drake, H. L. (1983). Properties of purified carbon monoxide dehydrogenase from Clostridium thermoaceticum a nickel, iron-sulfur protein. J. Biol. Chem. 258, 2364–2369.
Richard, R. M., and Ball, D. W. (2008). Ab initio calculations on the thermodynamic properties of azaborospiropentanes. J. Mol. Model. 14, 871–878.
Rospert, S., Böcher, R., Albracht, S. P. J., and Thauer, R. K. (1991a). Methyl-coenzyme M reductase preparations with high specific activity from H2-preincubated cells of Methanobacterium thermoautotrophicum. FEBS Lett. 291, 371–375.
Rospert, S., Breitung, J., Ma, K., Schworer, B., Zirngibl, C., Thauer, R. K., et al. (1991b). Methyl-coenzyme M reductase and other enzymes involved in methanogenesis from CO2 and H2 in the extreme thermophile Methanopyrus kandleri. Arch. Microbiol. 156, 49–55.
Rouviére, P. E., and Wolfe, R. S. (1988). Novel biochemistry of methanogenesis. J. Biol. Chem. 263, 7913–7916.
Scheller, S., Goenrich, M., Boecher, R., Thauer, R. K., and Jaun, B. (2010). The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature 465, 606–608.
Scherer, P. A., and Thauer, R. K. (1978). Purification and properties of reduced ferredoxin: CO2 oxidoreductase from Clostridium pasteurianum, a molybdenum iron-sulfur-protein. Eur. J. Biochem. 85, 125–135.
Tanner, R. S., Mcinerney, M. J., and Nagle, D. P. Jr. (1989). Formate auxotroph of Methanobacterium thermoautotrophicum Marburg. J. Bacteriol. 171, 6534–6538.
Terlesky, K. C., and Ferry, J. G. (1988). Ferredoxin requirement for electron transport from the carbon monoxide dehydrogenase complex to a membrane-bound hydrogenase in acetate-grown Methanosarcina thermophila. J. Biol. Chem. 263, 4075–4079.
Thauer, R. K. (1998). Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiology 144, 2377–2406.
Thauer, R. K. (2012). The Wolfe cycle comes full circle. Proc. Natl. Acad. Sci. U.S.A. 109, 15084–15085.
Thauer, R. K., Kaster, A. K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591.
Wang, M., Tomb, J. F., and Ferry, J. G. (2011). Electron transport in acetate-grown Methanosarcina acetivorans. BMC Microbiol. 11:165. doi: 10.1186/1471-2180-11-165
Wasserfallen, A., Nolling, J., Pfister, P., Reeve, J., and Conway De Macario, E. (2000). Phylogenetic analysis of 18 thermophilic Methanobacterium isolates supports the proposals to create a new genus, Methanothermobacter gen. nov., and to reclassify several isolates in three species, Methanothermobacter thermautotrophicus comb. nov., Methanothermobacter wolfeii comb. nov., and Methanothermobacter marburgensis sp. nov. Int. J. Syst. Evol. Microbiol. 50(Pt 1), 43–53.
Wuebbles, D. J., and Hayhoe, K. (2000). “Atmospheric methane: trends and impacts,” in Non-CO2 Greenhouse Gases: Scientific Understanding, Control and Implementation, eds J. van Ham, A. P. M. Baede, L. A. Meyer, and R. Ybema (Dordrecht: Kluwer Academic Publishers), 1–43.
Keywords: carbon monoxide, enzyme catalysis, metalloenzymes, nickel, electron transfer, EPR, methanogenesis, enzyme activation
Citation: Zhou Y, Dorchak AE and Ragsdale SW (2013) In vivo activation of methyl-coenzyme M reductase by carbon monoxide. Front. Microbiol. 4:69. doi: 10.3389/fmicb.2013.00069
Received: 28 January 2013; Paper pending published: 15 February 2013;
Accepted: 11 March 2013; Published online: 01 April 2013.
Edited by:
Partha Basu, Duquesne University, USAReviewed by:
Rudolf Kurt Thauer, Max Planck Institute for Terrestrial Microbiology, GermanyJohn Leigh, University of Washington, USA
Copyright: © 2013 Zhou, Dorchak and Ragsdale. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Stephen W. Ragsdale, Department of Biological Chemistry, University of Michigan Medical School, University of Michigan, 5301 Medical Science Research Building III, 1150 West Medical Center Drive, Ann Arbor, MI 48109-5606, USA. e-mail: sragsdal@umich.edu