 Marc G. Dumont
Marc G. Dumont Claudia Lüke
Claudia Lüke Yongcui Deng
Yongcui Deng Peter Frenzel
Peter Frenzel- 1Department of Biogeochemistry, Max Planck Institute for Terrestrial Microbiology, Marburg, Germany
- 2Department of Microbiology, Radboud University, Nijmegen, Netherlands
- 3Key Laboratory of Virtual Geographic Environment, Ministry of Education, Nanjing Normal University, Nanjing, China
The classification of high-throughput sequencing data of protein-encoding genes is not as well established as for 16S rRNA. The objective of this work was to develop a simple and accurate method of classifying large datasets of pmoA sequences, a common marker for methanotrophic bacteria. A taxonomic system for pmoA was developed based on a phylogenetic analysis of available sequences. The taxonomy incorporates the known diversity of pmoA present in public databases, including both sequences from cultivated and uncultivated organisms. Representative sequences from closely related genes, such as those encoding the bacterial ammonia monooxygenase, were also included in the pmoA taxonomy. In total, 53 low-level taxa (genus-level) are included. Using previously published datasets of high-throughput pmoA amplicon sequence data, we tested two approaches for classifying pmoA: a naïve Bayesian classifier and BLAST. Classification of pmoA sequences based on BLAST analyses was performed using the lowest common ancestor (LCA) algorithm in MEGAN, a software program commonly used for the analysis of metagenomic data. Both the naïve Bayesian and BLAST methods were able to classify pmoA sequences and provided similar classifications; however, the naïve Bayesian classifier was prone to misclassifying contaminant sequences present in the datasets. Another advantage of the BLAST/LCA method was that it provided a user-interpretable output and enabled novelty detection at various levels, from highly divergent pmoA sequences to genus-level novelty.
1. Introduction
High-throughput sequencing (HTS) technologies have aided our ability to investigate the diversity of microorganisms in environmental samples either by shotgun metagenomic or amplicon sequencing approaches. Many bioinformatic tools necessary to process and interpret the large volume of data obtained by HTS have been developed. For example, there are several choices of pipelines available to analyze 16S rRNA amplicon sequencing data such as RDP (Cole et al., 2005), mothur (Schloss et al., 2009) and QIIME (Caporaso et al., 2010). Similar strategies targeting genes encoding enzymes responsible for important biogeochemical or bioremediation processes are becoming more common, but the methods for analyzing the data are not as well established as for 16S rRNA.
The analysis of HTS amplicon data can be performed using taxonomy-dependent or independent approaches. The taxonomy-independent approach includes methods to compare sequence alignments and analyze operational taxonomic units (OTUs) based on sequence dissimilarity (Schloss and Handelsman, 2004; Cai and Sun, 2011). This approach is valuable for estimating ecological parameters, such as richness and diversity; however, the information on its own does not indicate how the sequences are related to those of cultivated organisms or those from other studies. Taxonomy-based methods classify sequences according to their relatedness to those of pure cultures and uncultivated organisms. This approach is necessary to incorporate knowledge of the physiological characteristics of different taxa, to identify novel sequence types and to compare results between published studies. Common methods for classification include naïve Bayesian classifiers (Wang et al., 2007), k-nearest neighbor (Cole et al., 2005) and BLAST (Altschul et al., 1990).
In general, the analysis of OTUs and phylogenetic trees calculated from individual datasets of protein-encoding genes can be performed with the same tools designed for the analysis of 16S rRNA sequences. In contrast, classifiers must be tailor made for each gene by establishing a taxonomy with representative sequences and choosing an appropriate classification algorithm. The objective of this study was to establish a robust and easily applied approach to classifying HTS amplicon sequences of pmoA, a key gene of methane-oxidizing bacteria. The method should also allow for novelty detection and be easily performed by a microbial ecologist with only fundamental knowledge of bioinformatics. We test a naïve Bayesian classifier and BLAST combined with the lowest common ancestor approach of MEGAN using previously published pmoA pyrosequencing data (Lüke and Frenzel, 2011; Deng et al., 2013). Previous studies have also compared both approaches for the classification of SSU rRNA (Lanzén et al., 2012) and fungal LSU rRNA sequences (Porter and Golding, 2012).
2. pmoA Taxonomy
An accurate taxonomic system for the gene sequences is a necessary prerequisite for classification. Since the classification of unknown sequences obtained by HTS can only be as accurate as the taxonomy, the analysis of database sequences and assignment of taxa is the critical step in the development of a classifier. In general, pmoA has been shown to be a good phylogenetic marker for methanotrophs (Degelmann et al., 2010), with some exceptions of divergent additional copies of the gene in some organisms (Dunfield et al., 2002; Stoecker et al., 2006; Baani and Liesack, 2008). Here we describe the taxonomy of pmoA genes (Table 1); earlier versions were described previously (Lüke and Frenzel, 2011; Deng et al., 2013).
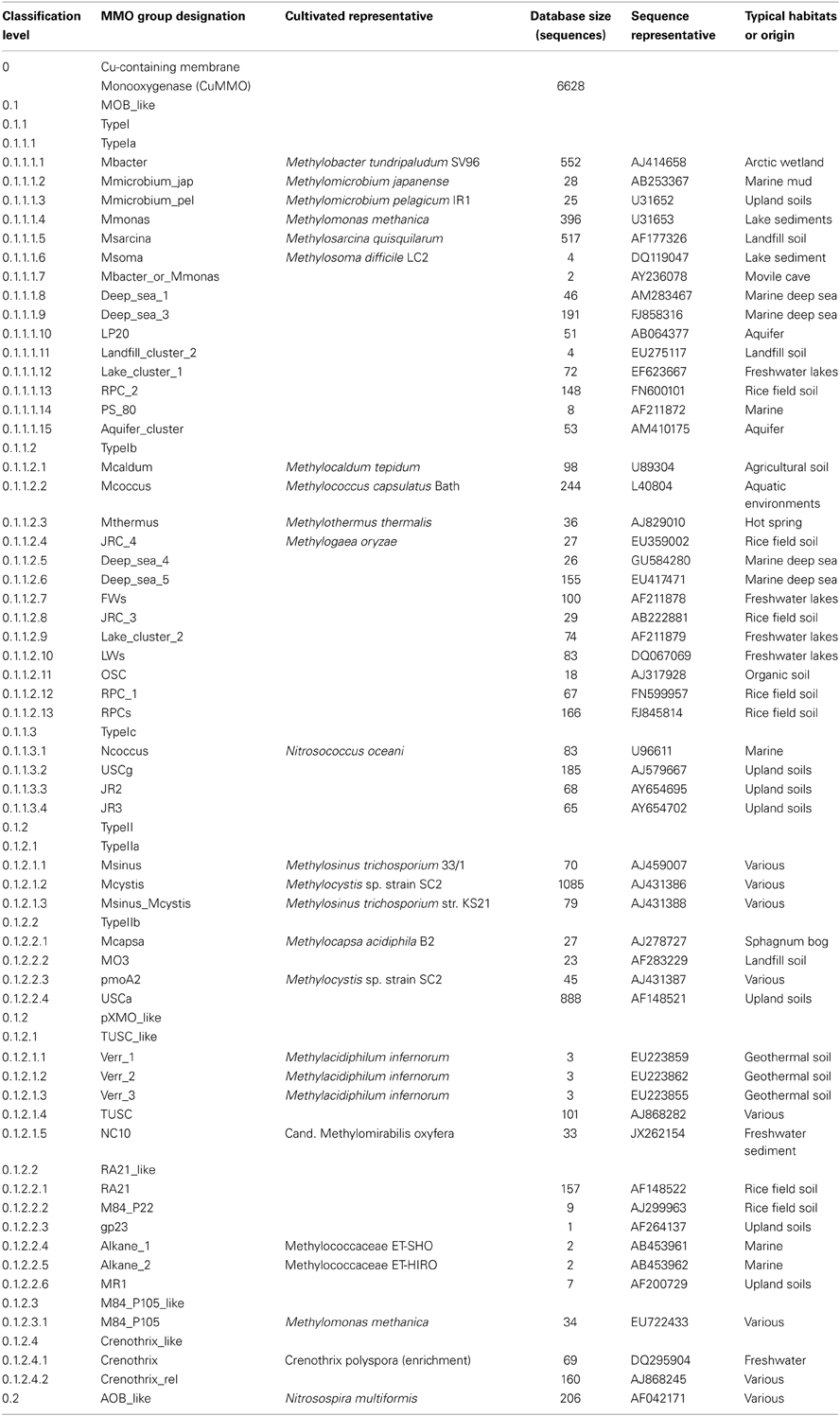
Table 1. Description of the pmoA database.
2.1. Overall Taxonomic System
The pmoA gene encodes the β-subunit of the particulate methane monooxygenase (pMMO), which belongs to the class of copper-containing membrane-bound monooxygenase (CuMMO) enzymes. In addition to the pMMO, this group includes the bacterial ammonia monooxygenase (Holmes et al., 1995), the thaumarchaeal ammonia monooxygenase (Pester et al., 2011), alkane monooxygenases and various uncharacterized enzymes encoded by genes detected in environmental surveys (Coleman et al., 2012). For our classifier we compiled a database of pmoA and related gene sequences obtained primarily from public databases. We focused on building a taxonomic structure for pmoA, but also included sequences of related genes that are often co-amplified with common pmoA primers, such as the bacterial amoA. Related sequences that are not co-amplified, such as the thaumarchaeal amoA, were not included.
Currently, our curated database includes 6628 reference sequences corresponding to 53 low-level taxa (Table 1). The assignment to taxa was determined by the phylogenetic analysis of the pmoA and related gene fragments using both the nucleotide and inferred protein sequences. Sequences were imported into ARB (Ludwig et al., 2004) and alignments of either 408 nucleotide or 136 amino acid residues were used to generate neighbor-joining (NJ) and maximum-likelihood (ML) trees. For ML trees, sequences were exported and uploaded to the RAxML web-server (Stamatakis et al., 2005). Tree topologies were compared and taxa were assigned according to groups of sequences that consistently clustered together in the analyses (Lüke and Frenzel, 2011). At the highest level, the sequences were categorized as MOB_like or AOB_like, depending on apparent relatedness to sequences from methane-oxidizing and ammonia-oxidizing bacteria respectively. The classifier currently contains 53 low-level taxa within the MOB_like group (Table 1). Taxa comprising cultivated methanotrophs were referred to as the respective genera or species (e.g., Mbacter, for Methylobacter-like pmoA). Taxa lacking isolates were named according to representative clones or to the environment in which they were predominantly or initially found (e.g., Aquifer_cluster or upland soil cluster—USC) (Lüke and Frenzel, 2011).
2.2. Type I and II pmoA Sequences
The MOB_like sequences were assigned to either Type I, Type II or pXMO_like. The Type I sequences were further divided into Type Ia, b, or c. Type Ia are pmoA sequences affiliated to the classic Type I methanotrophs (i.e., not Type X). Type Ib (also referred to elsewhere as Type X) are those of Methylococcus and closely related genera. Type Ic are all other Type I-related sequences with a more ambiguous affiliation. Type II sequences were divided into Type IIa and b. Type IIa was used for the primary pmoA sequences of the Methylocystaceae. Type IIb was used to group all other Type II-related (i.e., Alphaproteobacteria) sequences, including those from the Beijerinckiaceae (Theisen et al., 2005; Dunfield et al., 2010; Vorobev et al., 2011) and the alternate pMMO2 identified in some Methylocystis species (Dunfield et al., 2002; Baani and Liesack, 2008).
2.3. pXMO: Divergent pmoA Sequences
We use pXMO as the third category of pmoA-related sequences. The original description of pXMO was for the unusual pMMO-like enzyme identified in some Type I methanotrophs (Tavormina et al., 2011). Here we use pXMO_like to encompass all the divergent sequence types for which the substrate or biological function has not been clearly identified by biochemical or genetic tests. For example, we include the three verrucomicrobial pmoA-like sequences in this category until it is determined which, if not all, catalyze the oxidation of methane. The original pxmA genes identified in Methylomonas spp. (Tavormina et al., 2011) are classified in the M84_P105 low-level taxon. We have also included the pmoA sequences from the nitrite-dependent anaerobic methane oxidizers belonging to the NC10 phylum (Ettwig et al., 2009, 2010) into the pXMO_like category; it should be noted that these NC10 pmoA sequences are typically retrieved only after using specific primers and a special PCR program designed for their amplification (Luesken et al., 2011) and therefore are unlikely to be obtained in HTS pmoA surveys using the traditional pmoA primer sets.
2.4. Bacterial Ammonia Monooxygenase
Bacterial ammonia monooxygenase (amoA) genes were included since they are commonly co-amplified with pmoA genes in environmental PCR surveys. The amoA sequences of betaproteobacterial ammonia oxidizers were designated AOB_like, without making further lower-level distinctions. In contrast, the amoA sequences of Nitrosococcus were classified as “Ncoccus” within the MOB_like group since they are more closely related to pmoA than to other amoA genes.
3. Software and Associated Files
Mothur version 1.29.2 (Schloss et al., 2009) and MEGAN version 4.70.4 (Huson et al., 2011) were both downloaded from the author's webpages. Standalone BLAST 2.2.26+ (Camacho et al., 2009) was obtained from NCBI. These software programs can be installed on various platforms, but all analyses in this study were performed on a quad-core Apple MacBook Pro with a 2.2 GHz Intel Core i7 processor and 16 Gb of memory.
3.1. Naïve Bayesian Classifier
The training set for the naïve Bayesian classifier consists of two files: the database sequences in “fasta” format, and a taxonomy file indicating the taxonomy of each sequence. The taxonomy file was formatted to be compatible with mothur; both files are available in the supplement. The training set files were generated by exporting sequence information from ARB (Ludwig et al., 2004) and formatting the entries using standard spreadsheet and text-editing programs.
3.2. Blast and Megan
The BLAST database was generated from the taxonomy using the makeblastdb program included with BLAST 2.2.26+ package. The input was a fasta file with the sequence name header including the sequence accession number and the taxon in square brackets; as for the naïve Bayesian classifier, these files were made using common spreadsheet and text-editing software. makeblastdb outputs three files (.nsq,.nin, and.nhr); all files are provided in the supplementary material.
A Newick format tree corresponding to the pmoA taxonomy was written for MEGAN; the tree file (pmoa.megan.2013.tre) and a corresponding map file (pmoa.megan.2013.map) are provided in the supplement. The pmoA taxonomy is loaded into MEGAN by the option “use alternative taxonomy” and selecting the pmoa.megan.2013.tre file.
4. pmoA Amplification and Sequencing
Two primer sets are typically used to amplify pmoA sequences from environmental samples (McDonald et al., 2008). The A189f/A682r primer pair offers broad specificity covering many CuMMO monooxygenases (Holmes et al., 1995). The A189f/mb661r combination was designed to be more specific for pmoA (Costello and Lidstrom, 1999) and does not generally amplify amoA or pxmA-like sequences. For NGS amplicon sequencing, adaptors and barcodes are incorporated into the primers, or ligated onto the PCR products, in a manner compatible with the sequencing platform (Binladen et al., 2007; Berry et al., 2011).
Two previously described HTS pmoA amplicon datasets were used in this study to test the classification methods (Lüke and Frenzel, 2011; Deng et al., 2013). Both studies used the A189f/A682r and A189f/mb661r primer sets. The Lüke and Frenzel (2011) study focused on rice field soil samples from Italy and China and the sequences were analyzed in the original study by constructing phylogenetic trees and calculating OTUs from sequence alignments. The Deng et al. (2013) study focused on peatland samples from both submerged (hollow) and elevated (hummock) sites in the Qinghai-Tibetan plateau, and were analyzed in the original study by sequence similarity and an earlier implementation of the naïve Bayesian classifier. The basic steps for classifying pmoA sequence data are summarized in Figure 1 and a detailed protocol is included in the supplement.

Figure 1. Overview of the basic procedure for classifying pmoA sequences obtained by high-throughput sequencing. The manuscript section where a procedure is described is indicated and detailed instructions are available in the supplementary materials.
5. Raw Sequence Processing
Raw sequence data must first undergo basic processing. In this study we use mothur, but other software can be used for this purpose, such as QIIME (Caporaso et al., 2010), RDP (Cole et al., 2009) and Funframe (Weisman et al., 2013); each of these have unique features that might be beneficial for a particular dataset or objective. The basic necessary steps are the sorting of sequences according to barcodes, trimming and quality filtering. For data analyzed here we use a minimum sequence length of 300 bp and remove sequences with ambiguities or stretches of more than eight homopolymers. Chimeric sequences were identified in mothur using the uchime method (Edgar et al., 2011). Chimeras were detected de novo by using the “self” option in mothur, meaning that the pmoA pyrosequencing dataset was used as a reference. As an example, in the HYa-1 dataset (Deng et al., 2013), 1058 chimeras were identified in 7658 sequences. These sequences were removed using the remove.seqs command in mothur (see supplementary methods).
NGS sequence technologies have relatively large error rates compared with Sanger sequencing and there are various approaches available to denoise pyrosequencing data (Reeder and Knight, 2010; Quince et al., 2011; Rosen et al., 2012). If not removed, these errors generate false OTUs (Schloss et al., 2011). Another problem is that they often cause frameshift errors in protein-coding genes, making it difficult to infer amino acid sequences. There are methods available to specifically correct frameshift errors in functional gene pyrosequencing datasets, such as the FrameBot tool (http://fungene.cme.msu.edu/FunGenePipeline/) and HMM-FRAME (Weisman et al., 2013). Correcting the frameshift errors in pyrosequencing datasets is particularly important for calculating OTUs or phylogenetic distances based on amino acid sequences. In general we did not find denoising necessary for classifying our pmoA sequences obtained by pyrosequencing and therefore do not discuss the methods here; however, a study on fungal LSU rRNA amplicon sequencing reported that the BLAST/LCA method was less sensitive to sequence error than the naïve Bayesian classification approach (Porter and Golding, 2012).
6. Classification of pmoA Sequences
6.1. Naïve Bayesian Classifier
A naïve Bayesian classifier is the basis of the RDP classifier for 16S rRNA sequence data (Wang et al., 2007). In this study we have adopted the 16S rRNA classifier implemented in mothur, but replaced the 16S rRNA taxonomy with that of pmoA. The process in mothur is invoked with classify.seqs command with the option of the “wang” method. Other variables include the kmer (word) size and the cutoff value for bootstrap confidence estimates. In general we found that the default kmer size of 8 performed well and used a cutoff value of 80%.
6.2. BLAST/LCA
Different versions of BLAST were tested. Nucleotide BLAST types include MEGABLAST, BLASTN and discontiguous (DC)-MEGABLAST, in decreasing order of sensitivity for obtaining matches and alignments against distantly related sequences (McGinnis and Madden, 2004). In most cases the results of the different methods were nearly identical, except in some cases that DC-MEGABLAST would produce hits (alignments) with distantly related novel pmoA sequences that were not aligned by the other two methods. We found that the additional computation time required for DC-MEGABLAST was not compensated by the added sensitivity since this could be easily recovered by a reanalysis of the sequences that did not produce hits with MEGABLAST, as described in section 7.1.1. Protein BLAST approaches were also tested and classification results were similar to nucleotide BLAST, but with greater sensitivity for matching distantly related novel sequence types to the database. The protein BLAST searches were more vulnerable to the effect of frameshift errors in the query sequences because they cause breaks in the alignment that strongly affect the bit score of a match. In contrast, the insertion of gap during nucleotide BLAST adds a small penalty to the bit score and does not terminate the alignment. Therefore, in general we use MEGABLAST, which is the default algorithm for the blastn program.
BLAST has been shown to be relatively poor at identifying the most similar sequence in a dataset (Koski and Golding, 2001; Cole et al., 2005). We did not observe, nor do we foresee, this to be a problem for the classification since it is only necessary to identify the most similar taxon, which are all highly distinct (>5% nucleic acid identity) from one another.
6.2.1. BLAST interpretation by lowest common ancestor in MEGAN
MEGAN was developed for the classification of metagenomic sequence data by reading the output of BLAST queries (Huson et al., 2011; Mitra et al., 2010). It has been adapted for other purposes, for example it can read the log file of a sina alignment of 16S rRNA (Pruesse et al., 2012) and thereby used to analyze 16S rRNA sequence data (Mitra et al., 2011). Here we show that it could be adapted for the analysis of the pmoA BLAST queries against our taxonomy database, as has been demonstrated previously for SSU rRNA (Lanzén et al., 2012) and fungal LSU rRNA (Porter and Golding, 2012). A simple modification is possible in MEGAN to change the default NCBI taxonomy to a custom taxonomy, in this case pmoA. MEGAN parses the BLAST output file and collects only the top hits from each taxon and the associated alignment. In addition to summarizing the results, this has the added benefit of reducing the file size compared with the original BLAST output.
MEGAN uses a lowest common ancestor (LCA) algorithm (Huson et al., 2007) based on BLAST bit scores to classify the sequences. A sequence is classified at a particular level only when the bit score to the taxon is higher by a given margin than to those of any another taxon. The margin of difference can be adjusted in the LCA parameters of MEGAN with the option of “top percent.” The greater the margin, the greater is the minimum distance between the assigned taxon and any other taxon. The BLAST/LCA method provides several valuable benefits for the classification in comparison to simply classifying based on top hit, for example in novelty detection as discussed below.
6.3. Comparison of the Naïve Bayesian and BLAST/LCA Classifications
We found good agreement in the classification of the rice paddy soil datasets using the naïve Bayesian and BLAST/LCA approaches (Figure 2). Subsamples from each classification were analyzed in ARB by NJ and in general confirmed the assignments (results not shown). Some minor differences between the methods were also observed. For example, seven sequences in the China (old) A189f-A682r dataset were classified as gp23 whereas these seven sequences did not produce significant hits using BLAST/LCA. A close inspection indicated that the seven sequences were either highly divergent pmoA or non-specific PCR products related to an alpha-glucan branching protein or a peptidase. An analysis of the Riganqiao samples also showed a tendency for the naïve Bayesian classifier to assign contaminant sequences to gp23 (not shown). This is likely a result of gp23 only being represented by a single sequence in the database and being relatively divergent from other pmoA taxa (Lüke and Frenzel, 2011). This erroneous classification of non-target sequences as gp23 using the naïve Bayesian classifier could be reduced by increasing the kmer size to 10, but at a cost of decreased sensitivity in classifying bona fide pmoA sequences. In spite of this, both the naïve Bayesian and BLAST/LCA methods identified genuine gp23 sequences present in the China (young) A189f-A682r dataset (Figure 2). Previous studies also reported higher accuracies of classifications obtained by BLAST/LCA for 16S rRNA sequences (Lanzén et al., 2012), fungal LSU rRNA sequences (Porter and Golding, 2012) and rRNA internal transcribed spacer sequences (Porter and Golding, 2011). However, one clear advantage of the naïve Bayesian classifier was speed; on our system it could classify thousands of pmoA sequences per second compared to approximately 200 sequences per min for the MEGABLAST query.
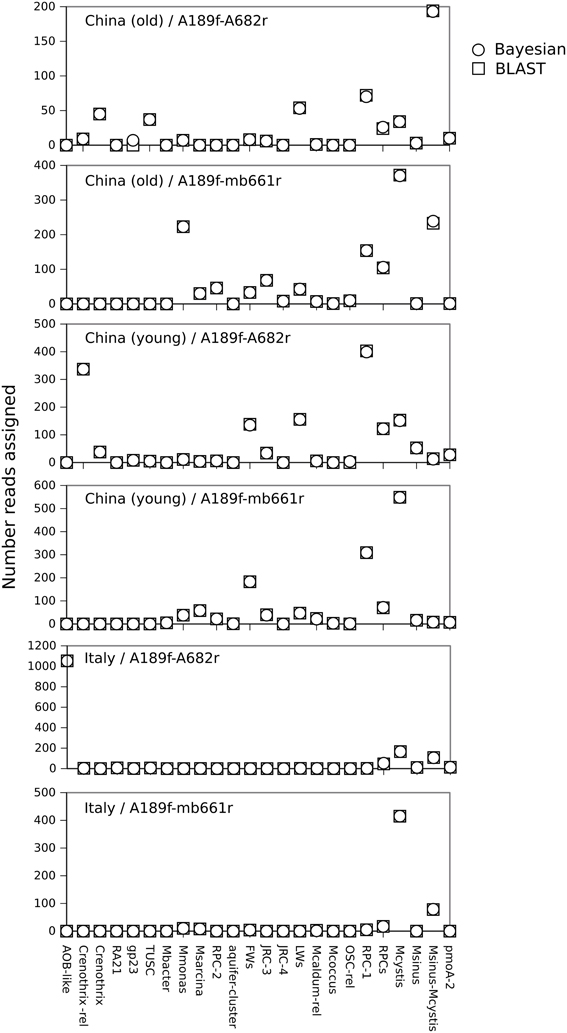
Figure 2. Comparison of classifications of paddy soil pmoA sequence data (Lüke and Frenzel, 2011) using the naïve Bayesian and BLAST/LCA methods. The datasets were generated from three different soils (young Chinese, old Chinese, and Italian) and with two different PCR primer combinations (A189f/A682r, A189f/mb661r) as indicated. The number of sequences assigned to each taxon is plotted. Only pmoA taxa detected in at least one dataset are shown.
7. Novelty Detection
Novel sequences can be identified by the naïve Bayesian classifier if they cannot be classified. For example, a novel taxon within the Type Ib's should be returned as “unclassified Type Ib.” It is possible to adjust this further by specifying a percent cutoff value for a bootstrapped analysis. In general, we found this method to be unreliable as even contaminant sequences were classified as gp23 with >80% bootstrap values, as discussed above.
7.1. Novelty Detection using BLAST/LCA in MEGAN
The BLAST/LCA procedure offers approaches for novelty detection at various levels, which are described individually in the following sections.
7.1.1. Identification of highly divergent CuMMO sequences
Deeply-branching novel sequences did not produce hits to the database, which is the first indication that it is potentially a novel sequence clade. This occurs in BLAST queries when the sequence does not contain a match of the minimum word size, which is 28 nucleotides in MEGABLAST. For example, this was the case in the Riganqiao dataset with the novel clade that was termed HY-3 (Deng et al., 2013). In contrast to MEGABLAST, these sequences could be identified as pmoA by a translated BLAST query (TBLASTX). The difference is that protein sequences are more conserved than nucleotide sequences and BLASTX uses a word size of 3 amino acids compared with a minimum word size of 7 nucleotides for BLASTN. Therefore, the first step in novelty detection is to select the sequences that do not produce MEGABLAST hits and to query them against the database using TBLASTX; in our experience, highly divergent novel clusters will produce significant hits (>50 bits) with TBLASTX, whereas unrelated contaminant sequences will not.
7.1.2. Identification of moderately divergent CuMMO sequences
Moderately divergent novel sequence clades can be identified by a relatively low MEGABLAST bit score. The bit score cutoffs can be adjusted in MEGAN and here we used a threshold of 150 bits to identify new clades. An example of a novel sequence that could be identified in this manner was the I141NRXW sequence from the paddy soil (Lüke and Frenzel, 2011), which had a top score of 89.8 bits. In comparison, the sequence with the next lowest bit score in that dataset had a value of 374, and maximum bit scores approached 900.
7.1.3. Lowest common ancestor classification of sequences
The next level of novelty can be identified using the lowest common ancestor (LCA) algorithm in MEGAN. For the HYa-1 dataset, assignments to higher nodes could be seen at the Type II (15 sequences), Type Ib (9 sequences) and Type IIa (151 sequences) taxonomic levels using a margin of 5% for the LCA calculation (Figure 3A). In contrast, classifications to the other lowest-level taxa were stable even using an LCA margin greater than 25%. The inability to classify sequences to the lowest levels indicates that the sequence may represent a new taxon branching from the LCA node. For example, the sequences assigned to the Type II node had similar bit scores to both Methylocystis and pmoA2 clades and a NJ analysis of the sequences also suggested that it was new lineage (termed HY-4) at the root of the Type II's (Figure 4). The assignments at the node of the Type IIa branch were the result of an inability to distinguish between the Methylocystis and Methylosinus-Methylocystis taxa, which might indicate intermediate sequence types or the existence of a bush-like continuum in this region of the tree (Lüke and Frenzel, 2011).
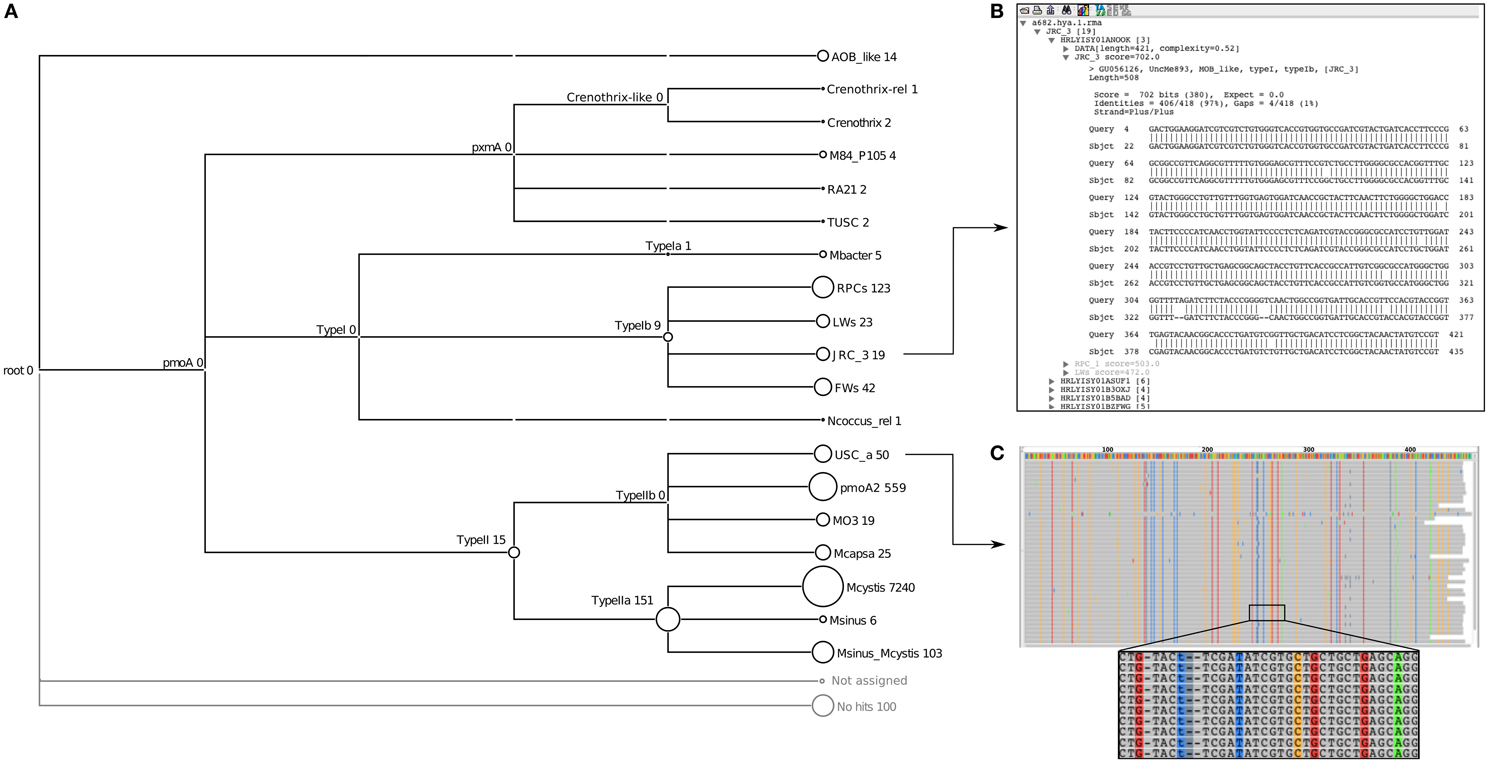
Figure 3. Examples of MEGAN visualizations of assignments for the pmoA data of the HYa-1 sample obtained from the Riganqiao pmoA pyrosequencing study (Deng et al., 2013). The tree shows the summary of taxa identified and their abundances (A); the circles at the nodes are proportional to the number of reads assigned to that taxon. Individual assignments can be inspected by right-clicking on a node and selecting “inspect,” as shown for the JRC-3 clade (B). Summary alignments can also be visualized, as shown for USCα (C); in this case it quickly shows that the best alignment to USCα have approximately 40 conserved mismatches, suggesting it is a novel pmoA cluster most closely related to USCα.
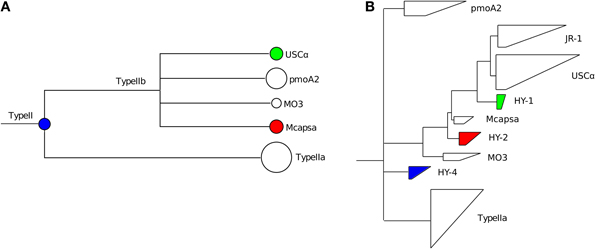
Figure 4. Analysis of selected Type II pmoA sequences from the HYa-1 sample (Deng et al., 2013). The sequences chosen for analysis are color-coded in the partial MEGAN tree (A). The USCα and Methylocapsa-assigned sequences were selected since the alignments showed conserved mismatches to the reference database (as shown for USCα sequences in Figure 3C). The sequences were imported into an ARB pmoA database, quality filtered by removing sequences with frameshifts, translated to amino acid sequences and added to the PmoA tree by parsimony and then reanalyzed by neighbor-joining. The positions of the sequences analyzed in ARB are shown (B). The new clades were named HY-1, HY-2 (Deng et al., 2013) and HY-4 (this study).
In addition to biological diversity, there could also be technical errors that impede classification, such as sequencing noise or chimerism. Furthermore, falsely assigned sequences in the taxonomy file would also result in sequences failing to be classified at the lowest level. Both of these situations can generally be detected by visually analyzing the BLAST alignments, as described in the following section.
7.1.4. Lowest-level diversity: examination of hits and alignments
The final level of novelty can be detected by examining the hits and alignments against the database. BLAST alignments of individual reads (Figure 3B) or summary alignments for groups of sequences (Figure 3C) can be examined in MEGAN. Examining individual reads gives an impression of how closely related a sequence is to members of the assigned taxon compared to the next-nearest taxon. In the example shown (Figure 3B), the top hit had 97% identity to a JRC_3 (702 bits) and the next best hit of only 89% identity (272 bits) to RPC_1. In this example, it is evident that the sequence is genuinely JRC_3. In contrast, the hits to Methylocapsa had maximum identities of only 95% (not shown), suggesting that these might represent a closely related novel cluster. An analysis by NJ of these sequences indicated that indeed they formed a new branch close to Methylocapsa (termed HY-2) (Figure 4).
The second option is to invoke the alignment of the hits to a taxon. Here it is possible to see evidence of novelty within a group of sequences classified to a particular taxon. For example, in some cases numerous conserved mismatches against the top database reference can suggest that the sequences belong to a divergent clade mostly related to the assigned taxon. For example, about 40 conserved mismatches were present within the assignment to USCα in the HYa-1 dataset (Figure 3C). A closer analysis by NJ of these sequences indicated they were a novel lineage most closely related to USCα (termed HY-1) (Figure 4).
8. Data Comparisons and Downstream Analysis
MEGAN has several built-in functions that offer possibilities to visualize and analyze the results. Comparisons between samples can be easily made using various visualization options. Trends in the data can be observed and demonstrated, such as the relative coverage of the two primer sets and the elevated abundance of RPCs in hollow soils (Figure 5). MEGAN can also calculate matrices of pairwise distances using six ecological measures: Euclidian, Goodall, Chi-Square, Kulczynski, Bray-Curtis, and Hellinger (Mitra et al., 2010). Data can also be easily exported into statistical software programs. Of course, the classification of sequence data to taxa is only one step in the analysis of HTS amplicon data of protein-coding genes and should be complemented by classification-independent analyses.
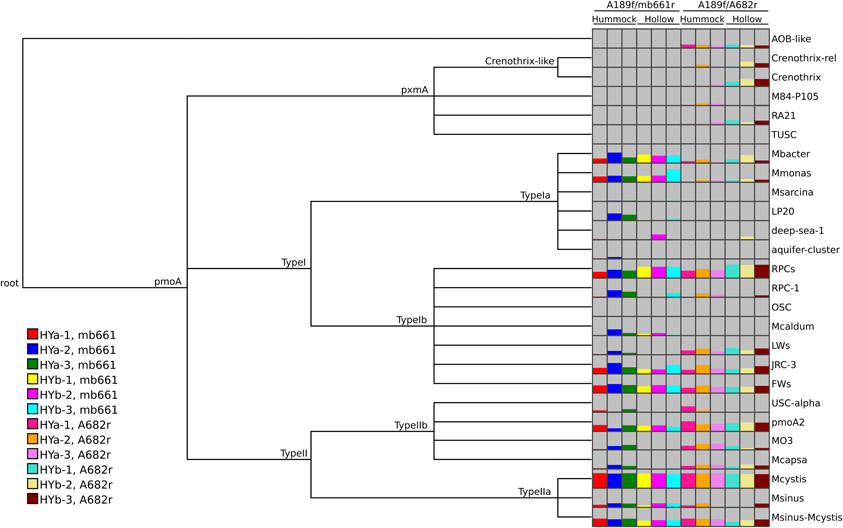
Figure 5. MEGAN comparison view of pmoA classifications from the Riganqiao pmoA pyrosequencing datasets (Deng et al., 2013). The pmoA datasets were obtained from triplicate samples from hummock (HYa) and hollow (HYb) sites. PCRs were performed with two primer combinations (A189f/A682r or A189f/mb661r), as indicated. The option to subsample datasets (3309 sequences) was chosen for the comparison. Assignments to internal nodes are not shown. MEGAN only shows taxa detected in at least one sample. The height of the bars was scaled to the number of reads assigned in each dataset and color-coded as indicated in the legend. The labeling at the top of the columns was added.
9. Conclusions
Although the naïve Bayesian and BLAST/LCA methods provided similar classifications of the high-throughput pmoA sequence data examined in this study, the BLAST/LCA approach had several advantages, such as being less sensitive to false classification of contaminant sequences and offering several options for novelty detection at various levels of sequence divergence. The BLAST/LCA method has another advantage that a researcher can visually interpret the calculations, in the form of alignments, therefore enabling the results to be verified and judged.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00034/abstract
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1006/jmbi.1990.9999
Baani, M., and Liesack, W. (2008). Two isozymes of particulate methane monooxygenase with different methane oxidation kinetics are found in Methylocystis sp strain SC2. Proc. Natl. Acad. Sci. U.S.A. 105, 10203–10208. doi: 10.1073/pnas.0702643105
Berry, D., Ben Mahfoudh, K., Wagner, M., and Loy, A. (2011). Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl. Environ. Microbiol. 77, 7846–7849. doi: 10.1128/AEM.05220-11
Binladen, J., Gilbert, M. T. P., Bollback, J. P., Panitz, F., Bendixen, C., Nielsen, R., et al. (2007). The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS ONE 2:e197. doi: 10.1371/journal.pone.0000197
Cai, Y., and Sun, Y. (2011). ESPRIT-Tree: hierarchical clustering analysis of millions of 16S rRNA pyrosequences in quasilinear computational time. Nucleic Acids Res. 2011, 1–10. doi: 10.1093/nar/gkr349
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. Bioinformatics 10, 421. doi: 10.1186/1471-2105-10-421
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cole, J. R., Chai, B., Farris, R. J., Wang, Q., Kulam, S. A., McGarrell, D. M., et al. (2005). The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 33, D294–D296. doi: 10.1093/nar/gki038
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., et al. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145. doi: 10.1093/nar/gkn879
Coleman, N. V., Le, N. B., Ly, M. A., Ogawa, H. E., McCarl, V., Wilson, N. L., et al. (2012). Hydrocarbon monooxygenase in Mycobacterium: recombinant expression of a member of the ammonia monooxygenase superfamily. ISME J. 6, 171–182. doi: 10.1038/ismej.2011.98
Costello, A. M., and Lidstrom, M. E. (1999). Molecular characterization of functional and phylogenetic genes from natural populations of methanotrophs in lake sediments. Appl. Environ. Microbiol. 65, 5066–5074.
Degelmann, D. M., Borken, W., Drake, H. L., and Kolb, S. (2010). Different atmospheric methane-oxidizing communities in European beech and Norway spruce soils. Appl. Environ. Microbiol. 76, 3228–3235. doi: 10.1128/AEM.02730-09
Deng, Y., Cui, X., Lüke, C., and Dumont, M. G. (2013). Aerobic methanotroph diversity in Riganqiao peatlands on the Qinghai-Tibetan Plateau. Environ. Microbiol. Rep. 5, 566–574. doi: 10.1111/1758-2229.12046
Dunfield, P. F., Belova, S. E., Vorob'ev, A. V., Cornish, S. L., and Dedysh, S. N. (2010). Methylocapsa aurea sp. nov., a facultative methanotroph possessing a particulate methane monooxygenase, and emended description of the genus Methylocapsa. Int. J. Syst. Evol. Microbiol. 60, 2659–2664. doi: 10.1099/Ijs.0.020149-0
Dunfield, P. F., Yimga, M. T., Dedysh, S. N., Berger, U., Liesack, W., and Heyer, J. (2002). Isolation of a Methylocystis strain containing a novel pmoA-like gene. FEMS Microbiol. Ecol. 41, 17–26. doi: 10.1111/j.1574-6941.2002.tb00962.x
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Ettwig, K. F., Butler, M. K., Le Paslier, D., Pelletier, E., Mangenot, S., Kuypers, M. M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548. doi: 10.1038/nature08883
Ettwig, K. F., van Alen, T., van de Pas-Schoonen, K. T., Jetten, M. S. M., and Strous, M. (2009). Enrichment and molecular detection of denitrifying methanotrophic bacteria of the NC10 phylum. Appl. Environ. Microbiol. 75, 3656–3662. doi: 10.1128/aem.00067-09
Holmes, A. J., Costello, A., Lidstrom, M. E., and Murrell, J. C. (1995). Evidence that particulate methane monooxygenase and ammonia monooxygenase may be evolutionarily related. FEMS Microbiol. Lett. 132, 203–208. doi: 10.1111/j.1574-6968.1995.tb07834.x
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Huson, D. H., Mitra, S., Ruscheweyh, H. J., Weber, N., and Schuster, S. C. (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560. doi: 10.1101/gr.120618.111
Koski, L. B., and Golding, G. B. (2001). The closest BLAST hit is often not the nearest neighbor. J. Mol. Evol. 52, 540–542. doi: 10.1007/s002390010184
Lanzén, A., Jørgensen, S. L., Huson, D. H., Gorfer, M., Grindhaug, S. H., Jonassen, I., et al. (2012). CREST – Classification resources for environmental sequence tags. PLoS ONE 77:e49334. doi: 10.1371/journal.pone.0049334
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Luesken, F. A., Zhu, B., van Alen, T. A., Butler, M. K., Rodriguez Diaz, M., Song B., et al. (2011). pmoA primers for detection of anaerobic methanotrophs. Appl. Environ. Microb. 77, 3877–3880. doi: 10.1128/AEM.02960-10
Lüke, C., and Frenzel, P. (2011). Potential of pmoA amplicon pyrosequencing for methanotroph diversity studies. Appl. Environ. Microb. 77, 6305–6309. doi: 10.1128/Aem.05355-11
McDonald, I. R., Bodrossy, L., Chen, Y., and Murrell, J. C. (2008). Molecular ecology techniques for the study of aerobic methanotrophs. Appl. Environ. Microb. 74, 1305–1315. doi: 10.1128/Aem.02233-07
McGinnis, S., and Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25. doi: 10.1093/nar/gkh435
Mitra, S., Gilbert, J. A., Field, D., and Huson, D. H. (2010). Comparison of multiple metagenomes using phylogenetic networks based on ecological indices. ISME J. 4, 1236–1242. doi: 10.1038/ismej.2010.51
Mitra, S., Stark, M., and Huson, D. H. (2011). Analysis of 16S rRNA environmental sequences using MEGAN. BMC Genomics 12. doi: 10.1186/1471-2164-12-s3-s17
Pester, M., Schleper, C., and Wagner, M. (2011). The Thaumarchaeota: an emerging view of their phylogeny and ecophysiology. Curr. Opin. Microbiol. 14, 300–306. doi: 10.1016/j.mib.2011.04.007
Porter, T. M., and Golding, G. B. (2011). Are similarity- or phylogeny-based methods more appropriate for classifying internal transcribed spacer (ITS) metagenomic amplicons? New Phytol. 192, 775–782. doi: 10.1111/j.1469-8137.2011.03838.x
Porter, T. M., and Golding, G. B. (2012). Factors that affect large subunit ribosomal DNA amplicon sequencing studies of fungal communities: classification method, primer choice, and error. PLoS ONE 7:e35749. doi: 10.1371/journal.pone.0035749
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Quince, C., Lanzen, A., Davenport, R. J., and Turnbaugh, P. J. (2011). Removing noise from pyrosequenced amplicons. BMC Bioinform. 12:38. doi: 10.1186/1471-2105-12-38
Reeder, J., and Knight, R. (2010). Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat. Methods 7:668–669. doi: 10.1038/nmeth0910-668b
Rosen, M. J., Callahan, B. J., Fisher, D. S., and Holmes, S. P. (2012). Denoising PCR-amplified metagenome data. BMC Bioinform. 13:283. doi: 10.1186/1471-2105-13-283
Schloss, P. D., and Handelsman, J. (2004). Status of the microbial census. Microbiol. Mol. Biol. Rev. 68, 686–691. doi: 10.1128/mmbr.68.4.686-691.2004
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-09
Schloss, P. D., Gevers D., and Westcott S. L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6:e27310. doi: 10.1371/journal.pone.0027310
Stamatakis, A., Ludwig, T., and Meier, H. (2005). RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463. doi: 10.1093/bioinformatics/bti191
Stoecker, K., Bendinger, B., Schoning, B., Nielsen, P. H., Nielsen, J. L., Baranyi, C., et al. (2006). Cohn's Crenothrix is a filamentous methane oxidizer with an unusual methane monooxygenase. Proc. Natl. Acad. Sci. U.S.A. 103, 2363–2367. doi: 10.1073/pnas.0506361103
Tavormina, P. L., Orphan, V. J., Kalyuzhnaya, M. G., Jetten, M. S. M., and Klotz, M. G. (2011). A novel family of functional operons encoding methane/ammonia monooxygenase-related proteins in gammaproteobacterial methanotrophs. Environ. Microbiol. Rep. 3, 91–100. doi: 10.1111/j.1758-2229.2010.00192.x
Theisen, A. R., Ali, M. H., Radajewski, S., Dumont, M. G., Dunfield, P. F., McDonald, I. R., et al. (2005). Regulation of methane oxidation in the facultative methanotroph Methylocella silvestris BL2. Mol. Microbiol. 58, 682–692. doi: 10.1111/j.1365-2958.2005.04861.x
Vorobev, A. V., Baani, M., Doronina, N. V., Brady, A. L., Liesack, W., Dunfield, P. F., et al. (2011). Methyloferula stellata gen. nov., sp nov., an acidophilic, obligately methanotrophic bacterium that possesses only a soluble methane monooxygenase. Int. J. Syst. Evol. Microbiol. 61, 2456–2463. doi: 10.1099/ijs.0.028118-0
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/aem.00062-07
Keywords: pmoA, methanotroph, pyrosequencing, diversity, NGS data analysis
Citation: Dumont MG, Lüke C, Deng Y and Frenzel P (2014) Classification of pmoA amplicon pyrosequences using BLAST and the lowest common ancestor method in MEGAN. Front. Microbiol. 5:34. doi: 10.3389/fmicb.2014.00034
Received: 30 August 2013; Paper pending published: 12 October 2013;
Accepted: 19 January 2014; Published online: 18 February 2014.
Edited by:
Kevin John Purdy, University of Warwick, UKReviewed by:
Anthony Yannarell, University of Illinois at Urbana-Champaign, USAMarco J. L. Coolen, Woods Hole Oceanographic Institution, USA
Copyright © 2014 Dumont, Lüke, Deng and Frenzel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc G. Dumont, Department of Biogeochemistry, Max Planck Institute for Terrestrial Microbiology, Karl-von-Frisch-Str., 10, 35043 Marburg, Germany e-mail: dumont@mpi-marburg.mpg.de