 Shannon M. Soucy
Shannon M. Soucy Matthew S. Fullmer
Matthew S. Fullmer R. Thane Papke
R. Thane Papke Johann Peter Gogarten
Johann Peter Gogarten- Department of Molecular and Cell Biology, University of Connecticut, Storrs, CT, USA
This research uses inteins, a type of mobile genetic element, to infer patterns of gene transfer within the Halobacteria. We surveyed 118 genomes representing 26 genera of Halobacteria for intein sequences. We then used the presence-absence profile, sequence similarity and phylogenies from the inteins recovered to explore how intein distribution can provide insight on the dynamics of gene flow between closely related and divergent organisms. We identified 24 proteins in the Halobacteria that have been invaded by inteins at some point in their evolutionary history, including two proteins not previously reported to contain an intein. Furthermore, the size of an intein is used as a heuristic for the phase of the intein's life cycle. Larger size inteins are assumed to be the canonical two domain inteins, consisting of self-splicing and homing endonuclease domains (HEN); smaller sizes are assumed to have lost the HEN domain. For many halobacterial groups the consensus phylogenetic signal derived from intein sequences is compatible with vertical inheritance or with a strong gene transfer bias creating these clusters. Regardless, the coexistence of intein-free and intein-containing alleles reveal ongoing transfer and loss of inteins within these groups. Inteins were frequently shared with other Euryarchaeota and among the Bacteria, with members of the Cyanobacteria (Cyanothece, Anabaena), Bacteriodetes (Salinibacter), Betaproteobacteria (Delftia, Acidovorax), Firmicutes (Halanaerobium), Actinobacteria (Longispora), and Deinococcus-Thermus-group.
Introduction
Inteins are self-splicing genetic parasites located in highly conserved sites of slowly evolving genes. They are found in all three domains of life and in viruses (Perler et al., 1997; Pietrokovski, 2001; Gogarten et al., 2002; Swithers et al., 2009). Similar to group I introns, inteins are often associated with a homing endonuclease (HEN). An important difference between inteins and introns is the timing of the splicing activity, which occurs immediately after transcription in introns and after translation in inteins (Hirata et al., 1990; Kane et al., 1990). The association with a HEN domain enables a cyclic invasion pattern, called the homing cycle (Goddard and Burt, 1999; Gogarten and Hilario, 2006). The homing cycle consists of three phases: intein invasion, intein fixation, and eventually loss of the intein enabling invasion to occur again. During invasion and fixation the intein splicing domains are associated with a HEN domain forming a canonical intein (hereafter referred to as a large intein); however, during the loss phase the function of the HEN is often disrupted and begins to degrade, generating a mini-intein. Simulations have shown that intein-containing and intein-free alleles can coexist in well mixed populations under some sets of parameters (Yahara et al., 2009; Barzel et al., 2011). Also, inteins with functioning HEN domains were inferred to have persisted in some eukaryotic lineages for several 100 million years (Butler et al., 2006; Gogarten and Hilario, 2006).
Inteins do not have an apparatus to penetrate the cell envelope. Therefore, they must rely on mechanisms in place within the population for insertion into the cell such as: conjugation, mating, generalized DNA uptake, and viruses or gene transfer agents (Lang et al., 2012). The faster-than-Mendelian inheritance of the large inteins (Gimble and Thorner, 1992), along with a nearly neutral fitness burden, enables these mobile elements to persist in organisms over evolutionary time as long as there are new populations to invade (Goddard and Burt, 1999; Gogarten and Hilario, 2006). Furthermore, the size of the intein (mini or large) provides information about the genomic mobility of the element as mini inteins are rarely integrated into the recipient's genome; whereas large inteins are more frequently integrated due to the activity of the HEN. The conservation of the recognition site provides an invasion target even in distantly related strains and species. Also, inteins have a higher substitution rate relative to their extein hosts (Swithers et al., 2013). This substitution rate gives rise to many evolutionarily informative sites when comparing a large collection of homologous inteins. In this work, we take advantage of these traits and survey the distribution of inteins in the Halobacteria, a highly recombinant class of halophilic Archaea (Williams et al., 2012) known to contain several intein alleles (Perler, 2002). We make use of 118 halobacterial genomes (Supplementary Table 1) and the previously reported and newly discovered intein alleles to survey networks of gene transfer within and outside the Halobacteria based on the presence-absence profile of the inteins, their sequence similarity, and the phylogenies reconstructed from intein sequences.
Materials and Methods
Halobacterial Intein Sequence Retrieval and Alignment
Position specific scoring matrices (PSSMs) were created using the collection of all inteins from InBase, the Intein database and registry (Perler, 2002). A custom database was created with all inteins, and each intein was used as a seed to create a PSSM using the custom database. These PSSMs were then used as a seed for PSI-BLAST (Altschul, 1997) searches against each of the halobacterial genomes available from NCBI as of June 2013 as well as a private collection sequenced by our collaborators. To remove false positives, a size exclusion step was then performed on each protein sequence as an intein domain adds 100–700 aa to invaded protein sequences. Inteins were then aligned using Muscle (Edgar, 2004) with default parameters in the SeaView version 4.0 software package (Gouy et al., 2010). Insertions, which passed the size exclusion step, but did not contain splicing domains, were removed and the previous steps were repeated using the resulting dataset on a collection of private genomes from the Papke lab. Prottest 3.2 (Guindon et al., 2010; Darriba et al., 2011) was used to determine an appropriate substitution model for the intein sequences, the WAG model was favored and used for all subsequent trees for consistency. Once the collection of halobacterial inteins was complete, sequences were re-aligned using SATé (Liu et al., 2012) to generate a final alignment using MAFFT (Katoh and Standley, 2013) to align, Muscle (Edgar, 2004) to merge, RAXML(Stamatakis, 2014) for tree estimation, and a WAG model for each allele.
To determine the relationship among all halobacterial inteins, the inteins were aligned using Muscle (Edgar, 2004). Subsequently a tree was built using PhyML v3.0 (Guindon et al., 2010) using a WAG substitution model with a Gamma shape parameter and the proportion of invariant sites estimated from the data.
Intein Retrieval Outside the Halobacteria
Each halobacterial intein was used as a BLAST (Altschul et al., 1990) query against the non-redundant database on NCBI. Any match with an e-value better than 0.000001 was aligned to the dataset to which its query belonged. Sequences were then filtered based on the protein annotation and goodness of fit to the existing alignment. As an additional filtering step each match was used as a query against the non-redundant database and the majority BLAST hit annotations were used to verify the protein identity, as annotations are not always reliable. Remaining sequences were aligned using Clustal Omega 1.1.0 (Sievers et al., 2011) with the profile alignment option in SeaView 4.0 (Gouy et al., 2010). Maximum-likelihood trees were built using PhyML (Guindon et al., 2010) with the WAG model, and rates estimated from the data.
To assess the relative contribution of different genera represented in each intein allele sequence data set, a stacked column graph was created. Sequence density was calculated for each intein allele by dividing the number of intein sequences in each genus by the number of total intein sequences in that allele.
Symbiotic State Assignment
Intein sequence length was used to determine symbiotic state. For each intein allele the length of the intein sequence was determined. A cutoff length for mini-intein assignment was based on the presence of a gap in intein lengths greater than 100 amino acids within an allele. The third intein state “no-intein” was assigned where the intein was clearly absent from the orthologous protein containing an intein in any of the halobacterial genomes examined. Additionally, once an intein was noted as a mini-intein the alignment was analyzed to ensure the gaps in these sequences correspond to the location of the HEN domain.
Ribosomal Protein Reference Tree
Alignments of 55 ribosomal protein for 21 Halobacteria (Williams et al., 2012) were used to find orthologous proteins in the genomes used in this work. In-house python scripts (data file 1) were used to concatenate the alignments, and PhyML v3.0 (Guindon et al., 2010) was used to build a tree. The tree used the WAG substitution model with the Gamma shape parameter and the proportion of invariant sites and base frequencies estimated from the data.
Bayesian Clustering with Intein Sequences
A concatenation of an intein presence-absence matrix and alignments for each intein allele were generated using in-house python scripts (data file 1). MrBayes version 3.2.1 (Ronquist et al., 2012) was then used to perform a clustering analysis using a partition allowing for character states in the presence-absence matrix and sequence information for each intein allele. The prior for the character portion of the data matrix used a symmetrical Dirichlet distribution with an exponential (1.0), and variable rates so each column was considered independent of the others. The likelihood for the character portion of the alignment used variable coding and 5 beta categories. The prior for the protein sequences in the alignment used a fixed WAG substitution model, with state frequencies estimated from the data, and the likelihood settings used a Gamma shape parameter and the proportion of invariant sites estimated from the data.
Results
Halobacterial Inteins
The intein content of a collection of halobacterial genomes was analyzed using an intein-allele-specific PSSM. This survey revealed 13 genes in the Halobacteria invaded by inteins at 24 distinct positions (intein alleles) (Table 1). Seven of these intein alleles were not previously reported in the Halobacteria, and two of the seven have not previously been reported to harbor inteins: a DNA ligase gene involved in double strand break repair, and a deaminase gene involved in nucleotide metabolism (Table 1). To determine if vertical inheritance was accountable for the distribution of intein alleles, the presence–absence matrix of intein alleles was mapped onto a reference phylogeny (Figure 1). Clearly, intein presence-absence is not concordant with the ribosomal protein phylogeny, implicating abundant horizontal genetic transfer (HGT) in creating the observed distribution. The presence of multiple intein alleles in the majority of genomes (70%) might be interpreted to suggest that inteins could spread locally within a single genome.

Table 1. Exteins in the halobacteria.
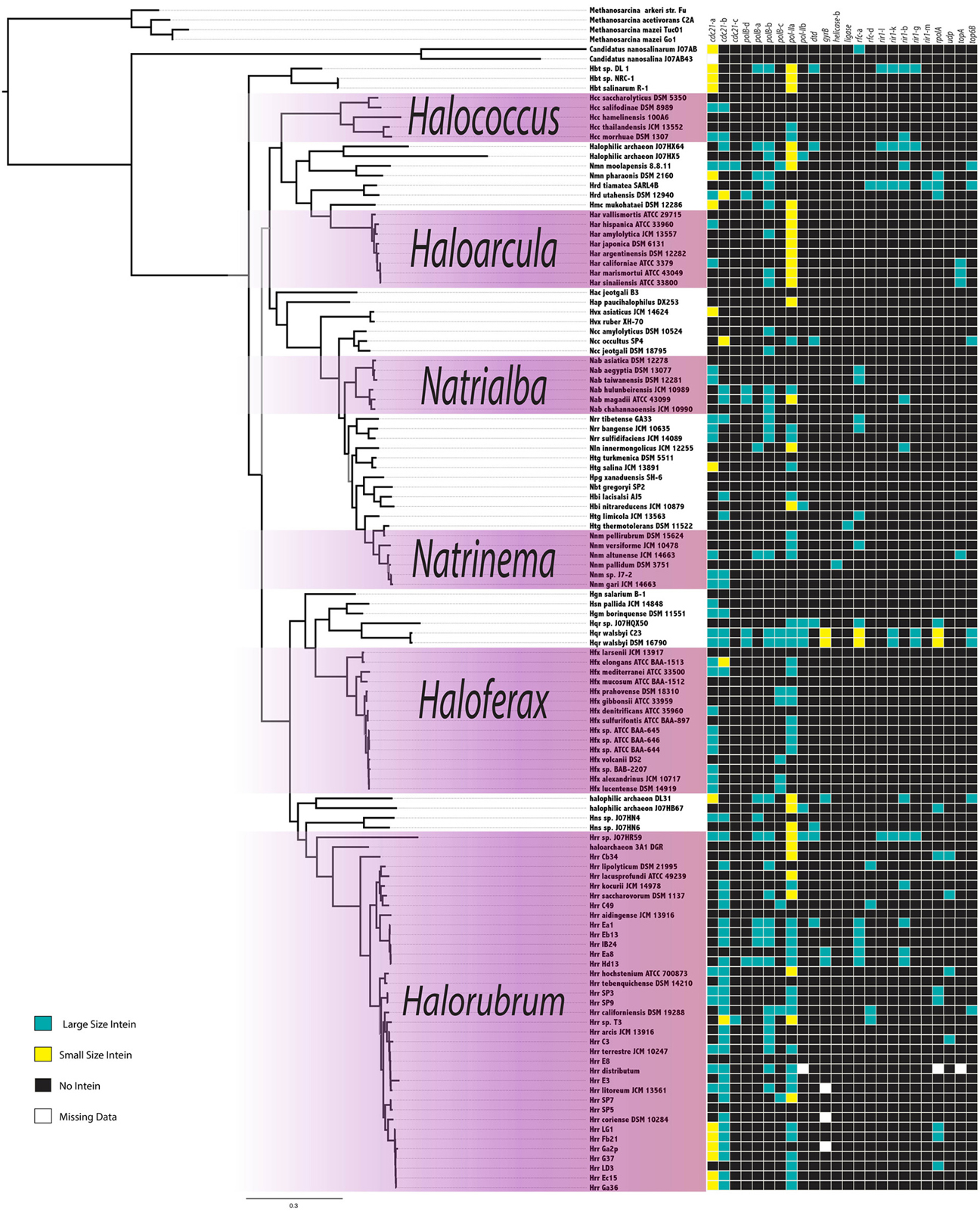
Figure 1. Intein Invasion Pattern in the Halobacteria. Intein pattern of presence-absence is mapped onto the tips of a ribosomal reference tree, teal boxes indicate the presence of a full size intein, yellow boxes indicate the presence of a mini-intein, black boxes indicate the absence of an intein, and white boxes indicate missing data. Purple shaded boxes indicate the genera with more than five species represented on the tree. Nodes with bootstrap support <70 are in gray.
Intein Propagation within the Halobacteria
To address the possibility of inteins moving locally within a genome, the phylogenetic relationships among all halobacterial intein sequences were analyzed (Figure 2). All of the intein alleles form highly supported clusters with others of the same type, with the exception of two sequences: the polB-c inteins of Haloferax larsenii and Haloferax elongans group inside the polB-b intein allele cluster; however, this node is poorly supported (59/100 bootstraps) indicating this relationship could be an artifact produced by poor resolution of the relationships that connect various intein alleles. Furthermore, there is poor support linking all of the intein allele clusters together (less than 70% bootstrap support), indicating sequence conversion (an intein invading an ectopic or atypical locus) between intein alleles, even within the same host protein, is uncommon. Among the inteins analyzed here, at most one invasion of an ectopic site is supported by the data, confirming that this type of event is rare (Perler et al., 1997; Gogarten et al., 2002). These data indicate that HGT is the only plausible explanation for the large number of different intein alleles in this class of organisms. Incongruence between the presence of inteins and ribosomal phylogeny also support this conclusion.
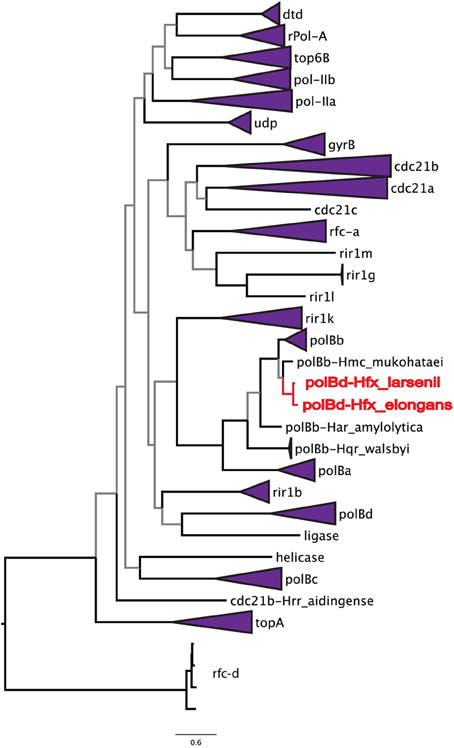
Figure 2. Relationships among intein alleles in the Halobacteria. This tree depicts the phylogenetic relationships among intein alleles in the Halobacteria. Inteins that clustered in concordance with the allele were collapsed to a single node, and labeled with the name of the intein allele. Two polB-d sequences did not group with the polB-d allele, and instead are located amongst the polB-c alleles these are indicated in red. Nodes with bootstrap support <70 are colored gray.
Bayesian Phylogenetic Analysis of Inteins
In an attempt to resolve the local events (transfers and vertical inheritance within the Halobacteria) that gave rise to the observed intein distribution in the Halobacteria, a Bayesian analysis based on the intein sequences for each allele and on the presence-absence pattern was performed (Figure 3). In this analysis two organisms may group together because they both inherited inteins from a common ancestor, or because an intein was recently transferred between them. The paucity of well-supported nodes (nodes with 0.95 or greater posterior probability were considered well-supported) in part reflects the extent to which our sample is biased toward very similar sequences (31% of halobacterial genomes in this study are from Halorubrum). Most of the well-supported clusters in the Bayesian tree also occur in the reference tree, suggesting these inteins may be the result of shared vertical inheritance. However, many of these clusters do not have identical intein profiles (clusters 1, 6, 8, and 10), thus HGT between close relatives is a better explanation than vertical inheritance for these clusters. Only three of the clusters, 2, 9, and 12, have branching orders that are different from those observed in the reference tree indicating HGT. Cluster 2 is made up of Natrinema spp. pellirubrum and versiforme which share only the pol-IIa intein. In the reference tree Nnm. versiforme groups with the rest of the Natrinema, and Nnm. pellirubrum groups with Haloterrigena thermotolerans. Natrinema sp. J7-2 is the only other member of the Natrinema that has an intein in the pol-IIa position, but the intein in this species is 14 aa shorter than the intein shared by Nnm. pellirubrum and Nnm. versiforme. Htg. thermotolerans shares no inteins with Nnm. pellirubrum. Cluster 9 is made up of Halorubrum spp. C49 and E3, which share only the cdc21-b intein. In the reference tree Hrr. E3 groups with Halorubrum litoreum and the two share the pol-IIa intein allele, but no others. Hrr. C49 groups with Halorubrum saccharovorum and they do not share any inteins. Cluster 12 is made up of Haloferax spp. denitrificans, lucentense, alexandrinus, and Haloferax sp. BAB2207, which all have an intein in the cdc21-a position. In the reference tree Hfx. lucentense, Hfx. sp. BAB2207, and Hfx. alexandrinus all group together, but Hfx. denitrificans groups with Haloferax sulfurifontis, and they do not share any inteins. The lack of shared inteins between clusters in the reference tree and differences among the inteins shared in these clusters cause these divergences in this tree as compared to the reference tree. This may indicate that the taxa in the Bayesian clusters are exchange partners, or that they share unsampled intermediate exchange partners. Additionally, the majority of clusters share 2 or fewer intein alleles between all members of the cluster (eight out of 12 clusters). The two clusters that share the most intein alleles between all members are Cluster 3, made up of Haloqudratum walsbyi strains DSM 16790 and C23 with 13 shared intein alleles, and cluster 7 made up of Halorubrum spp. strains SP3 and SP9 sharing 4 intein alleles. Both of these clusters have branching patterns identical to those on the reference tree, indicating that phylogenetic proximity plays a significant role in intein distribution.
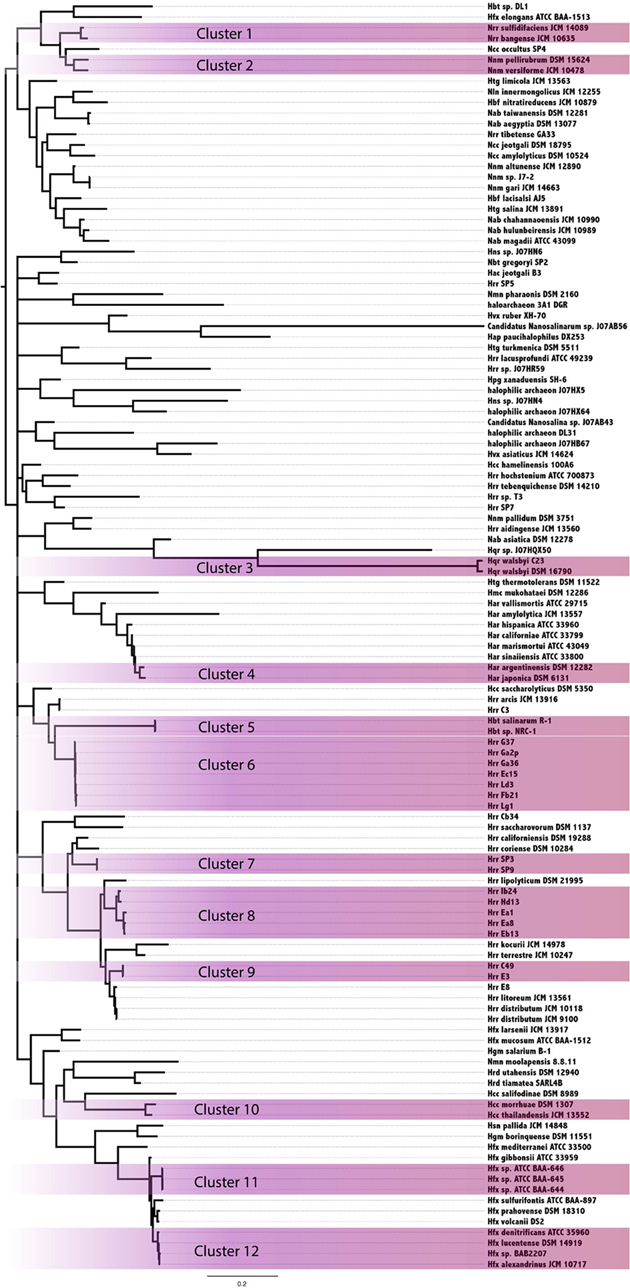
Figure 3. Clustering of Halobacteria based on intein sequences and distribution. Halobacteria were clustered based on intein sequences and the distribution in each genome. Clusters with posterior probability >95% are shaded purple.
Members of the Halorubrum genus, not surprisingly, were highly represented in the clusters (four of 12 total). All four of the clusters show a geographic bias. Clusters 6, 8, and 9 were all isolated from the Aran-Bidgol lake in Iran, and cluster 7 was isolated from the Sedom Ponds in Israel (Atanasova et al., 2012). Branch lengths in all of these clusters are very small, suggesting these populations are well mixed with respect to intein sequences. Geography does not seem to play a strong role in linking other well-supported clusters based on intein sequences. Furthermore, evidence of clustering based on geography in the Halorubrum is less interesting than the clear separation between groups isolated from the same location (cluster 6, 8, and 9). This separation of species of Halorubrum from the same location is echoed in the reference tree, and taken together with the short branch lengths in these clusters indicate that population structure plays a strong role in gene sharing at least for this location (see Fullmer et al., 2014 for in depth discussion). Increased geographical sampling could reveal similar trends in other locations.
Intein Homing in the Halobacteria
The existence of a singleton in an intein allele in the genomes analyzed could represent intein invasion from outside the Halobacteria; but could also be due to incomplete sampling. To investigate the phylogenetic distance of invasion events responsible for the observed distribution of inteins, the halobacterial inteins were used as queries to search for homologous sequences in the non-redundant database (Altschul et al., 1990). Intein sequences that matched the alleles in the Halobacteria were found in other Euryarchaeota (but not Crenarchaeota), and Bacteria (Table 2). To ascertain whether homing occurred between the Halobacteria and organisms outside the Halobacteria, a maximum likelihood tree was built for each intein allele. The tree topologies were evaluated with respect to the halobacterial inteins. If the halobacterial inteins in the tree were monophyletic it was assumed that except for the initial invasion gene flow for that intein allele occurred within the Halobacteria exclusively. If the halobacterial inteins were polyphyletic, invasion events that generated the observed distribution likely involved organisms outside the Halobacteria either as donors or as recipients. The majority of intein trees, 83%, were monophyletic, reinforcing the idea that recombination is more successful between closely related organisms (Gogarten et al., 2002; Zhaxybayeva et al., 2006; Andam et al., 2010; Papke and Gogarten, 2012; Williams et al., 2012). Interestingly, for trees where the Halobacteria were polyphyletic, the organisms interrupting the clade were Bacteria for two out of the four polyphyletic intein alleles. The sample size restricts building strong claims about HGT between the Halobacteria and the Bacteria. However, this claim is supported by previous evidence of gene exchange between the Bacteria and the Halobacteria (Ng et al., 2000; Khomyakova et al., 2011).
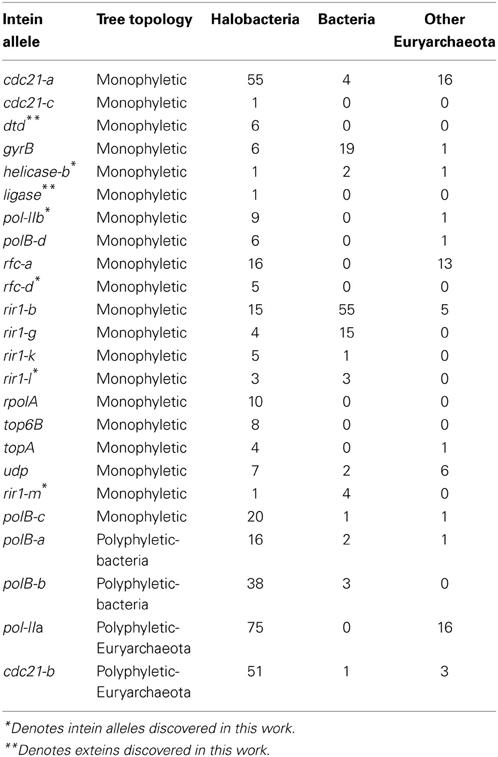
Table 2. Taxonomic distribution in each intein allele.
The tight clustering of halobacterial intein sequences and short branches between closely related strains indicate that in the majority cases inteins are inherited vertically or are transferred between closely related strains, and that successful invasion across large genetic distances is rare. Thus, intein alleles that are found in many different genera have been active for many generations, enabling invasion of many lineages, and accumulating examples of rare invasion events such as those that cross domain boundaries. Conversely, a lack of taxonomic diversity cannot be interpreted as a recent invasion as sampling limitations could be responsible for the paucity of samples in that intein allele. While many factors influence the success of intein transfer between divergent organisms, phylogenetic diversity of the organisms invaded by a particular intein allele also is a reflection of the time the intein allele has been present in a linage. Furthermore, a high density of intein sequences in a particular domain or group of genera can be used to determine the most likely reservoir for the circulating intein allele. A stacked column chart was used to quantify the representation of each of the genera in each of the intein alleles (Figure 4). Five intein alleles, cdc21b, pol-IIa, polBb, cdc21a, and rfc-d, show polarity in intein density favoring the Halobacteria (specifically Halorubrum) as the reservoir for the intein population. This is not surprising as the data indicate that the majority of intein transfer in the Halobacteria is within the class. Additionally, the diversity in five of the intein alleles, helicase-b, cdc21a, gyrB, rir1-b, and udp, suggests these intein populations may be more ancient than the others in this study as they have had time to accumulate rare, long distance transfers such that the diversity within them spans both class and domain boundaries. Interestingly, the helicase-b intein was only recently discovered in this study, though the diversity in the allele gives the impression that this intein has been around for a long time.
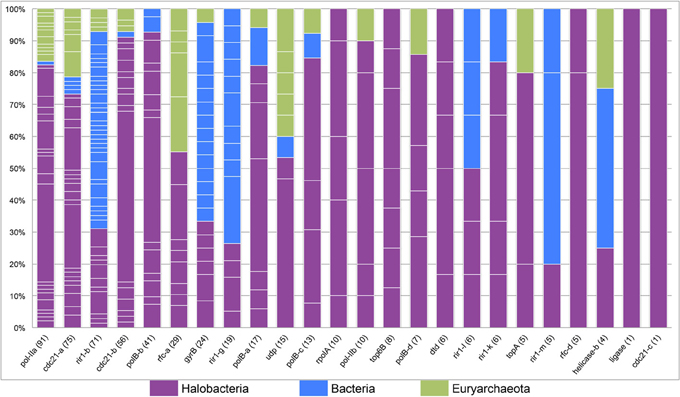
Figure 4. Phylogenetic diversity in halobacterial intein alleles. A stacked column graph depicts the representation of the Halobacteria (in purple), the Bacteria (in blue), and other Euryarchaeota (in green). Intein alleles are ordered by the number of intein sequences recovered for each allele, which is reported in parenthesis after the intein allele name on the x-axis. The number of genera for each intein allele is indicated by the number of breaks in the column (white lines) and the height of each of the fragments that make up a column indicate the proportion of sequences in that allele found in a particular genus.
Transfer of Inteins between Halobacterial and Non-Halobacterial Lineages
Not all inteins are transferred equally; the efficiency of intein invasion is affected largely by the state of the intein. The HEN domain in canonical inteins is required to induce a double strand break and the subsequent homologous repair that results in invasion (Pietrokovski, 2001). Thus, mini-inteins that have lost a functioning HEN domain are mainly transferred vertically (they may be transferred horizontally together with the host gene). If an intein containing allele has been fixed in a population, either a precise deletion of the mini intein encoding DNA could remove the intein from the population or homologous replacement by an intein-free allele transferred from outside the population. Thus, mini-inteins are maintained through strong purifying selection, because any mutation that decreases the self-splicing activity decreases the availability of the host protein (Barzel et al., 2011). The intein states were determined to infer patterns of homing in the Halobacteria. The size of inteins in each allele, along with the position of gaps in the alignment relative to the HEN domain were used as a heuristic for assigning mini-intein status. In most cases there was a clear separation in the distribution of intein lengths (at least 100 amino acids difference in length). The size of more populated intein alleles within the three genera of the Halobacteria with the largest number of available genomes, Haloarcula, Haloferax, and Halorubrum, were recorded in a matrix of intein alleles (Figure 5). Many intein alleles show a considerable size variation. This variability can be attributed to the accumulation of insertions and deletions in various lineages over time, which in some lineages leads to loss of the HEN domain. Notably, there is no variability in the size of intein sequences shared by the clusters recovered in the Bayesian analysis (orange boxes Figure 5) reinforcing the claim of ongoing gene exchange in these clusters.
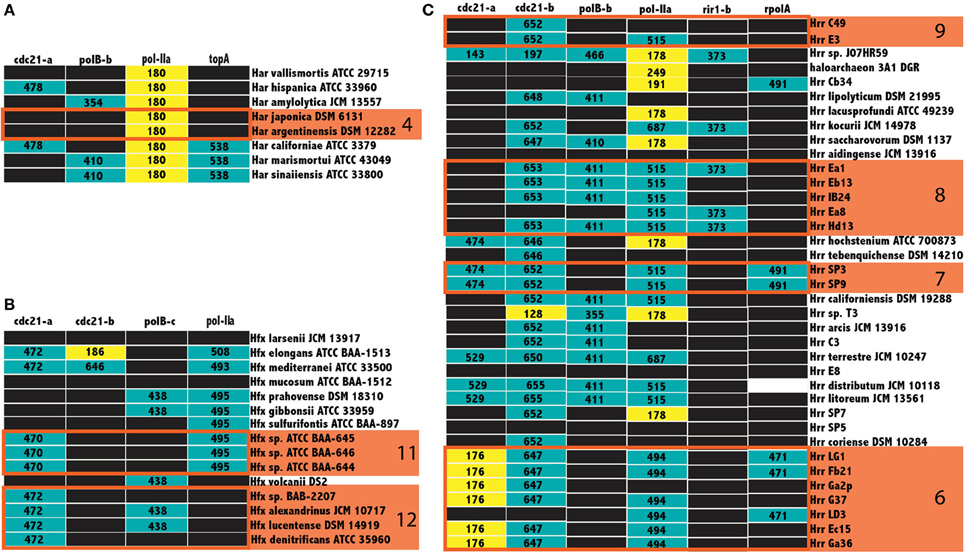
Figure 5. Intein size distributions in the Haloferax, Haloarcula, and Halorubrum. The size of inteins in the Haloarcula (A), Haloferax (B), and Halorubrum (C) are indicated in the column corresponding to the intein allele. Mini-inteins are colored yellow, large inteins are colored teal, black boxes indicate no intein, and white boxes indicate missing data, clusters from Figure 3 are indicated by numbered orange boxes. The cdc21-a, and b sequences for Halorubrum sp. J07HR59, though smaller than the rest, cannot be considered mini-inteins, as the intein sequences in these positions are not complete.
Invasion from outside the Halobacteria is one explanation for the polyphyletic topology observed in some halobacterial intein alleles. To determine when these homing events could have occurred, the state of each intein was determined and mapped onto polyphyletic intein allele trees: the results of that analysis are summarized in Table 3, with mini-inteins indicated with a star (*), and inteins that group within the Halobacteria indicated by a tilde (~) next to the name of the organism. Many of the intein sequences (5 out of 11) from taxa outside the Halobacteria that interrupt the clade are large-inteins, indicating that interactions between these taxa and the Halobacteria, though rare are ongoing (Table 3). Though the assignment of direction of transfers is extremely preliminary as limited sampling can affect the assignment of direction of transfer, there are some cases with an overwhelming signal where the majority of sequences originate from the Halobacteria, or the Bacteria in the case of rir1-m. The mixture of mini and large inteins represented in all of the intein alleles imply most of these inteins are active in the Halobacteria, and notably involve a wide distribution of taxonomic exchange partners.
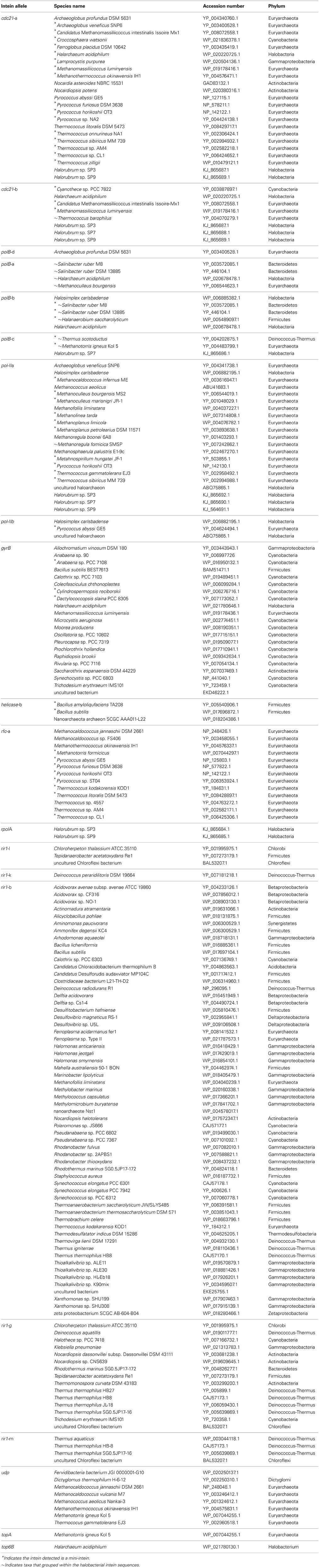
Table 3. Protein sequence identifiers for intein sequences.
Discussion
The importance of HGT throughout the tree of life demands the development of a system to monitor gene-flow within and between populations. This research provides fundamental evidence that mobile elements such as inteins can be used to uncover gene flow networks. Inteins have a unique combination of traits that make them ideal tools to study evolution in microbial populations. They have a naturally wide phylogenetic distribution, enabling detection of HGT between distantly related taxa. This is demonstrated in this work by the intein trees where the Halobacteria were polyphyletic (pol-IIa, polB-a, polB-b, and cdc21b) indicating intein transfer between the Halobacteria and the taxa that interrupt them, as well as by data from other studies where intein transfer has been detected across phyla and domains (Butler et al., 2006; Swithers et al., 2013). Inteins also have a high substitution rate relative to their extein hosts, and a propensity for accumulating insertions and deletions, which makes detection of transfers between close relatives (generally a difficult task) possible; for example, transfer within the Halorubrum clusters shown in Figure 3. Inteins can be associated with a HEN domain. If they are, they possess the ability to invade intein-free alleles following transfer; if they are not, they rely mainly on vertical inheritance together with the host gene, and the occasional transfer of the host gene. One intein allele, pol-IIa, is widely distributed in the Halobacteria and there are many examples of mini-intein sequences in this allele. These data suggest that invasion of this allele occurred early in the evolution of the Halobacteria, and that the intein may have been lost in some lineages, but retained as a mini intein in most of the genomes surveyed here. This could also be true for the cdc21-a intein; however, the distribution is not as diverse, and considerably fewer mini-inteins were detected. This is more suggestive of an intein that has been active in the Halobacteria for a long period of time, with the different intein states (empty target site, target site invaded by an intein with active HEN, target site occupied by an intein without functioning HEN; Yahara et al., 2009; Barzel et al., 2011) existing and co-existing in different halobacterial lineages.
The genomes analyzed in this work were cultured from salty water and soil samples around the world. The diverse background of the genomes may contribute to the spotty distribution of intein alleles (Figure 1). However, genomes isolated from the same location show variation as well (Figure 3) (Fullmer et al., 2014), reinforcing the notion that inteins are currently actively propagating in and being eliminated from halobacterial populations. Additionally, previous data have shown recombination occurs at a higher rate than mutation within the Halobacteria, and very little linkage between genes is detected in these genomes (Papke et al., 2004, 2007). These observations indicated gene flow as an important method for niche adaptation in these organisms. In Deep Lake, Antarctica the freezing temperatures limit the rate of replication to approximately 6 times per year and evolution in the halobacterial populations there mainly occurs through gene flow (Demaere et al., 2013). Recent whole genome comparisons revealed frequent gene transfer followed by homologous replacement of the transferred gene within the Halobacteria, hampering attempts to resolve the phylogeny within this group (Williams et al., 2012). Gene flow and recombination between populations and species make it difficult to resolve the species phylogeny among the different genera of Halobacteria (Papke et al., 2004). The use of gene concatenation in building reference trees, as exemplified by the ribosomal protein reference tree used in this work, has been pivotal in determining a branching order for the major clades of organisms, such as the Halobacteria, that participate in a large amount of recombination with close relatives. However, because genetic transfer and homologous recombination occur frequently between close relatives, the resulting phylogeny reflects both, shared ancestry and frequency of gene transfer. Therefore, determining the network of gene flow that overlays the vertical signal is important to the understanding of the evolution of these organisms. Inteins cannot penetrate the cell wall, and thus capitalize on existing gene flow in populations to efficiently invade when the opportunity presents itself. This trait can be exploited to keep track of successful homing events revealed by sequence similarity of inteins in distinct strains.
Halorubrum was the only genus in this study that had a large enough sample size to begin to uncover a signal reflecting population structure. Many of the Halorubrum genomes in this study were isolated from the same location, and this collection of genomes showed a clear signal for a structured population. Sixteen genomes from Aran-Bidgol were separated into four well-supported clusters. Three of the four clusters have branching orders identical to those in the reference tree, and the support values for those clusters could be attributed to both transfer within the group and a background phylogenetic signal or ancestral inheritance of similar intein alleles. However, only cluster 7 in the Halorubrum shares all intein alleles between all members of the cluster while the other clusters all contain intein alleles that are unique to certain members of the cluster, suggesting ongoing transfer of these inteins within the population. Additionally, three out of the twelve total clusters demonstrate unique branching orders compared to the reference tree, though only five of the clusters reflected in the reference tree have identical intein profiles. The lack of fixation for the intein alleles in the majority of clusters (seven out of twelve) indicates that a signal due to vertical inheritance may aid the formation of the clusters, but that HGT and its bias is the driving force for intein distribution. This analysis demonstrates the utility of intein sequences in distinguishing a population structure amongst genomes isolated from the same location, as demonstrated with the genomes isolated from Aran-Bidgol. These relationships are made evident through analyzing all of the signals from each of the intein alleles represented in the strains, and thus represent a collapsed view of the major gene sharing networks that have shaped the intein profiles of these strains over time. The collapsed networks indicate a higher rate of recombination within compared to between species and groups, a finding similar to the sexual outcrossing in fungal populations where inteins also thrive, as the semi-sexual lifestyle promotes intein homing (Giraldo-Perez and Goddard, 2013).
It is tempting to speculate that strains that harbor an abundance of intein alleles partake in more gene transfer than their counterparts without as many inteins; however, these two phenomena should not be expected to have a strict correlation as HGT between strains that possess only one intein each cannot produce hybrids with more than two inteins each. The number of inteins present in a group of different strains and species may be more reflective of transfers with divergent organisms than within-group transfer frequency.
The presented research demonstrates the utility of intein sequences to follow gene flow within and between populations. Improved reliability to assess the presence and activity of the HEN domain intein will provide a better distinction between vertical and horizontal inheritance of inteins. The overall utility of inteins improves as new intein alleles and new host proteins are reported, increasing the distribution of samples and improving statistical robustness of studies like the one done here. Prior to this work, nine proteins had been reported to contain inteins in the Halobacteria. This work established seven new intein alleles in the Halobacteria, including two proteins not previously reported to contain inteins. The presence of inteins is especially useful in populations where high rates of recombination and widely distributed populations may facilitate the maintenance of intein sequences over long periods of time (Gogarten and Hilario, 2006) and provide a means for distinguishing closely related partners involved in genetic transfers. The phylogenetic distribution of intein alleles, combined with the changing state within intein alleles, and the rapid substitution rate of inteins relative to the extein host sequences (Swithers et al., 2013) will provide a valuable tool to infer gene flow dynamics in and between sampled populations.
Author Contributions
Johann Peter Gogarten and Shannon M. Soucy participated in the design of this study and helped to draft the manuscript. Shannon M. Soucy performed the research and all authors contributed to data analysis. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The UConn Bioinformatics Facility provided computing resources for the analyses reported in this manuscript. The Halorubrum genomes provided by the Papke lab were sequenced in house by Andrea Makkay and Ryan Wheeler. We would like to thank them for their hard work, as well as acknowledge Dr. Elina Roine and Dennis Bamford (Helsinki University), and Dr. Antonio Ventosa (University of Sevilla) for supplying the sequenced strains. We would also like to recognize labs sequencing genomes and making them available in data repositories such as those hosted by the National Center for Biotechnology Information. This work was supported by the National Science Foundation Grant (DEB 0830024 and DEB0919290) and NASA Astrobiology: Exobiology and Evolutionary Biology Grants (NNX12AD70G and NNX13AI03G).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00299/abstract
References
Altschul, S. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andam, C. P., Williams, D., and Gogarten, J. P. (2010). Biased gene transfer mimics patterns created through shared ancestry. Proc. Natl. Acad. Sci. U.S.A. 107, 10679–10684. doi: 10.1073/pnas.1001418107
Atanasova, N. S., Roine, E., Oren, A., Bamford, D. H., and Oksanen, H. M. (2012). Global network of specific virus-host interactions in hypersaline environments. Environ. Microbiol. 14, 426–440. doi: 10.1111/j.1462-2920.2011.02603.x
Barzel, A., Obolski, U., Gogarten, J. P., Kupiec, M., and Hadany, L. (2011). Home and away- the evolutionary dynamics of homing endonucleases. BMC Evol. Biol. 11:324. doi: 10.1186/1471-2148-11-324
Butler, M. I., Gray, J., Goodwin, T. J., and Poulter, R. T. (2006). The distribution and evolutionary history of the PRP8 intein. BMC Evol. Biol. 6:42. doi: 10.1186/1471-2148-6-42
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
Demaere, M. Z., Williams, T. J., Allen, M. A., Brown, M. V., Gibson, J. A. E., Rich, J., et al. (2013). High level of intergenera gene exchange shapes the evolution of haloarchaea in an isolated Antarctic lake. Proc. Natl. Acad. Sci. U.S.A. 110, 16939–16944. doi: 10.1073/pnas.1307090110
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fullmer, M. S., Soucy, S. M., Swithers, K. S., Makkay, A. M., Wheeler, R., Ventosa, A., et al. (2014). Population and genomic analysis of the genus Halorubrum. Front. Microbiol. 5:140. doi: 10.3389/fmicb.2014.00140
Gimble, F. S., and Thorner, J. (1992). Homing of a DNA endonuclease gene by meiotic gene conversion in Saccharomyces cerevisiae. Nature 357, 301–306. doi: 10.1038/357301a0
Giraldo-Perez, P., and Goddard, M. R. (2013). A parasitic selfish gene that affects host promiscuity. Proc. Biol. Sci. 280:20131875. doi: 10.1098/rspb.2013.1875
Goddard, M. R., and Burt, A. (1999). Recurrent invasion and extinction of a selfish gene. Proc. Natl. Acad. Sci. 96, 13880–13885. doi: 10.1073/pnas.96.24.13880
Gogarten, J. P., and Hilario, E. (2006). Inteins, introns, and homing endonucleases: recent revelations about the life cycle of parasitic genetic elements. BMC Evol. Biol. 6:94. doi: 10.1186/1471-2148-6-94
Gogarten, J. P., Senejani, A. G., Zhaxybayeva, O., Olendzenski, L., and Hilario, E. (2002). Inteins: structure, function, and evolution. Annu. Rev. Microbiol. 56, 263–287. doi: 10.1146/annurev.micro.56.012302.160741
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hirata, R., Ohsumk, Y., Nakano, A., Kawasaki, H., Suzuki, K., and Anraku, Y. (1990). Molecular structure of a gene, VMA1, encoding the catalytic subunit of H(+)-translocating adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J. Biol. Chem. 265, 6726–6733.
Kane, P. M., Yamashiro, C. T., Wolczyk, D. F., Neff, N., Goebl, M., and Stevens, T. H. (1990). Protein splicing converts the yeast TFP1 gene product to the 69-kD subunit of the vacuolar H(+)-adenosine triphosphatase. Science 250, 651–657. doi: 10.1126/science.2146742
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Khomyakova, M., Bükmez, Ö., Thomas, L. K., Erb, T. J., and Berg, I. A. (2011). A methylaspartate cycle in haloarchaea. Science 331, 334–337. doi: 10.1126/science.1196544
Lang, A. S., Zhaxybayeva, O., and Beatty, J. T. (2012). Gene transfer agents: phage-like elements of genetic exchange. Nat. Rev. Microbiol. 10, 472–482. doi: 10.1038/nrmicro2802
Liu, K., Warnow, T. J., Holder, M. T., Nelesen, S. M., Yu, J., Stamatakis, A. P., et al. (2012). SATe-II: very fast and accurate simultaneous estimation of multiple sequence alignments and phylogenetic trees. Syst. Biol. 61, 90–106. doi: 10.1093/sysbio/syr095
Ng, W. V., Kennedy, S. P., Mahairas, G. G., Berquist, B., Pan, M., Shukla, H. D., et al. (2000). Genome sequence of Halobacterium species NRC-1. Proc. Natl. Acad. Sci. U.S.A. 97, 12176–12181. doi: 10.1073/pnas.190337797
Papke, R. T., and Gogarten, J. P. (2012). Ecology. How bacterial lineages emerge. Science 336, 45–46. doi: 10.1126/science.1219241
Papke, R. T., Koenig, J. E., Rodríguez-Valera, F., and Doolittle, W. F. (2004). Frequent recombination in a saltern population of halorubrum. Science 306, 1928–1929. doi: 10.1126/science.1103289
Papke, R. T., Zhaxybayeva, O., Feil, E. J., Sommerfeld, K., Muise, D., and Doolittle, W. F. (2007). Searching for species in haloarchaea. Proc. Natl. Acad. Sci. U.S.A. 104, 14092–14097. doi: 10.1073/pnas.0706358104
Perler, F. B. (2002). InBase: the Intein Database. Nucleic Acids Res. 30, 383–384. doi: 10.1093/nar/30.1.383
Perler, F. B., Olsen, G. J., and Adam, E. (1997). Compilation and analysis of intein sequences. Nucleic Acids Res. 25, 1087–1093. doi: 10.1093/nar/25.6.1087
Pietrokovski, S. (2001). Intein spread and extinction in evolution. Trends Genet. 17, 465–472. doi: 10.1016/S0168-9525(01)02365-4
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi: 10.1038/msb.2011.75
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Swithers, K. S., Senejani, A. G., Fournier, G. P., and Gogarten, J. P. (2009). Conservation of intron and intein insertion sites: implications for life histories of parasitic genetic elements. BMC Evol. Biol. 9:303. doi: 10.1186/1471-2148-9-303
Swithers, K. S., Soucy, S. M., Lasek-Nesselquist, E., Lapierre, P., and Gogarten, J. P. (2013). Distribution and evolution of the mobile vma-1b intein. Mol. Biol. Evol. 30, 2676–2687. doi: 10.1093/molbev/mst164
Williams, D., Gogarten, J. P., and Papke, R. T. (2012). Quantifying homologous replacement of loci between haloarchaeal species. Genome Biol. Evol. 4, 1223–1244. doi: 10.1093/gbe/evs098
Yahara, K., Fukuyo, M., Sasaki, A., and Kobayashi, I. (2009). Evolutionary maintenance of selfish homing endonuclease genes in the absence of horizontal transfer. Proc. Natl. Acad. Sci. U.S.A. 106, 18861–18866. doi: 10.1073/pnas.0908404106
Keywords: gene symbiosis, genome as an ecosystem, inteins, mobile genetic elements, gene flow, horizontal gene transfer, halobacteria
Citation: Soucy SM, Fullmer MS, Papke RT and Gogarten JP (2014) Inteins as indicators of gene flow in the halobacteria. Front. Microbiol. 5:299. doi: 10.3389/fmicb.2014.00299
Received: 02 January 2014; Accepted: 30 May 2014;
Published online: 26 June 2014.
Edited by:
Jesse Dillon, California State University, Long Beach, USAReviewed by:
Julie L. Meyer, University of Florida, USAKenneth Mills, College of the Holy Cross, USA
Copyright © 2014 Soucy, Fullmer, Papke and Gogarten. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johann Peter Gogarten, Microbiology Program, Department of Molecular and Cell Biology, University of Connecticut, 91 N. Eagleville Rd., Storrs, CT 06269-3125, USA e-mail: gogarten@uconn.edu; jpgogarten@gmail.com