 Julie Haendiges
Julie Haendiges Ruth Timme
Ruth Timme Marc W. Allard
Marc W. Allard Robert A. Myers1
Robert A. Myers1 Eric W. Brown
Eric W. Brown Narjol Gonzalez-Escalona
Narjol Gonzalez-Escalona- 1Department of Health and Mental Hygiene, Baltimore, MD, USA
- 2Center for Food and Applied Nutrition, Food and Drug Administration, College Park, MD, USA
Vibrio parahaemolyticus is the leading cause of foodborne illnesses in the US associated with the consumption of raw shellfish. Previous population studies of V. parahaemolyticus have used Multi-Locus Sequence Typing (MLST) or Pulsed Field Gel Electrophoresis (PFGE). Whole genome sequencing (WGS) provides a much higher level of resolution, but has been used to characterize only a few United States (US) clinical isolates. Here we report the WGS characterization of 34 genomes of V. parahaemolyticus strains that were isolated from clinical cases in the state of Maryland (MD) during 2 years (2012–2013). These 2 years saw an increase of V. parahaemolyticus cases compared to previous years. Among these MD isolates, 28% were negative for tdh and trh, 8% were tdh positive only, 11% were trh positive only, and 53% contained both genes. We compared this set of V. parahaemolyticus genomes to those of a collection of 17 archival strains from the US (10 previously sequenced strains and 7 from NCBI, collected between 1988 and 2004) and 15 international strains, isolated from geographically-diverse environmental and clinical sources (collected between 1980 and 2010). A WGS phylogenetic analysis of these strains revealed the regional outbreak strains from MD are highly diverse and yet genetically distinct from the international strains. Some MD strains caused outbreaks 2 years in a row, indicating a local source of contamination (e.g., ST631). Advances in WGS will enable this type of analysis to become routine, providing an excellent tool for improved surveillance. Databases built with phylogenetic data will help pinpoint sources of contamination in future outbreaks and contribute to faster outbreak control.
Introduction
Vibrio parahaemolyticus is a natural inhabitant of temperate and tropical coastal waters and is the leading cause of seafood-borne gastroenteritis in the United States (US) (Scallan et al., 2011). Cases of illness are usually associated with eating raw or undercooked seafood. Strains of V. parahaemolyticus carrying genes for thermostable direct hemolysin (tdh) and/or thermostable direct hemolysin-related hemolysin (trh) are considered pathogenic (Turner et al., 2013) and typically represent <1% of environmental V. parahaemolyticus strains (DePaola et al., 2000). However, this frequency may depend on the location, sample sources, and detection methods (Kaysner et al., 1990; Alam et al., 2002; Cook et al., 2002; Hervio-Heath et al., 2002; Martinez-Urtaza et al., 2008). During the last two decades V. parahaemolyticus infections and outbreaks have increased throughout the world. Most of these new cases belong to a pandemic clonal complex, known as CC3, first identified in in February of 1996 in India (Martinez-Urtaza et al., 2005; Nair et al., 2007; Gonzalez-Escalona et al., 2008; Haendiges et al., 2014).
The emergence of CC3 caused public health concerns about the potential worldwide spread of virulent V. parahaemolyticus, which previously had been restricted to particular regions. Other clonal complexes of V. parahaemolyticus, specifically CC36 and CC34, have been observed among coastal US strains (Gonzalez-Escalona et al., 2008), and strains of Sequence Type (ST) 36, the ancestral type of CC36, have also been detected on the western coast of Canada (Banerjee et al., 2014). Although infections in the US are typically caused by strains from CC36, which is endemic to the West Coast (Abbott et al., 1989; Gonzalez-Escalona et al., 2008), a Maryland outbreak in August 2012 (Haendiges et al., 2014) was caused by strains belonging to CC3.
Foodborne illnesses due to V. parahaemolyticus are uncommonly reported in the US and in the state of Maryland. Forty-six cases of V. parahaemolyticus gastroenteritis associated illnesses were reported between 2012 and 2013. From those cases, 34 strains were isolated. During the summer of 2012 a multistate outbreak associated with harvested shellfish was reported on the East Coast of the US (Newton et al., 2014), caused by V. parahaemolyticus strains belonging to CC36 (Martinez-Urtaza et al., 2013), subsequently shown to be ST36. That outbreak affected 28 persons in nine states (Newton et al., 2014), and the isolated strains were of the O4:K12 or O4:K (unknown) serotypes. Usually strains from this serotype/ST isolated in the US are from the West Coast (Gonzalez-Escalona et al., 2008; Newton et al., 2014). Another outbreak, involving 104 cases, occurred in the summer of 2013 affecting people in 13 US states and were caused by the same ST36 (Newton et al., 2014). An outbreak in Spain, during the summer of 2012, was attributed to strains that were ST36 as well (Martinez-Urtaza et al., 2013), associated with cooked seafood that had been cooled with untreated local seawater (Martinez-Urtaza et al., 2013); contrarily to the infections in the US which are often linked to the consumption of contaminated oysters (Newton et al., 2014).
The numbers of V. parahaemolyticus infections appears to have increased approximately 4 times in US in the last decade (Mead et al., 1999; Scallan et al., 2011). Scallan et al. (2011) reported that the domestically acquired foodborne average for V. parahaemolyticus infections annually were around 35,000 in the period 2000–2008, while Mead et al. (1999) reported around 8000 infections annually in the period 1992–1997. The increasing number of infections could have detrimental impacts on public health and economic growth, particularly in regions where seafood harvest and consumption are important. However, active surveillance depends upon having effective ways to identify and monitor the nature or identity of the V. parahaemolyticus strains causing outbreaks.
Various typing methods have been used to distinguish bacterial isolates for epidemiological investigations (Foxman et al., 2005). Pulsed Field Gel Electrophoresis (PFGE) has been a favored method for genotyping V. parahaemolyticus isolates (Marshall et al., 1999) and it is considered the “gold standard” for outbreak investigations (Parsons et al., 2007; Wagley et al., 2008; Dauros et al., 2011; Banerjee et al., 2014; Haendiges et al., 2014; Ma et al., 2014; Pazhani et al., 2014). While useful for short-term epidemiology, PFGE does not provide details of the genetic relationships among isolates (e.g., evolutionary relationships) (Foxman et al., 2005). Another common method for characterizing V. parahaemolyticus isolates is Multilocus Sequence Typing (MLST). This method is based on direct sequence analysis of housekeeping genes, making MLST better for long term evolutionary studies (Gonzalez-Escalona et al., 2008; Harth et al., 2009; Yan et al., 2011; Gavilan et al., 2013; Martinez-Urtaza et al., 2013; Turner et al., 2013). A public database was established to archive V. parahaemolyticus sequences (http://pubmlst.org/vparahaemolyticus) (Gonzalez-Escalona et al., 2008).
In the last 5 years, scientists have begun using genomic techniques to analyze historical collections of pathogens and outbreak isolates, providing new insights for outbreak investigations. Whole genome sequencing (WGS) and WGS-SNP data analyses allow us to better understand population dynamics and mechanisms contributing to increased virulence among foodborne bacterial pathogens, including outbreaks of Salmonella Montevideo in 2010 (Bakker et al., 2011; Allard et al., 2012), Vibrio cholerae in Haiti in 2010 (Chin et al., 2011), E. coli O104:H4 (in Germany in 2011, Rasko et al., 2011) and Salmonella Enteritidis in the US in 2010 (Allard et al., 2013). In the current project, we utilize a similar approach to explore the diversity and relationships among V. parahaemolyticus isolates causing outbreaks in Maryland.
Specifically, in order to better understand potential changes in V. parahaemolyticus populations in the state of Maryland and to investigate whether the spikes in cases during the summers of 2012 and 2013 were related to this recently introduced East Coast clone, ST36, we used WGS to compare genomes from those outbreaks with those of the other V. parahaemolyticus strains causing illnesses in recent years (2012–2013). We began by sequencing the 34 strains from MD, then 10 additional historical V. parahaemolyticus strains from different sources (clinical and environmental) from the East and West Coasts of US were also sequenced and used for phylogenetic comparative analysis. Additional available genomes from global V. parahaemolyticus strains were also used in the phylogenetic comparative analysis.
Materials and Methods
Bacterial Strains and Media
The V. parahaemolyticus isolates sequenced for this project are listed, along with their assigned CFSAN numbers, in Tables 1, 2. Table 1 lists the 34 clinical isolates from Maryland, and Table 2 lists the national and international V. parahaemolyticus isolates used for comparison. All isolates were retrieved from storage (−80°C freezer), transferred to Luria-Bertani (LB) medium with 3% NaCl and incubated at 250 rpm at 37°C.
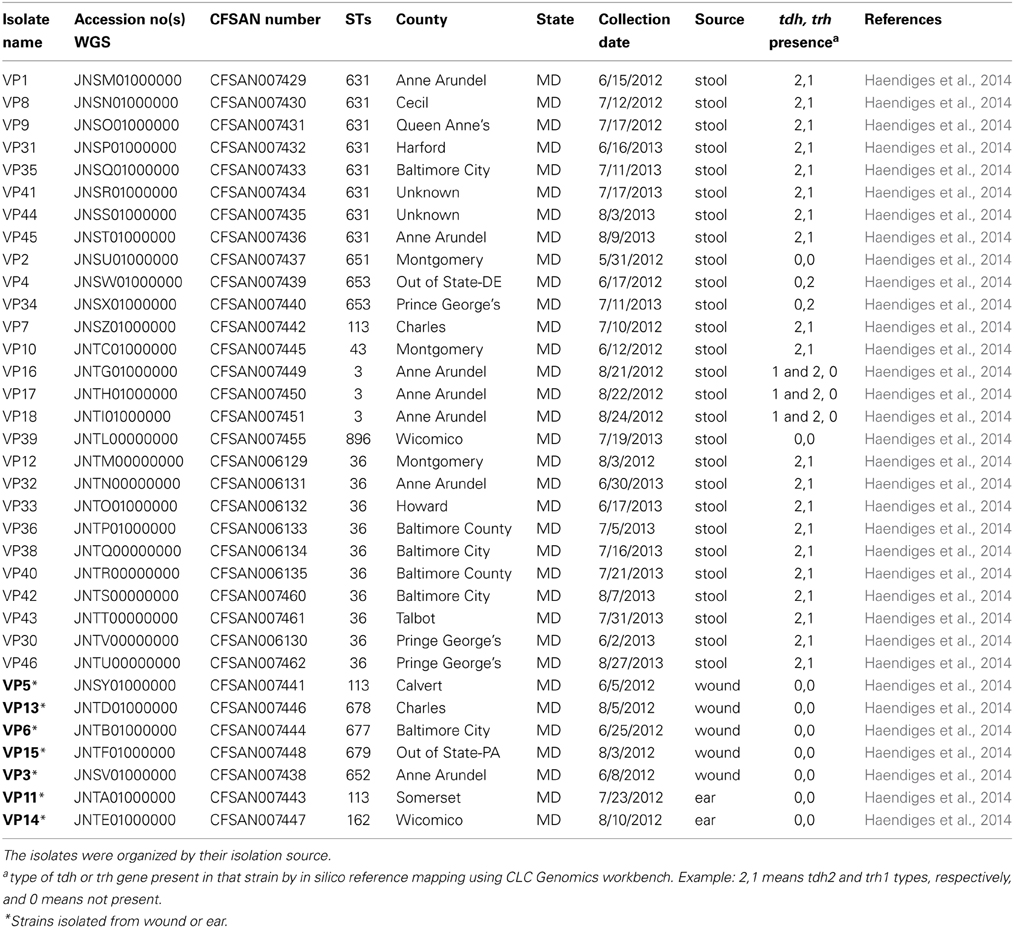
Table 1. Clinical V. parahaemolyticus isolates sequenced in this study and source information.
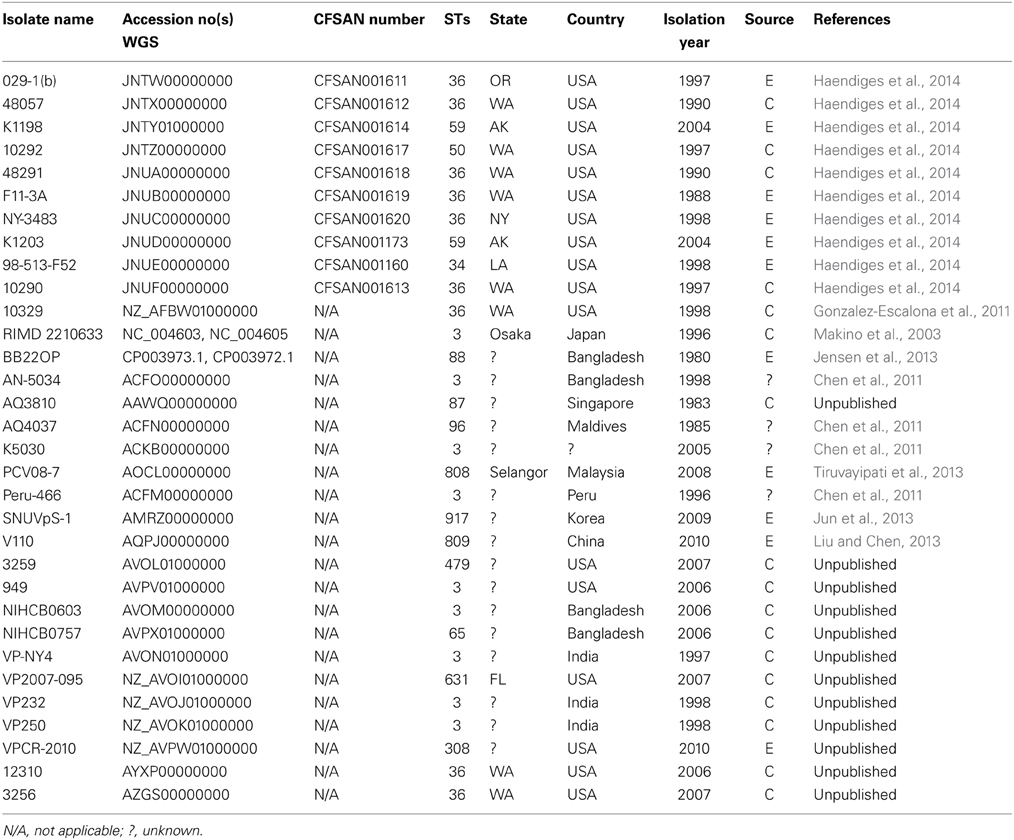
Table 2. Locally and globally diverse V. parahaemolyticus isolates genomes used for phylogenomic analyses available at the National Center for Biotechnology Information.
DNA Extraction and Quantification
Genomic DNA from each isolate was isolated from overnight cultures using the DNeasy Blood and Tissue Kit (QIAGEN, Valencia, CA). The quality of the DNA was checked using a NanoDrop 1000 (Thermo Scientific, Rockford, IL) and the concentration was determined using a Qubit double-stranded DNA HS assay kit and a Qubit 2.0 fluorometer (Life Technologies, Grand Island, NY), according to each manufacturer's instructions.
Whole Genome Sequencing, Contig Assembly, and Annotation
The genomes of the historical isolates were sequenced using 200 bp reads chemistry and the 36 MD outbreak isolates were sequenced using 300 bp reads chemistry, using an Ion Torrent (Thermo Scientific) sequencer, according to manufacturer's instructions, at approximately 20X coverage (Haendiges et al., 2014). Genomic sequence contigs were de novo assembled using CLC Genomics Workbench (QIAGEN). These draft genomes were annotated using the NCBI Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP, http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html) (Klimke et al., 2009).
In silico MLST Phylogenetic Analysis
The initial analysis and identification of the isolates was performed using an in silico V. parahaemolyticus MLST approach, based on information available at the V. parahaemolyticus MLST website (http://pubmlst.org/vparahaemolyticus). Seven loci (dnaE, gyrB, recA, dtdS, pntA, pyrC, and tnaA), described previously for V. parahaemolyticus (Gonzalez-Escalona et al., 2008), were used for MLST analysis. The same V. parahaemolyticus MLST database was also used to assign numbers for alleles and STs.
Assignment to Clonal Complexes
The program goeBURST v1.2.1 was used to identify CCs among the isolates (http://goeburst.phyloviz.net) (Francisco et al., 2009). To qualify as members of the same CC, isolates must share at least 5 of the 7 alleles used in the V. parahaemolyticus MLST scheme (Feil et al., 2004). Those STs that differ by two alleles are considered double locus variants (DLV), and those which differ at only a single locus are single locus variants (SLV).
Phylogenomic Analysis and Targeted SNP Analysis
We performed two phylogenetic analyses using the software kSNP v2 (Gardner and Hall, 2013) (kmer length = 31). The first one included a broad SNP analysis of all the genomes included in the analysis, including local, national and international isolates, and was performed to provide an evolutionary context for all the V. parahaemolyticus isolates used for the analysis. The second was conducted to provide a more detailed phylogenetic relationship among the isolates found in each individual cluster and were targeted for more refined SNP analyses. Our goal was to recover variations unique to each respective lineage. To do this, we created a matrix that included only core SNPs (i.e., SNPs that were present in all genomes) for each lineage, using the reference-free SNP- finding program, kSNP v2 (Gardner and Hall, 2013) (kmer length = 31). Maximum-likelihood phylogenies for each core SNP matrix were then constructed within the kSNP, labeled with the number of unique SNPs present in every descendant of that node. Draft assemblies were used to detect SNPs using the program, kSNP. Filtering was applied to eliminate detectable horizontal gene transfer such as mobile elements and paralogs, but we were not able to filter for genome recombination. Although important, reconstructing the reticulate evolutionary history of V. parahaemolyticus was outside the scope of this research effort.
Using the kSNP approach offers several advantages. First, the kSNP approach to gathering reference-free SNPs for downstream phylogenetic analysis has been effective for microbial-scale phylogenomic analyses (Sahl et al., 2013; Timme et al., 2013), Second, these kSNP analyses are not as vulnerable to assembly errors as other methods because the putative SNPs are extracted from kmers (in our present analysis, one SNP per 25 mer), effectively eliminating any influence of assembly bias. Third, the algorithm behind SNP identification is especially conservative about common pyrosequencing errors, such as the homopolymeric runs commonly observed in data from 454 and Ion Torrent sequencing procedures: if an insertion or deletion is present in a region, the resulting 25-mer will no longer match the other homologous kmer regions, thus that SNP will be missing in that taxon. Additionally, kSNP omits kmers where all SNPs are in the center of a homopolymer repeat (Gardner and Slezak, 2010). These features make our approach conservative and prevent repeat regions from becoming a factor in our analyses: any k-mers comprising such regions will fail to be unique, and therefore will not be used.
Nucleotide Sequence Accession Numbers
The draft genome sequences for all 46 V. parahaemolyticus strains in our analyses are available in GenBank under the accession numbers listed in Tables 1, 2.
Results and Discussion
Maryland V. parahaemolyticus Outbreak Strains and in silico MLST
Our MLST analyses provide a broad look at the diversity of V. parahaemolyticus clinical samples collected during 2010–2013 in the state of Maryland. These 34 V. parahaemolyticus strains isolated from patients—stool (80%), ear (6%), and wound (14%) were sequenced using Ion Torrent. A quarter of these strains (25%) came from an outbreak in the summer of 2013. All the reported cases occurred during the warmer months between June and August (Table 1).
In silico MLST (Gonzalez-Escalona et al., 2008) identified 13 different STs, showing that the sporadic outbreaks in Maryland were caused by highly diverse V. parahaemolyticus strains. Seven STs were novel (STs: 651, 652, 653, 677, 678, 679, and 896) and represented strains that have only been found (so far) in Maryland (Table 1). The most prevalent STs were ST36 (28%), ST631 (22%), ST3 (8%), ST113 (8%), and ST653 (6%). The rest of the STs were represented by a single strain.
Some of the identified STs were isolated from more than one location, more than one person, and in different years (Table 1 and Figure 1); most of the illnesses associated with these isolates occurred in MD coastal counties (Figure 1). Interestingly, most of the cases on 2013 (88%) were caused by ST631 (31%) and ST36 (56%). This suggests these two STs are either more prevalent and/or have replaced the endemic V. parahaemolyticus populations in this region. Additional surveys of pathogenic V. parahaemolyticus in MD could confirm or clarify our theories.
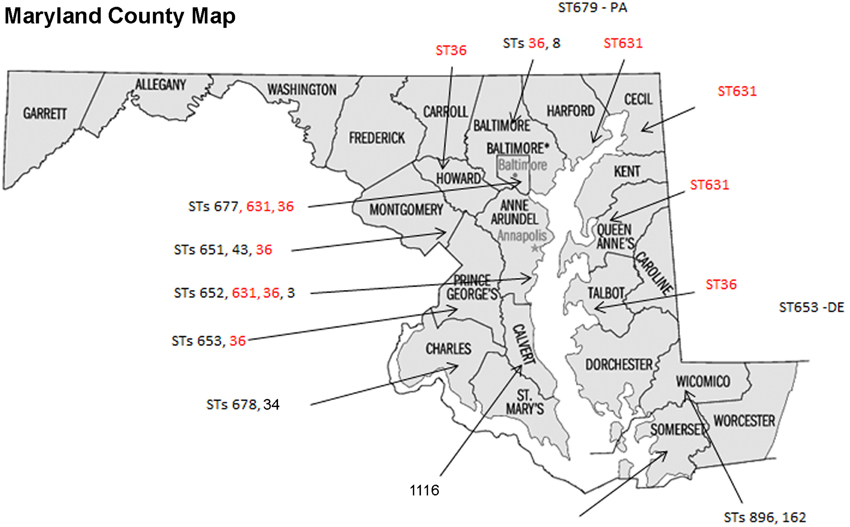
Figure 1. Sequence types (STs) isolation locations on the state of Maryland, USA. STs found in more than one location are in red fonts.
Our eBURST analysis (goeBURST) showed that the MD isolates do not form any clonal complexes and were not related to each other (Figure 2). Therefore, the outbreaks during this period in MD were caused by V. parahaemolyticus belonging to different populations. However, some findings suggested we had reached the limits of the MLST method: the ST36 observed in MD could not be distinguished from ST36 from the West Coast by this method. It is at this point we must turn to the higher discriminatory power of WGS analyses.

Figure 2. goeBURST population snapshot using the in silico MLST analysis of the strains sequenced in this study indicating the absence of any clonal complex and the relative frequency of each ST. Font sizes varied according to the frequency of the ST.
Phylogenomic Analysis
Using 7 V. parahaemolyticus genomes available from NCBI (www.ncbi.nlm.nih.gov/genome/?term=vibrio parahaemolyticus) and the 10 genomes from historical US V. parahaemolyticus strains (Table 2), we determined the phylogenetic relationships among the MD strains using a whole-genome SNP analysis (Figure 3). As can be seen from Figure 3, V. parahaemolyticus is a highly diverse microorganism, documented by the high number of SNPs defining each branch, and a low-resolution tree is obtained when using this entire genome database. Nevertheless, a similar clustering to the one achieved by MLST analysis can still be observed for the Maryland V. parahaemolyticus strains, where the same STs were also grouping at the genome level. Five clusters of related strains were identified, cluster I (ST36), cluster II (ST653), cluster III (ST113), cluster IV (ST631), and cluster V (ST3).
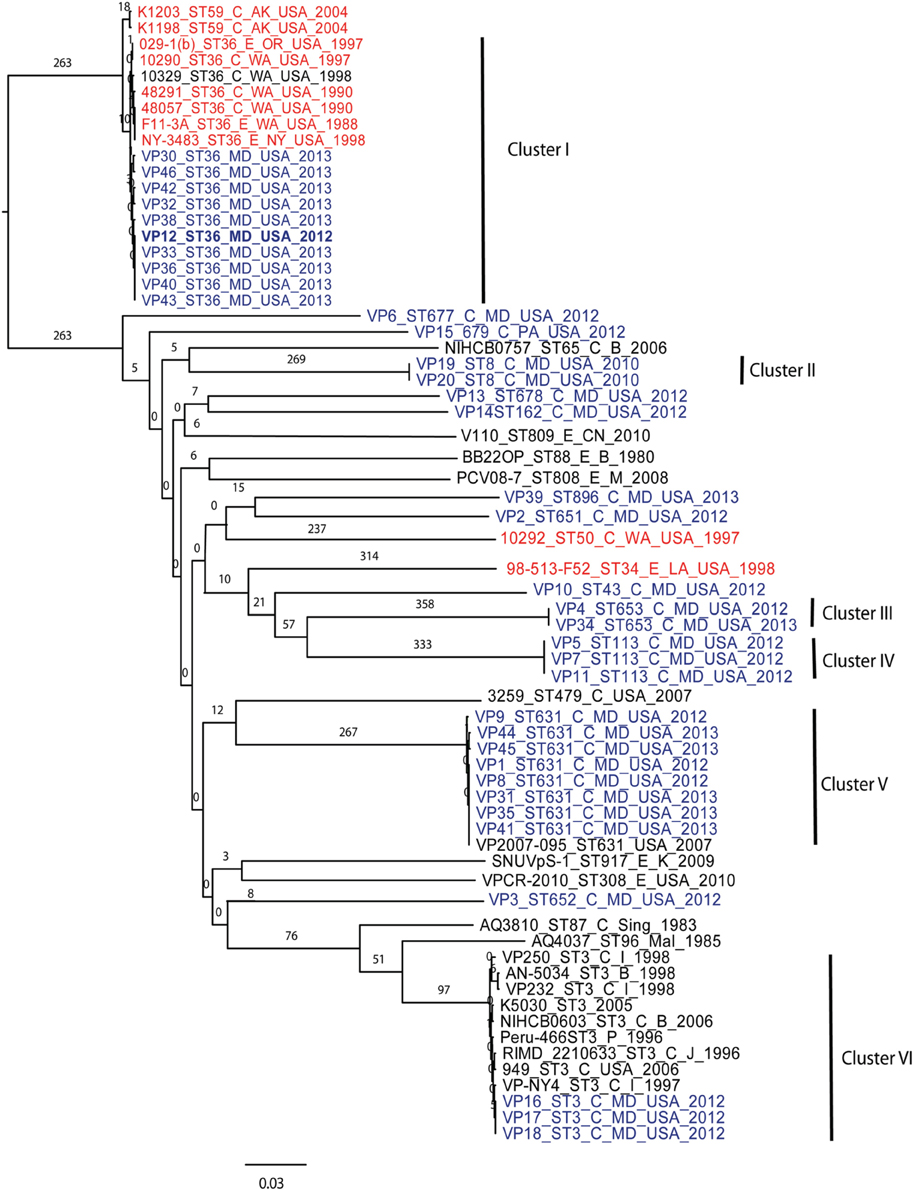
Figure 3. Phylogenetic clustering analysis of the outbreak strains of V. parahaemolyticus, Maryland, 2012–2013, by whole-genome SNP analysis. A k-SNP (Timme et al., 2013) analysis was performed. The maximum-likelihood tree shows V. parahaemolyticus MD outbreak isolates (in blue fonts), historical US strains sequenced in this study (in red fonts) and other related but unassociated V. parahaemolyticus strains (retrieved from NCBI, black fonts). Maximum-likelihood phylogeny was constructed from a 46,963 SNP matrix. Samples are annotated as follows: strain name, sequence type (ST), source (C, clinical; E, environmental), country of isolation (B, Bangladesh; CN, China; M, Malaysia; Mal, Maldives; USA, United States of America; Sing, Singapore; I, India, P, Peru; and K, Korea), and year when the samples were collected. All gene alignments of the SNPs observed in the entire dataset as well as in each individual cluster are available on request from the authors.
Besides Maryland ST36 strains (which appear in blue text on the Figure 3), Cluster I also contained strains that were isolated on the West Coast (1988–2004) (represented by the red text in Figure 3) and are part of CC36 (Gonzalez-Escalona et al., 2008). In this broad phylogeny, little differentiation can be observed among these ST36 strains. The same can be observed for most other clusters. But this broad phylogeny does allow us to confirm the diversity of populations causing the outbreaks in MD during that 2 year period.
Given these patterns, we then used a cluster-based analysis that enabled us to identify relationships among strains that would not have been found by examining the entire dataset (Figure 4). Each individual cluster (when more than three strains) was analyzed separately. This type of analysis helped us to clearly distinguish strains in ST36 from MD (2012–2013) from those originating on the West Coast (1988–1998). These groups differed by at least 150 SNPs (Figure 4A). Interestingly, MD strains were similar to ST36, also from the West Coast, of US but from a later time point: 2006–2007 (strains 12310 and 3256). This clearly indicates that ST36 strains have evolved in the last decade and the new ST36 strains from the West Coast are more similar to the ST36 from MD (2012–2013) than the historical ST36 from the West Coast. Also, it seems that the outbreak in 2013 in MD of ST36 was also composed by strains from at least three different subgroups within lineage ST36. One subgroup contained the strain responsible for the cases in 2012 and most 2013 cases (VP43, 12, 42, 33, 36, 40, and 38), and the other two subgroups were composed of one (VP32) and two strains (VP30 and VP46). These differences in subgroups within lineage ST36 could have arisen from adaptations to surviving in different oysters/habitats and/or to the introduction of new variants.
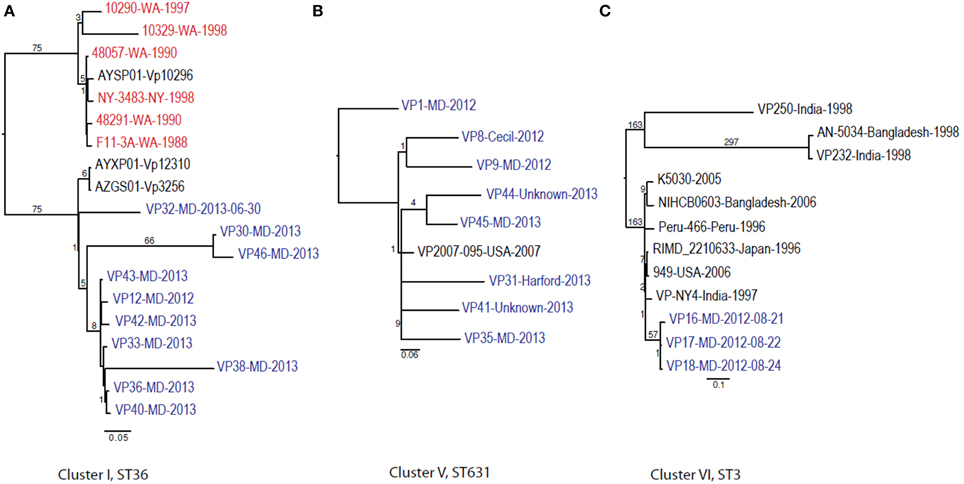
Figure 4. Maximum-likelihood trees for each individual V. parahaemolyticus outbreak cluster identified in Figure 3 and other related (if any) but unassociated V. parahaemolyticus strains (retrieved from NCBI). Maximum-likelihood phylogeny was constructed from a 347, 931, and 88 core SNP matrix for clusters I, V, and VI, respectively. (A) Cluster I, (B) cluster V, and (C) cluster VI. Samples are annotated as in Figure 3. All gene alignments of the SNPs observed in the entire dataset as well as in each individual cluster are available on request from the authors.
This is why WGS-SNP analyses provide clearer insights than earlier methods: many of these strains were unable to be distinguished from one another by MLST and PFGE analysis. The general understanding of V. parahaemolyticus evolution suggests that the typical mechanism is recombination, with a recombination ratio estimated at 2.5:1 and 8.8:1 by allele and site, respectively (Gonzalez-Escalona et al., 2008). Example of this are observed among strains belonging to CC345, where all known variants (SLV) of the ancestral ST type of the CC (ST345) have arisen by recombination, mainly in the recA allele (González-Escalona et al., in press).
Cluster II and Cluster III were composed by two and three strains each, respectively; no other genomes have been reported for those STs, therefore we could not use a targeted SNP analysis to further analyze those strains. Nevertheless, these clustered with other US strains (Figure 3) indicating that they are probably strains endemic to the US. Strains belonging to ST113 were isolated from wound, ear, and stool in MD, suggesting that Clusters II and III are composed of local strains. ST113 was first described for a V. parahaemolyticus strain isolated from oysters in Louisiana state in 2007 (Johnson et al., 2009).
The second most numerous ST observed among the MD outbreak strains was Cluster IV (ST631): these strains were highly similar, suggesting that they came from the same region 2 years in a row. In our analyses, the Cluster IV MD strains also clustered with a strain isolated in FL in 2007 (Figure 4B). If we fit this data together with the fact that ST631 had initially been reported through a strain isolated in Prince Edward Island (Canada) in 2009 (Banerjee et al., 2014), we have reason to believe that ST631 is a regional ST, endemic to the East Coast.
Finally, Cluster V (ST3), included the MD 3 strains recovered from an outbreak that caused six illnesses during the summer of 2012 (Haendiges et al., 2014). These strains had previously been characterized by Haendiges et al. (2014), using a different phylogenetic tool (BIGSdb genome comparator tool). Due to homopolymer errors (inherent to Ion Torrent sequencing), no further resolution could be observed among strains belonging to Cluster V, so we could not determine whether the strains causing the outbreak were the same or not. The other analyses performed in our study were not impacted by such errors, since the miscalls were pruned from the analysis and only true high quality SNPs were included. With this targeted SNP based analysis, these strains were undistinguishable and can be clearly separated from other ST3 strain which genome were available at NCBI by at least 57 unique SNPs and can conclusively be called as being undistinguishable and probably the same strain and coming from the same source (Figure 4C). Therefore, this targeted SNP analysis can be used for outbreak detection caused by a specific V. parahaemolyticus strain.
Presence and Absence of tdh and trh Genes and their Relation to Etiology
There are five different subtypes of tdh genes that differ by sequence (tdh1 through tdh5); these subtypes share 96–98% identity (Nishibuchi and Kaper, 1990). In the case of trh genes there are two recognized subtypes (trh1 and 2), which share 84% identity (Kishishita et al., 1992). Most of the clinical MD V. parahaemolyticus analyzed carried tdh and/or trh genes (72%) (Table 1). Ten isolates (28%) were negative for tdh and trh, three (8%) were only tdh positive, four (11%) were only trh positive, and the remaining 19 (53%) isolates contained both genes.
It was unusual to find that 28% of these clinical V. parahaemolyticus did not carry tdh or trh, since these strains are usually considered non-pathogenic (Nishibuchi and Kaper, 1995; Blackstone et al., 2003; DePaola et al., 2003). There have been some recent reports of V. parahaemolyticus without either of these two pathogenicity markers causing sporadic cases of illness (Jones et al., 2012; Banerjee et al., 2014) and having these 10 genomes may help researchers discover what makes these strains pathogenic. Jones et al. (2012) reported that 27% of their clinical isolates collected across US from July 2006 to November 2007 were tdh/trh negative while Banerjee et al. (2014) reported the same genotype for 4% of the clinical V. parahaemolyticus collected in Canada during 2000–2009. Furthermore, the majority of the tdh/trh negative strains (70%) were isolated from either wound or ear infections (Table 1). This further indicates that these two markers may not be necessary for human illness to occur (Broberg et al., 2011), although it is possible that these patients had some kind of immune deficiencies that could make them more prone to infections (chronic liver disease), as observed for V. vulnificus (Kim et al., 2014). The actual percentage of tdh/trh negative clinical strains is currently hard to estimate and might depend on several factors including sample size, V. parahaemolyticus endemic populations, type of infection, as well as errors caused by sporadic outbreaks that often go unnoticed or underreported (Scallan et al., 2011).
Most of the tdh and/or trh positive strains carried tdh2 and trh1 combination. Most clinical US strains carry both tdh/trh (Okuda et al., 1997a; Jones et al., 2012; Paranjpye et al., 2012; Turner et al., 2013) while pandemic strains carried tdh1 and tdh2 (Okuda et al., 1997b; Matsumoto et al., 2000; Makino et al., 2003; Gonzalez-Escalona et al., 2005). Among the MD V. parahaemolyticus strains, only three were both tdh1 and two positive and these were identified as belonging to the pandemic clonal complex and were ST3 (Haendiges et al., 2014). Only two MD strains carried exclusively trh2. A study of environmental V. parahaemolyticus in Norway also identified strains containing only the trh gene that were also trh2, however those strains belonged different STs than the one observed in this study (Ellingsen et al., 2013). The availability of these genomes would help in future analysis in determining which other genes besides tdh and trh are necessary for pathogenesis in this marine bacterium.
Distribution of T3SS and other Genomic Regions in MD V. parahaemolyticus Clinical Strains
We performed an in silico analysis of each strain's genome using CLC Genome Workbench software (QIAGEN) for 24 known regions described for pandemic strains, including both T3SS, T3SS1 and 2α (Boyd et al., 2008) and well as for T3SS2β (Park et al., 2000), containing the ure genes and trh gene and described previously for strains carrying both tdh and trh genes (Supplementary Table 1). All strains carried NK, T3SS-1, Osmotolerance (chromosome I), Gametolysin, Osmotolerance (chromosome II), CPS, Type I secretion, Type I pilus, Multidrug efflux, and Ferric uptake. Most strains lacked all the pathogenicity islands described for the pandemic strain RIMD2210633 (VPaI1-7). Four other MD strains carried either VpaI1 (VP10) or VpaI2 (VP2, and VP14), although carrying a sequence different from that of the pandemic strain. Of the nine MD strains lacking tdh or trh gene, six carried a T6SS different from the pandemic, for the presence/absent of the other elements or PIs were very similar to the strains carrying tdh, trh, or tdh/trh strains. Seven of these strains were isolated from wound or ear infections. Since tdh/trh and T3SS2s were described as needed for producing infections at the intestinal level [because tdh/trh causes intestinal fluid secretion as well as cytotoxicity in a variety of cell types (Raimondi et al., 2000) while TTSS2 has a role in enterotoxicity (Park et al., 2004)], then these strains could have caused infections by a rather opportunistic way than by an actual infection as observed for gastroenteritis. These strains still possess TSS1 which is known for being involved in the cytotoxicity (Park et al., 2004). A more detailed study could reveal additional elements we did were not able to assess in this study; such analyses will certainly shed more light on the infection mechanisms of these unusual V. parahaemolyticus strains.
Conclusions
Our study used WGS to provide a high resolution investigation of genetic diversity and relationships of clinical V. parahaemolyticus strains isolated in the state of Maryland during 2012–2013 period in comparison to those isolated from other geographical locations. Having an archive of V. parahaemolyticus genomes allowed us to perform a phylogenetic analysis of strains and revealed that strains causing outbreaks in MD were highly diverse, genetically distinct, and clearly different from other regional and global strains. Despite the geographic breadth of our strains, we are aware that our study captured only a fraction of the actual number of V. parahaemolyticus strains causing illness in the state of Maryland, due in part to the sporadic nature of outbreaks, the low number of strains recovered per outbreak, and the fact that many outbreaks are never investigated (Scallan et al., 2011).
We suggest using a similar approach during future outbreaks to analyze the identity of suspect V. parahaemolyticus strains and their relationship to strains causing previous outbreaks. Our WGS analysis of V. parahaemolyticus strains highlights the need for a genome database for V. parahaemolyticus to improve traceback and responses to outbreaks. We have created a new BioProject (PRJNA245882) at NCBI, spearheaded by FDA-CFSAN and the state of Maryland's DHMH, in which we are going to continually deposit new available genomes of V. parahaemolyticus causing outbreaks that would improve detection of new outbreak strains, track the emergence of new clonal strains in geographical regions where these strains are not endemic, and will be used in source tracking. We suggest and encourage the scientific community to deposit further V. parahaemolyticus genomes from their respective countries in centralize databases, such as NCBI, in order to improve source tracking.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by the FDA Foods Program Intramural Funds. The authors thank Dr. Lili Fox Vélez for her editorial assistance on this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2015.00125/abstract
References
Abbott, S. L., Powers, C., Kaysner, C. A., Takeda, Y., Ishibashi, M., Joseph, S. W., et al. (1989). Emergence of a restricted bioserovar of Vibrio parahaemolyticus as the predominant cause of Vibrio-associated gastroenteritis on the West Coast of the United States and Mexico. J. Clin. Microbiol. 27, 2891–2893.
Alam, M. J., Tomochika, K. I., Miyoshi, S. I., and Shinoda, S. (2002). Environmental investigation of potentially pathogenic Vibrio parahaemolyticus in the Seto-Inland Sea, Japan. FEMS Microbiol. Lett. 208, 83–87. doi: 10.1111/j.1574-6968.2002.tb11064.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Allard, M. W., Luo, Y., Strain, E., Li, C., Keys, C. E., Son, I., et al. (2012). High resolution clustering of Salmonella enterica serovar Montevideo strains using a next-generation sequencing approach. BMC Genomics 13:32. doi: 10.1186/1471-2164-13-32
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Allard, M. W., Luo, Y., Strain, E., Pettengill, J., Timme, R., Wang, C., et al. (2013). On the evolutionary history, population genetics and diversity among isolates of Salmonella Enteritidis PFGE pattern JEGX01.0004. PLoS ONE 8:e55254. doi: 10.1371/journal.pone.0055254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bakker, H. C., Switt, A. I., Cummings, C. A., Hoelzer, K., Degoricija, L., Rodriguez-Rivera, L. D., et al. (2011). A whole-genome single nucleotide polymorphism-based approach to trace and identify outbreaks linked to a common Salmonella enterica subsp. enterica serovar Montevideo pulsed-field gel electrophoresis type. Appl. Environ. Microbiol. 77, 8648–8655. doi: 10.1128/AEM.06538-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Banerjee, S. K., Kearney, A. K., Nadon, C. A., Peterson, C. L., Tyler, K., Bakouche, L., et al. (2014). Phenotypic and genotypic characterization of Canadian clinical isolates of Vibrio parahaemolyticus collected from 2000 to 2009. J. Clin. Microbiol. 52, 1081–1088. doi: 10.1128/JCM.03047-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blackstone, G. M., Nordstrom, J. L., Vickery, M. C., Bowen, M. D., Meyer, R. F., and DePaola, A. (2003). Detection of pathogenic Vibrio parahaemolyticus in oyster enrichments by real time PCR. J. Microbiol. Methods 53, 149–155. doi: 10.1016/S0167-7012(03)00020-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boyd, E. F., Cohen, A. L., Naughton, L. M., Ussery, D. W., Binnewies, T. T., Stine, O. C., et al. (2008). Molecular analysis of the emergence of pandemic Vibrio parahaemolyticus. BMC Microbiol. 8:110. doi: 10.1186/1471-2180-8-110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Broberg, C. A., Calder, T. J., and Orth, K. (2011). Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect. 13, 992–1001. doi: 10.1016/j.micinf.2011.06.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y., Stine, O. C., Badger, J. H., Gil, A. I., Nair, G. B., Nishibuchi, M., et al. (2011). Comparative genomic analysis of Vibrio parahaemolyticus: serotype conversion and virulence. BMC Genomics 12:294. doi: 10.1186/1471-2164-12-294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chin, C.-S., Sorenson, J., Harris, J. B., Robins, W. P., Charles, R. C., Jean-Charles, R. R., et al. (2011). The Origin of the Haitian Cholera Outbreak Strain. N. Engl. J. Med. 364, 33–42. doi: 10.1056/NEJMoa1012928
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cook, D. W., Bowers, J. C., and DePaola, A. (2002). Density of total and pathogenic (tdh+) Vibrio parahaemolyticus in Atlantic and Gulf coast molluscan shellfish at harvest. J. Food Prot. 65, 1873–1880.
Dauros, P., Bello, H., Dominguez, M., Hormazabal, J. C., and Gonzalez, G. (2011). Characterization of Vibrio parahaemolyticus strains isolated in Chile in 2005 and in 2007. J. Infect. Dev. Ctries. 5, 502–510.
DePaola, A., Kaysner, C. A., Bowers, J., and Cook, D. W. (2000). Environmental investigations of Vibrio parahaemolyticus in oysters after outbreaks in Washington, Texas, and New York (1997 and 1998). Appl. Environ. Microbiol. 66, 4649–4654. doi: 10.1128/AEM.66.11.4649-4654.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DePaola, A., Nordstrom, J. L., Bowers, J. C., Wells, J. G., and Cook, D. W. (2003). Seasonal abundance of total and pathogenic Vibrio parahaemolyticus in Alabama oysters. Appl. Environ. Microbiol. 69, 1521–1526. doi: 10.1128/AEM.69.3.1521-1526.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ellingsen, A. B., Olsen, J. S., Granum, P. E., Rorvik, L. M., and Gonzalez-Escalona, N. (2013). Genetic characterization of trh positive Vibrio spp. isolated from Norway. Front. Cell. Infect. Microbiol. 3:107. doi: 10.3389/fcimb.2013.00107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Foxman, B., Zhang, L., Koopman, J. S., Manning, S. D., and Marrs, C. F. (2005). Choosing an appropriate bacterial typing technique for epidemiologic studies. Epidemiol. Perspect. Innov. 2:10. doi: 10.1186/1742-5573-2-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Francisco, A. P., Bugalho, M., Ramirez, M., and Carrico, J. A. (2009). Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics 10:152. doi: 10.1186/1471-2105-10-152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gardner, S., and Slezak, T. (2010). Scalable SNP analyses of 100+ bacterial or viral genomes. J. Forensic Res. 1, 107. doi: 10.4172/2157-7145.1000107
Gardner, S. N., and Hall, B. G. (2013). When whole-genome alignments just won't work: kSNP v2 software for alignment-free SNP discovery and phylogenetics of hundreds of microbial genomes. PLoS ONE 8:e81760. doi: 10.1371/journal.pone.0081760
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gavilan, R. G., Zamudio, M. L., and Martinez-Urtaza, J. (2013). Molecular epidemiology and genetic variation of pathogenic Vibrio parahaemolyticus in Peru. PLoS Negl. Trop. Dis. 7:e2210. doi: 10.1371/journal.pntd.0002210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez-Escalona, N., Cachicas, V., Acevedo, C., Rioseco, M. L., Vergara, J. A., Cabello, F., et al. (2005). Vibrio parahaemolyticus diarrhea, Chile, 1998 and 2004. Emerg. Infect. Dis. 11, 129–131. doi: 10.3201/eid1101.040762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
González-Escalona, N., Gavilan, R. G., Brown, E. W., and Martinez-Urtaza, J. (in press). Transoceanic spreading of pathogenic strains of Vibrio parahaemolyticus with distinctive genetic signatures in the recA gene. PLOS ONE doi: 10.1371/journal.pone.0117485
Gonzalez-Escalona, N., Martinez-Urtaza, J., Romero, J., Espejo, R. T., Jaykus, L. A., Depaola, A., et al. (2008). Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 190, 2831–2840. doi: 10.1128/JB.01808-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez-Escalona, N., Strain, E. A., De Jesus, A. J., Jones, J. L., and DePaola, A. (2011). Genome sequence of the clinical O4:K12 serotype Vibrio parahaemolyticus strain 10329. J. Bacteriol. 193, 3405–3406. doi: 10.1128/JB.05044-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haendiges, J., Rock, M., Myers, R. A., Brown, E. W., Evans, P., and Gonzalez-Escalona, N. (2014). Pandemic Vibrio parahaemolyticus, Maryland, USA, 2012. Emerg. Infect. Dis. 20, 718–720. doi: 10.3201/eid2004.130818
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harth, E., Matsuda, L., Hernandez, C., Rioseco, M. L., Romero, J., Gonzalez-Escalona, N., et al. (2009). Epidemiology of Vibrio parahaemolyticus outbreaks, southern Chile. Emerg. Infect. Dis. 15, 163–168. doi: 10.3201/eid1502.071269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hervio-Heath, D., Colwell, R. R., Derrien, A., Robert-Pillot, A., Fournier, J. M., and Pommepuy, M. (2002). Occurrence of pathogenic vibrios in coastal areas of France. J. Appl. Microbiol. 92, 1123–1135. doi: 10.1046/j.1365-2672.2002.01663.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jensen, R. V., Depasquale, S. M., Harbolick, E. A., Hong, T., Kernell, A. L., Kruchko, D. H., et al. (2013). Complete genome sequence of prepandemic Vibrio parahaemolyticus BB22OP. Genome Announc. 1:e00002-12. doi: 10.1128/genomeA.00002-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johnson, C. N., Flowers, A. R., Young, V. C., Gonzalez-Escalona, N., DePaola, A., Noriea, N. F. III, et al. (2009). Genetic relatedness among tdh+ and trh+ Vibrio parahaemolyticus cultured from Gulf of Mexico oysters (Crassostrea virginica) and surrounding water and sediment. Microb. Ecol. 57, 437–443. doi: 10.1007/s00248-008-9418-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, J. L., Ludeke, C. H., Bowers, J. C., Garrett, N., Fischer, M., Parsons, M. B., et al. (2012). Biochemical, serological, and virulence characterization of clinical and oyster Vibrio parahaemolyticus isolates. J. Clin. Microbiol. 50, 2343–2352. doi: 10.1128/JCM.00196-12
Jun, J. W., Kim, J. H., Choresca, C. H., Shin, S. P., Han, J. E., and Park, S. C. (2013). Draft genome sequence of Vibrio parahaemolyticus SNUVpS-1 isolated from Korean seafood. Genome Announc. 1:e00132-12. doi: 10.1128/genomeA.00132-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaysner, C. A., Abeyta, C. Jr., Stott, R. F., Lilja, J. L., and Wekell, M. M. (1990). Incidence of urea-hydrolyzing Vibrio parahaemolyticus in Willapa Bay, Washington. Appl. Environ. Microbiol. 56, 904–907.
Kim, H. Y., Ayrapetyan, M., and Oliver, J. D. (2014). Survival of Vibrio vulnificus genotypes in male and female serum, and production of siderophores in human serum and seawater. Foodborne Pathog. Dis. 11, 119–125. doi: 10.1089/fpd.2013.1581
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kishishita, M., Matsuoka, N., Kumagai, K., Yamasaki, S., Takeda, Y., and Nishibuchi, M. (1992). Sequence variation in the thermostable direct hemolysin-related hemolysin (trh) gene of Vibrio parahaemolyticus. Appl. Environ. Microbiol. 58, 2449–2457.
Klimke, W., Agarwala, R., Badretdin, A., Chetvernin, S., Ciufo, S., Fedorov, B., et al. (2009). The national center for biotechnology information's protein clusters database. Nucleic Acids Res. 37, D216–D223. doi: 10.1093/nar/gkn734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, M., and Chen, S. (2013). Draft genome sequence of Vibrio parahaemolyticus V110, Isolated from Shrimp in Hong Kong. Genome Announc. 1:e00300-13. doi: 10.1128/genomeA.00300-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ma, C., Deng, X., Ke, C., He, D., Liang, Z., Li, W., et al. (2014). Epidemiology and etiology characteristics of foodborne outbreaks caused by Vibrio parahaemolyticus during 2008-2010 in Guangdong province, China. Foodborne Pathog. Dis. 11, 21–29. doi: 10.1089/fpd.2013.1522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Makino, K., Oshima, K., Kurokawa, K., Yokoyama, K., Uda, T., Tagomori, K., et al. (2003). Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet 361, 743–749. doi: 10.1016/S0140-6736(03)12659-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marshall, S., Clark, C. G., Wang, G., Mulvey, M., Kelly, M. T., and Johnson, W. M. (1999). Comparison of molecular methods for typing Vibrio parahaemolyticus. J. Clin. Microbiol. 37, 2473–2478.
Martinez-Urtaza, J., Baker-Austin, C., Jones, J. L., Newton, A. E., Gonzalez-Aviles, G. D., and DePaola, A. (2013). Spread of Pacific Northwest Vibrio parahaemolyticus strain. N. Engl. J. Med. 369, 1573–1574. doi: 10.1056/NEJMc1305535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martinez-Urtaza, J., Lozano-Leon, A., Varela-Pet, J., Trinanes, J., Pazos, Y., and Garcia-Martin, O. (2008). Environmental determinants of the occurrence and distribution of Vibrio parahaemolyticus in the rias of Galicia, Spain. Appl. Environ. Microbiol. 74, 265–274. doi: 10.1128/AEM.01307-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martinez-Urtaza, J., Simental, L., Velasco, D., DePaola, A., Ishibashi, M., Nakaguchi, Y., et al. (2005). Pandemic Vibrio parahalemolyticus O3: K6, Europe. Emerg. Infect. Dis. 11, 1319–1320. doi: 10.3201/eid1108.050322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsumoto, C., Okuda, J., Ishibashi, M., Iwanaga, M., Garg, P., Rammamurthy, T., et al. (2000). Pandemic spread of an O3:K6 clone of Vibrio parahaemolyticus and emergence of related strains evidenced by arbitrarily primed PCR and toxRS sequence analyses. J. Clin. Microbiol. 38, 578–585.
Mead, P. S., Slutsker, L., Dietz, V., McCaig, L. F., Bresee, J. S., Shapiro, C., et al. (1999). Food-related illness and death in the United States. Emerg. Infect. Dis. 5, 607–625. doi: 10.3201/eid0505.990502
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nair, G. B., Ramamurthy, T., Bhattacharya, S. K., Dutta, B., Takeda, Y., and Sack, D. A. (2007). Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin. Microbiol. Rev. 20, 39–48. doi: 10.1128/CMR.00025-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Newton, A. E., Garrett, N., Stroika, S. G., Halpin, J. L., Turnsek, M., and Mody, R. K. (2014). Notes from the field: increase in Vibrio parahaemolyticus infections associated with consumption of atlantic coast shellfish - 2013. MMWR Morb. Mortal. Wkly. Rep. 63, 335–336.
Nishibuchi, M., and Kaper, J. B. (1990). Duplication and variation of the thermostable direct haemolysin (tdh) gene in Vibrio parahaemolyticus. Mol. Microbiol. 4, 87–99. doi: 10.1111/j.1365-2958.1990.tb02017.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishibuchi, M., and Kaper, J. B. (1995). Thermostable direct hemolysin gene of Vibrio parahaemolyticus: a virulence gene acquired by a marine bacterium. Infect. Immun. 63, 2093–2099.
Okuda, J., Ishibashi, M., Abbott, S. L., Janda, J. M., and Nishibuchi, M. (1997a). Analysis of the thermostable direct hemolysin (tdh) gene and the tdh-related hemolysin (trh) genes in urease-positive strains of Vibrio parahaemolyticus isolated on the West Coast of the United States. J. Clin. Microbiol. 35, 1965–1971.
Okuda, J., Ishibashi, M., Hayakawa, E., Nishino, T., Takeda, Y., Mukhopadhyay, A. K., et al. (1997b). Emergence of a unique O3:K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from Southeast Asian travelers arriving in Japan. J. Clin. Microbiol. 35, 3150–3155.
Paranjpye, R., Hamel, O. S., Stojanovski, A., and Liermann, M. (2012). Genetic diversity of clinical and environmental Vibrio parahaemolyticus strains from the Pacific Northwest. Appl. Environ. Microbiol. 78, 8631–8638. doi: 10.1128/AEM.01531-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, K. S., Iida, T., Yamaichi, Y., Oyagi, T., Yamamoto, K., and Honda, T. (2000). Genetic characterization of DNA region containing the trh and ure genes of Vibrio parahaemolyticus. Infect. Immun. 68, 5742–5748. doi: 10.1128/IAI.68.10.5742-5748.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, K. S., Ono, T., Rokuda, M., Jang, M. H., Okada, K., Iida, T., et al. (2004). Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect. Immun. 72, 6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parsons, M. B., Cooper, K. L., Kubota, K. A., Puhr, N., Simington, S., Calimlim, P. S., et al. (2007). PulseNet USA standardized pulsed-field gel electrophoresis protocol for subtyping of Vibrio parahaemolyticus. Foodborne Pathog. Dis. 4, 285–292. doi: 10.1089/fpd.2007.0089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pazhani, G. P., Bhowmik, S. K., Ghosh, S., Guin, S., Dutta, S., Rajendran, K., et al. (2014). Trends in the epidemiology of pandemic and non-pandemic strains of Vibrio parahaemolyticus isolated from diarrheal patients in Kolkata, India. PLoS Negl. Trop. Dis. 8:e2815. doi: 10.1371/journal.pntd.0002815
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raimondi, F., Kao, J. P., Fiorentini, C., Fabbri, A., Donelli, G., Gasparini, N., et al. (2000). Enterotoxicity and cytotoxicity of Vibrio parahaemolyticus thermostable direct hemolysin in in vitro systems. Infect. Immun. 68, 3180–3185. doi: 10.1128/IAI.68.6.3180-3185.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rasko, D. A., Webster, D. R., Sahl, J. W., Bashir, A., Boisen, N., Scheutz, F., et al. (2011). Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 365, 709–717. doi: 10.1056/NEJMoa1106920
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sahl, J. W., Gillece, J. D., Schupp, J. M., Waddell, V. G., Driebe, E. M., Engelthaler, D. M., et al. (2013). Evolution of a pathogen: a comparative genomics analysis identifies a genetic pathway to pathogenesis in Acinetobacter. PLoS ONE 8:e54287. doi: 10.1371/journal.pone.0054287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M. A., Roy, S. L., et al. (2011). Foodborne illness acquired in the United States–major pathogens. Emerg. Infect. Dis. 17, 7–15. doi: 10.3201/eid1701.P11101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Timme, R. E., Pettengill, J. B., Allard, M. W., Strain, E., Barrangou, R., Wehnes, C., et al. (2013). Phylogenetic diversity of the enteric pathogen Salmonella enterica subsp. enterica inferred from genome-wide reference-free SNP characters. Genome Biol. Evol. 5, 2109–2123. doi: 10.1093/gbe/evt159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tiruvayipati, S., Bhassu, S., Kumar, N., Baddam, R., Shaik, S., Gurindapalli, A. K., et al. (2013). Genome anatomy of the gastrointestinal pathogen, Vibrio parahaemolyticus of crustacean origin. Gut Pathog. 5:37. doi: 10.1186/1757-4749-5-37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turner, J. W., Paranjpye, R. N., Landis, E. D., Biryukov, S. V., Gonzalez-Escalona, N., Nilsson, W. B., et al. (2013). Population structure of clinical and environmental Vibrio parahaemolyticus from the Pacific Northwest coast of the United States. PLoS ONE 8:e55726. doi: 10.1371/journal.pone.0055726
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagley, S., Koofhethile, K., Wing, J. B., and Rangdale, R. (2008). Comparison of V. parahaemolyticus isolated from seafoods and cases of gastrointestinal disease in the UK. Int. J. Environ. Health Res. 18, 283–293. doi: 10.1080/09603120801911064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, Y., Cui, Y., Han, H., Xiao, X., and Wong, H. (2011). Extended MLST-based population genetics and phylogeny of Vibrio parahaemolyticus with high levels of recombination. Int. J. Food Microbiol. 145, 106–112. doi: 10.1016/j.ijfoodmicro.2010.11.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: NGS, WGS, Vibrio parahaemolyticus, clinical, phylogenetic analysis, phylogeny, SNPs
Citation: Haendiges J, Timme R, Allard MW, Myers RA, Brown EW and Gonzalez-Escalona N (2015) Characterization of Vibrio parahaemolyticus clinical strains from Maryland (2012–2013) and comparisons to a locally and globally diverse V. parahaemolyticus strains by whole-genome sequence analysis. Front. Microbiol. 6:125. doi: 10.3389/fmicb.2015.00125
Received: 16 December 2014; Accepted: 02 February 2015;
Published online: 19 February 2015.
Edited by:
Pendru Raghunath, Dr. VRK Women's Medical College, IndiaReviewed by:
Adrian Canizalez-Roman, Autonomous University of Sinaloa, MexicoJaime Martinez-Urtaza, University of Bath, UK
Copyright © 2015 Haendiges, Timme, Allard, Myers, Brown and Gonzalez-Escalona. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Narjol Gonzalez-Escalona, Center for Food and Applied Nutrition, Food and Drug Administration, 5100 Paint Branch Parkway, College Park, MD 20740, USA e-mail: narjol.gonzalez-escalona@fda.hhs.gov