 Vikas Sharma
Vikas Sharma Philippe Colson
Philippe Colson Olivier Chabrol2
Olivier Chabrol2 Patrick Scheid
Patrick Scheid Pierre Pontarotti
Pierre Pontarotti Didier Raoult
Didier Raoult- 1Unité de Recherche sur les Maladies Infectieuses et Tropicales Emergentes UM63 CNRS 7278, IRD 198, INSERM U1095, Faculté de Médecine, Aix-Marseille University, Marseille, France
- 2I2M UMR 7373, CNRS équipe Evolution Biologique et Modélisation, Aix-Marseille University, Marseille, France
- 3Institut Hospitalo-Universitaire Méditerranée Infection, Pôle des Maladies Infectieuses et Tropicales Clinique et Biologique, Fédération de Bactériologie-Hygiène-Virologie, Centre Hospitalo-Universitaire Timone, Assistance Publique-Hôpitaux de Marseille, Marseille, France
- 4Medical Parasitology Laboratory, Laboratory Department I (Medicine), Diagnostics, Central Institute of the Bundeswehr Medical Service, Koblenz, Germany
Nucleocytoplasmic large DNA viruses, or representatives of the proposed order Megavirales, belong to families of giant viruses that infect a broad range of eukaryotic hosts. Megaviruses have been previously described to comprise a fourth monophylogenetic TRUC (things resisting uncompleted classification) together with cellular domains in the universal tree of life. Recently described pandoraviruses have large (1.9–2.5 MB) and highly divergent genomes. In the present study, we updated the classification of pandoraviruses and other reported giant viruses. Phylogenetic trees were constructed based on six informational genes. Hierarchical clustering was performed based on a set of informational genes from Megavirales members and cellular organisms. Homologous sequences were selected from cellular organisms using TimeTree software, comprising comprehensive, and representative sets of members from Bacteria, Archaea, and Eukarya. Phylogenetic analyses based on three conserved core genes clustered pandoraviruses with phycodnaviruses, exhibiting their close relatedness. Additionally, hierarchical clustering analyses based on informational genes grouped pandoraviruses with Megavirales members as a super group distinct from cellular organisms. Thus, the analyses based on core conserved genes revealed that pandoraviruses are new genuine members of the ‘Fourth TRUC’ club, encompassing distinct life forms compared with cellular organisms.
Introduction
Defining microbes has been a long journey. During the 18th century, Pasteur first described these organisms as microscopic organisms invisible to the naked eye, but visible under the microscope (Raoult, 2013). At the beginning of the 20th century, Chatton classified these microbes as eukaryotes and prokaryotes based on the presence or absence of a nucleus, and C. Woese subsequently classified these organisms into three domains of life, Archaea, Bacteria, and Eukarya, based on ribosomal DNA (Woese et al., 1990; Raoult, 2013). Although recently revealed as the most abundant biological entities in the biosphere, infecting cellular organisms from these three domains, viruses have been excluded from this classification of the living world because these organisms lack ribosomal DNA (Suttle, 2005; Raoult and Forterre, 2008; Raoult, 2013, 2015). Indeed, viruses were defined as invisible under a light microscope, and these organisms were initially referred to as inframicrobes. However, giant viruses of amoeba, first described in 2003 with the discovery of Mimivirus as a bona fide microbe, have suggested that this paradigm should be revisited (Raoult et al., 2004). Mimivirus, which was isolated through co-culturing with Acanthamoeba polyphaga, was initially considered to be a small bacterium due to its large size (750 nm in diameter) and Gram-positive staining (Raoult et al., 2007). Mimivirus has a large genome comprising 1.2 megabase pairs (Mbp) that was until recently the largest amongst viruses and even larger than those of small bacteria, such as Mycoplasma genitalium (Raoult et al., 2004). Moreover, this microbe has a repertoire of almost 1,000 genes encoding unexpected proteins, previously considered to be a trademark of cellular organisms, including components of the translation apparatus. Subsequently, several dozens of giant viruses of amoeba have been identified and classified in the family Mimiviridae, founded by Mimivirus (La Scola et al., 2008; Fischer et al., 2010; Arslan et al., 2011; Yoosuf et al., 2012; Santini et al., 2013; Yutin et al., 2013), or family Marseilleviridae, founded by Marseillevirus (Boyer et al., 2009; Thomas et al., 2011; Pagnier et al., 2013; Aherfi et al., 2014). These viruses are commonly observed in the biosphere, consistent with the frequent isolation of these organisms from environmental water, and sediment/soil samples in several countries and the concurrent detection of sequences related to the genomes of these microbes in various environmental metagenomes (Ghedin and Claverie, 2005; Monier et al., 2008; Kristensen et al., 2010; Williamson et al., 2012). Moreover, mimiviruses and marseilleviruses have been recently identified in human samples, associated with pneumonia and adenitis, respectively, (Popgeorgiev et al., 2013; Saadi et al., 2013). Giant viruses of amoeba have been associated with nucleocytoplasmic large DNA viruses (NCLDVs), a monophyletic group of viruses first identified in 2001, encompassing asfarviruses, poxviruses, phycodnaviruses, ascoviruses, and iridoviruses (Iyer et al., 2001). The reclassification of all NCLDV families in a new viral order, called the Megavirales, has recently been proposed (Colson et al., 2012, 2013). Phylogenetic and phyletic analyses have revealed a common origin for these viruses dating back to the early stage of eukaryotic evolution (Yutin et al., 2009; Boyer et al., 2010; Koonin and Yutin, 2010; Yutin and Koonin, 2012).
Subsequent to the identification of Mimivirus, the idea of a fourth domain of life emerged based on the phylogeny of the conserved genes shared by this virus and members from Eukarya, Archaea, and Bacteria (Raoult et al., 2004). Subsequently, in 2010, phylogenetic and phyletic analyses of informational genes involved in nucleotide metabolism and DNA processing and shared by cellular organisms and giant viruses suggested that the Megavirales members comprise a fourth branch of life with an origin as ancient as that of the three cellular branches (Boyer et al., 2010). This four branch topology was criticized based on the argument that the single-matrix evolutionary models used by Boyer et al. (2010) for their phylogeny reconstructions were not adequate with respect to the substantial compositional heterogeneity bias and homoplasy detected in the informational genes used (Williams et al., 2011). Accordingly, alternative trees were proposed by Williams et al. (2011) using models presented as more appropriate, but these phylogeny reconstructions did not show a monophyly for Eukarya. Besides, it was proposed that the informational genes present in Megavirales members might have been acquired from eukaryotic hosts through horizontal gene transfer (Moreira et al., 2005; Moreira and Brochier-Armanet, 2008; Yutin et al., 2014). In contrast, other findings have strengthened the fourth branch of life assumption. Thus, phylogeny reconstructions describing the evolution of proteomes and conserved protein domain structures of cellular organisms and giant viruses suggested that megaviruses comprise a fourth super group that is distinct from the eukaryotic, archaeal, and bacterial groups in the universal tree of life (Nasir et al., 2012). In addition, RNA polymerase (RNAP) beta subunit homologs were detected in the Global Ocean Sampling (GOS) expedition metagenomic database, representing novel branches, apart from those encompassing Bacteria, Archaea, and Eukarya and large viruses (only poxviruses being analyzed) and were considered to be possibly derived from unknown viruses (Wu et al., 2011). Moreover, we recently extended our 2010 study using DNA-dependent RNA polymerase subunit 2 (RNAP2) from a comprehensive, representative, and unbiased set of species from cellular organisms selected by using TimeTree, and from the Megavirales representatives, and the results strongly supported a four branch topology (Sharma et al., 2014). The name ‘TRUC’ (an acronym for “Things Resisting Uncompleted Classification”) corresponds to a new classification that has been recently created to accommodate the division of currently known microbes in four branches, i.e., bacteria, archaea, eukaryotes, and giant viruses (Raoult, 2013). The redefinition of microbes as TRUCs emphasizes that the three domains of life topology is based on ribosomal DNA and is thus associated with cellular organisms, while neglecting giant viruses.
In 2013, two new giant viruses, Pandoravirus dulcis, and P. salinus, were isolated from Acanthamoeba present in the mud of a pond in Australia and a marine sediment layer in Chile, respectively (Philippe et al., 2013). These viruses became the record holders of size of virions (ovoids in shape and ≈1 μm long) and contained the largest genome sizes (1.9 and 2.5 Mbp, respectively) identified to date. The genome from another Pandoravirus strain named Pandoravirus inopinatum has been recently sequenced. It is 2.2 Mbp large, being the second largest viral genome known to date, and is predicted to encode 1,902 proteins (Antwerpen et al., 2015). P. inopinatum was first found as an endocytobiont of Acanthamoeba strains isolated from the contact lens storage cases of a patient presenting keratitis, demonstrating that humans are exposed to these giant viruses (Scheid et al., 2014; Antwerpen et al., 2015). Notably, similar to Mimivirus, which has long been considered a bacterium instead of a virus (Raoult et al., 2007), Pandoravirus-like particles have been initially classified in 2008 as “extraordinary” endocytobionts of Acanthamoeba sp. and were described subsequently in greater details (Scheid et al., 2008, 2010, 2014; Scheid, 2014; Antwerpen et al., 2015). Noteworthy, Pandoravirus-related sequences have been recently detected in metagenomes generated from various soil samples worldwide (Kerepesi and Grolmusz, 2015).
Pandoraviruses have been identified as highly divergent viral entities. Indeed, only 16% of the predicted genes from P. salinus have significant matches in the NCBI GenBank sequence database, and more than half of these genes belong to families of paralogs (Philippe et al., 2013). In addition, only 14 of the 31 class I-III core genes of Megavirales members and 17 of the 49 inferred Megavirales ancestral genes have been identified in pandoraviral genomes (Philippe et al., 2013; Yutin and Koonin, 2013). Moreover, the size and shape of pandoraviruses are unique among viruses, and no capsid protein-encoding gene has been identified (Philippe et al., 2013). In the present study, we aimed to demonstrate that the giant pandoraviruses are members of the fourth TRUC.
Materials and methods
Viral Genes Used for the Analyses
In the present study, we used the approach previously described by Boyer et al. (2010). The genes considered herein were identified from clusters of orthologous groups of proteins (COGs) function categories associated with nucleotide transport and metabolism and information storage and processing (F, J, A, K, L, and B). Notably, these clusters included the genes encoding ribonucleotide reductase (RNR) and thymidylate synthase (TS), which are both key enzymes involved in the RNA–DNA shift; DNA polymerase family B (DNAPol); topoisomerase II A (TopoIIA); the Flap endonuclease (FEN); the processing factor Proliferating Cell Nuclear Antigen (PCNA); the RNAP2; the transcription factor II B (TFIIB); four amino-acyl tRNA synthetases; and the putative elongation factor EF-1. The presence of these genes was assessed in the genomes of pandoraviruses. Despite having tremendous gene contents, pandoraviruses lack many ancestral genes and have been described to share only 17 of the 49 conserved genes assigned to the putative common ancestor of the Megavirales members using maximum likelihood (ML) evolutionary reconstruction (Yutin et al., 2009) and 5 of the 12 genes involved in DNA processing. Therefore, the gene markers used in the present study included RNAP1, RNAP2, DNApol, RNR, tyrosyl-tRNA synthetase, and TFIIB. The viral orthologous sequences were collected using the OrthoMCL procedure (Li et al., 2003) with gene repertoires from 317 DNA viral genomes encoding proteomes comprising more than 100 protein sequences directly downloaded from the NCBI GenBank sequence database1. Recently available sequences from P. inopinatum were collected from the NCBI GenBank protein sequence database2. Comparative analysis of the gene repertoires from the three Pandoravirus isolates was performed using OrthoMCL and through the strategy of reciprocal best BLASTp hits (using as thresholds an e-value <1e-3, an amino acid identity >30% and a sequence coverage >70%) to identify the sets of bona fide orthologous genes (Jordan et al., 2002; Li et al., 2003).
Search for Homologous Sequences in Cellular Life Forms
Protein BLAST searches were performed using Standalone Blast 2.2.27 (Altschul et al., 1990) and giant viral protein sequences as queries against the NCBI GenBank non-redundant protein sequence database (nr) to collect homologous sequences from the members of the three cellular domains of life (Bacteria, Archaea, and Eukarya). The number of target sequences was limited to 20,000 per query.
Sequence Selection Criteria for Cellular Domains
Conserved genes might possess multiple homologs in sequence databases, and random selection among these genes for phylogeny reconstruction might result in the selection of strongly biased non-representative sequence sets. Hence, we selected homologous sequences from various species corresponding to representatives from different phyla of Bacteria, Archaea, and Eukarya using TimeTree, as previously described (Sharma et al., 2014). TimeTree is a public knowledge-base of divergence times among organisms estimated from molecular data in published studies (Hedges et al., 2006). This resource facilitated here the selection of sequences from species that diverged approximately 500 million years ago, which allowed obtaining an appropriate set of members from the three cellular domains of life through a good equilibrium between comprehensiveness and representativeness. In addition, the genomes of most of these cellular organisms are available and have been annotated. The Taxon filter program was subsequently used to filter out taxon and gi identification numbers from the results of the BLAST analysis, which facilitated the downloading of selected protein sequences directly from the NCBI GenBank non-redundant database. Partial or identical sequences were removed through clustering using the CD-HIT suite program (Huang et al., 2010).
Multiple Sequence Alignments and Phylogeny Reconstructions
The obtained sequences were aligned using the Muscle program (Edgar, 2004). Alignment quality was manually analyzed, and phylogenetic reconstructions were performed using ML inference, including the WAG model, and confidence values were calculated using the Shimodaira-Hasegawa (SH) test through the FastTree program (Price et al., 2010). Phylogenetic trees were constructed using the FigTree software program3.
Phyletic Pattern Analysis with Clusters of Orthologous Groups of Protein
Clusters of orthologous groups of proteins corresponding only to the selected functional COG categories [J, A, K, L, B, and F], encoding proteins involved in information storage and processing and nucleotide transport and metabolism, were used for the analysis. BLASTp searches for the selected COG categories, with e-values <1e-3, were performed against the members from Bacteria, Archaea, Eukarya, and Megavirales. Using the BLASTp output results, presence (1) and absence (0) matrices were constructed. A hierarchical clustering dendrogram was constructed using the Pearson distance method with R package ‘amap’4.
Results
A comprehensive search for the 12 informational genes used in a previous study to delineate a fourth TRUC showed that only four genes were conserved in pandoraviruses (Boyer et al., 2010). These genes encode RNAP1 and RNAP2, a DNApol, a RNR, and a tyrosyl-tRNA synthetase.
Phylogenetic Analyses
Informational Genes Showing Vertical Evolution
RNA polymerase is a multi-subunit enzyme that uses a DNA template for the synthesis of RNA to complete transcription (Werner and Grohmann, 2011). Both RNAP 1 and 2 subunits are universal informational genes conserved in all cellular life forms and Megavirales members (Sharma et al., 2014). Phylogenetic analyses based on RNAP1/2 showed that P. salinus, P. dulcis, and P. inopinatum were clustered with Emiliana huxlei virus, a coccolithovirus from the family Phycodnaviridae (Figures 1 and 2). Notably, newly sequenced mimiviruses or marseilleviruses, including LBA111 virus (Saadi et al., 2013), Insectomime virus (Boughalmi et al., 2013), and Tunisvirus (Aherfi et al., 2014), were identified as new bona fide members of the ‘Fourth TRUC’ club. Similarly, two hidden Megavirales members, recently misclassified as Hydra magnipapillata, which is a freshwater cnidarian, and a Marine Group II euryarchaeota, which is a marine archaea, were also identified as members of the ‘Fourth TRUC’ club (Sharma et al., 2014). Furthermore, RNAP1/2-based phylogeny reconstructions showed Megavirales and the three other cellular TRUCs as clearly distinct monophylogenetic groups.
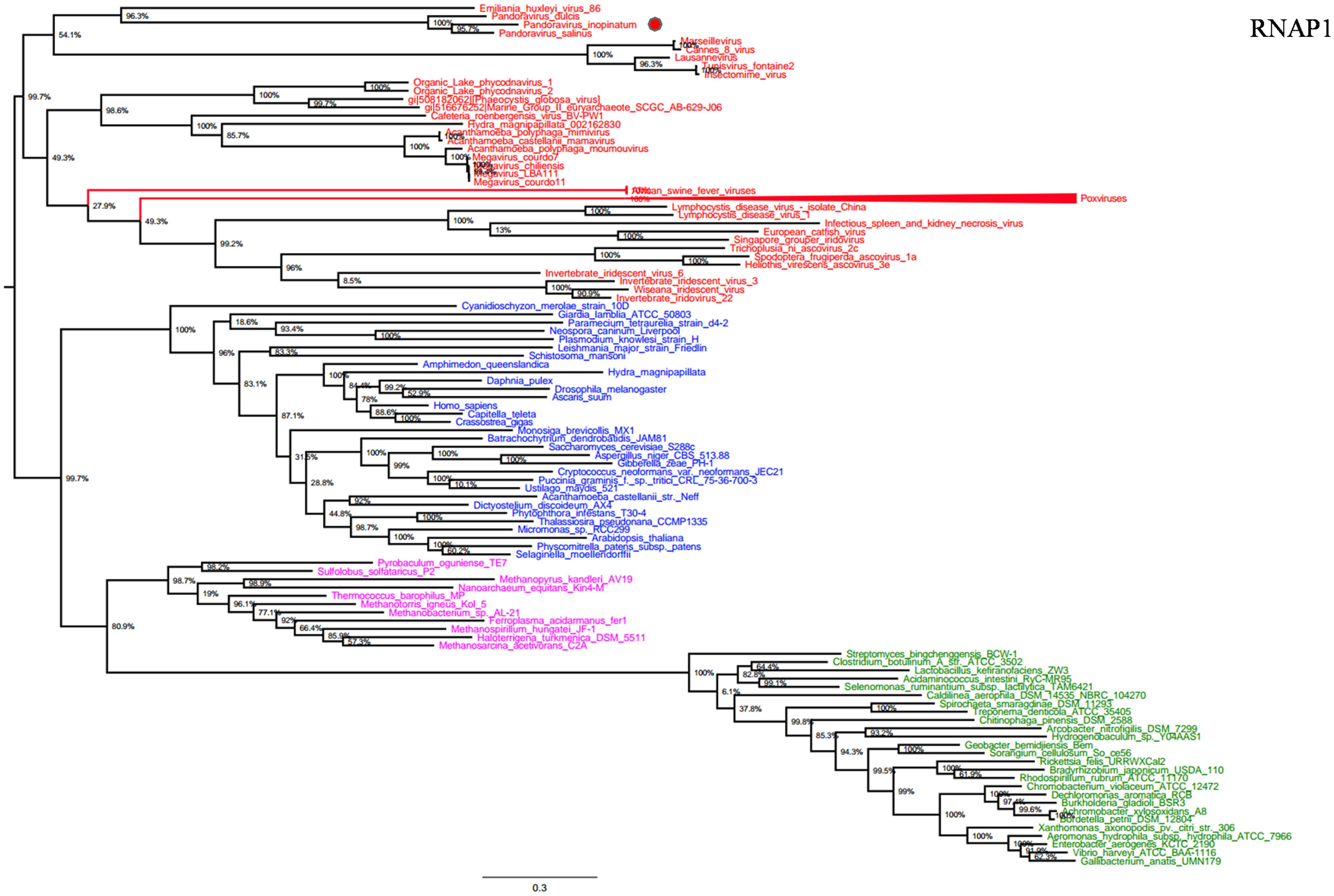
FIGURE 1. RNA polymerase subunit 1 (RNAP1) phylogenetic tree. RNAP1 Maximum-Likelihood (ML) tree was constructed using aligned sequences from Megavirales (red), Bacteria (green), Archaea (pink), and Eukarya (blue). The confidence values were computed through SH-like support using the FastTree program. The scale bar represents the number of estimated changes per position.
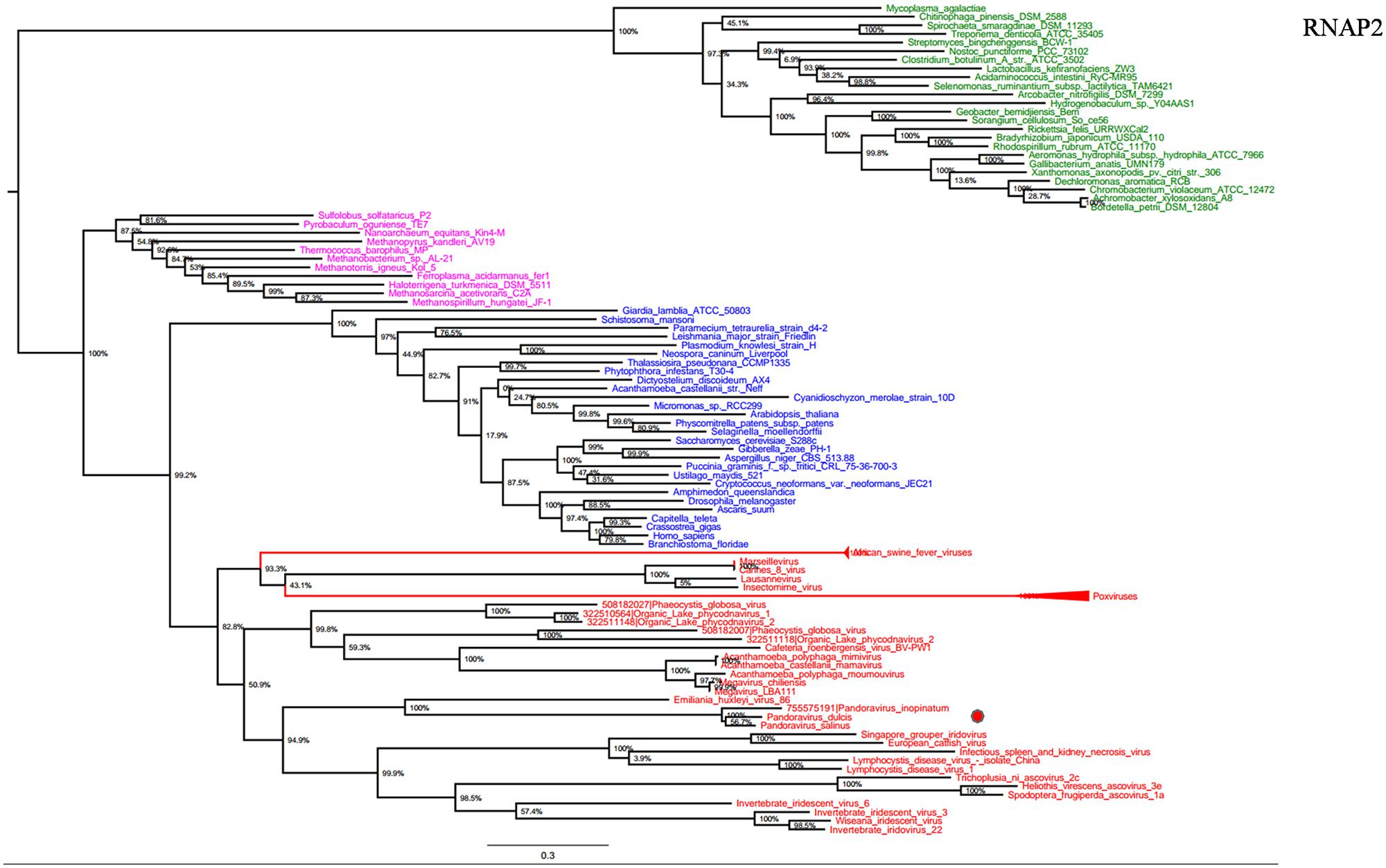
FIGURE 2. RNAP2 phylogenetic tree. RNAP1 ML tree was constructed using aligned sequences from Megavirales (red), Bacteria (green), Archaea (pink), and Eukarya (blue). The confidence values were computed through SH-like support using the FastTree program. The scale bar represents the number of estimated changes per position.
DNA polymerase family B is another highly conserved informational gene involved in DNA replication. Phylogenetic analyses based on DNApol showed that Megavirales members form a monophylogenetic group, with the exception of African swine fever viruses, the sole members of the family Asfarviridae (Dixon et al., 2000; Figure 3). Sequences from the three Pandoravirus genomes are located in the same position that in the RNAP trees, being clustered with Emiliana huxlei virus, a phycodnavirus. This result is congruent with those from previously described phylogeny reconstructions (Philippe et al., 2013; Yutin and Koonin, 2013) and suggests that pandoraviruses might be derived from phycodnaviruses, as previously pointed out based on phylogenomic analyses (Yutin and Koonin, 2013).
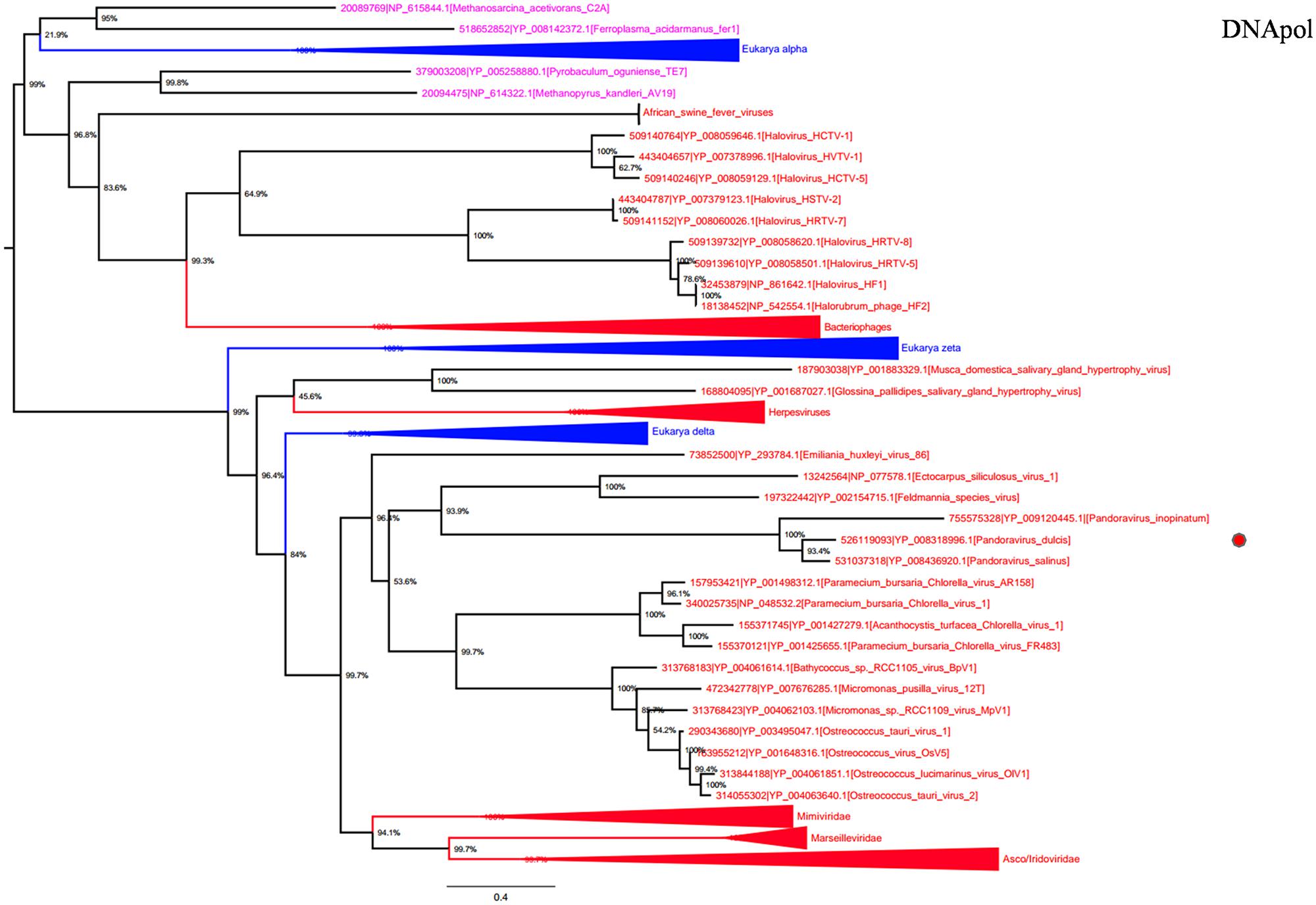
FIGURE 3. DNA polymerase family B phylogenetic tree. DNA polymerase family B ML tree was constructed using aligned sequences from Megavirales (red), Bacteria (green), Archaea (pink), and Eukarya (blue). The confidence values were computed through SH-like support using the FastTree program. The scale bar represents the number of estimated changes per position.
Although absent from bacteria, transcription factor TFIIB, one of the transcription factors comprising the RNA polymerase II preinitiation complex, is present in eukaryotes, archaea, and some Megavirales members but absent from Pandoravirus strains. Phylogenetic analysis based on this gene showed that the other megaviruses form a monophyletic group distinct from eukaryotic and archaeal lineages (Supplementary Figure S1).
Informational Genes Showing Horizontal Evolution
Ribonucleotide reductase is central to the biosynthesis of DNA precursors and catalyzes the reduction of RNA precursors into dNTP (Crona et al., 2013). This gene is present in all cellular organisms and Megavirales members. Among pandoraviruses, only P. inopinatum was found to lack this gene. Phylogenetic analysis showed that Pandoravirus RNRs were clustered with those of eukaryotes including the soil-living amoeba Dictyostelium discoideum AX4, and deeply branched inside the eukaryotic clade (Figure 4). Interestingly, this amoeba was previously identified, unlike most eukaryotes, as encoding both a class I and II RNRs (Crona et al., 2013). These results suggest that pandoraviruses might have acquired this gene through an ancient transfer from an eukaryotic host. In addition, giant viral RNR sequences were scattered in the tree, which may suggest several independent horizontal gene transfer events involving members of the fourth TRUC. Beside this most parsimonious explanation, other evolutionary scenarios including homoplasy, and divergent evolution could not be ruled out.
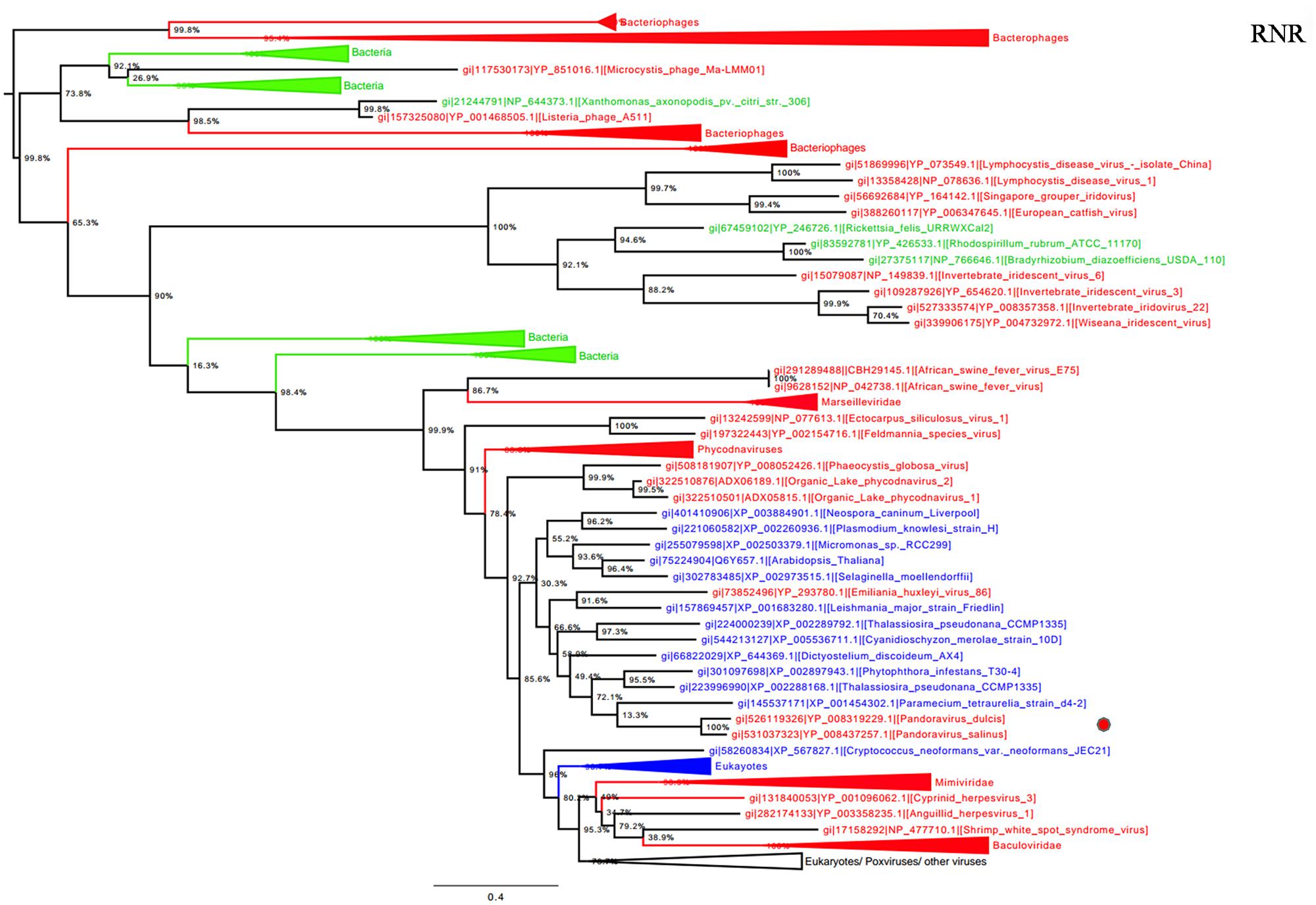
FIGURE 4. Ribonucleotide reductase (RNR) small subunit phylogenetic tree. RNR ML tree was constructed using aligned sequences from Megavirales (red), Bacteria (green), Archaea (pink), and Eukarya (blue). The confidence values were computed through SH-like support using the FastTree program. The scale bar represents the number of estimated changes per position.
Otherwise, Pandoravirus genomes harbor several genes involved in translation, a feature shared with other giant viruses. Notably, amino-acyl tRNA synthetases are conserved in some Megavirales members including pandoraviruses. Tyrosyl-tRNA synthetase is present in P. salinus and P. dulcis but absent in P. inopinatum. Phylogeny reconstruction based on this protein showed that pandoraviruses were clustered with Acanthamoeba castellanii str. Neff, as observed previously (Philippe et al., 2013), suggesting that these viruses have acquired this gene from their hosts, whereas family Mimiviridae forms a separate monophylogenetic cluster (Figure 5). We did not find a clustering between an A. castellanii gene and a megaviral gene in phylogenies built here apart from tyrosyl-tRNA from this amoeba and from pandoraviruses. Moreover, phylogeny based on tyrosyl-tRNA synthetase provided a complex evolutionary scenario, as Eukarya and Archaea members were scattered in the tree, suggesting that horizontal gene transfers occurred several times within different phyla.
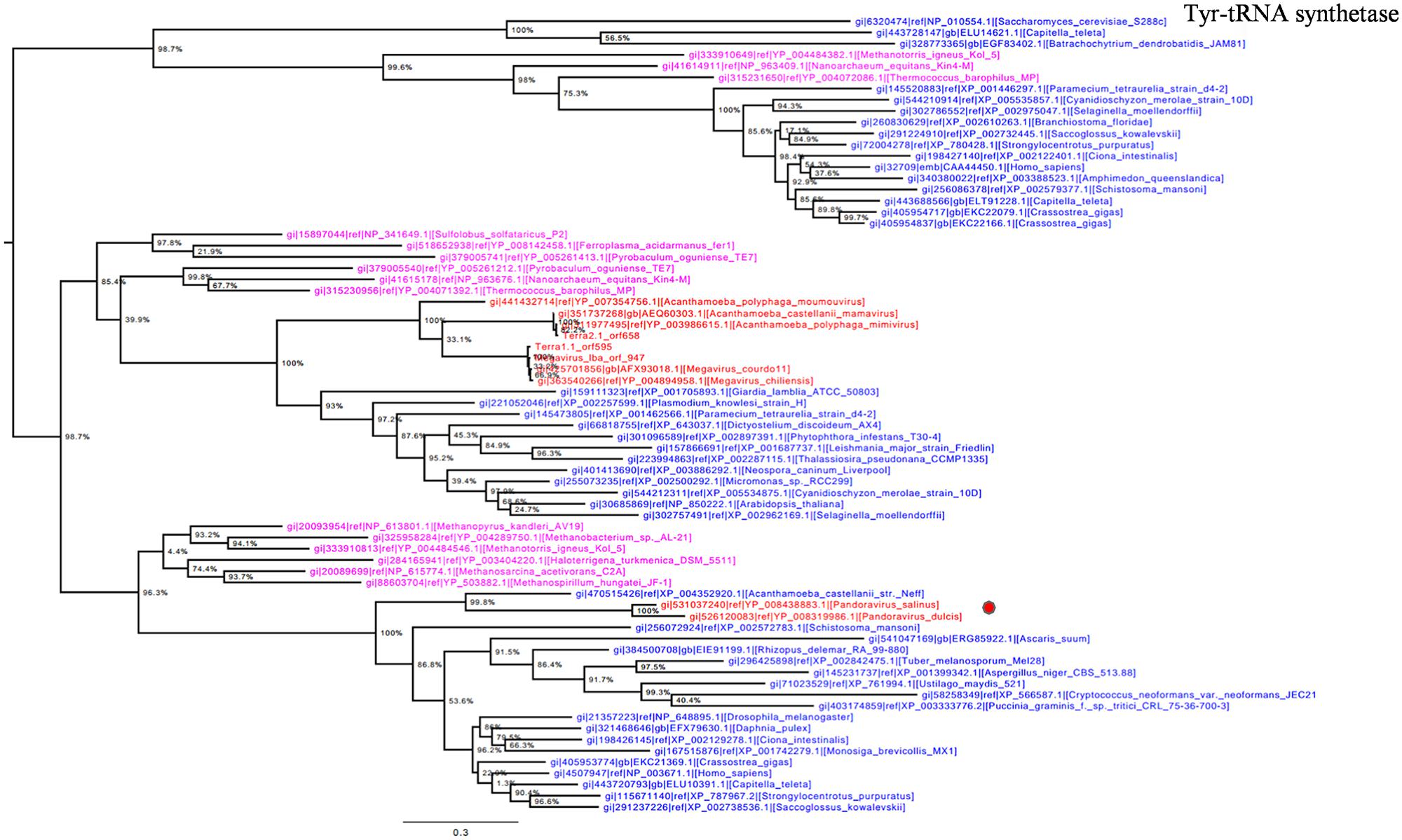
FIGURE 5. Tyrosyl-tRNA synthetase phylogenetic tree. Tyrosyl t-RNA ML tree was constructed using aligned sequences from Megavirales (red), Bacteria (green), Archaea (pink), and Eukarya (blue). The confidence values were computed through SH-like support using the FastTree program. The scale bar represents the number of estimated changes per position.
Phyletic Pattern Analyses
The hierarchical clustering dendrogram based on the presence/absence matrix using 737 COGs comprised of informational genes sharply delineated four clades. Pandoraviruses were embedded in the Megavirales branch. This finding is consistent with those from phylogenetic analyses, indicating that pandoraviruses are new genuine Megavirales members (Figure 6). Six COGs were present in P. salinus and P. dulcis and absent in other megaviruses. These COGs are widespread throughout the genomes of the members of the three cellular TRUCs, and only three of these genomes harbor less than two of these COGs and are considered to be short-sized genomes [491–1,485 kilobase pairs (kbp)]. Conversely, 70 COGs were absent from the P. salinus and P. dulcis genomes but present in at least one other Megavirales member, and these COGs were more abundant among mimiviruses, and secondarily, marseilleviruses, and phycodnaviruses. In addition, these COGs are widely represented in cellular organisms, the lowest number being 24. Moreover, the genome of P. inopinatum encodes 32 COGs among which 14 are shared with P. dulcis and 10 with P. salinus. Consistently, phyletic analyses considering similar gene contents and sizes showed that pandoraviruses clustered with Megavirales.
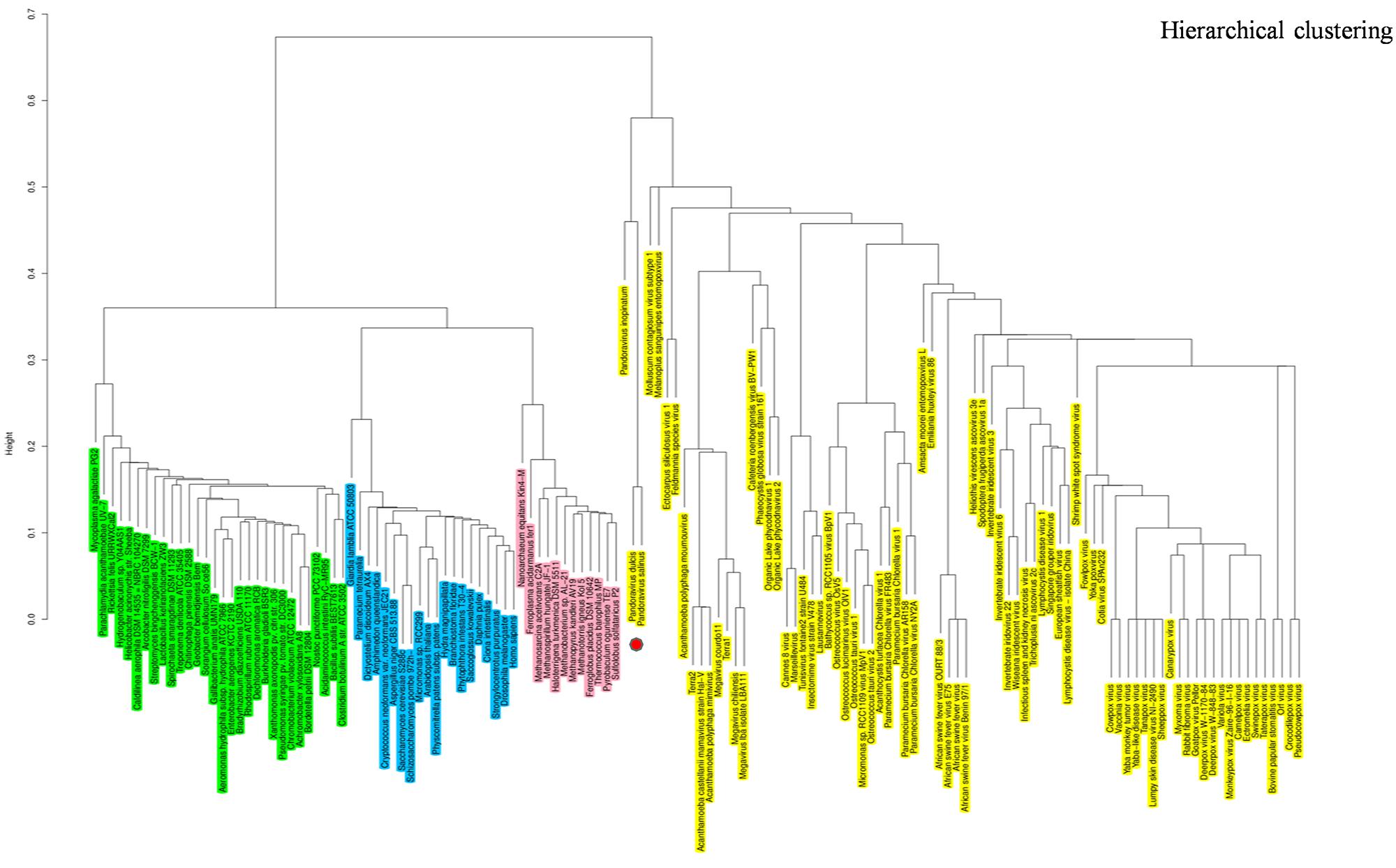
FIGURE 6. Hierarchical clustering by phyletic pattern. Dendrogram was constructed based on the presence/absence of informational clusters of orthologous groups of proteins (COGs) in Megavirales (yellow), Bacteria (green), Archaea (pink), and Eukarya (blue).
Comparative analysis of Pandoravirus genomes
Comparative analyses performed using OrthoMCL with gene repertoires from P. salinus (2,556 predicted genes), P. dulcis (1,502 predicted genes; Philippe et al., 2013) and P. inopinatum (1,902 predicted genes; Antwerpen et al., 2015) identified 1,103 COGs, out of which 681 were core genes, being shared by all three pandoraviruses. In addition, P. inopinatum was found to share 179 genes with P. salinus and 73 with P. dulcis. Besides, the number of pairs of genes involved in reciprocal best BLASTp hits was 562 for P. salinus and P. inopinatum, 510 for P. dulcis and P. inopinatum, and 668 for P. salinus and P. dulcis; while 375 of these bona fide orthologs shared by P. salinus and P. dulcis were also shared by these two viruses and P. inopinatum.
Discussion
Herein, we used the informational genes shared by Megavirales members and cellular organisms to demonstrate that P. salinus, P. dulcis and P. inopinatum are three new genuine members of the fourth TRUC. Thus, these giant viruses, the largest identified so far, were clustered with other megaviruses based on both phylogenetic and phyletic analyses conducted using a previously published strategy, which showed that Megavirales members comprise a monophyletic group apart from Bacteria, Archaea, and Eukarya (Boyer et al., 2010). In addition, in the present study, we used relevant criteria for the selection of cellular organisms, as recently reported (Sharma et al., 2014), to obtain a better understanding of the origin and evolution of Megavirales members.
RNA polymerase 1 and 2 subunits are universal genes, conserved in giant viruses and cellular organisms. Moreover, these genes are refractory to lateral gene transfer and, in viruses, these genes are resistant to recombination with the host genome (Case et al., 2007; Adékambi et al., 2009). The ubiquity of RNAP facilitated the construction of a phylogenetic tree comprising all studied organisms, confirming that pandoraviruses, and other giant viruses, comprised a fourth branch in the tree of life (Boyer et al., 2010; Sharma et al., 2014). Moreover, the phylogenies of other informational genes (including DNApol and TFIIB) with vertical evolution and poor evidence of recombination established a clear evolutionary picture, in which pandoraviruses, and other giant viruses, are new members of a ‘Fourth TRUC’ club. Phylogenetic analyses based on these genes and phyletic analysis using a set of informational COGs revealed that Megavirales members comprise a monophylogenetic super phylum, consistent with previous findings (Boyer et al., 2010; Colson et al., 2011). It has been suggested that Megavirales representatives evolved a majority of their genes via horizontal gene transfer and therefore do not represent a fourth TRUC (Williams et al., 2011). In contrast, phylogenomic analyses using conserved structural domains have also revealed large to medium DNA viruses as ancient biological entities distinct from Bacteria, Archaea, and Eukarya (Nasir et al., 2012). We also conducted a phylogenetic analysis based on RNR small subunit, which plays an important role in nucleotide metabolism, and an aminoacyl-tRNA synthetase, and the results suggested complex evolutionary histories with multiple horizontal gene transfers among life forms from different TRUC, as previously reported (Wolf et al., 1999; Moreira et al., 2005; Lundin et al., 2010).
Previous studies have suggested that Megavirales members originate from an ancestral virus, and phylogenetic analyses based on certain informational genes represent these organisms apart from Bacteria, Archaea, and Eukarya (Yutin et al., 2009; Boyer et al., 2010; Koonin and Yutin, 2010; Colson et al., 2012; Yutin and Koonin, 2013; Sharma et al., 2014). The finding that phylogenetic reconstructions, based on RNAP1 and RNAP2 genes, display the same topologies, and are consistent with the ribosomal tree of life, suggests that both genes followed the same monophylogenetic evolutionary history. The phylogenetic and phyletic analyses performed herein obtained the same results in favor of Megavirales as a fourth super-group, supporting the results of previous studies, and provide insight into the vertical and horizontal evolution of pandoraviruses.
Pandoraviruses are mosaic organisms, similar to other microbes, and they are highly divergent from other Megavirales members. A small proportion of the pandoravirus gene complement is indeed shared with other known giant viruses, and these organisms even lack a majority of the Megavirales core genes (Philippe et al., 2013), while only five of the 12 informational genes previously identified by Boyer et al. (2010) were detected in the Pandoravirus genome. Notwithstanding, some genes allow the construction of phylogenies showing Megavirales as a monophyletic group and suggest that early representatives may have harbored these bona fide ancient genes, then lost genes and acquired new ones from their hosts and other sources. Moreover, phylogenetic analyses based on genes with vertical evolution and phyletic analysis of the presence/absence of conserved informational COGs strongly suggest that pandoraviruses are new genuine members of the ‘Fourth TRUC’ club. Interestingly, pandoraviruses were found to be taxonomically the closest to coccolithoviruses, which belong to the family Phycodnaviridae. This is congruent with results from previous studies obtained using genes from P. salinus and P. dulcis (Philippe et al., 2013; Yutin and Koonin, 2013). It has been further hypothesized that pandoraviruses were highly derived phycodnaviruses that might have experienced considerable genome expansion from smaller Megavirales representatives (Yutin and Koonin, 2013).
Conclusion
Phylogenetic and phyletic analyses using informational genes suggest that pandoraviruses are new bona fide members of the ‘Fourth TRUC’ club, in addition to newly sequenced Megavirales representatives. The monophylogeny of Megavirales suggests a common origin and evolution from ancestral DNA viruses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
VS was financially supported through a grant from the Infectiopole Sud foundation.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00423/abstract
Footnotes
- ^ ftp://ftp.ncbi.nih.gov/genomes/Viruses/
- ^ http://www.ncbi.nlm.nih.gov/genbank/
- ^ http://tree.bio.ed.ac.uk/software/figtree/
- ^ http://www.r-project.org/
References
Adékambi, T., Drancourt, M., and Raoult, D. (2009). The rpoB gene as a tool for clinical microbiologists. Trends Microbiol. 17, 37–45. doi: 10.1016/j.tim.2008.09.008
Aherfi, S., Boughalmi, M., Pagnier, I., Fournous, G., La Scola, B., Raoult, D., et al. (2014). Complete genome sequence of Tunisvirus, a new member of the proposed family Marseilleviridae. Arch. Virol. 159, 2349–2358. doi: 10.1007/s00705-014-2023-5
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Antwerpen, M. H., Georgi, E., Zoeller, L., Woelfel, R., Stoecker, K., and Scheid, P. (2015). Whole-genome sequencing of a Pandoravirus isolated from keratitis-inducing Acanthamoeba. Genome Announc. 3:e00136-15. doi: 10.1128/genomeA.00136-15
Arslan, D., Legendre, M., Seltzer, V., Abergel, C., and Claverie, J.-M. (2011). Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. U.S.A. 108, 17486–17491. doi: 10.1073/pnas.1110889108
Boughalmi, M., Pagnier, I., Aherfi, S., Colson, P., Raoult, D., and La Scola, B. (2013). First isolation of a marseillevirus in the diptera syrphidae eristalis tenax. Intervirology 56, 386–394. doi: 10.1159/000354560
Boyer, M., Madoui, M.-A. A., Gimenez, G., La Scola, B., and Raoult, D. (2010). Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4th domain of life including giant viruses. PLoS ONE 5:e15530. doi: 10.1371/journal.pone.0015530
Boyer, M., Yutin, N., Pagnier, I., Barrassi, L., Fournous, G., Espinosa, L., et al. (2009). Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc. Natl. Acad. Sci. U.S.A. 106, 21848–21853. doi: 10.1073/pnas.0911354106
Case, R. J., Boucher, Y., Dahllöf, I., Holmström, C., Doolittle, W. F., and Kjelleberg, S. (2007). Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl. Environ. Microbiol. 73, 278–288. doi: 10.1128/AEM.01177-06
Colson, P., De Lamballerie, X., Fournous, G., and Raoult, D. (2012). Reclassification of giant viruses composing a fourth domain of life in the new order Megavirales. Intervirology 55, 321–332. doi: 10.1159/000336562
Colson, P., De Lamballerie, X., Yutin, N., Asgari, S., Bigot, Y., Bideshi, D. K., et al. (2013). “Megavirales,” a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 158, 2517–2521. doi: 10.1007/s00705-013-1768-6
Colson, P., Gimenez, G., Boyer, M., Fournous, G., and Raoult, D. (2011). The giant cafeteria roenbergensis virus that infects a widespread marine phagocytic protist is a new member of the fourth domain of life. PLoS ONE 6:e18935. doi: 10.1371/journal.pone.0018935
Crona, M., Avesson, L., Sahlin, M., Lundin, D., Hinas, A., Klose, R., et al. (2013). A rare combination of ribonucleotide reductases in the social amoeba Dictyostelium discoideum. J. Biol. Chem. 288, 8198–8208. doi: 10.1074/jbc.M112.442434
Dixon, L. K., Costa, J. V., Escribano, J. M., Rock, D. L., Vinuela, E., and Wilkinson, P. J. (2000). “Family asfarviridae,” in Virus taxonomy. Seventh report of the International Committee on Taxonomy of Viruses, eds M. H. V. van Regenmortel, C. M. Fauquet, D. H. L. Bishop, E. B. Carstens, M. K. Estes, and S. M. Lemon (San Diego, CA: Academic Press), 159–165.
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5:113. doi: 10.1186/1471-2105-5-113
Fischer, M. G., Allen, M. J., Wilson, W. H., and Suttle, C. A. (2010). Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. U.S.A. 107, 19508–19513. doi: 10.1073/pnas.1007615107
Ghedin, E., and Claverie, J.-M. (2005). Mimivirus relatives in the Sargasso sea. Virol. J. 2, 62. doi: 10.1186/1743-422X-2-62
Hedges, S. B., Dudley, J., and Kumar, S. (2006). TimeTree: a public knowledgebase of divergence times among organisms. Bioinformatics 22, 2971–2972. doi: 10.1093/bioinformatics/btl505
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. doi: 10.1093/bioinformatics/btq003
Iyer, L. M., Aravind, L., and Koonin, E. V. (2001). Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 75, 11720–11734. doi: 10.1128/JVI.75.23.11720-11734.2001
Jordan, I. K., Rogozin, I. B., Wolf, Y. I., and Koonin, E. V. (2002). Essential genes are more evolutionarily conserved than are non-essential genes in bacteria. Genome Res. 12, 962–968. doi: 10.1101/gr.87702
Kerepesi, C., and Grolmusz, V. (2015). Nucleotide sequences of giant viruses found in soil samples of the Mojave desert, the prairie, the tundra and the Antarctic dry valleys. arXiv: 1503.05575v1. doi: 10.1126/science.1101485.31.48
Koonin, E. V., and Yutin, N. (2010). Origin and evolution of eukaryotic large nucleo-cytoplasmic DNA viruses. Intervirology 53, 284–292. doi: 10.1159/000312913
Kristensen, D. M., Mushegian, A. R., Dolja, V. V., and Koonin, E. V. (2010). New dimensions of the virus world discovered through metagenomics. Trends Microbiol. 18, 11–19. doi: 10.1016/j.tim.2009.11.003
La Scola, B., Desnues, C., Pagnier, I., Robert, C., Barrassi, L., Fournous, G., et al. (2008). The virophage as a unique parasite of giant Mimivirus. Nature 455, 100–104. doi: 10.1038/nature07218
Li, L. L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lundin, D., Gribaldo, S., Torrents, E., Sjöberg, B.-M., and Poole, A. M. (2010). Ribonucleotide reduction - horizontal transfer of a required function spans all three domains. BMC Evol. Biol. 10:383. doi: 10.1186/1471-2148-10-383
Monier, A., Claverie, J.-M., and Ogata, H. (2008). Taxonomic distribution of large DNA viruses in the sea. Genome Biol. 9, R106. doi: 10.1186/gb-2008-9-7-r106
Moreira, D., and Brochier-Armanet, C. (2008). Giant viruses, giant chimeras: the multiple evolutionary histories of Mimivirus genes. BMC Evol. Biol. 8:12. doi: 10.1186/1471-2148-8-12
Moreira, D., López-García, P., Ogata, H., Abergel, C., Raoult, D., and Claverie, J. M. (2005). Comment on the 1.2-megabase genome sequence of Mimivirus. Science 308, 1114. doi: 10.1126/science.1110820
Nasir, A., Kim, K. M., and Caetano-Anolles, G. (2012). Giant viruses coexisted with the cellular ancestors and represent a distinct supergroup along with superkingdoms Archaea, Bacteria and Eukarya. BMC Evol. Biol. 12:156. doi: 10.1186/1471-2148-12-156
Pagnier, I., Reteno, D. G. I., Saadi, H., Boughalmi, M., Gaia, M., Slimani, M., et al. (2013). A decade of improvements in mimiviridae and marseilleviridae isolation from amoeba. Intervirology 56, 354–363. doi: 10.1159/000354556
Philippe, N., Legendre, M., Doutre, G., Couté, Y., Poirot, O., Lescot, M., et al. (2013). Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 341, 281–286. doi: 10.1126/science.1239181
Popgeorgiev, N., Boyer, M., Fancello, L., Monteil, S., Robert, C., Rivet, R., et al. (2013). Marseillevirus-like virus recovered from blood donated by asymptomatic humans. J. Infect. Dis. 208, 1042–1050. doi: 10.1093/infdis/jit292
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Raoult, D. (2013). TRUC or the need for a new microbial classification. Intervirology 56, 349–353. doi: 10.1159/000354269
Raoult, D. (2015). How the virophage compels the need to readdress the classification of microbes. Virology 477, 119–124. doi: 10.1016/j.virol.2014.11.014
Raoult, D., Audic, S., Robert, C., Abergel, C., Renesto, P., Ogata, H., et al. (2004). The 1.2-megabase genome sequence of Mimivirus. Science 306, 1344–1350. doi: 10.1126/science.1101485
Raoult, D., and Forterre, P. (2008). Redefining viruses: lessons from Mimivirus. Nat. Rev. Microbiol. 6, 315–319. doi: 10.1038/nrmicro1858
Raoult, D., La Scola, B., and Birtles, R. (2007). The discovery and characterization of Mimivirus, the largest known virus and putative pneumonia agent. Clin. Infect. Dis. 45, 95–102. doi: 10.1086/518608
Saadi, H., Pagnier, I., Colson, P., Cherif, J. K., Beji, M., Boughalmi, M., et al. (2013). First isolation of Mimivirus in a patient with pneumonia. Clin. Infect. Dis. 57, e127–e134. doi: 10.1093/cid/cit354
Santini, S., Jeudy, S., Bartoli, J., Poirot, O., Lescot, M., Abergel, C., et al. (2013). Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 110, 10800–10805. doi: 10.1073/pnas.1303251110
Scheid, P. (2014). Relevance of free-living amoebae as hosts for phylogenetically diverse microorganisms. Parasitol. Res. 113, 2407–2414. doi: 10.1007/s00436-014-3932-7
Scheid, P., Balczun, C., and Schaub, G. A. (2014). Some secrets are revealed: parasitic keratitis amoebae as vectors of the scarcely described pandoraviruses to humans. Parasitol. Res. 113, 3759–3764. doi: 10.1007/s00436-014-4041-3
Scheid, P., Hauröder, B., and Michel, R. (2010). Investigations of an extraordinary endocytobiont in Acanthamoeba sp.: development and replication. Parasitol. Res. 106, 1371–1377. doi: 10.1007/s00436-010-1811-4
Scheid, P., Zöller, L., Pressmar, S., Richard, G., and Michel, R. (2008). An extraordinary endocytobiont in Acanthamoeba sp. isolated from a patient with keratitis. Parasitol. Res. 102, 945–950. doi: 10.1007/s00436-007-0858-3
Sharma, V., Colson, P., Giorgi, R., Pontarotti, P., and Raoult, D. (2014). DNA-dependent RNA polymerase detects hidden giant viruses in published databanks. Genome Biol. Evol. 6, 1–22. doi: 10.1093/gbe/evu128
Thomas, V., Bertelli, C., Collyn, F., Casson, N., Telenti, A., Goesmann, A., et al. (2011). Lausannevirus, a giant amoebal virus encoding histone doublets. Environ. Microbiol. 13, 1454–1466. doi: 10.1111/j.1462-2920.2011.02446.x
Werner, F., and Grohmann, D. (2011). Evolution of multisubunit RNA polymerases in the three domains of life. Nat. Rev. Microbiol. 9, 85–98. doi: 10.1038/nrmicro2507
Williams, T. A., Embley, T. M., and Heinz, E. (2011). Informational gene phylogenies do not support a fourth domain of life for nucleocytoplasmic large DNA viruses. PLoS ONE 6:e21080. doi: 10.1371/journal.pone.0021080
Williamson, S. J., Allen, L. Z., Lorenzi, H. A., Fadrosh, D. W., Brami, D., Thiagarajan, M., et al. (2012). Metagenomic exploration of viruses throughout the indian ocean. PLoS ONE 7:e42047. doi: 10.1371/journal.pone.0042047
Woese, C. R., Kandler, O., and Wheelis, M. L. (1990). Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. U.S.A. 87, 4576–4579. doi: 10.1073/pnas.87.12.4576
Wolf, Y. I., Aravind, L., Grishin, N. V., and Koonin, E. V. (1999). Evolution of aminoacyl-tRNA synthetases–analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 9, 689–710.
Wu, D., Wu, M., Halpern, A., Rusch, D. B., Yooseph, S., Frazier, M., et al. (2011). Stalking the fourth domain in metagenomic data: searching for, discovering, and interpreting novel, deep branches in marker gene phylogenetic trees. PLoS ONE 6:e18011. doi: 10.1371/journal.pone.0018011
Yoosuf, N., Yutin, N., Colson, P., Shabalina, S. A., Pagnier, I., Robert, C., et al. (2012). Related giant viruses in distant locations and different habitats: Acanthamoeba polyphaga moumouvirus represents a third lineage of the Mimiviridae that is close to the Megavirus lineage. Genome Biol. Evol. 4, 1324–1330. doi: 10.1093/gbe/evs109
Yutin, N., Colson, P., Raoult, D., and Koonin, E. V. (2013). Mimiviridae: clusters of orthologous genes, reconstruction of gene repertoire evolution and proposed expansion of the giant virus family. Virol. J. 10, 106. doi: 10.1186/1743-422X-10-106
Yutin, N., and Koonin, E. V. (2012). Hidden evolutionary complexity of Nucleo-Cytoplasmic Large DNA viruses of eukaryotes. Virol. J. 9, 161. doi: 10.1186/1743-422X-9-161
Yutin, N., and Koonin, E. V. (2013). Pandoraviruses are highly derived phycodnaviruses. Biol. Dir. 8, 25. doi: 10.1186/1745-6150-8-25
Yutin, N., Wolf, Y. I., and Koonin, E. V. (2014). Origin of giant viruses from smaller DNA viruses not from a fourth domain of cellular life. Virology, 466–467, 38–52. doi: 10.1016/j.virol.2014.06.032
Keywords: giant virus, Pandoravirus, nucleocytoplasmic large DNA viruses, Megavirales, informational genes, domains of life, TRUC, phylogeny
Citation: Sharma V, Colson P, Chabrol O, Scheid P, Pontarotti P and Raoult D (2015) Welcome to pandoraviruses at the ‘Fourth TRUC’ club. Front. Microbiol. 6:423. doi: 10.3389/fmicb.2015.00423
Received: 17 February 2015; Paper pending published: 28 March 2015;
Accepted: 21 April 2015; Published online: 18 May 2015
Edited by:
Bruno Pozzetto, University Jean Monnet of Saint-Etienne, FranceReviewed by:
Patrick Forterre, Université Paris-Sud, FranceKiran Kondabagil, Indian Institute of Technology Bombay, India
Copyright © 2015 Sharma, Colson, Chabrol, Scheid, Pontarotti and Raoult. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Didier Raoult, Unité de Recherche sur les Maladies Infectieuses et Tropicales Emergentes UM63 CNRS 7278, IRD 198, INSERM U1095, Faculté de Médecine, Aix-Marseille University, 27 Boulevard Jean Moulin, 13385 Marseille, France, didier.raoult@gmail.com