 Lisa Ufarté
Lisa Ufarté- 1Université de Toulouse, Institut National des Sciences Appliquées (INSA), Université Paul Sabatier (UPS), Institut National Polytechnique (INP), Laboratoire d’Ingénierie des Systèmes Biologiques et des Procédés (LISBP), Toulouse, France
- 2INRA - UMR792 Ingénierie des Systèmes Biologiques et des Procédés, Toulouse, France
- 3CNRS, UMR5504, Toulouse, France
The rapid expansion of new sequencing technologies has enabled large-scale functional exploration of numerous microbial ecosystems, by establishing catalogs of functional genes and by comparing their prevalence in various microbiota. However, sequence similarity does not necessarily reflect functional conservation, since just a few modifications in a gene sequence can have a strong impact on the activity and the specificity of the corresponding enzyme or the recognition for a sensor. Similarly, some microorganisms harbor certain identified functions yet do not have the expected related genes in their genome. Finally, there are simply too many protein families whose function is not yet known, even though they are highly abundant in certain ecosystems. In this context, the discovery of new protein functions, using either sequence-based or activity-based approaches, is of crucial importance for the discovery of new enzymes and for improving the quality of annotation in public databases. This paper lists and explores the latest advances in this field, along with the challenges to be addressed, particularly where microfluidic technologies are concerned.
Introduction
The implications of the discovery of new protein functions are numerous, from both cognitive and applicative points of view. Firstly, it improves understanding of how microbial ecosystems function, in order to identify biomarkers and levers that will help optimize the services rendered, regardless of the field of application. Next, the discovery of new enzymes and transporters enables expansion of the catalog of functions available for metabolic pathway engineering and synthetic biology. Finally, the identification and characterization of new protein families, whose functions, three-dimensional structure and catalytic mechanism have never been described, furthers understanding of the protein structure/function relationship. This is an essential prerequisite if we are to draw full benefit from these proteins, both for medical applications (for example, designing specific inhibitors) and for relevant integration into biotechnological processes.
Many reviews have been published on functional metagenomics these last 10 years. Many of them focus on the strategies of library creation and on bio-informatic developments (Di Bella et al., 2013; Ladoukakis et al., 2014), while others describe the various approaches set up to discover novel targets [like therapeutic molecules (Culligan et al., 2014)] for a specific application. In particular several review papers have been written on the numerous activity-based metagenomics studies carried out to find new enzymes for biotechnological applications, without necessarily finding new functions or new protein families (Ferrer et al., 2009; Steele et al., 2009). The present review focuses on all the functional metagenomics approaches, sequence- or activity-based, allowing the discovery of new functions and families from the uncultured fraction of microbial ecosystems, and makes a recent overview on the advances of microfluidics for ultra-fast microbial screening of metagenomes.
Sampling Strategies
The literature describes a wide variety of microbial environments sampled in the search for new enzymes. A large number of studies look at ecosystems with high taxonomic and functional diversity, such as soils or natural aquatic environments that are either undisturbed or exposed to various pollutants (Gilbert et al., 2008; Brennerova et al., 2009; Zanaroli et al., 2010). Extreme environments enable the discovery of enzymes that are naturally adapted to the constraints of certain industrial processes, such as glycoside hydrolases and halotolerant esterases (Ferrer et al., 2005; LeCleir et al., 2007), thermostable lipases (Tirawongsaroj et al., 2008), or even psychrophilic DNA-polymerases (Simon et al., 2009). Other microbial ecosystems, such as anaerobic digesters including both human and/or animal intestinal microbiota and industrial remediation reactors, are naturally specialized in metabolizing certain substrates. These are ideal targets for research into particular functions, such as the degrading activity of lignocellulosic plant biomass (Warnecke et al., 2007; Tasse et al., 2010; Hess et al., 2011; Bastien et al., 2013) or dioxygenases for the degradation of aromatic compounds (Suenaga et al., 2007).
Some studies refer to enrichment steps that occur before sampling, with the aim of increasing the relative abundance of micro-organisms that have the target function. This enrichment can be done by modifying the physical and chemical conditions of the natural environment (van Elsas et al., 2008) or by incorporating the substrate to be metabolized in vivo (Hess et al., 2011) or in vitro, in reactors (DeAngelis et al., 2010) or mesocosms (Jacquiod et al., 2013). Through stable isotopic probing and cloning of the DNA of micro-organisms able to metabolize a specifically labeled substrate for the creation of metagenome libraries, it is possible to increase the frequency of positive clones by several orders of magnitude (Chen and Murrell, 2010). These approaches require functional and taxonomic controls at the different stages of enrichment, which are often sequential, to prevent the proliferation of populations dependent on the activity of the populations preferred at the outset. These kinds of checks are difficult to do in vivo, where there would actually be an increased risk of selecting populations able to metabolize only the degradation products of the initial substrate, to the detriment of those able to attack the more resistant original substrate with its more complex structure.
Functional Screening: New Challenges for the Discovery of Functions
Two complementary approaches can be used to discover new functions and protein families within microbial communities. The first involves the analysis of nucleotide, ribonucleotide or protein sequences, and the other the direct screening of functions before sequencing (Figure 1).
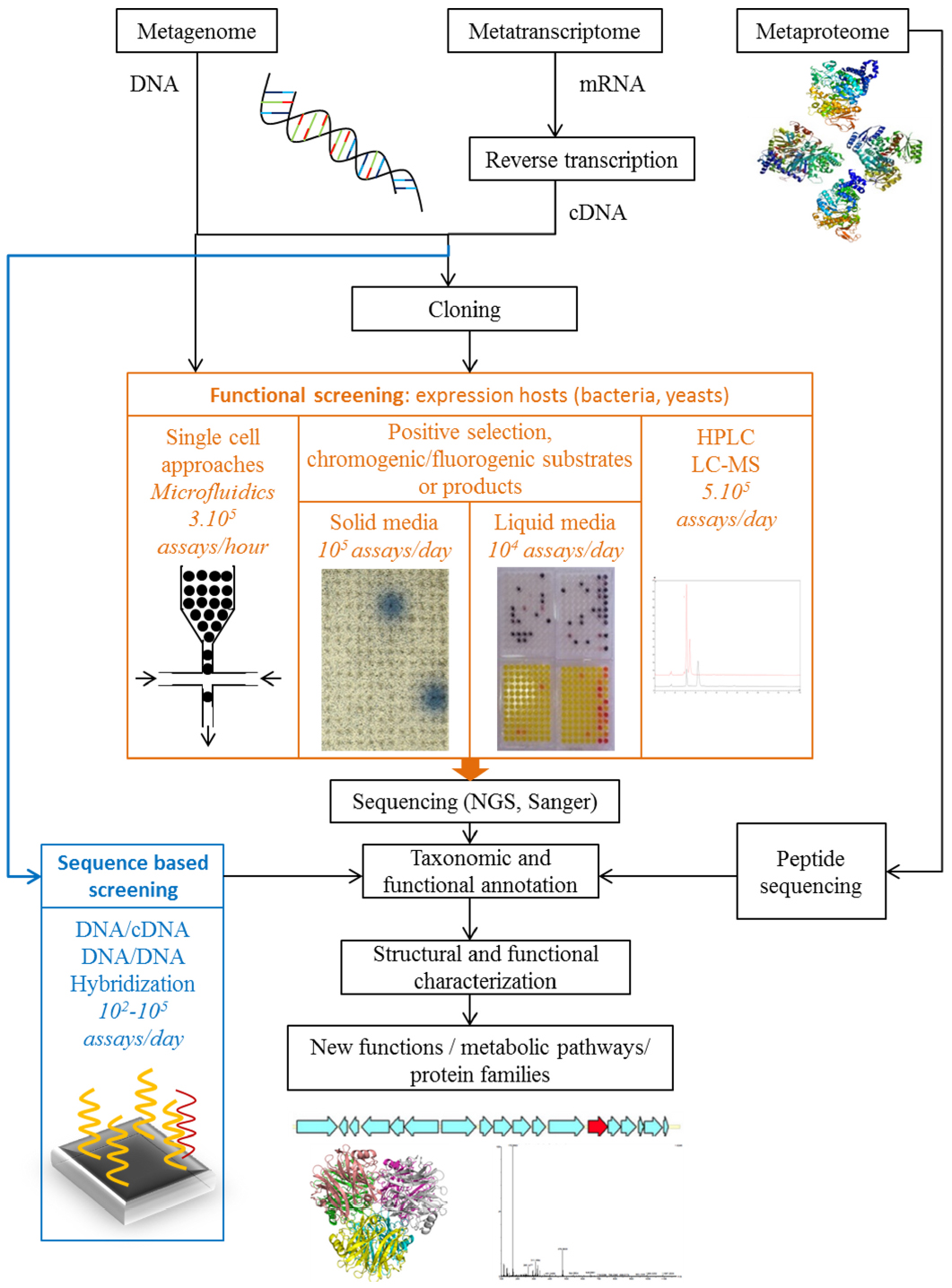
Figure 1. Strategies for the functional exploration of metagenomes, metatranscriptomes and metaproteomics to discover new functions and protein families.
The Sequence, Marker of Originality
There have been a number of large-scale random metagenome sequencing projects (Yooseph et al., 2007; Vogel et al., 2009; Gilbert et al., 2010; Qin et al., 2010; Hess et al., 2011) over the past few years, resulting in catalogs listing millions of genes from different ecosystems, the majority of which are recorded in the GOLD1 (RRID:nif-0000-02918), MG-RAST2 (RRID:OMICS_01456) and EMBL-EBI3 (RRID:nlx_72386) metagenomics databases. At the same time, the obstacles inherent to metatranscriptomic sampling (fragility of mRNA, difficulty with extraction from natural environments, separation of other types of RNA) have been removed, opening a window into the functional dynamics of ecosystems according to biotic or abiotic constraints (Saleh-Lakha et al., 2005; Warnecke and Hess, 2009; Schmieder et al., 2012). Metatranscriptomes sequencing has thus enabled the identification of new gene families, such as those found in microbial communities (prokaryotes and/or eukaryotes) expressed specifically in response to variations in the environment (Bailly et al., 2007; Frias-Lopez et al., 2008; Gilbert et al., 2008) and new enzyme sequences belonging to known carbohydrate active enzymes families (Poretsky et al., 2005; Tartar et al., 2009; Damon et al., 2012).
Regardless of the origin of the sequences (DNA or cDNA, with or without prior cloning in an expression host), the advances made with automatic annotation, most notably thanks to the IMG-M (RRID:nif-0000-03010) and MG-RAST (RRID:OMICS_01456) servers (Markowitz et al., 2007; Meyer et al., 2008), now make it possible to quantify and compare the abundance of the main functional families in the target ecosystems (Thomas et al., 2012), identified through comparison of sequences with the general functional databases: KEGG (RRID:nif-0000-21234) (Kanehisa and Goto, 2000), eggNOG (RRID:nif-0000-02789) (Muller et al., 2010), and COG/KOG (RRID:nif-0000-21313) (Tatusov et al., 2003). They also enable research into specific protein families, thanks to motif detection using Pfam (RRID:nlx_72111) (Finn et al., 2010), TIGRFAM (RRID:nif-0000-03560) (Selengut et al., 2007), CDD (RRID:nif-0000-02647) (Marchler-Bauer et al., 2009), Prosite (RRID:nif-0000-03351) (Sigrist et al., 2010), and HMM model construction (Hidden Markov Models; Söding, 2005). Other servers can be used to interrogate databases specialized in specific enzymatic families (Table 1).
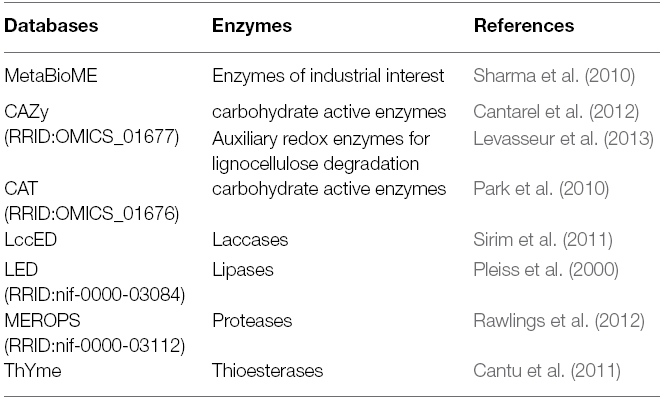
Table 1. Examples of databases specialized in enzymatic functions of biotechnological interest.
Finally, the performance of methods used to assemble next generation sequencing reads is set to open up access to a plethora of complete genes to feed expert databases, which currently only contain a tiny percentage of genes from uncultivated organisms—less than 1% for the CAZy database (RRID:OMICS_01677), for example—while the majority of metagenomic studies published target ecosystems with a high number of plant polysaccharide degradation activities by carbohydrate active enzymes (André et al., 2014).
Even based on a large majority of truncated genes, metagenomes and metatranscriptomes functional annotation enables in silico estimations of the functional diversity of the ecosystem and identification of the most original sequences within a known protein family. It is then possible to use PCR (Polymerase Chain Reaction) to capture those sequences specifically, and test their function experimentally to assess their applicative value. In this way, the sequencing of the rumen metagenome (268 Gb) enabled identification of 27,755 coding genes for carbohydrate active enzymes, and isolation of 51 active enzymes belonging to known families specifically involved in lignocellulose degradation (Hess et al., 2011).
PCR, and more generally DNA/DNA or DNA/cDNA hybridization, also make it possible to directly capture coding genes for protein families that are abundant and/or expressed in the target ecosystem, but with no need for a priori large-scale sequencing. This strategy requires the conception of nucleic acid probes or PCR primers using consensus sequences specific to known protein families. There are plenty of examples of the discovery of enzymes in metagenomes using these approaches, for instance bacterial laccases (Ausec et al., 2011), dioxygenases (Zaprasis et al., 2009), nitrites reductases (Bartossek et al., 2010), hydrogenases (Schmidt et al., 2010), hydrazine oxidoreductases (Li et al., 2010), or chitinases (Hjort et al., 2010) from various ecosystems. The Gene-Targeted-metagenomics approach (Iwai et al., 2009) combines PCR screening and amplicon pyrosequencing to generate primers in an iterative manner and increase the structural diversity of the target protein families, for example the dioxygenases from the microbiota of contaminated soil. Elsewhere, the use of high-density functional microarrays considerably multiplies the number of probes and is therefore a low-cost way of obtaining a snapshot of the abundance and diversity of sequences within specific protein families and even, where the DNA or cDNA has been cloned (He et al., 2010; Weckx et al., 2010), directly capturing targets of interest while rationalizing sequencing. Using a similar strategy, the solution hybrid selection method enables the selection of fragments of coding DNA for specific enzymatic families using 31-mers capture probes. Applied to the capture of cDNA, this method provides access to entire genes which can be then cloned and their activity tested (Bragalini et al., 2014). Solution hybrid selection can therefore be used to explore the taxonomic and functional diversity of all protein families. More especially, this approach opens the way for the selection and characterization of families that are highly represented in a microbiome but whose function remains unknown, in order to further the understanding of ecosystemic functions and discover novel biocatalysts.
Metaproteomics has recently proved its worth in identifying new protein families and/or functions. Paired with genomic, metagenomic and metatranscriptomic data (Erickson et al., 2012), it provides access to excellent biomarkers of the functional state of the ecosystem. Recent developments, such as high-throughput electrospray ionization paired with mass spectrometry, enable full metaproteome analysis after separation of proteins by liquid chromatography. It is thus possible to highlight hundreds of proteins with no associated function and new enzyme families playing a key functional role in the ecosystem (Ram et al., 2005).
This latter example illustrates the need for research and/or experimental proof of function for proteins where the function remains unknown (products of orphan genes or, on the contrary, genes highly prevalent in the microbial realm but that have never been characterized) or poorly annotated. In fact, annotation errors, which are especially common for multi-modular proteins such as carbohydrate active enzymes, are spread at an increasing rate as a result of the explosion in the number of functional genomics and meta-genomic, -transcriptomic and -proteomic projects. New annotation strategies, most notably based on the prediction of the three-dimensional structure of proteins, are also worth exploring (Uchiyama and Miyazaki, 2009). However, at the present time, it is very difficult to predict the specificity of substrate and the mechanism of action (and therefore the function of the protein) on the basis of sequence or even structure, especially where there is no homologue characterized from a structural and functional point of view. Functional screening can address this challenge.
Activity Screening: Speeding up the Discovery of Biotechnology Tools
There are three prerequisites for this approach: (i) the cloning of DNA or cDNA in an expression vector for the creation of, respectively, metagenomic or metatranscriptomic libraries, (ii) heterologous expression of cloned genes in a microbial host, iii) the conception of efficient phenotypic screens to isolate the clones of interest that produce the target activity, also referred to as “hits.”
Using this approach, the functions of a protein can be accessed without any prior information on its sequence. It is therefore the only way of identifying novel protein families that have known functions or previously unseen functions (as long as an adequate screen can be developed). Finally, it helps to rationalize sequencing efforts and focus them only on the hits: for example, those that are of biotechnological interest. The expression potential of the selected heterologous host, the size of the DNA inserts and the type of vectors all determine the success of functional screening. Short fragments of metagenomic DNA (smaller than 15 kb, and most often between 2 and 5 kb), or cDNA for the metatranscriptomic libraries, cloned in plasmids under the influence of a strong expression promoter, enable the overexpression of a single protein, and the easy recovery and sequencing of the hits’ DNA (Uchiyama and Miyazaki, 2009). On the other hand, fragments of bacterial DNA measuring between 15 and 40 kb, 25 and 45 kb or even 100 and 200 kb, cloned respectively in cosmids, fosmids or bacterial artificial chromosomes, can be used to explore a functional diversity of several Gb per library and, above all, provide access to operon-type multigene clusters, coding for complete catabolic or anabolic pathways This is of major interest for the discovery of cocktails of synergistic activities that degrade complex substrates such as plant cell walls for biorefineries. This strategy also ensures high reliability for the taxonomic annotation of inserts, and can even be used to identify the mobile elements responsible for the plasticity of the bacterial metagenome, mediated by horizontal gene transfers (Tasse et al., 2010). However, it requires sensitive activity screens, since the target genes are only weakly expressed, controlled by their own native promoters.
Escherichia coli, whose transformation efficiency is exceptionally high, even for fosmids or bacterial artificial chromosomes, remains the host of choice in the immense majority of studies published. The first exhaustive functional screening study of a fosmid library revealed that E. coli can be used to express genes from bacteria that are very different from a taxonomical point of view, including a large number of Bacteroidetes and Gram-positive bacteria (Tasse et al., 2010), contrary to what had been predicted by in silico detection of expression signals compatible with E. coli (Gabor et al., 2004). However, the value of developing shuttle vectors to screen metagenomic libraries in hosts with different expression and secretion potentials, for example Bacillus, Sphingomonas, Streptomyces, Thermus, or the α-, β- and γ–proteobacteria (Taupp et al., 2011; Ekkers et al., 2012) must not be underestimated, if we are to unlock the functional potential of varied taxons and increase the sensitivity of screens. Finally, it is still very difficult to get access to the uncultivated fraction of eukaryotic microorganisms, due to the lack of screening hosts with sufficient transformation efficiency for the creation of large clone libraries (and thus the exploration of a vast array of sequences) and compatible with the post-translational modifications required to obtain functional recombinant proteins from eukaryotes. Thus, at the present time, only a few studies have been published on the enzyme activity-based screening of metatranscriptomic libraries (making it possible to do away with introns) of eukaryotes from soil, rumen and the gut of the termite (Bailly et al., 2007; Findley et al., 2011, Sethi et al., 2013).
Regardless of the type of library screened, the functional exploration of hundreds of thousands of clones is required, whereas the hit rate rarely exceeds 6‰ (Duan et al., 2009; Bastien et al., 2013). This requires very high throughput primary screens, in a solid medium before or after the automated organization of libraries in 96- or 384-well micro-plate format, in a liquid medium after enzymatic cell lysis and/or thawing and freezing (Bao et al., 2011), or using UV-inducible auto-lytic vectors (Li et al., 2007). This stage is very often followed by medium or low throughput characterization of the properties of the hits obtained, particularly to assess their biotechnological interest (Tasse et al., 2010).
Two generic strategies, used at throughputs exceeding 400,000 tests per week, have been and continue to be applied widely. Positive selection on a medium containing, for example, substrates to be metabolized as the sole source of carbon, can be used to isolate enzymes (Henne et al., 1999), complete catabolic pathways (Cecchini et al., 2013), or membrane transporters (Majerník et al., 2001). This approach also helps easily identify antibiotic resistant genes (Diaz-Torres et al., 2006). The use of chromogenic (Beloqui et al., 2010; Bastien et al., 2013; Nyyssönen et al., 2013), fluorescent (LeCleir et al., 2007), or opalescent substrates or reagents, such as insoluble polymers or proteins (Mayumi et al., 2008; Waschkowitz et al., 2009), or simply the observation of an original clone phenotype, has already enabled the isolation of several 100 catabolic enzymes, like the numerous hydrolases of very varied taxonomic origin (Simon and Daniel, 2009), some of which were coded by genes that are very abundant in the target ecosystem (Jones et al., 2008; Gloux et al., 2011), but also, although much less frequently, new oxidoreductases (Knietsch et al., 2003). Novel enzymes (laccases, esterases and oxygenases in particular) from microbial communities of very diverse origins (soil, water, activated sludge, digestive tracts) have been highlighted for their capacity to degrade pollutants such as nitriles (Robertson and Steer, 2004), lindane (Boubakri et al., 2006), styrene (Van Hellemond et al., 2007), naphthalene (Ono et al., 2007), aliphatic and aromatic carbohydrates (Uchiyama et al., 2004; Brennerova et al., 2009; Lu et al., 2012), organophosphorus (Kambiranda et al., 2009; Math et al., 2010), or plastic materials (Mayumi et al., 2008).
The discovery of proteins involved in prokaryote-eukaryote interactions (Lakhdari et al., 2010) or anabolic pathways is rarer, since it often requires the development of complex screens and lower throughputs. Nonetheless, a few examples of simple screens, based on the aptitude of metagenomic clones to inhibit the growth of a strain by producing antibacterial activity or to complement an auxotrophic strain for a specific compound, have enabled the identification of new pathways for the synthesis of antimicrobials (Brady and Clardy, 2004) or biotin (Entcheva et al., 2001). Nano-technologies, and in particular the latest developments focused on the medium-throughput screening of libraries obtained by combinatorial protein engineering, enable the design of custom microarrays and covered with one to several 100 specific enzymatic substrates, the processing of which may be followed by fluorescence, chemiluminescence, immunodetection, surface plasmon resonance or mass spectrometry (André et al., 2014). Nanostructure-initiator mass spectrometry technology, combining fluorescence and mass spectrometry, is the first example of a functional metagenomic application for the discovery of anabolic enzymes, namely sialyltransferases (Northen et al., 2008).
The Immense Challenges of Ultra-fast Screening (Figure 2)
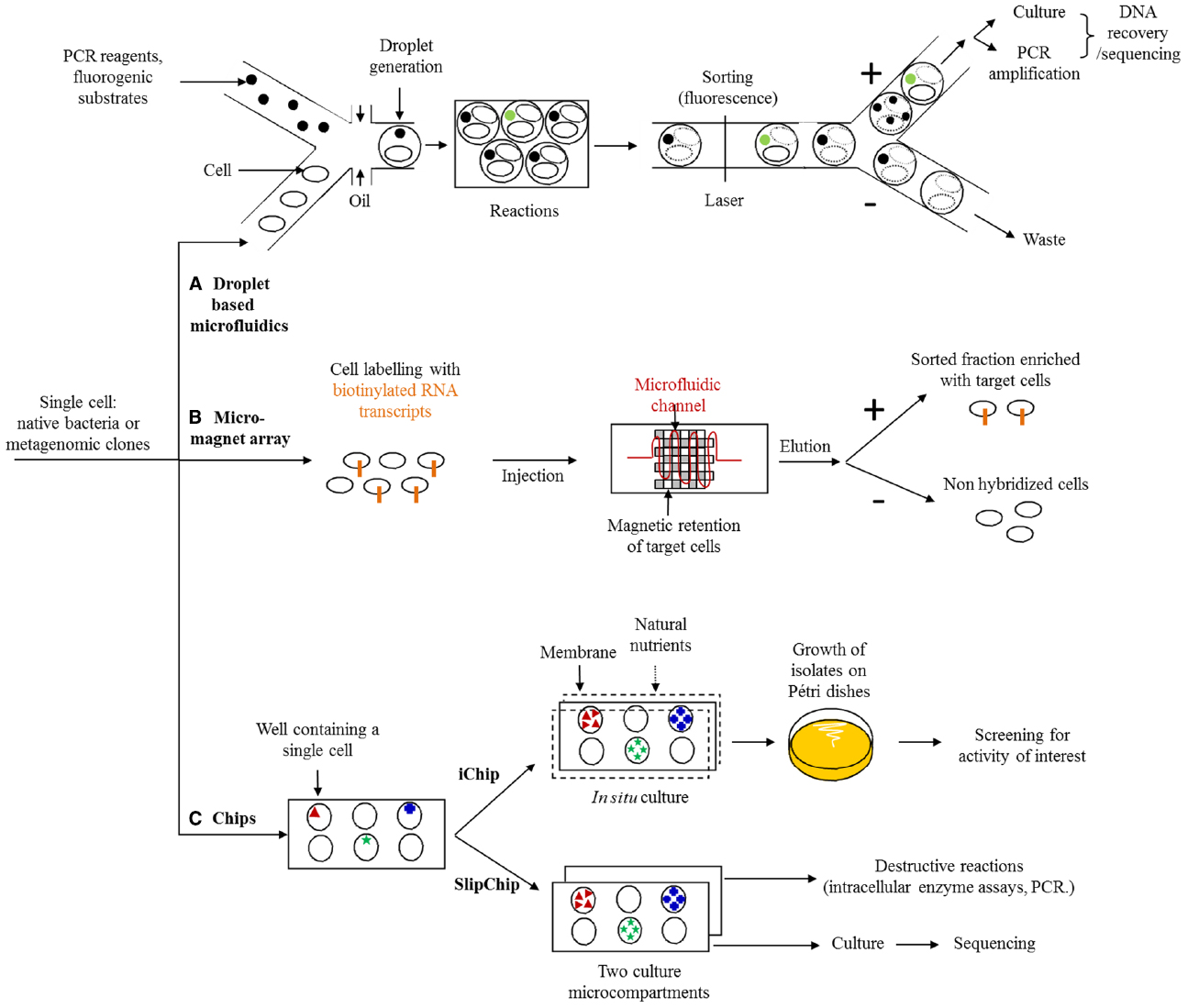
Figure 2. Microfluidic strategies for new enzyme screening. (A) Droplet based microfluidics: single cells are encapsulated with probes or fluorogenic substrates to create microdroplets, where reactions happen (substrate degradation, PCR). The hits are sorted using fluorescence detection. Non-lysed cells are cultured and DNA fragments from lysed cells are amplified. Both methods allow the recovery and sequencing of DNA. (B) Micro-magnet array: target cells are labeled with biotinylated RNA transcripts probes and injected inside the microchannel. Target cells are captured in the channel thanks to magnetic forces while non-targets cells pass through the device. (C) Chips: the chip wells are filled with a single cell. The iChip is covered by membranes, and reintroduced into original environment, where natural nutrients flow through membranes. Colonies are further isolated on Petri dishes to be screened for the activity of interest. The SlipChip is composed of two culture microcompartments which are further separated for destructive and non-destructive assays.
Microfluidic technologies are of undeniable interest when it comes to reaching screening rates of a million clones per day. The substrate induced gene-expression screening method has been developed to use fluorescence-activated cell sorting to isolate plasmidic clones containing genes (or fragments of genes) that induce the expression of a fluorescent marker in response to a specific substrate. However, this technique is only suited to small substrates that are non-lethal and internalizable for the host strain (Uchiyama and Watanabe, 2008). Finally, the advances made over the past few years in cellular compartmentalization (Nawy, 2013), selective sorting, based on sequence detection (Pivetal et al., 2014; Lim et al., 2015) or specific metabolites (Kürsten et al., 2014) and the control of reaction kinetics (Mazutis et al., 2009) in microfluidic circuits should allow for a huge acceleration in the discovery of new proteins and metabolic pathways expressed in prokaryotes and eukaryotes in an intercellular, membrane or extracellular manner.
The very first examples of metagenome functional exploration applications have already been used to establish the proof of concept regarding the effectiveness of microfluidics in the discovery of new bioactive molecules and new enzymes. For example, droplet-based microfluidics technology was recently used by the teams of A. Griffiths and A. Drevelle to isolate new strains producing cellobiohydrolase and cellulase activities at a rate of 300,000 cells sorted per hour, using just a few microliters of reagent, i.e., 250,000 times less than with the conventional technologies mentioned above (Najah et al., 2014). Here, soil bacteria and a fluorescent substrate were co-encapsulated in micro-droplets in order to sort cells on the basis of the extracellular activity only. In fact, the strategy used, which requires the seeding of cells on a defined medium after sorting, is not compatible with the detection of intracellular enzymes, which require a lethal lysis step to convert the substrate. Applying a similar principle, the ultra-rapid sorting of eukaryote cells encapsulated with their substrate now also makes it possible to select yeast clones presenting extracellular enzymatic activities (Sjostrom et al., 2014). This technology should, in the short term, make it possible to explore the functional diversity of uncultivated eukaryotes at a very high throughput, by directly sorting fungal populations or libraries of metatranscriptomic clones. In the latter case, access to the sequence involved in the target activity will be easy, since the libraries are built using hosts whose culture is well managed, with insertion of the metatranscriptomic cDNA fragment into a specific region of the genome. Where sorting is done without cloning of the metagenome or metatranscriptome, only microorganisms capable of growth on a defined medium can be recovered, which hugely limits access to functional diversity.
To increase the proportion of cultivable organisms, Kim Lewis’ team recently used the iChip to simultaneously isolate and cultivate soil bacteria thanks to the delivery of nutrients from the original medium, into which the iChip is introduced, via semi-permeable membranes. This method enables an increase in cultivable organisms ranging from 1 to 50%. Using colonies cultivated in the chip, the clones isolated in a Petri dish were screened for the production of antimicrobial compounds (Ling et al., 2015). A novel antibiotic was thus identified, together with its biosynthesis pathway, after sequencing and functional annotation of the complete genome.
It is quite another matter when it comes to selecting, on the basis of intracellular activity, completely uncultivable organisms or metagenomic clones containing DNA inserts of several dozen kbp, which are difficult to amplify using PCR. In this case, to liberate the enzymes in question, we are required to include a cellular lysis step, preventing seeding after sorting. On the other hand, this approach is compatible with the sorting of plasmid clone libraries, where the metagenomic or metatranscriptomic inserts can easily be amplified using PCR, on the basis of just a few dozen lysed cells. For libraries with large DNA inserts, the barriers are now being broken down, most notably thanks to the development of the SlipChips microfluidic approach (Ma et al., 2014), which uses two culture microcompartments, where the content of one can be lysed for the detection of enzymatic activities, for example, and the other is used as a backup replicate for the culture and recovery of subsequent DNA for sequencing. In spite of these recent, highly encouraging developments, the proof of concept has not yet been established for the identification of new functions and intracellular metabolic pathways.
Conclusion
The rapid expansion of meta-omic technologies over the past decade has shed light on the functions of the uncultivated fraction of microbial ecosystems. A huge number of enzymes have been discovered, in particular through experimental approaches to functional metagenomes exploration. Where their performance can be rapidly assessed within the framework of a known process, or where they catalyze new, previously undescribed reactions, many of them have provided new tools for industrial biotechnologies. However, several challenges still need to be addressed to speed up the rate at which new functions are discovered and to make optimal use of the functional diversity that so far remains unexplored. Firstly, while the uncultivated prokaryote fraction of microbial communities is still extensively studied, the functions of the eukaryote fraction are relatively unexplored from an experimental angle, even though they play a fundamental role for numerous ecosystems. Secondly, in the majority of cases, the functions discovered using meta-omic approaches play a catabolic role, mainly involved in the deconstruction of plant biomass or in bioremediation. It is thus necessary to develop functional screens to access anabolic functions and enrich the catalog of reactions available for synthetic biology. Finally, there are very few studies aimed at identifying the role of protein families that are highly prevalent in the target ecosystem but that have not yet been characterized, even though some of them could be considered as biomarkers of the functional state of the microbial community. Indeed, sequence-based functional metagenomic projects continuously highlight many sequences annotated as domains of unknown function in the Pfam database (RRID: nlx_72111) (Bateman et al., 2010; Finn et al., 2014), some with 3D structures solved thanks to structural genomics initiatives, and available in the Protein Data Bank (RRID: nif-0000-00135). With the goal of characterizing these new protein families and identifying previously unseen functions from the selection the most prevalent protein families (those containing the highest number of homologous sequences without any associated function) in the target ecosystem, the integration of structural, biochemical, genomic and meta-omic data is now also possible (Ladevèze et al., 2013). It allows to benefit from the huge amount of long scaffolds now available in sequence databases, and to access the genomic context of the targeted genes in order to facilitate functional assignation. In the next few years, these strategies should enhance our understanding of how microbial ecosystems function and, at the same time, enable greater control over them.
Author Contributions
LU, GPV, EL contributed equally to this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was funded by the Ministry of Education and Research (Ministère de l’Enseignement supérieur et de la Recherche, MESR), the Agence Nationale de la Recherche (Grant Number ANR 2011-Nano 007 03) and the INRA metaprogramme M2E (project Metascreen).
Footnotes
- ^ http://www.genomesonline.org/cgi-bin/GOLD/index
- ^ http://metagenomics.anl.gov/
- ^ http://www.ebi.ac.uk/metagenomics
References
André, I., Potocki-Véronèse, G., Barbe, S., Moulis, C., and Remaud-Siméon, M. (2014). CAZyme discovery and design for sweet dreams. Curr. Opin. Chem. Biol. 19, 17–24. doi: 10.1016/j.cbpa.2013.11.014
Ausec, L., van Elsas, J. D., and Mandic-Mulec, I. (2011). Two- and three-domain bacterial laccase-like genes are present in drained peat soils. Soil Biol. Biochem. 43, 975–983. doi: 10.1016/j.soilbio.2011.01.013
Bailly, J., Fraissinet-Tachet, L., Verner, M.-C., Debaud, J.-C., Lemaire, M., Wésolowski-Louvel, M., et al. (2007). Soil eukaryotic functional diversity, a metatranscriptomic approach. ISME J. 1, 632–642. doi: 10.1038/ismej.2007.68
Bao, L., Huang, Q., Chang, L., Zhou, J., and Lu, H. (2011). Screening and characterization of a cellulase with endocellulase and exocellulase activity from yak rumen metagenome. J. Mol. Catal. B Enzym. 73, 104–110. doi: 10.1016/j.molcatb.2011.08.006
Bartossek, R., Nicol, G. W., Lanzen, A., Klenk, H.-P., and Schleper, C. (2010). Homologues of nitrite reductases in ammonia-oxidizing archaea: diversity and genomic context. Environ. Microbiol. 12, 1075–1088. doi: 10.1111/j.1462-2920.2010.02153.x
Bastien, G., Arnal, G., Bozonnet, S., Laguerre, S., Ferreira, F., Fauré, R., et al. (2013). Mining for hemicellulases in the fungus-growing termite Pseudacanthotermes militaris using functional metagenomics. Biotechnol. Biofuels 6, 78. doi: 10.1186/1754-6834-6-78
Bateman, A., Coggill, P., and Finn, R. D. (2010). DUFs: families in search of function. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1148–1152. doi: 10.1107/S1744309110001685
Beloqui, A., Polaina, J., Vieites, J. M., Reyes-Duarte, D., Torres, R., Golyshina, O. V., et al. (2010). Novel hybrid esterase-haloacid dehalogenase enzyme. Chembiochem 11, 1975–1978. doi: 10.1002/cbic.201000258
Boubakri, H., Beuf, M., Simonet, P., and Vogel, T. M. (2006). Development of metagenomic DNA shuffling for the construction of a xenobiotic gene. Gene 375, 87–94. doi: 10.1016/j.gene.2006.02.027
Brady, S. F., and Clardy, J. (2004). Palmitoylputrescine, an antibiotic isolated from the heterologous expression of DNA extracted from bromeliad tank water. J. Nat. Prod. 67, 1283–1286. doi: 10.1021/np0499766
Bragalini, C., Ribiere, C., Parisot, N., Vallon, L., Prudent, E., Peyretaillade, E., et al. (2014). Solution hybrid selection capture for the recovery of functional full-length eukaryotic cDNAs from complex environmental samples. DNA Res. 21, 685–694. doi: 10.1093/dnares/dsu030
Brennerova, M. V., Josefiova, J., Brenner, V., Pieper, D. H., and Junca, H. (2009). Metagenomics reveals diversity and abundance of meta-cleavage pathways in microbial communities from soil highly contaminated with jet fuel under air-sparging bioremediation. Environ. Microbiol. 11, 2216–2227. doi: 10.1111/j.1462-2920.2009.01943.x
Cantarel, B. L., Lombard, V., and Henrissat, B. (2012). Complex carbohydrate utilization by the healthy human microbiome. PLoS ONE 7:e28742. doi: 10.1371/journal.pone.0028742
Cantu, D. C., Chen, Y., Lemons, M. L., and Reilly, P. J. (2011). ThYme: a database for thioester-active enzymes. Nucleic Acids Res. 39, D342–D346. doi: 10.1093/nar/gkq1072
Cecchini, D. A., Laville, E., Laguerre, S., Robe, P., Leclerc, M., Doré, J., et al. (2013). Functional metagenomics reveals novel pathways of prebiotic breakdown by human gut bacteria. PLoS ONE 8:e72766. doi: 10.1371/journal.pone.0072766
Chen, Y., and Murrell, J. C. (2010). When metagenomics meets stable-isotope probing: progress and perspectives. Trends Microbiol. 18, 157–163. doi: 10.1016/j.tim.2010.02.002
Culligan, E. P., Sleator, R. D., Marchesi, J. R., and Hill, C. (2014). Metagenomics and novel gene discovery: promise and potential for novel therapeutics. Virulence 5, 399–412. doi: 10.4161/viru.27208
Damon, C., Lehembre, F., Oger-Desfeux, C., Luis, P., Ranger, J., Fraissinet-Tachet, L., et al. (2012). Metatranscriptomics reveals the diversity of genes expressed by eukaryotes in forest soils. PLoS ONE 7:e28967. doi: 10.1371/journal.pone.0028967
DeAngelis, K. M., Gladden, J. M., Allgaier, M., D’haeseleer, P., Fortney, J. L., Reddy, A., et al. (2010). Strategies for enhancing the effectiveness of metagenomic-based enzyme discovery in lignocellulolytic microbial communities. BioEnergy Res. 3, 146–158. doi: 10.1007/s12155-010-9089-z
Diaz-Torres, M. L., Villedieu, A., Hunt, N., McNab, R., Spratt, D. A., Allan, E., et al. (2006). Determining the antibiotic resistance potential of the indigenous oral microbiota of humans using a metagenomic approach. FEMS Microbiol. Lett. 258, 257–262. doi: 10.1111/j.1574-6968.2006.00221.x
Di Bella, J. M., Bao, Y., Gloor, G. B., Burton, J. P., and Reid, G. (2013). High throughput sequencing methods and analysis for microbiome research. J. Microbiol. Methods 95, 401–414. doi: 10.1016/j.mimet.2013.08.011
Duan, C.-J., Xian, L., Zhao, G.-C., Feng, Y., Pang, H., Bai, X.-L., et al. (2009). Isolation and partial characterization of novel genes encoding acidic cellulases from metagenomes of buffalo rumens. J. Appl. Microbiol. 107, 245–256. doi: 10.1111/j.1365-2672.2009.04202.x
Ekkers, D. M., Cretoiu, M. S., Kielak, A. M., and van Elsas, J. D. (2012). The great screen anomaly—a new frontier in product discovery through functional metagenomics. Appl. Microbiol. Biotechnol. 93, 1005–1020. doi: 10.1007/s00253-011-3804-3
Entcheva, P., Liebl, W., Johann, A., Hartsch, T., and Streit, W. R. (2001). Direct cloning from enrichment cultures, a reliable strategy for isolation of complete operons and genes from microbial consortia. Appl. Environ. Microbiol. 67, 89–99. doi: 10.1128/AEM.67.1.89-99.2001
Erickson, A. R., Cantarel, B. L., Lamendella, R., Darzi, Y., Mongodin, E. F., Pan, C., et al. (2012). Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS ONE 7:e49138. doi: 10.1371/journal.pone.0049138
Ferrer, M., Beloqui, A., Timmis, K. N., and Golyshin, P. N. (2009). Metagenomics for mining new genetic resources of microbial communities. J. Mol. Microbiol. Biotechnol. 16, 109–123. doi: 10.1159/000142898
Ferrer, M., Golyshina, O. V., Chernikova, T. N., Khachane, A. N., Martins Dos Santos, V. A. P., Yakimov, M. M., et al. (2005). Microbial enzymes mined from the Urania deep-sea hypersaline anoxic basin. Chem. Biol. 12, 895–904. doi: 10.1016/j.chembiol.2005.05.020
Findley, S. D., Mormile, M. R., Sommer-Hurley, A., Zhang, X.-C., Tipton, P., Arnett, K., et al. (2011). Activity-based metagenomic screening and biochemical characterization of bovine ruminal protozoan glycoside hydrolases. Appl. Environ. Microbiol. 77, 8106–8113. doi: 10.1128/AEM.05925-11
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., et al. (2010). The Pfam protein families database. Nucleic Acids Res. 38, D211–D222. doi: 10.1093/nar/gkp985
Frias-Lopez, J., Shi, Y., Tyson, G. W., Coleman, M. L., Schuster, S. C., Chisholm, S. W., et al. (2008). Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U.S.A. 105, 3805–3810. doi: 10.1073/pnas.0708897105
Gabor, E. M., Alkema, W. B. L., and Janssen, D. B. (2004). Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ. Microbiol. 6, 879–886. doi: 10.1111/j.1462-2920.2004.00640.x
Gilbert, J. A., Field, D., Huang, Y., Edwards, R., Li, W., Gilna, P., et al. (2008). Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS ONE 3:e3042. doi: 10.1371/journal.pone.0003042
Gilbert, J. A., Field, D., Swift, P., Thomas, S., Cummings, D., Temperton, B., et al. (2010). The taxonomic and functional diversity of microbes at a temperate coastal site: a “Multi-Omic” study of seasonal and diel temporal variation. PLoS ONE 5:e15545. doi: 10.1371/journal.pone.0015545
Gloux, K., Berteau, O., El oumami, H., Beguet, F., Leclerc, M., and Dore, J. (2011). A metagenomic β-glucuronidase uncovers a core adaptive function of the human intestinal microbiome. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1), 4539–4546. doi: 10.1073/pnas.1000066107
He, S., Kunin, V., Haynes, M., Martin, H. G., Ivanova, N., Rohwer, F., et al. (2010). Metatranscriptomic array analysis of “Candidatus Accumulibacter phosphatis”-enriched enhanced biological phosphorus removal sludge: metatranscriptomic array analysis of EBPR sludge. Environ. Microbiol. 12, 1205–1217. doi: 10.1111/j.1462-2920.2010.02163.x
Henne, A., Daniel, R., Schmitz, R. A., and Gottschalk, G. (1999). Construction of environmental DNA libraries in Escherichia coli and screening for the presence of genes conferring utilization of 4-hydroxybutyrate. Appl. Environ. Microbiol. 65, 3901–3907.
Hess, M., Sczyrba, A., Egan, R., Kim, T.-W., Chokhawala, H., Schroth, G., et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467. doi: 10.1126/science.1200387
Hjort, K., Bergström, M., Adesina, M. F., Jansson, J. K., Smalla, K., and Sjöling, S. (2010). Chitinase genes revealed and compared in bacterial isolates, DNA extracts and a metagenomic library from a phytopathogen-suppressive soil. FEMS Microbiol. Ecol. 71, 197–207. doi: 10.1111/j.1574-6941.2009.00801.x
Iwai, S., Chai, B., Sul, W. J., Cole, J. R., Hashsham, S. A., and Tiedje, J. M. (2009). Gene-targeted-metagenomics reveals extensive diversity of aromatic dioxygenase genes in the environment. ISME J. 4, 279–285. doi: 10.1038/ismej.2009.104
Jacquiod, S., Franqueville, L., Cécillon, S., Vogel, T. M., and Simonet, P. (2013). Soil bacterial community shifts after chitin enrichment: an integrative metagenomic approach. PLoS ONE 8:e79699. doi: 10.1371/journal.pone.0079699
Jones, B. V., Begley, M., Hill, C., Gahan, C. G. M., and Marchesi, J. R. (2008). Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. U.S.A. 105, 13580–13585. doi: 10.1073/pnas.0804437105
Kambiranda, D. M., Asraful-Islam, S. M., Cho, K. M., Math, R. K., Lee, Y. H., Kim, H., et al. (2009). Expression of esterase gene in yeast for organophosphates biodegradation. Pestic. Biochem. Physiol. 94, 15–20. doi: 10.1016/j.pestbp.2009.02.006
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Knietsch, A., Waschkowitz, T., Bowien, S., Henne, A., and Daniel, R. (2003). Construction and screening of metagenomic libraries derived from enrichment cultures: generation of a gene bank for genes conferring alcohol oxidoreductase activity on Escherichia coli. Appl. Environ. Microbiol. 69, 1408–1416. doi: 10.1128/AEM.69.3.1408-1416.2003
Kürsten, D., Kothe, E., Wetzel, K., Bergmann, K., and Köhler, J. M. (2014). Micro-segmented flow and multisensor-technology for microbial activity profiling. Environ. Sci. Process. Impacts 16, 2362–2370. doi: 10.1039/C4EM00255E
Ladevèze, S., Tarquis, L., Cecchini, D. A., Bercovici, J., André, I., Topham, C. M., et al. (2013). Role of glycoside phosphorylases in mannose foraging by human gut bacteria. J. Biol. Chem. 288, 32370–32383. doi: 10.1074/jbc.M113.483628
Ladoukakis, E., Kolisis, F. N., and Chatziioannou, A. A. (2014). Integrative workflows for metagenomic analysis. Front. Cell Dev. Biol. 2:70. doi: 10.3389/fcell.2014.00070
Lakhdari, O., Cultrone, A., Tap, J., Gloux, K., Bernard, F., Ehrlich, S. D., et al. (2010). Functional metagenomics: a high throughput screening method to decipher microbiota-driven NF-κB modulation in the human gut. PLoS ONE 5:e13092. doi: 10.1371/journal.pone.0013092
LeCleir, G. R., Buchan, A., Maurer, J., Moran, M. A., and Hollibaugh, J. T. (2007). Comparison of chitinolytic enzymes from an alkaline, hypersaline lake and an estuary. Environ. Microbiol. 9, 197–205. doi: 10.1111/j.1462-2920.2006.01128.x
Levasseur, A., Drula, E., Lombard, V., Coutinho, P. M., and Henrissat, B. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6, 41. doi: 10.1186/1754-6834-6-41
Li, M., Hong, Y., Klotz, M. G., and Gu, J.-D. (2010). A comparison of primer sets for detecting 16S rRNA and hydrazine oxidoreductase genes of anaerobic ammonium-oxidizing bacteria in marine sediments. Appl. Microbiol. Biotechnol. 86, 781–790. doi: 10.1007/s00253-009-2361-5
Li, S., Xu, L., Hua, H., Ren, C., and Lin, Z. (2007). A set of UV-inducible autolytic vectors for high throughput screening. J. Biotechnol. 127, 647–652. doi: 10.1016/j.jbiotec.2006.07.030
Lim, S. W., Tran, T. M., and Abate, A. R. (2015). PCR-activated cell sorting for cultivation-free enrichment and sequencing of rare microbes. PLoS ONE 10:e0113549. doi: 10.1371/journal.pone.0113549
Ling, L. L., Schneider, T., Peoples, A. J., Spoering, A. L., Engels, I., Conlon, B. P., et al. (2015). A new antibiotic kills pathogens without detectable resistance. Nature 517, 455–459. doi: 10.1038/nature14098
Lu, Z., Deng, Y., Van Nostrand, J. D., He, Z., Voordeckers, J., Zhou, A., et al. (2012). Microbial gene functions enriched in the Deepwater Horizon deep-sea oil plume. ISME J. 6, 451–460. doi: 10.1038/ismej.2011.91
Ma, L., Datta, S. S., Karymov, M. A., Pan, Q., Begolo, S., and Ismagilov, R. F. (2014). Individually addressable arrays of replica microbial cultures enabled by splitting SlipChips. Integr. Biol. 6, 796–805. doi: 10.1039/C4IB00109E
Majerník, A., Gottschalk, G., and Daniel, R. (2001). Screening of environmental DNA libraries for the presence of genes conferring Na+(Li+)/H+ antiporter activity on Escherichia coli: characterization of the recovered genes and the corresponding gene products. J. Bacteriol. 183, 6645–6653. doi: 10.1128/JB.183.22.6645-6653.2001
Marchler-Bauer, A., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., DeWeese-Scott, C., Fong, J. H., et al. (2009). CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 37, D205–D210. doi: 10.1093/nar/gkn845
Markowitz, V. M., Ivanova, N. N., Szeto, E., Palaniappan, K., Chu, K., Dalevi, D., et al. (2007). IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res. 36, D534–D538. doi: 10.1093/nar/gkm869
Math, R. K., Asraful Islam, S. M., Cho, K. M., Hong, S. J., Kim, J. M., Yun, M. G., et al. (2010). Isolation of a novel gene encoding a 3,5,6-trichloro-2-pyridinol degrading enzyme from a cow rumen metagenomic library. Biodegradation 21, 565–573. doi: 10.1007/s10532-009-9324-5
Mayumi, D., Akutsu-Shigeno, Y., Uchiyama, H., Nomura, N., and Nakajima-Kambe, T. (2008). Identification and characterization of novel poly (DL-lactic acid) depolymerases from metagenome. Appl. Microbiol. Biotechnol. 79, 743–750. doi: 10.1007/s00253-008-1477-3
Mazutis, L., Baret, J.-C., and Griffiths, A. D. (2009). A fast and efficient microfluidic system for highly selective one-to-one droplet fusion. Lab Chip. 9, 2665–2672. doi: 10.1039/b903608c
Meyer, F., Paarmann, D., D’Souza, M., Olson, R., Glass, E. M., Kubal, M., et al. (2008). The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386
Muller, J., Szklarczyk, D., Julien, P., Letunic, I., Roth, A., Kuhn, M., et al. (2010). eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 38, D190–D195. doi: 10.1093/nar/gkp951
Najah, M., Calbrix, R., Mahendra-Wijaya, I. P., Beneyton, T., Griffiths, A. D., and Drevelle, A. (2014). Droplet-based microfluidics platform for ultra-high-throughput bioprospecting of cellulolytic microorganisms. Chem. Biol. 21, 1722–1732. doi: 10.1016/j.chembiol.2014.10.020
Nawy, T. (2013). Lab-On-A-Chip: receptive cells feel the squeeze. Nat. Methods 10, 198–198. doi: 10.1038/nmeth.2395
Northen, T. R., Lee, J.-C., Hoang, L., Raymond, J., Hwang, D.-R., Yannone, S. M., et al. (2008). A nanostructure-initiator mass spectrometry-based enzyme activity assay. Proc. Natl. Acad. Sci. U.S.A. 105, 3678–3683. doi: 10.1073/pnas.0712332105
Nyyssönen, M., Tran, H. M., Karaoz, U., Weihe, C., Hadi, M. Z., Martiny, J. B. H., et al. (2013). Coupled high-throughput functional screening and next generation sequencing for identification of plant polymer decomposing enzymes in metagenomic libraries. Front. Microbiol. 4:282. doi: 10.3389/fmicb.2013.00282
Ono, A., Miyazaki, R., Sota, M., Ohtsubo, Y., Nagata, Y., and Tsuda, M. (2007). Isolation and characterization of naphthalene-catabolic genes and plasmids from oil-contaminated soil by using two cultivation-independent approaches. Appl. Microbiol. Biotechnol. 74, 501–510. doi: 10.1007/s00253-006-0671-4
Park, B. H., Karpinets, T. V., Syed, M. H., Leuze, M. R., and Uberbacher, E. C. (2010). CAZymes Analysis Toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 20, 1574–1584. doi: 10.1093/glycob/cwq106
Pivetal, J., Toru, S., Frenea-Robin, M., Haddour, N., Cecillon, S., Dempsey, N. M., et al. (2014). Selective isolation of bacterial cells within a microfluidic device using magnetic probe-based cell fishing. Sens. Actuators B Chem. 195, 581–589. doi: 10.1016/j.snb.2014.01.004
Pleiss, J., Fischer, M., Peiker, M., Thiele, C., and Schmid, R. D. (2000). Lipase engineering database. J. Mol. Catal. B Enzym. 10, 491–508. doi: 10.1016/S1381-1177(00)00092-8
Poretsky, R. S., Bano, N., Buchan, A., LeCleir, G., Kleikemper, J., Pickering, M., et al. (2005). Analysis of microbial gene transcripts in environmental samples. Appl. Environ. Microbiol. 71, 4121–4126. doi: 10.1128/AEM.71.7.4121-4126.2005
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
Ram, R. J., Verberkmoes, N. C., Thelen, M. P., Tyson, G. W., Baker, B. J., Blake, R. C., et al. (2005). Community proteomics of a natural microbial biofilm. Science 308, 1915–1920. doi: 10.1126/science.1109070
Rawlings, N. D., Barrett, A. J., and Bateman, A. (2012). MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–D350. doi: 10.1093/nar/gkr987
Robertson, D. E., and Steer, B. A. (2004). Recent progress in biocatalyst discovery and optimization. Curr. Opin. Chem. Biol. 8, 141–149. doi: 10.1016/j.cbpa.2004.02.010
Saleh-Lakha, S., Miller, M., Campbell, R. G., Schneider, K., Elahimanesh, P., Hart, M. M., et al. (2005). Microbial gene expression in soil: methods, applications and challenges. J. Microbiol. Methods 63, 1–19. doi: 10.1016/j.mimet.2005.03.007
Schmidt, O., Drake, H. L., and Horn, M. A. (2010). Hitherto unknown [Fe-Fe]-hydrogenase gene diversity in anaerobes and anoxic enrichments from a moderately acidic fen. Appl. Environ. Microbiol. 76, 2027–2031. doi: 10.1128/AEM.02895-09
Schmieder, R., Lim, Y. W., and Edwards, R. (2012). Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 28, 433–435. doi: 10.1093/bioinformatics/btr669
Selengut, J. D., Haft, D. H., Davidsen, T., Ganapathy, A., Gwinn-Giglio, M., Nelson, W. C., et al. (2007). TIGRFAMs and genome properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res. 35, D260–D264. doi: 10.1093/nar/gkl1043
Sethi, A., Slack, J. M., Kovaleva, E. S., Buchman, G. W., and Scharf, M. E. (2013). Lignin-associated metagene expression in a lignocellulose-digesting termite. Insect Biochem. Mol. Biol. 43, 91–101. doi: 10.1016/j.ibmb.2012.10.001
Sharma, V. K., Kumar, N., Prakash, T., and Taylor, T. D. (2010). MetaBioME: a database to explore commercially useful enzymes in metagenomic datasets. Nucleic Acids Res. 38, D468–D472. doi: 10.1093/nar/gkp1001
Sigrist, C. J. A., Cerutti, L., de Castro, E., Langendijk-Genevaux, P. S., Bulliard, V., Bairoch, A., et al. (2010). PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 38, D161–D166. doi: 10.1093/nar/gkp885
Simon, C., and Daniel, R. (2009). Achievements and new knowledge unraveled by metagenomic approaches. Appl. Microbiol. Biotechnol. 85, 265–276. doi: 10.1007/s00253-009-2233-z
Simon, C., Herath, J., Rockstroh, S., and Daniel, R. (2009). Rapid identification of genes encoding DNA polymerases by function-based screening of metagenomic libraries derived from glacial ice. Appl. Environ. Microbiol. 75, 2964–2968. doi: 10.1128/AEM.02644-08
Sirim, D., Wagner, F., Wang, L., Schmid, R. D., and Pleiss, J. (2011). The Laccase Engineering Database: a classification and analysis system for laccases and related multicopper oxidases. Database 2011, bar006. doi: 10.1093/database/bar006
Sjostrom, S. L., Bai, Y., Huang, M., Liu, Z., Nielsen, J., Joensson, H. N., et al. (2014). High-throughput screening for industrial enzyme production hosts by droplet microfluidics. Lab Chip 14, 806–813. doi: 10.1039/C3LC51202A
Söding, J. (2005). Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960. doi: 10.1093/bioinformatics/bti125
Suenaga, H., Ohnuki, T., and Miyazaki, K. (2007). Functional screening of a metagenomic library for genes involved in microbial degradation of aromatic compounds. Environ. Microbiol. 9, 2289–2297. doi: 10.1111/j.1462-2920.2007.01342.x
Steele, H. L., Jaeger, K.-E., Daniel, R., and Streit, W. R. (2009). Advances in recovery of novel biocatalysts from metagenomes. J. Mol. Microbiol. Biotechnol. 16, 25–37. doi: 10.1159/000142892
Tartar, A., Wheeler, M. M., Zhou, X., Coy, M. R., Boucias, D. G., and Scharf, M. E. (2009). Parallel metatranscriptome analyses of host and symbiont gene expression in the gut of the termite Reticulitermes flavipes. Biotechnol. Biofuels 2, 25. doi: 10.1186/1754-6834-2-25
Tasse, L., Bercovici, J., Pizzut-Serin, S., Robe, P., Tap, J., Klopp, C., et al. (2010). Functional metagenomics to mine the human gut microbiome for dietary fiber catabolic enzymes. Genome Res. 20, 1605–1612. doi: 10.1101/gr.108332.110
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Taupp, M., Mewis, K., and Hallam, S. J. (2011). The art and design of functional metagenomic screens. Curr. Opin. Biotechnol. 22, 465–472. doi: 10.1016/j.copbio.2011.02.010
Thomas, T., Gilbert, J., and Meyer, F. (2012). Metagenomics—a guide from sampling to data analysis. Microb. Inform. Exp. 2, 3. doi: 10.1186/2042-5783-2-3
Tirawongsaroj, P., Sriprang, R., Harnpicharnchai, P., Thongaram, T., Champreda, V., Tanapongpipat, S., et al. (2008). Novel thermophilic and thermostable lipolytic enzymes from a Thailand hot spring metagenomic library. J. Biotechnol. 133, 42–49. doi: 10.1016/j.jbiotec.2007.08.046
Uchiyama, T., Abe, T., Ikemura, T., and Watanabe, K. (2004). Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat. Biotechnol. 23, 88–93. doi: 10.1038/nbt1048
Uchiyama, T., and Miyazaki, K. (2009). Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr. Opin. Biotechnol. 20, 616–622. doi: 10.1016/j.copbio.2009.09.010
Uchiyama, T., and Watanabe, K. (2008). Substrate-induced gene expression (SIGEX) screening of metagenome libraries. Nat. Protoc. 3, 1202–1212. doi: 10.1038/nprot.2008.96
van Elsas, J. D., Costa, R., Jansson, J., Sjöling, S., Bailey, M., Nalin, R., et al. (2008). The metagenomics of disease-suppressive soils—experiences from the METACONTROL project. Trends Biotechnol. 26, 591–601. doi: 10.1016/j.tibtech.2008.07.004
Van Hellemond, E. W., Janssen, D. B., and Fraaije, M. W. (2007). Discovery of a novel styrene monooxygenase originating from the metagenome. Appl. Environ. Microbiol. 73, 5832–5839. doi: 10.1128/AEM.02708-06
Vogel, T. M., Simonet, P., Jansson, J. K., Hirsch, P. R., Tiedje, J. M., van Elsas, J. D., et al. (2009). TerraGenome: a consortium for the sequencing of a soil metagenome. Nat. Rev. Microbiol. 7, 252–252. doi: 10.1038/nrmicro2119
Warnecke, F., and Hess, M. (2009). A perspective: metatranscriptomics as a tool for the discovery of novel biocatalysts. J. Biotechnol. 142, 91–95. doi: 10.1016/j.jbiotec.2009.03.022
Warnecke, F., Luginbühl, P., Ivanova, N., Ghassemian, M., Richardson, T. H., Stege, J. T., et al. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565. doi: 10.1038/nature06269
Waschkowitz, T., Rockstroh, S., and Daniel, R. (2009). Isolation and Characterization of metalloproteases with a novel domain structure by construction and screening of metagenomic libraries. Appl. Environ. Microbiol. 75, 2506–2516. doi: 10.1128/AEM.02136-08
Weckx, S., Van der Meulen, R., Allemeersch, J., Huys, G., Vandamme, P., Van Hummelen, P., et al. (2010). Community dynamics of bacteria in sourdough fermentations as revealed by their metatranscriptome. Appl. Environ. Microbiol. 76, 5402–5408. doi: 10.1128/AEM.00570-10
Yooseph, S., Sutton, G., Rusch, D. B., Halpern, A. L., Williamson, S. J., Remington, K., et al. (2007). The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol. 5:e16. doi: 10.1371/journal.pbio.0050016
Zanaroli, G., Balloi, A., Negroni, A., Daffonchio, D., Young, L. Y., and Fava, F. (2010). Characterization of the microbial community from the marine sediment of the Venice lagoon capable of reductive dechlorination of coplanar polychlorinated biphenyls (PCBs). J. Hazard. Mater. 178, 417–426. doi: 10.1016/j.jhazmat.2010.01.097
Keywords: metagenomics, discovery of new functions, proteins, high throughput screening, microbial ecosystems, microbial ecology, biotechnologies
Citation: Ufarté L, Potocki-Veronese G and Laville É (2015) Discovery of new protein families and functions: new challenges in functional metagenomics for biotechnologies and microbial ecology. Front. Microbiol. 6:563. doi: 10.3389/fmicb.2015.00563
Received: 17 April 2015; Accepted: 21 May 2015;
Published: 05 June 2015.
Edited by:
Eamonn P. Culligan, University College Cork, IrelandReviewed by:
Marc Strous, University of Calgary, CanadaLukasz Jaroszewski, Sanford-Burnham Institute for Medical Research, USA
Copyright © 2015 Ufarté, Potocki-Veronese and Laville. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Élisabeth Laville, Equipe de Catalyse et Ingénierie Moléculaire Enzymatiques, Laboratoire d’Ingénierie des Systèmes Biologiques et des Procédés, INSA - UMR INRA 792 - UMR CNRS 5504, 135 Avenue de Rangueil, 31077 Toulouse cedex 4, France, laville@insa-toulouse.fr