 Sergio López-Madrigal
Sergio López-Madrigal Amparo Latorre
Amparo Latorre Andrés Moya
Andrés Moya Rosario Gil
Rosario Gil- 1Institut Cavanilles de Biodiversitat i Biologia Evolutiva, Universitat de València, València, Spain
- 2Área de Genómica y Salud de la Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunitat Valenciana (FISABIO) – Salud Pública, València, Spain
Many insect species establish mutualistic symbiosis with intracellular bacteria that complement their unbalanced diets. The betaproteobacterium “Candidatus Tremblaya” maintains an ancient symbiosis with mealybugs (Hemiptera: Pseudococcidae), which are classified in subfamilies Phenacoccinae and Pseudococcinae. Most Phenacoccinae mealybugs have “Candidatus Tremblaya phenacola” as their unique endosymbiont, while most Pseudococcinae mealybugs show a nested symbiosis (a bacterial symbiont placed inside another one) where every “Candidatus Tremblaya princeps” cell harbors several cells of a gammaproteobacterium. Genomic characterization of the endosymbiotic consortium from Planococcus citri, composed by “Ca. Tremblaya princeps” and “Candidatus Moranella endobia,” unveiled several atypical features of the former's genome, including the concerted evolution of paralogous loci. Its comparison with the genome of “Ca. Tremblaya phenacola” PAVE, single endosymbiont of Phenacoccus avenae, suggests that the atypical reductive evolution of “Ca. Tremblaya princeps” could be linked to the acquisition of “Ca. Moranella endobia,” which possess an almost complete set of genes encoding proteins involved in homologous recombination. In order to test this hypothesis, we performed comparative genomics between “Ca. Tremblaya phenacola” and “Ca. Tremblaya princeps” and searched for the co-occurrence of concerted evolution and homologous recombination genes in endosymbiotic consortia from four unexplored mealybug species, Dysmicoccus boninsis, Planococcus ficus, Pseudococcus longispinus, and Pseudococcus viburni. Our results support a link between concerted evolution and nested endosymbiosis.
Introduction
The advances in genome sequencing and the development of metagenomic methods have been critical for our knowledge of the bacterial world. Now that complete genomes from closely related species or even from different strains of the same species are available, numerous studies have focused on the diversity of gene repertoire and genome rearrangements (Abby and Daubin, 2007). Horizontal gene transfer (HGT), transposition and intragenomic recombination are known to be important sources of evolutionary novelties, being responsible for bacterial huge metabolic diversity and adaptive potential, which are remarkable among free-living bacteria (Casjens, 1998; Rocha, 2008). However, the analysis of bacteria that have acquired an intracellular host-dependent life-style revealed important constrains to these evolutionary mechanisms.
During the last 15 years, the complete genomes of many endosymbionts (i.e., obligate symbiotic bacteria that live inside eukaryotic cells) have become available. The best studied cases of endosymbiosis involve mutualistic associations with insects. Comparative genomics has allowed the identification of several commonalities among them, which are related with the stage of integration of the bacteria with their respective hosts (Moya et al., 2008; McCutcheon and Moran, 2012). Generally, intracellular bacteria have smaller genomes than their free-living relatives, mostly due to a reduction in their gene content (McCutcheon and Moran, 2012). Gene losses affect loci performing functions that are unnecessary in an intracellular environment or that can be provided by the host. Thus, highly reduced genomes (i.e., those from endosymbionts that have maintained a long relationship with their hosts) have typically lost most genes involved in DNA recombination and repair, present almost no gene duplications, lack transposable elements and prophages and present high levels of structural stability.
Many insects maintain obligate mutualistic symbiosis with more than one bacterial species, so that two evolutionary outcomes are possible: complementation through the establishment of a bacterial consortium or replacement of one endosymbiont by another (Moya et al., 2009). Mealybugs (Hemiptera: Pseudococcidae) are phloem-feeding insects that have been classified in subfamilies Phenacoccinae and Pseudococcinae (Hardy et al., 2008), and present an intricate variety of endosymbiotic relationships. Based on phylogenetic analysis, it has been suggested that a betaproteobacterial ancestor of “Ca. Tremblaya” infected a mealybug ancestor before the split of the two subfamilies. In subfamily Phenacoccinae, “Ca. Tremblaya phenacola” is the obligate endosymbiont in most tested mealybug species, excluding the tribe Rhizoecini and the genus Rastrococcus, where it has been replaced by different Bacteroidetes (Gruwell et al., 2010; Husnik et al., 2013). In subfamily Pseudococcinae, the obligate endosymbiont “Ca. Tremblaya princeps” has been classified in up to six different clusters (A–F) (Thao et al., 2002). Except for the Ferrisia and Maconellicoccus clusters (B and F, respectively), where no additional endosymbiont has been reported, “Ca. Tremblaya princeps” has been recurrently infected by different gammaproteobacteria, establishing nested endosymbiotic consortia in which each “Ca. Tremblaya princeps” cell contains several cells of the corresponding gammaproteobacterium (von Dohlen et al., 2001; Thao et al., 2002; McCutcheon and von Dohlen, 2011; Gatehouse et al., 2012; Koga et al., 2013).
Early approaches to the genomic characterization of “Ca. Tremblaya princeps” revealed several atypical features for an obligate endosymbiont, including the presence of a 5.7-kb duplicated fragment involving the complete ribosomal operon and its closest genomic context (Baumann et al., 2002). Paralogous loci were detected in several strains from a diverse set of Pseudococcinae mealybug species, indicating that the duplication event occurred at early stages of “Ca. Tremblaya princeps” diversification. In spite of this, paralogous fragments have remained identical within each strain genome, suggesting that they have been affected by concerted evolution (Baumann et al., 2002). Concerted evolution is a molecular process driven by DNA recombination mechanisms that leads to homogenization of duplicated loci within a species. Consequently, paralogous loci are more closely related to each other than to the corresponding orthologous regions in another species, even though the duplication event preceded the speciation event (Liao, 1999).
The endosymbiotic system identified in Planococcus citri (cluster E), where “Ca. Tremblaya princeps” harbors “Ca. Moranella endobia,” has been extensively studied (von Dohlen et al., 2001; López-Madrigal et al., 2011, 2013a,b; McCutcheon and von Dohlen, 2011; Husnik et al., 2013). The complete sequencing of the 138.9-kb genome of “Ca. Tremblaya princeps” from P. citri confirmed the presence of the identical duplicated loci although no DNA repair and recombination genes were detected (López-Madrigal et al., 2011; McCutcheon and von Dohlen, 2011). In contrast, its gammaproteobacterial partner “Ca. Moranella endobia” (with a 538.2-kb genome) still retains a diverse set of genes involved in both the RecF and RecBCD recombination pathways, the two redundant mechanisms for this function that are nearly ubiquitous in free-living bacterial species (Rocha et al., 2005; Spies and Kowalczykowski, 2005; López-Madrigal et al., 2013a). Recent sequencing of the genome of “Ca. Tremblaya phenacola” PAVE, the sole obligatory endosymbiont of the mealybug Phenacoccus avenae, revealed that it also possess a tiny genome (171.5 kb), suggesting that a severe gene loss must have affected the common ancestor of both “Ca. Tremblaya” species at the beginning of the obligate intracellular symbiosis (Husnik et al., 2013). The genome of “Ca. Tremblaya princeps” is an almost perfect subset of that from “Ca. Tremblaya phenacola,” which has retained many essential genes involved in metabolic and informational functions that are absent in “Ca. Tremblaya princeps” and must be provided by its nested endosymbiont “Ca. Moranella endobia” (Husnik et al., 2013). However, “Ca. Tremblaya phenacola” PAVE also lacks all genes involved in DNA recombination. Therefore, the maintenance of homologous recombination (HR) pathways in “Ca. Moranella endobia” could be at the root of the concerted evolution noticed in the “Ca. Tremblaya princeps” genome. If this hypothesis is correct, we expect to find HR-related genes and signals of recent concerted evolution in additional endosymbiotic consortia from Pseudococcinae mealybugs.
We have checked for the co-occurrence of both features by analyzing the nested endosymbiotic systems from four unexplored mealybug species. The gray sugarcane mealybug Dysmicoccus boninsis, the long tailed mealybug Pseudococcus longispinus, and the obscure mealybug Pseudococcus viburni are phylogenetically distant members of the tribe Pseudococcini (Hardy et al., 2008), and their gammaproteobacterial endosymbionts have been independently acquired (López-Madrigal et al., 2014). The vine mealybug Planococcus ficus is a close relative of P. citri. Additionally, we explored the origin of the ribosomal operon duplication in “Ca. Tremblaya” and analyzed the susceptibility of both “Ca. Tremblaya princeps” and “Ca. Moranella endobia” to HR. Our results support a link between concerted evolution and nested endosymbiosis, suggesting a great impact of the gamma-endosymbionts on the reductive evolution of “Ca. Tremblaya princeps” genome, not only at the functional but also at the structural level.
Materials and Methods
Insect Sample Collection and DNA Extraction
Insects belonging to the species P. longispinus, P. viburni, and D. boninsis were field collected in the Botanical Garden of the Universitat de València (València, Spain. 39° 28′ 11.667″ N, 0° 22′ 34.637 W), with permission from the curator of the garden, Dr. Jaime Güemes. P. ficus was sampled from a population reared on Vitis vinifera at the Mediterranean Agroforestal Institute, Universitat Politècnica de València (València, Spain. 39° 29′ 1.699 N, 0° 20′ 28.978″ W). This study did not involve endangered or protected species. Insects were stored in absolute ethanol at −20°C. Total insect DNA (TDNA) was extracted from adult female insects, where endosymbiont populations are expected to reach a peak (Kono et al., 2008), using JETFLEX Genomic DNA Purification Kit (GENOMED).
DNA Amplification and Sequencing
PCR amplifications were performed on insect TDNA with appropriate primer pairs (see below), using 50–60 μmoles of each primer per 50 μl reaction, and the KAPATaq DNA Polymerase Kit (Kapa Biosystems). P. citri TDNA was used as a positive control. The thermal cycling protocol was as follows: an initial denaturation at 95°C for 5 min, followed by 35 cycles of 50 s at 95°C, 40 s at 55°C (or 52°C when indicated), and 2 min at 72°C, plus a final extension step of 7 min at 72°C. Amplicons were ABI sequenced at the sequencing facility of the Universitat de València.
Sequencing reads were quality surveyed and assembled with Staden Package (http://staden.sourceforge.net; Staden et al., 2000). Artemis software was used for sequence data management (http://www.sanger.ac.uk/resources/software/artemis/; Rutherford et al., 2000).
Molecular and Evolutionary Analysis
“Ca. Tremblaya princeps” genomic fragments leuA-rrs1 and prs-rrs2 were PCR amplified with proper combinations of the already described primers leuA, prs5/6 and U16S (Baumann et al., 2002). The same primers, as well as OR-leuAR2 (5′-TCAGTMATTAHGGCWACCTGCAC-3′), OR-prsR2 (5′-AATAGCYAAGCGGGTCAAGGC-3′) and OR-UF2 (5′-TGGCGCATGCTGTATGAGTTC-3′), were used to sequence the PCR products. tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/; Lowe and Eddy, 1997) and ARAGORN (http://mbio-serv2.mbioekol.lu.se/ARAGORN/; Laslett and Canback, 2004) were used for the prediction of tRNA genes. All other genes were annotated by BLAST searches (http://blast.ncbi.nlm.nih.gov/Blast.cgi/; Altschul et al., 1997). The newly obtained sequences have been deposited in the GenBank database (D. boninsis, KF591104 and KF591105; P. longispinus, KF591108, and KF591109; P. viburni, KF591110, and KF591111; P. ficus, KF591106, and KF591107).
The ancient state of sites under concerted evolution was inferred for the last common ancestor (LCA) of “Ca. Tremblaya princeps” strains from clusters C and E. Multiple alignment was done with ClustalW (Larkin et al., 2007). Analysis was performed by Maximum Likelihood (ML) with the DNAML program of the PHYLIP v3.69 package (Felsenstein, 2005), predefining the tree topology as already determined (Hardy et al., 2008).
The 16S rRNA gene (locus rrs) sequences from 19 gammaproteobacterial endosymbionts of mealybugs were retrieved from GenBank and aligned with ClustalW (Larkin et al., 2007). Later edition with Gblocks 0.91b (Castresana, 2000) yielded a total of 1389 unambiguously aligned sites. Phylogenetic analyses were performed by ML, Maximum Parsimony (MP) and Bayesian inference (BI) using RAxML v8 (Stamatakis, 2014), DNAPARS from PHYLIP v3.69 package (Felsenstein, 2005), and MrBayes 3.2 (Ronquist et al., 2012), respectively. A separate general time-reversible evolutionary model with gamma-distributed rates and a proportion of invariant sites (GTR+I+G) was applied for ML and BI phylogenetic reconstructions, according to inferences by JModelTest 2 (Guindon and Gascuel, 2003; Darriba et al., 2012). ML and MP reconstructions included a 1000-replications bootstrap analysis. BI reconstruction was generated from two runs of 150,000 generations. Likelihood settings were set to nst = 6, rates = invgamma and ngammacat = 4. Sampling was performed every 100 generations. First 3400 generations were discarded as “burn in.” The phylogenetic analysis Figure was prepared using FigTree v1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/) and Inkscape (https://inkscape.org/es/).
The complete genomes of “Ca. Tremblaya princeps” PCVAL and “Ca. Moranella endobia” PCVAL were scanned with plugin “Find Repeats” from Unipro UGENE v1.12.2 (Okonechnikov et al., 2012) to identify DNA repeats. Direct and inverted repeats (DR and IR, respectively) of at least 20 nucleotides in length were analyzed. GC content of independent repeats was calculated with MEGA5 (Tamura et al., 2011).
Gene Screening
Complete sequences of HR-related genes recA (encoding the recombination protein RecA), recG (encoding the ATP-dependent DNA helicase RecG, EC:3.6.4.12), ruvA (encoding the Holliday junction DNA helicase RuvA, EC:3.6.4.12), ruvB (encoding the Holliday junction DNA helicase RuvB, EC:3.6.4.12), ruvC (encoding the crossover junction endodeoxyribonuclease RuvC, EC:3.1.22.4), and priA (encoding the primosomal protein N', EC:3.6.4.-) were retrieved from GenBank for a set of selected betaproteobacteria (Burkholderia glumae BGR1, B. multivorans ATCC 17616, B. pseudomallei 1106a, and B. thailandensis E264) and gammaproteobacteria (Escherichia coli K-12 MG1655, Salmonella enterica Typhimurium LT2, Serratia proteomaculans 568, Sodalis glossinidius, Dickeya dadantii 3937, Yersinia pestis Angola, and “Ca. Moranella endobia” PCVAL). Multiple alignments were performed with ClustalW (Larkin et al., 2007) in order to identify conserved motifs where degenerate primers for PCR amplification and sequencing could be designed (Table S1). The annealing temperature in the PCR amplifications was 52°C. Most of the primer pairs (named as BG) are expected to amplify both beta- and gammaproteobacterial homologs of the targeted locus. Primers G-ruvAF/R were designed on a multiple alignment including gammaproteobacterial homologs only, in order to obtain a ruvA sequence larger than the one amplified by BG-ruvAF/R. BLAST searches against the non-redundant protein database (http://blast.ncbi.nlm.nih.gov/Blast.cgi/; Altschul et al., 1997) were performed in order to identify the putative taxonomic origin of the obtained sequences.
Results and Discussion
The Ancestral Duplicated Ribosomal Genomic Region in “Ca. Tremblaya”
Reductive evolution in obligate endosymbiont genomes is mostly due to the loss of genes that become redundant and/or unnecessary in the intracellular niche (McCutcheon and Moran, 2012). However, even though “Ca. Tremblaya princeps” from P. citri (cluster E) displays one of the most reduced genomes known so far, it presents an identical 5702-bp redundant sequence. It includes the complete ribosomal operon (rrs, rrl, and rrf) and its closest genomic context (the 3′ region of leuA, encoding the alpha-isopropylmalate synthase, EC 2.3.3.13; rpsO, encoding the ribosomal protein S15; and the 5′ region of rsmH, encoding the 16S rRNA m4C1402 methyltransferase, EC 2.1.1.199) (Figure 1). Detection of this duplicated region also in “Ca. Tremblaya princeps” strains from Dysmicoccus brevipes (cluster A), Melanococcus albizziae (cluster C), Maconellicoccus australiensis, and Maconellicoccus hirsutus (cluster F) led authors to suggest that a segmental duplication occurred at early stages of “Ca. Tremblaya princeps” diversification (Baumann et al., 2002). In order to study the origin of such duplication event, we performed a comparative analysis between the complete genomes of “Ca. Tremblaya princeps” PCVAL (López-Madrigal et al., 2011) and “Ca. Tremblaya phenacola” PAVE (Husnik et al., 2013). The analysis revealed the presence of an identical 386-bp inverted duplication in the latter. It is mostly composed by the remnants of the degraded ribosomal operon, including the 3′ end of a pseudogenized 23S rRNA gene (rrl, not annotated originally in the genome), the 5S rRNA gene (rrf) and the corresponding intergenic sequence. It also includes the TPPAVE_188 pseudogene, which is a truncated paralog of rsmH (Figure 1). This result suggests that the segmental duplication took place before the split of the two “Ca. Tremblaya” lineages. Moreover, the original copy of the ribosomal operon has undergone massive decay in “Ca. Tremblaya phenacola,” while the two identical copies preserved in “Ca. Tremblaya princeps” have evolved in a concerted manner.
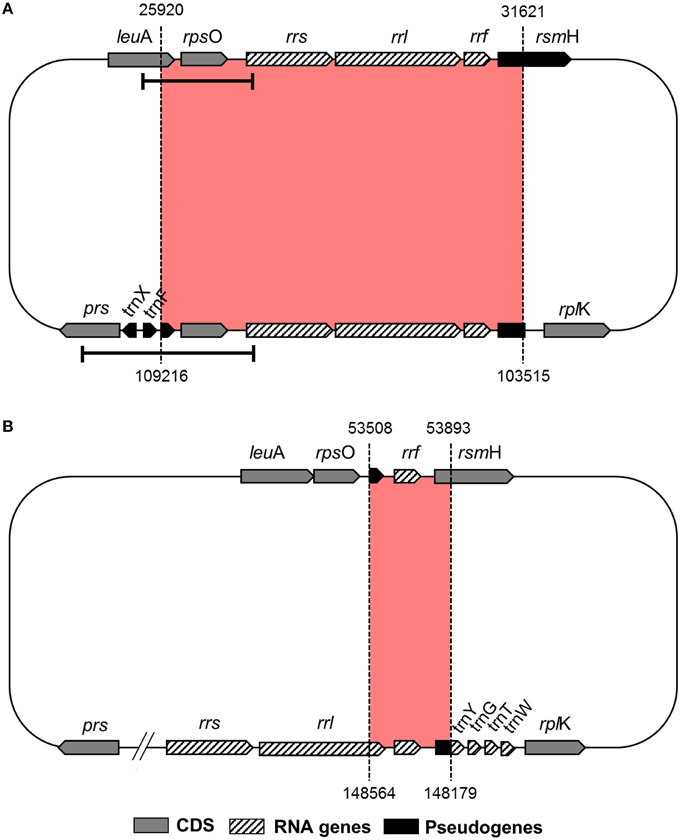
Figure 1. Duplicated genomic regions in Pseudococcidae beta-endosymbionts. (A) “Ca. Tremblaya princeps” PCVAL; (B) “Ca. Tremblaya phenacola” PAVE. Bars indicate the “Ca. Tremblaya princeps” fragments amplified in this work.
Co-Occurrence of Concerted Evolution and HR-Related Genes in Pseudococcinae Endosymbiotic Systems
Since both “Ca. Tremblaya” have a common evolutionary origin and “Ca. Tremblaya phenacola” has remained alone in the bacteriocytes of Phenacoccinae mealybugs (Gruwell et al., 2010; Koga et al., 2013), the massive decay of the paralogous loci in “Ca. Tremblaya phenacola” suggests that a link might exist between nested endosymbiosis and concerted evolution in “Ca. Tremblaya princeps.” The drastic reduction of the identical paralogous loci in “Ca. Tremblaya phenacola” PAVE co-occurs with additional genomic features that indicate a conventional reductive evolution (i.e., lower GC-content, high gene density; Husnik et al., 2013). No DNA repair and recombination genes were found in the genomes of “Ca. Tremblaya phenacola” PAVE or “Ca. Tremblaya princeps” from P. citri (López-Madrigal et al., 2011; McCutcheon and von Dohlen, 2011; Husnik et al., 2013), as it is typical for most endosymbionts with reduced genomes. In contrast, an almost complete set of HR-related loci were annotated in the genome of “Ca. Moranella endobia,” thus suggesting these genes to be responsible for the concerted evolution affecting “Ca. Tremblaya princeps” (López-Madrigal et al., 2013a). In order to test this hypothesis, we searched for the co-occurrence of signs of concerted evolution and the presence of HR-related genes in the endosymbiotic consortia from four unexplored mealybug species belonging to subfamily Pseudococcinae (D. boninsis, P. longispinus, P. viburni, and P. ficus).
Concerted Evolution in “Ca. Tremblaya Princeps”
To search for signals of concerted evolution, we focused on the molecular analysis of the 5′-flanking regions of the duplicated ribosomal operons (leuA-rrs1 and prs-rrs2) in the four “Ca. Tremblaya princeps” strains under study. The obtained amplicons include the 3′-end of leuA, an almost complete prs (encoding the phosphoribosylpyrophosphate synthetase, EC 2.7.6.1), the complete sequence of rpsO, several tRNA genes and the 5′-end of rrs (Figures 1, 2). The alignment of the amplified sequences revealed the existence of identical paralogous fragments ranging from 870 bp in “Ca. Tremblaya princeps” strain PLON (beta-endosymbiont of P. longispinus) to 899 bp in strain DBON (beta-endosymbiont of D. boninsis). Comparative analyses with available orthologous sequences of “Ca. Tremblaya princeps” strains from D. brevipes, P. citri, and M. albizziae (Baumann et al., 2002) showed that the length of these regions under concerted evolution remains relatively homogeneous (702–899 bp) among “Ca. Tremblaya princeps” lineages from clusters A, E, and C. In agreement with their close evolutionary relationship, identical duplicated loci start at orthologous positions for all available members of cluster A (nucleotide 25915/109218 in “Ca. Tremblaya princeps” PCVAL) and cluster E (nucleotides 25920/109216 in strain PCVAL; Figure 1), respectively. In contrast, identical loci are drastically reduced in strains from M. australiensis and M. hirsutus (cluster F; Baumann et al., 2002), whose initial nucleotides are orthologous of sites 26557/108579 and 26387/108749 in strain PCVAL, respectively. As above indicated, no nested intracellular bacteria have been reported in cluster F (Thao et al., 2002). However, no microscopic exploration of endosymbiotic systems from cluster F has been performed and, therefore, the presence of an undetected gamma-endosymbionts cannot be ruled out.
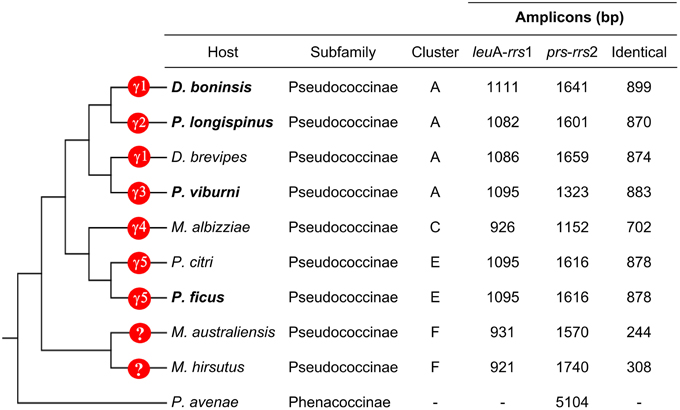
Figure 2. Characteristics of the leuA-rrs1 and prs-rrs2 regions. Host species from which sequences have been obtained in this work are in bold. The phylogenetic relationship among the insect hosts (Hardy et al., 2008), as well as the presence of gammaproteobacteria in the corresponding endosymbiotic systems are indicated. γ1 to γ5 represent polyphyletic bacterial lineages (see Section Genetic Screening of HR-related Genes).
P. citri and P. ficus are so closely related that they have been considered as cryptic species (Kol-Maimon et al., 2014). The comparison of the identical paralogous regions in the genomes of their “Ca. Tremblaya princeps” strains revealed homogenization of polymorphisms within each strain. Four indels and (at least) 15 nucleotide substitutions were detected (Table 1). In order to characterize the mutations leading to these homogenized polymorphic sites, their ancestral state in the LCA of “Ca. Tremblaya princeps” of clusters C and E was inferred with over 95% probability (Table 1). These data suggest ongoing homogenization by concerted evolution between the duplicated copies, at least in “Ca. Tremblaya princeps” from cluster E.
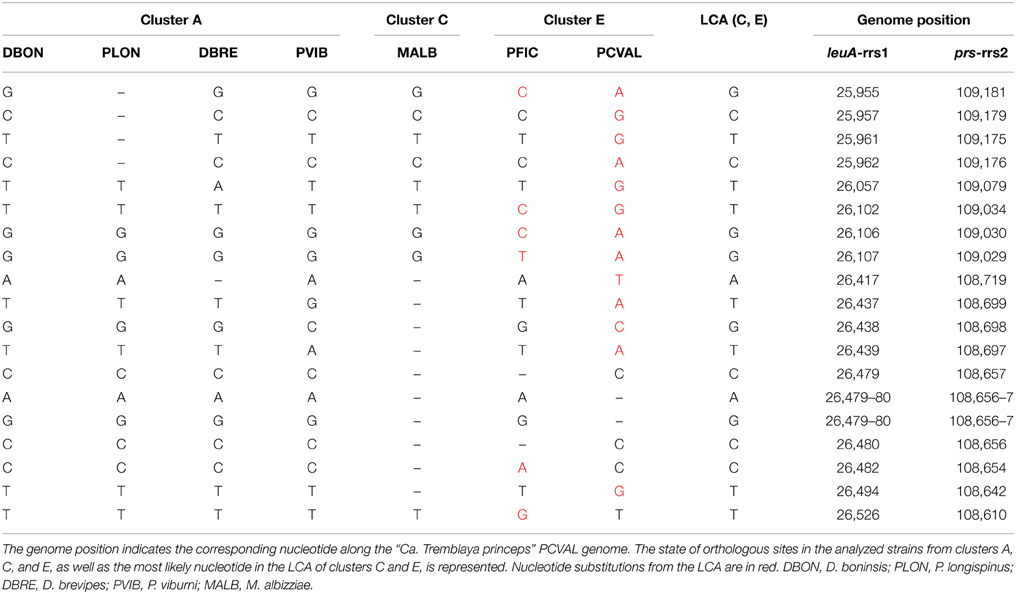
Table 1. Homogenized polymorphisms in “Ca. Tremblaya princeps” strains from P. ficus (PFIC) and P. citri (PCVAL).
Genetic Screening of HR-Related Genes
In order to explore the HR potential of the endosymbiotic consortia from the four analyzed Pseudococcinae species, we investigated the presence of a set of HR-related genes already identified in the genome of “Ca. Moranella endobia” from P. citri. Screened loci include recA, recG, ruvA, ruvB, ruvC, and priA. Most of them (recA, recG, ruvA, ruvB, ruvC) are common elements of both RecF and RecBCD pathways (Rocha et al., 2005; Spies and Kowalczykowski, 2005). RecG may functionally replace RuvABC (Meddows et al., 2004). In contrast, PriA is exclusively involved in the RecBCD pathway (Ng and Marians, 1996) and has been proposed to catalyze the assembly of the “Ca. Moranella endobia” incomplete primosome (López-Madrigal et al., 2013a).
The results are presented in Table 2. The GenBank accession numbers for all newly amplified sequences are also indicated. BLAST searches against the non-redundant protein database suggest a gammaproteobacterial origin for the loci detected in P. ficus, P. longispinus, and P. viburni. They show best similarity hits with homologs from bacteria of genus “Ca. Moranella”, Sodalis and Pectobacterium, respectively. Identical best similarity hits were observed when using their16S rRNA genes (AF476108, KF742539, JN182341) as query sequences. These results indicate that the internalization of the corresponding gamma-endosymbiont made recurrently available an HR machinery to the long-term endosymbiont “Ca. Tremblaya princeps.”
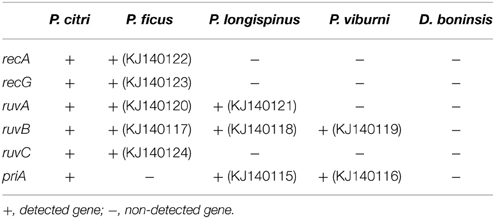
Table 2. Genetic screening of selected homologous recombination genes.
Although all primer combinations successfully amplified their target when applied to P. citri as a positive control, none of the analyzed consortia gave positive results for all screened genes. Negative results should be interpreted with caution, since they do not necessarily imply the absence of undetected loci. Degenerate primers were designed on gene regions encoding highly conserved motifs among beta and gammaproteobacterial homologs of the analyzed genes (Table S1). However, although highly conserved between distantly related bacteria, motifs acting as primer templates are not directly involved in protein functionality. Therefore, it is possible that non-synonymous substitutions affecting the target sequence lead to false negative results. Nevertheless, in accordance with the close evolutionary relationship between P. citri and P. ficus, five of the six screened loci were detected in the latter. Only priA could not be detected. PriA is needed for the assembly of the primosome, which is already incomplete in “Ca. Moranella endobia” PCVAL, due to the loss of dnaT and priC (López-Madrigal et al., 2013a). Thus, its absence suggests a relatively recent inactivation of the RecBCD pathway in the nested endosymbiont of “Ca. Tremblaya princeps” strains from cluster E. In contrast, as revealed by the very recent homogenization of polymorphisms (Table 1), the RecF pathway appears to be still acting on this cluster. Nevertheless, RecF function is expected to be attenuated because none of the components of the RecFOR complex, which enhances RecA loading onto SSB-coated single stranded DNA (Morimatsu and Kowalczykowski, 2003; Handa et al., 2009), is present in “Ca. Moranella endobia” PCVAL. Furthermore, recA mutations known to bypass the RecFOR complex deficiency (i.e., recA441, recA730, recA803; Lavery and Kowalczykowski, 1992) were not detected in that genome.
As for the endosymbiotic consortia involving the three “Ca. Tremblaya princeps” strains from cluster A under study (D. boninsis, P. longispinus, and P. viburni), our results suggest that both RecF and RecBCD pathways are currently inactive. Different patterns of conservation of HR-related genes were observed, which is consistent with the independent evolutionary origin of the gamma-endosymbionts (Gatehouse et al., 2012; López-Madrigal et al., 2014). Cluster A represents a very wide clade, including betaproteobacterial endosymbionts from mealybugs of the tribe Pseudococcini and the southern Africa group (Thao et al., 2002; Hardy et al., 2008). Moreover, Pseudococcus is a polyphyletic genus, and the two species analyzed in this work are phylogenetically distant, belonging to different clades of the tribe Pseudococcini. In order to place the three gamma-endosymbionts of these insects in the phylogenetic tree of those already described for mealybugs, we performed a phylogenetic analysis based on 16S rDNA sequences (Figure 3). According to our results, only the gamma-endosymbiont of D. boninsis groups with the other nested endosymbionts of “Ca. Tremblaya princeps” strains from cluster A, showing a long co-evolutionary history with its symbiotic partner. In contrast, the gamma-endosymbionts of P. longispinus and P. viburni group neither with any other cluster nor between them. The present analysis suggests the replacement of the ancestral gamma-endosymbiont in these two Pseudococcus species, and reveals two independent events of HR-related genes acquisition by the corresponding “Ca. Tremblaya princeps” strains. As expected for recently acquired obligate symbionts, these gamma-endosymbionts appear to be less affected by reductive evolution than that of D. boninsis, where none of the screened genes had been detected. Nevertheless, even if HR pathways appear to be currently inactive in the analyzed members of cluster A, this is not inconsistent with the observed signs of concerted evolution in the corresponding “Ca. Tremblaya princeps” strains (Figure 2). Signs of concerted evolution do not necessarily co-exist with functional HR pathways, since repeated identical sequences are expected to last on the genome over a certain time after the inactivation of such pathways. The presence of identical paralogous loci has also been noticed in the genome of “Ca. Portiera aleyrodidarum,” obligate endosymbiont of the whitefly Bemisia tabaci, where HR pathways have been recently lost (Sloan and Moran, 2013).
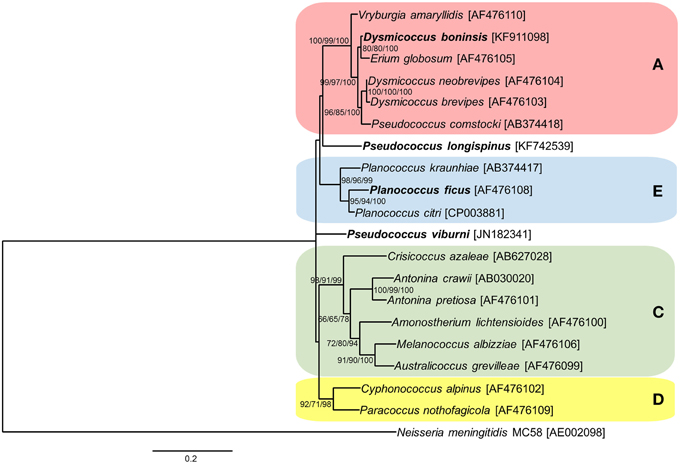
Figure 3. Phylogenetic relationships among gamma-endosymbionts of Pseudococcinae mealybugs. Already defined clusters A (red), C (green), D (yellow), and E (blue) are represented. Species used in this work are in bold. The betaproteobacterium Neisseria meningitidis MC58 was used as outgroup. ML, MP, and BI analysis gave essentially the same results. ML and MP bootstrap values, and Bayesian posterior probabilities over 50% are indicated. Scale bar represents substitutions per site.
Susceptibility to Homologous Recombination of Nested Endosymbionts from P. citri
Due to the reductive genome evolution in obligatory endosymbionts, genetic essentiality in their functional networks is typically higher than that observed in free-living bacteria (Thomas et al., 2009). Therefore, HR events and associated genome deletions or rearrangements could dramatically risk the stability of bacterial consortia involving tiny genomes. Repeat sequences ranging from 18 to 24 bp are thought to be long enough to promote HR events (Shen and Huang, 1986; Aras et al., 2003; Sloan and Moran, 2013). Therefore, in order to analyze the susceptibility to HR of both “Ca. Tremblaya princeps” and “Ca. Moranella endobia” from P. citri we performed a comprehensive search for direct (DR) and inverted (IR) repeats with at least 20 bp in length in both genomes (Table S2). Sixteen DRs (TDR01 to 16) and 12 IRs (TIR01 to 12) were found in “Ca. Tremblaya princeps.” Except for TIR12 (i.e., the duplicated region containing the ribosomal operon), all other repeats seem to have been randomly generated. As for “Ca. Moranella endobia”, 24 DRs (MDR01 to 24) and 16 IRs (MIR01 to 16) were found. Several of them appear to be consequence of ancestral duplications. Thus, MDR01, MDR02, and MDR11 map on a functional pdxJ (locus MPC_094 in the genome) and its pseudogenized copy (MPC_306), while MDR07, MDR14-16, MDR19, MDR22, and MDR23 are linked to a duplication including genes secE (MPC_278) and tuf (MPC_279). Additionally, seven DRs and five IRs map on several tRNA loci, which mostly display highly similar anticodon sequences and whose relative orientation along the genome is consistent with an ancestral proliferation process (Withers et al., 2006). Conservation of these repeats might be linked to mutational constraints, since 36–71% of their sequences correspond to tRNA stem regions (Table S3).
“Ca. Tremblaya princeps” repeats abundance is likely linked to its high genomic GC-content (Figure 4). The molecular characterization of independently generated repeats identified in these genomes reveals that those of “Ca. Tremblaya princeps” are GC-enriched compared to the whole genome (GCrepeats = 67.7% versus GCgenome = 59%, SDG+C = 6.5), while no bias is observed in the case of “Ca. Moranella endobia” (GCrepeats = 42.4% versus GCgenome = 44%, SDG+C = 11.0).
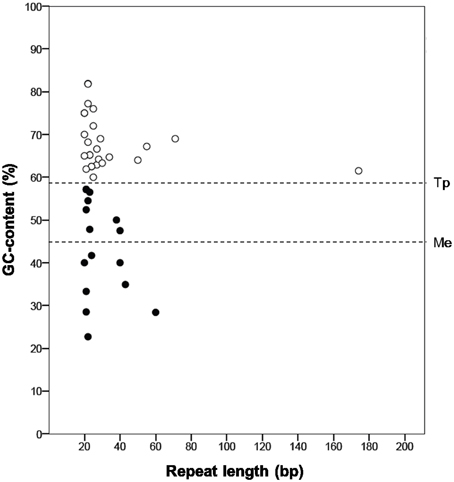
Figure 4. Molecular characterization of independent repeats. Those identified in the genomes of “Ca. Tremblaya princeps” (white circles) and “Ca. Moranella endobia” (black circles) are represented. The horizontal lines indicate the mean GC-content of each genome.
According to our results, sequence repeats are larger in “Ca. Tremblaya princeps” (mean length = 235.9 bp) than in Ca. Moranella endobia” (mean length = 127.6 bp). In addition, some of them (TDR8 and TDR12; TIR05 and TIR07) appear to derive from larger ancestral repeats. They are also more abundant in the former, where repeats density (abundance/kb) is 2.85 times larger than that of “Ca. Moranella endobia” (Table S2). Therefore, “Ca. Tremblaya princeps” must be more sensitive to HR than “Ca. Moranella endobia” (Rocha, 2003). In spite of this, HR events are not expected to be highly frequent in “Ca. Tremblaya princeps.” Recombination between DRs would cause DNA deletions or DNA duplications. Taking into account that the mean distance between DRs is about 50 kb (36% of the chromosome), further genome reduction would be strongly deleterious. On the other hand, recombination mediated by IRs would generate DNA inversions. Half of the IRs detected in the “Ca. Tremblaya princeps” genome map on relevant loci, including genes involved in translation (rplS, rpsF, rpmA) and essential amino acids biosynthesis (pheA, ilvI, aroB), whose functionality might be seriously compromised by HR events (Table S2). Thus, the apparent inactivation of the HR pathways in D. boninsis, P. longispinus, and P. viburni or its attenuation in P. ficus and P. citri may be helping to maintain the stability of the corresponding endosymbiotic systems.
In summary, our work reveals that the segmental duplication involving the ribosomal operon took place before the divergence between “Ca. Tremblaya princeps” and “Ca. Tremblaya phenacola.” Strikingly, there is a drastic reduction of the identical paralogous loci in the genome of “Ca. Tremblaya phenacola” PAVE. This is consistent with the apparently conventional reductive evolution undergone by this bacterium and suggest a link between concerted evolution and nested endosymbiosis. Results from the genetic screening indicate that independent internalization of different gamma-endosymbionts allowed the recurrent acquisition of HR capabilities by the corresponding endosymbiotic systems. Nevertheless, HR pathways appear to be currently attenuated or inactivated in the tested mealybug species, which could be enhancing the stability of these bacterial consortia. A metagenomic-based approach leading to the complete genomic characterization of the analyzed bacterial consortia would be useful in order to confirm our results.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Antonia Soto and Dr. Aleixandre Beltrà, from the Mediterranean Agroforestal Institute (Universitat Politècnica de València) for kindly supplying the biological samples. This work was supported by grants BFU2012-39816-C02-01 (co-financed by FEDER funds and Ministerio de Economía y Competitividad, Spain) to AL and PrometeoII/2014/065 (Conselleria d'Educació, Generalitat Valenciana, Spain) to AM. SL is a recipient of a fellowship from the Ministerio de Educación (Spain).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00642
References
Abby, S., and Daubin, V. (2007). Comparative genomics and the evolution of prokaryotes. Trends Microbiol. 15, 135–141. doi: 10.1016/j.tim.2007.01.007
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Aras, R. A., Kang, J., Tschumi, A. I., Harasaki, Y., and Blaser, M. J. (2003). Extensive repetitive DNA facilitates prokaryotic genome plasticity. Proc. Natl. Acad. Sci. U.S.A. 100, 13579–13584. doi: 10.1073/pnas.1735481100
Baumann, L., Thao, M. L., Hess, J. M., Johnson, M. W., and Baumann, P. (2002). The genetic properties of the primary endosymbionts of mealybugs differ from those of other endosymbionts of plant sap-sucking insects. Appl. Environ. Microbiol. 68, 3198–3205. doi: 10.1128/AEM.68.7.3198
Casjens, S. (1998). The diverse and dynamic structure of bacterial genomes. Annu. Rev. Genet. 32, 339–377. doi: 10.1146/annurev.genet.32.1.339
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
Felsenstein, J. (2005). Using the quantitative genetic threshold model for inferences between and within species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 1427–1434. doi: 10.1098/rstb.2005.1669
Gatehouse, L. N., Sutherland, P., Forgie, S. A., Kaji, R., and Christeller, J. T. (2012). Molecular and histological characterization of primary (betaproteobacteria) and secondary (gammaproteobacteria) endosymbionts of three mealybug species. Appl. Environ. Microbiol. 78, 1187–1197. doi: 10.1128/AEM.06340-11
Gruwell, M. E., Hardy, N. B., Gullan, P. J., and Dittmar, K. (2010). Evolutionary relationships among primary endosymbionts of the mealybug subfamily phenacoccinae (Hemiptera: Coccoidea: Pseudococcidae). Appl. Environ. Microbiol. 76, 7521–7525. doi: 10.1128/AEM.01354-10
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Handa, N., Morimatsu, K., Lovett, S. T., and Kowalczykowski, S. C. (2009). Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli. Genes Dev. 23, 1234–1245. doi: 10.1101/gad.1780709
Hardy, N. B., Gullan, P. J., and Hodgson, C. J. (2008). A subfamily-level classification of mealybugs (Hemiptera: Pseudococcidae) based on integrated molecular and morphological data. Syst. Entomol. 33, 51–71. doi: 10.1111/j.1365-3113.2007.00408.x
Husnik, F., Nikoh, N., Koga, R., Ross, L., Duncan, R. P., Fujie, M., et al. (2013). Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell 153, 1567–1578. doi: 10.1016/j.cell.2013.05.040
Koga, R., Nikoh, N., Matsuura, Y., Meng, X. Y., and Fukatsu, T. (2013). Mealybugs with distinct endosymbiotic systems living on the same host plant. FEMS Microbiol. Ecol. 83, 93–100. doi: 10.1111/j.1574-6941.2012.01450.x
Kol-Maimon, H., Ghanim, M., Franco, J. C., and Mendel, Z. (2014). Evidence for gene flow between two sympatric mealybug species (Insecta; Coccoidea; Pseudococcidae). PLoS ONE 9:e88433. doi: 10.1371/journal.pone.0088433
Kono, M., Koga, R., Shimada, M., and Fukatsu, T. (2008). Infection dynamics of coexisting beta- and gammaproteobacteria in the nested endosymbiotic system of mealybugs. Appl. Environ. Microbiol. 74, 4175–4184. doi: 10.1128/AEM.00250-08
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Lavery, P. E., and Kowalczykowski, S. C. (1992). Biochemical basis of the constitutive repressor cleavage activity of recA730 protein. A comparison to recA441 and recA803 proteins. J. Biol. Chem. 267, 20648–20658.
Liao, D. (1999). Concerted evolution: molecular mechanism and biological implications. Am. J. Hum. Genet. 64, 24–30. doi: 10.1086/302221
López-Madrigal, S., Balmand, S., Latorre, A., Heddi, A., Moya, A., and Gil, R. (2013a). How does Tremblaya princeps get essential proteins from its nested partner Moranella endobia in the mealybug Planoccocus citri? PLoS ONE 8:e77307. doi: 10.1371/journal.pone.0077307
López-Madrigal, S., Beltrà, A., Resurrección, S., Soto, A., Latorre, A., Moya, A., et al. (2014). Molecular evidence for ongoing complementarity and horizontal gene transfer in endosymbiotic systems of mealybugs. Front. Microbiol. 5:449. doi: 10.3389/fmicb.2014.00449
López-Madrigal, S., Latorre, A., Porcar, M., Moya, A., and Gil, R. (2011). Complete genome sequence of “Candidatus Tremblaya princeps” strain PCVAL, an intriguing translational machine below the living-cell status. J. Bacteriol. 193, 5587–5588. doi: 10.1128/JB.05749-11
López-Madrigal, S., Latorre, A., Porcar, M., Moya, A., and Gil, R. (2013b). Mealybugs nested endosymbiosis: going into the “matryoshka” system in Planococcus citri in depth. BMC Microbiol. 13:74. doi: 10.1186/1471-2180-13-74
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.0955
McCutcheon, J. P., and Moran, N. A. (2012). Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26. doi: 10.1038/nrmicro2670
McCutcheon, J. P., and von Dohlen, C. D. (2011). An interdependent metabolic patchwork in the nested symbiosis of mealybugs. Curr. Biol. 21, 1366–1372. doi: 10.1016/j.cub.2011.06.051.
Meddows, T. R., Savory, A. P., and Lloyd, R. G. (2004). RecG helicase promotes DNA double-strand break repair. Mol. Microbiol. 52, 119–132. doi: 10.1111/j.1365-2958.2003.03970.x
Morimatsu, K., and Kowalczykowski, S. C. (2003). RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol. Cell. 11, 1337–1347. doi: 10.1016/S1097-2765(03)00188-6
Moya, A., Gil, R., and Latorre, A. (2009). The evolutionary history of symbiotic associations among bacteria and their animal hosts: a model. Clin. Microbiol. Infect. 15(Suppl. 1), 11–13. doi: 10.1111/j.1469-0691.2008.02689.x
Moya, A., Peretó, J., Gil, R., and Latorre, A. (2008). Learning how to live together: genomic insights into prokaryote-animal symbioses. Nat. Rev. Genet. 9, 218–229. doi: 10.1038/nrg2319
Ng, J. Y., and Marians, K. J. (1996). The ordered assembly of the FX174-type primosome I. Isolation and identification of intermediate protein-DNA complexes. J. Biol. Chem. 271, 15642–15648. doi: 10.1074/jbc.271.26.15642
Okonechnikov, K., Golosova, O., and Fursov, M. (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. doi: 10.1093/bioinformatics/bts091
Rocha, E. P. C. (2003). DNA repeats lead to the accelerated loss of gene order in bacteria. Trends Genet. 19, 600–603. doi: 10.1016/j.tig.2003.09.011
Rocha, E. P. C. (2008). The organization of the bacterial genome. Annu. Rev. Genet. 42, 211–233. doi: 10.1146/annurev.genet.42.110807.091653
Rocha, E. P. C., Cornet, E., and Michel, B. (2005). Comparative and evolutionary analysis of the bacterial homologous recombination systems. PLoS Genet. 1:e15. doi: 10.1371/journal.pgen.0010015
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M. A., et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. doi: 10.1093/bioinformatics/16.10.944
Shen, P., and Huang, H. V. (1986). Homologous recombination in Escherichia coli: dependence on substrate length and homology. Genetics 112, 441–457.
Sloan, D. B., and Moran, N. A. (2013). The evolution of genomic instability in the obligate endosymbionts of whiteflies. Genome Biol. Evol. 5, 783–793. doi: 10.1093/gbe/evt044
Spies, M., and Kowalczykowski, S. C. (2005). “Homologous recombination by the RecBCD and the RecF pathways,” in The Bacterial Chromosome, ed N. P. Higgins (Washington, DC: ASM Press), 389–403.
Staden, R., Beal, K. F., and Bonfield, J. K. (2000). The Staden package, 1998. Methods Mol. Biol. 132, 115–130. doi: 10.1385/1-59259-192-2:115
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Thao, M., Gullan, P., and Baumann, P. (2002). Secondary (γ-Proteobacteria) endosymbionts infect the primary (β-Proteobacteria) endosymbionts of mealybugs multiple times and coevolve with their hosts. Appl. Environ. Microbiol. 68, 3190–3197. doi: 10.1128/AEM.68.7.3190
Thomas, G. H., Zucker, J., Macdonald, S. J., Sorokin, A., Goryanin, I., and Douglas, A. E. (2009). A fragile metabolic network adapted for cooperation in the symbiotic bacterium Buchnera aphidicola. BMC Syst. Biol. 3:24. doi: 10.1186/1752-0509-3-24
von Dohlen, C. D., Kohler, S., Alsop, S. T., and McManus, W. R. (2001). Mealybug β-proteobacterial endosymbionts contain γ-proteobacterial symbionts. Nature 412, 433–436. doi: 10.1038/35086563
Keywords: mealybugs, nested endosymbiosis, homologous recombination, concerted evolution
Citation: López-Madrigal S, Latorre A, Moya A and Gil R (2015) The link between independent acquisition of intracellular gamma-endosymbionts and concerted evolution in Tremblaya princeps. Front. Microbiol. 6:642. doi: 10.3389/fmicb.2015.00642
Received: 30 March 2015; Accepted: 12 June 2015;
Published: 25 June 2015.
Edited by:
Joerg Graf, University of Connecticut, USACopyright © 2015 López-Madrigal, Latorre, Moya and Gil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosario Gil, Institut Cavanilles de Biodiversitat i Biologia Evolutiva, Universitat de València, C/Catedrático José Beltrán 2, 46980 Paterna, Valencia, Spain, rosario.gil@uv.es