 Dongfang Hu
Dongfang Hu Lin Lv†
Lin Lv†- Department of Animal Science and Technology, Shandong Agricultural University, Tai’an, China
Classical swine fever virus (CSFV) causes an economically important and highly contagious disease of pigs worldwide. C-strain vaccination is one of the most effective ways to contain this disease. Since 2014, sporadic CSF outbreaks have been occurring in some C-strain vaccinated provinces of China. To decipher the disease etiology, 25 CSFV E2 genes from 169 clinical samples were cloned and sequenced. Phylogenetic analyses revealed that all 25 isolates belonged to subgenotype 2.1. Twenty-three of the 25 isolates were clustered in a newly defined subgenotype, 2.1d, and shared some consistent molecular characteristics. To determine whether the complete E2 gene was under positive selection pressure, we used a site-by-site analysis to identify specific codons that underwent evolutionary selection, and seven positively selected codons were found. Three positively selected sites (amino acids 17, 34, and 72) were identified in antigenicity-relevant domains B/C of the amino-terminal half of the E2 protein. In addition, another positively selected site (amino acid 200) exhibited a polarity change from hydrophilic to hydrophobic, which may change the antigenicity and virulence of CSFV. The results indicate that the circulating CSFV strains in Shandong province were mostly clustered in subgenotype 2.1d. Moreover, the identification of these positively selected sites could help to reveal molecular determinants of virulence or pathogenesis, and to clarify the driving force of CSFV evolution in East China.
Introduction
Classical swine fever (CSF), previously known as hog cholera, is an economically important, highly contagious disease of pigs that is classified as a notifiable disease by the Office International des Epizooties (Jiang et al., 2013). CSF is characterized by fever and hemorrhage with an acute or chronic course (Luo et al., 2011). CSF was first recognized in Tennessee, USA, in 1810, and then rapidly spread throughout the world (Edwards et al., 2000). As a result of systemic immunizations with live attenuated vaccines and/or strict epidemiological surveillance, CSF had been controlled and successfully eradicated from domestic pigs in some countries and regions, such as Australia, New Zealand, North America, and Western Europe (Paton and Greiser-Wilke, 2003; Ji et al., 2015). However, it still significantly affects swine production in Asia, South America, Eastern Europe, and parts of the former Soviet Union (Ji et al., 2015).
The causative agent, CSFV, is a member of the genus Pestivirus within the family Flaviviridae (Lowings et al., 1996). The positive-sense, single-stranded RNA CSFV genome is 12.3 kb in length, and it comprises one large ORF that is flanked by two NTRs (Rumenapf et al., 1991; Tautz et al., 2015). The ORF codes a 3898-AA polyprotein that is co- and post-translationally processed by cellular and viral proteases into four structural (C, Erns, E1, and E2) and eight non-structural proteins in the order NH2–(Npro-C-Erns-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B)–COOH (Rumenapf et al., 1991; Chang et al., 2010). The E2 protein is the main immunogen of CSFV, and it induces the production of neutralizing antibodies that provide protection against lethal challenge (Beer et al., 2015); it also plays multiple roles in the viral life cycle, and it mediates the entry of the virus into host cells (Sanchez et al., 2008; Shen et al., 2011).
The various isolates of CSFV consist of one serotype, reflecting a narrow range of evolutionary divergence (Vanderhallen et al., 1999; Deng et al., 2005). Therefore, genetic typing of the virus has been used to understand the evolution and spread of viruses, and the origins of disease outbreaks (Deng et al., 2005). 5′-NTR (96 nt), partial E2 (190 nt), and NS5B (409 nt) sequence similarities are extensively used for genetic analyses and to study viral diversity (Lowings et al., 1996; Greiser-Wilke et al., 1998; Paton et al., 2000). Recently, the full-length E2 coding sequence (1,119 nt) was also demonstrated to be reliable in detailed phylogenetic analyses (Postel et al., 2012; Beer et al., 2015). Analyses using these three or four regions have similarly classified CSFV into three genotypes, each with three to four subgenotypes (Lowings et al., 1996; Paton et al., 2000; Deng et al., 2005). Thus far, subgenotypes 1.1–1.4, 2.1–2.3, and 3.1–3.4 can be differentiated (Lowings et al., 1996; Paton et al., 2000; Postel et al., 2013).
Determining the selection pressures that have shaped the genetic variation of viruses is a major part of many molecular evolution studies (Kosakovsky Pond and Frost, 2005). A powerful method for studying adaptive molecular evolution is the use of a codon substitution model to identify AA sites where the dN exceeds the dS in a maximum likelihood context (Anisimova et al., 2001; Shen et al., 2011). Estimates of dN that are significantly different from dS provide convincing evidence for non-neutral evolution (Kosakovsky Pond and Frost, 2005). In viruses, the AAs at the interacting sites between envelope proteins and host molecules are continuously evolving under positive selection (Shen et al., 2011).
Since late 2014 in many regions of Shandong province in East China, a CSF epidemic, which is characterized by abortions and stillbirths of sows, as well as fever, anorexia, skin hemorrhages, and high-mortality among nursery pigs, has been occurring in many pig herds that were immunized with attenuated CSFV vaccines (the C strain, Hog Cholera Lapinized Virus). Most pigs in Shandong are vaccinated according to the following schemes: sows and boars are vaccinated simultaneously three times per year. Piglets are vaccinated first via an intramuscular injection at 21–28 days of age, and they receive a second vaccination at 7–8 weeks of age. Replacement gilts and boars are then vaccinated at 12–16 weeks, followed by a supplementary immunization before estrus (unpublished data). Here, we conducted a molecular epidemiological survey of 25 CSFV isolates and showed that the circulating CSFV strains in Shandong province were mostly clustered in subgenotype 2.1d. The selection pressures that act on the E2 gene of these new isolates and 120 reference strains were further analyzed to obtain insights into the driving forces of CSFV evolution in swine populations under regular vaccination programs.
Materials and Methods
Sample Preparation and Virus Isolation
A total of 169 tissue specimens, including the spleen, lymph nodes, tonsils, brain, lungs, and kidneys, were collected from clinically ill nursery pigs from different pig herds of various sizes in Shandong province from December 2013 to June 2015. The tissue samples were collected in accordance with the guidelines of the Shandong Agricultural University Animal Care and Use Committee (SDAUA-2013-001) and dissected for cryopreservation and fixed in 10% neutral formalin for virus detection and histological examination, respectively. Tissue samples were homogenized in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA), and then the tissue homogenates were centrifuged at 10,000 × g (4°C) for 10 min. Then, the suspension was passed through a 0.22-μm filter (EMD Millipore, Billerica, MA, USA) and transferred to PK-15 cell monolayers. Then, the cells were incubated at 37°C in 5% CO2 for 3–5 days, and the cultures were harvested and stored at –80°C as viral stocks.
Histological Examination and Polymerase Chain Reaction (PCR) Detection
The formalin-fixed samples were processed and embedded in paraffin. Thin sections of the fixed tissues were stained with H&E and examined microscopically. Viral DNA and RNA of the harvested cultures were extracted using the EasyPure viral DNA/RNA kit (TransGen, Beijing, China) according to the manufacturer’s instructions for the detection of suspected viruses. Four major pathogens, including CSFV, PRRSV, PRV, and PCV2 were detected by PCR or reverse transcription (RT)-PCR (Hu et al., 2015).
E2 Gene Amplification and Sequencing
Primers based on the published sequence of the CSFV Shimen strain (GenBank accession no. AF092448) were designed to amplify the complete E2 gene (forward primer: GTAAATATGTGTGTGTTAGACCAGA, reverse primer: GTGTGGGTAATTRAGTTCCCTATCA; Zhang et al., 2015). The viral RNA of CSFV-positive cultures was extracted, and the complete E2 gene was amplified using the EasyScript One-Step RT-PCR SuperMix (TransGen, Beijing, China). Briefly, 6 μL of RNA template, 25 μL of Reaction Mix, 1 μL of Enzyme Mix, and 16 μL of RNase-free water were mixed with 1 μL of each primer (10 μM). One-step RT-PCR was performed using the following conditions: 45°C for 25 min, 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 2 min, followed by a final extension at 72°C for 7 min. PCR/RT-PCR products were analyzed by 1% agarose gel electrophoresis. Target fragments were excised from the gels for purification using the Gel Extraction Kit (Tiangen, Beijing, China). Purified PCR products were cloned into the pMD18-T vector (TaKaRa, Beijing, China). Recombinant clones and the forward and reverse primers were sent to Sangon Bioscience (Shanghai, China) for sequencing.
Phylogenetic Analysis of the E2 Gene
The E2 gene sequences that were amplified from the clinical samples (Table 1) were aligned with 120 sequences in GenBank (Supplementary Table S1), and phylogenetic trees were constructed using MEGA 6.0 software1 by the maximum likelihood method based on the Tamura–Nei model (Tamura and Nei, 1993; Tamura et al., 2013). Bootstrap values were estimated for 1,000 replicates. Trees were determined based on the full-length E2 sequence (1,119 nt) and a partial E2 sequence (190 nt) (Lowings et al., 1996).
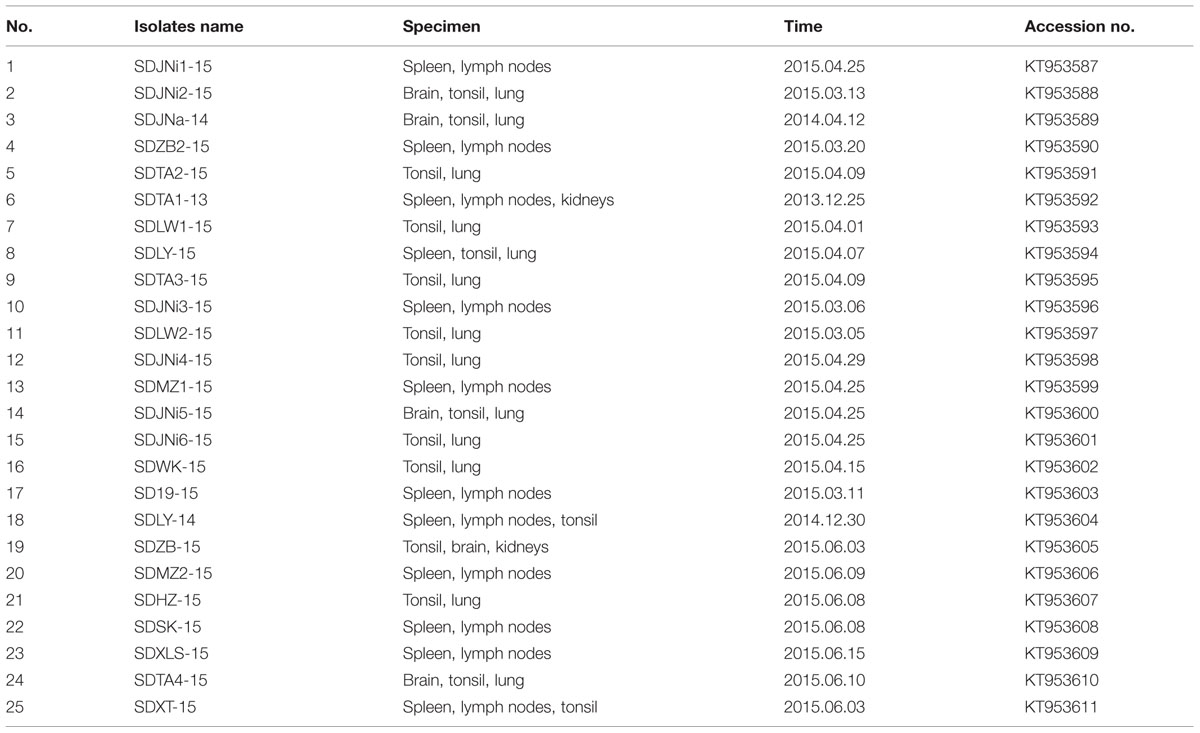
TABLE 1. Characteristics of the 25 new isolates and GenBank accession no. of E2 genes (1,119 nt).
Identities and AA Substitution Analysis of the E2 Gene/Protein
The nt and AA sequence identities of the 25 new CSFV isolates and eight representative CSFV isolates, including Shimen (AF092448, 1.1), SXCDK (GQ923951, 2.1a), HEBZ (GU592790, 2.1b), GDPY2008 (HQ697223, 2.1c), SDQS (JQ001834, 2.1d), LAL290 (KC851953, 2.2), Novska (HQ148061, 2.3), and TWN (AY646427, 3.4), were calculated using the MegAlign module (Clustal W method) of the Lasergene package (DNASTAR Inc., Madison, WI, USA). The AA substitutions of the new isolates were compared with those of the representative CSFV isolates, which included three genotypes (1.1–1.4, 2.1–2.3, and 3.4).
Selection Pressure Analysis of the E2 Gene
An analysis of the selection pressure acting on the codons of the E2 envelope protein, including the 25 new isolates and 120 reference strains, was conducted using the HyPhy open-source software package available at the datamonkey web-server2 (Delport et al., 2010). The level of positive selection was estimated using five different approaches: single likelihood ancestor counting (SLAC), fixed effects likelihood (FEL), internal fixed effects likelihood (IFEL), mixed effects model of evolution (MEME), and fast unbiased Bayesian approximation (FUBAR) (Sharma et al., 2013). The best nucleotide substitution model for different datasets, as determined via the available tool on the datamonkey server, was used in the analysis.
Results
Gross and Histological Lesions of CSF-Suspected Cases
Systematic necropsies were performed on pigs with clinical signs of CSF, including fever, anorexia, diffuse hemorrhage of the skin (Figure 1A), and conjunctivitis. Obvious hemorrhagic spots were found on the surface of the epicardium (Figure 1B). Scattered hemorrhagic infarcts were observed on the edge of the spleen (Figure 1C). Multiple lymph nodes were hemorrhagic and turgid (Figure 1D). The renal cortex were densely covered with petechial hemorrhages (Figure 1E). A mixture of small and large hemorrhagic spots, as well as ulcers, was seen on the surface of the gastric mucosa (Figure 1F). Histological examination mainly confirmed viral encephalitis, hemorrhages of many tissues, and necrotic foci of lymphoid tissues. The brain tissue exhibited typical viral encephalitis with lymphocyte infiltration around the small blood vessels (Figure 1G), as well as the proliferation of glial cells (Figure 1H). The histological structure of the spleen was disordered and characterized by necrosis, hemorrhage, and depletion of lymphocytes (Figure 1I). The lymph nodes showed hemorrhagic necrotizing lymphadenitis with necrotic lymphocytes and hyperplastic reticular cells (Figure 1J). The glomerulus and mesenchyme were hemorrhagic (Figure 1K).
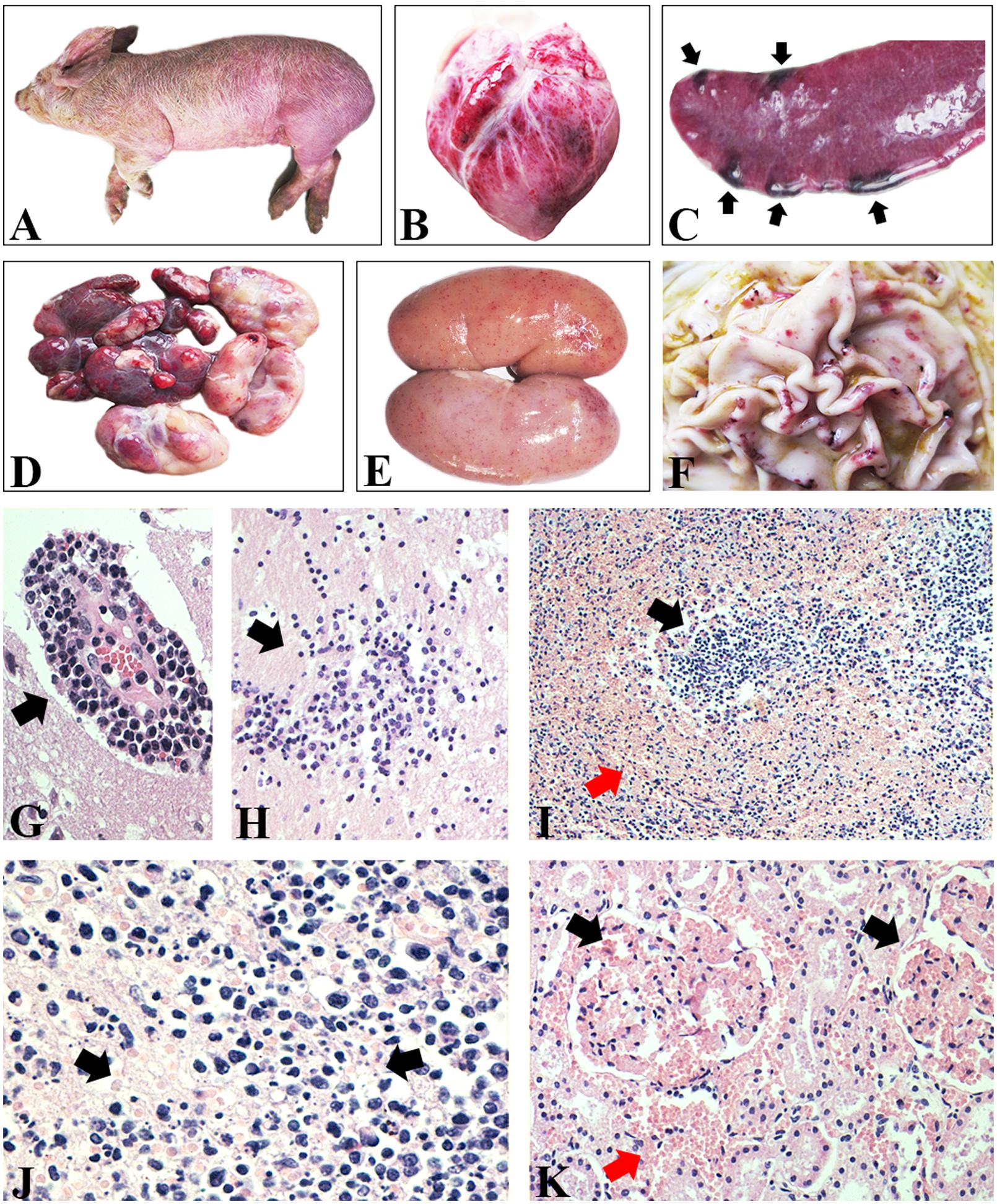
FIGURE 1. Gross and histological lesions of CSF-suspected pigs. (A) Diffuse hemorrhage of skin. (B) Epicardium hemorrhage. (C) Infarcts scattered on the edge of spleen. (D) Lymph nodes were hemorrhagic and turgid. (E) Renal cortex was densely covered with petechial hemorrhages. (F) Hemorrhagic spots and ulcer on the surface of gastric mucosa. (G) Lymphocyte infiltration around the small blood vessels in brain. H&E stain, ×400. (H) Proliferation of glial cells in brain. H&E stain, ×200. (I) Histological structure of spleen was disordered and characterized by necrotic lymphocytes and hemorrhage. H&E stain, ×100. (J) Lymphoid nodules showed necrotic lymphocytes and hyperplastic reticular cells and hemorrhage. H&E stain, ×400. (K) The glomerulus and mesenchyme were hemorrhagic. H&E stain, ×200.
Pathogens Detected in the Clinical Samples
The PCR/RT-PCR results showed that 25 of the 169 tissue specimens collected from different herds were positive for CSFV. Among the 25 samples, 12 samples were positive for PCV2, five for PRV, and four for PRRSV (data not shown). All 25 amplified E2 genes were sequenced and submitted to GenBank (Table 1).
Phylogenetic Analysis of the E2 Gene
A total of 145 full-length E2 gene (1,119 nt) sequences and 145 corresponding partial E2 gene (190 nt) sequences, including the sequences of the 25 new isolates, were used to construct phylogenetic trees (Figure 2). The analysis resulted in a classification of all 145 CSFVs into three main groups (genotypes 1–3) containing eight subgroups (1.1–1.4, 2.1–2.3, and 3.4; Figure 2).
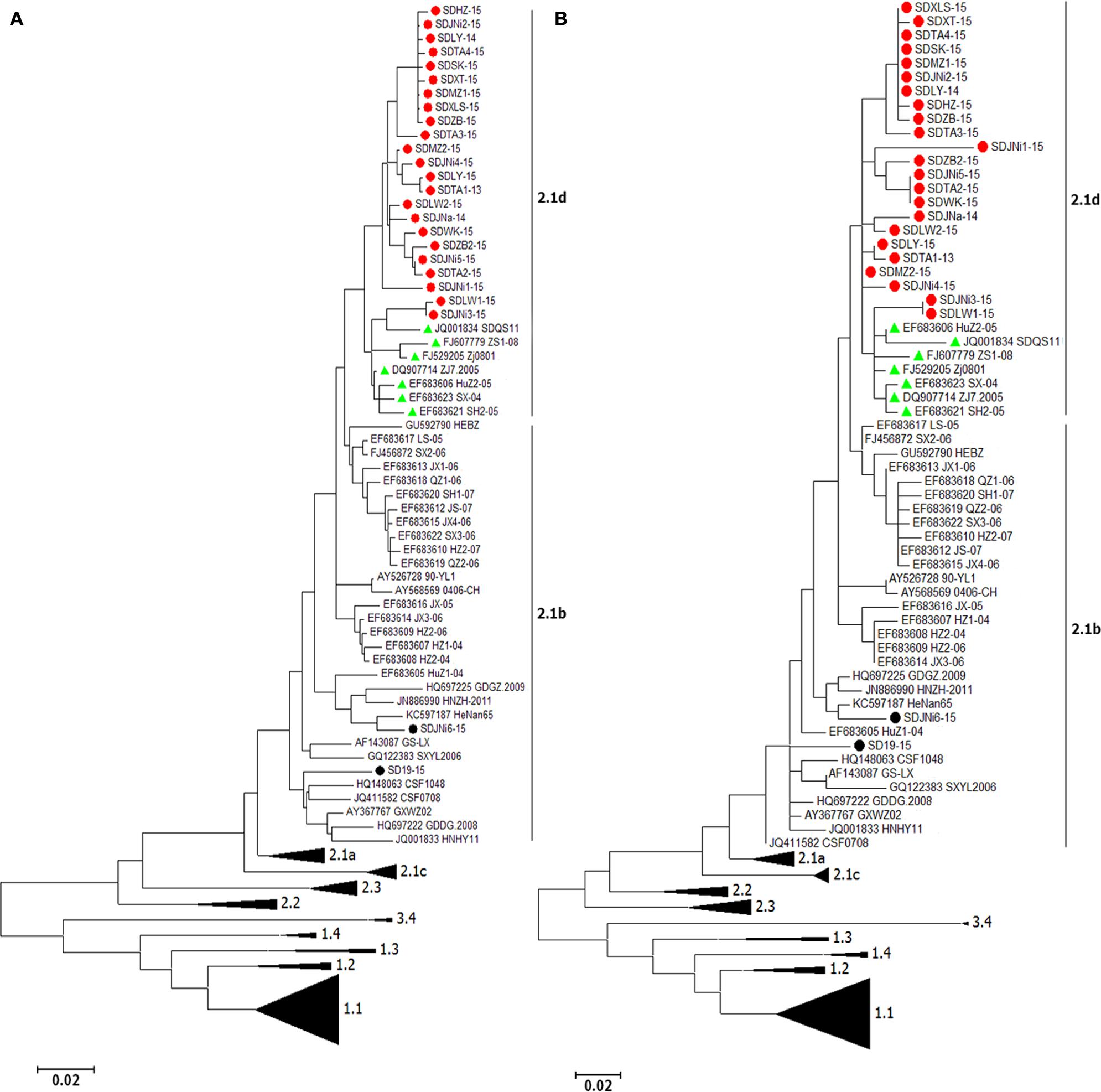
FIGURE 2. Phylogenetic analysis of different genomic regions of the 25 new isolates and 122 reference strains. (A) Full-length E2 gene sequences. (B) Partial E2 gene sequences. 23 newly isolated strains (indicated by  ) accompanied with seven references strains (indicated by
) accompanied with seven references strains (indicated by  ) were clustered into subgenotype 2.1d. The other two newly isolated strains (labeled by
) were clustered into subgenotype 2.1d. The other two newly isolated strains (labeled by  ) belonged to subgenotype 2.1b.
) belonged to subgenotype 2.1b.
Of the 25 new isolates, 21 isolates that were isolated in 2015 (SDHZ-15, SDJNi2-15, SDLY-15, SDTA4-15, SDSK-15, SDXT-15, SDMZ1-15, SDXLS-15, SDZB-15, SDTA3-15, SDMZ2-15, SDJNi4-15, SDLY-15, SDLW2-15, SDWK-15, SDZB2-15, SDJNi5-15, SDTA2-15, SDJNi1-15, SDLW1-15, and SDJNi3-15), two previously isolated strains (SDTA1-13 in 2013 and SDJNa-14 in 2014), and seven previously sequenced isolates [SDQS11 (JQ001834), ZS1-08 (FJ607779), Zj0801 (FJ529205), ZJ7.2005 (DQ907714), HuZ2-05 (EF683606), SX-04 (EF683623), and SH2-05(EF683621)] belonged to the new subgenotype 2.1d (Zhang et al., 2015). The remaining two new isolates, SD19-15 and SDJNi6-15, were clustered in subgenotype 2.1b. Phylogenetic trees based on the two different gene sequences, including the 145 full-length E2 gene sequences (Figure 2A) and 145 partial E2 gene sequences (Figure 2B), produced similar results. It is evident that all of the recently isolated CSFV strains in Shandong province were surprisingly divergent from the Shimen reference strain and the vaccine strain HCLV, and that the subgenotype 2.1 CSFV strains (mainly subgenotype 2.1d) predominated in more recent CSF epidemics in Shandong province in East China.
Site Mutation Analysis of the E2 Gene
The E2 gene of the 25 new isolates is 1,119 nt long, encoding a 373-AA protein. When compared with each of the eight reference strains (Table 2), the 25 newly isolated strains shared the lowest nt identities (81.7–82.5%) and AA identities (88.5–90.1%) with the TWN strain (subgenotype 3.4). The new isolates shared the highest nt and AA sequence similarities with 2.1 reference strains. When compared with each of the four subgenotypes of genotype 2.1, the two new isolates, SD19-15 and SDJNi6-15, had the highest nt identity (94.2%) and AA identities (96.2 and 97.1%, respectively) with the 2.1b reference strain HEBZ, while the other 23 new isolates shared the highest nt identities (95.9–97.5%) and AA identities (96.2–98.4%) with the 2.1d reference strain SDQS (Table 2, Supplementary Tables S2 and S3). In addition, the 25 new isolates had greater similarities to subgenotype 2.1b isolates than to either subgenotype 2.1a or 2.1c isolates, indicating a high similarity between subgenotypes 2.1b and 2.1d; these results are in accordance with the report by Zhang et al. (2015).
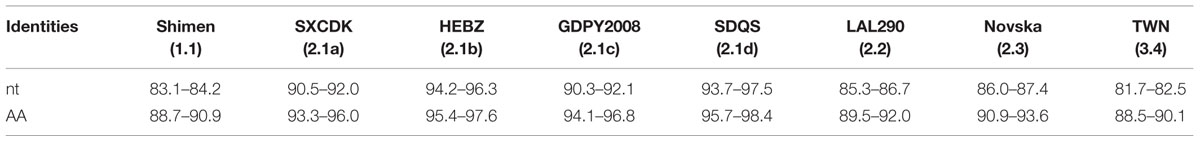
TABLE 2. Nucleotide (nt) and AA identities of E2 gene between the 25 new isolates and other eight representative CSFV isolates (%).
Compared with the reference strains, the two 2.1b new isolates, SD19-15 and SDJNi6-15, showed no characteristic AA substitutions, while the other 23 new isolates, which belonged to the 2.1d subgenotype, had some unique characteristics (Figure 3). Compared with all of the other isolates, the new 2.1d isolates, as well as four of the 2.1d reference strains (DQ907714, FJ529205, FJ607779, and JQ001834) showed consistent AA substitutions, including an R at position 31 (R31), S34, I56, K303, and A331. The subgenotype 2.1d isolates also showed unique AA substitutions, including G/D/N36S, D97N, K/N159R, and V/M/I168A. In addition, some subgenotype 2.1d isolates had two AA substitutions at positions 200 (Q200L) and 205 (R205K) compared with subgenotype 2.1a, 2.1b, and 2.1c isolates.

FIGURE 3. Amino acid (AA) sequences alignments of E2 genes of the 25 new isolates and 29 representative CSFV isolates. Consistent molecular characteristics of 2.1d strains were indicated by blue boxes  . Unique AA substitutions of 2.1d strains compared with all other strains were indicated by red boxes
. Unique AA substitutions of 2.1d strains compared with all other strains were indicated by red boxes  . Unique AA substitutions of 2.1d strains compared with subgenotype 2.1a, 2.1b, and 2.1c strains were indicated by black boxes
. Unique AA substitutions of 2.1d strains compared with subgenotype 2.1a, 2.1b, and 2.1c strains were indicated by black boxes  .
.
Selection Pressure Analysis
A selection pressure analysis of the E2 gene of 145 global CSFV strains revealed seven positively selected sites (AAs 17, 34, 72, 168, 200, 240, and 283) by at least two methods (Table 3). The detected positively selected sites were diverse, and most of the sites were hydrophilic sites (Table 4). There were no regular changes in polarity of the positively selected AAs, but a change from a polar AA (Q) at position 200 to non-polar AAs (V, P, and L) was observed (Table 4).
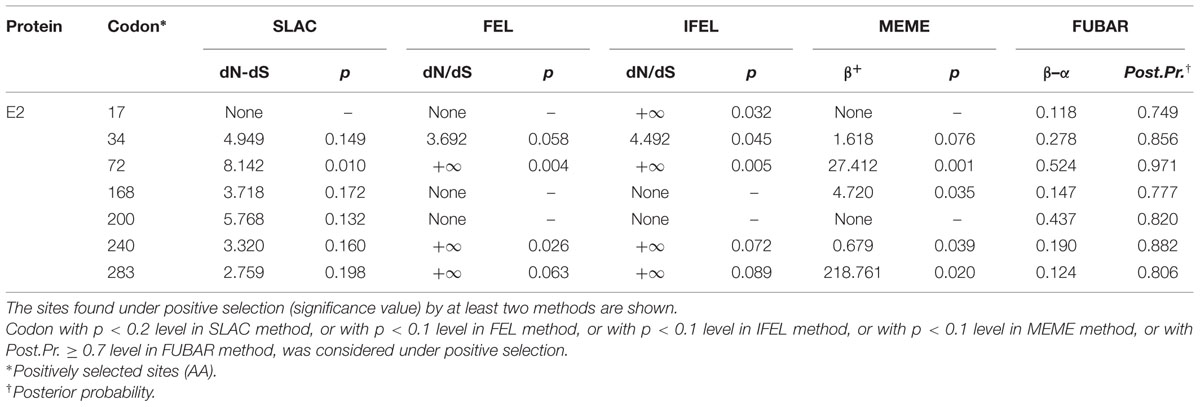
TABLE 3. Selection pressure analysis of E2 protein (373 codons) of CSFV using SLAC, FEL, IFEL, MEME, and FUBAR methods (www.datamonkey.org).
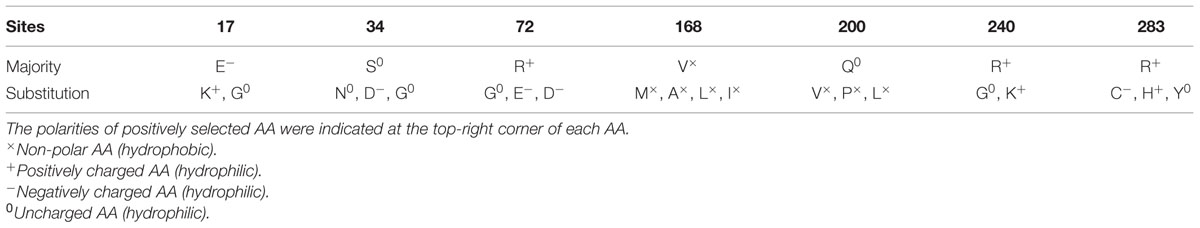
TABLE 4. Positions and polarities of the positively selected amino acid (AA).
Discussion
In China, a nationwide policy of biannual vaccinations of pigs in the spring and autumn has been performed using the C-strain vaccine, and large-scale outbreaks of CSF have rarely occurred since its introduction (Shen et al., 2011). Some of the cases that occurred were acute, but many cases of CSF were seen as subclinical, causing reproductive failure, neonatal death, or chronic infection in nursery pigs (Luo et al., 2011; Ji et al., 2015). However, in 2014, pigs in some herds in China that were immunized with attenuated CSFV vaccines showed CSF-suspected symptoms (Zhang et al., 2015), and subsequently a similar epidemic unexpectedly occurred in Shandong province, which caused heavy economic losses. To identify the pathogeneses and pathogens, specimens were collected and systemic examinations were performed, and the CSFV infection status was confirmed.
To further study the molecular epidemiology of CSF, 25 isolated CSFV strains were obtained, and their genetic diversity was analyzed. The full-length E2 gene sequence (1,119 nt), which provides better resolution for phylogenetic analysis than 5′-NTR, partial E2 gene, and NS5B sequences (Blacksell et al., 2004; Sarma et al., 2011; Zhang et al., 2015), was sequenced and examined in this study. Both the full-length E2 sequence and partial E2 sequence showed similar results, as the CSFV isolates could be divided into three genotypes (1, 2, and 3) as well as 11 subgenotypes [1.1–1.4, 2.1 (2.1a, 2.1b, 2.1c, and 2.1d), 2.2, 2.3, and 3.4]. Compared with representative strains of subgenotypes 1.1, 2.1, 2.2, 2.3, and 3.4, the 25 isolates all belonged to subgenotype 2.1, and most of the strains (92%, 23/25) were clustered in the newly defined subgenotype 2.1d (Figure 2, Table 2). High sequence variability is found in mainland China where CSFV subgenotype 1.1, 2.1, 2.2, and 2.3 strains are found, and subgenotype 2.1b has been shown to be the predominant strains within the last 10 years (Tu et al., 2001; Chen et al., 2010a; Beer et al., 2015). In this study, CSF cases caused by a new subgenotype, 2.1d, of CSFV in Shandong province were diagnosed following outbreaks in other provinces (Zhang et al., 2015), and the earliest discovered CSFV isolate, SDTA1-13, which was identified as subgenotype 2.1d in this study, was first isolated in 2013. The results indicate that the new strains may have emerged over a short period of time and spread to several provinces in China, which is worthy of attention because all of the new strains were isolated from CSFV-immunized pigs (Zhang et al., 2015). The pathogenicity, antigenicity, and virulence of the newly defined 2.1d isolates remain unclear, but we speculate that the unique molecular characteristics of the 2.1d isolates may contribute to the adaptive evolution of CSFV under C-strain vaccination, and may be responsible for the unsatisfactory immunoprotection of C-strain vaccinations.
To further study the molecular characteristics of CSFV strains, a selection pressure analysis of E2 AA sequences was performed, and the results showed that the protein mainly underwent purifying selection pressures. RNA viruses are known to have significantly greater mutation rates per site per round of replication than DNA viruses, a difference that is attributed to the error-prone nature of viral RNA-dependent RNA polymerases, and most mutations in coding regions are deleterious (Weiss, 2002; Hughes and Hughes, 2007). A mechanism to decrease the accumulation of deleterious mutations is essential for RNA viruses to remain stable, and purifying selection provides a useful tool to purge such mutations (Domingo and Holland, 1997). In addition, purifying selection was reportedly more effective in RNA viruses than in DNA viruses (Hughes and Hughes, 2007). Seven positively selected sites were observed in the E2 protein, which is the main immunogen of CSFV. E2 is a type I transmembrane protein with a transmembrane domain in its carboxyl-terminus that is anchored in the viral envelope (Li et al., 2013). The amino-terminal half of the E2 protein, which is an extracellular motif that contains four antigenic domains (A, B, C, and D), was more variable than the carboxyl-terminal half (van Rijn et al., 1994). E2 has a unique architecture consisting of two immunoglobulin-like domains (I and II). Domains D/A map to domain II (AAs 91–168) in the E2 crystal structure, and domains B/C correspond to domain I (AAs 1–90) (Li et al., 2013). Among the detected seven positively selected sites, AAs 17, 34, and 72 belonged to domains B/C. AA 168 belonged to domains D/A. Moreover, the other three sites (AAs 200, 240, and 283) are located in the carboxyl-terminal half of the E2 protein. Domains B/C, which form an independent antigenic unit, are responsible for antigenic specificity among various CSFVs, and the D/A domains of various CSFVs are relatively conserved (van Rijn et al., 1994; Chang et al., 2010). It has been reported that single mutations in the E2 B/C domains could lead to variations in viral neutralization (Chen et al., 2010b). The three positively selected sites found in domains B/C of the amino-terminal half of the E2 protein, which mediates viral entry into target cells, suggest that these changes could be associated with viral escape from neutralizing antibodies, and they could explain the lower severity of the clinical signs that developed in most of the affected animals. The positively selected AA 200 is reportedly necessary for the attenuation of the highly virulent Brescia strain, but the mechanisms mediating this attenuation remain unknown (Risatti et al., 2007; Tang et al., 2008). In this study, we observed a polarity change of AA 200 from hydrophilic to hydrophobic, which may contribute to a change of the antigenicity and virulence of CSFV. The other three positively selected sites (AAs 168, 240, and 283) found in this study are the first to be reported, and their biological significance needs to be further characterized. Understanding the functional importance of these positively selected AAs could help to predict possible changes in virulence, which will aid the study of the mechanism of immune evasion, and prevent CSF in the future.
Conclusion
The 25 CSFV isolates from East China were clustered in subgroup 2.1, and most of the isolates, together with some previously sequenced strains, formed the newly defined subgenotype 2.1d, indicating that 2.1d CSFV strains may be predominant epidemic strains in Shandong province. The selection pressure analysis revealed that the envelope protein-encoding E2 gene had undergone positive selection, and several positively selected sites were identified, which could help to identify the molecular determinants of virulence or pathogenesis, and to clarify the driving force of CSFV evolution in East China. Empirical studies are required to assess the antigenicity and virulence of the 2.1d CSFV strains, as well as the influence of the positively selected AAs identified in this study on CFSV virulence or pathogenesis.
Author Contributions
DH and SL contributed to conception and design of the study. YX contributed to design of the study. LL contributed to acquisition and analysis of data. JG and TC contributed to acquisition of data. DH and LL drafted the manuscript. YX and SL critically revised the manuscript.
Funding
This research was partially supported by the Open Fund of the State Key Laboratory of Veterinary Etiological Biology (SKLVEB2015KFKT0015).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00085
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AA, amino acid; CSFV, classical swine fever virus; dN, non-synonymous substitution rate; dS, synonymous rate; H&E, hematoxylin and eosin; nt, nucleotide; NTRs, non-translated regions; ORF, open reading frame; PCV2, porcine circovirus type 2; PRRSV, porcine reproductive and respiratory virus; PRV, porcine pseudorabies virus.
Footnotes
References
Anisimova, M., Bielawski, J. P., and Yang, Z. (2001). Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol. Biol. Evol. 18, 1585–1592. doi: 10.1093/oxfordjournals.molbev.a003945
Beer, M., Goller, K. V., Staubach, C., and Blome, S. (2015). Genetic variability and distribution of Classical swine fever virus. Anim. Health Res. Rev. 16, 33–39. doi: 10.1017/S1466252315000109
Blacksell, S. D., Khounsy, S., Boyle, D. B., Greiser-Wilke, I., Gleeson, L. J., Westbury, H. A., et al. (2004). Phylogenetic analysis of the E2 gene of classical swine fever viruses from Lao PDR. Virus Res. 104, 87–92. doi: 10.1016/j.virusres.2004.02.041
Chang, C. Y., Huang, C. C., Lin, Y. J., Deng, M. C., Chen, H. C., Tsai, C. H., et al. (2010). Antigenic domains analysis of classical swine fever virus E2 glycoprotein by mutagenesis and conformation-dependent monoclonal antibodies. Virus Res. 149, 183–189. doi: 10.1016/j.virusres.2010.01.016
Chen, N., Li, D., Yuan, X., Li, X., Hu, H., Zhu, B., et al. (2010a). Genetic characterization of E2 gene of classical swine fever virus by restriction fragment length polymorphism and phylogenetic analysis. Virus Genes 40, 389–396. doi: 10.1007/s11262-010-0465-8
Chen, N., Tong, C., Li, D., Wan, J., Yuan, X., Li, X., et al. (2010b). Antigenic analysis of classical swine fever virus E2 glycoprotein using pig antibodies identifies residues contributing to antigenic variation of the vaccine C-strain and group 2 strains circulating in China. Virol. J. 7, 378. doi: 10.1186/1743-422X-7-378
Delport, W., Poon, A. F., Frost, S. D., and Kosakovsky Pond, S. L. (2010). Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455–2457. doi: 10.1093/bioinformatics/btq429
Deng, M. C., Huang, C. C., Huang, T. S., Chang, C. Y., Lin, Y. J., Chien, M. S., et al. (2005). Phylogenetic analysis of classical swine fever virus isolated from Taiwan. Vet. Microbiol. 106, 187–193. doi: 10.1016/j.vetmic.2004.12.014
Domingo, E., and Holland, J. J. (1997). RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51, 151–178. doi: 10.1146/annurev.micro.51.1.151
Edwards, S., Fukusho, A., Lefevre, P. C., Lipowski, A., Pejsak, Z., Roehe, P., et al. (2000). Classical swine fever: the global situation. Vet. Microbiol. 73, 103–119. doi: 10.1016/S0378-1135(00)00138-3
Greiser-Wilke, I., Depner, K., Fritzemeier, J., Haas, L., and Moennig, V. (1998). Application of a computer program for genetic typing of classical swine fever virus isolates from Germany. J. Virol. Methods 75, 141–150. doi: 10.1016/S0166-0934(98)00109-8
Hu, D., Zhang, Z., Lv, L., Xiao, Y., Qu, Y., Ma, H., et al. (2015). Outbreak of variant pseudorabies virus in Bartha-K61–vaccinated piglets in central Shandong Province, China. J. Vet. Diagn. Invest. 27, 600–605. doi: 10.1177/1040638715593599
Hughes, A. L., and Hughes, M. A. (2007). More effective purifying selection on RNA viruses than in DNA viruses. Gene 404, 117–125. doi: 10.1016/j.gene.2007.09.013
Ji, W., Guo, Z., Ding, N. Z., and He, C. Q. (2015). Studying classical swine fever virus: making the best of a bad virus. Virus Res. 197, 35–47. doi: 10.1016/j.virusres.2014.12.006
Jiang, D. L., Gong, W. J., Li, R. C., Liu, G. H., Hu, Y. F., Ge, M., et al. (2013). Phylogenetic analysis using E2 gene of classical swine fever virus reveals a new subgenotype in China. Infect. Genet. Evol. 17, 231–238. doi: 10.1016/j.meegid.2013.04.004
Kosakovsky Pond, S. L., and Frost, S. D. (2005). Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208–1222. doi: 10.1093/molbev/msi105
Li, Y., Wang, J., Kanai, R., and Modis, Y. (2013). Crystal structure of glycoprotein E2 from bovine viral diarrhea virus. Proc. Natl. Acad. Sci. U.S.A. 110, 6805–6810. doi: 10.1073/pnas.1300524110
Lowings, P., Ibata, G., Needham, J., and Paton, D. (1996). Classical swine fever virus diversity and evolution. J. Gen. Virol. 77(Pt 6), 1311–1321. doi: 10.1099/0022-1317-77-6-1311
Luo, T. R., Liao, S. H., Wu, X. S., Feng, L., Yuan, Z. X., Li, H., et al. (2011). Phylogenetic analysis of the E2 gene of classical swine fever virus from the Guangxi Province of Southern China. Virus Genes 42, 347–354. doi: 10.1007/s11262-011-0578-8
Paton, D. J., and Greiser-Wilke, I. (2003). Classical swine fever–an update. Res. Vet. Sci. 75, 169–178. doi: 10.1016/S0034-5288(03)00076-6
Paton, D. J., Mcgoldrick, A., Greiser-Wilke, I., Parchariyanon, S., Song, J. Y., Liou, P. P., et al. (2000). Genetic typing of classical swine fever virus. Vet. Microbiol. 73, 137–157. doi: 10.1016/S0378-1135(00)00141-3
Postel, A., Schmeiser, S., Bernau, J., Meindl-Boehmer, A., Pridotkas, G., Dirbakova, Z., et al. (2012). Improved strategy for phylogenetic analysis of classical swine fever virus based on full-length E2 encoding sequences. Vet. Res. 43, 50. doi: 10.1186/1297-9716-43-50
Postel, A., Schmeiser, S., Perera, C. L., Rodriguez, L. J., Frias-Lepoureau, M. T., and Becher, P. (2013). Classical swine fever virus isolates from Cuba form a new subgenotype 1.4. Vet. Microbiol. 161, 334–338. doi: 10.1016/j.vetmic.2012.07.045
Risatti, G. R., Holinka, L. G., Fernandez Sainz, I., Carrillo, C., Lu, Z., and Borca, M. V. (2007). N-linked glycosylation status of classical swine fever virus strain Brescia E2 glycoprotein influences virulence in swine. J. Virol. 81, 924–933. doi: 10.1128/JVI.01824-06
Rumenapf, T., Meyers, G., Stark, R., and Thiel, H. J. (1991). Molecular characterization of hog cholera virus. Arch. Virol. Suppl. 3, 7–18. doi: 10.1007/978-3-7091-9153-8_2
Sanchez, O., Barrera, M., Rodriguez, M. P., Frias, M. T., Figueroa, N. E., Naranjo, P., et al. (2008). Classical swine fever virus E2 glycoprotein antigen produced in adenovirally transduced PK-15 cells confers complete protection in pigs upon viral challenge. Vaccine 26, 988–997. doi: 10.1016/j.vaccine.2007.11.014
Sarma, D. K., Mishra, N., Vilcek, S., Rajukumar, K., Behera, S. P., Nema, R. K., et al. (2011). Phylogenetic analysis of recent classical swine fever virus (CSFV) isolates from Assam, India. Comp. Immunol. Microbiol. Infect. Dis. 34, 11–15. doi: 10.1016/j.cimid.2009.09.005
Sharma, S., Joshi, G., Dash, P. K., Thomas, M., Athmaram, T. N., Kumar, J. S., et al. (2013). Molecular epidemiology and complete genome characterization of H1N1pdm virus from India. PLoS ONE 8:e56364. doi: 10.1371/journal.pone.0056364
Shen, H., Pei, J., Bai, J., Zhao, M., Ju, C., Yi, L., et al. (2011). Genetic diversity and positive selection analysis of classical swine fever virus isolates in south China. Virus Genes 43, 234–242. doi: 10.1007/s11262-011-0625-5
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tang, F., Pan, Z., and Zhang, C. (2008). The selection pressure analysis of classical swine fever virus envelope protein genes Erns and E2. Virus Res. 131, 132–135. doi: 10.1016/j.virusres.2007.08.015
Tautz, N., Tews, B. A., and Meyers, G. (2015). The molecular biology of pestiviruses. Adv. Virus Res. 93, 47–160. doi: 10.1016/bs.aivir.2015.03.002
Tu, C., Lu, Z., Li, H., Yu, X., Liu, X., Li, Y., et al. (2001). Phylogenetic comparison of classical swine fever virus in China. Virus Res. 81, 29–37. doi: 10.1016/S0168-1702(01)00366-5
van Rijn, P. A., Miedema, G. K., Wensvoort, G., Van Gennip, H. G., and Moormann, R. J. (1994). Antigenic structure of envelope glycoprotein E1 of hog cholera virus. J. Virol. 68, 3934–3942.
Vanderhallen, H., Mittelholzer, C., Hofmann, M. A., and Koenen, F. (1999). Classical swine fever virus is genetically stable in vitro and in vivo. Arch. Virol. 144, 1669–1677. doi: 10.1007/s007050050622
Weiss, R. A. (2002). Virulence and pathogenesis. Trends Microbiol. 10, 314–317. doi: 10.1016/S0966-842X(02)02391-0
Keywords: classical swine fever virus, genetic diversity, phylogenetic analysis, positive selection, subgenotype 2.1d
Citation: Hu D, Lv L, Gu J, Chen T, Xiao Y and Liu S (2016) Genetic Diversity and Positive Selection Analysis of Classical Swine Fever Virus Envelope Protein Gene E2 in East China under C-Strain Vaccination. Front. Microbiol. 7:85. doi: 10.3389/fmicb.2016.00085
Received: 10 December 2015; Accepted: 18 January 2016;
Published: 05 February 2016.
Edited by:
Akio Adachi, Tokushima University Graduate School, JapanReviewed by:
Stefan Vilcek, University of Veterinary Medicine and Pharmacy in Košice, SlovakiaMyung-Hee Kwon, Ajou University School of Medicine, South Korea
Copyright © 2016 Hu, Lv, Gu, Chen, Xiao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yihong Xiao, xiaoyihong01@163.com; Sidang Liu, liusid@sdau.edu.cn
†These authors have contributed equally to this work.