 Alexander B. Chase
Alexander B. Chase Philip Arevalo
Philip Arevalo Martin F. Polz
Martin F. Polz Renaud Berlemont
Renaud Berlemont Jennifer B. H. Martiny
Jennifer B. H. Martiny- 1Department of Ecology and Evolutionary Biology, University of California, Irvine, Irvine, CA, USA
- 2Parsons Laboratory for Environmental Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
- 3Department of Biological Sciences, California State University Long Beach, Long Beach, CA, USA
Assigning ecological roles to bacterial taxa remains imperative to understanding how microbial communities will respond to changing environmental conditions. Here we analyze the genus Curtobacterium, as it was found to be the most abundant taxon in a leaf litter community in southern California. Traditional characterization of this taxon predominantly associates it as the causal pathogen in the agricultural crops of dry beans. Therefore, we sought to investigate whether the abundance of this genus was because of its role as a plant pathogen or another ecological role. By collating >24,000 16S rRNA sequences with 120 genomes across the Microbacteriaceae family, we show that Curtobacterium has a global distribution with a predominant presence in soil ecosystems. Moreover, this genus harbors a high diversity of genomic potential for the degradation of carbohydrates, specifically with regards to structural polysaccharides. We conclude that Curtobacterium may be responsible for the degradation of organic matter within litter communities.
Introduction
Traditional ecological characterization of microorganisms often narrowly defines their roles in terms of interspecies interactions. Such limited classification of interactions ignores the dynamic alterations of life cycles indicative of microorganisms in changing environmental conditions (Redman et al., 2001; Kogel et al., 2006; Newton et al., 2010). Depending on the environment, microbes can transition from symbiont to pathogen (Johnson et al., 1997) or drastically alter their life history strategy altogether. For instance, endophytic fungi transition to decomposers after the leaves fall off its host plant (Osono, 2006; Korkama-Rajala et al., 2008). Such flexibility in ecological roles may also explain why Curtobacterium, a bacterial genus traditionally viewed as a plant pathogen (Hsieh et al., 2005), was recently found to be the dominant bacterium in the leaf litter of a Mediterranean-like grassland community (Matulich et al., 2015).
Members of the Curtobacterium genus are Gram-positive, obligately aerobic chemoorganotrophs in the family Microbacteriaceae, phylum Actinobacteria (Evtushenko and Takeuchi, 2006). The habitat of Curtobacterium is described mainly in association with plants and especially, the phyllosphere (Komagata et al., 1965; Behrendt et al., 2002). Indeed, most studies investigating Curtobacterium focus on its role as an economically important plant pathogen (Huang et al., 2009; Osdaghi et al., 2015b). The best-studied pathovar, C. flaccumfaciens pv. flaccumfaciens, is the causal agent of bacterial wilt in dry beans worldwide with reports on five continents (Wood and Easdown, 1990; Harveson et al., 2006; EPPO, 2011; Soares et al., 2013; Osdaghi et al., 2015a). The disease harbors a high degree of genetic and phenotypic diversity (Hedges, 1926; Conner et al., 2008) even within a single host (Agarkova et al., 2012).
Although economically important, C. flaccumfaciens is the only species of Curtobacterium associated with plant pathogenesis (Young et al., 1996), and there is evidence that other Curtobacterium species perform other ecological roles. For instance, isolates have been identified as endophytic symbionts (Sturz et al., 1997, 1999; Elbeltagy et al., 2000; Araújo et al., 2001; Bulgari et al., 2009). Similar to other beneficial endophytes (Benhamou et al., 2000; Taghavi et al., 2009), Curtobacterium can elicit plant defense responses (Bulgari et al., 2011) and reduce disease symptoms (Lacava et al., 2007). The genus has also been found to associate with roots and promote plant growth (Sturz et al., 1997). Even the presence of C. flaccumfaciens in the rhizosphere induced a systematic resistance in cucumber plants to pathogens (Raupach and Kloepper, 1998) and promoted plant growth (Raupach and Kloepper, 2000). Curtobacterium can also be found in soil (Ohya et al., 1986; Aizawa et al., 2007; Kim et al., 2008) with an ability to persist on plant debris (Silva Junior et al., 2012), although as a non-spore forming bacterium, the genus might be assumed to be a poor survivor in soil (Vidaver, 1982).
Our previous work in a Mediterranean-like grassland community revealed that a Curtobacterium taxon (defined by ≥ 97% similarity in 16S rRNA sequence) was the most abundant bacterium in leaf litter, the top layer of soil. The leaf litter community at this site is dominated by bacteria with a bacteria to fungi biomass ratio up to 30:1 (Alster et al., 2013). The community is highly diverse, but uneven; three phyla (Actinobacteria, Bacteroidetes, and Proteobacteria) made up 95% of total bacterial abundance (Matulich et al., 2015). Further analysis revealed that Curtobacterium constituted ~18% of 16S rRNA sequences amplified directly from 177 litter samples over a two-year period (Matulich et al., 2015). This high abundance was further supported by sequenced metagenomes from the same grassland. These samples suggested that >8% of the reads fall within Microbacteriaceae (Berlemont et al., 2014), most likely an underestimate due to lack of representation of Curtobacterium in genomic databases.
Given its dominance in grassland litter, this current study investigates the potential for Curtobacterium to play ecological roles other than a plant pathogen, and in particular, as a decomposer. We asked: (1) What is the geographic and habitat distribution of the genus? (2) Is the phylogenetic diversity of Curtobacterium related to its habitat distribution? and (3) What is the genus' genomic potential to degrade recalcitrant carbohydrates? To address these questions, we isolated and sequenced 14 Curtobacterium strains from grassland litter. Then, we combined our genome sequences with publically-available sequences from a variety of habitats and locations, collating >24,000 Microbacteriaceae 16S rRNA sequences. Finally, we investigated the genomic diversity of Curtobacterium with regards to its ability to degrade carbohydrates, an important attribute for litter decomposition. We searched for glycoside hydrolases (GHs), enzymes that target specific glycosidic bonds of carbohydrates (including cellulose and xylan in plant cell walls). We conclude that the genus Curtobacterium is cosmopolitan in terrestrial ecosystems and may be, at an intrageneric level, involved in a variety of ecological roles including decomposition of organic matter.
Materials and Methods
Geographic Distribution
To investigate the geographic extent of Curtobacterium, we searched for Curtobacterium sequences within the open reference dataset of the Earth Microbiome Project (EMP) (Gilbert et al., 2014). We obtained 41 unique Curtobacterium OTUs with metadata from 14,096 uploaded samples.
To gather additional Curtobacterium sequences, we used BLAST to search for sequences similar to eight Curtobacterium 16S rRNA gene sequences from the GreenGenes “Core Set” database (DeSantis et al., 2006) against the GenBank nr database (Benson et al., 2008). Additional sequences were identified using the keyword search: “Microbacteriaceae Curtobacterium 16S ribosomal RNA gene.” After removing redundant entries and 16S rRNA sequences that could not be identified to the genus level, 11,484 unique sequences remained.
We extracted metadata from either corresponding GenBank files, the EMP 10k merged mapping file, or manually reviewed the published literature to identify the isolation source and location of all retrieved Curtobacterium sequences. Each sequence was assigned to one of seven ecosystems: animal microbiome, aquatic, artificial, atmosphere, human microbiome, ice, or terrestrial. Terrestrial samples were further divided into six categories: plant, plant roots, plant seeds, rock, sediment, and soil.
The geographic distribution of the EMP and GenBank sequences were plotted using the R library “rworldmap” (South, 2011). For samples with minimal location data (mainly from the GenBank dataset), we used a publicly available dataset from Google Developers1 to assign approximate longitude and latitude coordinates based on the state, providence, and/or country of origin.
Phylogenetic Diversity
To establish a robust phylogenetic distribution, we downloaded 16S rRNA gene sequences from the SILVA SSU r123 database (Pruesse et al., 2007) on August 6, 2015. Sequences were obtained using SILVA's assigned taxonomy, yielding 1519 Curtobacterium sequences and 24,835 Microbacteriaceae sequences. Due to variability in taxonomic nomenclature by various databases, we confirmed all taxonomic assignments of all downloaded sequences. First, we assigned taxonomy with QIIME v1.6 (Caporaso et al., 2010) using the UCLUST consensus taxonomy assigner (Edgar, 2010) against the GreenGenes reference database (May 2013 revision; DeSantis et al., 2006). Next, we compared these taxonomic assignments to those assigned using the RDP Classifier (Wang et al., 2007). After removing sequences incorrectly assigned to Curtobacterium and/or Microbacteriaceae and other low quality sequences (<80% identity, <700 bp), 12,469 sequences remained.
To select a subset of this sequence diversity for phylogenetic analysis, we clustered the filtered sequences and the sequences of our litter isolates (see below) using QIIME v1.9. We defined OTUs at 97% identity with UCLUST using the optimal flag for OTU picking, and selected representative sequences for each OTU. The representative sequences were assigned a taxonomic designation at the genus level using a combination of UCLUST, BLAST, and the RDP Classifier. Specifically, genera designations for the representative sequences were only assigned when at least two of the aforementioned taxonomic designations matched at the genus level. We aligned the sequences using the Infernal Alignment Tool (Nawrocki et al., 2009). Gaps common in >90% of aligned sequences were manually removed, resulting in a 1900 bp alignment. OTU representative sequences that contained >25% gap regions were also removed. As a result, the sequences obtained from the EMP database were too short (~100–250 bp; mean size = 134 bp) to integrate in the phylogeny with the full 16S rRNA gene obtained from other datasets. A maximum likelihood tree with 100 bootstrap replications was constructed with RAxML v8.0, using the GTR + Gamma distribution model (Stamatakis, 2014). The tree was visualized using the Interactive Tree of Life (iTOL; Letunic and Bork, 2007).
The pipeline above was modified slightly to investigate the phylogenetic diversity within the Curtobacterium genus. This analysis incorporated all available 16S rRNA genes (n = 1532) from GenBank, SILVA, and litter isolates assigned to Curtobacterium. OTUs were clustered at 99% similarity to provide finer taxonomic resolution and included a sister genus, Frigoribacterium, as an outgroup.
Genomic Analysis of Litter Isolates
Isolation and Identification of Litter Isolates
Bacteria from litter were isolated from two grassland global change experiments. Isolates from the Loma Ridge Global Change Experiment (LRGCE) (in Irvine, California, USA [33° 44′ N, 117° 42′ W]; Potts et al., 2012) were previously identified and presented in Mouginot et al. (2014). Briefly, leaf litter particles were suspended in saline and inoculated onto nutrient-limited media plates made from Loma Ridge litter leachate and incubated at room temperature. For this study, additional strains were isolated from the Boston-Area Climate Experiment (BACE) [42° 23′ N, 71° 12′ W] (Tharayil et al., 2011) using the same protocol on Boston litter leachate media. Individual colonies were streaked onto LB plates three times to ensure clonal isolation.
To identify Curtobacterium isolates, the 16S rRNA gene was PCR amplified and sequenced. Individual colonies were boiled for 1 min in 50 μL of sterile dH2O prior to PCR amplification. Next, 3.0 μL of the boiled bacterial colony was added to the PCR cocktail containing 0.3 μL of Taq polymerase (5 units/μL), 15.0 μL of Premix F (Epicentre, Madison, WI), and 50 μM of each primer in a final volume of 30 μL. We amplified 1500 bp of the 16S rRNA gene using the pA (5′-AGAGTTTGATCCTGGCTCAG-3′) and pH (5′-AAGGAGGTGATCCAGCCGCA-3′) primers (Edwards et al., 1989). Forward and reverse strands were trimmed and merged using Geneious v6.1 (Drummond et al., 2011) under the default parameters. Isolate identity was tentatively assigned using the best-identified match with the blastn alignment (Altschul et al., 1997) within GenBank. In total, 34 Microbacteriaceae isolates were identified, including 17 Curtobacterium isolates.
Whole Genome Analysis
This Whole Genome Shotgun project including the genome sequences of 14 Curtobacterium, 1 Frigoribacterium, and 1 Plantibacter isolates deposited at GenBank under BioProject PRJNA342146 with accessions MJGI00000000-MJGX00000000. Paired-end 100 bp × 100 bp whole genome sequencing libraries with a mean gap size of 400 bp were prepared from genomic DNA using the Nextera DNA Library Preparation Kit (Illumina Inc., San Diego, CA, USA). Genomes were sequenced on an Illumina HiSeq 2500 apparatus (Illumina Inc., San Diego, CA, USA) at the Whitehead Institute Genome Technology Core (Cambridge, MA). After quality trimming and removal of short (<30 bp) reads, an initial de novo assembly was performed in CLC Genomics Workbench (CLC Bio, Cambridge, MA, USA) using the default parameters.
Genomes (fully assembled and whole genome shotgun assembly) belonging to the Microbacteriaceae were retrieved from the Pathosystems Resource Integration Center (PATRIC) database (Wattam et al., 2013). To annotate these downloaded genomes and our isolate genomes, we first assigned open reading frames (ORFs) sequences as called by Prodigal v2.6 (Hyatt et al., 2010). Genomic ORFs were searched against the Pfam database (Finn et al., 2014) for the presence of protein families using HMMer (Johnson et al., 2010). We identified the GH families as in Berlemont and Martiny (2013) and compiled the number of occurrences of each GH family in each genome. To create a phylogeny of the whole genome sequences, the 16S rRNA region of each genome was predicted using Barrnap2. The resulting sequences were used for phylogenetic reconstruction as described above.
Results
Geographic Distribution of Curtobacterium
We isolated 17 Curtobacterium strains from two invasive grassland sites. Although similar in their vegetation, LRGCE and BACE sites are 4130 km apart across the North American continent. Yet, from these sites, Curtobacterium strains comprised 10 and 15% of culturable isolates in LRGCE and BACE, respectively. Beyond these two terrestrial sites, data collected from a wide array of studies and isolation sources reveal that Curtobacterium is an abundant and globally distributed taxon. In total, we obtained 3360 16S rRNA sequences with corresponding metadata from GenBank and the EMP databases. The genus was found on all continents, ranging from the Arctic to the Antarctic (Figure 1). The majority of sequences were isolated from North America (61.6%), while there was a lack of representation in the Southern hemisphere, most likely due to sampling effort. Australia, South America, Africa, and Antarctica accounted for only 15.3% of all sequences.
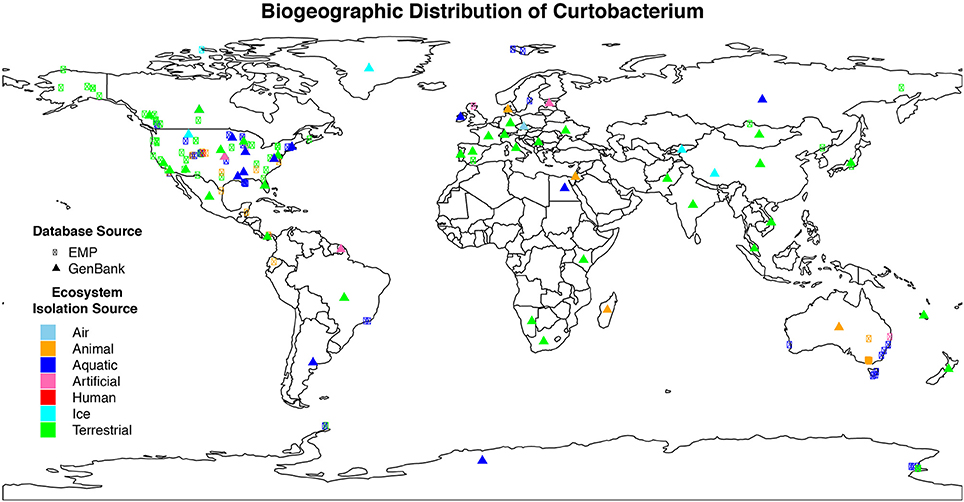
Figure 1. Geographic distribution of Curtobacterium compiled from various isolation sources. Colors indicate the different ecosystems from which the sequence was isolated. The symbol shape indicates the dataset from which the sequence originated. Sequences obtained from GenBank (triangle symbols) were mostly approximations as detailed GPS coordinates were not available.
Curtobacterium has been identified in all designated ecosystems, including animal microbiome, aquatic, artificial, atmosphere, human microbiome, ice, and terrestrial (Supplementary Table 1). The human and animal microbiome comprised 26.9 and 12.9% of all obtained Curtobacterium sequences, respectively. Curtobacterium sequences from humans were comprised almost exclusively of samples originating from skin, while those from animals were primarily collected from the gut. Most Curtobacterium sequences (32.6%) from the EMP dataset were from human microbiome samples, reflecting the emphasis on humans in this dataset. In contrast, only 10.8% of Curtobacterium sequences retrieved from GenBank were associated with the human microbiome. After excluding human microbiome samples, over 63% of all sequences originated from terrestrial ecosystems. Specifically, 14% of all sequences were extracted from a plant source and 21% from soil. Sequences from the GenBank database revealed a stronger association with 70.1% of sequences being classified into a terrestrial ecosystem (Supplementary Table 1). Terrestrial samples from the GenBank database included 58.9% from plants and 28.4% from soil.
Phylogenetic Diversity
The Microbacteriaceae sequences clustered into 971 OTUs at a 97% similarity level. Considering only OTUs with more than 10 sequence representatives, the remaining 183 OTUs represented 19 genera (Figure 2). The 10 Curtobacterium OTUs form a well-supported (bootstrap support of 89%) monophyletic clade. Their closest relatives belong to the Rathayibacter and Pseudoclavibacter genera. The 17 Curtobacterium litter isolates from the Loma Ridge and Boston sites clustered together into five OTUs. Two Curtobacterium OTUs contained only one litter isolate despite being in the top 25 of the most abundant OTUs in the SILVA database.
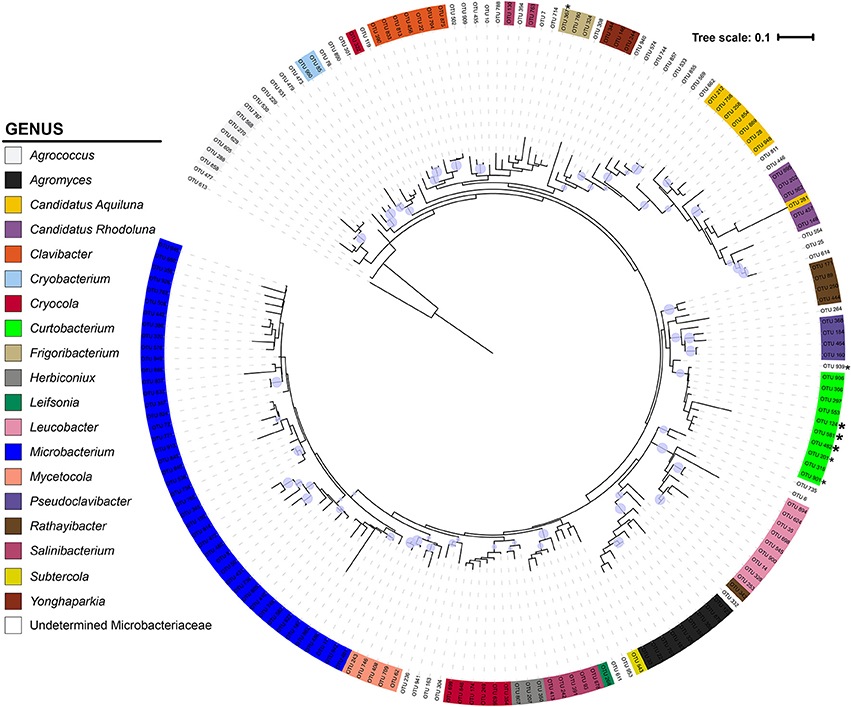
Figure 2. Phylogeny of Microbacteriaceae constructed from the 16S rRNA gene (maximum likelihood tree with 100 bootstraps and a GTR + GAMMA distribution). The tree is color-coded by genus using the taxonomic designation assigned from a combination of SILVA, BLAST, and RDP. The circles represent nodes with at least 70% support and the diameter of the circle represents the support level. * = 1 litter isolate within the OTU; * = >4 litter isolates in OTU.
To examine Curtobacterium diversity at a finer genetic resolution, we clustered the 1074 total sequences retrieved from GenBank and SILVA with our 22 isolates at a 99% similarity level. This yielded 100 Curtobacterium and 7 Frigoribacterium OTUs, a sister genus. Excluding singletons, the remaining 52 OTUs represented 1014 Curtobacterium sequences with 764 of those sequences containing metadata originating from GenBank entries. Of these sequences, 582 (74%) sequences were isolated from a terrestrial ecosystem. Due to some OTUs containing many sequences without habitat data, distribution of ecosystem preference across phylogeny was not possible. However, there were OTUs detected solely in one ecosystem (e.g., OTU 25 was only found in terrestrial ecosystems), while others OTUs were detected in a variety of ecosystems (e.g., OTU 38 was found in all seven assigned ecosystems). At the level of 99% sequence similarity, most (10 out of 18) of the litter isolates clustered into one abundant OTU (86). This abundant OTU contained over 202 sequences isolated from all seven assigned ecosystems (Figure 3).
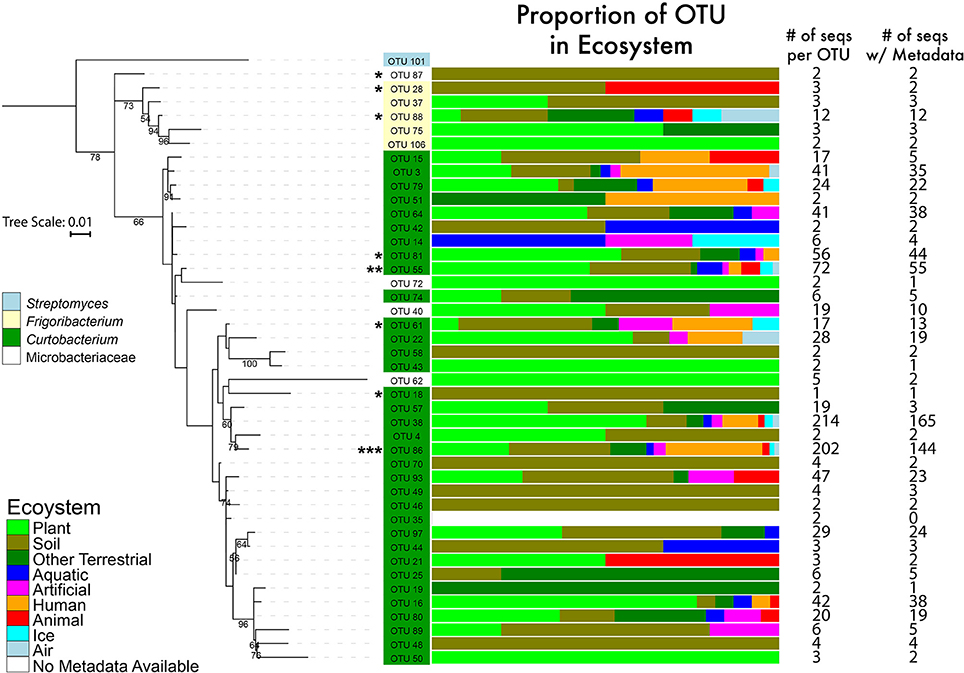
Figure 3. Phylogenetic tree of the genus Curtobacterium with 16S rRNA gene (maximum likelihood tree with 100 bootstraps and a GTR + GAMMA distribution). The numbers represent the support level of each node with at least 50% support. Bar graphs are color coded to show the percentages of the OTUs with sequences isolated from various ecosystems. Numbers in the columns represent the number of sequences incorporated into each branch for its respective OTU. * = 1–4 litter isolates within the OTU; ** = 5 isolates; *** = 10 isolates.
Genomic Potential for Carbohydrate Degradation
Full genomes were used to compare the genomic diversity of glycoside hydrolases within Microbacteriaceae. We included 14 Curtobacterium, 1 Frigoribacterium, and 1 Plantibacter isolates from our leaf litter sites at LRGCE and BACE. The Curtobacterium assemblies produced an average genome size of 3.76 Mbp in an average of 78 contigs (mean maximum contig length of 582,567 bp), with an average GC content of 70.47% (Table 1).
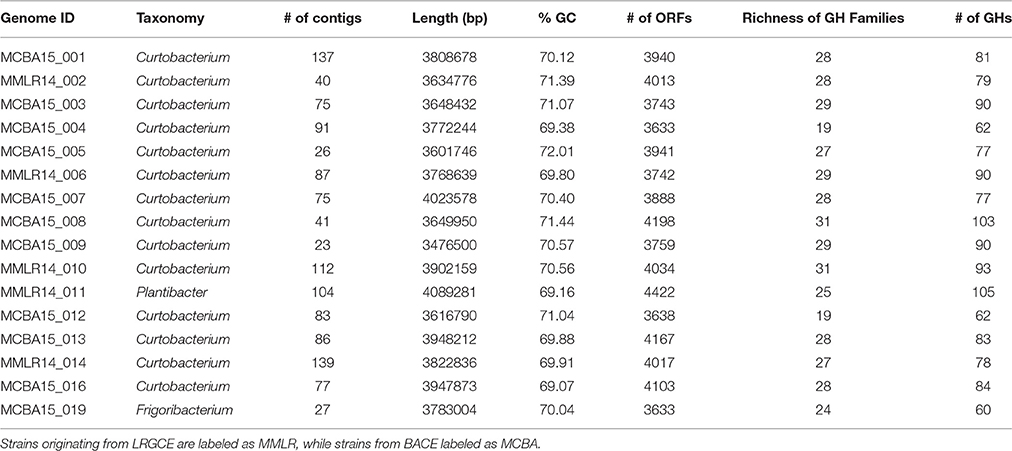
Table 1. General characteristics of the litter isolates.
Combining these genomes with the 104 publicly available genomes retrieved from the PATRIC database reveal that strains within Microbacteriaceae contain many diverse GH families. Across the 120 genomes, we identified 7355 potential glycoside hydrolases (GHs) and carbohydrate binding modules (CBMs) representing 63 GH/CBM families (Supplementary Table 2). The most common and ubiquitous families belonged to those targeting starch (GH13, CBM48) and oligosaccharides (GH1, 2, and 3). These GH families were present in most genomes with 92.5% of the genomes containing at least one copy of GH13. GHs that targeted more recalcitrant carbohydrates such as fructan, dextran, mixed polysaccharides, animal polysaccharides, plant polysaccharides, cellulose, chitin, and xylan were also detected in a variety of genomes across Microbacteriaceae, albeit at a lower frequency (Table 2).
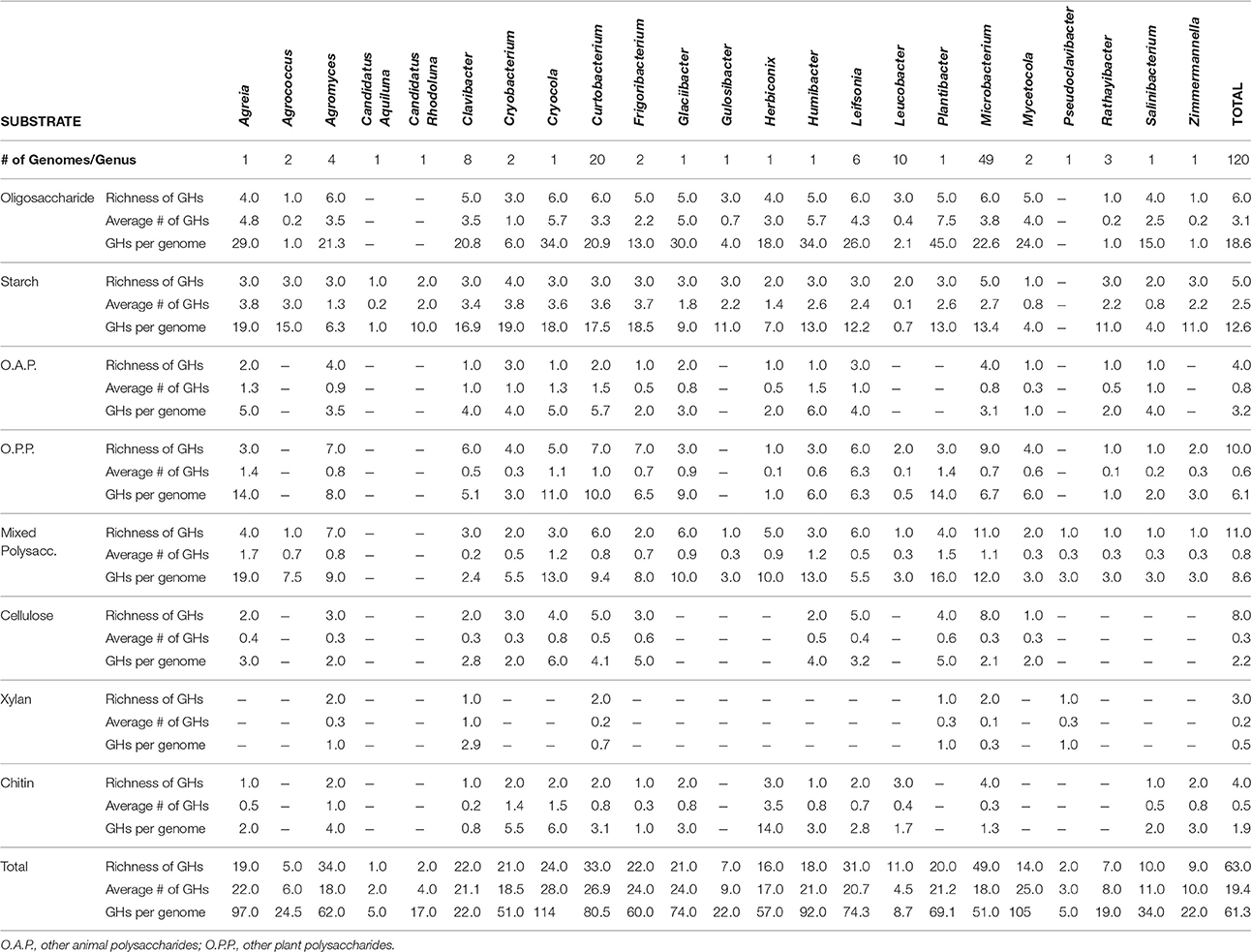
Table 2. Breakdown by genus of the distribution of GHs and CBMs by targeted substrate.
GH content was highly variable across genera. Some genera appeared not able to process any structural polysaccharides (cellulose, chitin, or xylan) and constrained to the targeting of oligosaccharides and starch (see Table 2). Others, like Pseudoclavibacter, lacked any of the identified GH families that process simpler substrates such as starch, and, presumably, are only capable of processing more complex carbohydrates. A few genera had the genomic potential to digest all identified substrates. Specifically, Curtobacterium appeared capable of targeting all substrates at a frequency almost double the family average, particularly with regard to structural polysaccharides. Individual strains with the potential to breakdown and digest all three structural polysaccharides appeared to be restricted within the genera Curtobacterium (N = 11 genomes; including 8 litter isolates), Clavibacter (N = 6 genomes), and Microbacterium (N = 6 genomes).
The average richness of GH families present in a Microbacteriaceae genome was 19.3 GH/CBM families (Table 2). However, GH/CBM richness varied widely across genomes; a Leucobacter genome contained only 1 GH family while one Microbacterium species, Microbacterium sp. SUBG005 (accession number JNNT00000000), had 35 GH families. The litter isolates belonging to Curtobacterium had an above average richness of 27.2 GH families with a range of 19–31 GH families. Further, most genomes harbored multiple copies of each protein family. For example, a Microbacterium genome had as many as 24 copies of the GH13 family. Due to the multiple GH copies, genomes varied in the total number of GHs present (mean number of GHs = 61.3), ranging from 3 GH proteins in a Leucobacter strain to 135 GH proteins in an Agromyces strain. On average, the Curtobacterium litter isolates encoded 82.1 GH proteins, almost 1.5 times the family average (Figure 4).
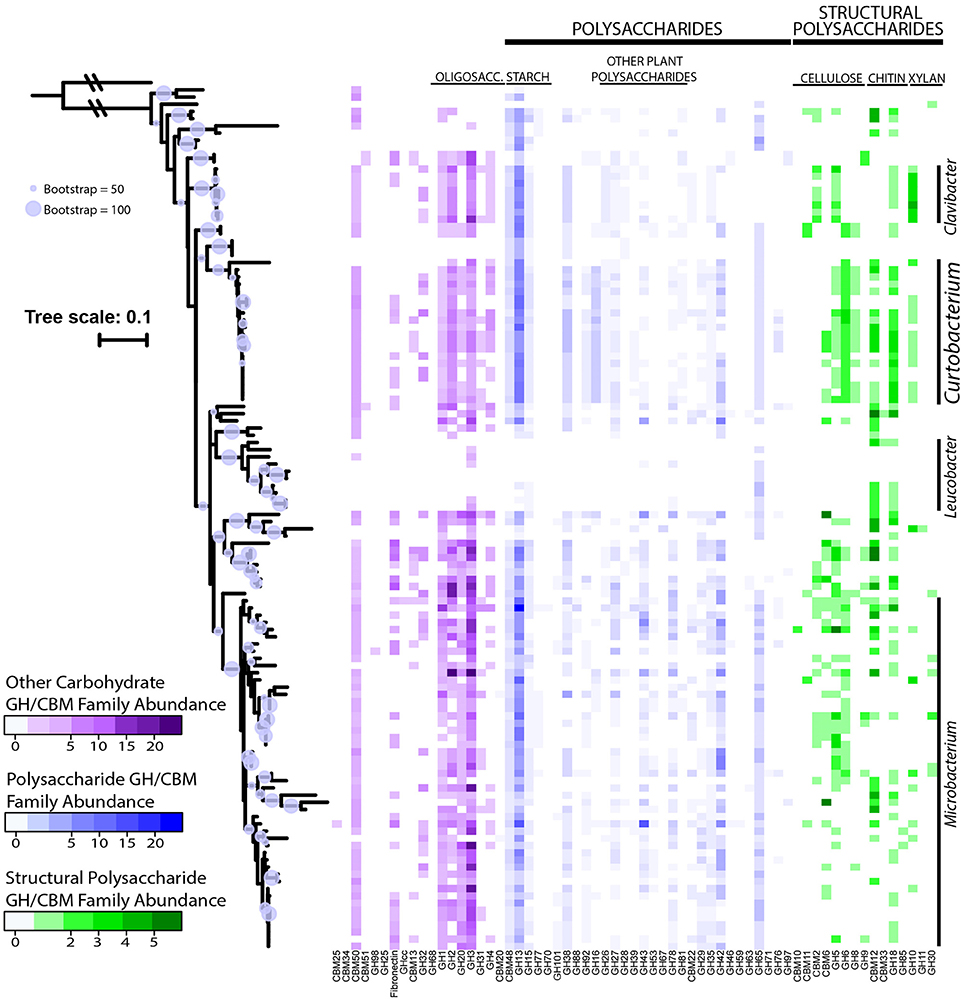
Figure 4. Abundance of GH and CBM families grouped and colored by substrate category across downloaded Microbacteriaceae genomes from the PATRIC database. Phylogenetic tree constructed from the 16S rRNA gene sequence (maximum likelihood tree with 100 bootstraps and a GTR + GAMMA distribution). The circles represent nodes with at least 50% support and the diameter of each circle represents the support level. Genera with more than 5 strains are denoted on the right.
We examined the potential for each individual genome to target multiple polysaccharides. Almost all genomes within the family had the potential to process oligosaccharides or starch with the exception of 2 genomes, a Leucobacter and Pseudoclavibacter strain. Further, a majority of the genomes (103 genomes or 85.8%) within Microbacteriaceae were capable of processing at least one structural polysaccharide. Specifically, the frequency to be able to target cellulose, chitin, and xylan occurred in 64.2, 64.2, and 29.2% of the genomes, respectively.
Discussion
In this study, we present the first global survey of Curtobacterium and show that it is ubiquitous in a variety of ecosystems (Figure 1) although it is most abundant in terrestrial ecosystems, and a majority of sequences are associated with plants and soil. This observation is in accordance with past studies of Curtobacterium that attribute its habitat to plants and the related phyllosphere (Komagata et al., 1965; Behrendt et al., 2002). However, Curtobacterium is primarily known as a plant pathogen and yet, the highest proportion of Curtobacterium strains resided in soil systems, suggesting that this genus may be capable of reproducing in soil.
We also provide a well-supported phylogeny of all known Microbacteriaceae genera. We built upon previous Microbacteriaceae phylogenetic analyses (see Evtushenko and Takeuchi, 2006) to incorporate all available Microbacteriaceae 16S rRNA sequences, providing the most comprehensive phylogenetic analysis of Microbacteriaceae to date (Figure 2). To explore diversity within Curtobacterium, we constructed a genus-specific tree to investigate the possibility of clade-specific habitat preference. Due to differences in sequencing platforms and targeted regions of the 16S rRNA gene, there may be habitat specialization at finer clade levels than we are able to differentiate here. In particular, the shorter sequenced reads (e.g., from the EMP dataset) are limited in their phylogenetic resolution and cannot resolve intrageneric patterns. Further, many GenBank sequences lacked metadata altogether or were limited in their details to allow for finer habitat designations (e.g., which part of the plant or the layer of soil from which a strain was isolated). Although we did not detect any clade-specific patterns of habitat preferences, most clades contained a majority of plant and soil isolated sequences (Figure 3), indicating that the genus as a whole may be adapted to plant or soil habitats.
Curtobacterium falls within the Actinobacteria phylum, which is known to play a crucial role in the recycling of organic material by decomposition and humus formation (Goodfellow and Williams, 1983). This characterization is supported by a comprehensive analysis into the distribution of GHs across all bacteria, which showed that Actinobacteria has the highest genomic potential for being cellulose degraders (Berlemont and Martiny, 2015). Therefore, we concentrated on these GH proteins, as they are responsible for the breakdown of large carbohydrates that may prove advantageous in decomposition of plant debris. For instance, an increase in diversity and abundance of GHs with the potential for cellulose utilization generally corresponds to better cellulose degradation (Fontes and Gilbert, 2010; Wilson, 2011; Berlemont and Martiny, 2015). Previously, Curtobacterium isolates collected from a neutral garden soil were shown to rapidly degrade cellulose fibers (Lednická et al., 2000). Indeed, our results provide a genomic underpin for Curtobacterium to be a degrader. The genus has an elevated richness and abundance of GHs relative to other Microbacteriaceae genera. While there is large variation within the family with respect to GH richness and substrate degradation, Curtobacterium is one of only three genera with the potential ability to target all identified carbohydrate substrates. Moreover, out of these three genera, Curtobacterium has the highest abundance of GHs, suggesting an increased ability to utilize and degrade a wide range of carbohydrates. This variability in carbon usage within Curtobacterium suggests that alternative, intrageneric ecological roles have yet to be identified.
We conclude that Curtobacterium may be a dominant player in the functional breakdown of dead organic material in leaf litter communities based on its dominance in two grassland litter microbial communities, its high representation in soils, and its genomic potential for being a degrader. This work supports previous studies that show that Curtobacterium has the capability to survive on litter (Silva Junior et al., 2012) and thrive as a cellulytic bacterium (Lednická et al., 2000). The conclusion also aligns with culture work that finds that coryneform bacterium, such as Curtobacterium, are in high abundance on grasses (Behrendt et al., 2002). Despite the focus in the literature on its role as a crop plant pathogen, future research into the contribution of Curtobacterium to the recycling of nutrients in terrestrial ecosystems warrants further attention.
Author Contributions
AC and JM: Developed and designed the study; AC: Preformed data analysis with JM contributing to data interpretation; RB: Assisted in analysis of carbohydrate data; PA and MP: Prepared, assembled, and annotated the genome libraries; AC: Wrote the manuscript with input from all authors.
Funding
Funding was provided by the US Department of Energy, Office of Science, Office of Biological and Environmental Research (BER), under Award Number DE-PS02-09ER09-25 and by the U.S. National Science Foundation (DEB-1457160) to JM. This work was supported by the U.S. Department of Energy (DE-SC0008743) to MP.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Adam Martiny and Travis Huxman for their guidance and helpful comments on earlier revisions. We thank Kristin Dolan for use of the Loma Ridge isolates, Jeff Dukes for supplying litter from BACE, and Sean Gibbons and Jack Gilbert for help with the open reference database for the EMP. We also thank Richard Puxty, Claudia Weihe, and Michaeline Nelson for their input and assistance with labwork and computational methods.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01874/full#supplementary-material
Footnotes
References
Agarkova, I. V., Lambrecht, P. A., Vidaver, A. K., and Harveson, R. M. (2012). Genetic diversity among Curtobacterium flaccumfaciens pv. flaccumfaciens populations in the American high plains. Can. J. Microbiol. 58, 788–801. doi: 10.1139/w2012-052
Aizawa, T., Ve, N. B., Kimoto, K., Iwabuchi, N., Sumida, H., Hasegawa, I., et al. (2007). Curtobacterium ammoniigenes sp. nov., an ammonia-producing bacterium isolated from plants inhabiting acidic swamps in actual acid sulfate soil areas of Vietnam. Int. J. Syst. Evol. Microbiol. 57(Pt 7), 1447–1452. doi: 10.1099/ijs.0.64731-0
Alster, C. J., German, D. P., Lu, Y., and Allison, S. D. (2013). Microbial enzymatic responses to drought and to nitrogen addition in a southern California grassland. Soil Biol. Biochem. 64, 68–79. doi: 10.1016/j.soilbio.2013.03.034
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Araújo, W. L., Maccheroni, W. Jr, Aguilar-Vildoso, C. I., Barroso, P. A., Saridakis, H. O., and Azevedo, J. L. (2001). Variability and interactions between endophytic bacteria and fungi isolated from leaf tissues of citrus rootstocks. Can. J. Microbiol. 47, 229–236. doi: 10.1139/w00-146
Behrendt, U., Ulrich, A., Schumann, P., Naumann, D., and Suzuki, K. (2002). Diversity of grass-associated Microbacteriaceae isolated from the phyllosphere and litter layer after mulching the sward; polyphasic characterization of Subtercola pratensis sp. nov., Curtobacterium herbarum sp. nov. and Plantibacter flavus gen. nov., sp. nov. Int. J. Syst. Evol. Microbiol. 52(Pt 5), 1441–1454. doi: 10.1099/00207713-52-5-1441
Benhamou, N., Gagné, S., Le Quéré, D., and Dehbi, L. (2000). Bacterial-mediated induced resistance in cucumber: beneficial effect of the endophytic bacterium Serratia plymuthica on the protection against infection by Pythium ultimum. Phytopathology 90, 45–56. doi: 10.1094/PHYTO.2000.90.1.45
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Wheeler, D. L. (2008). GenBank. Nucleic Acids Res. 36(Suppl. 1), D25–D30. doi: 10.1093/nar/gkm929
Berlemont, R., Allison, S. D., Weihe, C., Lu, Y., Brodie, E. L., Martiny, J. B., et al. (2014). Cellulolytic potential under environmental changes in microbial communities from grassland litter. Front. Microbiol. 5:639. doi: 10.3389/fmicb.2014.00639
Berlemont, R., and Martiny, A. C. (2013). Phylogenetic distribution of potential cellulases in bacteria. Appl. Environ. Microbiol. 79, 1545–1554. doi: 10.1128/AEM.03305-12
Berlemont, R., and Martiny, A. C. (2015). Genomic potential for polysaccharide deconstruction in bacteria. Appl. Environ. Microbiol. 81, 1513–1519. doi: 10.1128/AEM.03718-14
Bulgari, D., Casati, P., Brusetti, L., Quaglino, F., Brasca, M., Daffonchio, D., et al. (2009). Endophytic bacterial diversity in grapevine (Vitis vinifera L.) leaves described by 16S rRNA gene sequence analysis and length heterogeneity-PCR. J. Microbiol. 47, 393–401. doi: 10.1007/s12275-009-0082-1
Bulgari, D., Casati, P., Crepaldi, P., Daffonchio, D., Quaglino, F., Brusetti, L., et al. (2011). Restructuring of endophytic bacterial communities in grapevine yellows-diseased and recovered Vitis vinifera L. plants. Appl. Environ. Microbiol. 77, 5018–5022. doi: 10.1128/AEM.00051-11
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Conner, R. L., Balasubramanian, P., Erickson, R. S., Huang, H. C., and Mündel, H.-H. (2008). Bacterial wilt resistance in kidney beans. Can. J. Plant Sci. 88, 1109–1113. doi: 10.4141/CJPS08074
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Drummond, A., Ashton, B., Buxton, S., Cheung, M., Cooper, A., Duran, C., et al. (2011). Geneious, Version 5.4. Auckland: Biomatters Ltd.
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edwards, U., Rogall, T., Blöcker, H., Emde, M., and Böttger, E. C. (1989). Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 17, 7843–7853. doi: 10.1093/nar/17.19.7843
Elbeltagy, A., Nishioka, K., Suzuki, H., Sato, T., Sato, Y.-I., Morisaki, H., et al. (2000). Isolation and characterization of endophytic bacteria from wild and traditionally cultivated rice varieties. Soil Sci. Plant Nutr. 46, 617–629. doi: 10.1080/00380768.2000.10409127
EPPO (2011). Curtobacterium flaccumfaciens pv. flaccumfaciens. EPPO Bull. 41, 320–328. doi: 10.1111/j.1365-2338.2011.02496.x
Evtushenko, L. I., and Takeuchi, M. (2006). “The family Microbacteriaceae,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York, NY: Springer), 1020–1098.
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Fontes, C. M., and Gilbert, H. J. (2010). Cellulosomes: highly efficient nanomachines designed to deconstruct plant cell wall complex carbohydrates. Annu. Rev. Biochem. 79, 655–681. doi: 10.1146/annurev-biochem-091208-085603
Gilbert, J. A., Jansson, J. K., and Knight, R. (2014). The Earth Microbiome project: successes and aspirations. BMC Biol. 12:69. doi: 10.1186/s12915-014-0069-1
Goodfellow, M., and Williams, S. (1983). Ecology of actinomycetes. Annu. Rev. Microbiol. 37, 189–216. doi: 10.1146/annurev.mi.37.100183.001201
Harveson, R., Schwartz, H., Vidaver, A., Lambrecht, P. A., and Otto, K. (2006). New outbreaks of bacterial wilt of dry bean in Nebraska observed from field infections. Plant Dis. 90, 681–681. doi: 10.1094/PD-90-0681A
Hedges, F. (1926). Bacterial wilt of Beans (Bacterium flaccum-faciens Hedges), including comparisons with Bacterium phaseoli. Phytopathology 16, 1–22.
Hsieh, T., Huang, H., Mündel, H. H., Conner, R., Erickson, R., and Balasubramanian, P. (2005). Resistance of common bean (Phaseolus vulgaris) to bacterial wilt caused by Curtobacterium flaccumfaciens pv. flaccumfaciens. J. Phytopathol. 153, 245–249. doi: 10.1111/j.1439-0434.2005.00963.x
Huang, H., Erickson, R., Balasubramanian, P., Hsieh, T., and Conner, R. (2009). Resurgence of bacterial wilt of common bean in North America. Can. J. Plant Pathol. 31, 290–300. doi: 10.1080/07060660909507603
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Johnson, L. S., Eddy, S. R., and Portugaly, E. (2010). Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11:431. doi: 10.1186/1471-2105-11-431
Johnson, N., Graham, J. H., and Smith, F. (1997). Functioning of mycorrhizal associations along the mutualism–parasitism continuum. New Phytol. 135, 575–585. doi: 10.1046/j.1469-8137.1997.00729.x
Kim, M. K., Kim, Y.-J., Kim, H.-B., Kim, S.-Y., Yi, T.-H., and Yang, D.-C. (2008). Curtobacterium ginsengisoli sp. nov., isolated from soil of a ginseng field. Int. J. Syst. Evol. Microbiol. 58(Pt 10), 2393–2397. doi: 10.1099/ijs.0.65591-0
Kogel, K.-H., Franken, P., and Hückelhoven, R. (2006). Endophyte or parasite–what decides? Curr. Opin. Plant Biol. 9, 358–363. doi: 10.1016/j.pbi.2006.05.001
Komagata, K., Iizuka, H., and Takahashi, M. (1965). Taxonomic evaluation of nitrate respiration and carbohydrate fermentation in aerobic bacteria. J. Gen. Appl. Microbiol. 11, 191–201. doi: 10.2323/jgam.11.191
Korkama-Rajala, T., Müller, M. M., and Pennanen, T. (2008). Decomposition and fungi of needle litter from slow-and fast-growing Norway spruce (Picea abies) clones. Microb. Ecol. 56, 76–89. doi: 10.1007/s00248-007-9326-y
Lacava, P. T., Li, W., Arauújo, W. L., Azevedo, J. L., and Hartung, J. S. (2007). The endophyte Curtobacterium flaccumfaciens reduces symptoms caused by Xylella fastidiosa in Catharanthus roseus. J. Microbiol. 45, 388.
Lednická, D., Mergaert, J., Cnockaert, M. C., and Swings, J. (2000). Isolation and identification of cellulolytic bacteria involved in the degradation of natural cellulosic fibres. Syst. Appl. Microbiol. 23, 292–299. doi: 10.1016/S0723-2020(00)80017-X
Letunic, I., and Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Matulich, K. L., Weihe, C., Allison, S. D., Amend, A. S., Berlemont, R., Goulden, M. L., et al. (2015). Temporal variation overshadows the response of leaf litter microbial communities to simulated global change. ISME J. 9, 2477–2489. doi: 10.1038/ismej.2015.58
Mouginot, C., Kawamura, R., Matulich, K. L., Berlemont, R., Allison, S. D., Amend, A. S., et al. (2014). Elemental stoichiometry of Fungi and Bacteria strains from grassland leaf litter. Soil Biol. Biochem. 76, 278–285. doi: 10.1016/j.soilbio.2014.05.011
Nawrocki, E. P., Kolbe, D. L., and Eddy, S. R. (2009). Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337. doi: 10.1093/bioinformatics/btp157
Newton, A. C., Fitt, B. D., Atkins, S. D., Walters, D. R., and Daniell, T. J. (2010). Pathogenesis, parasitism and mutualism in the trophic space of microbe–plant interactions. Trends Microbiol. 18, 365–373. doi: 10.1016/j.tim.2010.06.002
Ohya, H., Komai, Y., and Yamaguchi, M. (1986). Occurrence of Curtobacterium sp. possessing ω-cyclohexyl fatty acids in soil with zinc added. Arch. Microbiol. 145, 9–12. doi: 10.1007/BF00413020
Osdaghi, E., Pakdaman Sardrood, B., Bavi, M., Akbari Oghaz, N., Kimiaei, S., and Hadian, S. (2015a). First report of Curtobacterium flaccumfaciens pv. flaccumfaciens causing cowpea bacterial wilt in Iran. J. Phytopathol. 163, 653–656. doi: 10.1111/jph.12300
Osdaghi, E., Taghavi, S. M., Fazliarab, A., Elahifard, E., and Lamichhane, J. R. (2015b). Characterization, geographic distribution and host range of Curtobacterium flaccumfaciens: an emerging bacterial pathogen in Iran. Crop Prot. 78, 185–192. doi: 10.1016/j.cropro.2015.09.015
Osono, T. (2006). Role of phyllosphere fungi of forest trees in the development of decomposer fungal communities and decomposition processes of leaf litter. Can. J. Microbiol. 52, 701–716. doi: 10.1139/w06-023
Potts, D., Suding, K., Winston, G., Rocha, A., and Goulden, M. (2012). Ecological effects of experimental drought and prescribed fire in a southern California coastal grassland. J. Arid Environ. 81, 59–66. doi: 10.1016/j.jaridenv.2012.01.007
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Raupach, G., and Kloepper, J. (2000). Biocontrol of cucumber diseases in the field by plant growth-promoting rhizobacteria with and without methyl bromide fumigation. Plant Dis. 84, 1073–1075. doi: 10.1094/PDIS.2000.84.10.1073
Raupach, G. S., and Kloepper, J. W. (1998). Mixtures of plant growth-promoting rhizobacteria enhance biological control of multiple cucumber pathogens. Phytopathology 88, 1158–1164. doi: 10.1094/PHYTO.1998.88.11.1158
Redman, R. S., Dunigan, D. D., and Rodriguez, R. J. (2001). Fungal symbiosis from mutualism to parasitism: who controls the outcome, host or invader? New Phytol. 151, 705–716. doi: 10.1046/j.0028-646x.2001.00210.x
Silva Junior, T. A. F., Negrão, D. R., Itako, A. T., Soman, J. M., and Maringoni, A. C. (2012). Survival of Curtobacterium flaccumfaciens pv. flaccumfaciens in soil and bean crop debris. J. Plant Pathol. 94, 331–337. doi: 10.4454/JPP.FA.2012.025
Soares, R. M., Fantinato, G. G. P., Darben, L. M., Marcelino-Guimarães, F. C., Seixas, C. D. S., and Carneiro, G. E. D. S. (2013). First report of Curtobacterium flaccumfaciens pv. flaccumfaciens on soybean in Brazil. Trop. Plant Pathol. 38, 452–454. doi: 10.1590/S1982-56762013000500012
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sturz, A., Christie, B., Matheson, B., Arsenault, W., and Buchanan, N. (1999). Endophytic bacterial communities in the periderm of potato tubers and their potential to improve resistance to soil-borne plant pathogens. Plant Pathol. 48, 360–369. doi: 10.1046/j.1365-3059.1999.00351.x
Sturz, A., Christie, B., Matheson, B., and Nowak, J. (1997). Biodiversity of endophytic bacteria which colonize red clover nodules, roots, stems and foliage and their influence on host growth. Biol. Fertil. Soils 25, 13–19. doi: 10.1007/s003740050273
Taghavi, S., Garafola, C., Monchy, S., Newman, L., Hoffman, A., Weyens, N., et al. (2009). Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl. Environ. Microbiol. 75, 748–757. doi: 10.1128/AEM.02239-08
Tharayil, N., Suseela, V., Triebwasser, D. J., Preston, C. M., Gerard, P. D., and Dukes, J. S. (2011). Changes in the structural composition and reactivity of Acer rubrum leaf litter tannins exposed to warming and altered precipitation: climatic stress-induced tannins are more reactive. New Phytol. 191, 132–145. doi: 10.1111/j.1469-8137.2011.03667.x
Vidaver, A. K. (1982). The plant pathogenic corynebacteria. Annu. Rev. Microbiol. 36, 495–517. doi: 10.1146/annurev.mi.36.100182.002431
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2013). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591. doi: 10.1093/nar/gkt1099
Wilson, D. B. (2011). Microbial diversity of cellulose hydrolysis. Curr. Opin. Microbiol. 14, 259–263. doi: 10.1016/j.mib.2011.04.004
Keywords: decomposition, leaf litter, glycoside hydrolases, Microbacteriaceae, Actinobacteria
Citation: Chase AB, Arevalo P, Polz MF, Berlemont R and Martiny JBH (2016) Evidence for Ecological Flexibility in the Cosmopolitan Genus Curtobacterium. Front. Microbiol. 7:1874. doi: 10.3389/fmicb.2016.01874
Received: 30 September 2016; Accepted: 07 November 2016;
Published: 22 November 2016.
Edited by:
Martin Hartmann, Swiss Federal Institute for Forest, Snow and Landscape Research, SwitzerlandReviewed by:
Livio Antonielli, Austrian Institute of Technology, AustriaErick Cardenas Poire, University of British Columbia, Canada
Copyright © 2016 Chase, Arevalo, Polz, Berlemont and Martiny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander B. Chase, abchase@uci.edu