Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases
 Matthew E. R. Butchbach1,2,3,4*
Matthew E. R. Butchbach1,2,3,4*- 1Center for Applied Clinical Genomics, Nemours Biomedical Research, Nemours Alfred I. duPont Hospital for Children, Wilmington, DE, USA
- 2Center for Pediatric Research, Nemours Biomedical Research, Nemours Alfred I. duPont Hospital for Children, Wilmington, DE, USA
- 3Department of Biological Sciences, University of Delaware, Newark, DE, USA
- 4Department of Pediatrics, Thomas Jefferson University, Philadelphia, PA, USA
Proximal spinal muscular atrophy (SMA), a leading genetic cause of infant death worldwide, is an early-onset, autosomal recessive neurodegenerative disease characterized by the loss of spinal α-motor neurons. This loss of α-motor neurons is associated with muscle weakness and atrophy. SMA can be classified into five clinical grades based on age of onset and severity of the disease. Regardless of clinical grade, proximal SMA results from the loss or mutation of SMN1 (survival motor neuron 1) on chromosome 5q13. In humans a large tandem chromosomal duplication has lead to a second copy of the SMN gene locus known as SMN2. SMN2 is distinguishable from SMN1 by a single nucleotide difference that disrupts an exonic splice enhancer in exon 7. As a result, most of SMN2 mRNAs lack exon 7 (SMNΔ7) and produce a protein that is both unstable and less than fully functional. Although only 10–20% of the SMN2 gene product is fully functional, increased genomic copies of SMN2 inversely correlates with disease severity among individuals with SMA. Because SMN2 copy number influences disease severity in SMA, there is prognostic value in accurate measurement of SMN2 copy number from patients being evaluated for SMA. This prognostic value is especially important given that SMN2 copy number is now being used as an inclusion criterion for SMA clinical trials. In addition to SMA, copy number variations (CNVs) in the SMN genes can affect the clinical severity of other neurological disorders including amyotrophic lateral sclerosis (ALS) and progressive muscular atrophy (PMA). This review will discuss how SMN1 and SMN2 CNVs are detected and why accurate measurement of SMN1 and SMN2 copy numbers is relevant for SMA and other neurodegenerative diseases.
Introduction
Proximal spinal muscular atrophy (SMA) is a leading genetic cause of infant death worldwide, alongside cystic fibrosis. The incidence of SMA is 1 in 6000–10,000 live births (Pearn, 1978; Cuscó et al., 2002; Sugarman et al., 2012). The carrier frequency for SMA is 1:25–50 in most populations (Ben-Shachar et al., 2011; Su et al., 2011; Lyahyai et al., 2012; Sugarman et al., 2012) though it is lower for some ethnicities (Zaldívar et al., 2005; Labrum et al., 2007; Hendrickson et al., 2009; Sangaré et al., 2014).
SMA is an early-onset neurodegenerative disease characterized by the loss of α-motor neurons in the anterior horn of the spinal cord, i.e., lower motor neurons (LMNs; Crawford and Pardo, 1996; Kolb and Kissel, 2015). This loss of α-motor neurons is associated with muscle weakness and atrophy. In SMA, those muscles that are proximally innervated are preferentially affected over distal muscles. SMA can be classified into five clinical grades based on age of onset and severity of the disease (Munsat and Davies, 1992; Russman, 2007; Table 1). Type 0 SMA infants present with very severe hypotonia and require respiratory support from birth. These SMA infants cannot survive beyond 6 months. Type I SMA [listed in the Online Inheritance in Man (OMIM) database under accession number 253300; http://www.omim.org/entry/253300] patients have an age of onset before 6 months. They show hypotonia and weakness in limbs; they are unable to sit independently. Type I SMA infants show a bell-shaped chest due to weakness in the intercostals muscles but sparing of the diaphragm; this bell-shaped chest results in abnormal breathing patterns. These patients typically live < 2 years. Type II SMA (http://www.omim.org/entry/253500) patients have an age of onset before 18 months. They are poor crawlers and weak sitters; most of these patients can rarely stand and only with support. Their legs are generally weaker than their arms. Due in part to better supportive care, these patients generally have a life expectancy into early adulthood. Type III SMA (http://www.omim.org/entry/253400) patients have an age of onset >18 months. These patients are able to walk with difficulty (waddling gait) and the legs are weaker than the arms. Type III patients usually have a normal lifespan. Adult-onset (type IV) SMA (http://www.omim.org/entry/271150) patients have an age of onset of 18–21 years. Type IV SMA exhibits as a slowly progressive limb weakness. The disease is fairly benign in these patients.
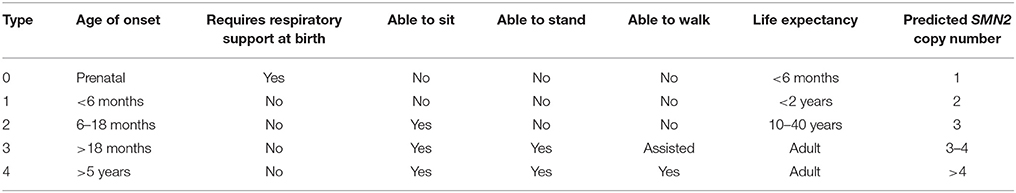
Table 1. Clinical classification of spinal muscular atrophy (SMA).
Genetics of SMA
SMA is an autosomal recessive disorder (Brandt, 1949). Linkage analysis (Brzustowicz et al., 1990; Gilliam et al., 1990; Melki et al., 1990a,b) along with genetic and physical mapping studies (reviewed in Morrison, 1996) identified the SMA locus on the long arm of chromosome 5, specifically in the 5q13 region. There is a 500 kilobase (kb) inverted segmental duplication within this region of chromosome 5 that is unique to humans (Courseaux et al., 2003; Schmutz et al., 2004). Four protein-coding genes have been identified within this region (Figure 1): SMN1 [survival motor neuron 1, telomeric SMN (SMNT; Lefebvre et al., 1995)], NAIP [neuronal apoptosis inhibitor protein (Roy et al., 1995)], GTF2H2A [general transcription factor IIH, p44 (Bürglen et al., 1997; Carter et al., 1997)], and SERF1A [small EDRK-rich factor 1A, H4F5A (Scharf et al., 1998)]. The duplicated genes are either identical to their partner gene (SERF1B), different in a small number of nucleotides [SMN2 or centromeric SMN (SMNC)] or are pseudogenes (ΨGTF2H2B and ΨNAIPΔ5).
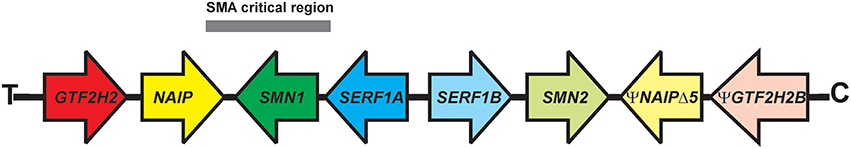
Figure 1. Organization of the inverted duplication locus on 5q13. 4 protein-coding genes—GTF2H2 (general transcription factor IIH), NAIP (neuronal apoptosis inhibitory protein), SMN1 (survival motor neuron 1), and SERF1A (small EDRK-rich factor 1A)—are present within the 500-kilobase inverted duplication on the long arm of chromosome 5 (5q13.2). The duplicated genes are ΨGTF2H2B (GTF2H2 pseudogene), ΨNAIPΔ5 (NAIP pseudogene with loss of exon 5), SMN2, and SERF1B. The SMA critical region is under the gray bar. C, centromeric end; T, telomeric end.
In more than 95% of cases, proximal SMA results from the loss of SMN1 but retention of SMN2, regardless of clinical grade (Lefebvre et al., 1995). Large-scale deletions in chromosome 5q13 that include SMN1, NAIP, SERF1A, and GTF2H2A are observed in patients with type I SMA (Wirth et al., 1995; Burlet et al., 1996; Rodrigues et al., 1996; Velasco et al., 1996; Bürglen et al., 1997; Carter et al., 1997). Smaller deletions only involving SMN1 have also been observed in type I SMA patients demonstrating that SMN1 is the most likely causative gene for SMA. In addition, the identification of intragenic SMA mutations in SMN1 (Lefebvre et al., 1995; see Burghes and Beattie, 2009 for a comprehensive listing of SMA-associated point mutations in SMN1) provides additional evidence to support SMN1 as the gene responsible for SMA. To date, no intragenic mutations in the other genes within this segmental duplication have been associated with SMA.
As mentioned earlier, the SMN gene is duplicated in humans to give rise to SMN1 and SMN2. This duplication of SMN is unique to humans (Rochette et al., 2001). What is the difference between SMN1 and SMN2? The major difference between these two SMN genes is a C-to-T transition in exon 7(SMN2 c.850C>T; Lorson et al., 1999; Monani et al., 1999). This nucleotide change is translationally silent. This position on exon 7 is in the middle of an exonic splicing enhancer (ESS) sequence that regulated the inclusion of exon 7 in SMN transcripts (Figure 2). For SMN1, the C at this position promotes inclusion of exon 7 in SMN1-derived mRNAs which leads to the production of full-length SMN protein. Full-length SMN protein is able to form functional complexes. For SMN2, the T at this position disrupts this ESS which results in the exclusion of exon 7 (SMNΔ7) from the majority of SMN2-derived mRNAs. As a result, a truncated SMNΔ7 protein is produced by the majority (~90%) of SMN2-derived mRNAs; this SMNΔ7 protein is unstable and is unable to associate with itself (Lorson and Androphy, 2000; Burnett et al., 2009; Cho and Dreyfuss, 2010). The SMNΔ7 protein is still partially functional given that transgenic overexpression of SMNΔ7 in severe SMA mice partially ameliorates their phenotype since these mice die at 14–15 days as opposed to 5–8 days (Le et al., 2005). About 10% of the mRNAs from SMN2 contain exon 7 and these full-length mRNAs can produce some full-length, functional SMN protein.
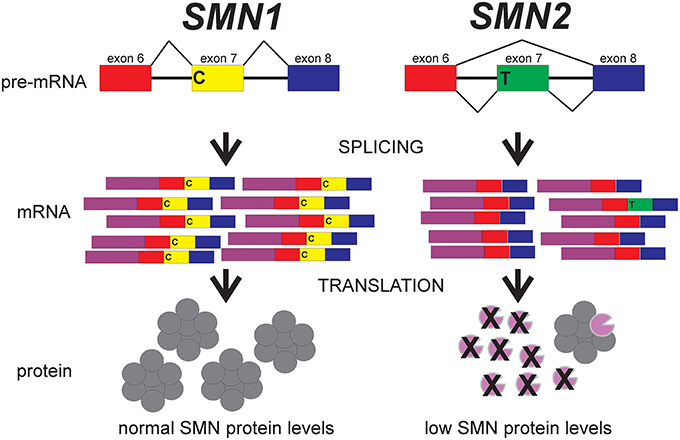
Figure 2. The effect of the C-to-T transition in exon 7 between SMN1 and SMN2 on splicing. Adapted from Butchbach and Burghes (2004).
Biology of SMN
SMN is a ubiquitously expressed protein whose expression is reduced in SMA. There is a strong inverse correlation between SMN protein levels and disease severity in SMA fibroblasts and lymphoblastoid cells as well (Coovert et al., 1997; Lefebvre et al., 1997; Kolb et al., 2006). Changes in SMN mRNA and protein levels observed in SMA patient-derived PBMCs mirror those observed in SMA cell lines (Sumner et al., 2006; Simard et al., 2007; Vezain et al., 2007; Tiziano et al., 2010; Crawford et al., 2012). SMN protein is present within the nuclei in discreet foci known as gems (Liu and Dreyfuss, 1996). In SMA fibroblasts, the number of SMN-positive subnuclear gems is higher in cells derived from mild SMA individuals than in those from children with more severe forms of SMA (Coovert et al., 1997).
SMN is required for the assembly of the small nuclear ribonucleoprotein (snRNP) complexes that mediate splicing (Pellizzoni, 2007; Burghes and Beattie, 2009). snRNP assembly is defective in SMN-deficient SMA cells (Wan et al., 2005). Since snRNP assembly is required for all cell types, why are motor neurons primarily affected in SMA? snRNP assembly is defective in tissues from mouse models for SMA and that the extent of reduced snRNP assembly correlates with phenotypic severity of these SMA mice (Gabanella et al., 2005; Zhang et al., 2008). snRNP assembly is more markedly reduced in SMA mouse neural tissues than in other tissues like the kidney (Gabanella et al., 2005) suggesting that motor neurons are more sensitive to deficits in snRNP assembly. SMN may also have a function that is unique to motor neurons. Axonal defects in Smn-knocked down zebrafish embryos (McWhorter et al., 2003) are corrected by overexpression of mutant SMNs which are incapable of snRNP assembly (Carrel et al., 2006).
SMN2 CNV in SMA
The number of SMN2 copies in the genome varies between 0 and 8. Numerous studies have demonstrated an inverse relationship between SMN2 copy number and disease severity among in SMA (Lefebvre et al., 1995, 1997; van der Steege et al., 1995; Coovert et al., 1997; McAndrew et al., 1997; Taylor et al., 1998; Feldkötter et al., 2002; Mailman et al., 2002; Anhuf et al., 2003; Gérard et al., 2004; Prior et al., 2005; Su et al., 2005; Swoboda et al., 2005; Arkblad et al., 2006; Scarciolla et al., 2006; Wirth et al., 2006; Gómez-Curet et al., 2007; Huang et al., 2007; Tiziano et al., 2007; Elsheikh et al., 2009; Wang et al., 2010a,b, 2014a; Alías et al., 2011; Amara et al., 2012; Crawford et al., 2012; Dobrowolski et al., 2012; Kirwin et al., 2013; Qu et al., 2014; Brkušanin et al., 2015; Fang et al., 2015; Stabley et al., 2015). Patients with milder forms of SMA have higher SMN2 copy numbers than severe SMA patients (Table 1).
Mice have only 1 SMN gene, mSmn, which is orthologous to SMN1 (DiDonato et al., 1997a; Viollet et al., 1997). Loss of mSmn leads to embryonic lethality (Schrank et al., 1997). Conditional loss of mSmn in specific cell types like neurons, myofibers and hepatocytes results in loss of those cells in vivo (Cifuentes-Diaz et al., 2002; Nicole et al., 2003; Vitte et al., 2004). Transgenic insertion of SMN2 rescues the embryonic lethality observed in mSmn nullizygous mice (Hsieh-Li et al., 2000; Monani et al., 2000; Michaud et al., 2010). While two copies of SMN2 rescues embryonic lethality in mSmn-deficient mice, these mice develop a very severe motor phenotype and die within 8 days after birth (Hsieh-Li et al., 2000; Monani et al., 2000). Those mSmn-deficient mice with 3-4 SMN2 copies exhibit a milder SMA phenotype than the two copy SMN2 SMA mice (Hsieh-Li et al., 2000; Michaud et al., 2010). If the SMN2 copy number is high (i.e., 8), then the resultant mSmn-deficient mice exhibit no signs of SMA and are phenotypically normal (Monani et al., 2000). SMN2 CNV, therefore, is a major modifier of disease severity in SMA in mice as well as in humans.
Gene conversion is one mechanism to account for increased SMN2 copy number in the absence of SMN1 in SMA (Burghes, 1997). In this scenario, the SMN1 gene actually contains part of SMN2, in particular within exon 7 (Wirth et al., 1995; Devriendt et al., 1996; Hahnen et al., 1996; van der Steege et al., 1996; Campbell et al., 1997; DiDonato et al., 1997b). It is hypothesized that type I SMA patients have deletions of SMN1 on both chromosomes. Type II SMA patients have an SMN1 deletion on one chromosome and a SMN1-to-SMN2 conversion on the other chromosome (three copies of SMN2). Type III SMA patients have SMN1-to-SMN2 converted genes on both chromosomes (four copies).
While the inverse relationship between SMN2 copy number and disease severity generally holds true in SMA, there are some exceptions. For example, there are cases of type II and III SMA patients who harbor only two copies of SMN2 instead of the predicted three or four copies (Prior et al., 2009; Bernal et al., 2010; Vezain et al., 2010). Sequencing of SMN2 in these cases revealed the presence of a rare single nucleotide variant (SMN2 c.859G>C) in exon 7 (Prior et al., 2009; Bernal et al., 2010; Vezain et al., 2010). This variant regulates the splicing of SMN2 pre-mRNAs so that a greater proportion of SMN2 transcripts contain exon 7. This variant may either create an additional SF2/ASF binding to promote exon 7 inclusion (Prior et al., 2009) or disrupt a hnRNPA1-dependent splicing silencer element (Vezain et al., 2010).
Intrafamilial variability in clinical presentation has been reported in SMA families with more than one affected sibling (Burghes et al., 1994; Cobben et al., 1995; Hahnen et al., 1995; McAndrew et al., 1997; Cuscó et al., 2006). Even though the siblings are haploidentical with respect to SMN2 copy number, they have differing clinical presentations. This would suggest that there are SMN2-independent modifiers of disease severity for SMA. Plastin-3 (PLS3) mRNA levels were higher in females with milder SMA than in discordant siblings with a more severe SMA clinical presentation (Oprea et al., 2008; Stratigopoulos et al., 2010; Bernal et al., 2011; Yanyan et al., 2014). In some families, however, female siblings with a more severe SMA phenotype show high PLS3 mRNA levels (Bernal et al., 2011). It is possible that the modifier property of PLS3 is age- and sex-dependent as well as incompletely penetrant; alternatively, PLS3 may not actually be a major modifier of SMA phenotype. There may be other non-SMN2 molecular modifiers of disease severity in SMA. It is important to identify and characterize these novel modifiers for the development of novel SMA biomarkers and targets for the development of therapeutic strategies for SMA as well as for the planning of current and future clinical trials in SMA (Wirth et al., 2013).
Measuring SMN1 and SMN2 CNVs
Because SMN2 copy number influences disease severity in SMA, there is prognostic value in accurate measurement of SMN2 copy number from patients being evaluated for SMA. Molecular diagnosis of SMA— i.e., loss of SMN1—has historically been made using a polymerase chain reaction (PCR)-based assay followed by digestion of the PCR product with specific restriction endonucleases (PCR-RFLP; Lefebvre et al., 1995; van der Steege et al., 1995). Numerous assays have since been developed to quantify SMN2 copy number in DNA samples from SMA patients. These assays include radioactive PCR (Coovert et al., 1997; McAndrew et al., 1997), fluorescent PCR (Taylor et al., 1998), quantitative (real-time) PCR (qPCR; Feldkötter et al., 2002; Anhuf et al., 2003; Gómez-Curet et al., 2007), competitive PCR/primer extension (Gérard et al., 2004), denaturing high performance liquid chromatography (Su et al., 2005), multiplex ligation-dependent probe amplification (MLPA; Arkblad et al., 2006; Scarciolla et al., 2006; Huang et al., 2007; Alías et al., 2011; Fang et al., 2015), quantitative capillary electrophoresis fragment analysis (Kirwin et al., 2013), short-amplicon melt profiling (Dobrowolski et al., 2012), fluorescent multiplex PCR/capillary electrophoresis (Wang et al., 2010a,b), and universal fluorescent triprobe ligation (Wang et al., 2014a). An important limitation of these established PCR-based copy number assays is the requirement for a parallel-run calibration curve to assign a breakpoint necessary that identifies placement of an ordinal SMN2 value. Additionally, these techniques cannot easily distinguish unit differences in SMN1 or SMN2 when the copy number is >3 (Gómez-Curet et al., 2007; Alías et al., 2011; Prior et al., 2011).
To overcome some of the limitations associated with the PCR-based assays described above, digital PCR (dPCR) distributed across a large number of partitions by limited dilution so that some partitions will lack the template DNA (Sykes et al., 1992; Vogelstein and Kinzler, 1999). The absolute abundance of the target gene can be measured by counting the number of positive partitions and the number of negative partitions. dPCR can reliably and accurately measure SMN1 and SMN2 copy numbers over a wide range, i.e., between 0 and 6 copies (Zhong et al., 2011; Stabley et al., 2015).
SMN1 and SMN2 CNVs in ALS
Amyotrophic lateral sclerosis (ALS) is a mostly adult-onset motor neuron disease characterized by a progressive loss of motor function leading to paralysis and respiratory failure (Boylan, 2015; Statland et al., 2015). Unlike SMA, ALS is caused by degeneration of LMNs as well as upper motor neurons (UMNs). ALS is usually fatal within 3–5 years after disease onset but there is considerable variability with respect to duration as well as phenotypic presentation (Swinnen and Robberecht, 2014). Most cases of ALS are sporadic in nature since there is no apparent family history. Approximately 10% of ALS is considered familial since either a causative gene has been identified or there is strong family history. With the recent advents of whole exome and whole genome sequencing, the genetic bases of almost 70% of familial ALS and 10% of sporadic ALS have been identified (Renton et al., 2014).
There are many case studies reporting the co-occurrence of SMA and ALS within a family (Appelbaum et al., 1992; Camu and Billiard, 1993; Orrell et al., 1997; Corcia et al., 2002a) which suggests that SMN1 deficiency may lead to ALS in addition to SMA. SMN1 deletions, however, have not been observed in either familial or sporadic ALS patients (Orrell et al., 1997; Corcia et al., 2002a). Furthermore, no intragenic point mutations in SMN1 have been reported in the ALS population (Blauw et al., 2012). The intrafamilial coexistence of SMA and ALS, therefore, occurs by chance.
Even though loss of SMN1 is not associated with ALS, CNVs in the SMN genes may modulate the clinical severity of ALS in addition to SMA. Multiple studies suggest that deletion of SMN2 leads to increased risk of the sporadic forms of amyotrophic lateral sclerosis (ALS) (Veldink et al., 2001, 2005; Kim et al., 2010; Corcia et al., 2012; Lee et al., 2012). Additionally, atypical SMN1 copy number—in other words, any number other than two—can affect the risk of ALS (Corcia et al., 2002b, 2006; Blauw et al., 2012; Wang et al., 2014b). Other studies, however, have shown no association between deletion of either SMN1 or SMN2 in ALS (Jackson et al., 1996; Moulard et al., 1998; Parboosingh et al., 1999; Crawford and Skolasky, 2002; Gamez et al., 2002). The discrepant results from these studies may be due, in part, to different assays used to assess SMN1 and SMN2 CNVs as some reports using quantitative PCR while others used MLPA or RFLP.
SMN and some ALS-associated proteins are involved in common biochemical pathways. Both familial and sporadic ALS have been linked to mutations in fused in sarcoma (FUS) (Kwiatkowski et al., 2009; Vance et al., 2009) (OMIM #608030) as well as in TAR DNA binding protein-43 kDa (TDP-43) (Kabashi et al., 2008; Sreedharan et al., 2008) (OMIM #612069). Both FUS and TDP-43 colocalize with SMN in subnuclear gems and ALS-associated mutations in FUS and TDP-43 reduce gem localization of SMN (Shan et al., 2010; Yamazaki et al., 2012; Gerbino et al., 2013; Groen et al., 2013; Ishihara et al., 2013; Sun et al., 2015). Gem localization of SMN, however, is not altered in other forms of sporadic ALS (Kariya et al., 2014). These mutant proteins also disrupt the SMN-mediated assembly of the splicing machinery by disrupting the interaction between SMN and U1-snRNPs (small nuclear ribonucleoprotein particles; Gerbino et al., 2013; Tsuiji et al., 2013; Sun et al., 2015; Yu et al., 2015). Additionally, ALS-associated FUS mutations disrupt the localization of SMN to axons (Groen et al., 2013). The SMN function, therefore, may be disrupted in certain forms of ALS.
Ectopic overexpression of SMN protects NSC34 motor neuron-like cells from cell death induced by ALS-associated mutant superoxide dismutase 1 (SOD1) (OMIM #105400) (Zou et al., 2007). The SOD1(G93A) transgenic mouse model for ALS that also harbors a knockout of 1 mSmn allele (SOD1(G93A)+∕−;mSmn+∕−) exhibits a more severe ALS phenotype than SOD1(G93A) ALS mice (SOD1(G93A)+∕−;mSmn+∕+) (Turner et al., 2009). Furthermore, ectopic overexpression of SMN in neurons and glia improves motor function of and delays motor neuron loss in SOD1(G93A) ALS mice (Turner et al., 2014). Strong transgenic overexpression of SMN2—in other words, eight copies of SMN2—delayed disease onset in the SOD1(G86R) mouse model for ALS (Kariya et al., 2012). Thesestudies suggest that increasing SMN expression may modulate disease severity in ALS. It will be interesting to determine the effect of SMN2 overexpression on disease severity in FUS- and TDP-43-associated ALS.
SMN1 and SMN2 CNVs in PMA
Progressive muscular atrophy (PMA) is an adult-onset motor neuron disease characterized by loss of LMNs (Rowland, 2010; Liewluck and Saperstein, 2015). It is a rare and sporadic disorder that is clinically distinct from ALS even though subclinical involvement of UMNs has been observed in many PMA patients. Those PMA patients exhibiting a more severe clinical presentation tend to harbor higher SMN1 copy numbers (Kuzma-Kozakiewicz et al., 2013). No relationship between SMN2 copy number and disease severity was noted in these PMA patients. Moulard et al. (1998) noted that the frequency of SMN2 deletion was higher in a small group of patients with sporadic LMN disease.
Conclusions
CNVs in the SMN genes can modulate disease severity in SMA as well as in other motor neurons. It is well-established that SMN2 copy number is inversely correlated with disease severity in SMA. Because of this relationship, SMN2 is a primary target for the development of therapeutics for SMA (Arnold and Burghes, 2013; Cherry et al., 2014). Numerous approaches including promoter activation, increased inclusion of exon 7 and protein stabilization are currently being developed to increase SMN2 expression. With respect to other motor neuron diseases, it is presently unclear whether increasing SMN1 or SMN2 expression would be beneficial or detrimental. On one hand, increasing SMN expression provides neuroprotective benefit to cell culture and transgenic mouse models for ALS; however, some genetic studies suggest that duplication of SMN1 increases the risk of sporadic ALS. Future studies will assess the relationship between SMN1 and SMN2 CNVs and disease risk and progression in ALS and PMA as well as in other disorders affecting motor neurons.
SMN2 copy number is becoming an inclusion criterion for many clinical trials for SMA. Additionally, SMN2 copy number can be used to help guide the type of care SMA patients will receive. It is, therefore, essential to be able to accurately and reliably measure SMN2 CNVs in SMA patient samples, especially for those individuals harboring more than three copies of SMN2. Newly developed technologies like dPCR offer a means to accurately determine SMN2 copy number over a wider range.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank the members of the Motor Neuron Diseases Research Laboratory for their input. This review was supported by the Nemours Foundation and an Institutional Development Award from the National Institute of General Medical Sciences of the National Institutes of Health (P20GM103464).
Abbreviations
ALS, amyotrophic lateral sclerosis; CNV, copy number variation; dPCR, digital polymerase chain reaction; FUS, fused in sarcoma; GTF2H2, general transcription factor IIH; LMN, lower motor neuron; MLPA, multiple ligation-dependent probe amplification; NAIP, neuronal apoptosis inhibitory protein; OMIM, Online Mendelian Inheritance in Man; PCR-RFLP, polymerase chain reaction-restriction fragment length polymorphism; PMA, progressive muscular atrophy; SERF1A, small EDRK-rich factor 1A; SMA, spinal muscular atrophy; SMNΔ7, survival motor neuron lacking exon 7; SMN1, survival motor neuron 1; SMN2, survival motor neuron 2; snRNP, small nuclear ribonucleoprotein particle; SOD1, superoxide dismutase 1; TDP-43, TAR DNA binding protein-43kDa; UMN, upper motor neuron.
References
Alías, L., Bernal, S., Barceló, M. J., Also-Rallo, E., Martínez-Hernández, R., Rodríguez-Alvarez, F. J., et al. (2011). Accuracy of marker analysis, quantitative real-time polymerase chain reaction and multiple ligation-dependent probe amplification to determine SMN2 copy number in patients with spinal muscular atrophy. Genet. Test. Mol. Biomarkers 15, 587–594. doi: 10.1089/gtmb.2010.0253
Amara, A., Adala, L., Ben Charfeddine, I., Mamaï, O., Mili, A., Ben Lazreg, T., et al. (2012). Correlation of SMN2, NAIP, p44, H4F5 and Occludin genes copy number with spinal muscular atrophy phenotype in Tunisian patients. Eur. J. Paediatr. Neurol. 16, 167–174. doi: 10.1016/j.ejpn.2011.07.007
Anhuf, D., Eggermann, T., Rudnik-Schöneborn, S., and Zerres, K. (2003). Determination of SMN1 and SMN2 copy number using TaqMan technology. Hum. Mutat. 22, 74–78. doi: 10.1002/humu.10221
Appelbaum, J. S., Roos, R. P., Salazar-Grueso, E. F., Buchman, A., Iannaccone, S., Glantz, R., et al. (1992). Intrafamilial heterogeneity in hereditary motor neuron disease. Neurology 42, 1488–1492. doi: 10.1212/WNL.42.8.1488
Arkblad, E. L., Darin, N., Berg, K., Kimber, E., Brandberg, G., Lindberg, C., et al. (2006). Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul. Disord. 16, 830–838. doi: 10.1016/j.nmd.2006.08.011
Arnold, W. D., and Burghes, A. H. M. (2013). Spinal muscular atrophy: development and implementation of potential therapeutics. Ann. Neurol. 74, 348–362. doi: 10.1002/ana.23995
Ben-Shachar, S., Orr-Urtreger, A., Bardugo, E., Shomrat, R., and Yaron, Y. (2011). Large-scale population screening for spinal muscular atrophy: clinical implications. Genet. Med. 13, 110–114. doi: 10.1097/GIM.0b013e3182017c05
Bernal, S., Alías, L., Barceló, M. J., Also-Rallo, E., Martínez-Hernández, R., Gámez, J., et al. (2010). The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J. Med. Genet. 47, 640–642. doi: 10.1136/jmg.2010.079004
Bernal, S., Also-Rallo, E., Martínez-Hernández, R., Alías, L., Rodríguez-Alvarez, F. J., Millán, J. M., et al. (2011). Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul. Disord. 21, 413–419. doi: 10.1016/j.nmd.2011.03.009
Blauw, H. M., Barnes, C. P., van Vught, P. W. J., van Rheenen, W., Verheul, M., Cuppen, E., et al. (2012). SMN1 gene duplications are associated with sporadic ALS. Neurology 78, 776–780. doi: 10.1212/WNL.0b013e318249f697
Boylan, K. (2015). Familial amyotrophic lateral sclerosis. Neurol. Clin. 33, 807–830. doi: 10.1016/j.ncl.2015.07.001
Brandt, S. (1949). Hereditary factors in infantile progressive muscular atrophy; study of 112 cases in 70 families. Am. J. Dis. Child. 78, 226–236. doi: 10.1001/archpedi.1949.02030050237007
Brkušanin, M., Kosać, A., Jovanović, V., Pešović, J., Brajušković, G., Dimitrijević, N., et al. (2015). Joint effect of the SMN2 and SERF1A genes on childhood-onset types of spinal muscular atrophy in Serbian patients. J. Hum. Genet. 60, 723–728. doi: 10.1038/jhg.2015.104
Brzustowicz, L. M., Lehner, T., Castilla, L. H., Penchaszadeh, G. K., Wilhelmsen, K. C., Daniels, R., et al. (1990). Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3. Nature 344, 540–541. doi: 10.1038/344540a0
Burghes, A. H. M., and Beattie, C. E. (2009). Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 10, 597–609. doi: 10.1038/nrn2670
Burghes, A. H. M., Ingraham, S. E., Kóte-Jarai, Z., Rosenfeld, S., Herta, N., Nadkarni, N., et al. (1994). Linkage mapping of the spinal muscular atrophy gene. Hum. Genet. 93, 305–312. doi: 10.1007/BF00212028
Burghes, A. H. M. (1997). When is a deletion not a deletion? When it is converted. Am. J. Hum. Genet. 61, 9–15. doi: 10.1086/513913
Bürglen, L., Seroz, T., Miniou, P., Lefebvre, S., Burlet, P., Munnich, A., et al. (1997). The gene encoding p44, a subunit of the transcription factor TFIIH, is involved in large-scale deletions associated with Werdnig-Hoffmann disease. Am. J. Hum. Genet. 60, 72–79.
Burlet, P., Bürglen, L., Clermont, O., Lefebvre, S., Viollet, L., Munnich, A., et al. (1996). Large scale deletions of the 5q13 region are specific to Werdnig-Hoffmann disease. J. Med. Genet. 33, 281–283. doi: 10.1136/jmg.33.4.281
Burnett, B. G., Muñoz, E., Tandon, A., Kwon, D. Y., Sumner, C. J., and Fischbeck, K. H. (2009). Regulation of SMN protein stability. Mol. Cell. Biol. 29, 1107–1115.
Butchbach, M. E. R., and Burghes, A. H. M. (2004). Perspectives on models of spinal muscular atrophy for drug discovery. Drug Discover. Today Dis. Models 1, 151–156. doi: 10.1016/j.ddmod.2004.07.001
Campbell, L., Potter, A., Ignatius, J., Dubowitz, V., and Davies, K. (1997). Genomic variation and gene conversion in spinal muscular atrophy: implications for disease process and clinical phenotype. Am. J. Hum. Genet. 61, 40–50. doi: 10.1086/513886
Camu, W., and Billiard, M. (1993). Coexistence of amyotrophic lateral sclerosis and Werdnig-Hoffmann disease within a family. Muscle Nerve 16, 569–570.
Carrel, T. L., McWhorter, M. L., Workman, E., Zhang, H., Wolstencroft, E. C., Lorson, C., et al. (2006). Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J. Neurosci. 26, 11014–11022. doi: 10.1523/JNEUROSCI.1637-06.2006
Carter, T. A., Bönnemann, C. G., Wang, C. H., Obici, S., Parano, E., De Fatima Bonaldo, M., et al. (1997). A multicopy transcription-repair gene, BTF2p44, maps to the SMA region and demonstrates SMA associated deletions. Hum. Mol. Genet. 6, 229–236. doi: 10.1093/hmg/6.2.229
Cherry, J. J., Kobayashi, D. T., Lynes, M. M., Naryshkin, N. N., Tiziano, F. D., Zaworksi, P. G., et al. (2014). Assays for the identification and prioritization of drug candidates for spinal muscular atrophy. Assay Drug Dev. Technol. 12, 315–341. doi: 10.1089/adt.2014.587
Cho, S., and Dreyfuss, G. (2010). A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 24, 438–442. doi: 10.1101/gad.1884910
Cifuentes-Diaz, C., Nicole, S., Velasco, M. E., Borra-Cebrian, C., Panozzo, C., Frugier, T., et al. (2002). Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum. Mol. Genet. 11, 1439–1447. doi: 10.1093/hmg/11.12.1439
Cobben, J. M., van der Steege, G., Grootscholten, P., de Visser, M., Scheffer, H., and Buys, C. H. (1995). Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am. J. Hum. Genet. 57, 805–808.
Coovert, D. D., Le, T. T., McAndrew, P. E., Strasswimmer, J., Crawford, T. O., Mendell, J. R., et al. (1997). The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 6, 1205–1214. doi: 10.1093/hmg/6.8.1205
Corcia, P., Camu, W., Halimi, J. M., Vourc'h, P., Antar, C., Vedrine, S., et al. (2006). SMN1 gene, but not SMN2, is a risk factor for sporadic ALS. Neurology 67, 1147–1150. doi: 10.1212/01.wnl.0000233830.85206.1e
Corcia, P., Ingre, C., Blasco, H., Press, P., Praline, J., Antar, C., et al. (2012). Homozygous SMN2 deletion is a protective factor in the Swedish ALS population. Eur. J. Hum. Genet. 20, 588–591. doi: 10.1038/ejhg.2011.255
Corcia, P., Khoris, J., Couratier, P., Mayeux-Portas, V., Bieth, E., de Toffol, B., et al. (2002a). SMN1 gene study in three families in which ALS and spinal muscular atrophy co-exist. Neurology 59, 1464–1466. doi: 10.1212/01.WNL.0000032500.73621.C5
Corcia, P., Mayeux-Portas, V., Khoris, J., de Toffol, B., Autret, A., Müh, J. P., et al. (2002b). Abnormal SMN1 gene copy number is a susceptibility factor for amyotrophic lateral sclerosis. Ann. Neurol. 51, 243–246. doi: 10.1002/ana.10104
Courseaux, A., Richard, F., Grosgeorge, J., Ortola, C., Viale, A., Turc-Carel, C., et al. (2003). Segmental duplications in euchromatic regions of human chromosome 5: a source of evolutionary instability and transcriptional innovation. Genome Res. 13, 369–381. doi: 10.1101/gr.490303
Crawford, T. O., and Pardo, C. A. (1996). The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 3, 97–110. doi: 10.1006/nbdi.1996.0010
Crawford, T. O., Paushkin, S. V., Kobayashi, D. T., Forrest, S. J., Joyce, C. L., Finkel, R. S., et al. (2012). Evaluation of SMN protein, transcript and copy number in the Biomarkers for Spinal Muscular Atrophy (BforSMA) clinical study. PLoS ONE 7:e33572. doi: 10.1371/journal.pone.0033572
Crawford, T. O., and Skolasky, R. L. Jr. (2002). The relationship of SMN to amyotrophic lateral sclerosis. Ann. Neurol. 52, 857–858. doi: 10.1002/ana.10378
Cuscó, I., Barceló, M. J., Rojas-García, R., Illa, I., Gámez, J., Cervera, C., et al. (2006). SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J. Neurol. 253, 21–25. doi: 10.1007/s00415-005-0912-y
Cuscó, I., Barceló, M. J., Soler, C., Parra, J., Baiget, M., and Tizzano, E. (2002). Prenatal diagnosis for risk of spinal muscular atrophy. Br. J. Obstet. Gynaecol. 109, 1244–1249. doi: 10.1016/S1470-0328(02)02983-X
Devriendt, K., Lammens, M., Schollen, E., Van Hole, C., Dom, R., Devlieger, H., et al. (1996). Clinical and molecular genetic features of congenital spinal muscular atrophy. Ann. Neurol. 40, 731–738. doi: 10.1002/ana.410400509
DiDonato, C. J., Chen, X. N., Noya, D., Korenberg, J. R., Nadeau, J. H., and Simard, L. R. (1997a). Cloning, characterization and copy number of the murine survival motor neuron gene: homolog of the spinal muscular atrophy-determining gene. Genome Res. 7, 339–352.
DiDonato, C. J., Ingraham, S. E., Mendell, J. R., Prior, T. W., Lenard, S., Moxley, R. T., et al. (1997b). Deletion and conversion in spinal muscular atrophy: is there a relationship to severity? Ann. Neurol. 41, 230–237. doi: 10.1002/ana.410410214
Dobrowolski, S. F., Pham, H. T., Pouch-Downes, F., Prior, T. W., Naylor, E. W., and Swoboda, K. J. (2012). Newborn screening for spinal muscular atrophy by calibrated short-amplicon melt profiling. Clin. Chem. 58, 1033–1039. doi: 10.1373/clinchem.2012.183038
Elsheikh, B., Prior, T., Zhang, X., Miller, R., Kolb, S. J., Moore, D., et al. (2009). An analysis of disease severity based on SMN2 copy number in adults with spinal muscular atrophy. Muscle Nerve 40, 652–656. doi: 10.1002/mus.21350
Fang, P., Li, L., Zhou, W. J., Wu, W. Q., Zhong, Z. Y., Yan, T. Z., et al. (2015). Molecular characterization and copy number of SMN1, SMN2 and NAIP in Chinese patients with spinal muscular atrophy and unrelated healthy controls. BMC Musculoskelet. Disord. 16:11. doi: 10.1186/s12891-015-0457-x
Feldkötter, M., Schwarzer, V., Wirth, R., Wienker, T. F., and Wirth, B. (2002). Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 70, 358–368. doi: 10.1086/338627
Gabanella, F., Carissimi, C., Usiello, A., and Pellizzoni, L. (2005). The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum. Mol. Genet. 14, 3629–3642. doi: 10.1093/hmg/ddi390
Gamez, J., Barceló, M. J., Muñoz, X., Carmona, F., Cuscó, I., Baiget, M., et al. (2002). Survival and respiratory decline are not related to homozygous SMN2 deletions in ALS patients. Neurology 59, 1456–1460. doi: 10.1212/01.WNL.0000032496.64510.4E
Gérard, B., Ginet, N., Matthijs, G., Evrard, P., Baumann, C., Da Silva, F., et al. (2004). Genotype determination at the survival motor neuron locus in a normal population and SMA carriers using competitive PCR and primer extension. Hum. Mutat. 16, 253–263. doi: 10.1002/1098-1004(200009)16:3<253::AID-HUMU8>3.0.CO;2-8
Gerbino, V., Carrì, M. T., Cozzolino, M., and Achsel, T. (2013). Mislocalised FUS mutants stall spliceosomal snRNPs in the cytoplasm. Neurobiol. Dis. 55, 120–128. doi: 10.1016/j.nbd.2013.03.003
Gilliam, T. C., Brzustowicz, L. M., Castilla, L. H., Lehner, T., Penchaszadeh, G. K., Daniels, R. J., et al. (1990). Genetic homogeneity between acute and chronic forms of spinal muscular atrophy. Nature 345, 823–825. doi: 10.1038/345823a0
Gómez-Curet, I., Robinson, K. G., Funanage, V. L., Crawford, T. O., Scavina, M., and Wang, W. (2007). Robust quantification of the SMN gene copy number by real-time TaqMan PCR. Neurogenetics 8, 271–278. doi: 10.1007/s10048-007-0093-1
Groen, E. J. N., Fumoto, K., Blokhuis, A. M., Engelen-Lee, J. Y., Zhou, Y., van den Heuvel, D. M. A., et al. (2013). ALS-associated mutations in FUS disrupt the axonal distribution and localization of SMN. Hum. Mol. Genet. 22, 3690–3704. doi: 10.1093/hmg/ddt222
Hahnen, E., Forkert, R., Marke, C., Rudnik-Schöneborn, S., Schonling, J., Zerres, K., et al. (1995). Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum. Mol. Genet. 4, 1927–1933. doi: 10.1093/hmg/4.10.1927
Hahnen, E., Schönling, J., Rudnik-Schöneborn, S., Zerres, K., and Wirth, B. (1996). Hybrid survival motor neuron genes in patients with autosomal recessive spinal muscular atrophy: new insights into molecular mechanisms responsible for the disease. Am. J. Hum. Genet. 59, 1057–1065.
Hendrickson, B. C., Donohoe, C., Akmaev, V. R., Sugarman, E. A., Labrousse, P., Boguslavskiy, L., et al. (2009). Differences in SMN1 allele frequencies among ethnic groups within North America. J. Med. Genet. 46, 641–644. doi: 10.1136/jmg.2009.066969
Hsieh-Li, H. M., Chang, J. G., Jong, Y. J., Wu, M. H., Wang, N. M., Tsai, C. H., et al. (2000). A mouse model for spinal muscular atrophy. Nat. Genet. 24, 66–70. doi: 10.1038/71709
Huang, C. H., Chang, Y. Y., Chen, C. H., Kuo, Y. S., Hwu, W. L., Gerdes, T., et al. (2007). Copy number analysis of survival motor neuron genes by multiplex ligation-dependent probe amplification. Genet. Med. 9, 241–248. doi: 10.1097/GIM.0b013e31803d35bc
Ishihara, T., Ariizumi, Y., Shiga, A., Kato, T., Tan, C. F., Sato, T., et al. (2013). Decreased number of gemini of coiled bodies and U12 snRNA level in amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 4136–4147. doi: 10.1093/hmg/ddt262
Jackson, M., Morrison, K. E., Al-Chalabi, A., Bakker, M., and Leigh, P. N. (1996). Analysis of chromosome 5q13 genes in amyotrophic lateral sclerosis: homozygous NAIP deletion in a sporadic case. Ann. Neurol. 39, 796–800. doi: 10.1002/ana.410390616
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kariya, S., Re, D. B., Jacquier, A., Nelson, K., Przedborski, S., and Monani, U. R. (2012). Mutant superoxide dismutase 1 (SOD1), a cause of amyotrophic lateral sclerosis, disrupts the recruitment of SMN, the spinal muscular atrophy protein to nuclear Cajal bodies. Hum. Mol. Genet. 21, 3421–3434. doi: 10.1093/hmg/dds174
Kariya, S., Sampson, J. B., Northrop, L. E., Luccarelli, C. M., Naini, A. B., Re, D. B., et al. (2014). Nuclear localization of SMN and FUS is not altered in fibroblasts from patients with sporadic ALS. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 581–587. doi: 10.3109/21678421.2014.907319
Kim, J., Lee, S. G., Choi, Y. C., Kang, S. W., Lee, J. B., Choi, J. R., et al. (2010). Association betwen survivor motor neuron 2 (SMN2) gene homozygous deletion and sporadic lower motor neuron disease in a Korean population. Ann. Clin. Lab. Sci. 40, 368–374.
Kirwin, S. M., Vinette, K. M. B., Gonzalez, I. L., Al Abdulwahed, H., Al-Sannaa, N., and Funanage, V. L. (2013). A homozygous double mutation in SMN1: a complicated genetic diagnosis of SMA. Mol. Genet. Genomic Med. 1, 113–117. doi: 10.1002/mgg3.10
Kolb, S. J., Gubitz, A. K., Olszewski, R. F. Jr., Ottinger, E., Sumner, C. J., Fischbeck, K. H., et al. (2006). A novel cell immunoassay to measure survival of motor neurons protein in blood cells. BMC Neurol. 6:6. doi: 10.1186/1471-2377-6-6
Kolb, S. J., and Kissel, J. T. (2015). Spinal muscular atrophy. Neurol. Clin. 33, 831–846. doi: 10.1016/j.ncl.2015.07.004
Kuzma-Kozakiewicz, M., Jedrzejowska, M., and Kazmierczak, B. (2013). SMN1 gene duplications are more frequent in patients with progressive muscular atrophy. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 457–462. doi: 10.3109/21678421.2013.771367
Kwiatkowski, T. J Jr., Bosco, D. A., LeClerc, A. L., Tamrazian, E., Vandenburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Labrum, R., Rodda, J., and Krause, A. (2007). The molecular basis of spinal muscular atrophy (SMA) in South African black patients. Neuromuscul. Disord. 17, 684–692. doi: 10.1016/j.nmd.2007.05.005
Le, T. T., Pham, L. T., Butchbach, M. E. R., Zhang, H. L., Monani, U. R., Coovert, D. D., et al. (2005). SMND7, the major product of the centromeric survival motor neuron gene (SMN2), extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 14, 845–857. doi: 10.1093/hmg/ddi078
Lee, J. B., Lee, K. A., Hong, J. M., Suh, G. I., and Choi, Y. C. (2012). Homozygous SMN2 deletion is a major risk factor among twenty-five Korean sporadic amyotrophic lateral sclerosis patients. Yonsei Med. J. 53, 53–57. doi: 10.3349/ymj.2012.53.1.53
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. doi: 10.1016/0092-8674(95)90460-3
Lefebvre, S., Burlet, P., Liu, Q., Bertrandy, S., Clermont, O., Munnich, A., et al. (1997). Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 16, 265–269. doi: 10.1038/ng0797-265
Liewluck, T., and Saperstein, D. S. (2015). Progressive muscular atrophy. Neurol. Clin. 33, 761–773. doi: 10.1016/j.ncl.2015.07.005
Liu, Q., and Dreyfuss, G. (1996). A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 15, 3555–3565.
Lorson, C. L., and Androphy, E. J. (2000). An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet. 9, 259–265. doi: 10.1093/hmg/9.2.259
Lorson, C. L., Hahnen, E., Androphy, E. J., and Wirth, B. (1999). A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci.U.S.A. 96, 6307–6311. doi: 10.1073/pnas.96.11.6307
Lyahyai, J., Sbiti, A., Barkat, A., Ratbi, I., and Sefiani, A. (2012). Spinal muscular atrophy carrier frequency and estimated prevalence of the disease in Moroccan newborns. Genet. Test. Mol. Biomarkers 16, 215–218. doi: 10.1089/gtmb.2011.0149
Mailman, M. D., Heinz, J. W., Papp, A. C., Snyder, P. J., Sedra, M. S., Wirth, B., et al. (2002). Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 4, 20–26. doi: 10.1097/00125817-200201000-00004
McAndrew, P. E., Parsons, D. W., Simard, L. R., Rochette, C., Ray, P. N., Mendell, J. R., et al. (1997). Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 60, 1411–1422. doi: 10.1086/515465
McWhorter, M. L., Monani, U. R., Burghes, A. H. M., and Beattie, C. E. (2003). Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 162, 919–931. doi: 10.1083/jcb.200303168
Melki, J., Abdelhak, S., Sheth, P., Bachelot, M. F., Burlet, P., Marcadet, A., et al. (1990a). Genes for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature 344, 767–768. doi: 10.1038/344767a0
Melki, J., Sheth, P., Abdelhak, S., Burlet, P., Bachelot, M. F., Lathrop, M. G., et al. (1990b). Mapping of acute (type I) spinal muscular atrophy to chromosome 5q12-q14. The French Spinal Muscular Atrophy Investigators. Lancet 336, 271–273. doi: 10.1016/0140-6736(90)91803-I
Michaud, M., Arnoux, T., Bielli, S., Durand, E., Rotrou, Y., Jablonka, S., et al. (2010). Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol. Dis. 38, 125–135. doi: 10.1016/j.nbd.2010.01.006
Monani, U. R., Lorson, C. L., Parsons, D. W., Prior, T. W., Androphy, E. J., Burghes, A. H. M., et al. (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183. doi: 10.1093/hmg/8.7.1177
Monani, U. R., Sendtner, M., Coovert, D. D., Parsons, D. W., Andreassi, C., Le, T. T., et al. (2000). The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn-/- mice and results in a mouse with spinal muscular atrophy. Hum. Mol.Genet. 9, 333–339. doi: 10.1093/hmg/9.3.333
Morrison, K. E. (1996). Advances in SMA research: review of gene deletions. Neuromuscul. Disord. 6, 397–408. doi: 10.1016/S0960-8966(96)00368-9
Moulard, B., Salachas, F., Chassande, B., Briolotti, V., Meininger, V., Malafosse, A., et al. (1998). Association between centromeric deletions of the SMN gene and sporadic adult-onset lower motor neuron disease. Ann. Neurol. 43, 640–644. doi: 10.1002/ana.410430513
Munsat, T. L., and Davies, K. E. (1992). International SMA Consortium meeting. Neuromuscul. Disord. 2, 423–428. doi: 10.1016/S0960-8966(06)80015-5
Nicole, S., Desforges, B., Millet, G., Lesbordes, J., Cifuentes-Diaz, C., Vertes, D., et al. (2003). Intact satellite cells lead to remarkable protection against Smn gene defect in differentiation skeletal muscle. J. Cell Biol. 161, 571–582. doi: 10.1083/jcb.200210117
Oprea, G. E., Kröber, S., McWhorter, M. L., Rossoll, W., Müller, S., Krawczak, M., et al. (2008). Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320, 524–527. doi: 10.1126/science.1155085
Orrell, R. W., Habgood, J. J., de Belleroche, J. S., and Lane, R. J. M. (1997). The relationship of spinal muscular atrophy to motor neuron disease. Investigation of SMN and NAIP gene deletions in sporadic and familial ALS. J. Neurol. Sci. 145, 55–61. doi: 10.1016/S0022-510X(96)00240-7
Parboosingh, J. S., Meininger, V., McKenna-Yasek, D., Brown, R. H. Jr., and Rouleau, G. A. (1999). Deletions causing spinal muscular atrophy do not predispose to amyotrophic lateral sclerosis. Arch. Neurol. 56, 710–712. doi: 10.1001/archneur.56.6.710
Pearn, J. (1978). Incidence, prevalence and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet. 15, 409–413. doi: 10.1136/jmg.15.6.409
Pellizzoni, L. (2007). Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 8, 340–345. doi: 10.1038/sj.embor.7400941
Prior, T. W., Krainer, A. R., Hua, Y., Swoboda, K. J., Snyder, P. C., Bridgeman, S. J., et al. (2009). A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 85, 408–413. doi: 10.1016/j.ajhg.2009.08.002
Prior, T. W., Nagan, N., Sugarman, E. A., Batish, S. D., and Braastad, C. (2011). Technical standards and guidelines for spinal muscular atrophy testing. Genet. Med. 13, 686–694. doi: 10.1097/GIM.0b013e318220d523
Prior, T. W., Swoboda, K. J., Scott, H. D., and Hejmanowski, A. Q. (2005). Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am. J. Med. Genet. 130A, 307–310. doi: 10.1002/ajmg.a.30251
Qu, Y., Ge, X., Bai, J., Wang, L., Cao, Y., Lu, Y., et al. (2014). Association of copy numbers of survival motor neuron gene 2 and neuronal apoptosis inhibitory protein gene with the natural history in a Chinese spinal muscular atrophy cohort. J. Child Neurol. 30, 429–436. doi: 10.1177/0883073814553271
Renton, A. E., Chiò, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Rochette, C. F., Gilbert, N., and Simard, L. R. (2001). SMN gene duplication and emergence of the SMN2 gene occured in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 108, 255–266. doi: 10.1007/s004390100473
Rodrigues, N. R., Owen, N., Talbot, K., Patel, S., Muntoni, F., Ignatius, J., et al. (1996). Gene deletions in spinal muscular atrophy. J. Med. Genet. 33, 93–96. doi: 10.1136/jmg.33.2.93
Rowland, L. P. (2010). Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve 41, 161–165. doi: 10.1002/mus.21565
Roy, N., Mahadevan, M. S., McLean, M., Shutler, G., Yaraghi, Z., Farahani, R., et al. (1995). The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 80, 167–178. doi: 10.1016/0092-8674(95)90461-1
Russman, B. S. (2007). Spinal muscular atrophy: clinical classification and disease heterogeneity. J. Child Neurol. 22, 946–951. doi: 10.1177/0883073807305673
Sangaré, M., Hendrickson, B., Sango, H. A., Chen, K., Nofziger, J., Amara, A., et al. (2014). Genetics of low spinal muscular atrophy carrier frequency in sub-Saharan Africa. Ann. Neurol. 75, 525–532. doi: 10.1002/ana.24114
Scarciolla, O., Stuppia, L., De Angelis, M. V., Murru, S., Palka, C., Giuliani, R., et al. (2006). Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics 7, 269–276. doi: 10.1007/s10048-006-0051-3
Scharf, J. M., Endrizzi, M. G., Wetter, A., Huang, S., Thompson, T. G., Zerres, K., et al. (1998). Identification of a candidate modifying gene for spinal muscular atrophy by comparative genomics. Nat. Genet. 20, 83–86. doi: 10.1038/1753
Schmutz, J., Martin, J., Terry, A., Couronne, O., Grimwood, J., Lowry, S., et al. (2004). The DNA sequence and comparative analysis of human chromosome 5. Nature 431, 268–274. doi: 10.1038/nature02919
Schrank, B., Götz, R., Gunnersen, J. M., Ure, J. M., Toyka, K. V., Smith, A. G., et al. (1997). Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. U.S.A. 94, 9920–9925. doi: 10.1073/pnas.94.18.9920
Shan, X., Chiang, P. M., Price, D. L., and Wong, P. C. (2010). Altered distributions of gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci.U.S.A. 107, 16325–16330. doi: 10.1073/pnas.1003459107
Simard, L. R., Bélanger, M. C., Morissette, S., Wride, M., Prior, T. W., and Swoboda, K. J. (2007). Preclinical validation of a multiplex real-time assay to quantify SMN mRNA in patients with SMA. Neurology 68, 451–456. doi: 10.1212/01.wnl.0000252934.70676.ab
Sreedharan, S., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Stabley, D. L., Harris, A. W., Holbrook, J., Chubbs, N. J., Lozo, K. W., Crawford, T. O., et al. (2015). SMN1 and SMN2 copy numbers in cell lines derived from patients with spinal muscular atrophy as measured by array digital PCR. Mol. Genet. Genomic Med. 3, 248–257. doi: 10.1002/mgg3.141
Statland, J. M., Barohn, R. J., McVey, A. L., Katz, J. S., and Dimachkie, M. M. (2015). Patterns of weakness, classification of motor neuron disease and clinical diagnosis of sporadic amyotrophic lateral sclerosis. Neurol. Clin. 33, 735–748. doi: 10.1016/j.ncl.2015.07.006
Stratigopoulos, G., Lanzano, P., Deng, L., Guo, J., Kaufmann, P., Darras, B., et al. (2010). Association of plastin 3 expression with disease severity in spinal muscular atrophy only in postpubertal females. Arch. Neurol. 67, 1252–1256. doi: 10.1001/archneurol.2010.239
Su, Y. N., Hung, C. C., Li, H., Lee, C. N., Cheng, W. F., Tsao, P. N., et al. (2005). Quantitative analysis of SMN1 and SMN2 genes based on DHPLC: a highly efficient and reliable carrier-screening test. Hum. Mutat. 25, 460–467. doi: 10.1002/humu.20160
Su, Y. N., Hung, C. C., Lin, S. Y., Chen, F. Y., Chern, J. P. S., Tsai, C., et al. (2011). Carrier screening for spinal muscular atrophy (SMA) in 107,611 pregnant women during the period 2005-2009: a prospective population-based cohort study. PLoS ONE 6:e17067. doi: 10.1371/journal.pone.0017067
Sugarman, E. A., Nagan, N., Zhu, H., Akmaev, V. R., Zhou, Z., Rohlfs, A. M., et al. (2012). Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of > 72400 specimens. Eur. J. Hum. Genet. 20, 27–32. doi: 10.1038/ejhg.2011.134
Sumner, C. J., Kolb, S. J., Harmison, G. G., Jeffries, N. O., Schadt, K., Finkel, R. S., et al. (2006). SMN mRNA and protein levels in peripheral blood. Biomarkers for SMA clinical trials. Neurology 66, 1067–1073. doi: 10.1212/01.wnl.0000201929.56928.13
Sun, S., Ling, S. C., Qiu, J., Albuquerque, C. P., Zhou, Y., Tokunaga, S., et al. (2015). ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun. 6, 6171. doi: 10.1038/ncomms7171
Swinnen, B., and Robberecht, W. (2014). The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 10, 661–670. doi: 10.1038/nrneurol.2014.184
Swoboda, K. J., Prior, T. W., Scott, C. B., McNaught, T. P., Wride, M. C., Reyna, S. P., et al. (2005). Natural history of denervation in SMA: relation to age, SMN2 copy number and function. Ann. Neurol. 57, 704–712. doi: 10.1002/ana.20473
Sykes, P. J., Neoh, S. H., Brisco, M. J., Hughes, E., Condon, J., and Morley, A. A. (1992). Quantitation of targets for PCR by use of limiting dilution. Biotechniques 13, 444–449.
Taylor, J. E., Thomas, N. H., Lewis, C. M., Abbs, S. J., Rodrigues, N. R., Davies, K. E., et al. (1998). Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy. Eur. J. Hum. Genet. 6, 467–474. doi: 10.1038/sj.ejhg.5200210
Tiziano, F. D., Bertini, E., Messina, S., Angelozzi, C., Pane, M., D'Amico, A., et al. (2007). The Hammersmith functional score correlates with the SMN2 copy number: a multicentric study. Neuromuscul. Disord. 17, 400–403. doi: 10.1016/j.nmd.2007.02.006
Tiziano, F. D., Pinto, A. M., Fiori, S., Lomastro, R., Messina, S., Bruno, C., et al. (2010). SMN transcript levels in leukocytes of SMA patients determined by absolute real-time PCR. Eur. J. Hum. Genet. 18, 52–58. doi: 10.1038/ejhg.2009.116
Tsuiji, H., Iguchi, Y., Furuya, A., Kataoka, A., Hatsuta, H., Atsuta, N., et al. (2013). Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol. Med. 5, 221–234. doi: 10.1002/emmm.201202303
Turner, B. J., Alfazema, N., Sheean, R. K., Sleigh, J. N., Davies, K. E., Horne, M. K., et al. (2014). Overexpression of survival motor neuron improves neuromuscular function and motor neuron survival in mutant SOD1 mice. Neurobiol. Aging 35, 906–915. doi: 10.1016/j.neurobiolaging.2013.09.030
Turner, B. J., Parkinson, N. J., Davies, K. E., and Talbot, K. (2009). Survival motor neuron deficiency enhances progression in an amyotrophic lateral sclerosis mouse model. Neurobiol. Dis. 34, 511–517. doi: 10.1016/j.nbd.2009.03.005
van der Steege, G., Grootscholten, P. M., Cobben, J. M., Zappata, S., Scheffer, H., den Dunnen, J. T., et al. (1996). Apparent gene conversions involving the SMN gene in the region of the spinal muscular atrophy locus at chromosome 5. Am. J. Hum. Genet. 59, 834–838.
van der Steege, G., Grootscholten, P. M., van der Vlies, P., Draaijers, T. G., Osinga, J., Cobben, J. M., et al. (1995). PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 345, 985–986. doi: 10.1016/S0140-6736(95)90732-7
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreeharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Velasco, E., Valero, C., Valero, A., Moreno, F., and Hernández-Chico, C. (1996). Molecular analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy (SMA) families and correlation between number of copies of cBCD541 and SMA phenotype. Hum. Mol. Genet. 5, 257–263. doi: 10.1093/hmg/5.2.257
Veldink, J. H., Kalmijn, S., Van der Hout, A. H., Lemmink, H. H., Groeneveld, G. J., Lummen, C., et al. (2005). SMN genotypes producing less SMN protein increase susceptibility to and severity of sporadic ALS. Neurology 65, 820–825. doi: 10.1212/01.wnl.0000174472.03292.dd
Veldink, J. H., van den Berg, L. H., Cobben, J. M., Stulp, R. P., De Jong, J. M. B. V., Vogels, O. J., et al. (2001). Homozygous deletion of the survival motor neuron 2 gene is a prognostic factor in sporadic ALS. Neurology 56, 753–757. doi: 10.1212/WNL.56.6.749
Vezain, M., Saugier-Veber, P., Melki, J., Toutain, A., Bieth, E., Husson, M., et al. (2007). A sensitive assay for measuring SMN mRNA levels in peripheral blood and in muscle samples of patients affected with spinal muscular atrophy. Eur. J. Hum. Genet. 15, 1054–1062. doi: 10.1038/sj.ejhg.5201885
Vezain, M., Saukkonen, A. M., Goina, E., Touraine, R., Manel, V., Toutain, A., et al. (2010). A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 31, E1110–E1125. doi: 10.1002/humu.21173
Viollet, L., Bertrandy, S., Beuno Brunialti, A. L., Lefebvre, S., Burlet, P., Clermont, O., et al. (1997). cDNA isolation, expression and chromosomal localization of the mouse survival motor neuron gene (Smn). Genomics 40, 185–188. doi: 10.1006/geno.1996.4551
Vitte, J. M., Davoult, B., Roblot, N., Mayer, M., Joshi, V., Courageot, S., et al. (2004). Deletion of murine Smn exon 7 directed to liver leads to severe defect of liver development associated with iron overload. Am. J. Pathol. 165, 1731–1741. doi: 10.1016/S0002-9440(10)63428-1
Vogelstein, B., and Kinzler, K. W. (1999). Digital PCR. Proc. Natl. Acad. Sci. U.S.A. 96, 9236–9241. doi: 10.1073/pnas.96.16.9236
Wan, L., Battle, D. J., Yong, J., Gubitz, A. K., Kolb, S. J., Wang, J., et al. (2005). The survival of motor neurons protein determines the capacity for snRNP assembly: biochemical deficiency in spinal muscular atrophy. Mol. Cell. Biol. 25, 5543–5551. doi: 10.1128/MCB.25.13.5543-5551.2005
Wang, C. C., Chang, J. G., Chen, Y. L., Jong, Y. J., and Wu, S. M. (2010a). Multi-exon genotyping of SMN gene in spinal muscular atrophy by universal fluorescent PCR and capillary electrophoresis. Electrophoresis 31, 2396–2404. doi: 10.1002/elps.201000124
Wang, C. C., Jong, Y. J., Chang, J. G., Chen, Y. L., and Wu, S. M. (2010b). Universal fluorescent multiplex PCR and capillary electrophoresis for evaluation of gene conversion between SMN1 and SMN2 in spinal muscular atrophy. Anal. Bioanal. Chem. 397, 2375–2383. doi: 10.1007/s00216-010-3761-1
Wang, C. C., Shih, C. J., Jong, Y. J., and Wu, S. M. (2014a). Universal fluorescent tri-probe ligation equipped with capillary electrophoresis for targeting SMN1 and SMN2 genes in diagnosis of spinal muscular atrophy. Anal. Chim. Acta 833, 40–47. doi: 10.1016/j.aca.2014.05.008
Wang, X. B., Cui, N. H., Gao, J. J., Qiu, X. P., and Zheng, F. (2014b). SMN1 duplications contribute to sporadic amyotrophic lateral sclerosis susceptibility: evidence from a meta-analysis. J. Neurol. Sci. 340, 63–68. doi: 10.1016/j.jns.2014.02.026
Wirth, B., Brichta, L., Schrank, B., Lochmüller, H., Blick, S., Baasner, A., et al. (2006). Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum. Genet. 119, 422–428. doi: 10.1007/s00439-006-0156-7
Wirth, B., Garbes, L., and Riessland, M. (2013). How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic applications. Curr. Opin. Genet. Dev. 23, 330–338. doi: 10.1016/j.gde.2013.03.003
Wirth, B., Hahnen, E., Morgan, K., DiDonato, C. J., Dadze, A., Rudnik-Schöneborn, S., et al. (1995). Allelic association and deletions in autosomal recessive proximal spinal muscular atrophy: association of marker genotype with disease severity and candidate cDNAs. Hum. Mol. Genet. 4, 1273–1284. doi: 10.1093/hmg/4.8.1273
Yamazaki, T., Chen, S., Yan, B., Haertlein, T. C., Carrasco, M. A., Tapia, J. C., et al. (2012). FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2, 799–806. doi: 10.1016/j.celrep.2012.08.025
Yanyan, C., Yujin, Q., Jinli, B., Yuwei, J., Hong, W., and Fang, S. (2014). Correlation of PLS3 expression with disease severity in children with spinal muscular atrophy. J. Hum. Genet. 59, 24–27. doi: 10.1038/jhg.2013.111
Yu, Y., Chi, B., Xia, W., Gangopadhyay, J., Yamazaki, T., Winkelbauer-Hurt, M. E., et al. (2015). U1 snRNP is mislocalized in ALS patient fibroblasts bearing NLS mutations in FUS and is required for motor neuron outgrowth in zebrafish. Nucleic Acids Res. 43, 3208–3218. doi: 10.1093/nar/gkv157
Zaldívar, T., Montejo, Y., Acevedo, A. M., Guerra, R., Vargas, J., Garofalo, N., et al. (2005). Evidence of reduced frequency of spinal muscular atrophy type I in the Cuban population. Neurology 65, 636–638. doi: 10.1212/01.wnl.0000172860.41953.12
Zhang, Z., Lotti, F., Dittmar, K., Younis, I., Wan, L., Kasim, M., et al. (2008). SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133, 585–600. doi: 10.1016/j.cell.2008.03.031
Zhong, Q., Bhattacharya, S., Kotsopoulos, S., Olson, J., Taly, V., Griffiths, A. D., et al. (2011). Multiplex digital PCR: breaking the one target per color barrier of quantitative PCR. Lab Chip 11, 2167–2174. doi: 10.1039/c1lc20126c
Keywords: spinal muscular atrophy, amyotrophic lateral sclerosis, progressive muscular atrophy, neurodegenerative disease, copy number variation, SMN1, SMN2
Citation: Butchbach MER (2016) Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. 3:7. doi: 10.3389/fmolb.2016.00007
Received: 14 December 2015; Accepted: 25 February 2016;
Published: 10 March 2016.
Edited by:
Megha Agrawal, University of Arkansas at Little Rock, USAReviewed by:
Alessandro Vercelli, University of Torino, ItalyMichal Mielcarek, Imperial College London, UK
Copyright © 2016 Butchbach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew E. R. Butchbach, butchbach@nemoursresearch.org