Intracellular accumulation of amyloid-beta – a predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease
Division of Molecular Psychiatry and Alzheimer Ph.D. Graduate School, Department of Psychiatry, University of Göttingen, Göttingen, Germany
Despite of long-standing evidence that beta-amyloid (Aβ) peptides have detrimental effects on synaptic function, the relationship between Aβ, synaptic and neuron loss is largely unclear. During the last years there is growing evidence that early intraneuronal accumulation of Aβ peptides is one of the key events leading to synaptic and neuronal dysfunction. Many studies have been carried out using transgenic mouse models of Alzheimer’s disease (AD) which have been proven to be valuable model systems in modern AD research. The present review discusses the impact of intraneuronal Aβ accumulation on synaptic impairment and neuron loss and provides an overview of currently available AD mouse models showing these pathological alterations.
Alzheimer’s disease (AD) represents the most frequent form of dementia and is characterized by two major neuropathological hallmarks: (i) extracellular plaques composed of the 40–42 residues Aβ peptide (Hardy and Allsop, 1991
) and (ii) neurofibrillary tangles, consisting of abnormal phosphorylated Tau protein (Braak and Braak, 1991
). There is increasing evidence that, in addition to the well-known extracellular amyloid deposition in the parenchyma, Aβ peptides accumulate inside neurons (Gouras et al., 2000
). It has been hypothesized that this initial accumulation is one of the earliest pathological events, thereby triggering the cascade leading to neurodegeneration (Wirths et al., 2004
). Since their initial generation in the mid 1990s, transgenic mice have been proven to represent valuable model systems reflecting various pathological subfields of AD like plaque deposition, inflammatory changes or behavioral abnormalities (reviewed in Games et al., 2006
; Duyckaerts et al., 2008
). In the present review, we summarize the current achievements of modeling early intraneuronal Aβ accumulation in transgenic mice with their resulting pathological consequences. Of special importance will be the critical discussion of this observation, because in all mouse models in which marked neuron loss has been so far reported, this was preceded by considerable amounts of intraneuronal Aβ peptides. In the present short review, we will discuss the view that intraneuronal accumulation of Aβ peptides leads to traffic problems accompanied by early axonopathy, synaptic loss and neuron death. Extracellular plaque pathology, as we and others believe, has a weaker impact on neurodegeneration.
The generation of Aβ peptides is due to enzymatic cleavage of the larger amyloid precursor protein (APP), which represents a type I membrane protein with a large N-terminal ectodomain and a short intracellular C-terminal domain. Alternative splicing of APP yields eight isoforms with lengths of 677–770 amino acid residues, of which APP695 is the primary transcript in neurons (Bayer et al., 2001
). APP can be processed by two different pathways: (i) non-amyloidogenic processing: Cleavage by α-secretase within the Aβ domain releases a secreted form of APP (sAPPα), thereby precluding the generation of toxic Aβ peptides. Different members of the ADAM protein family (a disintegrin and metalloprotease) have been demonstrated to possess α-secretase activity (Buxbaum et al., 1998
; Lammich et al., 1999
). (ii) Amyloidogenic processing: This APP processing pathway results in cleavage at the β-secretase site, liberating also a secreted form of APP (sAPPβ), and leading to the generation of a membrane-associated C-terminal fragment named C99. The β-site APP cleaving enzyme 1 (BACE) belongs to the family of aspartyl proteases and has been identified simultaneously by different research groups (Hussain et al., 1999
; Sinha et al., 1999
; Vassar et al., 1999
; Yan et al., 1999
). Subsequent cleavage of C99 by γ-secretase activity results in the generation of 40–42 residue Aβ peptides, as well as a short intracellular APP fragment named AICD. It has been shown that γ-secretase consists of a complex of different proteins including presenilin-1 (PS1) or presenilin-2 (PS2), as well as nicastrin, anterior pharynx defective (APH-1) and presenilin enhancer 2 (PEN-2) (reviewed in Selkoe and Wolfe, 2007
).
Mutations in either APP or in the presenilin genes have been linked to familiar, early onset forms of AD. These cases represent only a minor portion (∼5%), whereas the vast majority of AD cases develop sporadically. Most of the reported APP mutations are located near the secretase cleavage sites and lead to an overproduction of Aβ peptides. Some of these mutations (e.g. the Austrian mutation T714I), as well as a couple of PS1 mutations have a drastic effect on the Aβ42/Aβ40 ratio by strongly increasing Aβ42 production with concomitant suppression of Aβ40 secretion (Kumar-Singh et al., 2000
; Wolfe, 2007
).
The classical amyloid cascade hypothesis supports the idea that increased Aβ production and extracellular accumulation leads to progressive synaptic and neuronal injury ending up in widespread neuronal dysfunction and dementia (Hardy and Selkoe, 2002
). However, despite of intense research efforts during the last 20 years, the early molecular events leading to AD are still not clear. It has been shown that the loss of synaptic terminals correlates better with cognitive decline than extracellular plaque load or loss of neurons, leading to the concept that losing synapses is one of the key events leading to cognitive dysfunction in AD (Terry et al., 1991
). Neuropathological analyses confirmed a loss of synaptophysin-immunoreactive synaptic terminals, e.g., in the outer molecular layer of the dentate gyrus in AD (Masliah et al., 1994
), as well as a 25% loss of synaptophysin-immunoreactivity already in mild AD cases in the frontal cortex (Masliah et al., 2001
). Recently, it has been reported that mild AD patients had fewer synapses in the stratum radiatum of the CA1 subfield (55%) than control patients and that patients suffering from mild cognitive impairment (MCI) had a mean synaptic value which was 18% lower than in the control group without cognitive impairment. These results further strengthen the hypothesis that synaptic loss is one of the earliest events in AD pathology and support the concept that MCI is a transitional stage between no cognitive impairment and mild AD (Scheff et al., 2007
).
The occurrence and relevance of intraneuronal Aβ accumulations in AD have been a matter of controversial scientific debate. First reports showing that Aβ is initially deposited in neurons before occurring in the extracellular space date back roughly 20 years (Masters et al., 1985
; Grundke-Iqbal et al., 1989
). More recently it has been shown that neurons in AD-vulnerable regions accumulate Aβ42 and it has been further suggested that this accumulation precedes neurofibrillary tangle formation and extracellular Aβ deposition (Gouras et al., 2000
). Consecutively a variety of reports has been published demonstrating Aβ in neurons of AD (Mochizuki et al., 2000
; D’Andrea et al., 2001
, 2002
; Fernandez-Vizarra et al., 2004
) and Down syndrome (DS) patients (Gyure et al., 2001
; Busciglio et al., 2002
; Mori et al., 2002
). On the contrary, a more recent study described intracellular Aβ immunoreactivity during the entire life span in control subjects and DS patients, leading to the suggestion that this represents rather a feature of normal neuronal metabolism than a pathological alteration. As the authors found the strongest intraneuronal Aβ in brain structures that are not highly vulnerable to AD-associated changes, they believe that intraneuronal Aβ immunoreactivity is not a predictor of brain amyloidosis or neurofibrillary degeneration (Wegiel et al., 2007
). Aoki and colleagues investigated whether Aβ levels are changed in CA1 pyramidal neurons of AD hippocampus, using laser capture microdissection to isolate neurons and enzyme-linked immunosorbent assay for quantification. The results showed increased Aβ42 levels and an elevated Aβ42/Aβ40 ratio in neurons from sporadic as well as from familial AD cases, whereas Aβ40 levels remained unaffected (Aoki et al., 2008
). Recent reports have shown that modifications of the staining method have crucial influence on the detection of Aβ peptides in neurons. Whereas pretreatment of AD tissues using formic acid enhances the immunological detection of extracellular plaques, it might have an opposing effect on intracellular Aβ peptides. Heat-induced antigen retrieval has been proposed to have the most significant effect, whereas enzymatic treatment alone is not sufficient (D’Andrea et al., 2003
; Ohyagi et al., 2007
). Fixation might also be an important point, as intracellular Aβ detection in mice is well-documented (Wirths et al., 2001
, 2002
; Blanchard et al., 2003
; Oddo et al., 2003
; Casas et al., 2004
; Lord et al., 2006
; Oakley et al., 2006
; Knobloch et al., 2007b
), however, these tissues are usually fixed by transcardial perfusion in a narrow time frame, whereas human material as a general rule is subjected to much longer post-mortem intervals.
A second mechanism that contributes to intracellular Aβ accumulation is uptake from the extracellular space, in addition to intraneuronal Aβ production. It has been previously shown that cells that were treated with synthetic Aβ peptides, selectively accumulate Aβ42 and that this internalization could be prevented under conditions that block endocytosis (Knauer et al., 1992
). Uptake of Aβ1-42 is enhanced by integrin antagonists and blocked by NMDA receptor antagonists (Bi et al., 2002
). Cultured hippocampal slices were exposed to Aβ1-42 for 6 days in the presence or absence of soluble Gly-Arg-Gly-Asp-Ser-Pro, a peptide antagonist of Arg-Gly-Asp (RGD)-binding integrins, or the disintegrin echistatin. Aβ uptake, as assessed with immunocytochemistry, occurred in 42% of the slices incubated with Aβ peptide alone but in more than 80% of the slices co-treated with integrin antagonists. The selective NMDA receptor antagonist D-(-)-2-amino-5-phosphonovalerate completely blocked internalization of Aβ (Bi et al., 2002
). On the subcellular level, internalized Aβ42 peptides seem to accumulate especially in the endosomal/lysosomal system which leads to lysosomal permeability and membrane damage (Yang et al., 1998
; Ditaranto et al., 2001
). One possible internalization mechanism might be cell-surface receptor-mediated uptake via the α7 nicotinic acetylcholine receptor (Nagele et al., 2002
), but also passive diffusion of extracellular Aβ through the plasma membrane has been suggested (Li et al., 2007
).
A selective Aβ42 uptake was revealed in the CA1 subfield of rat organotypic slice cultures, whereas other hippocampal regions like CA3 and dentate gyrus remained almost unaffected (Bahr et al., 1998
). Primary neurons from APP transgenic Tg2576 mice accumulate Aβ peptides and have been shown to undergo selective reductions in synaptic levels of PSD-95 and the glutamate receptor subunit GluR1 compared to the wild-type situation (Almeida et al., 2005
). As Aβ has been implicated in the depression of AMPA receptor currents, thereby regulating synaptic activity (Kamenetz et al., 2003
), reduced levels of GluR1 provide a molecular basis for Aβ-induced AMPA current alterations (Almeida et al., 2005
). Wang et al. reported that Aβ1-42 inhibits alpha7nAChR-dependent calcium activation and acetylcholine release, two processes critically involved in memory and cognitive functions (Wang et al., 2000
). Internalization of Aβ1-42 may be facilitated by the high-affinity binding to the alpha7 receptor on neuronal cell surfaces, followed by endocytosis of the resulting complex (Nagele et al., 2002
). Dziewczapolski et al. have shown that, despite the presence of high amounts of APP and amyloid deposits, deleting the alpha7nAChR subunit in PDAPP mice leads to a protection from the dysfunction in synaptic integrity (pathology and plasticity) and learning and memory behavior. APP/alpha7nAChR KO mice express APP and Aβ at levels similar to APP mice, however they were able to solve a cognitive challenge such as the Morris water maze test significantly better than APP single transgenic mice, with performances comparable to control groups. Moreover, deleting the alpha7nAChR subunit protected the brain from loss of the synaptic markers synaptophysin and MAP2, reduced the gliosis, and preserved the capacity to elicit long-term potentiation (LTP) which is deficient in the single transgenic APP mice used. These results are consistent with the hypothesis that the alpha7nAChR plays a role in AD and suggest that interrupting alpha7nAChR function could be beneficial in the treatment of AD (Dziewczapolski et al., 2009
).
Reviewed in detail, there is increasing evidence that oxidative damage, induced by Aβ, is associated with mitochondrial dysfunction early in AD development (Reddy and Beal, 2008
). Aβ binds to alcohol dehydrogenase (ABAD) demonstrating a direct molecular link from Aβ to mitochondrial toxicity in AD patients and transgenic mice. The crystal structure of Aβ-bound ABAD showed substantial deformation of the active site inhibiting nicotinamide adenine dinucleotide (NAD) binding (Lustbader et al., 2004
). Of interest, intracellular Aβ is present in mitochondria in brains from APP transgenic mice and AD patients. It has been shown that Aβ progressively accumulated in mitochondria and was associated with diminished enzymatic activity of respiratory chain complexes (III and IV) and a reduction in the rate of oxygen consumption (Caspersen et al., 2005
). In addition, cytochrome c oxidase activity was found to be decreased in Tg2576 mice, suggesting that mutant APP and/or soluble Aβ impair mitochondrial metabolism in AD development and progression (Manczak et al., 2006
).
Treatment of cultured hippocampal neurons with Aβ resulted in rapid and severe impairment of mitochondrial transport without inducing apparent cell death and significant morphological changes, whereas stimulation of protein kinase A (PKA) by forskolin, cAMP analogs or neuropeptides effectively alleviated the impairment. In addition, Aβ inhibited mitochondrial transport by acting through glycogen synthase kinase 3β (GSK3β) (Rui et al., 2006
). Mitochondrial damage such as reduced mitochondrial membrane potential and ATP levels have been detected in the brains from APP and APP/PS1 transgenic mice before the onset of plaques arguing for a role of intracellular and/or soluble Aβ on mitochondrial dysfunction (Eckert et al., 2008
). Results from rat mitochondria demonstrated that Aβ is transported into mitochondria via the translocase of the outer membrane (TOM) machinery. Subcellular fractionation studies following the import experiments revealed Aβ association with the inner membrane fraction, and immunoelectron microscopy after import showed localization of Aβ to mitochondrial cristae. In addition, it could be demonstrated that extracellularly applied Aβ was taken up by human neuroblastoma cells which then colocalized with mitochondrial markers (Hansson Petersen et al., 2008
). It has also been reported that the receptor for advanced glycation end products (RAGE) contributes to transport of Aβ from the cell surface to the intracellular space. Mouse cortical neurons were treated with extracellular human Aβ showing detectable Aβ intracellularly in the cytosol and mitochondria by confocal and immunogold electron microscopy. Intraneuronal co-localization of Aβ and RAGE was also observed in the hippocampus of APP transgenic mice (Takuma et al., 2009
). Very recently, a drosophila model was described in which overexpression of Aβ, resulting in Aβ accumulation in the soma and axons of neurons, leads to a variety of mitochondrial abnormalities including depletion of presynaptic and axonal mitochondria, a decrease in axonal transport of mitochondria, as well as changes in mitochondrial size and number (Zhao et al., 2010
).
Aβ protein assembly and oligomer formation has been reviewed extensively in the past (for example, Klein et al., 2001
; Klein, 2002
; Roychaudhuri et al., 2009
) and is therefore not a focus of the present review. The formation of Aβ oligomers has been repeatedly reported to contribute significantly to the pathophysiological alterations underlying AD and it has been shown that soluble oligomeric Aβ42 and not plaque-associated Aβ correlates best with cognitive dysfunction (McLean et al., 1999
; Naslund et al., 2000
). Oligomers are formed preferentially intracellularly within neuronal processes and synapses rather than within the extracellular space (Walsh et al., 2000
; Takahashi et al., 2004
) and have been demonstrated to cause synaptic alterations (Lacor et al., 2004
, 2007
). These oligomers have been previously demonstrated to inhibit hippocampal LTP and disrupt synaptic plasticity (Lambert et al., 1998
; Walsh et al., 2002
) and have been shown to originate from intraneuronal rather than extracellular dimerization (Walsh et al., 2000
). By the use of antibodies raised against synthetic Aβ oligomer assemblies, these oligomers or Aβ-derived diffusible ligands (ADDLs) have detected in up to 70-fold higher concentrations in AD compared to control brains (Gong et al., 2003
). However, as these oligomers normally occur both inside neurons as well as in a secreted form, it is difficult to attribute the toxic actions to one of the different entities. In addition, it has been shown that Aβ-derived oligomers specifically bind to pyramidal neurons, promoting a rapid decrease in the membrane expression levels of memory-related receptors like NMDA or EphB2 (Lacor et al., 2007
). In hippocampal neuron cultures, ADDLs stimulated the excessive formation of reactive oxygen species (ROS) through a mechanism requiring NMDA receptor activation. Interestingly, the memory-preserving drug memantine, which represents a NMDA receptor antagonist, completely protected against these toxic effects, indicating that ADDLs bind to NMDA receptors and trigger neuronal damage through receptor-dependent calcium influx (De Felice et al., 2007
). In addition, concentrations of soluble Aβ clearly distinguished healthy controls from AD patients and represent a strong inverse correlate of synapse loss (Lue et al., 1999
; McLean et al., 1999
). In addition to ADDLs, globular Aβ species called globulomers have been found to be toxic (Barghorn et al., 2005
) and suppressed spontaneous synaptic activity by inhibition of P/Q-type calcium currents (Nimmrich et al., 2008
).
The data in AD mouse models regarding intraneuronal Aβ are much more consistent and an increasing consensus can be observed during the past years. Intraneuronal Aβ was first reported in familial PS1 transgenic mice that also showed neurodegeneration without the formation of plaque pathology (Chui et al., 1999
). During the last years, intraneuronal accumulation has been reported in several mouse models including APPSDLPS1M146L (Wirths et al., 2001
), APPSLPS1M146L (Wirths et al., 2002
), Tg2576 (Takahashi et al., 2002
), 3xTg-AD (Oddo et al., 2003
), 5xFAD (Oakley et al., 2006
), APPArc (Lord et al., 2006
; Knobloch et al., 2007b
), APPT714I mice (Van Broeck et al., 2008
), in APPSLPS1KIM233T, L235P mice (Casas et al., 2004
) in which it was recently shown to correlate with neuron loss (Christensen et al., 2008a
,b
; Breyhan et al., 2009
), as well as in TBA2 mice expressing pyroglutamate modified Aβ3-42 (Wirths et al., 2009
). These models differ in a variety of parameters including the expressed APP isoform, the APP mutations included, the expression level of APP, the promoter construct used to drive expression of the transgene, as well as in the fact whether they are single or multi-transgenic (co-expression of Presenilin-1 or Tau) (see Table 1
).
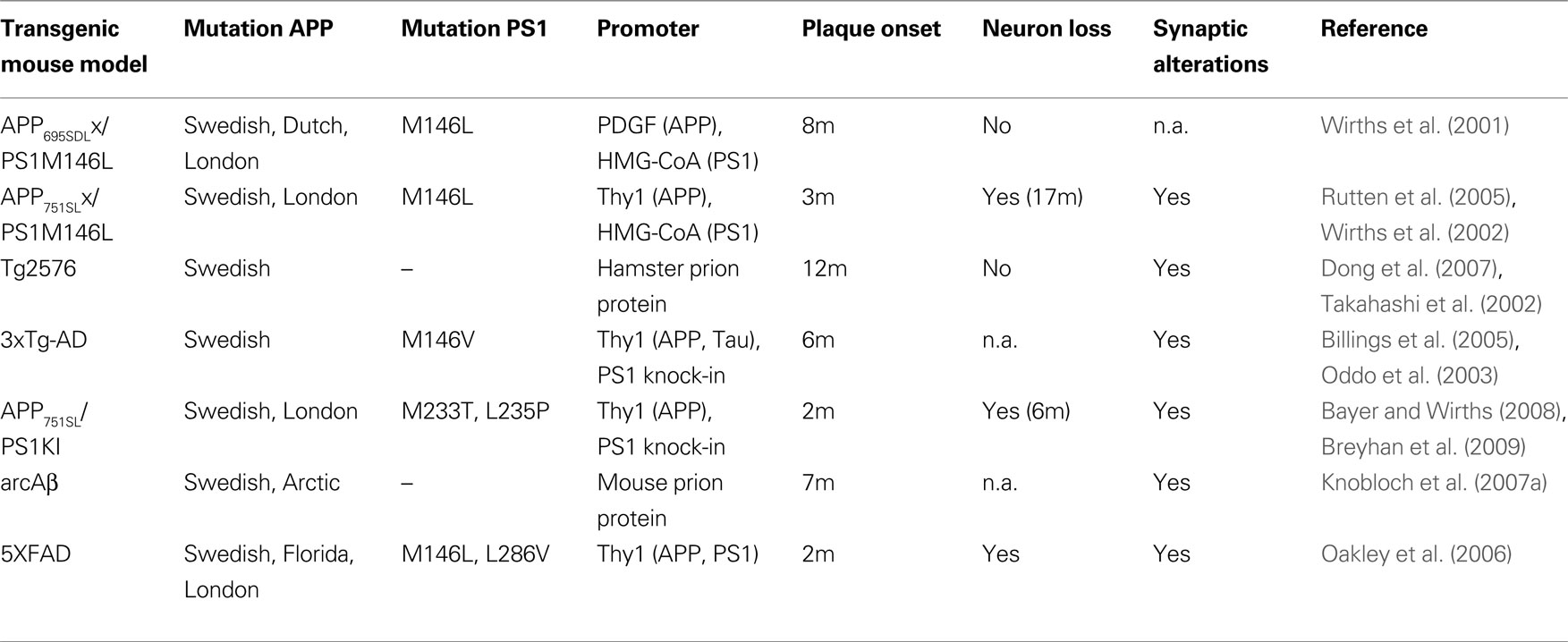
Table 1. Overview of transgenic AD mouse models in which intraneuronal Aβ accumulation has been reported. In addition, information on extracellular plaque onset, neuron loss and synaptic pathology are given (n.a., not analyzed).
Early intraneuronal Aβ accumulation preceding extracellular amyloid plaque pathology has been detected in a model expressing APP751 with the Swedish and London mutations (APP751SL), in combination with transgenic mutant human PS1 with the M146L mutation expressed under the control of the HMG-CoA-reductase promoter. Strong Aβ staining was identified in somatodendritic compartments of neurons in subiculum, CA1 region of the hippocampus, as well as in cortical areas already in young mice (Wirths et al., 2002
; Blanchard et al., 2003
). It has been shown that with incremental extracellular pathology an attenuated intraneuronal Aβ immunoreactivity was noted (Wirths et al., 2002
; Langui et al., 2004
). This finding corroborates an observation in DS patients, where consistently strong cellular Aβ staining was reported in young patients, with a progressive decline paralleling deposition and maturation of extracellular amyloid plaques (Mori et al., 2002
). A detailed stereological analysis revealed that both the single transgenic APP751SL mice and PS1M146L mice showed an age-related loss of synaptophysin-immunoreactive presynaptic boutons (SIPBs) within the stratum radiatum. Importantly, APP751SL/PS1M146L mice displayed the most severe age-related SIPB loss within stratum moleculare, stratum lacunosum and stratum radiatum, even in regions free of extracellular Aβ deposits (Rutten et al., 2005
).
Analysis of the subcellular localization of Aβ peptides by means of double-fluorescence experiments in these mice revealed that intracellular Aβ colocalized with lysosomal markers and less frequently with markers of the trans-Golgi network (TGN). Using electron microscopy, Aβ has been detected in the lumen of multivesicular bodies (MVBs) (Langui et al., 2004
), corroborating an earlier report in Tg2576 mice showing that Aβ42 is localized predominantly to MVBs within pre- and postsynaptic compartments (Takahashi et al., 2002
). In Tg2576 mice an altered synaptic morphology has been recognized which preceded extracellular amyloid deposition (Takahashi et al., 2002
) and it has been shown in a subsequent report that Aβ42 aggregates into oligomers within endosomal vesicles, as well as in neuronal processes (Takahashi et al., 2004
). This is particularly interesting as it has been hypothesized that Aβ oligomers have a significant impact on the pathological alterations underlying memory deficits in AD patients, as they have been demonstrated to disrupt synaptic plasticity and to inhibit hippocampal LTP (Lambert et al., 1998
; Walsh et al., 2002
). Using stereology at the light and electron microscopy level, synapse density was assessed in Tg2576 mice in hippocampus and entorhinal cortex. No overall decrease in the density of synaptophysin-positive boutons could be found on the light microscopy level, however, using electron microscopy, an overall decreases in synapse density in the outer molecular layer of the dentate gyrus at both 6–9 and 15–18 months of age, and in Layers II and III of the entorhinal cortex at 15–18 months of age was detected (Dong et al., 2007
).
A further interesting model expresses APP with the Swedish and London mutations on a homozygous mutant PS1 knock-in background (APP/PS1KI). These animals show early extracellular amyloid deposition at the age of 2 months, which is preceded by strong intraneuronal Aβ accumulation in hippocampus, cortex and motor neurons of the spinal cord (Figures 1
E,F). Staining with the β-sheet binding dye Thioflavin-S indicated that the intraneuronal Aβ is aggregated and is present in the form of β-pleated sheets. Further pathological features include severe axonal degeneration, as well as motor and cognitive deficits starting at the age of 6 months (Casas et al., 2004
; Wirths et al., 2007
, 2008
; Bayer and Wirths, 2008
). At this time point also synaptic alterations became evident. The levels of the postsynaptic marker protein PSD-95 were significantly decreased in whole brain lysates. In addition, analysis of synaptosome-enriched fractions revealed a significant decrease in the levels of the presynaptic markers SNAP25 and clathrin-light chain, as well as of the postsynaptic marker PSD-95. Recordings of field excitatory postsynaptic potentials (fEPSPs) disclosed a significant reduction of fEPSPs in 6-month-old APP/PS1KI compared to PS1KI or wild-type mice (Figures 1
A–D). Short-term synaptic plasticity was also affected, as 6-month-old APP/PS1KI mice disclosed unaltered paired pulse facilitation (PPF) ratios before and after LTP, in contrast to PS1KI and WT mice, which showed the expected lower PPF ratios after LTP induction (Breyhan et al., 2009
).
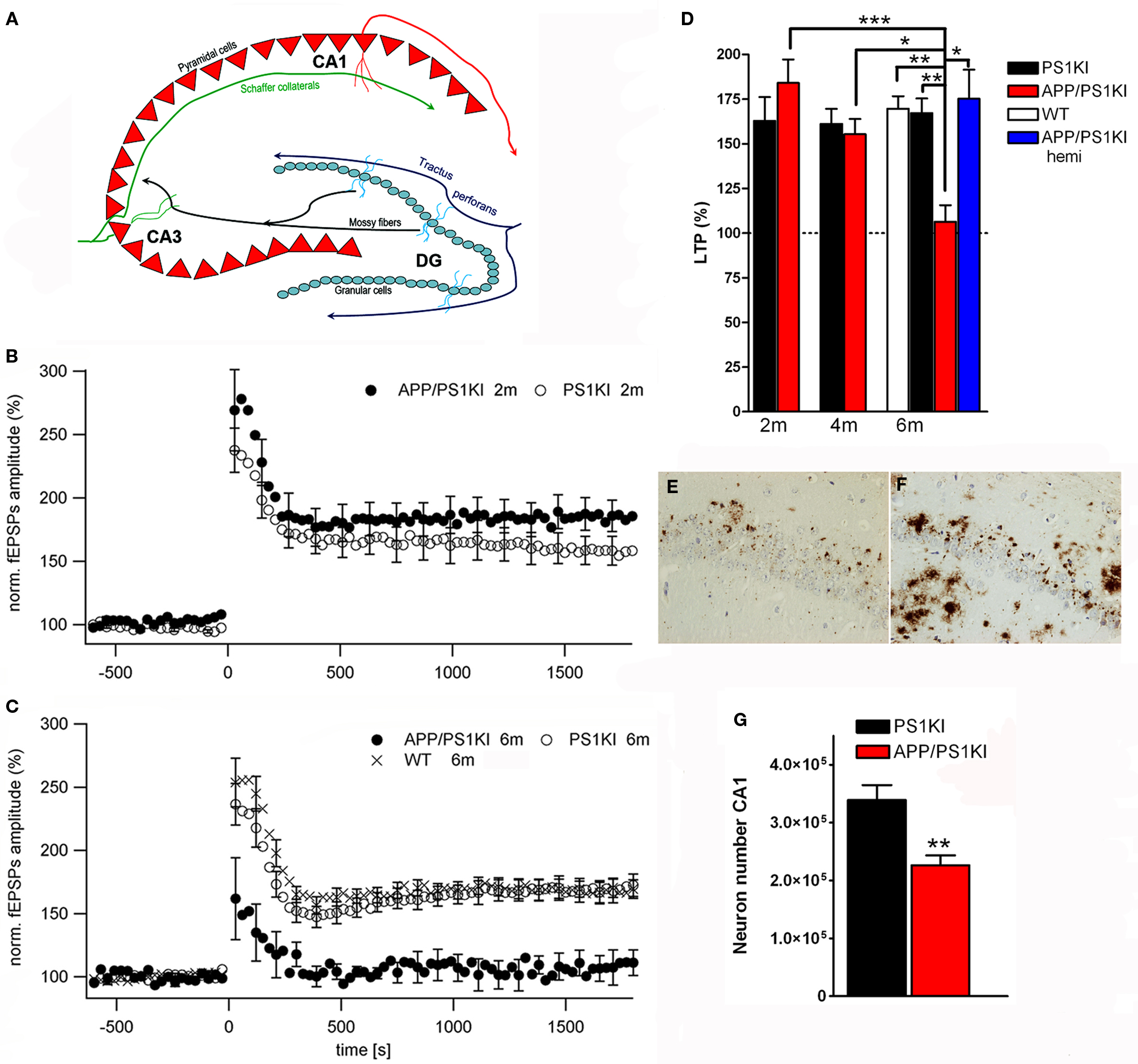
Figure 1. Intraneuronal Aβ accumulation and impaired LTP in APP/PS1KI mice. Schematic drawing of the hippocampus (A). At 6 months of age deficits in synaptic transmission in the form of altered LTP are detected in APP/PS1KI mice (B–D). Intraneuronal accumulation of Aβ peptides occurs already at 2 months of age in the CA1 region of the hippocampus (E) and precedes neuron loss which becomes evident at 6 months of age (F,G).
Mice expressing human APP with the Arctic mutation (E693G) have been recently developed (Lord et al., 2006
; Knobloch et al., 2007b
). This mutation is particularly interesting as it has been demonstrated that protofibrils are produced at a much higher rate and in larger quantities than wild-type Aβ (Nilsberth et al., 2001
). The available mouse models express APP with both the Swedish and the Artic mutation. A transgenic line named tg-APPArcSwe mice has been created under the control of the murine Thy1 promoter. Strong intraneuronal Aβ immunostaining was observed at 2 months of age, which preceded plaque formation occurring at 5–6 months of age (Lord et al., 2006
). Another mouse line named ArcAβ uses the murine prion protein promoter to express APP695 with the Swedish and Arctic mutations. These mice show intracellular Aβ staining already at 3 months of age, with increasing amounts during aging. Interestingly, these mice develop age-dependent cognitive impairments, as shown by deficits in behavior tasks like the Y-maze or a two-way active avoidance paradigm (Knobloch et al., 2007b
). Hippocampal LTP measurements disclosed a severe LTP impairment in young mice already at 3.5 and 7.5 months of age, which was obviously not due to developmental defects, as 1-month-old mice showed normal LTP and basal synaptic transmission. The mutations used in this model render Aβ to be more prone to oligomer formation, which was held responsible for this effect as it could be reversed by a single dose of an antibody directed against the Aβ sequence administered to the animals 48 h before LTP induction (Knobloch et al., 2007a
).
Recently a triple transgenic mouse model has been developed expressing both APP and Tau on a mutant PS1 knock-in background (3xTg-AD mice) (Oddo et al., 2003
). Intracellular Aβ is apparent between 3 and 4 months in these mice and precedes the deposition of extracellular Aβ peptides starting around the age of 6 months. At this time point synaptic plasticity was already strongly compromised in these mice, as shown by impaired LTP (Oddo et al., 2003
). Intracellular Aβ accumulation is functionally linked to cognitive impairment in these mice, as they develop deficits in long-term retention at the age of 4 months, a time point prior to plaque deposition where only intracellular Aβ is present (Billings et al., 2005
). Morphological alterations of hippocampal synapses have been characterized in 13-month-old 3xTg-AD mice and age-matched PS1KI control mice. The numeric density of synapses, the average synaptic contact area, as well as the synaptic surface density were not altered, however, 3xTg-AD mice disclosed a significant decrease in the fraction of perforated synapses, which is believed to represent a reliable indirect index of synaptic plasticity (Bertoni-Freddari et al., 2008
).
5XFAD mice express human APP with the Swedish, Florida (I716V) and London mutations, together with mutant PS1 (M146L, L286V) under the control of the murine Thy1 promoter (Oakley et al., 2006
). The earliest intracellular Aβ accumulation could be detected at 1.5 months of age, immediately preceding extracellular plaque deposition occurring at the age of 2 months. The intraneuronal Aβ is in an aggregated state, as shown by Thioflavin-S staining and memory impairment, as shown by a reduced Y-maze performance and deficits in trace fear conditioning, compared to wild-type control animals, became evident already at the age of 4–5 and 5–6 months respectively (Oakley et al., 2006
; Ohno et al., 2007
). Synaptic pathology in this model was demonstrated by reduced synaptophysin, syntaxin and PSD-95 levels in whole brain lysates. Synaptophysin levels start to decline already at 4 months of age and from 9 months on significantly reduced levels of syntaxin and PSD-95 were described (Oakley et al., 2006
).
Early pathological changes like impaired synaptic transmission have been also reported in other transgenic AD mouse models, where intraneuronal Aβ accumulation has not been reported. In APP transgenic mice harboring the Swedish and Indiana mutations under the control of the PGDFβ-Promoter, deficits in synaptic transmission became evident prior to extracellular plaque formation. Without showing direct experimental evidence, the authors speculated that this might be due to neurotoxic effects possibly induced by intraneuronal Aβ accumulation or diffusible Aβ oligomers (Hsia et al., 1999
). Another transgenic model, expressing APP with the London mutation, showed early behavioral changes, differential glutamate responses, as well as deficits in LTP, all occurring before extracellular plaque formation (Moechars et al., 1999
). Finally, single APP (Swedish) and double APP (Swedish) and PS1 (M146L) transgenic mice were reported to show a reduced spontaneous alternation performance in the Y-maze task, before substantial extracellular plaque pathology became evident. The authors suggested that at least some aspects of the behavioral phenotype might be due to pathological alterations preceding plaque formation (Holcomb et al., 1999
). In summary, we think that it would be worthwhile to examine the models with phenotypical changes prior to plaque formation on the occurrence of intraneuronal Aβ aggregates.
Despite of a plethora of transgenic AD mouse models expressing various APP isoforms and mutations, efforts modeling significant neuronal loss were, until recently, not successful (Irizarry et al., 1997a
,b
). First evidence for significant hippocampal neuron loss (−14%), was reported in the APP23 mouse model, where the number of CA1 neurons was inversely correlated with CA1 plaque load (Calhoun et al., 1998
).
Using unbiased stereologic methods, a loss of CA1-3 neurons in a magnitude of ∼30% was detected in 17-month-old APP/PS1 transgenic mice, compared to age-matched PS1 control animals. Interestingly, the plaque load was approximately 10% smaller than the level of hippocampal pyramidal cell loss in these mice, indicating a loss of neurons at sites of Aβ aggregation but additionally also distant from extracellular Aβ deposits (Schmitz et al., 2004
).
Another model showing massive hippocampal neuron loss is the abovementioned APP/PS1KI mouse model (Casas et al., 2004
). At the age of 10 months an extensive neuron loss (>50%) in the hippocampus was reported, that correlated with the accumulation of intraneuronal Aβ and Thioflavin-S positive intracellular material and was already detectable at the age of 6 months (Casas et al., 2004
). At this time point, a loss of 33% of CA1 pyramidal neurons compared to PS1KI littermates could be demonstrated (Figure 1
G), together with decreased CA1 volume (−30%) and an atrophy of the entire hippocampus of 18% (Breyhan et al., 2009
). A detailed stereological analysis of the frontal cortex revealed an early loss of cortical neurons starting at the age of 6 months which correlated with the transient intraneuronal Aβ accumulation in contrast to extracellular plaque pathology (Christensen et al., 2008b
). A related observation was made in distinct cholinergic brain stem nuclei (Mo5, 7N) in these mice, where neuronal loss at 6 or 12 months of age correlated with the presence of intraneuronal Aβ peptides (Christensen et al., 2008a
).
The 5XFAD mice underscore the potential influence of intraneuronal Aβ accumulation on the loss of neurons. Cresyl violet staining revealed a reduced number of cortical layer 5 neurons, a region with robust intracellular Aβ immunoreactivity. The same holds true for the subiculum where neurons where pale or entirely missing (Oakley et al., 2006
). However, unbiased stereological neuron quantifications are needed to disclose the onset and severity of neuron loss in this model.
Recently, a new mouse model expressing only N-truncated AβpE3-42 in neurons was generated and it was demonstrated for the first time that this peptide is neurotoxic in vivo inducing neuron loss and an associated neurological phenotype (Wirths et al., 2009
).
In summary, there is accumulating evidence demonstrating that intraneuronal Aβx-42 triggers early synaptic deficits and neuron loss. Extracellular Aβ deposition has been challenged in the past to be a correlate for the striking region specific neuron loss, like the layer two pyramidal neurons in the entorhinal cortex and the CA1 neurons in the hippocampus (reviewed in Morrison and Hof, 1997
). Among other interesting mouse models for AD, APP/PS1KI mice show early intraneuronal aggregation of full-length and N-terminal modified Aβx-42 peptides. Evidence has been demonstrated that these peptides are aggregated as shown by the OC antibody and Thioflavin-S staining. The time point of loss of LTP and disrupted PPF correlates with CA1 neuron loss, hippocampus atrophy and hippocampus-dependent deficits in learning and memory behavior in this model. This was accompanied by reduced levels of pre- and postsynaptic markers (Breyhan et al., 2009
). Aβ42 comprises 85% of the total Aβ in this model, consisting of full-length and heterogeneous N-terminal modified Aβ variants. The APP/PS1KI model is so far the model with the most aggressive pathology (Casas et al., 2004
). Together with the data obtained from other transgenic mouse models, these data demonstrate that both synaptic deficits and neuron loss are a consequence of intraneuronal accumulation of Aβ peptides.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Alzheimer Forschung Initiative e.V., Fritz Thyssen Foundation and the European Commission, Marie Curie Early Stage Training, MEST-CT-2005-020013 (NEURAD), Alzheimer Ph.D. Graduate School.
Bahr, B. A., Hoffman, K. B., Yang, A. J., Hess, U. S., Glabe, C. G., and Lynch, G. (1998). Amyloid beta protein is internalized selectively by hippocampal field CA1 and causes neurons to accumulate amyloidogenic carboxyterminal fragments of the amyloid precursor protein. J. Comp. Neurol. 397, 139–147.
Barghorn, S., Nimmrich, V., Striebinger, A., Krantz, C., Keller, P., Janson, B., Bahr, M., Schmidt, M., Bitner, R. S., Harlan, J., Barlow, E., Ebert, U., and Hillen, H. (2005). Globular amyloid beta-peptide oligomer – a homogenous and stable neuropathological protein in Alzheimer’s disease. J. Neurochem. 31, 31.
Blanchard, V., Moussaoui, S., Czech, C., Touchet, N., Bonici, B., Planche, M., Canton, T., Jedidi, I., Gohin, M., Wirths, O., Bayer, T. A., Langui, D., Duyckaerts, C., Tremp, G., and Pradier, L. (2003). Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp. Neurol. 184, 247–263.
Buxbaum, J. D., Liu, K. N., Luo, Y., Slack, J. L., Stocking, K. L., Peschon, J. J., Johnson, R. S., Castner, B. J., Cerretti, D. P., and Black, R. A. (1998). Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273, 27765–27767.
Casas, C., Sergeant, N., Itier, J. M., Blanchard, V., Wirths, O., van der Kolk, N., Vingtdeux, V., van de Steeg, E., Ret, G., Canton, T., Drobecq, H., Clark, A., Bonici, B., Delacourte, A., Benavides, J., Schmitz, C., Tremp, G., Bayer, T. A., Benoit, P., and Pradier, L. (2004). Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 165, 1289–1300.
Chui, D. H., Tanahashi, H., Ozawa, K., Ikeda, S., Checler, F., Ueda, O., Suzuki, H., Araki, W., Inoue, H., Shirotani, K., Takahashi, K., Gallyas, F., and Tabira, T. (1999). Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat. Med. 5, 560–564.
D’Andrea, M. R., Reiser, P. A., Polkovitch, D. A., Gumula, N. A., Branchide, B., Hertzog, B. M., Schmidheiser, D., Belkowski, S., Gastard, M. C., and Andrade-Gordon, P. (2003). The use of formic acid to embellish amyloid plaque detection in Alzheimer’s disease tissues misguides key observations. Neurosci. Lett. 342, 114–118.
De Felice, F. G., Velasco, P. T., Lambert, M. P., Viola, K., Fernandez, S. J., Ferreira, S. T., and Klein, W. L. (2007). Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601.
Fernandez-Vizarra, P., Fernandez, A. P., Castro-Blanco, S., Serrano, J., Bentura, M. L., Martinez-Murillo, R., Martinez, A., Rodrigo, J., Encinas, J. M., Munoz, P., Alonso, D., Gomez, M. B., Sanchez, J., Rios-Tejada, F., Salas, E., Lisazoain, I., Leza, J. C., Lopez, J. C., Manuel Encinas, J., Lorenzo, P., Pedrosa, J. A., Peinado, M. A., Richart, A., Santacana, M., Cuttitta, F., Uttenthal, L. O., Bosca, L., Rodriguez, I., and Ruiz-Cabello, J. (2004). Intra- and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer’s disease. Histol. Histopathol. 19, 823–844.
Gong, Y., Chang, L., Viola, K. L., Lacor, P. N., Lambert, M. P., Finch, C. E., Krafft, G. A., and Klein, W. L. (2003). Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A. 100, 10417–10422.
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., Leinonen, V., Ito, A., Winblad, B., Glaser, E., and Ankarcrona, M. (2008). The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U.S.A. 105, 13145–13150.
Hussain, I., Powell, D., Howlett, D. R., Tew, D. G., Meek, T. D., Chapman, C., Gloger, I. S., Murphy, K. E., Southan, C. D., Ryan, D. M., Smith, T. S., Simmons, D. L., Walsh, F. S., Dingwall, C., and Christie, G. (1999). Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 14, 419–427.
Kumar-Singh, S., De Jonghe, C., Cruts, M., Kleinert, R., Wang, R., Mercken, M., De Strooper, B., Vanderstichele, H., Lofgren, A., Vanderhoeven, I., Backhovens, H., Vanmechelen, E., Kroisel, P. M., and Van Broeckhoven, C. (2000). Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated A beta(42) in Alzheimer’s disease. Hum. Mol. Genet. 9, 2589–2598.
Lacor, P. N., Buniel, M. C., Furlow, P. W., Clemente, A. S., Velasco, P. T., Wood, M., Viola, K. L., and Klein, W. L. (2007). Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807.
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., Morgan, T. E., Rozovsky, I., Trommer, B., Viola, K. L., Wals, P., Zhang, C., Finch, C. E., Krafft, G. A., and Klein, W. L. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453.
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., Caspersen, C., Chen, X., Pollak, S., Chaney, M., Trinchese, F., Liu, S., Gunn-Moore, F., Lue, L. F., Walker, D. G., Kuppusamy, P., Zewier, Z. L., Arancio, O., Stern, D., Yan, S. S., and Wu, H. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452.
Moechars, D., Dewachter, I., Lorent, K., Reverse, D., Baekelandt, V., Naidu, A., Tesseur, I., Spittaels, K., Haute, C. V., Checler, F., Godaux, E., Cordell, B., and Van Leuven, F. (1999). Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 274, 6483–6492.
Nilsberth, C., Westlind-Danielsson, A., Eckman, C. B., Condron, M. M., Axelman, K., Forsell, C., Stenh, C., Luthman, J., Teplow, D. B., Younkin, S. G., Naslund, J., and Lannfelt, L. (2001). The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat. Neurosci. 4, 887–893.
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., Guillozet-Bongaarts, A., Ohno, M., Disterhoft, J., Van Eldik, L., Berry, R., and Vassar, R. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140.
Rutten, B. P., Van der Kolk, N. M., Schafer, S., van Zandvoort, M. A., Bayer, T. A., Steinbusch, H. W., and Schmitz, C. (2005). Age-related loss of synaptophysin immunoreactive presynaptic boutons within the hippocampus of APP751SL, PS1M146L, and APP751SL/PS1M146L transgenic mice. Am. J. Pathol. 167, 161–173.
Schmitz, C., Rutten, B. P., Pielen, A., Schafer, S., Wirths, O., Tremp, G., Czech, C., Blanchard, V., Multhaup, G., Rezaie, P., Korr, H., Steinbusch, H. W., Pradier, L., and Bayer, T. A. (2004). Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 164, 1495–1502.
Sinha, S., Anderson, J. P., Barbour, R., Basi, G. S., Caccavello, R., Davis, D., Doan, M., Dovey, H. F., Frigon, N., Hong, J., Jacobson-Croak, K., Jewett, N., Keim, P., Knops, J., Lieberburg, I., Power, M., Tan, H., Tatsuno, G., Tung, J., Schenk, D., Seubert, P., Suomensaari, S. M., Wang, S., Walker, D., John, V. (1999). Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402, 537–540.
Takuma, K., Fang, F., Zhang, W., Yan, S., Fukuzaki, E., Du, H., Sosunov, A., McKhann, G., Funatsu, Y., Nakamichi, N., Nagai, T., Mizoguchi, H., Ibi, D., Hori, O., Ogawa, S., Stern, D. M., Yamada, K., and Yan, S. S. (2009). RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc. Natl. Acad. Sci. U.S.A. 106, 20021–20026.
Van Broeck, B., Vanhoutte, G., Pirici, D., Van Dam, D., Wils, H., Cuijt, I., Vennekens, K., Zabielski, M., Michalik, A., Theuns, J., De Deyn, P. P., Van der Linden, A., Van Broeckhoven, C., and Kumar-Singh, S. (2008). Intraneuronal amyloid beta and reduced brain volume in a novel APP T714I mouse model for Alzheimer’s disease. Neurobiol. Aging 29, 241–252.
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., Teplow, D. B., Ross, S., Amarante, P., Loeloff, R., Luo, Y., Fisher, S., Fuller, J., Edenson, S., Lile, J., Jarosinski, M. A., Biere, A. L., Curran, E., Burgess, T., Louis, J. C., Collins, F., Treanor, J., Rogers, G., and Citron, M. (1999). Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741.
Wegiel, J., Kuchna, I., Nowicki, K., Frackowiak, J., Mazur-Kolecka, B., Imaki, H., Wegiel, J., Mehta, P. D., Silverman, W. P., Reisberg, B., Deleon, M., Wisniewski, T., Pirttilla, T., Frey, H., Lehtimaki, T., Kivimaki, T., Visser, F. E., Kamphorst, W., Potempska, A., Bolton, D., Currie, J. R., and Miller, D. L. (2007). Intraneuronal Abeta immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration. Acta Neuropathol. (Berl.) 113, 389–402.
Yan, R., Bienkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., Pauley, A. M., Brashier, J. R., Stratman, N. C., Mathews, W. R., Buhl, A. E., Carter, D. B., Tomasselli, A. G., Parodi, L. A., Heinrikson, R. L., and Gurney, M. E. (1999). Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 402, 533–537.