Synapses, synaptic activity and intraneuronal Aβ in Alzheimer’s disease
- Department of Neurology and Neuroscience, Weill Cornell Medical College, New York, NY, USA
β-Amyloid peptide accumulation plays a central role in the pathogenesis of Alzheimer’s disease. Aberrant β-amyloid buildup in the brain has been shown to be present both in the extracellular space and within neurons. Synapses are important targets of β-amyloid, and alterations in synapses better correlate with cognitive impairment than amyloid plaques or neurofibrillary tangles. The link between β-amyloid and synapses became even tighter when it was discovered that β-amyloid accumulates within synapses and that synaptic activity modulates β-amyloid secretion. Currently, a central question in Alzheimer’s disease research is what role synaptic activity plays in the disease process, and how specifically β-amyloid is involved in the synaptic dysfunction that characterizes the disease.
Synapses are considered the earliest site of pathology, and synaptic loss is the best pathological correlate of cognitive impairment in Alzheimer’s disease (AD) ( Hamos et al., 1989 ; DeKosky and Scheff, 1990 ; Terry et al., 1991 ; Selkoe, 2002 ; Coleman and Yao, 2003 ). Toxic effects of extracellular β-amyloid (Aβ) on synapses have been known for many years. Synapses and neurites are also frequently damaged near Aβ plaques (Dong et al., 2007 ; Meyer-Luehmann et al., 2009 ). The first direct association between Aβ peptide and synapse pathologies in the brain was provided by immunoelectron microscopy (IEM); this showed that aberrant Aβ42 accumulation within distal neurites and synapses was directly associated with pathology (Takahashi et al., 2002 ). Emerging studies have provided very interesting evidence that synaptic activity modulates Aβ homeostasis. Increased synaptic activation enhanced, while inhibition reduced, secretion of Aβ in cultured hippocampal slices and mouse brains ( Kamenetz et al., 2003 ; Cirrito et al., 2005 ; Cirrito et al., 2008 ). These were important discoveries and appeared to fit the scenario that brain areas with the highest synaptic activity, including hippocampus and entorhinal cortex, are among the most vulnerable to early AD pathology (Buckner et al., 2005 ). Extracellular Aβ is known to reduce synaptic plasticity, and alter synaptic function, structure and protein levels in AD model systems ( Selkoe, 2002 ; Trinchese et al., 2004a ; Almeida et al., 2005 ; Snyder et al., 2005 ; Hsieh et al., 2006 ; Shankar et al., 2008 ; Deshpande et al., 2009 ). Therefore, increased secretion of Aβ induced by synaptic activity could lead to damage and loss of synapses and to progressive accumulation of extracellular Aβ into amyloid plaques, which are important hallmarks of AD. From this point of view, synaptic activity could be detrimental and contribute to AD pathogenesis. In fact, aberrant hyperexcitability is present in brains of AD transgenic mice ( Palop et al., 2007 ; Busche et al., 2008 ), which could increase activity-induced release of Aβ and thereby aggravate pathology. Moreover, cerebral Aβ deposition is increased in human epilepsy (Mackenzie and Miller, 1994 ) and seizures are increased in human AD (Palop et al., 2007 ).
Increased cognitive activity is also able to increase Aβ secretion, since recovery of cognitive function after brain injury correlates with increased levels of extracellular Aβ in human brain (Brody et al., 2008 ). Levels of Aβ in the interstitial fluid have been shown to follow the circadian rhythm, being elevated when awake and reduced when asleep (Kang et al., 2009 ). These observations support that Aβ secretion could be a physiologic event occurring with normal brain activity. With activity-induced secretion, the concentration of extracellular Aβ remains in the picomolar range in vitro as well as in vivo ( Cirrito et al., 2003 ; Trinchese et al., 2004b ). It can be hypothesized that at this low concentration extracellular Aβ might be nontoxic. Indeed, recent studies reported that picomolar concentrations of Aβ enhanced synaptic plasticity and memory ( Puzzo et al., 2008 ; Garcia-Osta and Alberini, 2009 ).
Another set of studies supports the idea that cognitive activity may be protective against AD. Higher educational level or involvement in mentally stimulating activities correlated with a lower probability of developing AD (Stern, 2006 ). Experiments on environmental enrichment in AD transgenic mice demonstrated reduced plaque deposition and up-regulation of genes involved in memory formation and Aβ degradation (Lazarov et al., 2005 ). Moreover, recent cell biological studies provided evidence that synaptic activation reduces levels of intraneuronal Aβ and protects synapses in models of AD, despite a concomitant increase in Aβ secretion (Tampellini et al., 2009 ). Next to the well known presence of extracellular Aβ as plaques, AD pathology also includes a pool of Aβ accumulating within neurons ( Gouras et al., 2000 ; D’Andrea et al., 2001 ; Gyure et al., 2001 ; Wirths et al., 2001 ; Mori et al., 2002 ; Oddo et al., 2003 ; Cataldo et al., 2004 ), which is increasingly considered to play a critical role in the disease ( Gouras et al., 2005 ; LaFerla et al., 2007 ; Bayer and Wirths, 2010 ). Accumulation and oligomerization of Aβ42, the most pathologic Aβ isoform, occurs progressively within distal neurites and synaptic compartments, and is directly associated with subcellular pathology (Takahashi et al., 2002 , 2004 ) and with alterations of synaptic proteins (Almeida et al., 2005 ). Furthermore, intraneuronal Aβ accumulation is associated with the pre-plaque onset of physiological and behavioral abnormalities in AD transgenic models ( Oddo et al., 2003 ; Echeverria et al., 2004 ; Billings et al., 2005 ; Cruz et al., 2006 ; Knobloch et al., 2007 ; Lord et al., 2009 ). Clearance of intraneuronal Aβ by treatment with Aβ antibodies protected synapses (Tampellini et al., 2007 ) and correlated with memory improvement (Billings et al., 2005 ). Overall, cumulative evidence support a scenario where progressive accumulation of intraneuronal Aβ42 becomes extracellular not by secretion but rather following degeneration of neurites and synapses. Release of this intraneuronal Aβ42 into the extracellular space may then result in an abnormally high concentration of extracellular Aβ42, potentially leading to toxic spread of Aβ pathology to surrounding synapses (Figure 1 ). Given the emerging evidence of declining extracellular Aβ42 levels in cerebrospinal fluid (CSF) in patients as the earliest harbinger for the subsequent development of AD (Fagan et al., 2009 ), one can now even hypothesize that AD is characterized by decreased Aβ42 secretion as Aβ42 accumulates inside neurons.
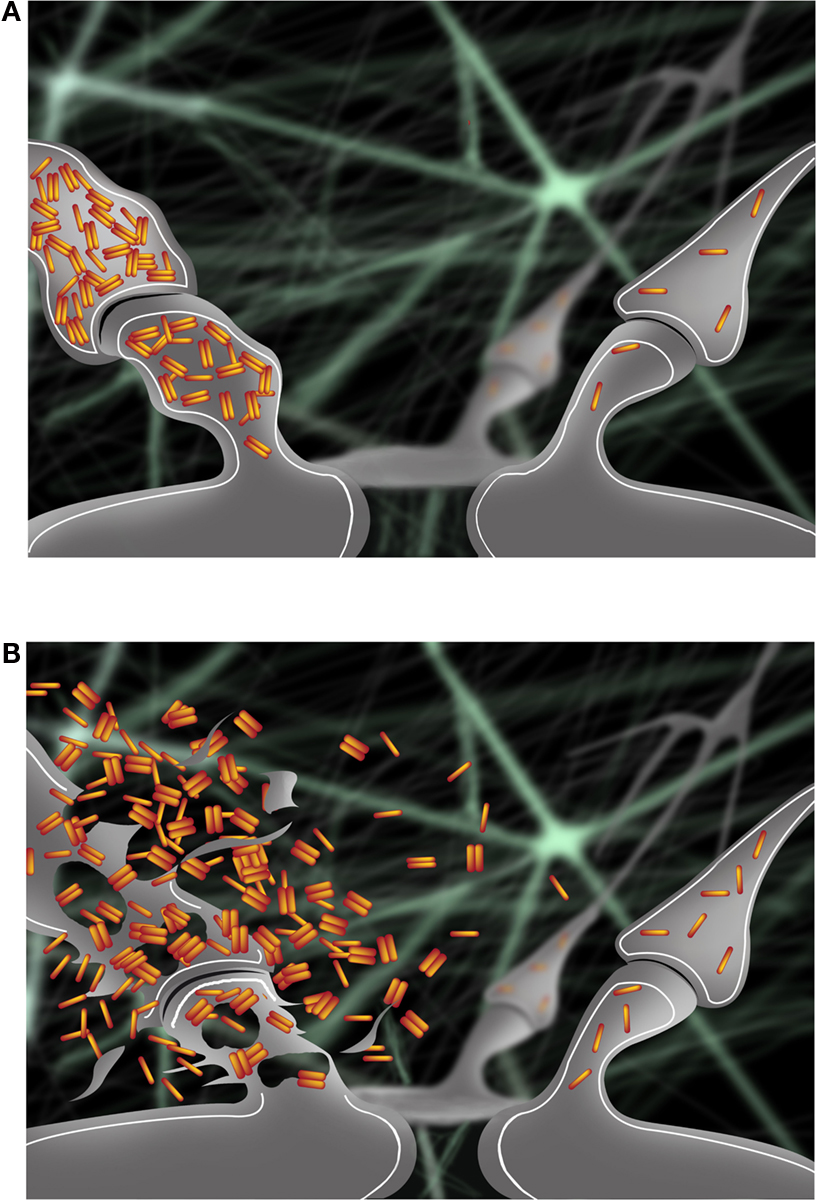
Figure 1. (A) Accumulation of intraneuronal Aβ42 at early stages of AD occurs progressively in a synapse (left) and is associated with pathological alterations compared to a normal synapse (right). (B) Intraneuronal Aβ42 is released in the extracellular space following degeneration of the synapse (left). Release of intraneuronal Aβ42 into the extracellular space may contribute to the toxic spread of Aβ pathology to a nearby synapse (right).
It is important to note that the extra- and intracellular pools of Aβ are related, although their relationship is complex and they can influence each other ( Yang et al., 1999 ; Oddo et al., 2006 ; Tampellini et al., 2009 ). Intraneuronal Aβ accumulation begins before plaques ( Gouras et al., 2000 ; Wirths et al., 2001 ; Mori et al., 2002 ; Oddo et al., 2003 ), and, after clearance by Aβ immunotherapy, reappears prior to plaques (Oddo et al., 2004 ). How specifically extracellular Aβ is toxic to neurons and synapses remains poorly understood, but appears to occur via APP and potentially intraneuronal Aβ. Addition of extracellular Aβ1–42 markedly increased intracellular Aβ (Yang et al., 1999 ; Tampellini et al., 2009 ). The role and extent of Aβ internalization from the extracellular space remain less clear (Yang et al., 1999 ; Saavedra et al., 2007 ; Kandimalla et al., 2009 ). Yang et al. (1999) showed minimal internalization of labeled Aβ1–42, whereas adding of unlabeled Aβ1–42 lead to a marked up-regulation of newly generated intracellular Aβ42. Extracellular Aβ1–42 was shown to induce cell death in wild type neurons but not in neurons lacking APP (Lorenzo et al., 2000 ) or in cells where the NPXY motif of APP was mutated (Shaked et al., 2006 ). Moreover, extracellular Aβ1–42 is unable to reduce levels of synaptic proteins in APP knockout neurons or when it is applied together with a γ-secretase inhibitor (Tampellini et al., 2009 ). Overall, these data suggest that extracellular Aβ1–42 synapto-toxicity requires γ-secretase cleavage of APP and also intraneuronal Aβ42 accumulation.
The precise molecular and cellular mechanism(s) whereby synaptic activity modulates Aβ and then Aβ first alters synapses are of major interest. The reduction of intracellular Aβ with synaptic activation is partially due to secretion of intracellular Aβ to the extracellular space (Tampellini et al., 2009 ). Aβ degradation is also involved in the reduction of the intraneuronal pool during activity. Synaptic activity failed to reduce the intraneuronal pool of Aβ42 in the presence of the neprilysin inhibitor thiorphan or in neprilysin knockout neurons. These data implicate neprilysin in the degradation of the most disease-linked Aβ isoform with activity. In contrast, levels of intraneuronal Aβ40 were still decreased, although to a lesser extent, with thiorphan treatment or in neprilysin knockout neurons, during synaptic activity (Tampellini et al., 2009 ). This might be explained by Aβ40 being more soluble and more abundantly secreted than Aβ42 (Kamenetz et al., 2003 ). The reduction of intraneuronal Aβ with synaptic activity, which correlated with synaptic improvement, supports the hypothesis that synaptic activity may also be protective in AD pathogenesis. Currently, AD patients are encouraged to increase their mental and social activities. We hypothesize that under physiological conditions of synaptic activity both extra- and intracellular pools of Aβ are efficiently cleared, but then with aging and other AD risk factors (e.g. apolipoprotein Eε4), Aβ clearance mechanisms become impaired. It is of considerable interest that levels of neprilysin have been reported to decrease with aging ( Iwata et al., 2002 ; Apelt et al., 2003 ). Because neprilysin localizes to synapses (Fukami et al., 2002 ), its age-dependent decline could explain the progressive synaptic accumulation of Aβ42 (Takahashi et al., 2002 , 2004 , 2008 ).
With synaptic activation, APP is anterogradely transported to synapses, followed by internalization and amyloidogenic processing at active synapses (Tampellini et al., 2009 ). The effect of synaptic activity on transport in neurites of protein cargoes is not uniform ( Perestenko and Henley, 2003 ; Bingol and Schuman, 2006 ; Cai et al., 2007 ; Kang et al., 2008 ). The synaptic activity-dependent increase of APP anterograde transport and β-cleavage support that Aβ production is locally enhanced at activated synapses (Cirrito et al., 2008 ; Tampellini et al., 2009 ). How Aβ starts to accumulate at synapses and contributes to pathogenesis remains unclear. The role of aging should not be underestimated in promoting AD pathogenesis. Mitochondrial dysfunction and oxidative stress, as well as reduced cellular degradation, appear to be important contributors to the neurobiology of aging. In fact, it was reported that elevated oxidative stress promoted Aβ pathology in AD transgenic mice (Li et al., 2004 ). On the other hand, once Aβ accumulation begins with aging, it is unclear how Aβ then contributes to progression of synaptic dysfunction. Cumulative data over the past two decades support that APP is trafficked via the Golgi apparatus to the plasma membrane where α-secretase cleavage can occur and thereby preclude Aβ formation. An additional pool of APP is re-internalized from the plasma membrane and then processed to Aβ peptides in endosomes (Rajendran et al., 2008 ; Thinakaran and Koo, 2008 ), followed by secretion of predominantly Aβ40 peptides. Evidence supports that relatively higher amounts of the more hydrophobic, aggregation-prone and AD-linked Aβ42 is retained within endosomes. It is this pool of Aβ42 that prominently accumulates and then oligomerizes with aging in distal neurites and synapses in the brain with AD pathogenesis (Takahashi et al., 2002 , 2004 ). At a basic level it would appear that aberrant Aβ42 accumulation in endosomes near synapses might be central to early synaptic dysfunction in AD. Indeed, Aβ accumulating primary neurons derived from APP-transgenic mice compared to wild type neurons were shown to have Aβ-dependent endocytic abnormalities specifically in the multivesicular body (MVB) sorting pathway but not in the internalization or recycling pathways (Almeida et al., 2006 ). Recent work has shown that intraneuronal Aβ can also be pathogenic to mitochondria. Because of the critical role that mitochondria play in cells in general, and particularly at active synapses where they are abundant, Aβ effects on mitochondria may be critical for the pathogenesis of AD ( Lin and Beal, 2006 ; Chen and Yan, 2007 ; Reddy, 2009 ). The pathogenic pathway from aberrant Aβ accumulation to synaptic damage likely includes altered signaling pathways, cellular ionic imbalance, and emerging apoptosis pathways at synapses that remain to be determined.
Elucidating the molecular and cellular mechanisms by which synaptic activity and Aβ homeostasis affect each other may provide new insights both in understanding the pathogenesis of AD and for the development of new therapies.
Conflict of Interest Statement
The authors declare that they have no commercial or financial relationship that could be construed as a potential conflict of interest.
Acknowledgments
The support of an Alzheimer’s Association New Investigator Award (D. Tampellini) and Zenith Award (G.K. Gouras), and National Institute of Health grants AG027140 and AG028174 (G.K. Gouras). We thank Mr. Matteo Tampellini for the preparation of the schematic figures.
References
Almeida, C. G., Takahashi, R. H., and Gouras, G. K. (2006). Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. 26, 4277–4288.
Almeida, C. G., Tampellini, D., Takahashi, R. H., Greengard, P., Lin, M. T., Snyder, E. M., and Gouras, G. K. (2005). Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 20, 187–198.
Apelt, J., Ach, K., and Schliebs, R. (2003). Aging-related down-regulation of neprilysin, a putative beta-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of beta-amyloid plaques. Neurosci. Lett. 339, 183–186.
Bayer, T. A., and Wirths, O. (2010). Intracellular accumulation of amyloid-beta – a predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front. Aging Neurosci. 2, 1–10.
Billings, L. M., Oddo, S., Green, K. N., McGaugh, J. L., and LaFerla, F. M. (2005). Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688.
Bingol, B., and Schuman, E. M. (2006). Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature 441, 1144–1148.
Brody, D. L., Magnoni, S., Schwetye, K. E., Spinner, M. L., Esparza, T. J., Stocchetti, N., Zipfel, G. J., and Holtzman, D. M. (2008). Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 321, 1221–1224.
Buckner, R. L., Snyder, A. Z., Shannon, B. J., LaRossa, G., Sachs, R., Fotenos, A. F., Sheline, Y. I., Klunk, W. E., Mathis, C. A., Morris, J. C., and Mintun, M. A. (2005). Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J. Neurosci. 25, 7709–7717.
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K. H., Haass, C., Staufenbiel, M., Konnerth, A., and Garaschuk, O. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689.
Cai, Q., Pan, P. Y., and Sheng, Z. H. (2007). Syntabulin-kinesin-1 family member 5B-mediated axonal transport contributes to activity-dependent presynaptic assembly. J. Neurosci. 27, 7284–7296.
Cataldo, A. M., Petanceska, S., Terio, N. B., Peterhoff, C. M., Durham, R., Mercken, M., Mehta, P. D., Buxbaum, J., Haroutunian, V., and Nixon, R. A. (2004). Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol. Aging 25, 1263–1272.
Chen, J. X., and Yan, S. D. (2007). Amyloid-beta-induced mitochondrial dysfunction. J. Alzheimer’s Dis. 12, 177–184.
Cirrito, J. R., Kang, J. E., Lee, J., Stewart, F. R., Verges, D. K., Silverio, L. M., Bu, G., Mennerick, S., and Holtzman, D. M. (2008). Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58, 42–51.
Cirrito, J. R., May, P. C., O’Dell, M. A., Taylor, J. W., Parsadanian, M., Cramer, J. W., Audia, J. E., Nissen, J. S., Bales, K. R., Paul, S. M., DeMattos, R. B., and Holtzman, D. M. (2003). In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J. Neurosci. 23, 8844–8853.
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., Schoepp, D. D., Paul, S. M., Mennerick, S., and Holtzman, D. M. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922.
Coleman, P. D., and Yao, P. J. (2003). Synaptic slaughter in Alzheimer’s disease. Neurobiol. Aging 24, 1023–1027.
Cruz, J. C., Kim, D., Moy, L. Y., Dobbin, M. M., Sun, X., Bronson, R. T., and Tsai, L. H. (2006). p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J. Neurosci. 26, 10536–10541.
D’Andrea, M. R., Nagele, R. G., Wang, H. Y., Peterson, P. A., and Lee, D. H. (2001). Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 38, 120–134.
DeKosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464.
Deshpande, A., Kawai, H., Metherate, R., Glabe, C. G., and Busciglio, J. (2009). A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J. Neurosci. 29, 4004–4015.
Dong, H., Martin, M. V., Chambers, S., and Csernansky, J. G. (2007). Spatial relationship between synapse loss and beta-amyloid deposition in Tg2576 mice. J. Comp. Neurol. 500, 311–321.
Echeverria, V., Ducatenzeiler, A., Dowd, E., Janne, J., Grant, S. M., Szyf, M., Wandosell, F., Avila, J., Grimm, H., Dunnett, S. B., Hartmann, T., Alhonen, L., and Cuello, A. C. (2004). Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the beta-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience 129, 583–592.
Fagan, A. M., Head, D., Shah, A. R., Marcus, D., Mintun, M., Morris, J. C., and Holtzman, D. M. (2009). Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann. Neurol. 65, 176–183.
Fukami, S., Watanabe, K., Iwata, N., Haraoka, J., Lu, B., Gerard, N. P., Gerard, C., Fraser, P., Westaway, D., St George-Hyslop, P., and Saido, T. C. (2002). Abeta-degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Abeta pathology. Neurosci. Res. 43, 39–56.
Garcia-Osta, A., and Alberini, C. M. (2009). Amyloid beta mediates memory formation. Learn. Mem. 16, 267–272.
Gouras, G. K., Almeida, C. G., and Takahashi, R. H. (2005). Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol. Aging 26, 1235–1244.
Gouras, G. K., Tsai, J., Naslund, J., Vincent, B., Edgar, M., Checler, F., Greenfield, J. P., Haroutunian, V., Buxbaum, J. D., Xu, H., Greengard, P., and Relkin, N. R. (2000). Intraneuronal Abeta42 accumulation in human brain. Am. J. Pathol. 156, 15–20.
Gyure, K. A., Durham, R., Stewart, W. F., Smialek, J. E., and Troncoso, J. C. (2001). Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch. Pathol. Lab Med. 125, 489–492.
Hamos, J. E., DeGennaro, L. J., and Drachman, D. A. (1989). Synaptic loss in Alzheimer’s disease and other dementias. Neurology 39, 355–361.
Hsieh, H., Boehm, J., Sato, C., Iwatsubo, T., Tomita, T., Sisodia, S., and Malinow, R. (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843.
Iwata, N., Takaki, Y., Fukami, S., Tsubuki, S., and Saido, T. C. (2002). Region-specific reduction of A beta-degrading endopeptidase, neprilysin, in mouse hippocampus upon aging. J. Neurosci. Res. 70, 493–500.
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., Sisodia, S., and Malinow, R. (2003). APP processing and synaptic function. Neuron 37, 925–937.
Kandimalla, K. K., Scott, O. G., Fulzele, S., Davidson, M. W., and Poduslo, J. F. (2009). Mechanism of neuronal versus endothelial cell uptake of Alzheimer’s disease amyloid beta protein. PLoS One 4, e4627. DOI:10.1371/journal.pone.0004627.
Kang, J. E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., Fujiki, N., Nishino, S., and Holtzman, D. M. (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007.
Kang, J. S., Tian, J. H., Pan, P. Y., Zald, P., Li, C., Deng, C., and Sheng, Z. H. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148.
Knobloch, M., Konietzko, U., Krebs, D. C., and Nitsch, R. M. (2007). Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol. Aging 28, 1297–1306.
LaFerla, F. M., Green, K. N., and Oddo, S. (2007). Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509.
Lazarov, O., Robinson, J., Tang, Y. P., Hairston, I. S., Korade-Mirnics, Z., Lee, V. M., Hersh, L. B., Sapolsky, R. M., Mirnics, K., and Sisodia, S. S. (2005). Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell 120, 701–713.
Li, F., Calingasan, N. Y., Yu, F., Mauck, W. M., Toidze, M., Almeida, C. G., Takahashi, R. H., Carlson, G. A., Flint Beal, M., Lin, M. T., and Gouras, G. K. (2004). Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J. Neurochem. 89, 1308–1312.
Lord, A., Englund, H., Soderberg, L., Tucker, S., Clausen, F., Hillered, L., Gordon, M., Morgan, D., Lannfelt, L., Pettersson, F. E., and Nilsson, L. N. (2009). Amyloid-beta protofibril levels correlate with spatial learning in Arctic Alzheimer’s disease transgenic mice. FEBS J. 276, 995–1006.
Lorenzo, A., Yuan, M., Zhang, Z., Paganetti, P. A., Sturchler-Pierrat, C., Staufenbiel, M., Mautino, J., Vigo, F. S., Sommer, B., and Yankner, B. A. (2000). Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer’s disease. Nat. Neurosci. 3, 460–464.
Mackenzie, I. R., and Miller, L. A. (1994). Senile plaques in temporal lobe epilepsy. Acta Neuropathol. 87, 504–510.
Meyer-Luehmann, M., Mielke, M., Spires-Jones, T. L., Stoothoff, W., Jones, P., Bacskai, B. J., and Hyman, B. T. (2009). A reporter of local dendritic translocation shows plaque- related loss of neural system function in APP-transgenic mice. J. Neurosci. 29, 12636–12640.
Mori, C., Spooner, E. T., Wisniewsk, K. E., Wisniewski, T. M., Yamaguch, H., Saido, T. C., Tolan, D. R., Selkoe, D. J., and Lemere, C. A. (2002). Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid 9, 88–102.
Oddo, S., Billings, L., Kesslak, J. P., Cribbs, D. H., and LaFerla, F. M. (2004). Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43, 321–332.
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., Metherate, R., Mattson, M. P., Akbari, Y., and LaFerla, F. M. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421.
Oddo, S., Caccamo, A., Smith, I. F., Green, K. N., and LaFerla, F. M. (2006). A dynamic relationship between intracellular and extracellular pools of Abeta. Am. J. Pathol. 168, 184–194.
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., Yoo, J., Ho, K. O., Yu, G. Q., Kreitzer, A., Finkbeiner, S., Noebels, J. L., and Mucke, L. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711.
Perestenko, P. V., and Henley, J. M. (2003). Characterization of the intracellular transport of GluR1 and GluR2 alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J. Biol. Chem. 278, 43525–43532.
Puzzo, D., Privitera, L., Leznik, E., Fa, M., Staniszewski, A., Palmeri, A., and Arancio, O. (2008). Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545.
Rajendran, L., Schneider, A., Schlechtingen, G., Weidlich, S., Ries, J., Braxmeier, T., Schwille, P., Schulz, J. B., Schroeder, C., Simons, M., Jennings, G., Knolker, H. J., and Simons, K. (2008). Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science 320, 520–523.
Reddy, P. H. (2009). Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 218, 286–292.
Saavedra, L., Mohamed, A., Ma, V., Kar, S., and de Chaves, E. P. (2007). Internalization of beta-amyloid peptide by primary neurons in the absence of apolipoprotein E. J. Biol. Chem. 282, 35722–35732.
Shaked, G. M., Kummer, M. P., Lu, D. C., Galvan, V., Bredesen, D. E., and Koo, E. H. (2006). Abeta induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597-624). FASEB J. 20, 1254–1256.
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., Brett, F. M., Farrell, M. A., Rowan, M. J., Lemere, C. A., Regan, C. M., Walsh, D. M., Sabatini, B. L., and Selkoe, D. J. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842.
Snyder, E. M., Nong, Y., Almeida, C. G., Paul, S., Moran, T., Choi, E. Y., Nairn, A. C., Salter, M. W., Lombroso, P. J., Gouras, G. K., and Greengard, P. (2005). Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 8, 1051–1058.
Stern, Y. (2006). Cognitive reserve and Alzheimer disease. Alzheimer’s Dis. Assoc. Disord. 20, S69–74.
Takahashi, R. H., Almeida, C. G., Kearney, P. F., Yu, F., Lin, M. T., Milner, T. A., and Gouras, G. K. (2004). Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 24, 3592–3599.
Takahashi, R. H., Capetillo-Zarate, E., Lin, M. T., Milner, T. A., and Gouras, G. K. (2008). Co-occurrence of Alzheimer’s disease beta-amyloid and tau pathologies at synapses. Neurobiol. Aging 31. doi: 10.1016/j.neurobiolaging.2008.07.021.
Takahashi, R. H., Milner, T. A., Li, F., Nam, E. E., Edgar, M. A., Yamaguchi, H., Beal, M. F., Xu, H., Greengard, P., and Gouras, G. K. (2002). Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 161, 1869–1879.
Tampellini, D., Magrane, J., Takahashi, R. H., Li, F., Lin, M. T., Almeida, C. G., and Gouras, G. K. (2007). Internalized antibodies to the Abeta domain of APP reduce neuronal Abeta and protect against synaptic alterations. J. Biol. Chem. 282, 18895–18906.
Tampellini, D., Rahman, N., Gallo, E. F., Huang, Z., Dumont, M., Capetillo-Zarate, E., Ma, T., Zheng, R., Lu, B., Nanus, D. M., Lin, M. T., and Gouras, G. K. (2009). Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J. Neurosci. 29, 9704–9713.
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., Hansen, L. A., and Katzman, R. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580.
Thinakaran, G., and Koo, E. H. (2008). Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619.
Trinchese, F., Liu, S., Battaglia, F., Walter, S., Mathews, P. M., and Arancio, O. (2004a). Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol. 55, 801–814.
Trinchese, F., Liu, S., Ninan, I., Puzzo, D., Jacob, J. P., and Arancio, O. (2004b). Cell cultures from animal models of Alzheimer’s disease as a tool for faster screening and testing of drug efficacy. J. Mol. Neurosci. 24, 15–21.
Wirths, O., Multhaup, G., Czech, C., Blanchard, V., Moussaoui, S., Tremp, G., Pradier, L., Beyreuther, K., and Bayer, T. A. (2001). Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 306, 116–120.
Keywords: Alzheimer disease, amyloid, amyloid precursor protein, synapse, synaptic plasticity, neprilysin, neuron, neurodegeneration
Citation: Tampellini D and Gouras GK (2010) Synapses, synaptic activity and intraneuronal Aβ in Alzheimer’s disease. Front. Ag. Neurosci. 2:13. doi: 10.3389/fnagi.2010.00013
Received: 08 January 2010;
Paper pending published: 16 February 2010;
Accepted: 12 March 2010;
Published online: 21 May 2010
Edited by:
P. Hemachandra Reddy, Oregon Health and Science University, USAReviewed by:
Peizhong Mao, Oregon Health and Science University, USAQitao Ran, University of Texas, USA; University of Texas Health Science Center at San Antonio, USA
P. Hemachandra Reddy, Oregon Health and Science University, USA
Copyright: © 2010 Tampellini and Gouras. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Gunnar K. Gouras, Laboratory of Alzheimer’s disease Neurobiology, Department of Neurology and Neuroscience, Weill Cornell Medical College, 525 East 68th Street, New York, NY 10065, USA. e-mail: gkgouras@med.cornell.edu