Institut de Neurociències, Departament Bioquímica i Biologia Molecular, Centro de Investigación Biomédica en Red Enfermedades Neurodegenerativas (CIBERNED), Universitat Autònoma de Barcelona, Bellaterra, Spain
Presenilins (PS) are the catalytic components of γ-secretase, an aspartyl protease that regulates through proteolytic processing the function of multiple signaling proteins. Specially relevant is the γ-secretase-dependent cleavage of the β-amyloid precursor protein (APP) since generates the β-amyloid (Aβ) peptides that aggregate and accumulate in the brain of Alzheimer’s disease (AD) patients. Abnormal processing and/or accumulation of Aβ disrupt synaptic and metabolic processes leading to neuron dysfunction and neurodegeneration. Studies in presenilin conditional knockout mice have revealed that presenilin-1 is essential for age-dependent Aβ accumulation and inflammation. By contrast, mutations in the presenilin genes responsible for early onset familial AD cause rapid disease progression and accentuate clinical and pathological features including inflammation. In addition, a number of loss of function mutations in presenilin-1 have been recently associated to non-Alzheimer’s dementias including frontotemporal dementia and dementia with Lewy bodies. In agreement, total loss of presenilin function in the brain results in striking neurodegeneration and inflammation, which includes activation of glial cells and induction of proinflammatory genes, besides altered inflammatory responses in the periphery. Interestingly, some non-steroidal anti-inflammatory drugs that slow cognitive decline and reduce the risk of AD, decrease amyloidogenic Aβ42 levels by modulating allosterically PS/γ-secretase. In this review, I present current evidence supporting a role of presenilin/γ-secretase signaling on gliogenesis and gliosis in normal and pathological conditions. Understanding the cellular mechanisms regulated by presenilin/γ-secretase during chronic inflammatory processes may provide new approaches for the development of effective therapeutic strategies for AD.
Inflammation is the main biological defense mechanism in response to external injury. Besides having multiple therapeutical benefits, inflammation has been extensively implicated in many pathological conditions including cancer, diabetes, heart infarct and brain diseases. Inflammatory responses are common in acute and chronic diseases of the central nervous system. For instance, inappropriate inflammation contributes directly to acute neurodegeneration during brain stroke, trauma and viral infection (Allan and Rothwell, 2003
). Similarly, chronic inflammatory responses are also common to many age-related neurodegenerative disorders including Alzheimer’s disease (AD). Neuroinflammatory responses, characterized by activation of glial cells, especially microglia and astrocytes, and increased production of inflammatory molecules are closely associated to neuropathological lesions in AD brain (Mrak and Griffin, 2005
). Moreover, inflammation is an important neuropathological feature that contributes to neuronal dysfunction and cognitive dysfunction during normal aging and age-related neurodegenerative conditions (Lynch, 2010
).
AD is the most common neurodegenerative disorder in the elderly. AD causes progressive cognitive impairment and behavioral disturbances leading finally to dementia. The classical neuropathological features of AD brain include neuronal loss and the presence of extracellular amyloid plaques containing β-amyloid (Aβ) peptides and intraneuronal neurofibrillary tangles (NFTs) composed of aggregated phosphorylated tau. Aβ peptides, the principal constituents of amyloid plaques, stimulate glial cells to undergo morphological and gene expression changes causing activation and/or secretion of proinflammatory molecules including cytokines, complement components, adhesion molecules and cyclooxygenase (COX) enzymes (Mrak and Griffin, 2005
). Once activated, microglia and astrocytes contribute to removal of Aβ from the brain parenchyma (Wyss-Coray et al., 2001
, 2003
). By contrast, long-term chronic inflammatory responses contribute actively to neuron dysfunction and further neurodegeneration in AD, which suggests that inflammation may have early benefits as well as long-term detrimental effects during progression of the disease.
The prevalent hypothesis of AD postulates that one of the initiating events of the disease is related to abnormal processing, aggregation and/or clearance of Aβ peptides (Hardy and Selkoe, 2002
). The initial accumulation of Aβ peptides and/or phosphorylated tau may occur when individuals are still cognitively normal, whereas accumulation of Aβ in plaques and the presence of NFTs accompanied by neuronal dysfunction and neurodegeneration correlate closely with severity of clinical symptoms (Jack et al., 2010
). At initial stages, accumulation of aggregated oligomeric Aβ species can interfere with neuronal and synaptic function without affecting neuronal viability. The key step regulating Aβ generation is the sequential proteolytic processing of amyloid precursor protein (APP) by β-secretase (BACE) and γ-secretase proteases. Presenilins (PS), the catalytic components of γ-secretase, are essential for Aβ generation, age-dependent Aβ accumulation and inflammatory responses during progression of the disease (Saura et al., 2005
). Therefore, modulation of γ-secretase by using specific inhibitors has been suggested as potential therapeutic strategy to reduce Aβ-induced pathology and clinical symptoms. Interestingly, some non-steroidal anti-inflammatory drugs (NSAIDs) that ameliorate symptoms and reduce the risk of AD were shown to lower the generation of Aβ42 by affecting the γ-secretase-dependent cleavage of APP (Weggen et al., 2007
). The therapeutic benefits of PS/γ-secretase inactivation may directly depend on the target selectivity and reduction of side effects on other important signaling pathways.
Here, I describe recent findings implicating PS/γ-secretase on inflammation, especially those related with cellular and genetic mechanisms regulated by PS during inflammatory responses in brain diseases. Finally, I discuss implications for development and ongoing therapeutic strategies based on γ-secretase targeting and modulation in AD.
PS are multifunctional proteins that contain the catalytic core of γ-secretase, an aspartyl protease that mediates the regulated intramembrane cleavage of multiple cell surface signaling proteins including among others APP, Notch receptors, ErbB4, E- and N-cadherins, p75 and EphB2 (Koo and Kopan, 2004
). PS are transmembrane proteins that undergo regulated endoproteolysis to generate N- and C-terminal fragments, which are the predominant PS species that accumulate in vivo within the enzymatic complex (Thinakaran et al., 1996
; Saura et al., 1999
). The functional active γ-secretase complex is composed of PS dimers and other components such as nicastrin, Pen-2 and Aph1 (De Strooper, 2003
; Cervantes et al., 2004
). These four components are essential for stabilization and function of γ-secretase. The mechanism of complex assembly involves stabilization of full-length presenilin by Aph-1/nicastrin, followed by binding of Pen-2, which facilitates endoproteolysis of presenilin and confers the γ-secretase activity (Takasugi et al., 2003
).
Two presenilin homologous presenilin-1 (PS1) and presenilin-2 (PS2) located on chromosomes 14q24.3 and 1q31-q42, respectively, are present in mammals. Gene expression studies in different species have revealed wide-spread expression of PS in different cell types and tissues. PS1 and PS2 are ubiquitously expressed in most tissues including the brain (Lee et al., 1996
; Lah et al., 1997
). During mouse development, PS1 mRNA expression is particularly high in neural organs, whereas PS2 is barely expressed. By contrast, PS1 and PS2 mRNAs are highly expressed in neurons of the hippocampus and entorhinal cortex in adult mouse brain (Lee et al., 1996
). In human brain, PS1 protein is highly expressed in pyramidal neurons of the hippocampus and neocortex, magnocellular neurons of the basal forebrain, brainstem motoneurons and some interneuron populations (Kim et al., 1997
; Lah et al., 1997
). Although PS are mainly expressed in neurons, PS1 and PS2 mRNAs are detected at low levels in white matter glial cells and cultured astrocytes (Lee et al., 1996
). However, PS1 expression is upregulated in reactive astrocytes and activated microglia under pathological conditions such as those occurring in AD, brain injury and hypoxia (Weggen et al., 1998
; Peers et al., 2004
; Nadler et al., 2008
).
γ-Secretase facilitates Notch signaling by mediating the ligand-induced proteolytic cleavage of Notch1 receptor that generates the Notch intracellular domain (NICD) (De Strooper et al., 1999
). NICD then translocates to the nucleus where binds to CBF1/RBPJk/Su(H)/Lag1 (CSL) family of transcriptional activators or repressors that participate in the transcriptional control of downstream target genes. The function of the Notch signaling pathway is specially relevant for controlling cell fate decisions during development. Notch signaling mediates neural stem cell survival, self-renewal, proliferation and cell fate specification during central nervous system development (Lathia et al., 2008
). In this way, Notch signaling maintains neural progenitor cells in a proliferative stage, inhibits neuronal differentiation and specifies radial glial cell fate (Gaiano and Fishell, 2002
). Activation of Notch-1 and Notch-3 in embryonic progenitors promotes radial glial fate and astrocyte fate postnatally (Gaiano et al., 2000
). Accordingly, PS1-deficient mice show disturbed neurogenesis and somite formation, premature differentiation of progenitor cells accompanied by severe brain morphological defects resembling Notch mutants (Handler et al., 2000
). Therefore, PS/γ-secretase-dependent Notch signaling promotes glial and astrocytic fates in the developing and adult brain (Figure 1
).
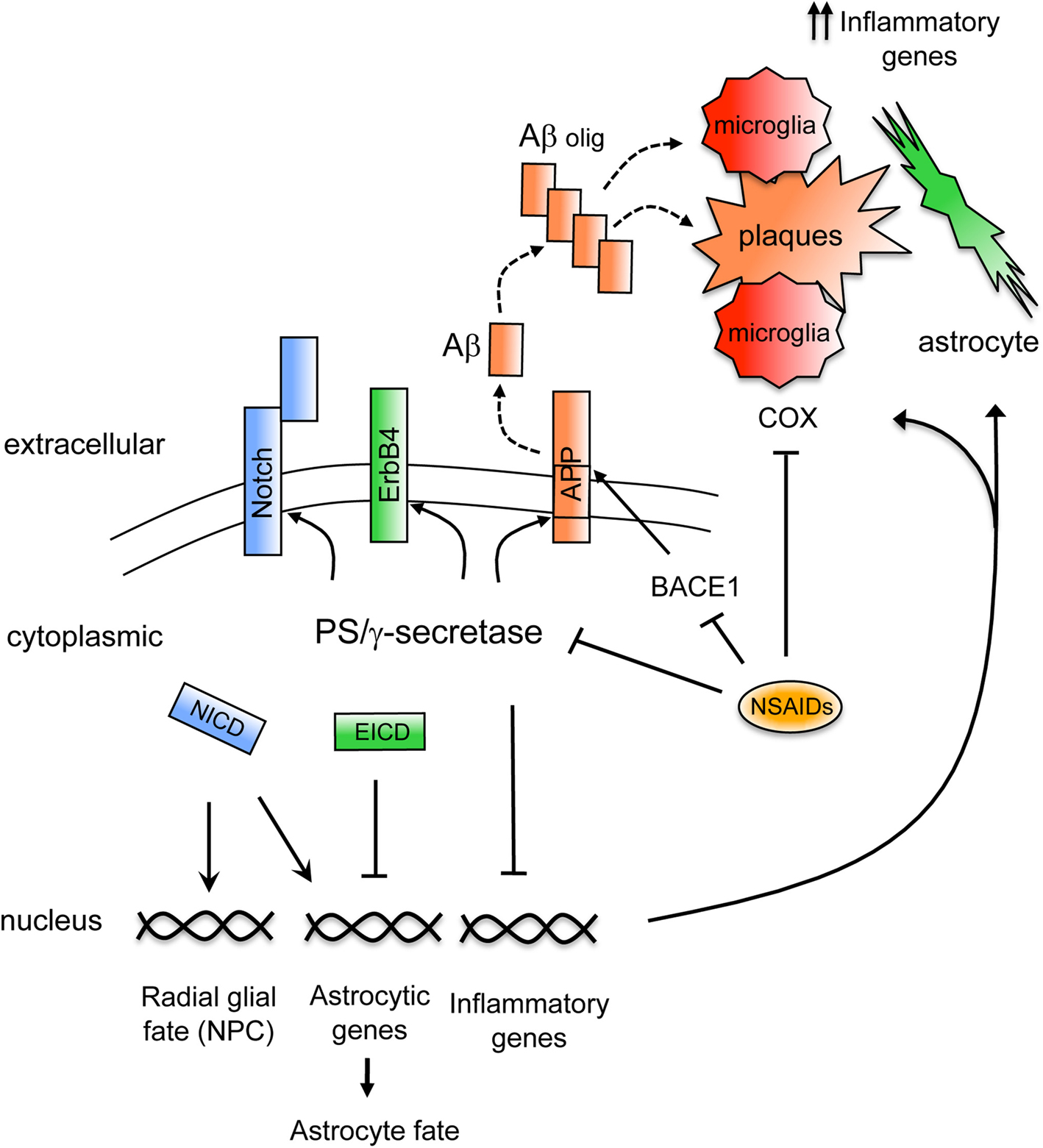
Figure 1. Mechanisms regulating brain inflammation by PS/γ secretase. Schematic model showing signaling mechanisms regulated by PS/γ-secretase on inflammation. The γ-secretase-dependent cleavage of APP generates the Aβ peptide that following oligomerization (Aβ olig) accumulates in plaques in AD brain (dashed lines). Aβ stimulates inflammatory cells that upon activation upregulate proinflammatory genes and secrete inflammatory molecules. Activated microglia and reactive astrocytes accumulate surrounding the amyloid plaques. Non-steroidal anti-inflammatory drugs (NSAIDs) reduce inflammation by acting through different molecular mechanisms including inhibition of COX enzymes in microglia or by reducing Aβ42 generation through γ-secretase or BACE1. On the other hand, the γ-secretase-dependent cleavage of Notch receptor generates the Notch intracellular domain (NICD), which promotes radial glial and astrocyte fates in neural precursor cells (NPC) by affecting target gene expression. By contrast, γ-secretase-dependent cleavage of ErbB4 generates the ErbB4 intracellular domain (EICD) that represses expression of astrocytic genes. In addition, PS downregulate the expression of inflammatory genes in glial cells by still unknown mechanisms.
By contrast, PS/γ-secretase represses the astrocytic fate of embryonic neural precursors in the developing brain by regulating the activation of the tyrosine kinase receptor ErbB4. Similarly to Notch processing, upon neuregulin binding and activation of ErbB4, γ-secretase releases the intracellular domain of ErbB4 (Ni et al., 2001
), which then binds to TAB2 and the corepressor N-CoR (Sardi et al., 2006
). This complex translocates to the nucleus and represses expression of astrocytic genes required for astrocyte fate, an observation that agrees with accelerated astrogenesis observed in ErbB4 knockout mice (Sardi et al., 2006
). Thus, PS/γ-secretase-dependent nuclear signaling and gene transcription by ErbB4 directly regulates glial fate specification (Figure 1
).
γ-Secretase plays a critical role on AD pathogenesis by mediating the generation of Aβ peptides. Aβ generation results from the extracellular cleavage of APP by BACE1 and after shedding of the soluble ectodomain (APPsβ), PS/γ-secretase cleaves the transmembrane domain of the APP C-terminal fragment to generate Aβ peptides of variable length (39–43 aa) (Selkoe, 2002
). PS/γ-secretase also cleaves APP on the ε-cleavage site to generate the APP intracellular domain (AICD), a cytosolic fragment that translocates to the nucleus, activates gene expression and regulates apoptotic cell death (Muller et al., 2008
). Presenilin is essential for generation and accumulation of Aβ, as evidenced by the lack of Aβ production in cells derived from PS1 and PS1/PS2 deficient mice (De Strooper et al., 1998
; Zhang et al., 2000
) and the absence of Aβ accumulation and amyloid plaques in the brain of APP transgenic mice lacking PS1 expression (Saura et al., 2005
) (Figure 2
).
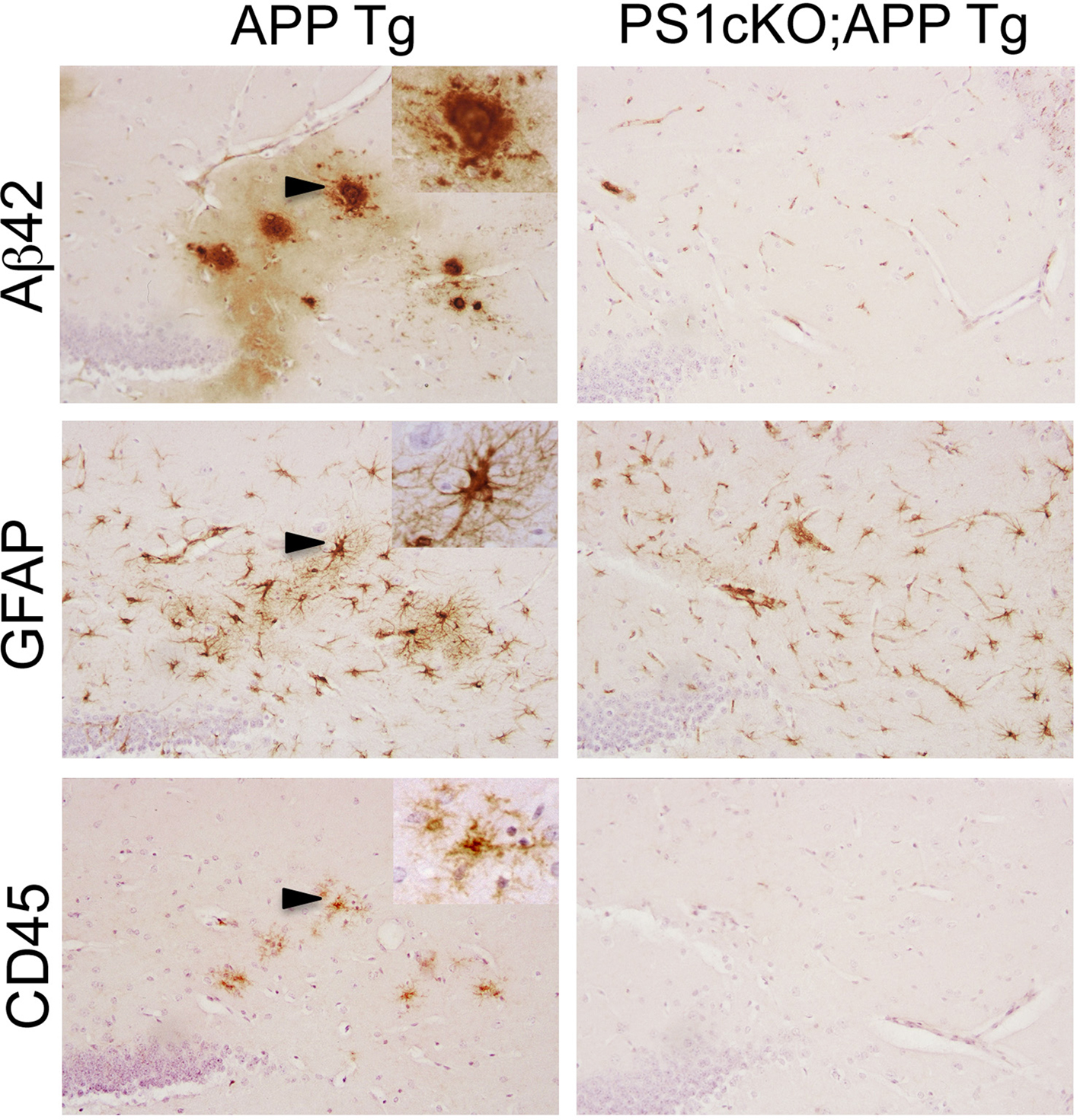
Figure 2. PS1 inactivation reduces brain inflammation in APP transgenic mice. The images show consecutive serial brain sections of 6 month-old APP (APPSw,Ind; J20) and PS1cKO;APP transgenic (Tg) mice stained with antibodies against Aβ42, GFAP and CD45. Reactive astrocytes labeled with a GFAP antibody and activated microglia stained with anti CD45 are found in the hippocampus of APP mice, but not in PS1cKO;APP mice. Activated microglia and astrocytes in APP brains are closely associated with Aβ42-containing plaques as observed in these three consecutive brain sections. Insets: Higher-power views of the lesions marked with arrowheads.
The first evidence linking PS and AD came from the discovery that autosomal dominant mutations in the PSEN1 gene were responsible for early-onset familial AD (Sherrington et al., 1995
). This was followed by the finding that inherited mutations in PSEN2 caused AD with variable penetrance and later onset (Levy-Lahad et al., 1995
; Rogaev et al., 1995
). A large number of mutations in PSEN1 (∼170) and PSEN2 (14) are currently known to cause the majority (∼30–50%) of familial AD cases1 (http://www.molgen.ua.ac.be/ADMutations
). Mutations in PS alter the cleavage of APP resulting in generation of distinct amyloidogenic Aβ peptides. Thus, several mutations in distinct domains of PS1 (EΔ9, A79V, I143T, L166P, A231V, L262F, L282V, G384A) or PS2 (N141I) decrease total Aβ or Aβ40 levels with little or unappreciable changes on the more amyloidogenic Aβ42 species (Kumar-Singh et al., 2006
; De Strooper, 2007
). Other studies have demonstrated increased Aβ42(43) peptides, and/or increased Aβ42/Aβ40 ratio in cells or transgenic mice expressing mutant PS1 or PS2 genes (Selkoe, 2002
). The concentration balance of different Aβ species seems to be important for aggregation and their toxic effects in AD brain. Notably, the age of onset of dementia in families with PS mutations correlates with increase of Aβ42/Aβ ratio and decrease Aβ40 levels (Kumar-Singh et al., 2006
). The fact that PS pathogenic mutations suppress the γ- and/or ε-secretase cleavage of several substrates including APP has lead to the hypothesis that these mutations may act through a loss of function mechanism (De Strooper, 2007
; Shen and Kelleher III, 2007
).
Mutations in the presenilin genes accelerate age of onset and cause earlier and severe progression of neurodegeneration than sporadic AD. The presence of some PS mutations results in quantitative differences in brain neuropathology compared to sporadic forms of AD (Shepherd et al., 2009
). Thus, despite similar disease duration, familial cases show similar or greater atrophy and neuronal loss, specially in the medial temporal lobes and frontal/temporal cortices, than sporadic AD cases (Gomez-Isla et al., 1999
; Gregory et al., 2006
; Shepherd et al., 2007
). Similarly, an increase of NFTs and higher NFT-associated neuritic pathology have been reported in PS-linked familial AD cases compared to sporadic AD (Gomez-Isla et al., 1999
; Sudo et al. 2005
; Woodhouse et al. 2009
). Several studies have also shown increased overall amyloid plaques, especially those containing higher deposition of Aβ42 in genetics forms of AD (Shepherd et al., 2009
). By contrast, other reports show similar amyloid plaque deposition in PSEN1 and sporadic AD cases (Nochlin et al., 1993
; Lippa et al., 1996
). The distinct effect of PS mutations on disease progression may reflect a differential effect of PS mutations on Aβ processing, tau phosphorylation and other signaling pathways (GSK3β, β-catenin, calsenilin, Notch, CREB, E/N-cadherin, etc…).
Genetic studies have identified PSEN1 mutations in families presenting clinical symptoms of frontotemporal dementia (FTD), atypical dementia with a FTD phenotype and dementia with Lewy bodies (Hutton, 2004
). For instance, the PSEN1 L424H mutation causes FTD characterized by AD neuropathology, whereas the PSEN1 V272A mutation is associated with subcortical dementia with Lewy body pathology (Zekanowski et al., 2006
; Gomez-Tortosa et al., 2010
). Several mutations in PSEN1 including L113P, G183V, L226F and V412I have been found in early-onset familial FTD (Raux et al., 2000
; Dermaut et al., 2004
; Bernardi et al., 2009
). These PSEN1 mutations cause severe neurodegeneration and dementia associated with accumulation of phosphorylated tau in the absence of Aβ pathology in the frontal cortex. Thus, the G183V mutation increases the levels of phosphorylated tau without affecting Aβ2 levels and deposition (Dermaut et al., 2004
). Consistent with a role of PS on tau phosphorylation, overexpression of mutant PS1 in mice results in activation of GSK-3β and enhanced phosphorylation of tau (Dewachter et al., 2008
). Interestingly, inactivation of both PS1 and PS2 in the adult mouse brain leads to increased phosphorylation of tau and age-dependent memory deficits and neurodegeneration (Saura et al., 2004
). This raises the possibility that some PS pathogenic mutations may cause memory deficits and neurodegeneration by affecting tau phosphorylation and/or aggregation.
Abundant inflammation correlates with the initial cognitive decline and severity of dementia in AD (Parachikova et al., 2007
). Reactive glial cells, especially activated microglia and astrocytes, and increased production of inflammatory molecules are associated to neuritic plaques (Holmes et al., 2009
). Activation of microglia is initiated by local binding of fibrillar Aβ to C1q receptors and a receptor complex formed by the β-class scavenger receptor CD36, the integrin-associated protein/CD47, and the α(6)β(1)-integrin (Eikelenboom and Veerhuis, 1996
; Bamberger et al., 2003
). The subsequent activation of intracellular signaling pathways in microglia leads to the proinflammatory response. Microglia activation induces secretion of cytokines and other neuroinflammatory molecules that promote stimulation and activation of astrocytes around the plaques. Once activated, microglia and astrocytes contribute actively to removal of Aβ from the brain parenchyma (Wyss-Coray et al., 2001
, 2003
), as revealed by accumulation of Aβ42 peptides in astrocytes in AD brain (Nagele et al., 2003
).
An increase of neuroinflammatory mediators resulting from enhanced amyloid pathology in familial AD or as a direct consequence of PS mutations may have detrimental consequences for neuronal function. Monocyte chemoattractant protein-1 (MCP-1), interleukin-6 (IL-6) and interleukin-8 (IL-8) are increased by familial AD-linked PS mutations (Sokolova et al., 2009
), and mutant PS1 enhances expression of microglial factors that promote differentiation of neural precursor cells into glial fibrillary acidic protein (GFAP)-positive cells (Choi et al., 2008
). Production of inflammatory cytokines like IL-1β, IL-6 and TNFα by activated microglia, but also by reactive astrocytes, negatively impact on neuronal function and behavior. For instance, secretion of toxic neuroinflammatory mediators such as reactive oxygen species (ROS), cytokines and chemokines directly contributes to neurite retraction, neuronal dysfunction and neuronal death in AD (Munch et al., 2003
; Sokolova et al., 2009
). Nonetheless, inflammatory cytokines have been suggested to be the primary contributors to synaptic plasticity deficits and behavioral changes during normal aging and age-dependent neurological diseases (Lynch, 2010
). Recent findings demonstrate that overexpression of IL-1β in the hippocampus is accompanied by microglial activation and increased expression of chemokines MCP-1 and MIP-1, which negatively affect hippocampal-dependent memory (Moore et al., 2009
). The negative impact of chronic inflammation on synaptic function during neurodegeneration has been corroborated in several studies in aged and AD transgenic mice (McGeer and McGeer, 2007
).
Besides the classic neuritic plaques, cotton wool and ‘inflammatory’ plaques are specific pathological hallmarks commonly observed in familial AD caused by PS mutations. Cotton wool plaques, which are composed mainly of Aβ42, are usually devoid of a compact amyloid core, neuritic changes and glial cell components (Mann et al., 2001
). Cotton wool plaques are mainly detected in neocortex, limbic system and striatum in AD cases caused by PSEN1 mutations in exons 4, 5, 6, 8, 9 and 12 and intron 8 (Mann et al., 2001
; Dumanchin et al., 2006
), whereas they are very rare in late-onset AD (Le et al., 2001
). By contrast, ‘inflammatory plaques’ are small dense plaques with a central core devoid of Aβ, phosphorylated tau, ApoE and α-synuclein. ‘Inflammatory plaques’ display a strong inflammatory response in the form of numerous activated microglia and astrocytes in the cortex of familial AD cases linked to PSEN1 and APP mutations (Shepherd et al., 2005
). The increase of inflammatory cells surrounding these plaques may contribute significantly to greater neuronal dysfunction and neurodegeneration in some genetic AD cases linked to PS mutations (Shepherd et al., 2009
). The variability on clinical symptoms and brain pathology in subjects carrying the same PS1 mutation (Martikainen et al., 2009
), suggests that other genetic, cellular or environmental factors may alter the pathological phenotypes in genetically predetermined familial AD cases.
Epidemiological studies indicate that head trauma is an important risk factor for developing AD. Levels of PS1 and nicastrin increase significantly in activated astrocytes and microglia following brain damage by head injury, brain stabbing and inflammatory insult (Nadler et al., 2008
). Indeed, chronic cortical gliosis and inflammatory mediators stimulate APP metabolism and elevate Aβ peptides by increasing expression of APP and PS1, which in turn affects glial activation and neuronal function (Bates et al., 2002
). Increased expression of mutant PS1 in astrocytes has been shown to contribute to astrocyte reactivity in response to ATP and glutamate through a mechanism involving increased Ca2+ oscillations (Johnston et al., 2006
). This result indicates that PS1 mutations enhance intracellular Ca2+ mobilization in astrocytes contributing to the accelerated pathogenesis in familial AD. Similarly, PS1 expression increases in astrocytes under hypoxia conditions contributing to enhancement of intracellular calcium, whereas inhibition of PS1 reverses the effects of hypoxia on calcium signaling (Peers et al., 2004
). Interestingly, hypoxia increases Aβ levels by affecting APP processing and/or Aβ clearance. The role of PS on brain hypoxia and its association to dementia is particularly intriguing since hypoxia, which can arise from cardiorespiratory diseases, predispose individuals to the development of dementias and especially to AD (Peers et al., 2009
).
Brain ischemia is known to contribute to AD dementia, whereas individuals with severe cognitive decline and diagnosis of AD are at increased risk for ischemic events in the brain. PS/γ-secretase seems to mediate the effect of Notch on ischemia events. In a mouse model of ischemic stroke, Notch increases the vulnerability of cells to undergo apoptosis, activates microglial cells and stimulates infiltration of proinflammatory leukocytes (Arumugam et al., 2006
). By contrast, γ-secretase inhibitors and expression of a Notch1 antisense reduce brain damage and improve neural function in that experimental model of ischemic stroke (Arumugam et al., 2006
). Given that PS/γ-secretase-mediated Notch signaling worsens brain damage during ischemic stroke, it has been suggested that agents that target Notch function may be useful for therapeutic interventions in some brain disorders (Lathia et al., 2008
). These findings indicate that PS may contribute to brain damage and/or repair by regulating glial activation and function.
Enhanced cell death and calcium signaling defects in peripheral lymphocytes have been demonstrated in AD patients, suggesting that immune dysfunction can contribute to the pathogenesis of AD. Defects on T-cell activation by a PS1 mutation that causes familial AD has been reported in mutant PS knockin mice (Morgan et al., 2007
). Lymphocytes from PS1 mutant transgenic mice show higher susceptibility to cell death as do peripheral cells from AD patients, which is likely caused by increased production of ROS and altered calcium levels (Eckert et al., 2001
). Notably, lack of presenilins in the brain leads to robust inflammatory responses both in the brain and periphery. Beginning at 6 months of age, and coinciding with the neurodegenerative process, leukocytes and inflammatory mediators are elevated in serum of mutant PS1/PS2 conditional double knockout (PS cDKO) mice (Jiang et al., 2009
). This finding indicates that inflammatory responses in the brain rapidly expand to systemic tissue under pathological conditions caused by loss of PS function in the brain.
Notch signaling plays a critical role on lymphocyte development by specifying T cell fate and differentiation. Notch signaling regulates CD4/CD8 T cell lineage commitment, whereas PS/γ-secretase-dependent Notch signaling promotes the selection and maturation of CD4 and CD8 T cells in immature thymocytes (Laky and Fowlkes, 2008
). A role of PS/γ-secretase on T cell fate is evident in PS-deficient mice, in which lack of presenilin expression in thymocyte precursors inefficiently generate CD4+ T cells by impairing TCR signaling (Laky and Fowlkes, 2007
). This has lead to the hypothesis that PS-dependent Notch signaling positively influences the selection and development of T cells by modifying TCR signaling. Similarly, inactivation of both PS in thymic cells results in a significant decrease of CD8+ T cells in the periphery (Maraver et al., 2007
). However, whereas PS facilitate CD4+ T cell proliferation and cytokine secretion, they are not required for cytokine-induced Th1/Th2 fate selection (Ong et al., 2008
). A direct role for PS proteins on B cell function has been also reported. Loss of PS function in B cells results in substantial deficit in both lipopolysaccharide and B cell antigen receptor-induced proliferation and signaling events, including a defect in anti-IgM-mediated calcium flux (Yagi et al., 2008
).
A mechanism that could mediate presenilin-dependent regulation of lymphocyte activation is CD46, a signaling receptor with complement and T cell regulatory functions in human. In response to infectious pathogens such as Neisseria gonorrhoeae and Neisseria meningitidis, processing of CD46 by PS/γ-secretase stimulates CD46-dependent T cell responses (Weyand et al., 2010
). Therefore, PS/γ-secretase seems to be particularly important in response to pathogens by mediating the cleavage of CD46 and regulating signaling pathways in immune cells. Interestingly, PS1+/−; PS2−/− mice develop an autoimmune disease characterized by dermatitis, glomerulonephritis, keratitis and vasculitis. At advancing ages, these mutant mice develop skin hyperplasia, increased CD4+/CD8+ T cell ratio and B-cell infiltrates in several tissues (Tournoy et al., 2004
). Together, compelling evidence suggests that PS/γ-secretase regulates maturation of T cells, whereas its inactivation in the periphery has functional detrimental consequences for immune system.
A number of recent studies using PS loss-of-function mutant mice has added a new view on the role of PS on neuroinflammation. Consistent with the idea that PS1/γ-secretase plays an essential role on AD pathogenesis, PS1 inactivation reduces significantly age-dependent Aβ accumulation and amyloid plaque deposition in APP transgenic mice (Dewachter et al., 2002
; Saura et al., 2005
). Interestingly, conditional deletion of PS1 in the adult forebrain of APP transgenic mice reduces dramatically amyloid-associated microgliosis and astrocytosis (Figure 2
), which is accompanied by reversal of hippocampal-dependent spatial and associative memory deficits (Saura et al., 2005
). Whereas these results suggest that PS1/γ-secretase is required for Aβ-induced neuroinflammatory responses, they also indicate that targeting PS1/γ-secretase may have therapeutic benefits in AD.
In contrast to the protective role of partial PS/γ-secretase inactivation on amyloid pathology and inflammation, genetic ablation of both PS causes progressive inflammatory responses in the cerebral cortex of PS cDKO mice. The early widespread inflammatory response observed in PS cDKO mice is independent of Aβ accumulation and appears before synapse loss and tau hyperphosphorylation (Beglopoulos et al., 2004
). Inflammatory changes are the first pathological hallmarks observed in PS cDKO mice and these are followed by synaptic and memory impairments and progressive neurodegeneration (Saura et al., 2004
). By 3 months of age, and long before activated inflammatory cells and neurodegeneration are apparent, several proinflammatory genes including complement component C1qα-β, cathepsins D and S, small inducible cytokine A27, MHC histocompatibility proteins and CD antigens are up-regulated in the cortex and hippocampus of PS cDKO mice (Dong et al., 2007
). Abundant reactive inflammatory cells, including activated microglia and reactive astrocytes, are observed in the hippocampus and neocortex of PS cDKO mutant mice by 6 months of age (Beglopoulos et al., 2004
; Saura et al., 2004
). Gene expression profile analyses in PS cDKO mice at that age have revealed an upregulation of multiple inflammatory genes including complement component C1qα-β, MHC histocompatibility proteins, cathepsins D, H, S and Z, chemokines, CD antigens and GFAP (Beglopoulos et al., 2004
; Dong et al., 2007
). Interestingly, the complement system, which targets toxic substances in the immune system, is directly involved in the inflammatory responses in AD and Down syndrome patients (Tenner, 2001
). Cathepsin S, a lysosomal cysteine protease that is secreted by activated microglia and macrophages and mediates major histocompatibility class II-dependent immunity, is increased in AD and Down syndrome brains (Lemere et al., 1995
). On the other hand, increased expression of GFAP, a marker of astrogliosis, correlates with progression of dementia and synaptic and tau pathologies in AD brain (Ingelsson et al., 2004
).
Interestingly, cognitive stimulation by long-term environmental enrichment improved memory and reduced brain degeneration and astrogliosis in PS cDKO mice. Environmental enrichment significantly reduced 34 genes related to immunity and inflammation that were upregulated in the hippocampus of PS cDKO mice (Dong et al., 2007
). These findings suggest that the therapeutic benefits of cognitive stimulation may be in part related to inhibition of expression of inflammatory genes. Together, the above evidence points to a role of PS on regulating directly or indirectly the expression of proinflammatory genes. Although the specific mechanisms by which PS repress expression of inflammatory genes are still unknown, it is tempting to speculate that PS/γ-secretase-dependent mechanisms, such as that involving repression of astrocytic genes by the ErbB4 intracellular domain (Sardi et al., 2006
), might be involved on transcriptional regulation of inflammatory genes during neurodegeneration (Figure 1
).
Epidemiological studies indicate that chronic treatment with NSAIDs delays the onset and reduces the risk of AD (Stewart et al., 1997
; McGeer and McGeer, 2007
). Prospective population-based cohort studies revealed that NSAID use for more than 2 years decreases by 60–80% the risk of developing AD but not vascular dementia (Stewart et al., 1997
; in t’ Veld et al., 2001
). Regular use of specific NSAIDs for 1 year or more than 5 years decreases the risk of AD significantly from 2% to 34%, respectively (Vlad et al., 2008
). For instance, ibuprofen and indomethacin, two NSAIDs that decrease Aβ42, showed a strong protective effect that increased with duration of use. By contrast, celecoxib and salicylates, two NSAIDs that do not affect Aβ42, did not show any protective effects (Vlad et al., 2008
). Randomized clinical studies with indomethacin for 6 months improved cognitive function in mild/moderate AD patients, whereas naproxen, rofecoxib, diclofenac or nimesulide revealed no cognitive benefits (McGeer and McGeer, 2007
). R-flurbiprofen, an enantiomer of flurbiprofen that selectively lower Aβ42 and lacks COX inhibitory activity, did not slow cognitive decline or improved activities of daily living in phase III trials (Green et al., 2009
). These differential effects of NSAIDs on AD may be caused by distinct activity of NSAIDs on COX-dependent or -independent mechanisms.
The molecular mechanisms by which NSAIDs reduce inflammation and ameliorate clinical symptoms in AD are largely unclear. The classical mechanism of anti-inflammatory action of NSAIDs involves reduction of prostaglandin synthesis through inhibition of COX enzymes (COX-1 and COX-2). NSAIDs have been also shown to activate the nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ) and inhibit expression of proinflammatory genes. Alternatively, NSAIDs inhibit Aβ42 generation by repressing BACE1 expression through PPARγ and by regulating the presenilin/γ-secretase-dependent cleavage of APP (Weggen et al., 2007
). In support of a role of some NSAIDs on protecting from Aβ pathology through PPARγ, it has been shown that NSAIDs repress BACE expression and negatively modulate Aβ generation (Sastre et al., 2006
), whereas acute treatments with ibuprofen or the PPARγ agonist pioglitazone reduce BACE1 expression and glial inflammation in APP transgenic mice (Heneka et al., 2005
). On the other hand, some NSAIDs affect Aβ levels by modulating γ-secretase activity, the non amyloidogenic α-secretase pathway and/or Aβ aggregation (Weggen et al., 2007
). For instance, NSAIDs bind to and inhibit formation of Aβ fibrils in vitro (Agdeppa et al., 2003
; Hirohata et al., 2005
), a mechanism that may explain the beneficial effects of chronic treatment of ibuprofen or R-flurbiprofen on reducing amyloid deposits in very old APP transgenic mice (Lim et al., 2000
; Kukar et al., 2007
). A subset of NSAIDs induce a shift on the γ-secretase proteolytic cleavage of APP causing a specific decrease on the production of Aβ42 while increasing shorter Aβ species (i.e. Aβ38) and without affecting Aβ40 (Weggen et al., 2001
). Indeed, the generation of Aβ42 and Aβ38 by γ-secretase is independent from each other and NSAID differentially affect the levels of these peptides. Thus, PS mutants that are insensitive to sulindac sulfide show a robust increase of Aβ38 (Page et al., 2008
). Ibuprofen, sulindac sulfide and indomethacin, selectively decrease Aβ42 peptides independently of COX inhibition, whereas R-flurbiprofen, which lacks COX inhibitory activity, decreases Aβ42 levels in cell culture and APP transgenic models (Weggen et al., 2001
). In addition, cell-based screenings identified a number of COX-2 selective NSAIDs that selectively raised Aβ42, and some of these also decreased Aβ38 (Kukar et al., 2005
). These Aβ42-raising NSAIDs seem to act independently of RhoA activity by modulating directly γ-secretase activity.
The mechanism of action of this subgroup of NSAIDs on γ secretase activity is independent of COX inhibition and most likely involves allosteric modulation of the γ-secretase complex and/or binding to APP. The fact that these compounds are effective in lowering or raising Aβ42 in vitro in cell-free assays supports the notion that they act directly on γ-secretase (Eriksen et al., 2003
; Kukar et al., 2005
). In support of this view, Aβ42-lowering NSAIDs change the conformation of presenilin-1 and alter the interaction of APP and presenilin/γ-secretase (Lleo et al., 2004
). This may explain why some Aβ42-lowering NSAIDs reduce Aβ42 generation without affecting the ε-cleavage of APP, Notch and ErbB4. Indeed, the selectivity of NSAID for targeting the γ-secretase cleavage of APP instead of other substrates may be explained by the specific binding of these compounds to APP (Kukar et al., 2008
). Several familial AD-linked PS mutations attenuate the inhibitory effect of NSAIDs on Aβ42 production. The insensitivity of PS mutants to Aβ42-lowering NSAIDs is mimicked by non-related structural γ-secretase inhibitors suggesting that the binding site of these compounds are located in close proximity within the γ-secretase complex (Czirr et al., 2007
).
Several studies have shown that long-term treatment with specific NSAIDs reduces Aβ pathology and inflammatory responses in APP transgenic mice. Ibuprofen markedly reduced soluble Aβ42, amyloid plaques and microglia activation in APP Tg2576 mice by a mechanism independent of PPARγ (Yan et al., 2003
). By contrast, acute treatment of old APPV717I transgenic mice with ibuprofen or the PPARγ agonist pioglitazone reduced glial inflammation and amyloid plaques by decreasing BACE1 expression (Heneka et al., 2005
). Other studies using NSAIDs revealed similar reductions in insoluble Aβ and inflammation, or unchanged Aβ40/42 ratio but decreased Aβ plaques in APP transgenic mice (Lim et al., 2000
; Kukar et al., 2007
; McGeer and McGeer, 2007
). A recent study, however, revealed no advantage of Aβ42-lowering NSAIDS for preventing from AD compared to classical NSAIDs (Szekely et al., 2008
). Interestingly, therapeutic benefits have been reported with triflusal, an anti-inflammatory drug that inhibits COX-1 and delays the conversion of amnestic mild cognitive impairment to AD dementia. Triflusal reduced proinflammatory cytokine levels (IL-1β and TNFα), dense-core plaques and neuritic dystrophy in APP Tg2576 transgenic mice (Coma et al., 2010
). In old APP Tg2576 mice, triflusal efficiently rescued spatial and associative memory deficits and expression of genes activated by CREB, a signaling pathway underlying the molecular basis of synaptic plasticity (Coma et al., 2010
). Despite the mechanism of action and the controversy in human trials, NSAIDs effectively reduces amyloid pathology and inflammation in AD transgenic mice. It is conceivable that the protective effect of NSAIDs on AD pathology may be mediated by multiple mechanisms including among others COX, PPARγ, γ-secretase or a combination of these pathways.
A large number of studies over the past years have revealed that PS regulate multiple signaling pathways in neurons and glia. For instance, the γ-secretase-dependent cleavage of APP generates the Aβ peptide, which plays a central role on AD pathogenesis by disrupting synaptic and metabolic processes essential for neuron survival. Several pathological hallmarks, including inflammation, are exacerbated by presenilin mutations that cause early-onset familial AD or FTD. These studies strongly suggest γ-secretase as potential therapeutic target to reduce Aβ-induced pathology and clinical symptoms in AD. This therapeutic approach was successful in mice, in which partial inactivation of PS efficiently reduced age-dependent Aβ accumulation and temporally rescued memory deficits in an AD mouse model (Dewachter et al., 2002
; Saura et al., 2005
). However, clinical treatment with the γ-secretase inhibitor LY450139 in AD patients, although was well tolerated and reduced plasma Aβ40 levels, did not result in significant differences in cognitive or functional measures. Similarly, NSAIDs that reduce Aβ-associated pathology and inflammation in AD transgenic mice revealed no advantage for preventing from AD compared to classical NSAIDs (Szekely et al., 2008
). Notably, targeting the Notch pathway by using γ-secretase inhibitors has been suggested as therapeutic approach in cancer disorders caused by aberrant Notch signaling such as T-cell acute lymphoblastic leukemia, breast cancer and ovarian carcinomas (Shih and Wang, 2007
). However, pharmacological inhibition of PS/γ-secretase during AD therapeutics may have detrimental effects not only for systemic immune function but also for epidermal and skin barrier homeostasis (Rocher-Ros et al., 2010
). Before clinical treatments, future studies should focus primarily on dissecting target specificity of γ-secretase modulators. Nonetheless, inactivation of PS function in the forebrain results in synaptic and cognitive dysfunction followed by neurodegeneration (Saura et al., 2004
). PS inactivation in brain also causes inflammatory responses in brain and periphery organs. In addition, compelling evidence indicate that PS/γ secretase regulates maturation of T cells by regulating the Notch signaling pathway, whereas its inactivation causes side effects on immune function. Taking into consideration that PS/γ-secretase plays an essential function on multiple signaling pathways, a better understanding of the molecular mechanisms regulated by PS in inflammation may lead to the development of novel therapeutic strategies for disorders of the nervous and immune systems.
All experiments shown in this study were performed in accordance with institutional and national guidelines following approval by the Animal Care and Ethical Committee of the Universitat Autònoma de Barcelona.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from Ministerio de Ciencia e Innovación of Spain (SAF2007-64115 and CIBERNED CB06/05/0042), Fundació Marató-TV3 (050710) and 7th Framework Programme of the European Union (MEMOSAD project, grant 200611).
Agdeppa, E. D., Kepe, V., Petri, A., Satyamurthy, N., Liu, J., Huang, S. C., Small, G. W., Cole, G. M., and Barrio, J. R. (2003). In vitro detection of (S)-naproxen and ibuprofen binding to plaques in the Alzheimer’s brain using the positron emission tomography molecular imaging probe 2-(1-[6-[(2-[(18)F]fluoroethyl)(methyl)amino]-2-naphthyl]ethylidene) malono nitrile. Neuroscience 117, 723–730.
Arumugam, T. V., Chan, S. L., Jo, D. G., Yilmaz, G., Tang, S. C., Cheng, A., Gleichmann, M., Okun, E., Dixit, V. D., Chigurupati, S., Mughal, M. R., Ouyang, X., Miele, L., Magnus, T., Poosala, S., Granger, D. N., and Mattson, M. P. (2006). Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat. Med. 12, 621–623.
Bernardi, L., Tomaino, C., Anfossi, M., Gallo, M., Geracitano, S., Costanzo, A., Colao, R., Puccio, G., Frangipane, F., Curcio, S. A., Mirabelli, M., Smirne, N., Iapaolo, D., Maletta, R. G., and Bruni, A. C. (2009). Novel PSEN1 and PGRN mutations in early-onset familial frontotemporal dementia. Neurobiol. Aging 30, 1825–1833.
Cervantes, S., Saura, C. A., Pomares, E., Gonzalez-Duarte, R., and Marfany, G. (2004). Functional implications of the presenilin dimerization: reconstitution of γ-secretase activity by assembly of a catalytic site at the dimer interface of two catalytically inactive presenilins. J. Biol. Chem. 279, 36519–36529.
Coma, M., Sereno, L., Da Rocha-Souto, B., Scotton, T. C., España, J., Sánchez, M. B., Rodríguez, M., Agulló, J., Guardia-Laguarta, C., Garcia-Alloza, M., Borreli, L. A., Clarimón, J., Lleó, A., Bacskai, B. J., Saura, C. A., Hyman, B., and Gómez-Isla, T. (2010). Triflusal reduces dense-core plaque load, associated axonal alterations and inflammatory changes, and rescues cognition in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. (in press). [Epub ahead of print].
Czirr, E., Leuchtenberger, S., Dorner-Ciossek, C., Schneider, A., Jucker, M., Koo, E. H., Pietrzik, C. U., Baumann, K., and Weggen, S. (2007). Insensitivity to Aβ42-lowering nonsteroidal anti-inflammatory drugs and γ-secretase inhibitors is common among aggressive presenilin-1 mutations. J. Biol. Chem. 282, 24504–24513.
Dermaut, B., Kumar-Singh, S., Engelborghs, S., Theuns, J., Rademakers, R., Saerens, J., Pickut, B. A., Peeters, K., Van Den Broeck, M., Vennekens, K., Claes, S., Cruts, M., Cras, P., Martin, J. J., Van Broeckhoven, C., and De Deyn, P. P. (2004). A novel presenilin 1 mutation associated with Pick’s disease but not β-amyloid plaques. Ann. Neurol. 55, 617–626.
Dewachter, I., Reverse, D., Caluwaerts, N., Ris, L., Kuiperi, C., Van den Haute, C., Spittaels, K., Umans, L., Serneels, L., Thiry, E., Moechars, D., Mercken, M., Godaux, E., and Van Leuven, F. (2002). Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J. Neurosci. 22, 3445–3453.
Dumanchin, C., Tournier, I., Martin, C., Didic, M., Belliard, S., Carlander, B., Rouhart, F., Duyckaerts, C., Pellissier, J. F., Latouche, J. B., Hannequin, D., Frebourg, T., Tosi, M., and Campion, D. (2006). Biological effects of four PSEN1 gene mutations causing Alzheimer disease with spastic paraparesis and cotton wool plaques. Hum. Mutat. 27, 1063.
Gomez-Isla, T., Growdon, W. B., McNamara, M. J., Nochlin, D., Bird, T. D., Arango, J. C., Lopera, F., Kosik, K. S., Lantos, P. L., Cairns, N. J., and Hyman, B. T. (1999). The impact of different presenilin 1 and presenilin 2 mutations on amyloid deposition, neurofibrillary changes and neuronal loss in the familial Alzheimer’s disease brain: evidence for other phenotype-modifying factors. Brain 122(Pt 9), 1709–1719.
Gomez-Tortosa, E., Barquero, S., Baron, M., Gil-Neciga, E., Castellanos, F., Zurdo, M., Manzano, S., Munoz, D. G., Jimenez-Huete, A., Rabano, A., Sainz, M. J., Guerrero, R., Gobernado, I., Perez-Perez, J., and Jimenez-Escrig, A. (2010). Clinical-genetic correlations in familial Alzheimer’s disease caused by presenilin 1 mutations. J. Alzheimers Dis. 19, 873–884.
Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Hanke, A., Dewachter, I., Kuiperi, C., O’Banion, K., Klockgether, T., Van Leuven, F., and Landreth, G. E. (2005). Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1-42 levels in APPV717I transgenic mice. Brain 128, 1442–1453.
Kukar, T., Murphy, M. P., Eriksen, J. L., Sagi, S. A., Weggen, S., Smith, T. E., Ladd, T., Khan, M. A., Kache, R., Beard, J., Dodson, M., Merit, S., Ozols, V. V., Anastasiadis, P. Z., Das, P., Fauq, A., Koo, E. H., and Golde, T. E. (2005). Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Aβ42 production. Nat. Med. 11, 545–550.
Kukar, T. L., Ladd, T. B., Bann, M. A., Fraering, P. C., Narlawar, R., Maharvi, G. M., Healy, B., Chapman, R., Welzel, A. T., Price, R. W., Moore, B., Rangachari, V., Cusack, B., Eriksen, J., Jansen-West, K., Verbeeck, C., Yager, D., Eckman, C., Ye, W., Sagi, S., Cottrell, B. A., Torpey, J., Rosenberry, T. L., Fauq, A., Wolfe, M. S., Schmidt, B., Walsh, D. M., Koo, E. H., and Golde, T. E. (2008). Substrate-targeting γ-secretase modulators. Nature 453, 925–929.
Kumar-Singh, S., Theuns, J., Van Broeck, B., Pirici, D., Vennekens, K., Corsmit, E., Cruts, M., Dermaut, B., Wang, R., and Van Broeckhoven, C. (2006). Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Aβ42 and decreased Aβ40. Hum. Mutat. 27, 686–695.
Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J., Pettingell, H., Yu, C., Jondro, P. D., Schmidt, S. D., Wang, K., Crowley, A. C., Fu, Y-H., Guenette, S. Y., Galas, D., Nemens, E., Wijsman, E. M., Bird, T. D., Schellenberg, G. D., and Tanzi, R. E. (1995). Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269, 973–977.
Lippa, C. F., Saunders, A. M., Smith, T. W., Swearer, J. M., Drachman, D. A., Ghetti, B., Nee, L., Pulaski-Salo, D., Dickson, D., Robitaille, Y., Bergeron, C., Crain, B., Benson, M. D., Farlow, M., Hyman, B. T., George-Hyslop, S. P., Roses, A. D., and Pollen, D. A. (1996). Familial and sporadic Alzheimer’s disease: neuropathology cannot exclude a final common pathway. Neurology 46, 406–412.
Mann, D. M., Takeuchi, A., Sato, S., Cairns, N. J., Lantos, P. L., Rossor, M. N., Haltia, M., Kalimo, H., and Iwatsubo, T. (2001). Cases of Alzheimer’s disease due to deletion of exon 9 of the presenilin-1 gene show an unusual but characteristic β-amyloid pathology known as ‘cotton wool’ plaques. Neuropathol. Appl. Neurobiol. 27, 189–196.
Munch, G., Gasic-Milenkovic, J., Dukic-Stefanovic, S., Kuhla, B., Heinrich, K., Riederer, P., Huttunen, H. J., Founds, H., and Sajithlal, G. (2003). Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Exp. Brain. Res. 150, 1–8.
Page, R. M., Baumann, K., Tomioka, M., Perez-Revuelta, B. I., Fukumori, A., Jacobsen, H., Flohr, A., Luebbers, T., Ozmen, L., Steiner, H., and Haass, C. (2008). Generation of Aβ38 and Aβ42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and γ-secretase modulation. J. Biol. Chem. 283, 677–683.
Rogaev, E. I., Sherrington, R., Rogaeva, E. A., Levesque, G., Ikeda, M., Liang, Y., Chi, H., Lin, C., Holamn, K., Tsuda, T., Mar, L., Sorbi, S., Nacmias, B., Piacentini, S., Amaducci, L., Chumakov, I., Cohen, D., Lannfelt, L., Fraser, P. E., Rommens, J. M., and St. George-Hyslop, P. H. (1995). Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376, 775–778.
Sastre, M., Dewachter, I., Rossner, S., Bogdanovic, N., Rosen, E., Borghgraef, P., Evert, B. O., Dumitrescu-Ozimek, L., Thal, D. R., Landreth, G., Walter, J., Klockgether, T., van Leuven, F., and Heneka, M. T. (2006). Nonsteroidal anti-inflammatory drugs repress β-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. U.S.A. 103, 443–448.
Saura, C. A., Chen, G., Malkani, S., Choi, S. Y., Takahashi, R. H., Zhang, D., Gouras, G. K., Kirkwood, A., Morris, R. G., and Shen, J. (2005). Conditional inactivation of presenilin-1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J. Neurosci. 25, 6755–6764.
Saura, C. A., Choi, S. Y., Beglopoulos, V., Malkani, S., Zhang, D., Shankaranarayana Rao, B. S., Chattarji, S., Kelleher, R. J. III, Kandel, E. R., Duff, K., Kirkwood, A., and Shen, J. (2004). Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42, 23–36.
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., Tsuda, T., Mar, L., Foncin, J-F., Bruni, A. C., Montesi, M. P., Sorbi, S., Rainero, I., Pinessi, L., Nee, L., Chumakov, I., Pollen, D. A., Roses, A. D., Fraser, P. E., Rommens, J. M., and St. George-Hyslop, P. H. (1995). Cloning of a novel gene bearing missense mutations in early onset familial Alzheimer’s disease. Nature 375, 754–760.
Szekely, C. A., Green, R. C., Breitner, J. C., Ostbye, T., Beiser, A. S., Corrada, M. M., Dodge, H. H., Ganguli, M., Kawas, C. H., Kuller, L. H., Psaty, B. M., Resnick, S. M., Wolf, P. A., Zonderman, A. B., Welsh-Bohmer, K. A., and Zandi, P. P. (2008). No advantage of Aβ42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology 70, 2291–2298.
Thinakaran, G., Borchelt, D. R., Lee, M. K., Slunt, H. H., Spitzer, L., Kim, G., Rotovitsky, T., Davenport, F., Nordstedt, C., Seeger, M., Hardy, J., Levey, A. I., Gandy, S. E., Jenkins, N. A., Copeland, N. G., Price, D. L., and Sisodia, S. S. (1996). Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190.
Weggen, S., Eriksen, J. L., Das, P., Sagi, S. A., Wang, R., Pietrzik, C. U., Findlay, K. A., Smith, T. E., Murphy, M. P., Bulter, T., Kang, D. E., Marquez-Sterling, N., Golde, T. E., and Koo, E. H. (2001). A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216.
Zekanowski, C., Golan, M. P., Krzysko, K. A., Lipczynska-Lojkowska, W., Filipek, S., Kowalska, A., Rossa, G., Peplonska, B., Styczynska, M., Maruszak, A., Religa, D., Wender, M., Kulczycki, J., Barcikowska, M., and Kuznicki, J. (2006). Two novel presenilin 1 gene mutations connected with frontotemporal dementia-like clinical phenotype: genetic and bioinformatic assessment. Exp. Neurol. 200, 82–88.