Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment?
- 1 Research and Development Department, Chiesi Farmaceutici, Parma, Italy
- 2 Department of Geriatrics, Center for Aging Brain, Memory Unit, University of Bari, Bari, Italy
- 3 Department of Medical Sciences, Geriatric Unit and Gerontology-Geriatrics Research Laboratory, IRCCS “Casa Sollievo della Sofferenza”, San Giovanni Rotondo, Foggia, Italy
Several epidemiological studies suggest that long-term use of non-steroidal anti-inflammatory drugs (NSAIDs) may protect subjects carrying one or more ε4 allele of the apolipoprotein E (APOE ε4) against the onset of Alzheimer’s disease (AD). The biological mechanism of this protection is not completely understood and may involve the anti-inflammatory properties of NSAIDs or their ability of interfering with the β-amyloid (Aβ) cascade. Unfortunately, long-term, placebo-controlled clinical trials with both non-selective and cyclooxygenase-2 (COX-2) selective inhibitors in mild-to-moderate AD patients produced negative results. A secondary prevention study with rofecoxib, a COX-2 selective inhibitor, in patients with mild cognitive impairment was also negative. A primary prevention study (ADAPT trial) of naproxen (a non-selective COX inhibitor) and celecoxib (a COX-2 selective inhibitor) in cognitively normal elderly subjects with a family history of AD was prematurely interrupted for safety reasons after a median period of treatment of 2 years. Although both drugs did not reduce the incidence of dementia after 2 years of treatment, a 4-year follow-up assessment surprisingly revealed that subjects previously exposed to naproxen were protected from the onset of AD by 67% compared to placebo. Thus, it could be hypothesized that the chronic use of NSAIDs may be beneficial only in the very early stages of the AD process in coincidence of initial Aβ deposition, microglia activation and consequent release of pro-inflammatory mediators. When the Aβ deposition process is already started, NSAIDs are no longer effective and may even be detrimental because of their inhibitory activity on chronically activated microglia that on long-term may mediate Aβ clearance. The research community should conduct long-term trials with NSAIDs in cognitively normal APOE ε4 carriers.
Introduction
Alzheimer’s disease (AD) is the most common form of dementia, affecting approximately 5% of the population over the age of 65. As the population ages, the social impact of AD is becoming more critical. Thus, there is an urgent need for effective pharmacological treatments. Cholinesterase inhibitors have consistently shown symptomatic benefits and are now recognized as the standard treatments in patients with mild-to-moderate AD (Doody, 2008 ). A non-competitive N-methyl-D-aspartate antagonist, memantine, is also available for the symptomatic treatment of moderately severe to severe patients (Doody, 2008 ). Unfortunately, neither class of drugs is able to halt or to slow the disease progression.
The basic pathological abnormalities in AD are amyloid plaques and neurofibrillary tangles. Their exact relationship is still unclear and how they may cause neuronal death is still an area of intense research effort (Small and Duff, 2008 ). Amyloid plaques consist of a proteinaceous core composed of 5–10 nm amyloid fibrils surrounded by dystrophic neurites, astrocytic processes and microglial cells. The β-amyloid peptide (Aβ) consists of 38–42 amino acids generated by the cleavage of amyloid precursor protein (APP). The main form of Aβ contains 40 amino acids (Aβ40). The 42-residue species (Aβ42) is formed in less amounts than Aβ40 but is more prone to aggregate into fibrils and makes up the major component of amyloid plaques. Amyloid fibrils present a dynamic nature with a mechanism of dissociation and re-association resulting in a recycling of molecules within the fibril population (Carulla et al., 2005 ). Plaques may not be the main contributor to neuronal death, as there are consistent evidences that soluble oligomeric forms of Aβ are strongly neurotoxic (Shankar et al., 2008 ). In the last 10 years, most of the pharmaceutical efforts have been directed against the production and the accumulation of Aβ, the major component of the amyloid plaques, with the hope of slowing the deterioration rate of patients (Opar, 2008 ).
Brain Inflammation in Alzheimer’s Disease
In the AD brain, degenerating neurons, deposits of aggregated Aβ and neurofibrillary tangles are sites of inflammation. Amyloid plaques are associated with activated microglia and reactive astrocytosis. These cellular events are accompanied by increased expression of members of the complement pathway (C1q, C3b, C3a, membrane attack complex), cytokines and chemokines (interleukin-1β, interleukin-6, tumor necrosis factor α and transforming growth factor β), and acute phase reactive proteins (α-2-macroglobulin and α1-antichymotrypsin) surrounding amyloid deposits. However, edema and leukocyte infiltration is not observed (Weggen et al., 2007 ). Aggregation of soluble Aβ in oligomeric species can result in complement activation through the binding of complement molecules C1q and C3b (Figure 1 ). This activation stimulates production of C3b, membrane attack complex (MAC) and C3a. MAC can have toxic effects, and can result in cell lysis. C3b can reinforce complement activation. C3a activates microglial cells which can clear Aβ through phagocytosis. Uncleared Aβ may trigger a feedback loop in which Aβ deposition may be enhanced through the up-regulation of cytokines, such as interleukin-1β and interleukin-6, that have been reported to increase expression of APP (Wyss-Coray and Mucke, 2002 ) and to stimulate the synthesis of Aβ40 and Aβ42 peptides (Blasko et al., 2000 ). Additionally, the inflammation-associated proteins α1-ACT and apolipoprotein (ApoE) may also promote Aβ deposition (Nilsson et al., 2004 ) (Figure 1 ).
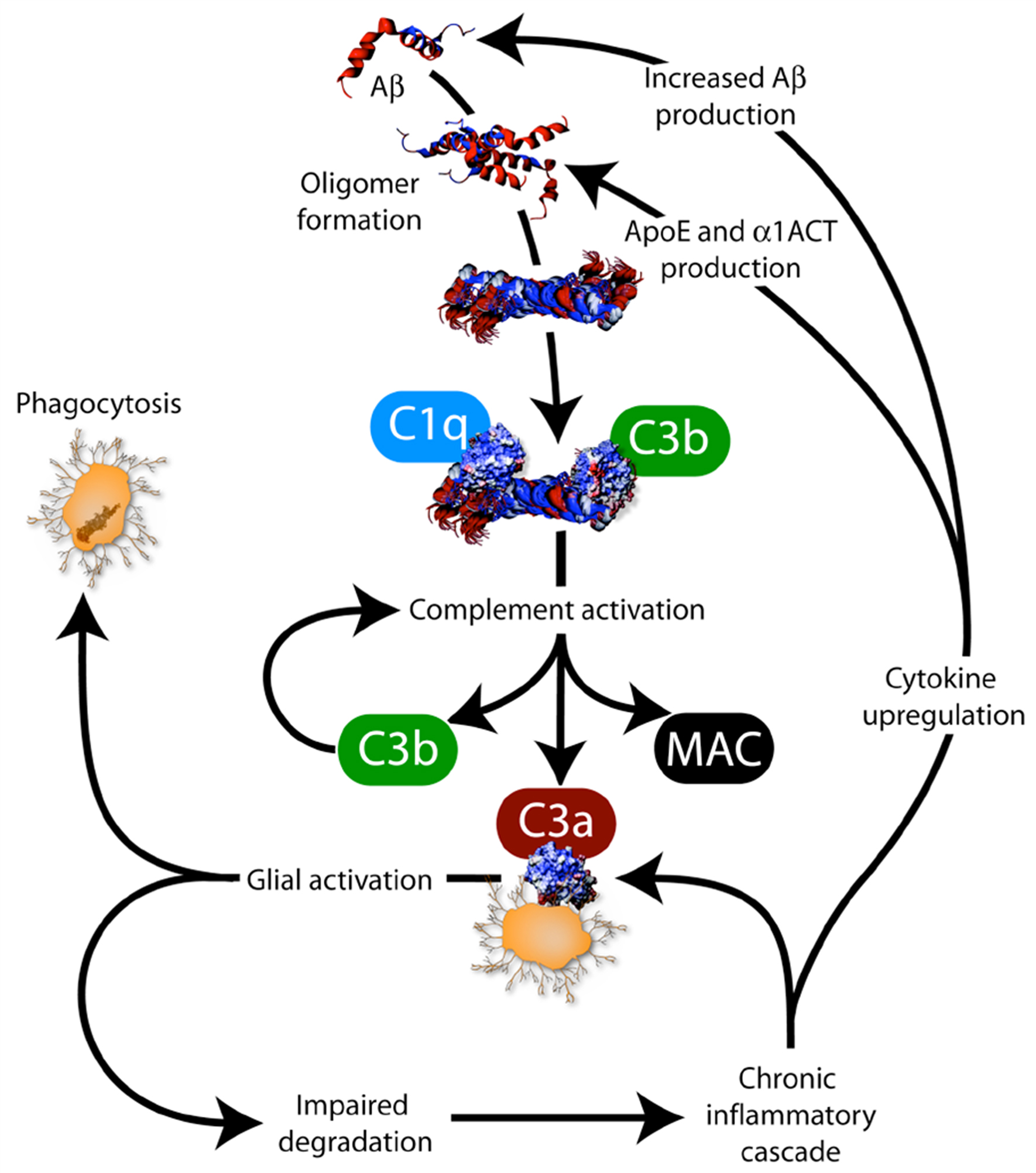
Figure 1. Interplay between amyloid pathology and inflammation. Aβ is β-amyloid, C1q, C3b, C3a and MAC (membrane attack complex) are complement factors, cytokines include interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor α (TNF-α) and TGF-β, ApoE is apolipoprotein E and α1-antichymotrypsin (α1-ACT) is an acute phase reactive protein (from Weggen et al., 2007 ).
Cyclooxygenase Inhibitors
The best-characterized action of NSAIDs is the inhibition of cyclooxygenase (COX), leading to marked reductions in the biosynthesis of pro-inflammatory prostaglandins (PGs). There are at least two COX isoforms, COX-1 and COX-2, and each NSAID varies in its ability to inhibit these isoforms. COX-3 was recently described as a splice variant of COX-1, but its functions are unknown. COX-1 is expressed in many tissues, and its metabolic products are considered to be involved in cellular “housekeeping” functions. COX-2 has lower expression levels in most tissues in healthy conditions, but increases during inflammatory response. The expression levels of COX-1 and COX-2 change in the different stages of AD pathology (Hoozemans et al., 2008 ). In an early stage, when low Aβ deposits are present and only very few neurofibrillary tangles are observed in the cortical areas, COX-2 is increased in neurons. The increased neuronal COX-2 expression parallels and co-localizes with the expression of cell cycle proteins, suggesting that COX-2 may be involved in regenerating pathways. COX-1 is primarily expressed in microglia, which are associated with fibrillar Aβ deposits. This suggests that COX-1 is involved in inflammatory pathway of the AD brain.
There are two classes of NSAIDs: non-selective COX-inhibitors and COX-2 selective inhibitors. Non-selective COX-inhibitors inhibit COX-1 and have variable activities against COX-2. Examples of non-selective COX-inhibitors are aspirin, ibuprofen, naproxen, piroxicam, indomethacin and sulindac. The discovery of COX-2 has led to the development of highly selective COX-2 inhibitors, such as celecoxib and rofecoxib, with the rationale that this isoform is inducible at the site of inflammation whereas COX-1 is expressed constitutively in several tissues including gastric epithelium. COX-2 selective inhibitors were thought to be safer for ulcerations of the gastro-intestinal mucosa observed with non-selective inhibitors. Meloxicam, nimesulide and etodolac were found as selective COX-2 inhibitors in pharmacological testing although they were not designed specifically as COX-2 inhibitors.
COX-2 inhibitors and some other NSAIDs have received attention because of their protective effects against colon cancer. In late 1999 celecoxib was approved by the FDA for the prevention of colon cancer in patients with familial adenomatous polyposis, which is a hereditary precancerous disease due to a loss of the adenomatous polyposis coli tumor suppressor gene. Nevertheless, in September 2004, Merck and Co announced the voluntary withdrawal of rofecoxib worldwide because of an increased risk of cardiovascular events. This decision and the unexpected findings of a colon cancer study which showed that celecoxib might also increase the chance of heart attack and stroke in some patients, raised serious concerns about the safety of selective COX-2 inhibitors (Finckh and Aronson, 2005 ). The mechanism with which COX-2 inhibitors may increase the risk of cardiovascular events is not clear but their depressive effects on prostaglandin I2 formation has been advocated as the cause of blood pressure elevation, acceleration of atherogenesis and exaggerated thrombotic response to the rupture of an atherosclerotic plaque (Fitzgerald, 2004 ). These findings represent a major drawback for the clinical use of COX-2 inhibitors in general (Couzin, 2004 ) and in December 2004 the FDA announced the suspension of a large, randomized, controlled, prevention trial of celecoxib in AD (Alzheimer’s Disease Anti-inflammatory Prevention Trial or ADAPT) (see paragraph 8). The ADAPT study was actually suspended prematurely because of an apparent increase in cardiovascular and cerebrovascular events in the naproxen arm compared to placebo. This event has revived the debate on the cardiovascular safety of NSAIDs, but this time, with a special emphasis on the impact of non-selective COX-inhibitors on the incidence of cardiovascular events. The published data are quite discordant and one cannot conclude that there is clear evidence to support a cardiovascular hazard from the administration of traditional NSAIDs like ibuprofen or naproxen (Maillard and Burnier, 2006 ).
Epidemiological Studies of NSAIDs in Alzheimer’s Disease
Several epidemiological studies, but not all studies, have evidenced a reduced prevalence of AD among users of NSAIDs (Table 1 ). The protective effect of NSAIDs is strongly dependent from the duration of treatment and on APOE genotype.
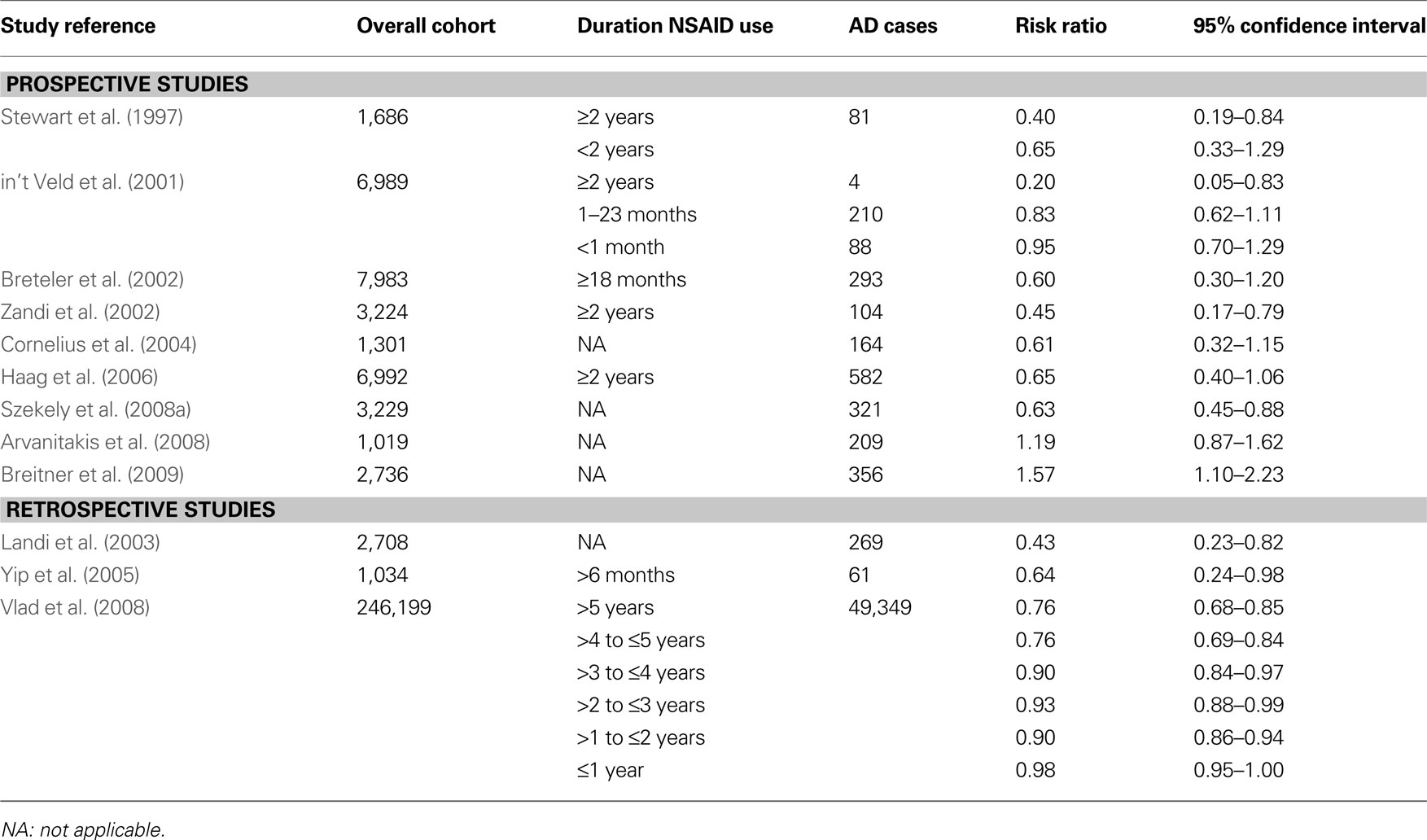
Table 1. Summary of the main prospective and retrospective epidemiological studies of NSAIDs and AD.
The Effect of Treatment Duration
The first study showing that increasing duration of NSAID use is associated with decreasing risk of AD was published by Stewart et al. (1997) . They examined data from a longitudinal study of 1,686 participants (the Baltimore < Longitudinal Study of Aging) and found that among subjects with two or more years of NSAID use, the relative risk was 0.40 compared to 0.65 for those with less than 2 years of NSAID use. This observation was later confirmed by several other studies. A population-based cohort study of 6,989 subjects found a relative AD risk of 0.95 in subjects with drug short-term use (≤1 month), 0.83 in those with intermediate-term use (1–24 months), and 0.20 in those with long-term use (≥24 months) (in t’ Veld et al., 2001 ). Pooled data from nine studies involving 14,654 subjects confirmed that the protective effects depend on the duration of NSAID use with relative risks of 0.95 among short-term (<1 month), 0.83 among intermediate-term (1–24 months) and 0.27 among long-term (>24 months) users (Etminan et al., 2003 ). A systematic review of seven prospective cohort studies found that those reporting duration of use of two or more years the combined risk estimate for developing AD was 0.42 compared to 0.74 for those whom any lifetime NSAID exposure was reported (Szekely et al., 2004 ). A large case-control study in 246,199 subjects found an adjusted odd ratio of 0.98 for ≤1 year of NSAID use and 0.76 for >5 years use (Vlad et al., 2008 ). For users of ibuprofen, the adjusted odd ratio decreased from 1.03 to 0.56.
The finding that increasing duration of NSAID use is associated with a decreasing risk of AD probably reflects the fact that the long-term users are taking NSAIDs at younger ages when the disease process is not yet started.
The Effect of the APOE Genotype
The protective effects of NSAIDs on AD onset are strongly dependent from the APOE genotype of the NSAID users. A prospective study (the Cardiovascular Health Cognition Study) followed for up to 10 years 3,229 elderly subjects (≥65 years) free of dementia at baseline. The study found that use of NSAIDs was associated with a lower risk of AD (adjusted hazard ratio of 0.63) (Szekely et al., 2008a ). AD risk reduction with NSAID was statistically significant in subjects having an APOE ε4 allele (adjusted hazard ratio of 0.34 compared to APOE ε4 carriers which did not take NSAIDs) but not in those without an APOE ε4 allele (adjusted hazard ratio of 0.88) (Szekely et al., 2008a ). Another prospective study (the Cache County Study) that evaluated for 8 years 3,383 elderly subjects cognitive normal at baseline, found that NSAID use prior to the age of 65 years in subjects with one or more ε4 alleles produced the greatest protective effects against cognitive decline evaluated with the Modified Mini-Mental State Examination test (0.40 vs 0.10 points per year) (Hayden et al., 2007 ). The MIRAGE Study that involved 691 AD patients and 973 family members, showed that the protective effects of NSAID use was more pronounced among APOE-ε4 carriers than non-carriers (adjusted odd ratios of 0.49 and 0.70, respectively) (Yip et al., 2005 ). In a cohort study (the Kungsholmen Project) that evaluated for up to 6 years 1,301 subjects free of dementia at baseline a reduction of risk of AD was seen only in ε4 carriers (relative risk = 0.47) but not in ε4 non-carriers (relative risk = 1.08) (Cornelius et al., 2004 ). Similarly, in the Rotterdam cohort study the risk reduction occurred mainly in ε4 carriers and not in ε4-non-carriers (relative risks 0.73 and 0.94, respectively) (in t’ Veld et al., 2001 ).
The reasons why NSAIDs users with one or more APOE ε4 allele have a greater AD risk reduction are unclear. In brain, apoE is mainly synthesized and secreted by astrocytes and microglia (Boyles et al., 1985 ). Astrocytes and microglia promote Aβ clearance and degradation (Koistinaho et al., 2004 ) via an apoE-dependent mechanism (Jiang et al., 2008 ). Transgenic AD mice expressing human APOE-ε4 have lower brain apoE levels and higher brain Aβ accumulation than APOE-ε4 non-carriers (Bales et al., 2009 ). Cognitively normal elderly men with APOE-ε4 have higher levels of brain fibrillar Aβ than ε4-non-carriers (Reiman et al., 2009 ). Thus, it can be hypothesized that the protective effects of NSAIDs are more evident in APOE-ε4 carriers simply because these subjects are more prone to Aβ brain deposition and accumulation and eventually to develop AD than ε4-non-carriers.
The Effect of Age
Several studies have suggested that the risk reduction with NSAIDs decreases with age (in t’ Veld et al., 1998; Zandi et al., 2002 ; Szekely et al., 2008a ). This finding may simply reflect the known tendency of those with APOE ε4 to develop AD at younger ages. A population-based cohort study (the Adult Changes in Thought study) in 2,736 elderly subjects (median 74.8 years at enrolment) without dementia at baseline followed for up to 12 years, unexpectedly found that NSAID users had a significant increased incidence of AD, with adjusted hazard ratios of 1.17 for moderate users and 1.57 for heavy users (Breitner et al., 2009 ). Data on NSAIDs use was particularly reliable because was based on computerized pharmacy dispensing records. To explain these unexpected findings, Authors hypothesized that NSAID exposure may delay the onset of AD with younger cohorts showing a reduced frequency of disease and older cohorts being enriched for cases that would otherwise have appeared earlier.
The Effect of NSAID Type
The type of NSAID also appears to affect the magnitude of the protective effect. Generally, the protective effects of NSAIDs are higher for users of non-aspirin compounds compared to those using aspirin. Users of acetaminophen are not protected (Stewart et al., 1997 ). In a cohort study of 1,301 dementia-free subjects at baseline and followed for 6 years, no subjects who used non-aspirin NSAIDs for around 3 years developed AD 3 years later (Cornelius et al., 2004 ). The reanalysis of the Rotterdam study indicates that risk decrease is restricted to NSAIDs that lower Aβ42 (ibuprofen, sulindac, flurbiprofen, indomethacin and diclofenac) (Breteler et al., 2002 ; Haag et al., 2006 ). In a cross-sectional retrospective study involving 2,708 community-dwelling elderly patients, a significantly decreased risk of cognitive impairment was found for patients using non-aspirin NSAIDs (Landi et al., 2003 ). The lowest adjusted odd ratio for a single non-aspirin NSAID drug was observed for diclofenac (0.20). A large case-control study evaluated the effects of type of NSAIDs used and the duration of the use and found the most significant protective effect is for the for the Aβ42-lowering ibuprofen with an odd ratio of 0.56 for greater than 5 year users (Vlad et al., 2008 ).However, the CHS Cognition Study did not find an arithmetic advantage for Aβ42-lowering NSAIDs compared to non-lowering ones (adjusted hazard ratios of 0.33 and 0.34, respectively), although only in the first group was the protective effect statistically significant. A pooled analysis of six prospective studies involving a total of 13,499 initially dementia-free participants did not find an apparent advantage in AD risk reduction for the subset of NSAIDs with Aβ42-lowering properties (Szekely et al., 2008b ).
It is important to point out that not all studies have found that NSAID use is associated to a decrease risk of AD. A meta-analysis of 25 case-control and cohort studies found that the benefit of NSAIDs in preventing dementia was 50% in studies with prevalent dementia cases, declined to 20% in studies with incident dementia cases, and was absent in studies where cognitive decline was used as endpoint (de Craen et al., 2005 ). Authors concluded that most of the reported beneficial effects of NSAIDs may result from recall bias, prescription bias, and publication bias. Another study in 1,019 older Catholic clergy followed for up to 12 years did not find a significant relationship between NSAID use and incident AD, change in cognitive performance or AD neuropathology (Arvanitakis et al., 2008 ).
Clinical Trials of Anti-Inflammatory Drugs in Alzheimer’s Disease
The hypothesis that the suppression of the inflammatory processes underlying the pathological cascade of AD will lessen the rate of disease progression and the evidence from epidemiological studies suggesting the protective effects of long-term use of NSAIDs against the onset of AD formed the rationale for a series of trials of different anti-inflammatory drugs in AD (Aisen and Davis, 1994 ).
NSAIDs
The initial pilot studies with NSAIDs produced encouraging results (Table 2 ). A first 6-month, double-blind, placebo-controlled study in 44 AD patients showed a significant (p = 0.003) slower cognitive decline in patients receiving indomethacin (100–150 mg) compared to those on placebo (Rogers et al., 1993 ). Unfortunately, 42% of patients abandoned the study due to adverse events (10 on indomethacin and 6 on placebo). Thus, the positive results of this study are of difficult interpretation. A subsequent 1-year double-blind, placebo-controlled, study of indomethacin (100 mg/day) given with omeprazole (20 mg/day) as gastroprotective agent, to 51 mild-to-moderate AD patients produced relatively low drop-out rates (7 out of 26 patients on indomethacin and 6 out of 25 patients on placebo) (de Jong et al., 2008 ). Unfortunately, the low number of subjects rendered the study inconclusive with non-significant trends in favor of indomethacin on two cognitive scales (Alzheimer’s Disease Assessment Scale Cognitive Subscale, ADAS-Cog and Mini-Mental State Examination, MMSE) at 12 months. In another small 6-month, double-blind, placebo-controlled study in 41 AD patients, arithmetic trends were observed in patients receiving diclofenac (50 mg/day) and misoprostol as gastroprotective agent (Scharf et al., 1999 ). Again, the withdrawal rate was high in the active treatment group (12 of 24 patients vs only 2 of 17 subjects on placebo), indicating that AD patients poorly tolerate standard prescription doses of NSAIDs. A small and short-term (3 months), placebo-controlled study of nimesulide, a preferential COX-2 inhibitor, in 40 AD patients did not show any significant differences at 12 weeks (Aisen et al., 2002 ).
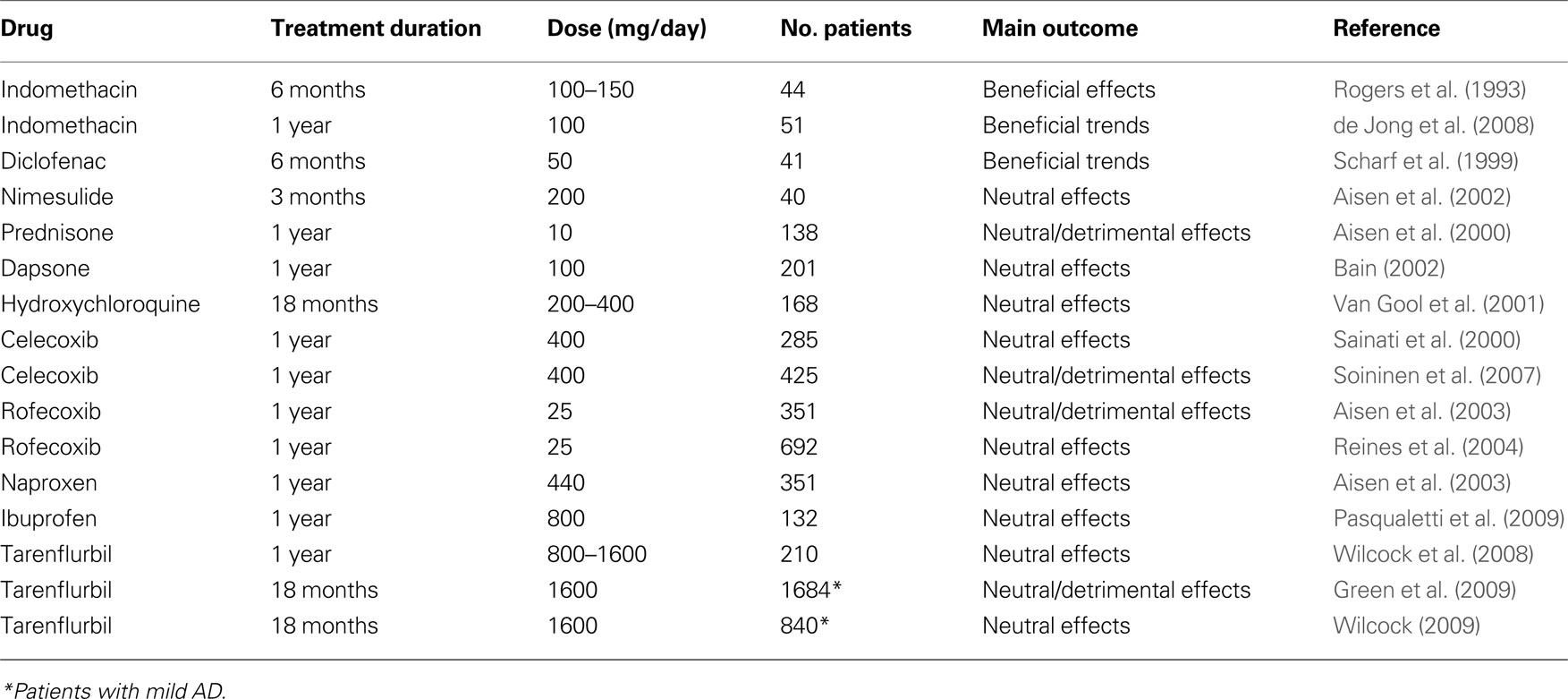
Table 2. Double-blind, randomized, placebo-controlled trials of anti-inflammatory drugs in patients with mild-to-moderate AD.
Studies suggesting that COX-2 may contribute to the neurodegeneration occurring in AD brains prompted the concept that inhibition of COX-2 might be a useful therapeutic target. COX-2 inhibitors were initially considered the preferred agents over classic NSAIDs, given their better tolerability at full anti-inflammatory doses. Unfortunately, a 1-year, double-blind placebo-controlled study with the COX-2 inhibitor celecoxib (200 mg b.i.d.) failed to demonstrate efficacy in slowing cognitive decline in 285 AD patients (Sainati et al., 2000 ) (Table 2 ). Another 1-year study with celecoxib (200 mg bid) in 425 mild-to-moderate AD patients did not show significant differences between the COX-2 inhibitor and placebo at the end of the 52-week treatment period on either cognitive (ADAS-Cog) or clinical global (Clinician Interview-Based Impression of Change Plus, or CIBIC-Plus) scales (Soininen et al., 2007 ). There were statistically significant differences in favor of placebo in two categories of the patient quality of life scale SF-36 as rated by the caregiver at week 52 (Role-Physical, p = 0.022 and Role-Emotional, p = 0.043). A 1-year, double-blind, placebo-controlled study in 351 AD patients comparing the ability of rofecoxib (25 mg once-a-day), another COX-2 inhibitor, and naproxen (220 mg twice-a-day) to slow cognitive deterioration produced negative results (Aisen et al., 2003 ) with cognitive decline (as assessed with ADAS-Cog) being actually faster in rofecoxib-treated patients compared to placebo (unadjusted p = 0.044). A further even larger study of rofecoxib (25 mg once-a-day) was recently completed in 692 patients and did not show any significant difference between rofecoxib and placebo after 1 year of treatment (Reines et al., 2004 ).
Long-term studies with non-selective COX-inhibitors were also negative (Table 2 ). A 1-year, placebo-controlled study in 132 mild-to-moderate AD patients evaluated the effects of ibuprofen (400 mg b.i.d. + esomeprazol 20 mg o.d.) on the cognitive decline (Pasqualetti et al., 2009 ). All patients were receiving stably therapy with donepezil. Seventy-seven percent of patients on ibuprofen and 70% of those on placebo completed the study. Overall, there were no effects of ibuprofen on the cognitive and clinical global scales. Interestingly, between AD patients carrying one or more ε4 allele, those receiving ibuprofen had lower cognitive and clinical decay than those that took placebo. The opposite pattern was observed in ε4 non-carriers.
A 1-year, placebo-controlled trial with tarenflurbil (R-flurbiprofen) in 210 patients with mild-to-moderate AD has produced, overall, negative results (Wilcock et al., 2008 ). The study tested two different doses of tarenflurbil (400 or 800 mg twice-a-day) and patients were allowed to take concomitantly cholinesterase inhibitors and/or memantine. A pre-specified subgroup analysis revealed that patients with mild AD in the 800 mg b.i.d. group had lower rates of decline than did those in the placebo group in activities of daily living (Alzheimer’s Disease Cooperative Study Activities of Daily Living or ADCS-ADL) and global function (CDR-SB). However, an accurate examination of the results reveals that the apparent positive effects of tarenflurbil at the dose of 800 mg b.i.d. in mildly affected patients were likely due to an anomalous deterioration rate observed in patients treated with placebo, rather than to the drug itself (Imbimbo, 2009 ). The apparently positive results of high doses of tarenflurbil in mildly affected AD patients prompted the execution of the largest ever study in patients with AD that involved 1,684 mildly affected patients that were treated with tarenflurbil (800 mg b.i.d.) for 18 months (Green et al., 2009 ). Unfortunately, the study came out completely negative with no statistically significant differences on the two primary cognitive and functional variables of the study (ADAS-Cog and ADCS-ADL). Paradoxically, patients on tarenflurbil had a significantly higher deterioration than those on placebo on the CDR-SB scale at the end of the 18-month treatment period (p = 0.004). Another 18-month study in 840 mild affected AD patients confirmed that tarenflurbil does not provide clinical benefit in this population (Wilcock et al., 2009 ).
Other Anti-Inflammatory Drugs
Studies with other anti-inflammatory agents produced completely negative results (Table 2 ). A 1-year, double-blind, placebo-controlled trial of low doses of prednisone (10 mg daily) in 138 AD patients did not show any significant benefit of the glucocorticoid treatment on the rate of cognitive decline (Aisen et al., 2000 ). Actually, patients treated with prednisone displayed a greater behavioral decline compared with those treated with placebo (p = 0.003 on the Brief Psychiatric Rating Scale) and a detrimental trend also on the global clinical status (p = 0.07 on the Clinical Dementia Rating Sum of Boxes, CDR-SB). An 18-month, randomized, double-blind, placebo-controlled study evaluated the ability of hydroxychloroquine, a potent anti-inflammatory drug widely used in the treatment of rheumatoid arthritis and able to cross the blood-brain barrier, to delay progression of AD (Van Gool et al., 2001 ). The study involved 168 patients and was completed by 92% of participants. Unfortunately, at the end of the 18-month treatment period there were no significant differences in any of the efficacy outcome measures (activities of daily living, cognitive function and behavioral abnormalities). A 1-year, randomized, double-blind, placebo-controlled study was carried out with dapsone, an old anti-leprosy agent with strong anti-inflammatory properties and for which a preventive role in AD was claimed (McGeer et al., 1992 ). Dapsone (100 mg/day) and placebo were administered orally, once daily for 52 weeks in 201 patients with mild-to-moderate AD. At the end of treatment there were no significant differences between dapsone and placebo on either cognitive or other measures of efficacy (Bain, 2002 ).
Preventive Trials of NSAIDs in Alzheimer’s Disease
Primary and secondary studies have been designed and conducted to verify if anti-inflammatory drugs are able to prevent the onset of AD in patients with mild cognitive impairment (MCI) (Table 3 ) or in healthy elderly subjects at risk of developing AD (Table 4 ).
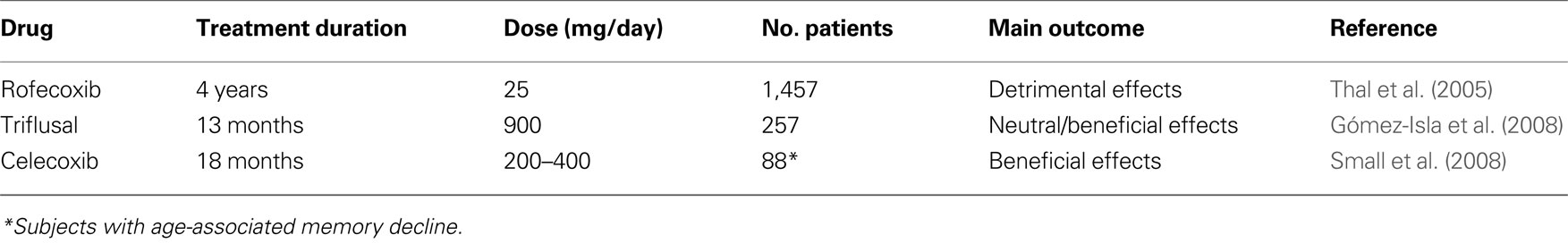
Table 3. Double-blind, randomized, placebo-controlled trials of NSAIDs in patients with MCI.
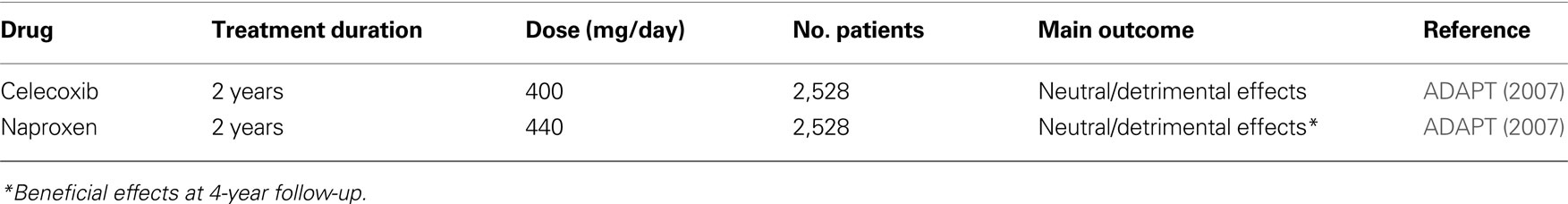
Table 4. Double-blind, randomized, placebo-controlled primary prevention studies of NSAIDs in AD.
A 4-year, double-blind, placebo-controlled study with rofecoxib in 1,457 patients with MCI showed that the annual conversion rate to AD was significantly higher in patients treated with rofecoxib (25 mg/day) than in those treated with placebo (6.4% versus 4.5%, p = 0.011) (Thal et al., 2005 ). Another double-blind, placebo-controlled trial in MCI patients was conducted with triflusal, a non-selective NSAID (Gómez-Isla et al., 2008 ). Subjects were randomly assigned to receive 900 mg/day of triflusal or placebo for 18 months. A slow rate of recruitment forced a premature cessation of the study. Two hundred and fifty-seven subjects were enrolled and followed-up for an average of 13 months. Compared to placebo, there was a significant lower rate of conversion to AD in the triflusal group (hazard ratio = 2.10; 95% confidence interval, 1.10–4.01; p = 0.024). Compared to placebo, there was also a trend in favor of triflusal regarding cognitive decline (ADAS-Cog), although the difference did not reach statistical significance, p = 0.096). Because the trial was prematurely halted, these results should be interpreted with caution and require further confirmation.
Another 18-month, double-blind, placebo-controlled study of celecoxib in 88 subjects with mild, self-reported, memory complaints but with normal memory performance score was recently published (Small et al., 2008 ) and reported significant differences in favor of the COX-2 inhibitor on executive functioning (p = 0.03) and language/semantic memory (p = 0.02). Significant differences in favor of celecoxib were also detected in regional brain metabolism measured with FDG-PET in prefrontal cortex. These apparently positive cognitive and metabolic results should be considered with caution because of the high drop-out rate (48 out of 88 subjects).
A primary prevention trial (ADAPT) was conducted in 2,528 cognitively normal individuals at risk for AD (age > 70 years and first degree relative with dementia). Subjects were randomized to receive naproxen (220 mg twice-a-day) or celecoxib (200 mg twice-a-day) or placebo (ADAPT Research Group, 2007 ). Initially, treatments were planned to last 7 years. The main endpoint of the study was the onset of AD after randomization. Enrolment began in early 2001. On December 17, 2004, treatments were suspended. Median follow-up times were 733 days for celecoxib-, 734 days for naproxen- and 735 days for placebo-assigned participants. Analyses that excluded the seven individuals with dementia that were erroneously enrolled in the study, showed increased hazard ratios for AD compared to placebo with both celecoxib (hazard ratio = 4.11, p = 0.22) and naproxen (hazard ratio = 3.57, p = 0.04). After 2 years of treatment, mean scores of the Modified MMSE of both drug-treated groups were worse than that of placebo (−0.69 points for celecoxib, p = 0.004 and −0.35 points for naproxen, p = 0.06) (ADAPT Research Group, 2008 ). Masked long-term follow-up of the enrolled subjects is ongoing. A recent analyses of the 4-year follow-up data surprisingly revealed that subjects previously exposed to naproxen were protected from the onset of AD by 67% compared to placebo (Breitner, 2009 ). The overall results of the ADAPT trial suggest that people who already had disease process ongoing in their brain (even if asymptomatic) at the beginning of the trial indeed worsened if they took NSAIDs, which accelerated their underlying disease. In contrast, people who had brain completely normal at baseline and took naproxen for 1–3 years appeared to fare better. At the long-term examination, they had a lower incidence of AD than did those on placebo. This apparent clinical protection was supported by favorable CSF biomarkers values (low tau and high Aβ42 levels) at 21–42 months after treatment.
Mechanisms for Protective Effects of NSAIDs in Alzheimer’s Disease
A number of non-mutually exclusive mechanisms have been proposed to account for the protective effects of NSAIDs in AD. These mechanisms are either related to the anti-inflammatory properties of NSAIDs or are supposed to directly target APP processing, Aβ production and Aβ aggregation (Figure 2 ).
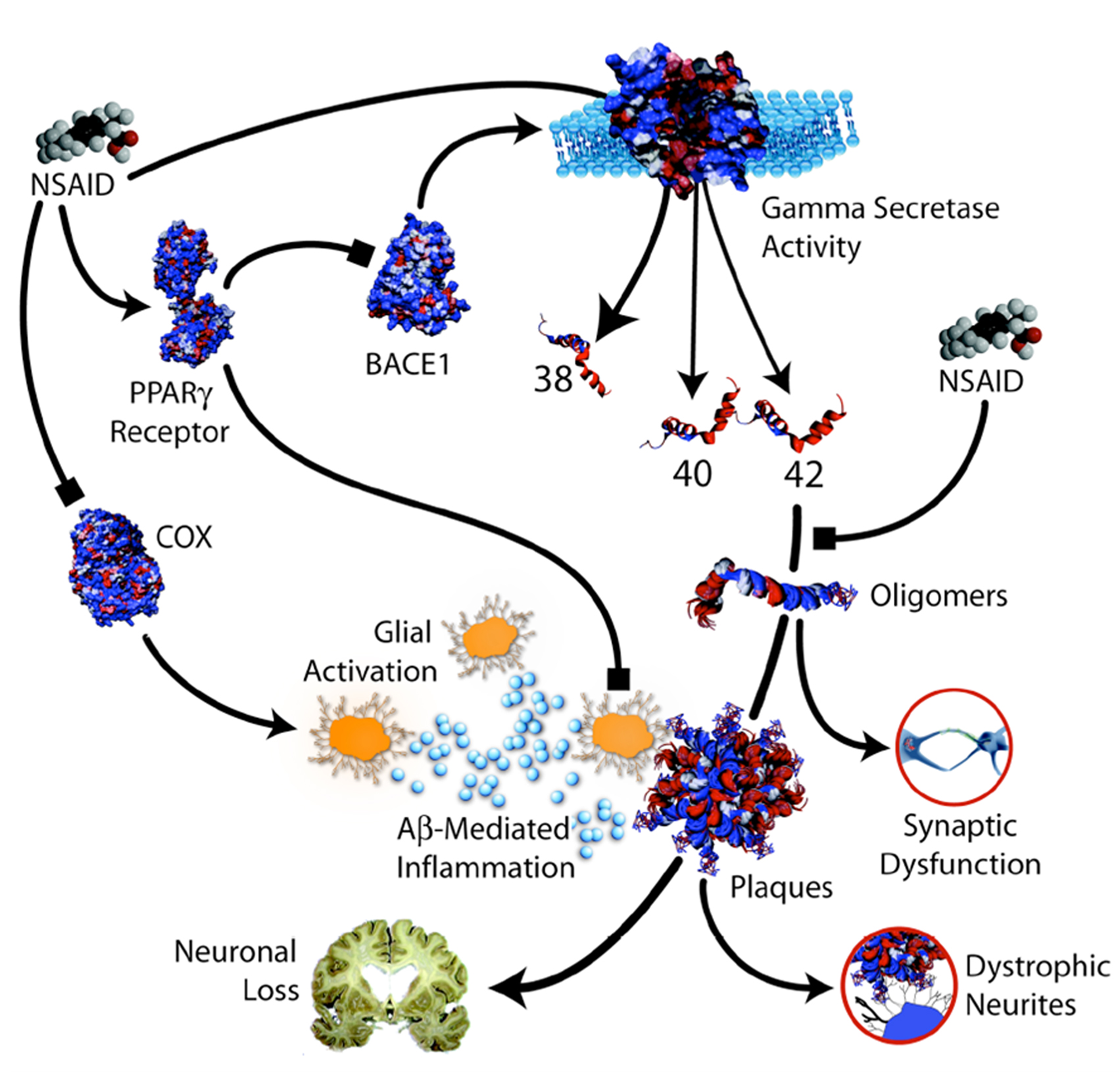
Figure 2. Potential protective mechanisms of NSAIDs in AD (from Weggen et al. , 2007 ).
NSAIDs exert their principal therapeutic effects, reducing fever, pain and inflammation by blocking the COX-mediated synthesis of inflammatory PGs (Hoozemans et al., 2008 ). It is believed that NSAIDs may reduce brain inflammation in AD through their inhibitory effects on COX activity. It has been proposed that the beneficial effects of NSAIDs on memory in transgenic mouse models of AD do not depend upon lowered levels of the inflammatory cytokines, tumor necrosis factor α and interleukin-1β but on the blockade of a COX-2-mediated prostaglandin E2 response (Kotilinek et al., 2008 ). Another mechanism that has been implicated in the anti-inflammatory action of NSAIDs is the inhibition of the activity of the nuclear factor κB (Sung et al., 2004 ), a transcription factor involved in the regulation of several cellular target genes and found to be increased in AD brains. Other studies have implicated peroxisome proliferator-activated receptor-γ (PPARγ), a nuclear receptor that controls expression of pro-inflammatory genes, as an alternative mediator of the anti-inflammatory properties of NSAIDs (Sastre et al., 2003 ). NSAIDs may stimulate PPARγ activity with the consequent inhibition of the inflammatory response. More recently, it has been shown that ibuprofen and naproxen are able to blocked neuronal cell cycle events and alterations in brain microglia of a transgenic hAPP mouse model of AD without altering APP processing and steady-state brain Aβ levels (Varvel et al., 2009 ).
The other hypothesis that has been proposed for explaining the beneficial effect of NSAIDs in AD is their activity on APP processing, Aβ production or Aβ aggregation. A number of mechanisms have been proposed for this action. First, some NSAIDs, including ibuprofen and indomethacin, were shown to selectively lower Aβ42 production in vitro and in APP-transgenic mouse models of AD (Weggen et al., 2001 ). In addition, it has also been shown that ibuprofen and indomethacin potently stimulate secretion of the neurotrophic and neuroprotective APP ectodomain in neuronal cell lines by up-regulation of the non-amyloidogenic α-secretase pathway (Avramovich et al., 2002 ). Furthermore, ibuprofen and indomethacin have been shown to reverse elevations in BACE1 expression and Aβ secretion after stimulation of cell lines with pro-inflammatory cytokines (Sastre et al., 2006 ). Ibuprofen can reduce expression of α1-antichymotrypsin (α1-ACT), an acute phase protein that accelerates amyloid pathology in APP-transgenic mice (Morihara et al., 2005 ). NSAIDs may also inhibit Aβ aggregation and fibril formation in vitro (Thomas et al., 2001 ). Finally, NSAIDs may act to inhibit the formation of Aβ oligomers and deposits through direct interaction with the Aβ peptide (Kukar et al., 2008 ). Human neuropathological studies have failed to show that NSAID treatment ameliorates brain plaque burden in AD patients (Alafuzoff et al., 2000 ; Halliday et al., 2000 ).
Other mechanisms possibly explaining the protective role of NSAIDs in AD include increase in the expression of neurotrophic factors (Zhao et al., 2008 ), regulation of glutamate homeostasis (Casper et al., 2000 ; Khansari and Halliwell, 2009 ), maintenance of synaptic plasticity (Stéphan et al., 2003 ; Kotilinek et al., 2008 ).
The Effects of NSAIDs on Microglia
The role of microglia in the AD pathogenesis is very much debated. Microglial activation adjacent to amyloid deposits in AD has suggested that microglia-induced neuroinflammatory responses mediate neurodegeneration (Wyss-Coray, 2006 ). It has been proposed that cytokines, chemokines and neurotoxins generated by Aβ-activated microglia cause neuronal damage (El Khoury and Luster, 2008 ). However, different reports showed that microglia or bone marrow-derived macrophages may have a function of stimulating Aβ clearance and antagonizing plaque growth (Simard et al., 2006 ; Bolmont et al., 2008 ; Meyer-Luehmann et al., 2008 ). Conversely, a recent paper has shown that amyloid plaque formation and maintenance in two transgenic mouse models of AD was not influenced by nearly complete ablation of microglia achieved with gene manipulation (Grathwohl et al., 2009 ). Although this work showed that microglia ablation does not cause an increase in plaque burden, it did not demonstrate that microglia and peripheral macrophages are not able to inhibit brain plaque formation or clear plaques in vivo. In addition, microglia ablation was induced too late in respect to the crucial phase of plaque seeding of the two AD transgenic mice. It could be that microglia may damage the AD brain before full plaque deposition as suggested by a recent paper (Varvel et al., 2009 ).
Although many animal studies support the notion that NSAIDs attenuate microglia activation and inhibit inflammatory mediators in transgenic mouse models of AD, data in humans are quite scanty. One study in 66 subjects reported that brains of non-demented arthritic patients chronically treated with NSAIDs has reduced number of activated microglial cells than non-demented control subjects (Mackenzie and Munoz, 1998 ). On the other hand, another study in 42 AD patients with a history of regular use of NSAIDs showed significant lower counts of brain reactive astrocytes but not of activated microglia (Alafuzoff et al., 2000 ). A further neuropathological study in 12 AD subjects failed to show significant differences in number of microglia and astrocytes in NSAID users and non-users (Halliday et al., 2000 ).
Nevertheless, the hypothesis that the beneficial effects of NSAIDs derive from their action on microglia has been recently reinforced in a study employing a reliable transgenic animal model of AD (R1.40 mouse) (Varvel et al., 2009 ). This study showed that alterations in brain microglia were coincident with the first evidence of neuronal cell cycle events, a early pathogenic process in AD (McShea et al., 1997 ; Yang et al., 2001 ). Interestingly, in R1.40 transgenic mice both microglia activation and increased neuronal cell cycle events occurred before the onset of Aβ deposition. Induction of systemic inflammation promoted the early appearance of neuronal cell cycle events in young transgenic mice, but not in non-transgenic controls. In addition, inhibition of neuroinflammation in young transgenic animals by chronic administration of ibuprofen and naproxen blocked alterations in brain microglia as well as neuronal cell cycle events in the absence of any effect on brain Aβ40 and Aβ42 levels. Notably, therapeutic NSAID treatment of older transgenic mice did not reverse the presence of existing cell cycle events, even after several months of treatment. These results provide a potential explanation for the failures of the prospective NSAID trials in AD patients.
The Effects of NSAIDs on Astrocytes
In the healthy brain, astrocytes support neurons metabolically and functionally. In the AD brain, astrocytes surround plaques and might play a critical role in Aβ deposition and clearance, neuronal calcium homeostasis and synaptic function and might contribute to cortical dysfunction (Kuchibhotla et al., 2009 ). Reactive astrocytes neglect their neurosupportive functions, thus rendering neurons vulnerable to neurotoxins including pro-inflammatory cytokines and reactive oxygen species (Fuller et al., 2009 ). NSAIDs may interact with astrocytes by modulating activation and migration. A short-term treatment with ibuprofen of adult APP-transgenic mice resulted in a significant reduction in the number of reactive astrocytes in the hippocampus and cortex (Heneka et al., 2005 ). The effects of NSAIDs on number of reactive astrocytes may be ascribed to the inhibitory effect on Aβ deposition (Richardson et al., 2002 ) but are also due to a direct modulating effect on astrocytes. In rat primary cultures, ibuprofen produced profound stellation of astrocytes and altered their migration (Lichtenstein et al., 2009 ). In mouse and human astrocyte cell culture ibuprofen reduced α1-antichymotrypsin release induced by LPS or IL-1β (Morihara et al., 2005 ). Ibuprofen significantly reduced NMDA-induced neuronal cell death in mixed cortical cultures containing mice neuronal and glial cells but not in near-pure neuronal cultures containing less than 5% astrocytes (Iwata et al., 2010 ). Thus, NSAIDs may protect against AD by direct and indirect interaction with reactive astrocytes.
Neuroprotective Effects of NSAIDs
Some reports show that NSAIDs may be neuroprotective. A study using rat cerebellar granule cell culture showed that sulindac sulphide, indomethacin, ibuprofen and R-flurbiprofen, at low concentrations (1 μM), depolarize mitochondria and inhibit mitochondrial Ca2+ overload, cytochrome c release and cell death induced by Aβ oligomers (Sanz-Blasco et al., 2008 ). Ibuprofen has been shown to promote neurite growth in neuroblastoma B104 cell line (Dill et al., 2010 ) and to promote significant axonal growth and functional recovery following spinal cord lesions in rodents (Fu et al., 2007 ) via a RhoA-PPARγ-mediated mechanism.
Why did Anti-Inflammatory Drugs Fail in Alzheimer’s Disease?
So far, clinical trials designed to inhibit brain inflammation in AD patients have failed. These failures have been ascribed to wrong timing of intervention, to the short duration of treatment, to the low doses used and to the wrong class of drugs. However, the real problem is that it is still not clear whether or not inflammatory mechanisms are actually causing damage in AD or whether they are merely present to remove the debris from other more primary pathological processes. The inhibitory effects of NSAIDs on activated microglia may play a negative role (Masters et al., 2008 ). For many years microglial activation has been considered to contribute to AD progression. Recent studies using in vivo imaging have demonstrated that microglia rapidly migrate to newly formed Aβ deposits in mouse models of AD but are not able to clear them but instead tend to stabilize the plaques avoiding further growth (Meyer-Luehmann et al., 2008 ; Bolmont et al., 2008 ). The initial inability of activate microglia of phagocyting fibrillary Aβ has been ascribed to the release of pro-inflammatory cytokines that follows microglial activation. Thus, initial neuroinflammation may, in part, underlie the accumulation of fibrillary Aβ-containing plaques within the AD brain (Koenigsknecht-Talboo and Landreth, 2005 ). At his initial stage of the disease process, the administration of NSAIDs could be beneficial for its ability to reduce microglia activation and cytokine release. However, chronic microglia activation may lead to plaque removal (Backsai et al., 2001 ). In addition, bone marrow-derived macrophages, the peripheral counterpart of microglia, may migrate in the brain and contribute to removal of plaques (Malm et al., 2005 ). Indeed, bone marrow-derived microglia, but not resident microglia, have been reported to prevent the formation and even eliminate brain amyloid deposits (Simard et al., 2006 ). Interestingly, the removal of brain soluble and insoluble Aβ by bone marrow-derived macrophages is dependent from APOE genotype with macrophages expressing apoE2 being more efficient at degrading Aβ than apoE3-expressing, apoE4-expressing, or apoE-deficient macrophages (Zhao et al., 2009 ). A study in mice lacking the chemokine receptor 2, a microglial cell-surface receptor that mediates recruitment of blood-derived microglia, supports the idea that bone marrow-derived macrophages infiltrate the brain and can clear Aβ from the brain (El Khoury et al., 2007 ). In addition, it has been recently demonstrated that microglia is able to internalize soluble Aβ, the most toxic species of Aβ, from the extracellular milieu through a macropinocytic mechanism into late endolysosomal compartments where they are subject to degradation (Mandrekar et al., 2009 ). Thus, the use of NSAIDs in advanced stages of AD may be detrimental by inhibiting microglia-mediated clearance of Aβ.
Recently, activated microglia has been shown to be involved in the regulation of several aspects of neurogenesis under certain experimental conditions both in vitro and in vivo (Ekdahl et al., 2009 ). Under physiological conditions, microglia remains in a resting state in the dentate gyrus (Olah et al., 2009 ). Under pathological conditions, there is accumulation of microglia with proneurogenic phenotype in the adult rat subventricular zone of the dentate gyrus of the hippocampus implying a supportive role of these cells for the continuous neurogenesis after damage (Thored et al., 2009 ). The function of neurogenesis in the human hippocampus remains elusive, but growing evidence suggests its importance for memory tasks (Shors et al., 2001 ). Neurogenesis is increased in AD hippocampus where it may represent a compensatory phenomenon to replace neurons lost during the disease (Jin et al., 2004 ). In a chronically altered environment, persistently activated microglia can display protective functions that favor brain repair processes (Cacci et al., 2008 ). Indeed, the use of COX-2 inhibitors has been shown to attenuate the proliferation of progenitors in the subgranular zone after transient forebrain ischemia in adult mouse (Sasaki et al., 2003 ). Thus, inhibition of activated microglia by anti-inflammatory drugs may be detrimental also by interfering with compensatory neurogenesis processes.
Conclusions
Despite epidemiological evidences and an apparently robust biochemical rationale, all large, long-term, prospective, randomized, placebo-controlled studies aimed to reduce inflammation in the brain of AD patients have produced negative results. The failures include both steroidal anti-inflammatory agents like prednisone, non-steroidal anti-inflammatory drugs like hydroxychloroquine or dapsone, non-selective COX-inhibitors like naproxen as well as COX-2 selective inhibitors like rofecoxib and celecoxib. Unfortunately, ibuprofen, the NSAID with the clearest findings in epidemiological studies (Vlad et al., 2008 ), the most promising and consistent results in pharmacological studies (Imbimbo, 2004 ) and one of the better tolerated agent in humans (McGettigan and Henry, 2006 ), has been tested only in a relatively small study in AD patients (Pasqualetti et al., 2009 ) but not in patients with MCI.
These negative results may be ascribed to low doses used insufficiently to suppress brain inflammatory activity. However, negative results were also obtained with a potent anti-inflammatory drug like prednisone, which paradoxically accelerated clinical deterioration of AD patients (Aisen et al., 2000 ). It has been argued that anti-inflammatory therapy cannot protect patients when dementia is fully established. However, a large trial in patients with MCI indicated that rofecoxib could even accelerate the conversion to AD (Thal et al., 2005 ). In addition, a primary prevention study of naproxen and celecoxib in elderly subjects with family history of AD has been interrupted prematurely with indications of negative or even detrimental effects of the two NSAIDs (ADAPT Research Group, 2007 , 2008 ).
The discovery that some NSAIDs selectively decrease Aβ42 by allosterically inhibiting the γ-secretase complex, has initially shed light on the apparent discrepancy between epidemiological studies and negative trials. The inhibition of Aβ42 production is independent from COX activity and depends on the chemical structure of the NSAIDs, some compounds being active (ibuprofen, sulindac sulphide, flurbiprofen, indomethacin, diclofenac) and others not (naproxen, aspirin, meloxicam, celecoxib, rofecoxib). Thus, the negative results of the AD trial with naproxen, dapsone, prednisone, celecoxib and rofecoxib might be explained by the fact these compounds do not lower Aβ42 production. The encouraging results observed in small AD trials with indomethacin and diclofenac would be in agreement with their inhibitory activity on Aβ42 production. The detrimental effects of rofecoxib observed in both AD and MCI would also agree with the ability of the compound to stimulate Aβ42 production. However, the recent negative results of two large 18-month studies with tarenflurbil (R-flurbiprofen) have questioned the hypothesis that Aβ42-lowering NSAIDs may be clinically efficacious in AD patients (Green et al., 2009 ; Wilcock et al., 2009 ). Nevertheless, tarenflurbil may not be the right Aβ42 lowering NSAID derivative because of its weak pharmacological activity and poor brain penetration (Imbimbo, 2009 ). In addition, it could be that the compound, even though it is the R enantiomer of flurbiprofen, still maintains a significant anti-COX activity at the high dose used in the study (800 mg b.i.d.). The occurrence of several gastro-intestinal adverse events in AD patients treated with tarenflurbil during the Phase III study, including eight cases of peptic ulcer (vs. only one in the placebo group) (Green et al., 2009 ), seems to confirm this hypothesis. Evidences or indications of detrimental effects compared to placebo have already been observed in other large, long-term, controlled studies with anti-inflammatory drugs, including prednisone (Aisen et al., 2000 ), rofecoxib (Aisen et al., 2003 ) and celecoxib (Soininen et al., 2007 ) in AD patients, rofecoxib in patients with MCI (Thal et al., 2005 ), celecoxib and naproxen in elderly subjects at risk of developing AD (ADAPT Research Group, 2007 , 2008 ).
These negative and even detrimental effects of NSAIDs in patients with AD or MCI and in subjects at risk of AD appear to be in contrast to the epidemiologic findings suggesting reduced AD incidence following prolonged NSAID use. A possible explanation is that the effects of NSAID exposures differ with the stage of brain disease progression. NSAIDs suppress brain inflammatory mechanisms, particularly the secretion by activated microglia of cytokines and chemokines that are believed to promote AD pathogenesis. However, it is likely that NSAID suppression of microglial activation might exert protective effects only when given years before the development of clinical symptoms. Once the pathological brain deposition of Aβ is started, microglia activation may be essential to boost the clearance of the excess of toxic amyloid. At this stage, when clinical symptoms start to become evident, NSAID suppression of microglial activity could be not beneficial or could even be detrimental by accelerating the disease process. This hypothesis appears to be consistent with data from both the Rotterdam (Szekely et al., 2004 ), the Cache County (in t’ Veld et al., 2001 ) and the US Veterans (Vlad et al., 2008 ) observational studies. All these studies suggest no protection with NSAIDs used in the 2 years preceding the onset of dementia. If timing of exposure determines whether NSAIDs produce benefit or harm, the negative results of the prematurely interrupted primary prevention study with naproxen and celecoxib (ADAPT study) should not be surprising because subjects were exposed to NSAIDs for only 2 years. According to this hypothesis, NSAIDs may prevent AD if given years before the typical onset age, but may increase risks when used later in life. Since the observational studies suggest that NSAIDs may have protective effects only in subjects with a normal brain, continued observations in the ADAPT cohort could show mitigation or even reversal of treatment effects that presently appear null or detrimental. Recent results at 4 years of the masked long-term follow-up of the ADAPT study, showing a robust reduction in AD risk in subjects exposed to naproxen (Breitner, 2009 ), seem to confirm this hypothesis. Thus, long-term trials with NSAIDs or NSAIDs-derivatives (Imbimbo et al., 2007 ) in cognitively normal APOE ε4 carriers appear rationale. On the other hand, we cannot exclude the possibility that most of the reported beneficial effects of NSAIDs in epidemiological studies may result from various forms of bias (de Craen et al., 2005 ) and that there is in fact no role for NSAIDs in AD prevention or treatment.
Epidemiological Glossary
Epidemiological study a statistical study on human populations, which attempts to link human health effects to a specified cause.
Cohort study epidemiological study in which individuals with differing exposures to a suspected factor are identified and then observed for the occurrence of certain health effects over some period.
Prospective study cohort study in which the subjects are identified and then followed forward in time.
Longitudinal study cohort study that involves repeated observations of the same items over long periods of time.
Case-control study epidemiological study which compares the frequency of a purported risk factor (generally called the “exposure”) in a group of people (the “cases”) affected by a medical condition of interest and a group of individuals (the “controls”) who are not affected but are otherwise similar.
Retrospective study a study that starts after the onset of disease and looks back to the postulated causal factors.
Primary prevention study a study including general or healthy populations.
Secondary prevention study a study including participants with specific disease.
Cross-sectional study a study which disease and exposure status are measured simultaneously in a given population.
Incident cases number of new cases of the disease occurring in the population in a given period of time.
Relative risk ratio of the probability of the event occurring in the exposed group versus a non-exposed group.
Odds ratio of the number of people incurring an event to the number of people who have non-events.
Odds ratio odds in favor of being exposed in subjects with the target disorder divided by the odds in favor of being exposed in control subjects.
Systematic review a study of the literature that tries to identify, appraise, select and synthesize all high quality research evidence relevant to that question.
Adjustment statistical technique that estimates the association between an exposure and an outcome while “controlling” (“adjusting”) for one or more possible confounding variables.
Hazard ratio a measure of the frequency of occurrence of an event in one group compared to that occurring in another group, over time.
Population-based study a study involving a subset (or sample) of individuals selected from a larger group (population).
Meta-analysis a systematic review that uses quantitative methods to summarize the results.
Prevalent cases number of cases of the disease in the population at a given time.
Randomized clinical trial a clinical study that compares the efficacy of two or more drugs or medical interventions in which the different treatments are randomly allocated to subjects.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
ADAPT Research, Group, Lyketsos, C. G., Breitner, J. C., Green, R. C., Martin, B. K., Meinert, C., Piantadosi, S., and Sabbagh, M. (2007). Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 68, 1800–1808.
ADAPT Research, Group, Martin, B. K., Szekely, C., Brandt, J., Piantadosi, S., Breitner, J. C., Craft, S., Evans, D., Green, R., and Mullan, M. (2008). Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch. Neurol. 65, 896–905.
Aisen, P. S., and Davis, K. L. (1994). Inflammatory mechanisms in Alzheimer’s disease. Implications for therapy. Am. J. Psychiatr. 151, 1105–1113.
Aisen, P. S., Davis, K. L., Berg, J. D., Schafer, K., Campbell, K., Thomas, R. G., Weiner, M. F., Farlow, M. R., Sano, M., Grundman, M., and Thal, L. J. (2000). A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology 54, 588–593.
Aisen, P. S., Schafer, K. A., Grundman, M., Pfeiffer, E., Sano, M., Davis, K. L., Farlow, M. R., Jin, S., Thomas, R. G., and Thal, L. J. (2003). Effects of rofecoxib or naproxen vs. placebo on Alzheimer disease progression. JAMA 289, 2819–2826.
Aisen, P. S., Schmeidler, J., and Pasinetti, G. M. (2002). Randomized pilot study of nimesulide treatment in Alzheimer’s disease. Neurology 58, 1050–1054.
Alafuzoff, I., Overmyer, M., Helisalmi, S., and Soininen, H. (2000). Lower counts of astroglia and activated microglia in patients with Alzheimer’s disease with regular use of non-steroidal anti-inflammatory drugs. J. Alzheimers Dis. 2, 37–46.
Arvanitakis, Z., Grodstein, F., Bienias, J. L., Schneider, J. A., Wilson, R. S., Kelly, J. F., Evans, D. A., and Bennett, D. A. (2008). Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology 70, 2219–2225.
Avramovich, Y., Amit, T., and Youdim, M. B. (2002). Non-steroidal anti-inflammatory drugs stimulate secretion of non-amyloidogenic precursor protein. J. Biol. Chem. 277, 31466–31473.
Backsai, B. J., Kajdasz, S. T., Christie, R. H., Carter, C., Games, D., Seubert, P., Schenk, D., and Hyman, B. T. (2001). Imaging of amyloid-b deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat. Med. 7, 369–372.
Bain, A. (2002). Alzheimer Disease. Phase 2 Trial Results Reported. Immune Network Ltd. Vancouver, Canada.
Bales, K. R., Liu, F., Wu, S., Lin, S., Koger, D., DeLong, C., Hansen, J. C., Sullivan, P. M., and Paul, S. M. (2009). Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779.
Blasko, I., Veerhuis, R., Stampfer-Kountchev, M., Saurwein-Teissl, M., Eikelenboom, P., and Grubeck-Loebenstein, B. (2000). Costimulatory effects of interferon-γ and interleukin-1β or tumor necrosis factor α on the synthesis of Aβ1-40 and Aβ1-42 by human astrocytes. Neurobiol. Dis. 7, 682–689.
Bolmont, T., Haiss, F., Eicke, D., Radde, R., Mathis, C. A., Klunk, W. E., Kohsaka, S., Jucker, M., and Calhoun, M. E. (2008). Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 28, 4283–4292.
Boyles, J. K., Pitas, R. E., Wilson, E., Mahley, R. W., and Taylor, J. M. (1985). Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Invest. 76, 1501–1513.
Breitner, J. C. (2009). “A bi-directional effect of naproxen on the pathogenesis of AD: clinical and biological evidence from a randomized primary prevention trial,” in Clinical Trials on Alzheimer’s Disease, Las Vegas. Abstract O11.
Breitner, J. C., Haneuse, S. J., Walker, R., Dublin, S., Crane, P. K., Gray, S. L., and Larson, E. B. (2009). Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 72, 1899–1905.
Breteler, M. M. B., in Het Veld, B., Hofman, A., and Stricher, B. H. (2002). Aβ-42 peptide lowering NSAIDs and Alzheimer’s disease. Neurobiol. Aging 23, S286.
Cacci, E., Ajmone-Cat, M. A., Anelli, T., Biagioni, S., and Minghetti, L. (2008). In vitro neuronal and glial differentiation from embryonic or adult neural precursor cells are differently affected by chronic or acute activation of microglia. Glia 56, 412–425.
Carulla, N., Caddy, G. L., Hall, D. R., Zurdo, J., Gairí, M., Feliz, M., Giralt, E., Robinson, C. V., and Dobson, C. M. (2005). Molecular recycling within amyloid fibrils. Nature 436, 554–558.
Casper, D., Yaparpalvi, U., Rempel, N., and Werner, P. (2000). Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci. Lett. 289, 201–204.
Cornelius, C., Fastbom, J., Winblad, B., and Viitanen, M. (2004). Aspirin, NSAIDs, risk of dementia, and influence of the apolipoprotein E ε4 allele in an elderly population. Neuroepidemiology 23, 135–143.
Couzin, J. (2004). Clinical trials. Nail-biting time for trials of COX-2 drugs. Science 306, 1673–1675.
de Craen, A. J., Gussekloo, J., Vrijsen, B., and Westendorp, R. G. (2005). Meta-analysis of nonsteroidal antiinflammatory drug use and risk of dementia. Am. J. Epidemiol. 161, 114–120.
de Jong, D., Jansen, R., Hoefnagels, W., Jellesma-Eggenkamp, M., Verbeek, M., Borm, G., and Kremer, B. (2008). No effect of one-year treatment with indomethacin on Alzheimer’s disease progression: a randomized controlled trial. PLoS ONE 3, e1475. doi: 10.1371/journal.pone.0001475
Dill, J., Patel, A. R., Yang, X. L., Bachoo, R., Powell, C. M., and Li, S. (2010). A molecular mechanism for ibuprofen-mediated RhoA inhibition in neurons. J. Neurosci. 30, 963–972.
Doody, R. S. (2008). Cholinesterase inhibitors and memantine: best practices. CNS Spectr. 13(10 Suppl. 16), 34–35.
Ekdahl, C. T., Kokaia, Z., and Lindvall, O. (2009). Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience 158, 1021–1029.
El Khoury, J., and Luster, A. D. (2008). Mechanisms of microglia accumulation in Alzheimer’s disease: therapeutic implications. Trends Pharmacol. Sci. 29, 626–632.
El Khoury, J., Toft, M., Hickman, S. E., Means, T. K., Terada, K., Geula, C., and Luster, A. D. (2007). Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438.
Etminan, M., Gill, S., and Samii, A. (2003). Effect of non-steroidal antiinflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ 327, 128–132.
Finckh, A., and Aronson, M. D. (2005). Cardiovascular risks of cyclooxygenase-2 inhibitors: where we stand now. Ann. Intern. Med. 142, 212–214.
Fu, Q., Hue, J., and Li, S. (2007). Nonsteroidal anti-inflammatory drugs promote axon regeneration via RhoA inhibition. J. Neurosci. 27, 4154–4164.
Fuller, S., Steele, M., and Münch, G. (2009). Activated astroglia during chronic inflammation in Alzheimer’s disease-Do they neglect their neurosupportive roles? Mutat. Res. [Epub ahead of print].
Gómez-Isla, T., Blesa, R., Boada, M., Clarimón, J., Del Ser, T., Domenech, G., Ferro, J. M., Gómez-Ansón, B., Manubens, J. M., Martínez-Lage, J. M., Muñoz, D., Peña-Casanova, J., and Torres, F. (2008). TRIMCI Study Group. A randomized, double-blind, placebo controlled-trial of triflusal in mild cognitive impairment: the TRIMCI study. Alzheimer Dis. Assoc. Disord. 22, 21–29.
Grathwohl, S. A., Kälin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., Odenthal, J., Radde, R., Eldh, T., Gandy, S., Aguzzi, A., Staufenbiel, M., Mathews, P. M., Wolburg, H., Heppner, F. L., and Jucker, M. (2009). Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363.
Green, R. C., Schneider, L. S., Amato, D. A., Beelen, A. P., Wilcock, G., Swabb, E. A., and Zavitz, K. H. (2009). Tarenflurbil phase 3 study group. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302, 2557–2564.
Haag, M. D., van Oijen, M., de Jong, F. J., Hofman, A., Stijnen, T., and Stricker, B. H. (2006). Amyloid β42-level lowering non-steroidal anti-inflammatory drugs and the risk of Alzheimer’s disease. Alzheimers Dement. 2(Suppl. 1), S43 (Abstract O2-06-04).
Halliday, G. M., Shepherd, C. E., McCann, H., Reid, W. G., Grayson, D. A., Broe, G. A., and Kril, J. J. (2000). Effect of anti-inflammatory medications on neuropathological findings in Alzheimer disease. Arch. Neurol. 57, 831–836.
Hayden, K. M., Zandi, P. P., Khachaturian, A. S., Szekely, C. A., Fotuhi, M., Norton, M. C., Tschanz, J. T., Pieper, C. F., Corcoran, C., Lyketsos, C. G., Breitner, J. C., and Welsh-Bohmer, K. A. (2007). Cache County Investigators. Does NSAID use modify cognitive trajectories in the elderly? The Cache County study. Neurology 69, 275–282.
Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Hanke, A., Dewachter, I., Kuiperi, C., O’Banion, K., Klockgether, T., Van Leuven, F., and Landreth, G. E. (2005). Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1-42 levels in APPV717I transgenic mice. Brain 128, 1442–1453.
Hoozemans, J. J., Rozemuller, J. M., van Haastert, E. S., Veerhuis, R., and Eikelenboom, P. (2008). Cyclooxygenase-1 and -2 in the different stages of Alzheimer’s disease pathology. Curr. Pharm. Des. 14, 1419–1427.
Imbimbo, B. P. (2004). The potential role of non-steroidal anti-inflammatory drugs in treating Alzheimer’s disease. Expert Opin. Investig. Drugs 13, 1469–1481.
Imbimbo, B. P. (2009). Why did tarenflurbil fail in Alzheimer’s disease. J. Alzheimers Dis. 17, 757–760.
Imbimbo, B. P., Del Giudice, E., Colavito, D., D’Arrigo, A., Dalle Carbonare, M., Villetti, G., Facchinetti, F., Volta, R., Pietrini, V., Baroc, M. F., Serneels, L., De Strooper, B., and Leon, A. (2007). CHF5074, a novel γ-secretase modulator, reduces brain β-amyloid pathology in a transgenic mouse model of Alzheimer’s disease without causing peripheral toxicity. J. Pharmacol. Exp. Ther. 323, 822–830.
in t’ Veld, B. A., Ruitenberg, A., Hofman, A., Launer, L. J., Van Duijn, C. M., Stijnen, T., Breteler, M. M., and Stricker, B. H. (2001). Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 345, 515–521.
Iwata, Y., Nicole, O., Zurakowski, D., Okamura, T., Jonas, R. A., Iwata, Y., Nicole, O., Zurakowski, D., Okamura, T., and Jonas, R. A. (2010). Ibuprofen for neuroprotection after cerebral ischemia. J. Thorac. Cardiovasc. Surg. 139, 489–493.
Jiang, Q., Lee, C. Y., Mandrekar, S., Wilkinson, B., Cramer, P., Zelcer, N., Mann, K., Lamb, B., Willson, T. M., Collins, J. L., Richardson, J. C., Smith, J. D., Comery, T. A., Riddell, D., Holtzman, D. M., Tontonoz, P., and Landreth, G. E. (2008). ApoE promotes the proteolytic degradation of Aβ. Neuron 58, 681–693.
Jin, K., Peel, A. L., Mao, X. O., Xie, L., Cottrell, B. A., Henshall, D. C., and Greenberg, D. A. (2004). Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 343–347.
Khansari, P. S., and Halliwell, R. F. (2009). Evidence for neuroprotection by the fenamate NSAID, mefenamic acid. Neurochem. Int. 55, 683–688.
Koenigsknecht-Talboo, J., and Landreth, G. E. (2005). Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 25, 8240–8249.
Koistinaho, M., Lin, S., Wu, X., Esterman, M., Koger, D., Hanson, J., Higgs, R., Liu, F., Malkani, S., Bales, K. R., and Paul, S. M. (2004). Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 10, 719–726.
Kotilinek, L. A., Westerman, M. A., Wang, Q., Panizzon, K., Lim, G. P., Simonyi, A., Lesne, S., Falinska, A., Younkin, L. H., Younkin, S. G., Rowan, M., Cleary, J., Wallis, R. A., Sun, G. Y., Cole, G., Frautschy, S., Anwyl, R., and Ashe, K. H. (2008). Cyclooxygenase-2 inhibition improves amyloid-β-mediated suppression of memory and synaptic plasticity. Brain 131, 651–664.
Kuchibhotla, K. V., Lattarulo, C. R., Hyman, B. T., and Bacskai, B. J. (2009). Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215.
Kukar, T. L., Ladd, T. B., Bann, M. A., Fraering, P. C., Narlawar, R., Maharvi, G. M., Healy, B., Chapman, R., Welzel, A. T., Price, R. W., Moore, B., Rangachari, V., Cusack, B., Eriksen, J., Jansen-West, K., Verbeeck, C., Yager, D., Eckman, C., Ye, W., Sagi, S., Cottrell, B. A., Torpey, J., Rosenberry, T. L., Fauq, A., Wolfe, M. S., Schmidt, B., Walsh, D. M., Koo, E. H., and Golde, T. E. (2008). Substrate-targeting γ-secretase modulators. Nature 453, 925–929.
Landi, F., Cesari, M., Onder, G., Russo, A., Torre, S., and Bernabei, R. (2003). Non-steroidal anti-inflammatory drug (NSAID) use and Alzheimer disease in community-dwelling elderly patients. Am. J. Geriatr. Psychiatry 11, 179–185.
Lichtenstein, M., Carriba, P., Baltrons, M. A., Wojciak-Stothard, B., Peterson, J. R., García, A., and Galea, E. (2009). “Secretase-independent and Rho GTPase-dependent regulation of cytoskeleton dynamics in astrocytes by ibuprofen and derivatives,” in XIII Congreso of the Sociedad Espanola de Neurociencia (Tarragona, Spain). September 16–19.
Mackenzie, I. R., and Munoz, D. G. (1998). Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology 50, 986–990.
Maillard, M., and Burnier, M. (2006). Comparative cardiovascular safety of traditional nonsteroidal anti-inflammatory drugs. Expert Opin. Drug. Saf. 5, 83–94.
Malm, T. M., Koistinaho, M., Parepalo, M., Vatanen, T., Ooka, A., Karlsson, S., and Koistinaho, J. (2005). Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to β-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol. Dis. 18, 134–142.
Mandrekar, S., Jiang, Q., Lee, C. Y., Koenigsknecht-Talboo, J., Holtzman, D. M., and Landreth, G. E. (2009). Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 29, 4252–4262.
Masters, C. L., Wyss-Coray, T., Giulio Pasinetti, G. M. (2008). Anti-inflammatory drugs fall short in Alzheimer’s disease. Nat. Med. 14, 916.
McGeer, P. L., Harada, N., Kimura, H., Mcgeer, E., and Schulzer, M. (1992). Prevalence of dementia amongst elderly Japanese with leprosy: apparent effect of chronic drug therapy. Dementia 3, 146–149.
McGettigan, P., and Henry, D. (2006). Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA 296, 1633–1644.
McShea, A., Harris, P. L., Webster, K. R., Wahl, A. F., and Smith, M. A. (1997). Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am. J. Pathol. 150, 1933–1939.
Meyer-Luehmann, M., Spires-Jones, T. L., Prada, C., Garcia-Alloza, M., de Calignon, A., Rozkalne, A., Koenigsknecht-Talboo, J., Holtzman, D. M., Bacskai, B. J., and Hyman, B. T. (2008). Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724.
Morihara, T., Teter, B., Yang, F., Lim, G. P., Boudinot, S., Boudinot, F. D., Frautschy, S. A., and Cole, G. M. (2005). Ibuprofen suppresses interleukin-1β induction of pro-amyloidogenic α1-antichymotrypsin to ameliorate β-amyloid (Aβ) pathology in Alzheimer’s models. Neuropsychopharmacology 30, 1111–1120.
Nilsson, L. N., Arendash, G. W., Leighty, R. E., Costa, D. A., Low, M. A., Garcia, M. F., Cracciolo, J. R., Rojiani, A., Wu, X., Bales, K. R., Paul, S. M., and Potter, H. (2004). Cognitive impairment in PDAPP mice depends on ApoE and ACT-catalyzed amyloid formation. Neurobiol. Aging 25, 1153–1167.
Olah, M., Ping, G., De Haas, A. H., Brouwer, N., Meerlo, P., Van Der Zee, E. A., Biber, K., and Boddeke, H. W. (2009). Enhanced hippocampal neurogenesis in the absence of microglia T cell interaction and microglia activation in the murine running wheel model. Glia 57, 1046–1061.
Opar, A. (2008). Mixed results for disease-modification strategies for Alzheimer’s disease. Nat. Rev. Drug Discov. 7, 717–718.
Pasqualetti, P., Bonomini, C., Dal Forno, G., Paulon, L., Sinforiani, E., Marra, C., Zanetti, O., and Rossini, P. M. (2009). A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer’s disease. Aging Clin. Exp. Res. 21, 102–110.
Reiman, E. M., Chen, K., Liu, X., Bandy, D., Yu, M., Lee, W., Ayutyanont, N., Keppler, J., Reeder, S. A., Langbaum, J. B., Alexander, G. E., Klunk, W. E., Mathis, C. A., Price, J. C., Aizenstein, H. J., DeKosky, S. T., and Caselli, R. J. (2009). Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 106, 6820–6825.
Reines, S. A., Block, G. A., Morris, J. C., Liu, G., Nessly, M. L., Lines, C. R., Norman, B. A., and Baranak, C. C. (2004). Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 62, 66–71.
Richardson, R. L., Kim, E. M., Shephard, R. A., Gardiner, T., Cleary, J., and O’Hare, E. (2002). Behavioural and histopathological analyses of ibuprofen treatment on the effect of aggregated Aβ1-42 injections in the rat. Brain Res. 954, 1–10.
Rogers, J., Kirby, L, C., Hempelman, S, R., Berry, D, L., Mcgeer, P, L., Kaszniak, A, W., Zalinski, J., Cofield, M., Mansukhani, L., Willson, P., and Kogan, F. (1993). Clinical trial of indomethacin in Alzheimer’s disease. Neurology 43, 1609–1611.
Sainati, S. M., Ingram, D. M., Talwalker, S., and Geis, G. (2000). “Results of a double-blind, randomized, placebo-controlled study of celecoxib in the treatment of progression of Alzheimer’s disease,” in 6th International Stockholm/Springfield Symposium on Advances in Alzheimer Therapy (Stockholm, Sweden) April 5–8, Abstract Book (p. 180).
Sanz-Blasco, S., Valero, R. A., Rodríguez-Crespo, I., Villalobos, C., and Núñez, L. (2008). Mitochondrial Ca2+ overload underlies Aβ oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 3: e2718. doi:10.1371/journal.pone.0002718.
Sasaki, T., Kitagawa, K., Sugiura, S., Omura-Matsuoka, E., Tanaka, S., Yagita, Y., Okano, H., Matsumoto, M., and Hori, M. (2003). Implication of cyclooxygenase-2 on enhanced proliferation of neural progenitor cells in the adult mouse hippocampus after ischemia. J. Neurosci. Res. 72, 461–471.
Sastre, M., Dewachter, I., Landreth, G. E., Willson, T. M., Klockgether, T., Van Leuven, F., and Heneka, M. T. (2003). Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-γ agonists modulate immunostimulated processing of amyloid precursor protein through regulation of β-secretase. J. Neurosci. 23, 9796–9804.
Sastre, M., Dewachter, I., Rossner, S., Bogdanovic, N., Rosen, E., Borghgraef, P., Evert, B. O., Dumitrescu-Ozimek, L., Thal, D. R., Landreth, G., Walter, J., Klockgether, T., van Leuven, F., and Heneka, M. T. (2006). Nonsteroidal anti-inflammatory drugs repress β-secretase gene promoter activity by the activation of PPARγ. Proc. Natl. Acad. Sci. U.S.A. 103, 443–448.
Scharf, S., Mander, A., Ugoni, A., Vajda, F., and Christophidis, N. P. (1999). A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 53, 197–201.
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., Brett, F. M., Farrell, M. A., Rowan, M. J., Lemere, C. A., Regan, C. M., Walsh, D. M., Sabatini, B. L., and Selkoe, D. J. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842.
Shors, T. J., Miesegaes, G., Beylin, A., Zhao, M., Rydel, T., and Gould, E. (2001). Neurogenesis in the adult is involved in the formation of trace memories. Nature 410, 372–376.
Simard, A. R., Soulet, D., Gowing, G., Julien, J. P., and Rivest, S. (2006). Bone marrow derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49, 489–502.
Small, G. W., Siddarth, P., Silverman, D. H., Ercoli, L. M., Miller, K. J., Lavretsky, H., Bookheimer, S. Y., Huang, S. C., Barrio, J. R., and Phelps, M. E. (2008). Cognitive and cerebral metabolic effects of celecoxib versus placebo in people with age-related memory loss: randomized controlled study. Am. J. Geriatr. Psychiatry 16, 999–1009.
Small, S. A., and Duff, K. (2008). Linking Aβ and tau in late-onset alzheimer’s disease: a dual pathway hypothesis. Neuron 60, 534–542.
Soininen, H., West, C., Robbins, J., and Niculescu, L. (2007). Long-term efficacy and safety of celecoxib in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 23, 8–21.
Stéphan, A., Laroche, S., and Davis, S. (2003). Learning deficits and dysfunctional synaptic plasticity induced by aggregated amyloid deposits in the dentate gyrus are rescued by chronic treatment with indomethacin. Eur. J. Neurosci. 17, 1921–1927.
Stewart, W. F., Kawas, C., Corrada, M., and Metter, E. J. (1997). Risk of Alzheimer’s disease and duration of NSAID use. Neurology 48, 626–632.
Sung, S., Yang, H., Uryu, K., Lee, E. B., Zhao, L., Shineman, D., Trojanowski, J. Q., Lee, V. M., and Praticò, D. (2004). Modulation of NF-κB activity by indomethacin influences Aβ levels but not APP metabolism in a model of Alzheimer disease. Am. J. Pathol. 165, 2197–2206.
Szekely, C. A., Breitner, J. C., Fitzpatrick, A. L., Rea, T. D., Psaty, B. M., Kuller, L. H., and Zandi, P. P. (2008a). NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology 70, 17–24.
Szekely, C. A., Green, R. C., Breitner, J. C., Østbye, T., Beiser, A. S., Corrada, M. M., Dodge, H. H., Ganguli, M., Kawas, C. H., Kuller, L. H., Psaty, B. M., Resnick, S. M., Wolf, P. A., Zonderman, A. B., Welsh-Bohmer, K. A., and Zandi, P. P. (2008b). No advantage of Aβ42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology 70, 2291–2298.
Szekely, C. A., Thorne, J. E., Zandi, P. P., Ek, M., Messias, E., Breitner, J. C., and Goodman, S. N. (2004). Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: a systematic review. Neuroepidemiology 23, 159–169.
Thal, L. J., Ferris, S. H., Kirby, L., Block, G. A., Lines, C. R., Yuen, E., Assaid, C., Nessly, M. L., Norman, B. A., Baranak, C. C., and Reines, S. A. (2005). Rofecoxib Protocol 078 study group. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 30, 1204–1215.
Thomas, T., Nadackal, T. G., and Thomas, K. (2001). Aspirin and non-steroidal anti-inflammatory drugs inhibit amyloid-β aggregation. Neuroreport 12, 3263–3267.
Thored, P., Heldmann, U., Gomes-Leal, W., Gisler, R., Darsalia, V., Taneera, J., Nygren, J. M., Jacobsen, S. E., Ekdahl, C. T., Kokaia, Z., and Lindvall, O. (2009). Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 57, 835–849.
Van Gool, W. A., Weinstein, H. C., Scheltens, P., Walstra, G. J., and Scheltens, P. K. (2001). Effect of hydroxychloroquine on progression of dementia in early Alzheimer’s disease: an 18 month randomised, double-blind, placebo-controlled study. Lancet 358, 455–460.
Varvel, N. H., Bhaskar, K., Kounnas, M. Z., Wagner, S. L., Yang, Y., Lamb, B. T., and Herrup, K. (2009). NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J. Clin. Invest. 119, 3692–3702.
Vlad, S. C., Miller, D. R., Kowall, N. W., and Felson, D. T. (2008). Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70, 1672–1677.
Weggen, S., Czirr, E., Leuchtenberger, S., and Eriksen, J. (2007). “Nonsteroidal anti-inflammatory drugs (NSAIDs) and derived Aβ42-lowering molecules for treatment and prevention of Alzheimer’s disease (AD),” in Pharmacological Mechanisms in Alzheimer’s Therapeutics, ed. C. A. Cuello (New York: Springer), 167–193.
Weggen, S., Eriksen, J. L., Das, P., Sagi, S. A., Wang, R., Pietrzik, C. U., Findlay, K. A., Smith, T. E., Murphy, M. P., Bulter, T., Kang, D. E., Marquez-Sterling, N., Golde, T. E., and Koo, E. H. (2001). A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216.
Wilcock, G. K., Black, S. E., Balch, A. H., Amato, D. A., Beelen, A. P., Schneider, L. S., Green, R. C., Swabb, E. A., and Zavitz, K. H. (2009). Safety and efficacy of tarenflurbil in subjects with mild Alzheimer’s disease: results from an 18-month international multi-center Phase 3 trial. Alzheimers Dement. 5(Suppl. 1), P86 (Abstract O1-04-07).
Wilcock, G. K., Black, S. E., Hendrix, S. B., Zavitz, K. H., Swabb, E. A., and Laughlin, M. A. (2008). Efficacy and safety of tarenflurbil in mild to moderate Alzheimer’s disease: a randomised phase II trial. Lancet Neurol. 7, 483–493.
Wyss-Coray, T. (2006). Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med. 12, 1005–1015.
Wyss-Coray, T., and Mucke, L. (2002). Inflammation in neurodegenerative disease-a double-edged sword. Neuron 35, 419–432.
Yang, Y., Geldmacher, D. S., and Herrup, K. (2001). DNA replication precedes neuronal cell death in Alzheimer’s disease. J. Neurosci. 21, 2661–2668.
Yip, A. G., Green, R. C., Huyck, M., Cupples, L. A., and Farrer, L. A. (2005). MIRAGE Study Group. Nonsteroidal anti-inflammatory drug use and Alzheimer’s disease risk: the MIRAGE Study. BMC Geriatr. 5, 2.
Zandi, P. P., Anthony, J. C., Hayden, K. M., Mehta, K., Mayer, L., and Breitner, J. C. (2002). Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology 59, 880–886.
Zhao, L., Lin, S., Bales, K. R., Gelfanova, V., Koger, D., Delong, C., Hale, J., Liu, F., Hunter, J. M., and Paul, S. M. (2009). Macrophage-mediated degradation of β-amyloid via an apolipoprotein E isoform-dependent mechanism. J. Neurosci. 29, 3603–3612.
Keywords: Alzheimer’s disease, non-steroidal anti-inflammatory drugs, β-amyloid, microglia
Citation: Imbimbo BP, Solfrizzi V and Panza F (2010) Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front. Ag. Neurosci. 2:19. doi: 10.3389/fnagi.2010.00019
Received: 24 February 2010;
Paper pending published: 08 April 2010;
Accepted: 23 April 2010;
Published online: 21 May 2010
Edited by:
Elena Galea, Universitat Autònoma de Barcelona, SpainReviewed by:
Mathieu Lichtenstein, Universitat Autònoma de Barcelona, SpainMerce Boada, Institut Català de Neurociències Aplicades, Spain
Elena Galea, Universitat Autònoma de Barcelona, Spain
Copyright: © 2010 Imbimbo, Solfrizzi and Panza. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Bruno P. Imbimbo, Research and Development Department, Chiesi Farmaceutici, Via Palermo 26/A, 43100 Parma, Italy. e-mail: b.imbimbo@chiesigroup.com