Staging anti-inflammatory therapy in Alzheimer’s disease
- 1 Institut de Neurociències, Universitat Autònoma de Barcelona, Barcelona, Spain
- 2 Institut Catalá de Recerca i Estudis Avançats, Barcelona, Spain
- 3 Institut d’Investigació Biomèdica de Bellvitge, Hospitalet de Llobregat, Barcelona, Spain
The use of non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease (AD) is controversial because conclusions from numerous epidemiological studies reporting delayed onset of AD in NSAID users have not been corroborated in clinical trials. The purpose of this personal view is to revise the case for NSAIDs in AD therapeutics in light of: (i) the last report from the only primary prevention trial in AD, ADAPT, which, although incomplete, points to significant protection in long-term naproxen users, and (ii) the recently proposed dynamic model of AD evolution. The model contends that there is a clinical silent phase in AD that can last up to 20 years, the duration depending on life style habits, genetic factors, or cognitive reserve. The failure of many purported disease-modifying drugs in AD clinical trials is forcing the view that treatments will only be efficacious if administered pre-clinically. Here we will argue that NSAIDs failed in clinical trials because they are disease-modifying drugs, and they should be administered in early stages of the disease. A complete prevention trial in cognitively normal individuals is thus called for. Further, the shift of anti-inflammatory treatment to early stages uncovers a knowledge void about the targets of NSAIDs in asymptomatic individuals. AD researchers have mostly relied on post-mortem analysis of Aβ plaque-laden brains from demented patients or animal models, thus drawing conclusions about AD pathogenesis based on late symptoms. We will discuss evidence in support that defective, not excessive, inflammation underlies AD pathogenesis, that NSAIDs are multifunctional drugs acting on inflammatory and non-inflammatory targets, and that astrocytes and microglia may play differing roles in disease progression, with an emphasis of ApoEε4 as a key, undervalued target of NSAIDs. According to a meta-analysis of epidemiological data, NSAIDs afford an average protection of 58%. If this figure is true, and translated into patient numbers, NSAID treatment may revive as a worth pursuing strategy to significantly reduce the socio-economical burden imposed by AD.
Introduction
A group of leading Alzheimer’s disease (AD) experts have recently integrated available information about the five best characterized biomarkers into a dynamic model of disease evolution overtime (Jack et al., 2010). This groundbreaking contribution provides a framework to select individuals for clinical trials, and decide upon outcome measurements. According to the model, AD progresses in a continuum where stages can be defined by biomarkers. There is a damaging phase wherein amyloid β(Aβ) and hyperphosphorylated tau accumulate (phase 1), followed by a phase of synaptic and metabolic alterations (phase 2), which leads to a final stage when clinical symptoms – cognitive impairment and brain atrophy – are detected (phase 3). This model fairly recapitulates the emerging view that there is a clinically silent phase in AD that can last up 20 years before dementia is manifest. Henceforth, any therapy for AD will need to be contrasted with this paradigm. This is the case of non-steroidal anti-inflammatory drugs (NSAIDs). The field of neuroinflammation in AD has taken several unexpected turns from the Rotterdam epidemiological study reporting, in 2001, a 80% decrease in the risk of developing AD in long-term users of NSAIDs, to the ensuing failure of some NSAIDs and derivatives like R-flurbiprofen in phase III clinical trials. In this review we will argue that wrong timing of drug administration, incomplete knowledge, and biased assumptions about of the role of neuroinflammation in neurodegeneration may have led to the current impasse. Revision of anti-inflammatory treatments in the light of the dynamic model of AD progression will thus provide new research and clinical directions.
Epidemiological Data and Clinical Trials
The epidemiological studies and clinical trials that, in striking number – over 40 –, have been developed to examine the benefits of NSAID in AD have been thoroughly described elsewhere (Imbimbo et al., 2010). The results are paradoxical: while the epidemiological data points to a reduced incidence of AD in NSAID users, most of the ensuing clinical trials in AD or mild cognitive impairment (MCI) have shown no effect or even detrimental effects. These results have casted serious doubts on epidemiological analyses and, therefore, on NSAID-based therapeutics for AD after the initial hype in the 90s. In hindsight, among the several explanations put forth to explain the discrepancy between epidemiological data and clinical trials – including wrong choice of NSAID or dosage in clinical trials, recall bias in epidemiological studies, or that arthritis, not NSAIDs, is the protective factor – a research group in Baltimore may have got it right. They argued that, since protection was only observed after sustained uptake well before the onset of AD, NSAIDs were preventive and would not work in clinical trials with patients. To test this idea, they launched in 2000 a primary prevention trial, the ADAPT trial, to test the effect of naproxen (a dual cycloxygenase type 1, COX1 and 2, COX2 inhibitor) and celecoxib (a selective COX2 inhibitor) in healthy volunteers, 70-years-old in average. Unfortunately, the ADAPT trial was cancelled after 2 years and a half over concerns of cardiovascular damage by COX2 inhibitors, although the group have kept reporting the progression of AD incidence overtime. At the time of cancellation, no protection was observed by NSAID, and even the cohort taking naproxen appeared to fare worse than the placebo group. However, 2 years later (i.e., 4 years after the start of the treatment) the data indicated (70% protection in naproxen users (Hayden et al., 2007). That is, naproxen conferred protection to people in the path to have dementia if they were at least 4 years shy of developing clinical signs. The evidence supports that NSAIDs need to be taken during preclinical phases of AD, and would validate the conclusions of epidemiological data. It is worth stressing that numerous clinical trials for AD with purported disease-modifying drugs have spectacularly failed in the last years (Sabbagh, 2009). These sobering results have prompted the view that disease-modifying drugs, including NSAIDs, will be efficacious only if administered preventively before neurodegeneration is well advanced. According to a meta-analysis of epidemiological data (Szekely et al., 2004), NSAIDs reduce AD incidence by an average of 58%. If this figure is translated into patient numbers, anti-inflammatory treatment arises as a worth pursuing strategy to significantly reduce the socio-economical burden caused by AD. But first, a complete prevention trial is necessary. The ADAPT trial illustrates the difficulties of primary prevention trial design, including selecting individuals on their way toward AD, tracking disease progression, and deciding upon most efficient treatment drugs and protocols. Considering that the average onset of AD is 65- to 70-years-old, and that NSAIDs work only if taken 4 years before the disease is clinically detected, participants in the prevention trial should be 60- to 65-years-old. Are currently available cognitive, neuroimaging, and blood tests valid to include individuals in the trial, and to measure outcomes? The experience accumulated by the Alzheimer’s Disease Cooperative Study (ADCS) dictates that Aβ42 contents in cerebrospinal fluid (CSF) can be used for subject selection ((193 pg/ml, cut-off value), while fluorodeoxyglucose (FDG) contents and hippocampal volume are appropriate surrogate markers for disease progression (Aisen, 2010; Jack et al., 2010). The former can be traced by positron emission tomography (PET), while the latter is routinely assessed with magnetic resonance (MRI). Importantly, participants should carry the allele ε4 of apolipoprotein E (ApoEε4) because, according to epidemiological data, NSAIDs are effective only in this subpopulation (In’t Veld et al., 2001; Yip et al., 2005; Hayden et al., 2007; Szekely et al., 2008), which brings up the next question about the target of NSAIDs.
Molecular, Cellular, and Functional Targets of NSAIDs
The quest for the underpinnings of NSAID-mediated protection is confounded by two factors. One, the realization that AD progresses in stages implies that NSAID actions may differ with the stage of disease progression. Two, NSAIDs may be multifunctional drugs targeting non-inflammatory molecules aside from cyclooxygenases (COXs).
In the model of AD progression, asymptomatic phases 1 and 2 are the target zones for preventive therapy (Figure 1), and presumably where NSAIDs are acting upon according to epidemiological data. Decreased CSF-Aβ42 defines phase 1, while increased CSF-tau, decreased PET-FDG, and decrease of hippocampal volume are signs of neuronal injury and dysfunction in phase 2 (Jack et al., 2010). The “inflammation” associated to these preclinical phases is not well characterized. In general, “neuroinflammation” has grown to be a too wide term encompassing a body of standard reactions against tissue damage or infection, with little consideration for specifics of disease, stage of disease progression, brain area, or cell type. In AD, researchers have mostly relied on post-mortem brains from demented patients, plaque-laden transgenic models, or cell cultures. The use of these materials has imposed a biased view of inflammation in AD pathogenesis, based on observations pertaining phase 3, which place microglia-derived neurotoxins as culprit of the disease (see below). A single study illustrates this misconception. Jacobsen et al. (2006) have reported dendrite damage, memory deficits and behavioral alterations in a transgenic-mouse model of AD months before Aβ plaques appear. Glial cells, the holders of the innate immune system, could not account for this early damage because reactive astrocytes and microglia were visible later, in parallel with plaque deposition. Whether a subtle earlier glia response went unnoticed, or if a second wave of damage followed the plaque-associated canonical inflammation was not determined. In order to understand the modus operandi of NSAIDs it is necessary to: (i) define stages 1–3 in animal models, and (ii) focus on asymptomatic or early stages in disease progression in mice and humans. Specifically, we have to clarify the role of glial cells on: (i) mitochondrial damage, production of reactive oxygen species (ROS), and oxidative lesions to proteins or nucleic acids, (ii) metabolic impairment, as defined by PET-FDG, and (iii) decreases in hippocampal volume, all preclinical or early signs of damage in humans (Fox et al., 1999; Nunomura et al., 2001; Petrie et al., 2009; Martinez et al., 2010). In AD transgenic mice, oxidative damage to lipids occurs before Aβ deposition (Pratico et al., 2001), and an interplay exists between Aβ production and oxidative damage (Tamagno et al., 2002; Lustbader et al., 2004). Whether glial cells reinforce or attenuate these cascades, and if astrocytes and microglia have different roles, remain unknown.
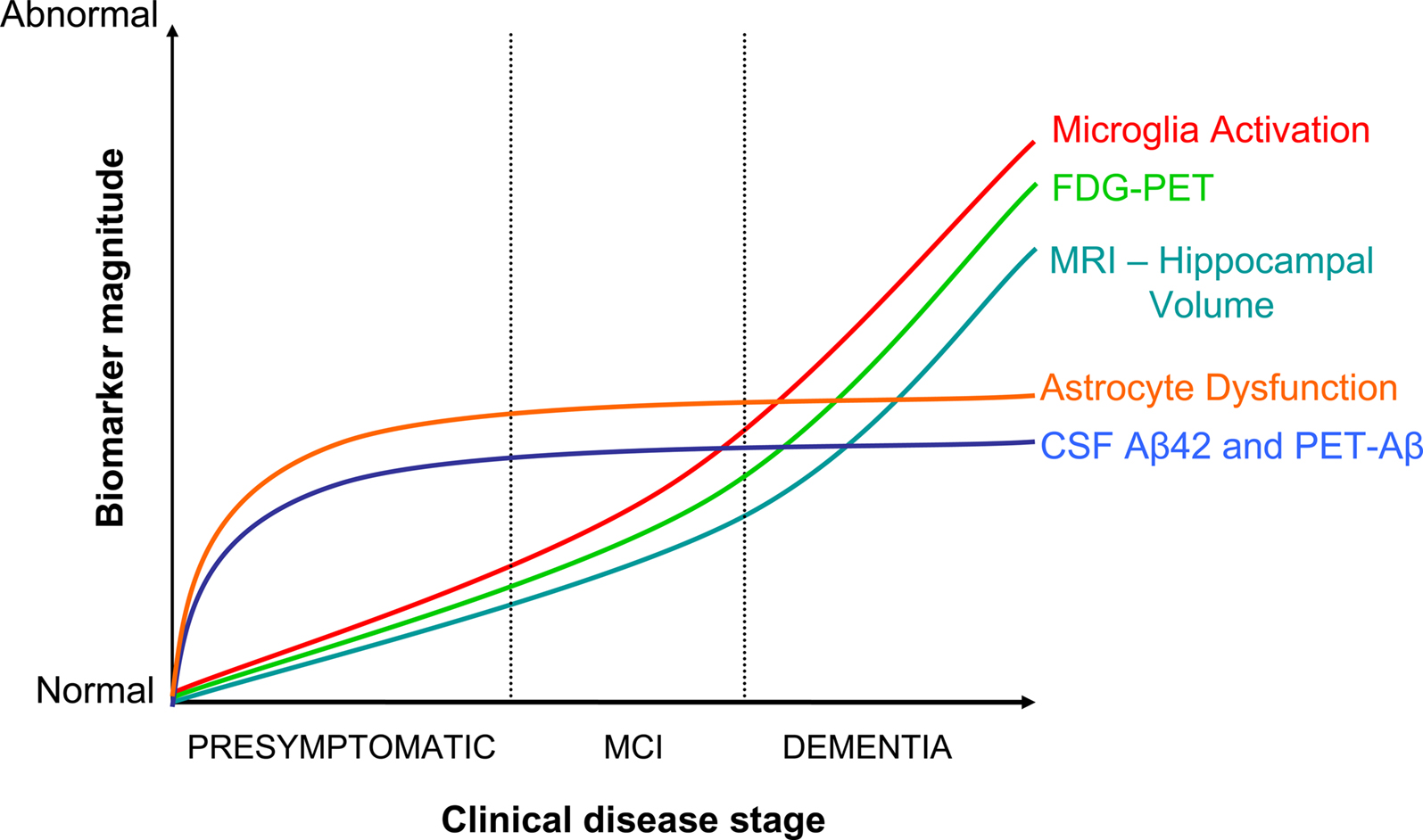
Figure 1. Integration of NSAID actions in the dynamic model of AD evolution. Adapted from Jack et al. (2010). Dysfunctional astrocytes would exert a primary role in early disease events by promoting Aβ accumulation, and by affecting neurovascular coupling, metabolic homeostasis, or synaptic plasticity. Microglia activation would be secondary to Aβ accumulation and neuronal damage. NSAID effects would depend on the stage of disease progression. Initially, the drugs would be beneficial by counteracting ApoEε4-mediated detrimental effects, whereas in advanced stages NSAID may offer no protection, or become detrimental by further blocking faulty microglia/myeloid cell attempts at Aβ clearance and tissue repair.
The possible multifunctional nature of NSAIDs is supported by cumulative evidence showing that the drugs, other than COX, can target γ-secretase (Weggen et al., 2003), Rho-GTPases (Fu et al., 2007), and peroxisome proliferator-activated receptors (PPAR) (Nicolakakis et al., 2008). The γ-secretase mediates production of Aβ, while Rho-GTPases regulate several phenomena relevant to AD including axon growth (Fu et al., 2007), tau phosphorylation (Sayas et al., 1999), and astrocyte motility (Lichtenstein et al., 2010). Finally, PPARs are powerful modulators of inflammation (Pascual et al., 2005), oxidative stress (Nunomura et al., 2001; Kang et al., 2005), and Aβ production via BACE (Sastre et al., 2006).
The real molecular target of NSAIDs has been arguably the most elusive question in anti-inflammatory therapeutics in AD. In the early 90s it was thought that NSAIDs prevented via COX2 the detrimental effects of chronic microglia activation. The failure of clinical trials with COX2 inhibitors and corticosteroids, together with the fortuitous finding that some NSAIDs modulated γ-secretase, thereby decreasing the production of Aβ42, prompted researchers and companies to modify the chemical structure of NSAIDs to increase specificity and potency toward γ-secretase. R-flurbiprofen was born (Kukar et al., 2007) but failed in Phase III clinical trial (Green et al., 2009), probably because, as we believe now, disease-modifying drugs are only effective pre-clinically. Of note, naproxen is not a γ-secretase modulator (Takahashi et al., 2003), suggesting that the protective effects revealed by the recent ADAPT trial follow-up are γ-secretase-independent. Whether NSAIDs act on PPAR receptors and/or Rho-GTPases in vivo is unknown, but it is desirable that they did, in view of the large number of possible beneficial actions (Table 1). Efforts to streamline NSAID specificity may thus render therapeutically weaker drugs, and should await further characterization of NSAID targets. Finally, COX1 should be redeemed back for now as a possible target in view of recent evidence indicating robust protective effects of triflusal in a AD mouse model (Choi et al., 2009; Coma et al., 2010).
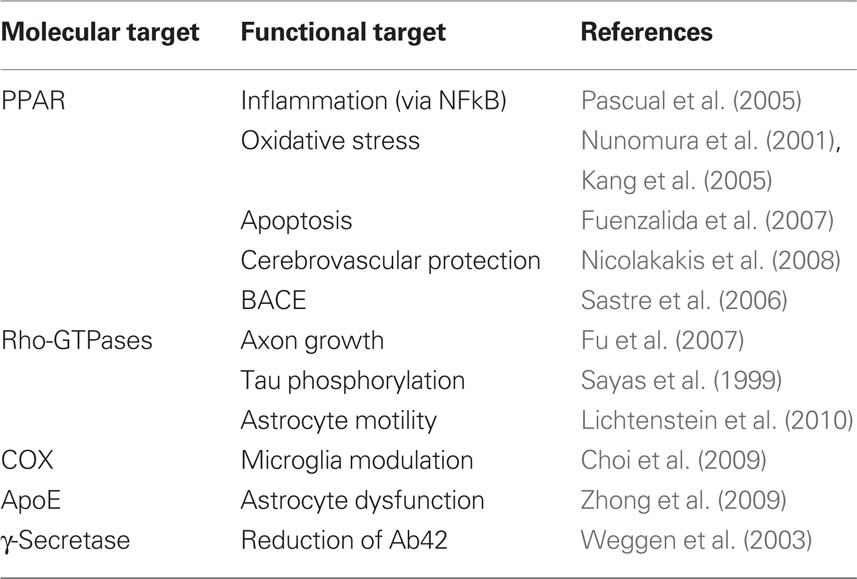
Table 1. Possible targets of NSAIDs.
The Role of ApoEξ4
The startling conclusion from the epidemiological data that NSAIDs are protective exclusively in ApoEε4 carriers (In’t Veld et al., 2001; Yip et al., 2005; Hayden et al., 2007; Szekely et al., 2008) places the lipoprotein as a possible target of NSAIDs. Although inheritance of ApoEε4 allele is the strongest known risk factor for the development of sporadic AD (Ertekin-Taner, 2010), the mechanisms underlying this correlation are not well understood. ApoEε4 carriers actually experience memory loss beginning in their fifties (Caselli et al., 2009), strongly indicating that ApoEε4-related damage is an early event in disease progression. ApoE controls cholesterol homeostasis. Most of the evidence indicates that ApoE promotes the clearance and/or degradation of Aβ via the physical interaction between the two proteins, and that ApoEε4 performs this function worse than ApoEε3 or ApoEε2 (Kim et al., 2009), thereby contributing to Aβ accumulation in the brain. Moreover, ApoE is immunomodulatory (Pocivavsek et al., 2009), and the allele matters: ApoEε4 is associated to greater production of pro-inflammatory cytokines than ApoEε3 in mice challenged with lipopolysaccharide (Vitek et al., 2009). This indicates that apoEε4 protein may alter inflammation partly by dose effects (i.e., a net decrease in ApoE production), and partly by being qualitatively different than ApoEε3.
Surprisingly, despite this evidence, ApoEε4 has not been at the center stage of AD therapeutics. The principal cells that secrete ApoE in the brain are astrocytes and, to a lesser extent, microglia. A recent structural analysis of ApoE isoforms has revealed that domain interaction and protein misfolding – caused by a difference in a single amino acid – distinguish ApoEε4 from ApoEε3 and ApoEε2 (Zhong et al., 2009). The introduction of an ApoEε4-like domain interaction in mice results in endoplasmic reticulum (ER) stress in astrocytes, associated to severe cognitive deficits (Zhong et al., 2009). This indicates that astrocyte dysfunction affecting neurovascular coupling, synaptic plasticity, metabolic homeostasis – all phenomena controlled by astrocytes –, along with impaired clearance of Aβ and altered inflammation, may underlie ApoEε4-related damage in AD. Conversion of ApoEε4 into ApoEε3 by disrupting the domain interaction with small molecules is currently being pursued as a therapeutic approach (Ye et al., 2005). It is tempting to speculate that some NSAIDs may act as domain-interaction disruptors, and/or reverse early astrocyte alterations associated to ApoEε4 expression.
The Role of Microglia
The clustering of microglia around plaques in post-mortem AD brains (Haga et al., 1989) has contributed to three central tenets in the field of neuroinflammation and AD. One is that microglia, challenged by Aβ, release neurotoxic factors like ROS, reactive nitrogen species, and elements of the complement system, as well as inflammatory mediators including interleukin-1β (IL-1β), monocyte chemotactic protein-1 (MCP-1), interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), which engage other cells, including astrocytes and vascular cells, in a complex interplay of signaling loops that amplify the inflammatory reaction, and cause great damage to neurons. The second tenet is that pathology arises when microglia is chronically activated, either due to persistence of the inducers, or a failure in the resolution mechanisms. And the third tenet is that microglia contributes to removal of Aβ plaques by phagocytosis. The terms “classic” and “alternative” have been coined to define the pro-inflammatory versus the phagocytic status of microglia activation, the latter being associated to production of growth factors (De et al., 2003). Careful perusal of the evidence offers, however, the following, alternative angles to these tenets.
While a wealth of data from brains from diseased individuals and mouse models demonstrates that microglia express pro-inflammatory mediators, data is scant in support that these cause disease; information is also lacking about the dynamic interplay of functional microglia phenotypes overtime. The noxious role of microglia is largely based on: (i) immunohistochemical correlations (Griffin et al., 1995; Arends et al., 2000; Vehmas et al., 2003); and (ii) in vitro findings, cultures being a pathological condition because the mechanical stress implicit in the isolation procedure renders microglia overly reactive (Streit, 2010). Moreover, cultures are prepared from postnatal microglia, which may not recapitulate the decline of microglial functions in aging (Sastre and Gentleman, 2010). Alternatively, mitochondrial alterations and oxidative damage, the early signs of damage in AD, may be directly caused by soluble Aβ (Tamagno et al., 2002; Lustbader et al., 2004), and/or be secondary to vascular, metabolic, and synaptic alterations caused by ApoEε4-harboring dysfunctional astrocytes. As to the notion that sustained inflammation is detrimental, a recent study reports that chronic overexpression of IL-1( in the hippocampus of APP/PS1 transgenic animals results in decreased plaque burden and insoluble Aβ peptide, with no alterations in A( processing or APP expression (Shaftel et al., 2007). The increased number of peri-plaque microglia points to increased Aβ phagocytosis as the underlying mechanism, consistent with the observation that microglia expresses phagocytic markers (Jimenez et al., 2008). Thus, there is a basis to support that microglia are phagocytes, but the following points have to be made.
(i) Resident microglia has little phagocytic capacity toward Aβ according to animal models (Simard et al., 2006; Majumdar et al., 2008; Grathwohl et al., 2009). The phagocytic potential can be enhanced or restored with inflammatory agents such as IL-1β (Shaftel et al., 2007), lipopolysaccharide (Herber et al., 2004), or granulocyte colony stimulating factor (G-CSF) (Sanchez-Ramos et al., 2009). That is, “classic” inflammation activates the “alternative” phenotype. It follows that inhibition of the former may cause inhibition of the latter. A proportion of brain phagocytes are blood or bone-marrow derived monocytes, which infiltrate the brain and cluster around Aβ plaques (Malm et al., 2005; Simard et al., 2006; Butovsky et al., 2007; Town et al., 2008). Interestingly, monocyte recruitment depends on the chemoattractant MCP-1 (El et al., 2007), pointing to a cross-talk between cerebral and peripheral immune systems.
(ii) Ultrastructural analysis of post-mortem brains from AD patients reveals no sign of Aβ fibers in the lysosomal system of peri-plaque microglia (Wisniewski et al., 1992), despite the fact that they express the phagocyte marker human leukocyte antigen type DR (HLA-DR) (McGeer et al., 1987). This suggests defective activation. Currently, researchers are pursuing various means to activate Aβ phagocytosis in brain by resident or infiltrated cells. Strategies include immunotherapy (Schenk et al., 2004), or infusion of Aβ-specific T-cells (Ethell et al., 2006).
(iii) Microglia or myeloid cells can also contribute to Aβ removal through the release of proteolytic enzymes (Jiang et al., 2008), or the component system element C3 (Wyss-Coray et al., 2002). Activation of the complement system would thus have a protective rather than the allegedly detrimental role in AD.
Overall, it appears that the inflammatory activation of microglia detected in clinical stages of AD, or in plaque-laden transgenic-mouse brains, is destined, however inefficiently, to remove Aβ and to promote repair implicating the peripheral immune system.
Conclusions and Perspectives of Anti-Inflammatory Treatment in Alzheimer’s Disease
Surveillance, coordinated recruitment of immune cells, removal of damaging elements, and tissue repair, that is, the set of sequential reactions known as “inflammation”, are functions of the innate immune system in the brain, as elsewhere. We postulate that a defective rather than an excessive inflammatory response contributes to AD pathogenesis. Our view has three core ideas (Figure 1):
(i) One is that ApoEε4 plays a key role in early disease pathogenesis by damaging astrocytes, the principal holders of the lipoprotein. Consequences of astrocyte dysfunction would be: (a) Aβ build-up due to impaired clearance/degradation, (b) alterations of functions depending on astrocytes (neurovascular coupling, metabolic homeostasis, and synaptic activity), which will be further impaired by excess Aβ, and (c) impaired release of growth factors (e.g., fibroblast growth factor type 2), cytokines (e.g., IL-6) or chemokines (e.g., MCP-1), thus hindering neuronal protection and the full completion of inflammatory cascades.
(ii) The second idea is that microglia becomes progressively activated overtime, although sub-optimally, in response to fibrillar Aβ accumulation and, in later stages of the disease, due to signals released by dying cells.
(iii) NSAID actions depend on the stage of disease progression. Early administration of NSAIDs would delay disease progression by multitargeted actions on ApoEε4, Aβ production, oxidative damage, tau hyperphosphorylation, or synaptic plasticity (Table 1). Available knowledge is incomplete to understand how NSAIDs may regulate overtime the complex spectrum of microglia phenotypes, but we posit that late administration of NSAIDs in the course of the disease – as in clinical trials – may interfere with the cross-talk between brain and peripheral immune systems, thus hampering efforts to remove Aβ and cellular debris prior to tissue repair and regeneration.
In summary, the motto for NSAID therapeutics in AD should be “the earlier the better”. In order to test these ideas necessary efforts are: (i) a prevention trial with a non-selective NSAID in ApoEε4 carriers, (ii) to define the dynamic evolution of inflammation in AD with an emphasis on preclinical stages; (iii) to define the particulars of microglia and astrocytes, e.g., regulation and role of “classic” and “alternative” phenotypes (in both glia types); (iv) to define the biological role of ApoE in glia, and (v) to characterize the molecular targets of NSAIDs and their effect on different glia phenotypes.
The slow progression of AD, while complicating our understanding of the disease, gives opportunities for intervention. The duration of the clinically silent phases seems to be influenced by genetic and lifestyle factors, and by cognitive reserve, an indication of brain resiliency. Future research will clarify whether routinary protocols for AD prevention should include NSAIDs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aisen, P. (2010). Treating before symptoms-ADCS invites ideas for clinical trials in very early AD. Alzforum http://www.alzforum.org/res/for/journal/detail.asp?liveID=180.
Arends, Y. M., Duyckaerts, C., Rozemuller, J. M., Eikelenboom, P., and Hauw, J. J. (2000). Microglia, amyloid and dementia in alzheimer disease. A correlative study. Neurobiol. Aging 21, 39–47.
Butovsky, O., Kunis, G., Koronyo-Hamaoui, M., and Schwartz, M. (2007). Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer’s disease model. Eur. J. Neurosci. 26, 413–416.
Caselli, R. J., Dueck, A. C., Osborne, D., Sabbagh, M. N., Connor, D. J., Ahern, G. L., Baxter, L. C., Rapcsak, S. Z., Shi, J., Woodruff, B. K., Locke, D. E., Snyder, C. H., Alexander, G. E., Rademakers, R., and Reiman, E. M. (2009). Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N. Engl. J. Med. 361, 255–263.
Choi, S. H., Aid, S., and Bosetti, F. (2009). The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol. Sci. 30, 174–181.
Coma, M., Sereno, L., Da Rocha-Souto, B., Scotton, T. C., Espana, J., Sanchez, M. B., Rodriguez, M., Agullo, J., Guardia-Laguarta, C., Garcia-Alloza, M., Borrelli, L. A., Clarimon, J., Lleo, A., Bacskai, B. J., Saura, C. A., Hyman, B. T., and Gomez-Isla, T. (2010). Triflusal reduces dense-core plaque load, associated axonal alterations and inflammatory changes, and rescues cognition in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 38, 482–491.
De, S. R., Jmone-Cat, M. A., Tirassa, P., and Minghetti, L. (2003). Apoptotic PC12 cells exposing phosphatidylserine promote the production of anti-inflammatory and neuroprotective molecules by microglial cells. J. Neuropathol. Exp. Neurol. 62, 208–216.
El, K.J., Toft, M., Hickman, S. E., Means, T. K., Terada, K., Geula, C., and Luster, A. D. (2007). Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438.
Ertekin-Taner, N. (2010). Genetics of Alzheimer disease in the pre- and post-GWAS era. Alzheimer’s Res. Ther. 2, 3.
Ethell, D. W., Shippy, D., Cao, C., Cracchiolo, J. R., Runfeldt, M., Blake, B., and Arendash, G. W. (2006). Abeta-specific T-cells reverse cognitive decline and synaptic loss in Alzheimer’s mice. Neurobiol. Dis. 23, 351–361.
Fox, N. C., Warrington, E. K., and Rossor, M. N. (1999). Serial magnetic resonance imaging of cerebral atrophy in preclinical Alzheimer’s disease. Lancet 353, 2125.
Fu, Q., Hue, J., and Li, S. (2007). Nonsteroidal anti-inflammatory drugs promote axon regeneration via RhoA inhibition. J. Neurosci. 27, 4154–4164.
Fuenzalida, K., Quintanilla, R., Ramos, P., Piderit, D., Fuentealba, R. A., Martinez, G., Inestrosa, N. C., and Bronfman, M. (2007). Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J. Biol. Chem. 282, 37006–370015.
Grathwohl, S. A., Kalin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., Odenthal, J., Radde, R., Eldh, T., Gandy, S., Aguzzi, A., Staufenbiel, M., Mathews, P. M., Wolburg, H., Heppner, F. L., and Jucker, M. (2009). Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363.
Green, R. C., Schneider, L. S., Amato, D. A., Beelen, A. P., Wilcock, G., Swabb, E. A., and Zavitz, K. H. (2009). Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302, 2557–2564.
Griffin, W. S., Sheng, J. G., Roberts, G. W., and Mrak, R. E. (1995). Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. J. Neuropathol. Exp. Neurol. 54, 276–281.
Haga, S., Akai, K., and Ishii, T. (1989). Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol. 77, 569–575.
Hayden, K. M., Zandi, P. P., Khachaturian, A. S., Szekely, C. A., Fotuhi, M., Norton, M. C., Tschanz, J. T., Pieper, C. F., Corcoran, C., Lyketsos, C. G., Breitner, J. C., and Welsh-Bohmer, K. A. (2007). Does NSAID use modify cognitive trajectories in the elderly? The Cache County study. Neurology 69, 275–282.
Herber, D. L., Roth, L. M., Wilson, D., Wilson, N., Mason, J. E., Morgan, D., and Gordon, M. N. (2004). Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp. Neurol. 190, 245–253.
Imbimbo, B. P., Solfrizzi, V., and Panza, F. (2010). Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front. Ag. Neurosci. 2, 19. doi: 10.3389/fnagi.2010.00019.
In’t Veld, B. A., Ruitenberg, A., Hofman, A., Launer, L. J., van Duijn, C. M., Stijnen, T., Breteler, M. M., and Stricker, B. H. (2001). Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 345, 1515–1521.
Jack, C. R. Jr., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., Petersen, R. C., and Trojanowski, J. Q. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128.
Jacobsen, J. S., Wu, C. C., Redwine, J. M., Comery, T. A., Arias, R., Bowlby, M., Martone, R., Morrison, J. H., Pangalos, M. N., Reinhart, P. H., and Bloom, F. E. (2006). Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5161–5166.
Jiang, Q., Lee, C. Y., Mandrekar, S., Wilkinson, B., Cramer, P., Zelcer, N., Mann, K., Lamb, B., Willson, T. M., Collins, J. L., Richardson, J. C., Smith, J. D., Comery, T. A., Riddell, D., Holtzman, D. M., Tontonoz, P., and Landreth, G. E. (2008). ApoE promotes the proteolytic degradation of Abeta. Neuron 58, 681–693.
Jimenez, S., Baglietto-Vargas, D., Caballero, C., Moreno-Gonzalez, I., Torres, M., Sanchez-Varo, R., Ruano, D., Vizuete, M., Gutierrez, A., and Vitorica, J. (2008). Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J. Neurosci. 28, 11650–11661.
Kang, K. W., Lee, S. J., and Kim, S. G. (2005). Molecular mechanism of nrf2 activation by oxidative stress. Antioxid. Redox. Signal 7, 1664–1673.
Kim, J., Basak, J. M., and Holtzman, D. M. (2009). The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303.
Kukar, T., Prescott, S., Eriksen, J. L., Holloway, V., Murphy, M. P., Koo, E. H., Golde, T. E., and Nicolle, M. M. (2007). Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neurosci. 8, 54.
Lichtenstein, M., Carriba, P., Wojciak-Stothard, B., Baltrons, M. A., Petersen, J., García, A., and Galea, E. (2010). Secretase-independent and RhoGTPase/ERK/PAK regulation of cytoskeleton dynamics and motility in astrocytes by NSAIDs and derivatives. J. Alzheimer’s Dis. doi: 10.3233/JAD-2010-101332.
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., Caspersen, C., Chen, X., Pollak, S., Chaney, M., Trinchese, F., Liu, S., Gunn-Moore, F., Lue, L. F., Walker, D. G., Kuppusamy, P., Zewier, Z. L., Arancio, O., Stern, D., Yan, S. S., and Wu, H. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452.
Majumdar, A., Chung, H., Dolios, G., Wang, R., Asamoah, N., Lobel, P., and Maxfield, F. R. (2008). Degradation of fibrillar forms of Alzheimer’s amyloid beta-peptide by macrophages. Neurobiol Aging 29, 707–715.
Malm, T. M., Koistinaho, M., Parepalo, M., Vatanen, T., Ooka, A., Karlsson, S., and Koistinaho, J. (2005). Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol. Dis. 18, 134–142.
Martinez, A., Portero-Otin, M., Pamplona, R., and Ferrer, I. (2010). Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol. 20, 281–297.
McGeer, P. L., Itagaki, S., Tago, H., and McGeer, E. G. (1987). Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 79, 195–200.
Nicolakakis, N., Aboulkassim, T., Ongali, B., Lecrux, C., Fernandes, P., Rosa-Neto, P., Tong, X. K., and Hamel, E. (2008). Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J. Neurosci. 28, 9287–9296.
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., Jones, P. K., Ghanbari, H., Wataya, T., Shimohama, S., Chiba, S., Atwood, C. S., Petersen, R. B., and Smith, M. A. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767.
Pascual, G., Fong, A. L., Ogawa, S., Gamliel, A., Li, A. C., Perissi, V., Rose, D. W., Wilson, T. M., Rosenfeld, M. G., and Glass, K. G. (2005). A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437, 759–763.
Petrie, E. C., Cross, D. J., Galasko, D., Schellenberg, G. D., Raskind, M. A., Peskind, E. R., and Minoshima, S. (2009). Preclinical evidence of Alzheimer changes: convergent cerebrospinal fluid biomarker and fluorodeoxyglucose positron emission tomography findings. Arch. Neurol. 66, 632–637.
Pocivavsek, A., Burns, M. P., and Rebeck, G. W. (2009). Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia 4, 444–453.
Pratico, D., Uryu, K., Leight, S., Trojanoswki, J. Q., and Lee, V. M. (2001). Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 21, 4183–4187.
Sabbagh, M. N. (2009). Drug development for Alzheimer’s disease: where are we now and where are we headed? Am. J. Geriatr. Pharmacother. 7, 167–185.
Sanchez-Ramos, J., Song, S., Sava, V., Catlow, B., Lin, X., Mori, T., Cao, C., and Arendash, G. W. (2009). Granulocyte colony stimulating factor decreases brain amyloid burden and reverses cognitive impairment in Alzheimer’s mice. Neuroscience 163, 55–72.
Sastre, M., Dewachter, I., Rossner, S., Bogdanovic, N., Rosen, E., Borghgraef, P., Evert, B. O., Dumitrescu-Ozimek, L., Thal, D. R., Landreth, G., Walter, J., Klockgether, T., van Leuven, F., Heneka, M. T. (2006). Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. U.S.A. 103, 443–448.
Sastre, M., and Gentleman, S. M. (2010). NSAIDs: how they work and their prospects as therapeutics in Alzheimer’s disease. Front. Ag. Neurosci. 2, 20. doi: 10.3389/fnagi.2010.00020.
Sayas, C. L., Moreno-Flores, M. T., Avila, J., and Wandosell, F. (1999). The neurite retraction induced by lysophosphatidic acid increases Alzheimer’s disease-like Tau phosphorylation. J. Biol. Chem. 274, 37046–37052.
Schenk, D., Hagen, M., and Seubert, P. (2004). Current progress in beta-amyloid immunotherapy. Curr. Opin. Immunol. 16, 599–606.
Shaftel, S. S., Kyrkanides, S., Olschowka, J. A., Miller, J. N., Johnson, R. E., and O’Banion, M. K. (2007). Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Invest. 117, 1595–1604.
Simard, A. R., Soulet, D., Gowing, G., Julien, J. P., and Rivest, S. (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49, 489–502.
Streit, W. J. (2010). Microglial activation and neuroinflammation in Alzheimer’s disease: a critical examination of recent history. Front. Ag. Neurosci. 2, 22. doi: 10.3389/fnagi.2010.00022.
Szekely, C. A., Breitner, J. C., Fitzpatrick, A. L., Rea, T. D., Psaty, B. M., Kuller, L. H., Zandi, P. P. (2008). NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology 70, 17–24.
Szekely, C. A., Thorne, J. E., Zandi, P. P., Ek, M., Messias, E., Breitner, J. C., and Goodman, S. N. (2004). Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: a systematic review. Neuroepidemiology 23, 159–169.
Takahashi, Y., Hayashi, I., Tominari, Y., Rikimaru, K., Morohashi, Y., Kan, T., Natsugari, H., Fukuyama, T., Tomita, T., and Iwatsubo, T. (2003). Sulindac sulfide is a noncompetitive gamma-secretase inhibitor that preferentially reduces Abeta 42 generation. J. Biol. Chem. 278, 18664–18670.
Tamagno, E., Bardini, P., Obbili, A., Vitali, A., Borghi, R., Zaccheo, D., Pronzato, M. A., Danni, O., Smith, M. A., Perry, G., and Tabaton, M. (2002). Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 10, 279–288.
Town, T., Laouar, Y., Pittenger, C., Mori, T., Szekely, C. A., Tan, J., Duman, R. S., and Flavell, R. A. (2008). Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 14, 681–687.
Vitek, M. P., Brown, C. M., and Colton, C. A. (2009). APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 9, 1350–1360.
Vehmas, A. K., Kawas, C. H., Stewart, W. F., and Troncoso, J. C. (2003). Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol. Aging 24, 321–331.
Weggen, S., Eriksen, J. L., Sagi, S. A., Pietrzik, C. U., Ozols, V., Fauq, A., Golde, T. E., Koo, E. H. (2003). Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J. Biol. Chem. 278, 31831–31837.
Wisniewski, H. M., Wegiel, J., Wang, K. C., and Lach, B. (1992). Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol. 84, 117–127.
Wyss-Coray, T., Yan, F., Lin, A. H., Lambris, J. D., Alexander, J. J., Quigg, R. J., Masliah, E. (2002). Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc. Natl. Acad. Sci. U.S.A. 99, 10837–10842.
Ye, S., Huang, Y., Mullendorff, K., Dong, L., Giedt, G., Meng, E. C., Cohen, F. E., Kuntz, I. D., Weisgraber, K. H., and Mahley, R. W. (2005). Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc. Natl. Acad. Sci. U.S.A. 102, 18700–18705.
Yip, A. G., Green, R. C., Huyck, M., Cupples, L. A., and Farrer, L. A. (2005). Nonsteroidal anti-inflammatory drug use and Alzheimer’s disease risk: the MIRAGE Study. BMC Geriatr. 5, 2.
Keywords: ibuprofen, naproxen, astrocytes, ApoE, microglia, biomarkers
Citation: Lichtenstein MP, Carriba P, Masgrau R, Pujol A and Galea E (2010) Staging anti-inflammatory therapy in Alzheimer’s disease. Front. Ag. Neurosci. 2:142. doi: 10.3389/fnagi.2010.00142
Received: 30 July 2010;
Accepted: 16 September 2010;
Published online: 25 October 2010.
Edited by:
Paul G. Luiten, University of Groningen, Netherlands AntillesReviewed by:
Ana I. Duarte, University of Coimbra, PortugalJose G. Castaño, Universidad Autónoma de Madrid, Spain
Copyright: © 2010 Lichtenstein, Carriba, Masgrau, Pujol and Galea. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Elena Galea, Institute of Neurociències, Edifici M, Universitat Autònoma de Barcelona, 08193 Bellaterra, Barcelona, Spain. e-mail: galea.inc@gmail.com