Current pathophysiological concepts in cerebral small vessel disease
 Fred Rincon1
Fred Rincon1  Clinton B. Wright2,3,4,5*
Clinton B. Wright2,3,4,5*- 1Department of Neurology, Thomas Jefferson University, Philadelphia, PA, USA
- 2Department of Neurosurgery, Thomas Jefferson University, Philadelphia, PA, USA
- 3Evelyn F. McKnight Brain Institute, Department of Neurology, University of Miami, Miami, FL, USA
- 4Department of Epidemiology and Public Health, University of Miami, Miami, FL, USA
- 5Neuroscience Program, University of Miami, Miami, FL, USA
The association between cerebral small vessel disease (SVD) – in the form of white matter lesions, infarctions, and hemorrhages – with vascular cognitive impairment (VCI), has mostly been deduced from observational studies. Pathological conditions affecting the small vessels of the brain and leading to SVD have suggested plausible molecular mechanisms involved in vascular damage and their impact on brain function. However, much still needs to be clarified in understanding the pathophysiology of VCI, the role of neurodegenerative processes such as Alzheimer’s disease, and the impact of aging itself. In addition, both genetic predispositions and environmental exposures may potentiate the development of SVD and interact with normal aging to impact cognitive function and require further study. Advances in technology, in the analysis of genetic and epigenetic data, neuroimaging such as magnetic resonance imaging, and new biomarkers will help to clarify the complex factors leading to SVD and the expression of VCI.
Introduction
Stroke remains the No. 1 cause of disability and the fourth leading cause of death in the US (Roger et al., 2012). Prevention strategies aimed at treating modifiable risk factors have been advocated by clinicians and epidemiologists (Rincon and Sacco, 2008). Among the important causes of stroke, hypertension-related small vessel disease (SVD) and cerebral amyloid angiopathy (CAA) are the most common forms, and have generated significant academic interest because of their sinister impact on brain function. Understanding the pathophysiological mechanisms involved in SVD and possible treatments has remained elusive (Pantoni, 2010). In this review, we sought to summarize recent advances in the understanding of the pathophysiological mechanisms of SVD.
Small Vessel Disease
The term SVD refers to the syndrome of clinical, cognitive, neuroimaging, and neuropathological findings thought to arise from damage to (a) small arteries, (b) arterioles, (c) capillaries, and (d) small veins and venules in the brain (Moody et al., 1995). SVD preferentially affects the vessels of the basal ganglia, peripheral white matter, leptomeningeal arteries, thalamic and cerebellar white matter vessels, and vessels of the brainstem. Cortical vessels are usually not involved in SVD (Thal et al., 2003).
Small vessel disease is an important clinico-pathological condition as it is the cause of 20% of strokes worldwide, and the most common cause of vascular and mixed dementia [vascular dementia (VaD) and Alzheimer’s disease (AD); Pantoni, 2010; Gorelick et al., 2011]. Dementia is currently a pressing public health problem as numbers of affected patients increase steadily. Vascular brain injury is the second most common cause of dementia after AD and a defining feature of vascular cognitive impairment (VCI; Rincon and Wright, 2013). AD commonly coexists with cerebrovascular disease in the elderly population. Though the risk factors for both SVD and AD overlap (Dichgans and Zietemann, 2012), the differentiation on clinical grounds is often difficult (Schneider et al., 2007).
Recently, studies have emphasized on the comorbidities associated with AD and VaD. Established risk factors for both VaD and AD are age, smoking, physical inactivity, obesity, diabetes mellitus, stroke, and peripheral arterial disease (Dichgans and Zietemann, 2012). Brains from AD patients exhibit more cerebrovascular lesions than non-AD patients (Jellinger and Attems, 2005). Pathological examination of brains from AD patients reveal higher prevalence of lacunes, white matter lesions (WMLs), microbleeds, and CAA (Jellinger and Attems, 2005). Pathological changes seen in AD have led authors to believe that vascular brain damage is an important component of AD pathophysiology (de la Torre, 2002). Almost all brains of AD patients have CAA (Jellinger, 2002). This suggests a common β-amyloid-based pathogenesis for the disease. However, despite this molecular relationship, CAA is a different entity from AD as less than 50% of CAA cases meet the pathologic criteria for AD and >75% of patients with AD have mild or no CAA at all (Vinters, 1987; Ellis et al., 1996).
Cerebral amyloid angiopathy-related impairments in cerebral perfusion may be responsible for subcortical WMLs and microscopic damage seen in the disease (Gurol et al., 2006; Holland et al., 2008; Viswanathan et al., 2008). Some studies have suggested that advanced CAA is associated with a larger burden of WMLs as compared to healthy controls or AD patients (Gurol et al., 2006; Holland et al., 2008). Interestingly, CAA induced cognitive impairment may be a reflection of WMLs independent of brain hemorrhages (Viswanathan et al., 2008). It appears that CAA-related WMLs spread through similar areas affected by hypertensive SVD. However, there is some suggestion that CAA induced WMLs may be preferentially seen in the posterior white matter (Zhu et al., 2012). In addition, pathologic studies have suggested that cortical microinfarcts are common in CAA (Soontornniyomkij et al., 2010). The presence of these microinfarcts may be unrelated to classic vascular risk factors. The pathophysiological mechanisms thought to be involved are impaired autoregulation, smooth muscle damage, and capillary occlusion (Shin et al., 2007; Smith et al., 2008; Thal et al., 2009).
Although pathological studies have shown a significantly higher prevalence of vascular pathology in AD patients (Jellinger and Attems, 2005) and despite stroke being a frequent occurrence in elderly AD patients (Honig et al., 2003), the pathophysiological mechanisms and impact of these cerebrovascular abnormalities on cognitive decline in AD remains unclear.
Diagnosis
Because small vessel damage cannot be readily visualized in vivo, the effect of SVD on the brain parenchyma is usually inferred from findings on computed tomography (CT) or magnetic resonance imaging (MRI), and these changes are considered the hallmarks of the disease. As such, SVD is often equated with brain parenchymal lesions. However, it may be beneficial to broaden the definition of this phenotype of SVD to include vascular damage prior to ischemic injury, and use other measures of vascular dysfunction – such as measures of cerebral autoregulation. To date such studies have been limited (Pantoni, 2010). This broader view of SVD allows consideration of plausible therapeutic interventions aimed at modulating the progression of disease before irreversible damage is done.
Consequences of SVD in the brain parenchyma include lesions located in the subcortical structures such as lacunar infarcts, WMLs, and deep hemorrhages (large sub-cortical hemorrhages and microbleeds; Pantoni, 2010; Table 1). The prevalence of silent brain infarcts has varied across study populations, across imaging techniques, and the definition of infarct used (Vermeer et al., 2007). Several large-population-based studies have reported prevalence estimates of 8–28%, with differences mainly explained by age (Vermeer et al., 2007). Microscopic damage, which escapes the resolution of most CT and MRI machines, is even more prevalent (Launer et al., 2011). Microscopic brain infarcts (MBI) have been seen in up to 68% of community-based participants (Launer et al., 2011). The therapeutic implications of SVD are important, as SVD is the cause of up to 25% of ischemic strokes and the majority of intracerebral hemorrhages (Petty et al., 2000). No specific therapy exists in the acute setting for strokes caused by SVD. Perhaps more importantly, there is no suggestion that cornerstone treatments of ischemic stroke such as aspirin, thrombolysis, or admission to a stroke unit or neuroscience center are associated with better outcomes in this subset of patients than for other stroke subtypes (Cocho et al., 2006; Pantoni, 2010). Beyond control of blood pressure and statin treatment (Dufouil et al., 2005; Mok et al., 2009), there appears to be no effective therapy to limit the extent and progression of SVD-related stroke. Thus, an opportunity exists to design novel prevention strategies to treat SVD if the pathophysiological factors that distinguish it from large vessel disease and cardiac causes of stroke can be identified. Indeed, chronic hypoperfusion, ischemia, and, finally, necrosis of brain tissue are often associated with SVD and include a number of types of injuries that serve as markers of disease severity, and relate to the risk of poor outcomes and prognosis in a number of clinical settings (Wright et al., 2008).
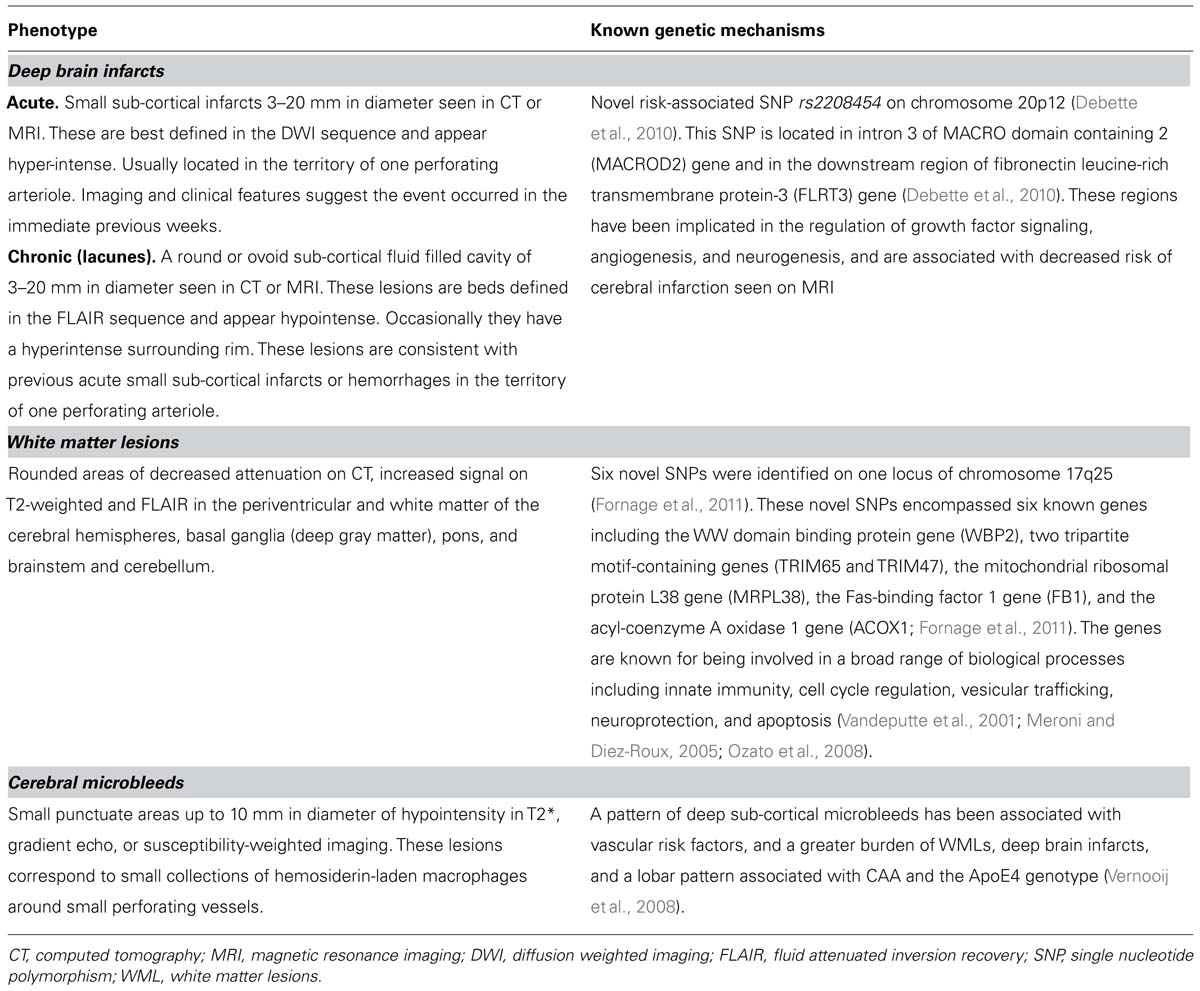
TABLE 1. Glossary of definitions of SVD phenotypes.
For the purpose of this review, we will concentrate on SVD caused by traditional vascular risk factors (sporadic SVD), rather than those caused by genetic abnormalities or associated with systemic conditions [i.e., cerebral autosomal dominant arteriopathy with sub-cortical infarcts and leukoencephalopathy (CADASIL), cerebral autosomal recessive arteriopathy with sub-cortical infarcts and leukoencephalopathy (CARASIL), collagen 4 AI gene (COL4AI) mutations, Fabry’s, hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS), and small vessel arteritis].
Deep Brain Infarcts
This term refers to small sub-cortical infarcts of 3–20 mm in diameter identified on either CT or MRI (Norrving, 2003; Wardlaw et al., 2013). Deep brain infarcts, often referred to as lacunar strokes in the clinical setting, account for 20–30% of all stroke sub-types, and have an incidence of about 33 per 100,000 persons/years (Sudlow and Warlow, 1997). Acutely, deep brain infarcts are better detected by MRI than by CT, and appear hyperintense on diffusion-weighted imaging (DWI), and within hours to days on T2-weighted imaging or fluid attenuated inversion recovery (FLAIR) sequences (Patel and Markus, 2011). Chronic deep brain infarcts appear hypo-intense on T1 and FLAIR, and often have a hyper-intense rim around them on the latter sequence (Patel and Markus, 2011). Once macrophages have removed infarcted tissue, irregular cavities are left with surrounding gliosis, and lipid-rich and hemosiderin-rich macrophages are left in surrounding gliotic tissue along with extravasated plasma proteins, fibrinoid necrosis, and vascular fragments (Fisher, 1968). Many risk factors associated with deep brain infarcts, such as older age, and particularly hypertension, but also diabetes mellitus, smoking, excess alcohol consumption, and dyslipidemia, are shared with those of superficial infarcts. Some epidemiological studies suggest these risk factors may not be as important as in infarcts related to atherosclerotic arteriopathy (Jackson and Sudlow, 2005; Jackson et al., 2010).
While CADASIL, CARASIL, Fabry’s disease, and a number of other genetic forms of SVD are known, the genetic underpinning of most SVD is poorly understood. However, recent studies are beginning to show that genetic factors likely play a role. A genetic mechanism leading to SVD has been suggested as underlying some deep brain infarcts (Jackson et al., 2010). For example, a recent population-based genome-wide association study (GWAS) on covert MRI-defined brain infarcts found the novel risk-associated single nucleotide polymorphism (SNP) rs2208454 on chromosome 20p12 (Debette et al., 2010). This SNP is located in intron 3 of MACRO domain containing 2 (MACROD2) gene and in the downstream region of fibronectin leucine-rich transmembrane protein-3 (FLRT3) gene (Debette et al., 2010). These regions have been implicated in the regulation of growth factor signaling, angiogenesis, and neurogenesis, and are associated with decreased risk of cerebral infarction seen on MRI (Debette et al., 2010).
Deep brain infarction is associated with classical clinical syndromes (also named lacunar syndromes; Donnan and Norrving, 2009) and are closely associated with radiological evidence of ischemia, although some authors have demonstrated that the clinical syndrome may not be entirely predictive of the lesion or location (Gan et al., 1997; Arboix et al., 2010). The presence of sub-clinical evidence of brain ischemia, or “silent” brain infarcts (Vermeer et al., 2007), makes it difficult to identify associated risk factors, underlying mechanisms, and potential therapies for intervention. Though deep brain infarcts have an overall better prognosis (Norrving, 2003), they have a higher rate of recurrence and affected individuals are at greater risk of developing cognitive impairment, depression, and long-term functional decline (Miyao et al., 1992; Samuelsson et al., 1996; Yamamoto et al., 2002; Vermeer et al., 2003, 2007; Baezner et al., 2008).
Recently, there has been a growing interest in the clinical significance of deep brain infarcts and WMLs as causes of VCI. It is important to note, that the classic lacunar syndromes did not include cognitive impairment as a feature or dedicated syndrome (Norrving, 2003). However, small deep brain infarcts are known to cause so-called “strategic infarct dementia” (Tatemichi et al., 1992) and the lesion burden has also been associated with dementia risk (Koga et al., 2009). In one study, the presence of thalamic lacunes was associated with poor global cognitive performance, low motor activity and executive function performance; and the presence of lacunes in the pallidum or putamen was associated with memory dysfunction (Benisty et al., 2009). Deep brain infarcts have been associated with a number of outcomes that are relevant to VCI, including cognitive decline, dementia, gait disturbance, urinary incontinence, and disability (Miyao et al., 1992; Samuelsson et al., 1996; Yamamoto et al., 2002; Vermeer et al., 2003, 2007; Baezner et al., 2008), making the term “silent infarct” an inappropriate and misleading term, given these poor outcomes (Pantoni, 2003; Hachinski, 2008).
White Matter Lesions
This phenotype of SVD represents a different entity than deep brain infarcts, however, they often coexist (Pantoni, 2010).
The prevalence of WMLs in the white population is 80% or greater in those 60 years old or older (de Leeuw et al., 2001), and seen more in women as compared to men (de Leeuw et al., 2001). Before the advent of MRI, WMLs were seen on CT imaging as x-ray attenuation in white matter areas and described in the literature by Hachinski et al. (1987) as “leukoaraiosis.” On MRI, WMLs are seen as white matter hyperintensities (WMH) on T2 and FLAIR sequences (Wardlaw et al., 2013). Such WMLs are seen in white matter tracts surrounding the ventricular system, though they are also seen in other areas and in the immediate subcortical white matter. Magnetic resonance-based diffusion tensor imaging (DTI) provides a measure the diffusion of water in white matter tracts, allowing researchers to examine the patency of axonal pathways in patients with SVD (de Laat et al., 2011). Studies using DTI have shown that white matter integrity is compromised immediately outside WMH lesions (Maillard et al., 2011), suggesting that visible lesions are indicative of wider injury. Additional promising MRI-based techniques for the study of WMLs in SVD include magnetization transfer (MTI) and high-field-MRI (Fazekas et al., 2005; Bastin et al., 2009; Kang et al., 2010), as the severity of tissue changes associated with incidental WMLs in these patients cannot be sufficiently determined by conventional MRI.
Population-based studies have demonstrated a strong association between both age and hypertension and WMLs (Enzinger et al., 2007). Similarly, pathological studies have shed some light on the association between ischemia and WMLs as well (Enzinger et al., 2007). Common pathological findings in WMLs are: mild perivascular alterations to large areas with variable loss of fibers, multiple small cavitations, and marked arteriolosclerosis (Fazekas et al., 1993). In addition, WMLs have a variety of pathological correlates depending on the severity of ischemic tissue damage: myelin pallor, gliosis, axonal loss, complete nerve fiber destruction (Fazekas et al., 1993), and, in the worst cases, blood–brain barrier disruption and loss of endothelium (Young et al., 2008). The tissue surrounding WMLs may be highly “active” with foam cells, activated astrocytes, and microglia (Fazekas et al., 1993). Up-regulation of inflammatory markers seen in these areas, including apolipoprotein E (ApoE), α2-microglobulin, and immunoglobulin G may also contribute to the pathophysiological processes leading to WMLs (Lammie, 2002; Nag, 2003). Another pathological process of deep small cerebral vessels particularly affecting the small veins of periventricular areas is known as venous collagenosis (Moody et al., 1995). This process, which has received limited attention compared to arteriolosclerosis in relation to the pathophysiology of SVD, is primarily associated with WMLs. Recent biological studies have demonstrated an association between alterations in RNA transcription in multiple genes involved in cell cycle, proteolysis, immunological modulation, and apoptosis and WMLs (Simpson et al., 2009). Genetic factors also appear to play a role in the development of WMLs, with reported heritability of up to 55–80% (Carmelli et al., 1998; Atwood et al., 2004).
The results of GWAS also provide powerful tools to identify genes related to complex multifactorial traits such as WMLs. In a recent meta-analysis of GWAS involving 9,361 individuals of European descent and belonging to seven community-based cohorts, six novel SNPs were identified on one locus of chromosome 17q25 (Fornage et al., 2011). These novel SNPs encompassed six known genes including the WW domain binding protein gene (WBP2), two tripartite motif-containing genes (TRIM65 and TRIM47), the mitochondrial ribosomal protein L38 gene (MRPL38), the Fas-binding factor 1 gene (FB1), and the acyl-coenzyme A oxidase 1 gene (ACOX1; Fornage et al., 2011). The genes are known for being involved in a broad range of biological processes including innate immunity, cell cycle regulation, vesicular trafficking, neuroprotection, and apoptosis (Vandeputte et al., 2001; Meroni and Diez-Roux, 2005; Ozato et al., 2008). This provides the first step toward characterization of biological mechanisms that influence the pathophysiology associated with WMLs (Fornage et al., 2011). The multiple pathophysiological processes involved in the genesis of WMLs underscore the complexity of this phenotype.
Though WMLs were historically considered incidental findings of doubtful clinical significance, recent epidemiological studies have demonstrated that WMLs are associated with cognitive decline (van Straaten et al., 2006; Frisoni et al., 2007; Pantoni, 2008; Wright et al., 2008). Moreover, the combination of WMLs in patients with deep brain infarcts is also associated with more cognitive decline (Miyao et al., 1992; van Swieten et al., 1996). WMLs are seen in at least 30% of patients with AD and in 60% of patients with dementia (Steingart et al., 1987). Some studies have found that a greater burden of WMLs is also associated with incontinence, gait dyspraxia, and incident falls (de Leeuw et al., 2001; Baezner et al., 2008; Srikanth et al., 2009). An appraisal of 16 studies confirmed the association between WMLs and cognitive decline in different patient settings: hospital-based to population-based (Pantoni et al., 2007). Finally, a large meta-analysis of 46 observational studies, demonstrated that WMLs are associated with greater risk of future stroke, dementia, and death (Debette and Markus, 2010). Effective therapies targeting the development of WMLs based on the understanding of the pathophysiology and plausible molecular targets are desperately needed.
Cerebral Microbleeds
This phenotype of SVD refers to small deep or superficial hemorrhages of 2–10 mm in diameter seen by MRI (Greenberg et al., 2009; Shoamanesh et al., 2011; Wardlaw et al., 2013). The T2* gradient echo sequence, and the newer susceptibility-weighted imaging (SWI), provide sensitive methods for detecting microbleeds (Tanaka et al., 1999). These lesions correspond to small collections of hemosiderin-laden macrophages around small perforating vessels (Greenberg et al., 2009; Shoamanesh et al., 2011; Wardlaw et al., 2013). Two types of cerebral microbleeds have been described in the literature (Vernooij et al., 2008). A pattern of deep sub-cortical microbleeds associated with vascular risk factors, and a greater burden of WMLs and deep brain infarcts, and a lobar pattern associated with CAA and the ApoE4 genotype (Vernooij et al., 2008). The prevalence of cerebral microbleeds in the general population is about 5% but could be as high as 23–44% in those patients that have suffered ischemic strokes, and 52–83% in those that have suffered intracranial hemorrhage (ICH; Cordonnier et al., 2007). Indeed, cerebral microbleeds are a strong predictor of future spontaneous and symptomatic ICH (Lee et al., 2004). The implications of SVD and cerebral microbleeds on clinical management in acute ischemic stroke are very important. Intravenous recombinant tissue plasminogen activator (i.v. rt-PA) is an effective therapy in the acute setting of stroke (The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995; Wardlaw et al., 2009; Lees et al., 2010). In addition to older age, hypertension, and hyperglycemia, the presence of SVD and microbleeds has been associated with a greater risk of ICH (Neumann-Haefelin et al., 2006; Palumbo et al., 2007; Charidimou et al., 2013). There is some data on the effect of SVD in candidates for the extended t-PA window (3–4.5 h). However, a history of prior stroke is considered to be a contraindication for thrombolysis in this time window, and as mentioned previously, patients with a prior stroke have a higher prevalence of cerebral microbleeds (Charidimou et al., 2013). Finally, the role of antiplatelet or anticoagulant therapy in patients with SVD and microbleeds deserves comment. The presence of SVD and older age are each associated with ICH risk during antiplatelet therapy, as reported by the Stroke Prevention in Reversible Ischemia Trial (SPIRIT; Gorter, 1999). In a systematic review, the burden of microbleeds in warfarin users with ICH compared to other groups shows that microbleeds increase the risk of warfarin-associated ICH (Lovelock et al., 2010). Therefore, in these patient population, conventional secondary prevention strategies using antiplatelet or anticoagulation therapy requires a thorough analysis of the risk benefit ratio.
The correlation of cerebral microbleeds and cognition is a matter of current research. A greater burden of cerebral microbleeds has been associated with cognitive impairment (Werring et al., 2004), and some studies in subjects with CADASIL have found a greater burden of cerebral microbleeds are associated with worse functional ability (Viswanathan et al., 2010). A recent MRI-based study confirmed that the presence of cerebral microbleeds was associated with global cognitive dysfunction (Yakushiji et al., 2008) in independent adults with no evidence of neurological dysfunction. The mechanisms underlying the pathological association between cerebral microbleeds cognitive dysfunction and overt VCI remain unclear (Charidimou and Werring, 2012).
Future Directions and Ongoing Research
Small vessel disease is an important cause of stroke and VCI. Single component clinical trials targeting classic risk factors for both VaD and AD are ongoing. The ongoing SPRINT Memory and cognition IN Decreased hypertension (SPRINT-MIND)1 study will attempt to determine if lower systolic blood pressure (SBP) goals influence the rate of incident dementia and MCI, global and domain-specific cognitive function, and small vessel ischemic disease. The Aspirin in Reducing Events in the Elderly (ASPREE) trial is evaluating the effect of daily aspirin on incident dementia and physical disability. The sub-study of the Secondary Prevention of Small Subcortical Strokes (SPS3) trial is looking at the rate of cognitive decline in patients treated with aspirin and/or clopidogrel (Benavente et al., 2011). The Efficacy and Safety Study of Nimodipine to Prevent Mild Cognitive Impairment After Acute Ischemic Strokes (NICE) is testing the hypothesis that the calcium channel blocker nimodipine may be associated with less cognitive decline and VaD. Additional ongoing trials are considering the effect of multi-component interventions. The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER) trial is using lifestyle counseling including nutritional guidance, increased physical activity, cognitive training, increased social activity, and intensive monitoring of vascular and metabolic risk factors to prevent VCI. The Austrian Polyintervention Study to Prevent Cognitive Decline After Ischemic Stroke (ASPIS) trial is using Intensive control and motivation for better compliance with medication, regular blood pressure measurements, diet changes, and physical activity versus standard stroke care to prevent cognitive decline. The Prevention of Dementia by Intensive Vascular Care (PREDIVA) trial uses intensive vascular care with visiting a practice nurse every 4 months to assess vascular risk factors, including hypertension, hypercholesterolemia, diabetes, overweight, smoking, and level of physical exercise; intervention: lifestyle and medical to prevent dementia and disability. Finally the Prevention of Decline in Cognition After Stroke Trial (PODCAST) is testing the hypothesis that intensive blood pressure (SBP < 125 mm Hg) and/or lipid-lowering [low density lipoprotein (LDL) < 2.0 mmol/L] versus moderate blood pressure (SBP < 140 mm Hg) and LDL (<3.0 mmol/L) is associated with less cognitive decline, AD, and/or VaD.
Novel molecular interventions using genetic approaches include targeting of proteins related to specific pathways of acute and chronic ischemia. Candidate genes include NOTCH3, HTRA1, and APOE ε4 (Dichgans and Zietemann, 2012). There is also interest in novel risk factors for dementia such as the role of free radical oxygen formation in mediating some of the deleterious effects of aging, hypertension and CAA-β-amyloid deposition on small vessels (Iadecola et al., 2009).
Conclusion
The association between cerebral SVD and VCI has been deduced mostly from case series and observational studies. Pathophysiological studies of conditions affecting the small vessels of the brain and leading to SVD have suggested plausible molecular mechanisms involved in vascular damage and their impact on brain function. Similarly, MRI technology has helped us to better define surrogates of disease that may be used as markers of disease onset, progression, and impact of future therapies. GWAS studies have also elucidated some potential molecular mechanisms associated in pathophysiology of certain phenotypes of SVD. However, there is much that still needs to be clarified in understanding the pathophysiology of VCI in relation to SVD. No specific therapy currently exists for SVD-related stroke, and, more importantly, standard treatments for acute ischemic stroke are not associated with better outcomes in patients with SVD. Effective therapies to limit or halt the progression of SVD are needed. Until more is known in reference to the pathophysiological mechanisms of SVD and results of ongoing clinical trials become available, treatment of vascular risk factures such as hypertension should be the focus of prevention strategies. Based on the results of recent clinical trials antiplatelets or combination antiplatelets should be used with caution in this patient population.
Future prevention strategies will depend primarily on the refinement of our understanding of the pathophysiology of this condition. The results of clinical trials targeting known risk factors for VCI are forthcoming.
Conflict of Interest Statement
Fred Rincon: None; Clinton B. Wright: royalties from UpToDate, Inc. for review articles on vascular dementia.
Acknowledgments
This study was supported by American Heart Association (12CRP12050342) to Fred Rincon; and National Institutes of Neurological Disorders and Stroke (K02 NS 059729) and National Heart Lung and Blood (R01 HL 108623) to Clinton B. Wright.
Footnotes
References
Arboix, A., Massons, J., Garcia-Eroles, L., Targa, C., Comes, E., and Parra, O. (2010). Clinical predictors of lacunar syndrome not due to lacunar infarction. BMC Neurol. 10:31. doi: 10.1186/1471-2377-10-31
Atwood, L. D., Wolf, P. A., Heard-Costa, N. L., Massaro, J. M., Beiser, A., D’Agostino, R. B., et al. (2004). Genetic variation in white matter hyperintensity volume in the Framingham Study. Stroke 35, 1609–1613. doi: 10.1161/01.STR.0000129643.77045.10
Baezner, H., Blahak, C., Poggesi, A., Pantoni, L., Inzitari, D., Chabriat, H., et al. (2008). Association of gait and balance disorders with age-related white matter changes: the LADIS study. Neurology 70, 935–942. doi: 10.1212/01.wnl.0000305959.46197.e6
Bastin, M. E., Clayden, J. D., Pattie, A., Gerrish, I. F., Wardlaw, J. M., and Deary, I. J. (2009). Diffusion tensor and magnetization transfer MRI measurements of periventricular white matter hyperintensities in old age. Neurobiol. Aging 30, 125–136. doi: 10.1016/j.neurobiolaging.2007.05.013
Benavente, O. R., White, C. L., Pearce, L., Pergola, P., Roldan, A., Benavente, M. F., et al. (2011). The Secondary Prevention of Small Subcortical Strokes (SPS3) study. Int. J. Stroke 6, 164–175. doi: 10.1111/j.1747-4949.2010.00573.x
Benisty, S., Gouw, A. A., Porcher, R., Madureira, S., Hernandez, K., Poggesi, A., et al. (2009). Location of lacunar infarcts correlates with cognition in a sample of non-disabled subjects with age-related white-matter changes: the LADIS study. J. Neurol. Neurosurg. Psychiatry 80, 478–483. doi: 10.1136/jnnp.2008.160440
Carmelli, D., DeCarli, C., Swan, G. E., Jack, L. M., Reed, T., Wolf, P. A., et al. (1998). Evidence for genetic variance in white matter hyperintensity volume in normal elderly male twins. Stroke 29, 1177–1181. doi: 10.1161/01.STR.29.6.1177
Charidimou, A., Kakar, P., Fox, Z., and Werring, D. J. (2013). Cerebral microbleeds and the risk of intracerebral haemorrhage after thrombolysis for acute ischaemic stroke: systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 84, 277–280. doi: 10.1136/jnnp-2012-303379
Charidimou, A., and Werring, D. J. (2012). Cerebral microbleeds and cognition in cerebrovascular disease: an update. J. Neurol. Sci. 322, 50–55. doi: 10.1016/j.jns.2012.05.052
Cocho, D., Belvis, R., Marti-Fabregas, J., Bravo, Y., Aleu, A., Pagonabarraga, J., et al. (2006). Does thrombolysis benefit patients with lacunar syndrome? Eur. Neurol. 55, 70–73. doi: 10.1159/000091982
Cordonnier, C., Al-Shahi Salman, R., and Wardlaw, J. (2007). Spontaneous brain microbleeds: systematic review, subgroup analyses and standards for study design and reporting. Brain 130, 1988–2003. doi: 10.1093/brain/awl387
Debette, S., Bis, J. C., Fornage, M., Schmidt, H., Ikram, M. A., Sigurdsson, S., et al. (2010). Genome-wide association studies of MRI-defined brain infarcts: meta-analysis from the CHARGE Consortium. Stroke 41, 210–217. doi: 10.1161/STROKEAHA.109.569194
Debette, S., and Markus, H. S. (2010). The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ 341, c3666. doi: 10.1136/bmj.c3666
de Laat, K. F., van Norden, A. G., Gons, R. A., van Oudheusden, L. J., van Uden, I. W., Norris, D. G., et al. (2011). Diffusion tensor imaging and gait in elderly persons with cerebral small vessel disease. Stroke 42, 373–379. doi: 10.1161/STROKEAHA.110.596502
de la Torre, J. C. (2002). Alzheimer disease as a vascular disorder: nosological evidence. Stroke 33, 1152–1162. doi: 10.1161/01.STR.0000014421.15948.67
de Leeuw, F. E., de Groot, J. C., Achten, E., Oudkerk, M., Ramos, L. M., Heijboer, R., et al. (2001). Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study. The Rotterdam Scan Study. J. Neurol. Neurosurg. Psychiatry 70, 9–14. doi: 10.1136/jnnp.70.1.9
Dichgans, M., and Zietemann, V. (2012). Prevention of vascular cognitive impairment. Stroke 43, 3137–3146. doi: 10.1161/STROKEAHA.112.651778
Donnan, G. A., and Norrving, B. (2009). Lacunes and lacunar syndromes. Handb. Clin. Neurol. 93, 559–575. doi: 10.1016/S0072-9752(08)93027-X
Dufouil, C., Chalmers, J., Coskun, O., Besancon, V., Bousser, M. G., Guillon, P., et al. (2005). Effects of blood pressure lowering on cerebral white matter hyperintensities in patients with stroke: the PROGRESS (Perindopril Protection Against Recurrent Stroke Study) Magnetic Resonance Imaging Substudy. Circulation 112, 1644–1650. doi: 10.1161/CIRCULATIONAHA.104.501163
Ellis, R. J., Olichney, J. M., Thal, L. J., Mirra, S. S., Morris, J. C., Beekly, D., et al. (1996). Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology 46, 1592–1596. doi: 10.1212/WNL.46.6.1592
Enzinger, C., Fazekas, F., Ropele, S., and Schmidt, R. (2007). Progression of cerebral white matter lesions – clinical and radiological considerations. J. Neurol. Sci. 257, 5–10. doi: 10.1016/j.jns.2007.01.018
Fazekas, F., Kleinert, R., Offenbacher, H., Schmidt, R., Kleinert, G., Payer, F., et al. (1993). Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology 43, 1683–1689. doi: 10.1212/WNL.43.9.1683
Fazekas, F., Ropele, S., Enzinger, C., Gorani, F., Seewann, A., Petrovic, K., et al. (2005). MTI of white matter hyperintensities. Brain 128, 2926–2932. doi: 10.1093/brain/awh567
Fisher, C. M. (1968). The arterial lesions underlying lacunes. Acta Neuropathol. 12, 1–15. doi: 10.1007/BF00685305
Fornage, M., Debette, S., Bis, J. C., Schmidt, H., Ikram, M. A., Dufouil, C., et al. (2011). Genome-wide association studies of cerebral white matter lesion burden: the CHARGE consortium. Ann. Neurol. 69, 928–939. doi: 10.1002/ana.22403
Frisoni, G. B., Galluzzi, S., Pantoni, L., and Filippi, M. (2007). The effect of white matter lesions on cognition in the elderly – small but detectable. Nat. Clin. Pract. Neurol. 3, 620–627. doi: 10.1038/ncpneuro0638
Gan, R., Sacco, R. L., Kargman, D. E., Roberts, J. K., Boden-Albala, B., and Gu, Q. (1997). Testing the validity of the lacunar hypothesis: the Northern Manhattan Stroke Study experience. Neurology 48, 1204–1211. doi: 10.1212/WNL.48.5.1204
Gorelick, P. B., Scuteri, A., Black, S. E., Decarli, C., Greenberg, S. M., Iadecola, C., et al. (2011). Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42, 2672–2713. doi: 10.1161/STR.0b013e3182299496
Gorter, J. W. (1999). Major bleeding during anticoagulation after cerebral ischemia: patterns and risk factors. Stroke Prevention In Reversible Ischemia Trial (SPIRIT). European Atrial Fibrillation Trial (EAFT) study groups. Neurology 53, 1319–1327. doi: 10.1212/WNL.53.6.1319
Greenberg, S. M., Vernooij, M. W., Cordonnier, C., Viswanathan, A., Al-Shahi Salman, R., Warach, S., et al. (2009). Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 8, 165–174. doi: 10.1016/S1474-4422(09)70013-4
Gurol, M. E., Irizarry, M. C., Smith, E. E., Raju, S., Diaz-Arrastia, R., Bottiglieri, T., et al. (2006). Plasma beta-amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology 66, 23–29. doi: 10.1212/01.wnl.0000191403.95453.6a
Hachinski, V. (2008). World Stroke Day 2008: “little strokes, big trouble”. Stroke 39, 2407–2420. doi: 10.1161/STROKEAHA.108.531681
Hachinski, V. C., Potter, P., and Merskey, H. (1987). Leuko-araiosis. Arch. Neurol. 44, 21–23. doi: 10.1001/archneur.1987.00520130013009
Holland, C. M., Smith, E. E., Csapo, I., Gurol, M. E., Brylka, D. A., Killiany, R. J., et al. (2008). Greenberg, spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke 39, 1127–1133. doi: 10.1161/STROKEAHA.107.497438
Honig, L. S., Tang, M. X., Albert, S., Costa, R., Luchsinger, J., Manly, J., et al. (2003). Stroke and the risk of Alzheimer disease. Arch. Neurol. 60, 1707–1712. doi: 10.1001/archneur.60.12.1707
Iadecola, C., Park, L., and Capone, C. (2009). Threats to the mind: aging, amyloid, and hypertension. Stroke 40, S40–S44. doi: 10.1161/STROKEAHA.108.533638
Jackson, C., and Sudlow, C. (2005). Are lacunar strokes really different? A systematic review of differences in risk factor profiles between lacunar and nonlacunar infarcts. Stroke 36, 891–901. doi: 10.1161/01.STR.0000157949.34986.30
Jackson, C. A., Hutchison, A., Dennis, M. S., Wardlaw, J. M., Lindgren, A., Norrving, B., et al. (2010). Differing risk factor profiles of ischemic stroke subtypes: evidence for a distinct lacunar arteriopathy? Stroke 41, 624–629. doi: 10.1161/STROKEAHA.109.558809
Jellinger, K. A. (2002). Alzheimer disease and cerebrovascular pathology: an update. J. Neural Transm. 109, 813–836. doi: 10.1007/s007020200068
Jellinger, K. A., and Attems, J. (2005). Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer disease. J. Neurol. Sci. 229–230, 37–41. doi: 10.1016/j.jns.2004.11.018
Kang, C. K., Park, C. A., Kim, K. N., Hong, S. M., Park, C. W., Kim, Y. B., et al. (2010). Non-invasive visualization of basilar artery perforators with 7T MR angiography. J. Magn. Reson. Imaging 32, 544–550. doi: 10.1002/jmri.22250
Koga, H., Takashima, Y., Murakawa, R., Uchino, A., Yuzuriha, T., and Yao, H. (2009). Cognitive consequences of multiple lacunes and leukoaraiosis as vascular cognitive impairment in community-dwelling elderly individuals. J. Stroke Cerebrovasc. Dis. 18, 32–37. doi: 10.1016/j.jstrokecerebrovasdis.2008.07.010
Lammie, G. A. (2002). Hypertensive cerebral small vessel disease and stroke. Brain Pathol. 12, 358–370.
Launer, L. J., Hughes, T. M., and White, L. R. (2011). Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann. Neurol. 70, 774–780. doi: 10.1002/ana.22520
Lee, S. H., Bae, H. J., Kwon, S. J., Kim, H., Kim, Y. H., Yoon, B. W., et al. (2004). Cerebral microbleeds are regionally associated with intracerebral hemorrhage. Neurology 62, 72–76. doi: 10.1212/01.WNL.0000101463.50798.0D
Lees, K. R., Bluhmki, E., von Kummer, R., Brott, T. G., Toni, D., Grotta, J. C., et al. (2010). Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet 375, 1695–1703. doi: 10.1016/S0140-6736(10)60491-6
Lovelock, C. E., Cordonnier, C., Naka, H., Al-Shahi Salman, R., Sudlow, C. L., Sorimachi, T., et al. (2010). Antithrombotic drug use, cerebral microbleeds, and intracerebral hemorrhage: a systematic review of published and unpublished studies. Stroke 41, 1222–1228. doi: 10.1161/STROKEAHA.109.572594
Maillard, P., Fletcher, E., Harvey, D., Carmichael, O., Reed, B., Mungas, D., et al. (2011). White matter hyperintensity penumbra. Stroke 42, 1917–1922. doi: 10.1161/STROKEAHA.110.609768
Meroni, G., and Diez-Roux, G. (2005). TRIM/RBCC, a novel class of “single protein RING finger” E3 ubiquitin ligases. Bioessays 27, 1147–1157. doi: 10.1002/bies.20304
Miyao, S., Takano, A., Teramoto, J., and Takahashi, A. (1992). Leukoaraiosis in relation to prognosis for patients with lacunar infarction. Stroke 23, 1434–1438. doi: 10.1161/01.STR.23.10.1434
Mok, V. C., Lam, W. W., Fan, Y. H., Wong, A., Ng, P. W., Tsoi, T. H., et al. (2009). Effects of statins on the progression of cerebral white matter lesion: post hoc analysis of the ROCAS (Regression of Cerebral Artery Stenosis) study. J. Neurol. 256, 750–757. doi: 10.1007/s00415-009-5008-7
Moody, D. M., Brown, W. R., Challa, V. R., and Anderson, R. L. (1995). Periventricular venous collagenosis: association with leukoaraiosis. Radiology 194, 469–476.
Nag, S. (2003). Blood–brain barrier permeability using tracers and immunohistochemistry. Methods Mol. Med. 89, 133–144.
Neumann-Haefelin, T., Hoelig, S., Berkefeld, J., Fiehler, J., Gass, A., Humpich, M., et al. (2006). Leukoaraiosis is a risk factor for symptomatic intracerebral hemorrhage after thrombolysis for acute stroke. Stroke 37, 2463–2466. doi: 10.1161/01.STR.0000239321.53203.ea
Norrving, B. (2003). Long-term prognosis after lacunar infarction. Lancet Neurol. 2, 238–245. doi: 10.1016/S1474-4422(03)00352-1
Ozato, K., Shin, D. M., Chang, T. H., and Morse, H. C. III. (2008). TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8, 849–860. doi: 10.1038/nri2413
Palumbo, V., Boulanger, J. M., Hill, M. D., Inzitari, D., and Buchan, A. M. (2007). Leukoaraiosis and intracerebral hemorrhage after thrombolysis in acute stroke. Neurology 68, 1020–1024. doi: 10.1212/01.wnl.0000257817.29883.48
Pantoni, L. (2003). Not-so-silent infarcts. Lancet Neurol. 2, 335. doi: 10.1016/S1474-4422(03)00406-X
Pantoni, L. (2008). Leukoaraiosis: from an ancient term to an actual marker of poor prognosis. Stroke 39, 1401–1403. doi: 10.1161/STROKEAHA.107.505602
Pantoni, L. (2010). Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 9, 689–701. doi: 10.1016/S1474-4422(10)70104-6
Pantoni, L., Poggesi, A., and Inzitari, D. (2007). The relation between white-matter lesions and cognition. Curr. Opin. Neurol. 20, 390–397. doi: 10.1097/WCO.0b013e328172d661
Patel, B., and Markus, H. S. (2011). Magnetic resonance imaging in cerebral small vessel disease and its use as a surrogate disease marker. Int. J. Stroke 6, 47–59. doi: 10.1111/j.1747-4949.2010.00552.x
Petty, G. W., Brown, R. D. Jr., Whisnant, J. P., Sicks, J. D., O’Fallon, W. M., and Wiebers, D. O. (2000). Ischemic stroke subtypes : a population-based study of functional outcome, survival, and recurrence. Stroke 31, 1062–1068. doi: 10.1161/01.STR.31.5.1062
Rincon, F., and Sacco, R. L. (2008). Secondary stroke prevention. J. Cardiovasc. Nurs. 23, 34–41; quiz 42–43. doi: 10.1097/01.JCN.0000305059.81000.d3
Rincon, F., and Wright, C. B. (2013). Vascular cognitive impairment. Curr. Opin. Neurol. 26, 29–36. doi: 10.1097/WCO.0b013e32835c4f04
Roger, V. L., Go, A. S., Lloyd-Jones, D. M., Benjamin, E. J., Berry, J. D., Borden, W. B., et al. (2012). Heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation 125, e2–e220. doi: 10.1161/CIR.0b013e31823ac046
Samuelsson, M., Soderfeldt, B., and Olsson, G. B. (1996). Functional outcome in patients with lacunar infarction. Stroke 27, 842–846. doi: 10.1161/01.STR.27.5.842
Schneider, J. A., Arvanitakis, Z., Bang, W., and Bennett, D. A. (2007). Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24
Shin, H. K., Jones, P. B., Garcia-Alloza, M., Borrelli, L., Greenberg, S. M., Bacskai, B. J., et al. (2007). Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain 130, 2310–2319. doi: 10.1093/brain/awm156
Shoamanesh, A., Kwok, C. S., and Benavente, O. (2011). Cerebral microbleeds: histopathological correlation of neuroimaging. Cerebrovasc. Dis. 32, 528–534. doi: 10.1159/000331466
Simpson, J. E., Hosny, O., Wharton, S. B., Heath, P. R., Holden, H., Fernando, M. S., et al. (2009). Microarray RNA expression analysis of cerebral white matter lesions reveals changes in multiple functional pathways. Stroke 40, 369–375. doi: 10.1161/STROKEAHA.108.529214
Smith, E. E., Vijayappa, M., Lima, F., Delgado, P., Wendell, L., Rosand, J., et al. (2008). Impaired visual evoked flow velocity response in cerebral amyloid angiopathy. Neurology 71, 1424–1430. doi: 10.1212/01.wnl.0000327887.64299.a4
Soontornniyomkij, V., Lynch, M. D., Mermash, S., Pomakian, J., Badkoobehi, H., Clare, R., et al. (2010). Cerebral microinfarcts associated with severe cerebral beta-amyloid angiopathy. Brain Pathol. 20, 459–467. doi: 10.1111/j.1750-3639.2009.00322.x
Srikanth, V., Beare, R., Blizzard, L., Phan, T., Stapleton, J., Chen, J., et al. (2009). Cerebral white matter lesions, gait, and the risk of incident falls: a prospective population-based study. Stroke 40, 175–180. doi: 10.1161/STROKEAHA.108.524355
Steingart, A., Hachinski, V. C., Lau, C., Fox, A. J., Fox, H., Lee, D., et al. (1987). Cognitive and neurologic findings in demented patients with diffuse white matter lucencies on computed tomographic scan (leuko-araiosis). Arch. Neurol. 44, 36–39. doi: 10.1001/archneur.1987.00520130028013
Sudlow, C. L., and Warlow, C. P. (1997). Comparable studies of the incidence of stroke and its pathological types: results from an international collaboration. International Stroke Incidence Collaboration. Stroke 28, 491–499. doi: 10.1161/01.STR.28.3.491
Tanaka, A., Ueno, Y., Nakayama, Y., Takano, K., and Takebayashi, S. (1999). Small chronic hemorrhages and ischemic lesions in association with spontaneous intracerebral hematomas. Stroke 30, 1637–1642. doi: 10.1161/01.STR.30.8.1637
Tatemichi, T. K., Desmond, D. W., Prohovnik, I., Cross, D. T., Gropen, T. I., Mohr, J. P., et al. (1992). Confusion and memory loss from capsular genu infarction: a thalamocortical disconnection syndrome? Neurology 42, 1966–1979. doi: 10.1212/WNL.42.10.1966
Thal, D. R., Capetillo-Zarate, E., Larionov, S., Staufenbiel, M., Zurbruegg, S., and Beckmann, N. (2009). Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol. Aging 30, 1936–1948. doi: 10.1016/j.neurobiolaging.2008.01.017
Thal, D. R., Ghebremedhin, E., Orantes, M., and Wiestler, O. D. (2003). Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J. Neuropathol. Exp. Neurol. 62, 1287–1301.
The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. (1995). Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 333, 1581–1587.
Vandeputte, D. A., Meije, C. B., van Dartel, M., Leenstra, S., IJlst-Keizers, H., Das, P. K., et al. (2001). GOA, a novel gene encoding a ring finger B-box coiled-coil protein, is overexpressed in astrocytoma. Biochem. Biophys. Res. Commun. 286, 574–579. doi: 10.1006/bbrc.2001.5431
van Straaten, E. C., Fazekas, F., Rostrup, E., Scheltens, P., Schmidt, R., Pantoni, L., et al. (2006). Impact of white matter hyperintensities scoring method on correlations with clinical data: the LADIS study. Stroke 37, 836–840. doi: 10.1161/01.STR.0000202585.26325.74
van Swieten, J. C., Staal, S., Kappelle, L. J., Derix, M. M., and van Gijn, J. (1996). Are white matter lesions directly associated with cognitive impairment in patients with lacunar infarcts? J. Neurol. 243, 196–200. doi: 10.1007/BF02444014
Vermeer, S. E., Longstreth, W. T. Jr., and Koudstaal, P. J. (2007). Silent brain infarcts: a systematic review. Lancet Neurol. 6, 611–619. doi: 10.1016/S1474-4422(07)70170-9
Vermeer, S. E., Prins, N. D., den Heijer, T., Hofman, A., Koudstaal, P. J., and Breteler, M. M. (2003). Silent brain infarcts and the risk of dementia and cognitive decline. N. Engl. J. Med. 348, 1215–1222. doi: 10.1056/NEJMoa022066
Vernooij, M. W., van der Lugt, A., Ikram, M. A., Wielopolski, P. A., Niessen, W. J., Hofman, A., et al. (2008). Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology 70, 1208–1214. doi: 10.1212/01.wnl.0000307750.41970.d9
Vinters, H. V. (1987). Cerebral amyloid angiopathy. A critical review. Stroke 18, 311–324. doi: 10.1161/01.STR.18.2.311
Viswanathan, A., Godin, O., Jouvent, E., O’Sullivan, M., Gschwendtner, A., Peters, N., et al. (2010). Impact of MRI markers in subcortical vascular dementia: a multi-modal analysis in CADASIL. Neurobiol. Aging 31, 1629–1636. doi: 10.1016/j.neurobiolaging.2008.09.001
Viswanathan, A., Patel, P., Rahman, R., Nandigam, R. N., Kinnecom, C., Bracoud, L., et al. (2008). Tissue microstructural changes are independently associated with cognitive impairment in cerebral amyloid angiopathy. Stroke 39, 1988–1992. doi: 10.1161/STROKEAHA.107.509091
Wardlaw, J. M., Murray, V., Berge, E., and Del Zoppo, G. J. (2009). Thrombolysis for acute ischaemic stroke. Cochrane Database Syst. Rev. 4, CD000213. doi: 10.1002/14651858.CD000213.pub2
Wardlaw, J. M., Smith, E. E., Biessels, G. J., Cordonnier, C., Fazekas, F., Frayne, R., et al. (2013). Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 12, 822–838. doi: 10.1016/S1474-4422(13)70124-8
Werring, D. J., Frazer, D. W., Coward, L. J., Losseff, N. A., Watt, H., Cipolotti, L., et al. (2004). Cognitive dysfunction in patients with cerebral microbleeds on T2*-weighted gradient-echo MRI. Brain 127, 2265–2275. doi: 10.1093/brain/awh253
Wright, C. B., Festa, J. R., Paik, M. C., Schmiedigen, A., Brown, T. R., Yoshita, M., et al. (2008). White matter hyperintensities and subclinical infarction: associations with psychomotor speed and cognitive flexibility. Stroke 39, 800–805. doi: 10.1161/STROKEAHA.107.484147
Yakushiji, Y., Nishiyama, M., Yakushiji, S., Hirotsu, T., Uchino, A., Nakajima, J., et al. (2008). Brain microbleeds and global cognitive function in adults without neurological disorder. Stroke 39, 3323–3328. doi: 10.1161/STROKEAHA.108.516112
Yamamoto, Y., Akiguchi, I., Oiwa, K., Hayashi, M., Kasai, T., and Ozasa, K. (2002). Twenty-four-hour blood pressure and MRI as predictive factors for different outcomes in patients with lacunar infarct. Stroke 33, 297–305.
Young, V. G., Halliday, G. M., and Kril, J. J. (2008). Neuropathologic correlates of white matter hyperintensities. Neurology 71, 804–811. doi: 10.1212/01.wnl.0000319691.50117.54
Keywords: stroke, leukoaraiosis, silent brain infarction, vascular cognitive impairment, Alzheimer’s disease
Citation: Rincon F and Wright CB (2014) Current pathophysiological concepts in cerebral small vessel disease. Front. Aging Neurosci. 6:24. doi: 10.3389/fnagi.2014.00024
Received: 20 August 2013; Accepted: 12 February 2014;
Published online: 24 March 2014.
Edited by:
Orly Lazarov, The University of Illinois at Chicago, USAReviewed by:
Jose G. Castaño, Universidad Autónoma de Madrid, SpainJunming Wang, University of Mississippi Medical Center, USA
Copyright © 2014 Rincon and Wright. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clinton B. Wright, Evelyn F. McKnight Brain Institute, Department of Neurology, University of Miami, CRB 1349, 1120 NW 14th Street, Miami, FL 33136, USA e-mail: cwright@med.miami.edu