Metals and cholesterol: two sides of the same coin in Alzheimer’s disease pathology
 Bruce X. Wong
Bruce X. Wong Ya Hui Hung
Ya Hui Hung Ashley I. Bush
Ashley I. Bush James A. Duce
James A. Duce- 1Oxidation Biology Unit, The Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, VIC, Australia
- 2School of Molecular and Cellular Biology, Faculty of Biological Sciences, University of Leeds, Leeds, North Yorkshire, UK
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disease. It begins years prior to the onset of clinical symptoms, such as memory loss and cognitive decline. Pathological hallmarks of AD include the accumulation of β-amyloid in plaques and hyperphosphorylated tau in neurofibrillary tangles. Copper, iron, and zinc are abnormally accumulated and distributed in the aging brain. These metal ions can adversely contribute to the progression of AD. Dysregulation of cholesterol metabolism has also been implicated in the development of AD pathology. To date, large bodies of research have been carried out independently to elucidate the role of metals or cholesterol on AD pathology. Interestingly, metals and cholesterol affect parallel molecular and biochemical pathways involved in AD pathology. The possible links between metal dyshomeostasis and altered brain cholesterol metabolism in AD are reviewed.
Introduction
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disease characterized by pathological hallmarks of extracellular β-amyloid (Aβ) plaques (Glenner and Wong, 1984a,b; Masters et al., 1985) and intracellular neurofibrillary tangles (Delacourte and Defossez, 1986; Kosik et al., 1986; Lee et al., 1991) in the brain. The rate of AD progression is variable, but on average, patients may live up to 10 years after diagnosis (Whitehouse, 1997). Approximately 8–10% of the population over the age of 65 have AD, and its prevalence doubles every 5 years thereafter (Cummings, 2004; Bertram and Tanzi, 2005). These data, coupled with ever increasing life expectancy, marks AD as one of the most significant health and socio-economic problems, particularly in industrialized nations.
As with most diseases, genetic and environmental factors can contribute to its development. AD can be broadly characterized as either familial or sporadic. Early-onset familial AD (FAD) are caused by mutations within three genes, which encode the amyloid precursor protein (APP) and presenilins 1 and 2 (PSEN1 and PSEN2; Holmes, 2002; Tanzi and Bertram, 2005; Bertram et al., 2007). These mutations are autosomal dominant, and symptoms of AD manifest prior to 65 years of age. FAD accounts for less than 5% of AD cases (Janssen et al., 2003; Raux et al., 2005). The disease etiology for late-onset sporadic AD is complex and multifactorial, which may involve age-related alterations in metabolism, repair mechanisms, immune response, and environmental factors such as life style, prior brain trauma, and oxidative stress (Muller-Spahn and Hock, 1999; Chen et al., 2009). Genome-wide association studies (GWAS) have identified candidate genes that significantly increase the risk of late-onset AD. By far, the strongest risk factor found is the ε4 allele of the apolipoprotein E (APOE) gene (Farrer et al., 1997). Possessing just a single ε4 allele increases the risk of developing AD by two- to fivefold, while having two alleles increases the risk to more than fivefold (Poirier et al., 1993; Strittmatter et al., 1993; Holmes, 2002; Poirier, 2003; Bertram et al., 2007; Coon et al., 2007).
Neuritic plaques are multi-cellular lesions containing Aβ peptides (especially the neurotoxic Aβ42 species), reactive astrocytes, activated microglia, and dystrophic neurites (Maulik et al., 2013). Aβ peptide is produced by the proteolytic cleavage of APP by β- and γ-secretases (see APP Processing and Aβ Generation). Interestingly, these plaques also have an enrichment of cholesterol (Panchal et al., 2010) and metals such as copper, iron, and zinc (Goodman, 1953; Connor et al., 1992; Bush et al., 1994c; Lovell et al., 1998; Suh et al., 2000; Collingwood et al., 2005; Stoltenberg et al., 2005; Miller et al., 2006; Baltes et al., 2011), which indicate a failure of cholesterol and metal regulatory systems in the brain. While the underlying etiology of AD is yet to be clearly established, mounting evidence derived from epidemiological, clinical and biochemical studies have independently implicated roles for metals and cholesterol in the pathogenesis of AD. This review presents an overview of the roles of metals and cholesterol in APP/Aβ metabolism and their relationship in the development of AD.
APP Processing and Aβ Generation
Amyloid precursor protein is a type I trans-membrane protein that can be post-translationally modified by N- and O-glycosylation, tyrosine sulfation, and phosphorylation (Weidemann et al., 1989; Walter et al., 2000). Full-length APP is sequentially processed via two pathways: the non-amyloidogenic and amyloidogenic. The cleavage by α- or β-secretases at the N-terminus of the Aβ domain generates soluble APP derivatives: sAPPα and sAPPβ, respectively, leaving behind membrane tethered C-terminal fragments (CTFα and CTFβ, respectively). Subsequent cleavage of these CTFs by the γ-secretase generates either p3 (from CTFα) or Aβ (from CTFβ), and liberation of the APP intracellular domain (AICD; reviewed in Zheng and Koo, 2011; Figure 1B).
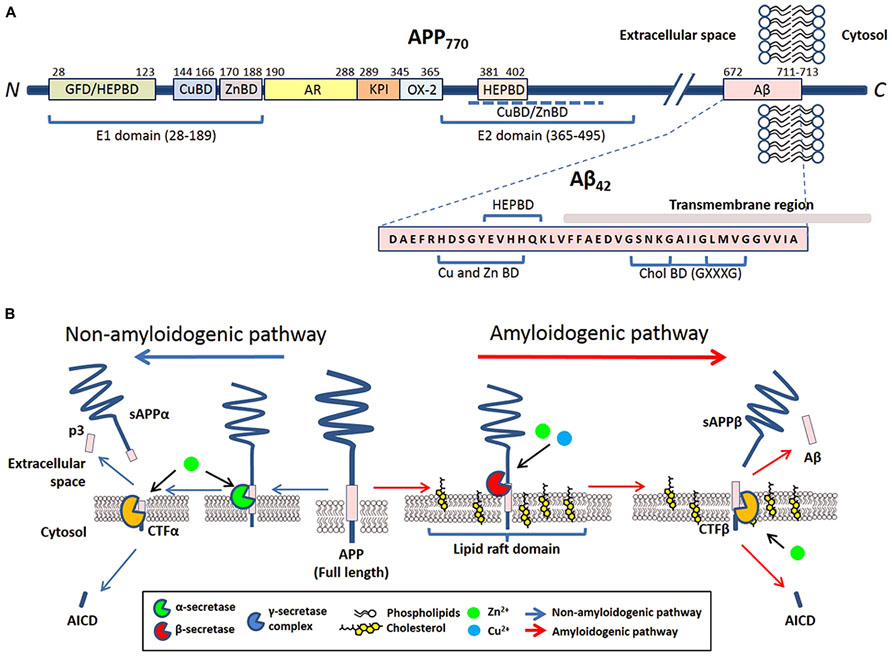
FIGURE 1. The involvement of metals and cholesterol in post-translational modification of APP. (A) Schematic of reported metal and cholesterol binding domains in APP770 and Aβ in relation to other recognized motifs. APP770 is the longest isoform of APP with the APP751 isoform lacking the OX-2 domain and the neuron prevalent isoform APP695 lacking both OX-2 and Kunitz-type protease inhibitor (KPI). Within the extracellular presented ectodomain of APP, the E1 region at the N-terminal contains a copper binding domain (CuBD) and zinc binding domain (ZnBD) that is C-terminally orientated compared to the growth factor domain (GFD) which incorporates a heparin binding domain (HEPBD). The E1 domain is followed by the acidic region (AR), KPI and OX-2 before the E2 domain of APP, containing a HEPBD and CuBD/ZnBD that is yet to be exactly mapped (Dahms et al., 2012). The E2 domain is followed by the Aβ peptide that is partially embedded into the transmembrane region. Aβ also has a recognized CuBD/ZnBD as well as a Cholesterol binding region (CholBD) that incorporates the GXXXG motifs. (B) Proteolytic processing of APP predominantly follows two pathways that are initiated by separate secretases. The non-amyloidogenic pathway (blue arrows) initiates with the cleavage of full-length APP by α-secretase within the Aβ sequence. Following further cleavage by the γ-secretase complex, this pathway results in the generation of soluble N-terminal APP fragment (sAPPα) and C-terminal fragments (p3 and AICD). The alternative amyloidogenic pathway (red arrows) involves sequential cleavage of APP by β-secretase followed by the γ-secretase complex, which results in the liberation of a soluble N-terminal sAPPβ fragment, Aβ peptide, and AICD. Copper and zinc affect the processing of APP and Aβ generation on neuronal membranes through their direct influence on the enzymatic activity of β-, α-, and γ-secretases. The influence of cholesterol is through its requirement in lipid raft domains, the location for amyloidogenic processing of APP.
The trans-membrane aspartyl protease β-site APP cleaving enzyme 1 (BACE1) is the major β-secretase in neurons (Sinha et al., 1999). This is the rate-limiting enzyme involved in the generation of Aβ (Vassar et al., 1999; Yan et al., 1999). In contrast, α-secretase cleavage of APP can be stimulated by a disintegrin and metalloproteinase (ADAM) family of proteases (reviewed in Lichtenthaler, 2011) and a number of other molecules (e.g., phorbol ester) or via protein kinase C activation, in which case the cleavage is regulated by tumor necrosis factor α-converting enzyme (TACE; Buxbaum et al., 1998; Blacker et al., 2002). Studies have indicated that in neurons, α-secretase activity is likely to be primarily mediated by ADAM10 (Kuhn et al., 2010). The mature γ-secretase is a polytopic complex consisting of four individual components: presenilin (PS); nicastrin (Nct); anterior pharynx defective 1 (Aph1); and presenilin enhancer 2 (Pen-2; reviewed in Edbauer et al., 2003; Iwatsubo, 2004). Presenilin, an aspartyl protease, is the main catalytic unit of the complex. Aβ of varying length is the result of hierarchical and site-specific cleavage of APP by β- and γ-secretase (Figure 1B). Other than APP, all three APP-cleaving secretases can digest other biological substrates required for multiple biological functions such as regulation of development, differentiation, and proliferation.
Metal and Cholesterol Modulation of APP and Aβ Metabolism
The dynamics of biological metal ions (e.g., copper, zinc, and iron) is critical for many physiological functions. Metal ions are key components in many enzymatic functions, which include catalysis, structural stability, transportation of oxygen, and cellular signaling. The passive flux of metals between the circulation and the brain is tightly regulated by the blood–brain barrier (BBB; Duce and Bush, 2010). The impact of metals on the brain causing neurodegeneration may be caused by increased toxic exposure, as well as a breakdown in the mechanisms that compartmentalize and regulate metal homeostasis.
The brain is the most cholesterol-rich organ in the body. Functionally, cholesterol plays a critical role in neuronal development and maintenance of synaptic plasticity. As a component of the plasma membrane, it regulates ion homeostasis, endocytosis, and intracellular signaling pathways. It also serves as a precursor for the production of steroid hormones, vitamin D, and oxysterols. Like metals, experimental work has shown compartmentalization between levels of cholesterol in the serum and brain that is regulated by BBB (Hung et al., 2013). Substantial evidence correlates cholesterol homeostasis dysregulation with AD. In cell culture systems, production of Aβ is linked to cholesterol levels. However, the exact influence of cholesterol in Aβ generation is still unclear.
Metals and APP
The APP sequence contains putative binding sites for copper (Hesse et al., 1994; Atwood et al., 2000; Simons et al., 2002; Barnham et al., 2003; Valensin et al., 2004) and zinc (Bush et al., 1993, 1994a,b,c). Copper binds to APP between residues 142 and 166 (White et al., 1999a; Barnham et al., 2003), a site where it can also catalytically reduce copper (Multhaup et al., 1996). Recently, two copper binding residues at histidine 149 and 151 have been identified as crucial for APP metabolism, protein folding and stability (Spoerri et al., 2012). The Aβ segment of APP is another region that directly interacts with copper and is explained in more detail in Section “Metal Modulation of Aβ Generation, Aggregation, and Cell Toxicity.” The N-terminal copper binding domain of APP has been found to play crucial roles in homodimerization (Hesse et al., 1994; Kaden et al., 2008), and an elevation in copper levels increases APP homodimerization (Noda et al., 2013). Zinc binds to a conserved region of amino acids between position 170 and 188 of APP (Bush et al., 1993, 1994a). The coordination binding involves two key cysteines at positions 186 and 187, as well as other potential ligands (e.g., C174, M170, D177, and E184). Similar to copper, the binding of zinc may also play an important functional role in homodimerization of APP (Scheuermann et al., 2001; Ciuculescu et al., 2005; Figure 1A).
A number of in vivo and in vitro studies highlight the reciprocal regulation between APP and metal ions. The regulation of APP gene expression is linked to altered cellular copper levels. Studies in the in vitro cell culture show that copper depletion by overexpressing copper transporter ATP7A result in down-regulation of APP gene expression and APP protein level; conversely, APP gene expression level is up-regulated under conditions of copper overload due to ATP7A-deficiency (Armendariz et al., 2004; Bellingham et al., 2004b). On the other hand, copper concentration is increased in brain and liver tissue as well as primary neuronal and skin fibroblast cells from APP and amyloid precursor-like protein 2 (APLP2) knockdown mice (White et al., 1999b; Bellingham et al., 2004a; Hung et al., 2009; Acevedo et al., 2011). The difference in copper level is even more pronounced in aged mice (Needham et al., 2014). In contrast, APP over-expressing transgenic mice have decreased copper in the brain (Maynard et al., 2002; Bayer et al., 2003; Phinney et al., 2003). Copper treatment stimulates the movement of APP from the trans-Golgi network to the plasma membrane and attenuates internalization of APP to BACE1-rich endosomes. However, copper treatment does not result in any detectable change in APP processing (Hung et al., 2009; Acevedo et al., 2011). In humans, low copper diet is associated with a significant decrease in APP expression in platelets from healthy postmenopausal women (Davis et al., 2000).
Iron regulates APP translation, which involves an iron response element (IRE) RNA stem loop in its 5′-untranslated region (UTR). The APP IRE is homologous with the canonical IRE RNA stem-loop that binds iron regulatory proteins (IRP1 and IRP2) to control intracellular iron homoeostasis by modulating ferritin mRNA translation and transferrin receptor mRNA stability (Rogers et al., 2002). IRP1, but not IRP2, selectively binds to the APP IRE in human neural cells (Cho et al., 2010). Intracellular metal chelation selectively down-regulates APP 5′-UTR translation, which is reversed by cytoplasmic labile iron (Venti et al., 2004). The regulation of APP by iron through the 5′-UTR indicates that iron has a role in APP metabolism.
In the brain, ferroportin (Fpn) is required for excess iron to exit the cell (Donovan et al., 2005; Ganz, 2005). Fpn channels transport iron through the plasma membrane where it is required to be converted to its ferric form before being released and loaded onto transferrin, the extracellular iron-transporting protein that transfers iron between cells (Swaiman and Machen, 1984). APP may play a role in the iron export mechanism of cells through the stabilization of Fpn (Duce et al., 2010). APP knockout mice exposed to dietary iron results in ferrous iron accumulation and oxidative stress in cortical neurons. Ablation of APP in HEK293T cells and primary neurons negates iron export, which can be restored by the addition of exogenous APP (Duce et al., 2010). This iron-export capability of APP requires tau to traffic endogenous APP to the cell surface (Lei et al., 2012).
Cholesterol and APP
Cholesterol is not symmetrically distributed laterally and between the two leaflets of the lipid membrane bilayer. The significance of this asymmetry is not yet known, although cholesterol has been implicated in cell membrane fluidity, integrity, and function (Wood et al., 1999; Hayashi et al., 2002). Patches of the membrane highly enriched with cholesterol and sphingolipid are termed lipid rafts (also known as detergent-resistant microdomains). Cholesterol provides structural stability in rafts by serving as a molecular spacer, filling in voids between raft proteins and other raft lipids such as sphingolipids and gangliosides (Xu and London, 2000; Ramstedt and Slotte, 2006). Therefore, modulation of cholesterol can result in dissociation, dysregulation, and/or inactivation of raft proteins. Indeed, APP processing and activity is influenced by its membrane domain localization.
Binding of cholesterol to APP occurs in the trans-membrane carboxyl-terminal region between amino acids 672 and 770 (or CTFβ) through interactions with membrane-buried GXXXG motifs (G, glycine; X, any amino acid; Barrett et al., 2012; Figure 1A). The GXXXG motif is involved with APP homodimerization (Kim et al., 2005; Munter et al., 2007; Kienlen-Campard et al., 2008; Miyashita et al., 2009; Sato et al., 2009). Competitive studies of C99 with cholesterol suggest that complexing of cholesterol:C99 at a 1:1 ratio is preferred over C99 homodimers under most physiological conditions (Song et al., 2013). The binding of cholesterol directly to APP and CTFβ may promote amyloidogenic processing by increasing the localization of APP/CTFβ to cholesterol-rich membrane domains and organelles, where γ- and β-secretases preferentially reside (Beel et al., 2010).
In cultured rat neuronal cells, up-regulation of APP gene expression reduces cholesterol biosynthesis while down-regulation of APP gene expression has the opposite effect (Pierrot et al., 2013). Membrane cholesterol content, however, is not affected. Sterol receptor element binding protein (SREBP) and rate limiting enzyme HMG-CoA reductase (HMGCR) control biosynthesis of cholesterol. The site-2 zinc metalloprotease (S2P) cleaves SREBP at Site-2 within the membrane-spanning domain (Brown and Goldstein, 1999). Interaction of APP with SREBP1 prevents S2P-mediated processing of mSREBP1 nuclear translation of its target genes including HMGCR (Pierrot et al., 2013). Interestingly, the APP/Aβ GXXXG motif is critical in the regulation of HMGCR. In contrast to neuronal cells, APP interaction with SREBP1 and resulting cholesterol biosynthesis is not detectable in astrocytes (Pierrot et al., 2013). APP expression associated reduction of cholesterol and oxysterol production is mediated via down-regulation of both HMGCR and 24-hydroxylase [required to convert cholesterol to 24S-hydroxycholesterol (24OHC)] activities, respectively. Since membrane cholesterol remains the same, it is suggested that APP controls cholesterol turnover (Pierrot et al., 2013).
Metal Modulation of APP Processing Enzymes
Metals can indirectly affect Aβ generation by altering secretase-dependent processing of APP. To date, all three secretases involved in APP cleavage are known to have interactions with different metal species. The α-secretase TACE contains a zinc ion in its catalytic domain (Cross et al., 2002). TACE enzymatic activity is controlled by a “cysteine-switch” motif mediated by an intramolecular bond between cysteine and a zinc atom in its catalytic site. Subsequently, it has been shown that other regions of the TACE prodomain are able to circumvent the “cysteine-switch” and inhibit enzymatic activity (Buckley et al., 2005). Correspondingly, the metalloprotease ADAM10 can be inhibited by its dominant-negative form that has a point mutation in its zinc-binding site (Lammich et al., 1999).
The major β-secretase BACE1, binds copper in its C-terminal domain, the same region that interacts with domain I of copper chaperone for superoxide dismutase-1 (CCS; Angeletti et al., 2005). The expression of BACE1 reduces superoxidase 1 (SOD1) activity. In contrast, in cells overexpressing both BACE1 and CCS, SOD1 activity is restored by CCS (Angeletti et al., 2005). An interaction between BACE1 and CCS has been demonstrated by co-immunoprecipitation from brain homogenates and their co-transport through the axon (Angeletti et al., 2005).
Presenilin, the active subunit of the γ-secretase, is also sensitive to metal levels. Neonatal cortical cultures exposed to zinc increases C-terminal fragmentation of PS1 by enhancing synthesis of the protein (Park et al., 2001). However, zinc induces oligomerization of an APP γ-secretase substrate and inhibits its processing, which supports a role for zinc dysregulation in Aβ processing (Hoke et al., 2005; Greenough et al., 2011).
Taken together, these results suggest a direct influence of metals on secretase enzymatic activity to process APP and therefore may have detrimental implications in AD pathology when metal homeostasis is altered.
Cholesterol Modulation of APP Processing
Previous studies show that full-length APP, Aβ, APP-CTFs, and PS1 are associated with lipid rafts (Lee et al., 1998; Simons et al., 2001; Hur et al., 2008). Studies with cultured cells demonstrate cholesterol depletion by β-cyclodextrin extraction or inhibition of cholesterol biosynthesis by statins (Simons et al., 1998; Wahrle et al., 2002), result in decreased Aβ production. Conversely, increasing cellular cholesterol levels enhance Aβ production and reduce α-secretase cleavage of APP (Bodovitz and Klein, 1996; Frears et al., 1999).
Since APP, β- and γ-secretases are associated with lipid raft domains, it is not surprising that altered cellular cholesterol content affects Aβ generation, aggregation, and clearance. The presence of lipid raft domains has been found in plasma membranes and endosomes. More recently, a study uncovered lipid raft-like domains in mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs), a sub-compartment of the ER connected to mitochondria (Area-Gomez et al., 2012). Lipid rafts are sensitive to altered cholesterol metabolism, and cholesterol depletion results in lipid raft destabilization (Eckert et al., 2010). As previously mentioned, cholesterol enriched in lipid rafts can influence dynamics of proteins within these rafts. Altered cholesterol levels affect lipid raft localization of APP and its derivatives together with secretases required for APP processing. Biochemical isolation of lipid rafts indicates that BACE1 and γ-secretase protein are localized within these lipid domains (Wahrle et al., 2002; Vetrivel et al., 2004; Kalvodova et al., 2005; Osenkowski et al., 2008), while the α-secretase ADAM10 is predominantly localized outside the lipid rafts (Kojro et al., 2001). Consistent with other lipid raft domains, MAMs have a high concentration of APP, PS1, and PS2 (catalytic subunits of γ-secretase) and γ-secretase activity. APP is believed to exist in either pool within plasma membranes (Ehehalt et al., 2003). Experimental evidence suggests that amyloidogenic processing of APP occurs in lipid rafts while the non-amyloidogenic processing occurs mainly in the non-raft regions. If this is the case, then cholesterol levels contribute to regulation of APP processing through these two pathways. The non-amyloidogenic pathway predominates, because only small amounts of APP appear to be present in lipid rafts under physiological conditions (Bouillot et al., 1996; Parkin et al., 1999). Increasing membrane cholesterol levels may increase overall percentage of lipid rafts, which favors APP and BACE1 interaction and increases Aβ generation. Several studies support this idea. Firstly, imaging of fluorescently tagged APP and BACE1 demonstrates that cholesterol loading does not increase Aβ production through BACE1 catalytic activity but rather by altering the accessibility of BACE1 to its substrate APP in lipid rafts (Marquer et al., 2011). Secondly, APP and BACE1 copatch at the plasma membrane upon antibody cross-linking, which increases Aβ production in a cholesterol-dependent manner (Ehehalt et al., 2003). Lastly, inhibition of γ-secretase activity leads to an accumulation of APP-CTFs in lipid rafts (Vetrivel et al., 2004).
Niemann–Pick type C disease (NP-C) is a lysosomal lipid storage disorder, characterized by accumulation of cholesterol and sphingolipids within the endosomal–lysosomal system. The majority of NP-C cases are caused by functional loss of NPC1 protein activity, due to genetic mutation. Neuronal degeneration underlies neurological symptoms in NP-C patients, which include cerebellar ataxia, dysphagia, dysarthria, and dementia. Altered cholesterol distribution within subcellular compartments has been implicated in the aberrant trafficking and processing of APP similar to that observed in AD (Runz et al., 2002; Vanier and Millat, 2003; Jin et al., 2004; Walkley and Suzuki, 2004; Vance, 2006; Kodam et al., 2010; Kosicek et al., 2010; Malnar et al., 2010, 2012). In cell models of NP-C, cholesterol overload due to NPC1 deficiency leads to increased APP lipid raft localization and internalization from the plasma membrane to BACE1-rich endosomes, where amyloidogenic processing occurs (Kosicek et al., 2010; Malnar et al., 2010). This can be corrected by cholesterol depletion in cultured cells using lipid-deficient serum, lovastatin treatment, or methyl-β-cyclodextrin treatment (Malnar et al., 2012). The cholesterol-dependent change in APP trafficking and lipid raft localization parallels previous studies of APP’s response to changes in cellular copper levels (Hung et al., 2009; Acevedo et al., 2011). Furthermore, cholesterol-dependent APP trafficking and metabolism may explain some of the metal changes observed in NP-C tissue samples (Hung et al., 2014). Taken together, these evidences suggest a synergistic interaction between copper and cholesterol pathways in the regulation of APP metabolism that may contribute to AD pathogenesis.
Altered intracellular cholesterol metabolism can also affect APP processing. Cultured cells exposed to a cholesterol transport inhibitor, U18666A, accumulate cholesterol in late endosomes and lysosomes, and results in a dose-dependent decrease in Aβ production (Runz et al., 2002; Davis, 2008). However, the inhibitor also increases accumulation of γ-secretase, CTFβ, and Aβ-related peptides in vesicular organelles (Runz et al., 2002; Jin et al., 2004). From these studies, it can be inferred that cholesterol is able to influence APP processing through re-internalization of surface APP, as well as redistribution of APP and its processing enzymes within subcellular compartments.
Metal Modulation of Aβ Generation, Aggregation, and Cell Toxicity
Aβ binds to zinc, copper, and iron to form various precipitous complexes, which are dependent on pH, buffer conditions, and initial peptide aggregation rate (Bush et al., 1994b; Huang et al., 1997; Garai et al., 2006; Tougu et al., 2008). Human Aβ binding of zinc, and both oxidized and reduced copper (Bush et al., 1994c; Atwood et al., 2000; Syme et al., 2004; Syme and Viles, 2006; Danielsson et al., 2007; Himes et al., 2008; Karr and Szalai, 2008; Shearer and Szalai, 2008; Hureau and Faller, 2009) is mediated by nitrogen ligands from histidine at positions 6, 13, and 14 together with an oxygen ligand (Curtain et al., 2001). Interestingly, rat and mouse have different amino acids at the metal ion coordination site, which could explain why these animals resist developing amyloid pathology compared to other mammals (Gaggelli et al., 2008). More details regarding the biophysical and biochemical binding of Aβ and the above mentioned metals have been reviewed (Faller and Hureau, 2009; Rozga et al., 2009).
Neurotoxic effects of Aβ depend on peptide aggregation, metal ion interaction, and generation of reactive oxygen species (ROS) with the subsequent formation of soluble covalently cross-linked oligomers. Both Cu:Aβ and Fe:Aβ complexes have been shown to exhibit cytotoxic effects (Schubert and Chevion, 1995; Liu et al., 2011; You et al., 2012), which can be rescued by chelation or competitive binding (Huang et al., 2004; Wu et al., 2008; Perrone et al., 2010). Interestingly, it has been shown that modifying copper binding histidine 6 or 13 to alanine induces significant cell toxicity in primary cortical cell cultures at levels similar to the wild-type peptide (Smith et al., 2010). However, modifying histidine 14 (a known ligand for copper and the cell plasma membrane), did not induce any measurable toxicity that correlates with the ability of the modified peptide to bind to cell membranes (Smith et al., 2010).
Under normal physiological conditions, non-toxic monomeric forms of Aβ are the predominant species (Haass et al., 1992; Vigo-Pelfrey et al., 1993; Shoji, 2002). However, pathological stimuli are thought to trigger complex conformational changes and assembly of Aβ peptides to form a heterogeneous mixture of oligomers and fibrils. This aggregation of Aβ is a critical event for neurotoxicity to occur. Soluble Aβ oligomers, and not fibrils, are currently considered the proximate neurotoxin in AD pathology (Dahlgren et al., 2002; Kayed et al., 2003; Cleary et al., 2005; Haass and Selkoe, 2007; Lesne et al., 2008; Roychaudhuri et al., 2009; Shankar and Walsh, 2009). However, as both Aβ oligomers and fibrils can interact synergistically with tau and cause mitochondrial function impairment in the P301L tau transgenic mouse model (Eckert et al., 2008), the distinction in all forms of neurotoxicity between Aβ species is not clear. Both copper and iron have been shown to modify Aβ and accelerate its aggregation in vitro (Mantyh et al., 1993; Atwood et al., 2000; Ali et al., 2005). Oxidation of the Aβ side-chain by copper leads to covalent oligomerization (Ciccotosto et al., 2004; Ali et al., 2005). Tyrosine at position 10 of Aβ is particularly susceptible to free radical attack. When complexed to Cu2+ or Fe3+ and in the presence of H2O2, Aβ forms dityrosine cross-linked oligomers, which are suggested to seed accelerated Aβ aggregation (Atwood et al., 1998, 2004; Barnham et al., 2004). Unlike zinc, copper mediates Aβ oligomer formation rather than amyloid fibrils, and thus Aβ:Cu oligomers are not recognized by the β-sheet marker, thioflavin T (Jiao and Yang, 2007; Tougu et al., 2009).
Investigations on metal-mediated modulation of Aβ have been carried out in APP transgenic models supplemented with either dietary copper or zinc. Administration of copper to APP23 mice, overexpressing human APP with the AD-related Swedish mutation, elevated copper levels in the brain compared to wild-type littermate controls, resulting in a lowering of soluble and insoluble Aβ (Bayer et al., 2003). Dietary zinc supplementation also reduced Aβ plaques in brains of Tg2576 (another transgenic mouse model carrying the Swedish-APP mutation) and TgCRND8 (a triple transgenic mouse model carrying APP with Swedish and Indiana mutations). However, AD-like spatial memory impairments are increased in the zinc-fed transgenic mice (Linkous et al., 2009). Conversely, decreased dietary zinc in a APP/PS1 transgenic mouse model of AD elevated plaque volume (Stoltenberg et al., 2007). Elevation of brain copper by crossing TgCRND8 with a transgenic mouse model deficient in the copper transporter, ATP7B, reduces plaque load as well as soluble and insoluble Aβ levels (Phinney et al., 2003). These evidences suggest that an intracellular shift in copper reduces Aβ aggregation.
Intracellular zinc export takes place through the zinc transporters (ZnT) protein family. Currently eight ZnTs are known, of which, ZnT-1 is the only member that exports zinc across the plasma membrane within the brain (Lovell et al., 2005). ZnT-3 transports zinc to glutamatergic vesicles in hippocampal granule, pyramidal, and interneuron cells (Cole et al., 1999; Linkous et al., 2008), ZnT-4 sequesters cytosolic zinc into acidic vesicles (Kelleher and Lonnerdal, 2002) and ZnT-6 sequesters zinc in the trans-Golgi network and vesicular compartments (Huang et al., 2002). The highest concentration of labile zinc is present in synaptic vesicles that are released during synaptic transmission of neocortical glutamatergic fibers. As mentioned, the activity of ZnT-3 is required for the passage and pooling of zinc within these pre-synaptic vesicles, making it available for an interaction with the Aβ that is predominantly located within the synapse. Crossing of ZnT-3 knockout mice with Tg2576 mice, reduces both cerebral plaque load (Lee et al., 2002) and amyloid angiopathy (Friedlich et al., 2004). This supports the theory that high concentrations of zinc in the synaptic cleft play a role in amyloid formation in AD.
Oxidative stress-induced damage of brain tissues is a major hallmark of AD. The redox chemistry involved in the production of toxic ROS from metal enriched Aβ complexes and general metal dyshomeostasis is implicated in this process. Binding of oxidized copper or iron to Aβ results in reduction of the metal valency state and subsequent production of H2O2 (Huang et al., 1999; Opazo et al., 2002; Tabner et al., 2002; Nelson and Alkon, 2005). This can be further exacerbated by the reaction of hydrogen peroxide with reduced metal to produce hydroxyl radicals through Fenton and Haber–Weiss reactions (Fenton, 1894; Haber and Weiss, 1934). Hydroxyl radicals are highly chemically reactive and contribute to generation of lipid peroxidation products, protein carbonyl modifications, and nucleic acid adducts such as 8-hydroxy guanosine, all of which feature strongly in AD neuropathology (Smith et al., 1996, 1997). Of note, evidence suggests that the biological reductants involved in Aβ redox cycling are most likely cholesterol and long chain fatty acids (Opazo et al., 2002; Barnham et al., 2004; Haeffner et al., 2005; Nelson and Alkon, 2005; Puglielli et al., 2005; Smith et al., 2006). This is consistent with experimental evidence demonstrating that toxicity associated with Aβ occurs on the plasma membrane (Ciccotosto et al., 2004). Additionally, the products of lipid oxidation such as oxysterols, 7β-hydroxycholesterol and 4-hydroxy-2-nonenal, which in turn increases Aβ cross-linking (Murray et al., 2005), are elevated in AD tissues and mouse models of the disease (Opazo et al., 2002; Haeffner et al., 2005; Nelson and Alkon, 2005; Puglielli et al., 2005; Smith et al., 2006).
Cholesterol Modulation of Aβ Generation, Aggregation, and Cell Toxicity
The majority of in vivo data provide support for an involvement of cholesterol in Aβ generation (Sparks et al., 1994; Bodovitz and Klein, 1996; Bouillot et al., 1996; Lee et al., 1998; Simons et al., 1998, 2001; Frears et al., 1999; Kojro et al., 2001; Wahrle et al., 2002; Ehehalt et al., 2003; Vetrivel et al., 2004; Kalvodova et al., 2005; Osenkowski et al., 2008). However, the impact of altering plasma cholesterol on brain Aβ generation remains unclear. Animal studies report no correlation (Parkin et al., 1999) or inverse correlation (Jin et al., 2004; Davis, 2008; Marquer et al., 2011) between dietary or peripheral cholesterol and Aβ. Several reasons can account for this disparity between studies, which include genetic background, the transgenes present, age, gender, and/or treatment conditions and environment. Another significant reason may be associated with the inherent selectivity of the BBB. Cholesterol in the brain is synthesized de novo and it is unclear to what extent peripheral or dietary cholesterol influences brain cholesterol levels due to limited BBB penetration. Moreover, most studies that examine the effects of high dietary cholesterol on Aβ levels fail to measure brain cholesterol levels in the same experimental settings. It is therefore uncertain if alteration of brain Aβ levels is due to cholesterol changes in the brain or some other indirect mechanism that is caused by the modulation of peripheral cholesterol. Effects of Aβ generation under in vivo paradigms and of cholesterol modulating genes on APP processing/Aβ generation have been reviewed recently in detail (Maulik et al., 2013). Results from these studies have shown strong evidence that modulating cholesterol synthesis (Crameri et al., 2006), intracellular trafficking (Burns et al., 2003; Bryleva et al., 2010; Kodam et al., 2010; Borbon and Erickson, 2011), uptake (Bales et al., 1997, 1999; Holtzman et al., 2000; Irizarry et al., 2000; Cao et al., 2006; Kim et al., 2009), and removal (Koldamova et al., 2005; Wahrle et al., 2005, 2008) causally influence APP processing and Aβ generation.
Cholesterol-rich lipid rafts may play a role in catalyzing the aggregation of Aβ to its neurotoxic oligomeric state. Aβ isolated from AD patients is associated with lipid rafts in a cholesterol-dependent manner and reducing cholesterol levels results in less aggregated Aβ peptides (Schneider et al., 2006). Cholesterol is likely to modulate Aβ aggregation through modifying raft composition. The ganglioside GM1, which is predominantly found in the central nervous system, can bind Aβ peptides in lipid rafts to form a complex that acts as an endogenous seed to promote amyloid oligomerization, aggregation, and subsequent fibril formation (Choo-Smith et al., 1997; Kakio et al., 2002; Kim et al., 2006; Okada et al., 2008; Matsuzaki et al., 2010). This has been shown to be a primary mediator of oxidative stress on plasma membrane (Zampagni et al., 2010). Some studies examining effects of cholesterol on Aβ toxicity in vitro provide evidence that decreasing cholesterol, sialic acid, and ganglioside synthesis is protective to PC12 cells, while increasing cholesterol levels lead to increased Aβ neurotoxicity (Wang et al., 2001; Lin et al., 2008). Interestingly, it has been observed that sustained ROS production is associated with Aβ toxicity when exogenous cholesterol is increased (Ferrera et al., 2008). Other studies disagree with these results. PC12 cells and cultured neurons with high cholesterol levels in the membrane are resistant to Aβ toxicity, while low cholesterol levels increase their susceptibility (Zhou and Richardson, 1996; Yip et al., 2001; Arispe and Doh, 2002; Sponne et al., 2004). These divergent results suggest a dynamic yet intricate correlation between cholesterol and Aβ peptide, such that cholesterol’s influence on physical properties of lipid rafts can modulate Aβ binding and aggregation to affect cell viability.
Metal Modulation of Aβ Degradation
The over-production of toxic Aβ is only one side of the equation that contributes to senile plaque production and AD pathology, with the other possible side, less frequently studied but equally important, involving a fault in the degradation and clearance regulatory pathways of Aβ (reviewed in Carson and Turner, 2002; Ling et al., 2003). Three proteases in the brain most frequently studied in Aβ degradation, are insulin-degrading enzyme (IDE), neprilysin (NEP), and plasmin. Of these three proteases, IDE and NEP are members of the zinc metallopeptidase family of proteins that have a zinc binding domain with common sequence homology that can be potentially altered with aberrant zinc metabolism (Vekrellis et al., 2000; Fan et al., 2009). Additionally, metal binding ligands of both enzymes are oxidatively modified in the AD brain by various ROS, such as hydroxyl radicals and products of ROS, such as 4-hydroxy-2-nonenal (Wang et al., 2003; Caccamo et al., 2005; Shinall et al., 2005). These data suggest that the generation of ROS, perhaps as a product of metal:Aβ redox cycling, may serve to inactivate proteases involved in Aβ degradation. Conversion of plasminogen to plasmin involves cleavage from either tissue-type (tPA) or urokinase-type plasminogen activator (Ledesma et al., 2003). Inhibition by tPA cleavage of plasminogen is again caused by increased redox cycling and production of ROS in the presence of copper/ascorbate (Lind et al., 1993). Plasmin itself may also be regulated by site-specific oxidation; in particular, modification of the histidine molecule that resides in its active site (Lind et al., 1993). Lastly, Aβ is a substrate for matrix metalloproteinase (MMP), and plasmin has been shown to activate MMP2 degradation of Aβ, a process that is inhibited in the presence of zinc but not copper (Crouch et al., 2009).
Cholesterol Modulation of Aβ Degradation
A number of recent studies have shown that cholesterol may be involved in Aβ clearance by regulating Aβ degrading enzymes. After synthesis, IDE is transported via the secretory pathway to the cell membrane where it either remains or is secreted. Given that a subset of IDE is localized in lipid rafts (Bulloj et al., 2008), it is possible that cholesterol levels or distribution can regulate the transport and release of this protease to influence Aβ degradation. Similar to IDE, the mature form of NEP also associates with lipid rafts (Sato et al., 2012). Contradictorily, targeting NEP chimeric proteins to lipid rafts fails to efficiently degrade Aβ in this fraction (Hama et al., 2004). It is of note that plasmin is also a raft protein (Ledesma et al., 2003). Mice deficient in seladin-1, which is required for cholesterol synthesis, present disorganized rafts and impaired plasmin function (Crameri et al., 2006; Stefani and Liguri, 2009). These evidence supports the notion that cholesterol, possibly through raft maintenance, is required for plasmin degradation of Aβ.
APOE Association with Metal and Cholesterol on its Role of Aβ Clearance
There is strong evidence that ApoE plays a central, if not direct, role in the pathogenesis of AD. The human APOE gene exists as three polymorphic alleles (ε2, ε3, and ε4), and individuals possessing the ε4 allele are at highest risk of developing AD (Bales et al., 2009; Reiman et al., 2009; Castellano et al., 2011). ApoE is well known for its involvement in the transportation of cholesterol. Together with a multitude of other apolipoproteins, lipoprotein receptors, and lipid transporters, ApoE controls cholesterol homeostasis in the brain (brain cholesterol homeostasis reviewed in Hung et al., 2013). Studies in human and transgenic mice demonstrate an isoform dependent (ε4 > ε3 > ε2) accumulation of Aβ levels and amyloid plaque load.
Currently, there is no clear evidence that ApoE affects APP processing and Aβ production in vitro and in vivo (Biere et al., 1995; Cedazo-Minguez et al., 2001; Irizarry et al., 2004). However, ApoE appears to play an important role in Aβ clearance through several possible mechanisms. In vitro studies with neuronal cells have shown that lipidated ApoE binds to soluble Aβ in an isoform-dependent manner (ε2 > ε3 > ε4) and is internalized into various brain cells for degradation by receptor-mediated endocytosis (Beffert et al., 1998, 1999; Yang et al., 1999; Cole and Ard, 2000; Yamauchi et al., 2000, 2002). ApoE may also facilitate removal of Aβ from the brain through the BBB (Cirrito et al., 2005; Zlokovic, 2008). ApoE may be able to facilitate the cellular degradation of Aβ in vitro, however, the mechanism and whether it is isoform-specific still requires clarification (Crameri et al., 2006; Bryleva et al., 2010).
There are very limited studies investigating metal interaction with ApoE and its relationship with APP or Aβ. ApoE protein binds copper, iron, and zinc, suggesting that ApoE has the ability to sequester metals. This may underlie its isoform-dependent antioxidant activity (ε2 > ε3 > ε4; Miyata and Smith, 1996). Interestingly, ApoE4 contains a cysteine to arginine substitution at positions 112 and 158. Since cysteine is believed to be involved in transition metal binding, reduced affinity of ApoE4 to metal may therefore relate to diminished antioxidant effects of the ApoE4 allele (Moir et al., 1999).
ATP binding cassette transporter A1 (ABCA1) is a cell surface membrane protein that promotes efflux of cellular cholesterol to acceptor molecules, including ApoE and ApoA1. The ApoE4 isoform has been found to reduce ABCA1-mediated cholesterol efflux in astrocytes and neurons in vitro (Michikawa et al., 2000; Gong et al., 2007). Studies involving ABCA1-deficient mice show poor lipidation with decreased levels of ApoE (70–80% reduction) and a concurrent increase in amyloid plaque burden (Wahrle et al., 2004, 2005; Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005). Conversely, ABCA1 overexpression in mice result in increased lipidation and ApoE levels, and decreased amyloid plaque formation (Wahrle et al., 2008). Interestingly, both ApoE and ABCA1 can be modulated by transcription factor liver-X-receptors (LXRs), which may be a key regulator in brain lipid homeostasis. Indeed, deficiencies in LXRα and/or β augment AD pathology (Zelcer et al., 2007), whereas treating AD mice with LXR agonists, including Bexarotene, result in reduced amyloid plaque burden and improved cognitive function (Eckert et al., 2007; Riddell et al., 2007; Vanmierlo et al., 2011; Cramer et al., 2012).
Two Sides of the Same Coin: Possible Crosstalk between Metals and Cholesterol in APP/Aβ Metabolism
Independently, there are large bodies of research detailing the influence of metals or cholesterol on the development, progression, and pathogenesis of AD. However, there are limited studies on the relationship between metals and cholesterol in AD pathology. The review thus far gives an overview of the impact of either metals or cholesterol on the amyloidogenic and non-amyloidogenic processing pathways of APP. Interestingly, there are many overlaps between these two factors impacting various processes in both of these pathways. The review will now examine interactions between metals and cholesterol with APP and Aβ.
Epidemiological studies have shown that dietary intake of trans- and saturated fats lead to an unfavorable cholesterol profile in AD patients and may associate with cognitive decline (Morris et al., 2003, 2004). A follow-up study indicates that higher copper intake is associated with an accelerated rate of cognitive decline and that the copper-dependent acceleration is lost in individuals who did not consume a high fat diet (Morris et al., 2006). In animal studies, cholesterol-fed rabbits have exacerbated neurodegeneration following consumption of trace amounts of copper (Sparks and Schreurs, 2003). Lowering cholesterol levels in patients by atorvastatin (a cholesterol lowering drug which inhibits HMGCR) saw an increase in circulating ceruloplasmin levels (Sparks et al., 2005), a ferroxidase involved in iron homeostasis and due to its role as a plasma copper transporter, a surrogate marker of plasma copper status. Interestingly, like copper, increased dietary cholesterol leads to dysregulation of iron regulatory proteins in rabbits and also iron accumulation in Aβ plaques (Ghribi et al., 2006). An epidemiological study in a large cohort of adults found that abnormally high dietary cholesterol and iron intakes increase the risk of AD (Mainous et al., 2005).
Based on evidence from the studies reviewed in Sections “Metals and APP” and “Cholesterol and APP,” it may be inferred that both metals and cholesterol are able to modulate APP metabolism/expression through a process that remains to be elucidated. Conversely, APP itself can regulate metal and cholesterol homeostasis. Therefore, APP may be envisioned as a key regulator linking both metal and cholesterol homeostasis, whereby unregulated metal or cholesterol leads to a downstream effect on APP that may ultimately cause an erroneous outcome in collateral systems.
One way in which copper, iron, and zinc can impact on the pathology of AD stems from their relative ease in switching oxidation states. This property makes it particularly useful for enzymatic reactions requiring electron transfer (Waldron et al., 2009). Metals can affect APP processing (Figure 1B) and Aβ degradation (Figure 2) by altering catalytic properties of secretases, which are metalloproteins (reviewed in Sections “Metal Modulation of APP Processing Enzymes” and “Metal Modulation of Aβ Degradation”). Cholesterol, on the other hand influences APP processing (Figure 1B) and Aβ degradation (Figure 2) through lipid raft association of substrates (APP and Aβ) and enzymes (APP processing secretases and Aβ degrading proteases; reviewed in sections “Cholesterol Modulation of APP Processing” and “Cholesterol Modulation of Aβ Degradation”). Cholesterol influences both the quantity and quality of the lipid raft domains. For example, cholesterol can modulate the order of raft components to provide the right environment for protein binding or function. In other words, metals modulate APP processing and Aβ degradation through the mechanistic action of the enzyme, whereas cholesterol does so through manipulation of the environment and presentation of the substrate and/or enzyme on the plasma membrane. Although this relationship between metals and cholesterol may not be mutually exclusive, a fault in either system may still lead to similar pathological outcomes in AD (Figure 2).
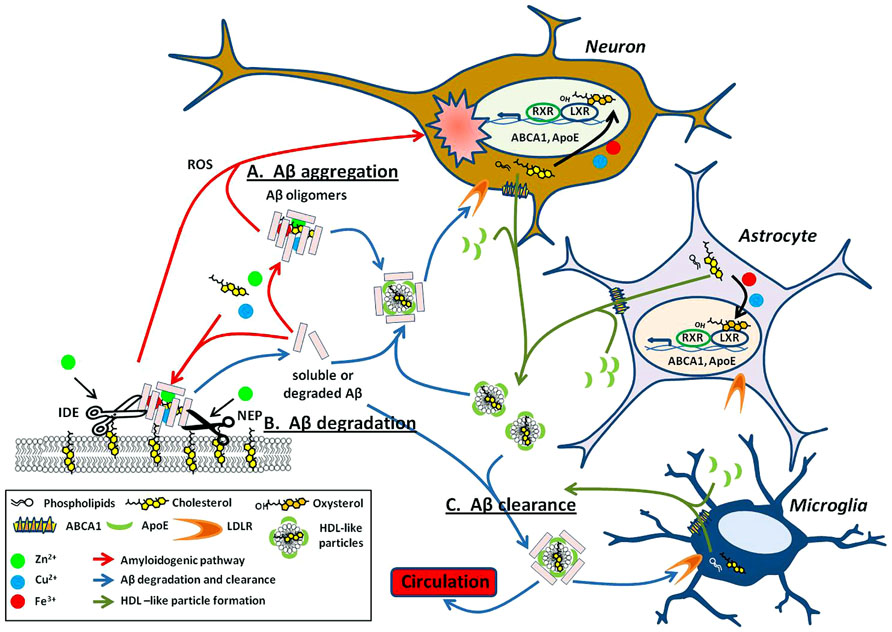
FIGURE 2. Metals and cholesterol implicated in Aβ processing and neurotoxicity. (A) Upon cleavage from APP, both metal and cholesterol bind to Aβ monomers promoting oligomerization of the peptide into multiple types of Aβ aggregates. These aggregates are either present within the extracellular space or bound to the surface of the plasma membrane (red arrows). Select Aβ aggregates are neurotoxic through multiple mechanisms such as their ability to generate reactive oxygen species (ROS) and may have implications in AD associated neuropathology. The ability of copper, iron, and cholesterol to promote redox cycling are acutely involved in the cytotoxicity caused by aggregated Aβ. (B) As with APP processing enzymes (Figure 1B), proteases that degrade Aβ, such as neprilysin (NEP) and insulin degrading enzyme (IDE) are dependent on metals for their catalytic activity. These proteases are also lipid raft associated and modulated by cholesterol levels in these domains. (C) Lipidated ApoE, produced mainly by astrocytes and microglia, binds soluble Aβ and facilitates its degradation through receptor-mediated endocytosis within neurons and microglial or clearance from the brain through the blood–brain barrier (BBB; blue arrows). Lipidated ApoE regulation is modulated by nuclear transcription factors LXRs. LXR heterodimerizes with RXR to transcriptionally regulate ABCA1 and ApoE. ABCA1 exports cellular cholesterol and phospholipids that in turn lipidate ApoE to form HDL-like particles (green arrows). These HDL-like particles are required for the clearance of Aβ. Intriguingly, the LXR agonist, oxysterols is elevated in the AD brain and may result from cholesterol oxidation by metals and 24S-hydroxylase. Evidence implies that the involvement of metal and cholesterol in the Aβ processing pathway is not just deleterious (as in A) but may also have importance in degradation and clearance of this potentially harmful peptide (B,C).
More recently, copper has been observed to directly influence the lipid raft protein, flotillin-2 (Hung et al., 2009). Flotillin-2 interacts with APP at the cell surface (Schneider et al., 2008). The endocytosis of APP to BACE1-rich endosomes, required for β-cleavage of APP, is sensitive to flotillin-2 depletion (Ehehalt et al., 2003; Schneider et al., 2008). Analogous to cholesterol depletion, elevated copper reduces flotillin-2 association with lipid rafts, thereby reducing endocytosis of APP and attenuating Aβ production (Hung et al., 2009).
Interestingly, both metals and cholesterol are able to catalyze the oligomeric aggregation of Aβ required for its cytotoxic effect (see “Metal Modulation of Aβ Generation, Aggregation, and Cell Toxicity” and “Cholesterol Modulation of Aβ Generation, Aggregation, and Cell Toxicity”; Figure 2). The mechanism of aggregated Aβ toxicity is still a matter of debate. However, elevation of ROS in both metal:Aβ or GM1:Aβ complexes suggests an involvement of metal as a mechanistic partner to redox cycle and generating harmful ROS products from both metal- and cholesterol-based aggregation of Aβ in vitro.
Oxysterols also play an important role in the regulation of cholesterol in the brain and the body. In the brain, oxysterols are produced by conversion of cholesterol to the oxidized species, 24OHC, by the enzyme 24S-hydroxylase. 24OHC represents one of the main forms of cholesterol that can be trafficked out of the brain to the circulatory system by its permeability across the BBB (Bjorkhem et al., 1998; Lund et al., 1999; Ehehalt et al., 2003; Lutjohann and von Bergmann, 2003; Schneider et al., 2008). Interestingly, APP has been shown recently to regulate 24S-hydroxylase levels (Pierrot et al., 2013). Oxysterols are agonists of LXRs, the latter of which form heterodimer complexes with retinoid x receptor (RXR) to transcriptionally regulate the production of a number of genes involved in the cholesterol regulatory pathway including ApoE, ABCA1, ABCG1, and SREBP1 (Bjorkhem, 2013). Through this pathway, oxysterols are able to regulate cholesterol efflux from cells via LXRs (Figure 2). In the progression of AD, levels of oxysterols are elevated, possibly due to effects of 24S-hydroxylase and non-enzymatic oxidation of cholesterol caused by elevated metal levels (Iuliano, 2011). This may be a way in which the brain is utilizing a feedback mechanism to clear excess cholesterol and Aβ peptides. Therefore, it is not surprising that elevated LXR-induced expression of ApoE4 (with defective Aβ and cholesterol clearance) compared to ApoE2, results in continued accumulation of neuritic plaques. The pathology of the disease continues to progress in a positive feedback loop of increased cholesterol, ROS, and Aβ generation (Figure 2).
Conclusion
Metal and cholesterol are intrinsically linked to the pathogenesis of AD. Despite large bodies of research examining the abnormalities of metals and cholesterol in AD, the reciprocal influence of these two factors in the cause and progression of the disease remains to be elucidated. This review presents an overview of how metals and cholesterol independently impact upon the amyloidogenic and non-amyloidogenic processing of APP. It highlights the close and complex relationship between metals and cholesterol in the maintenance of normal brain physiology and the progression of AD pathology, with respect to interactions with AD-related proteins APP and Aβ. In the scheme of APP processing and Aβ metabolism, a disturbance to one homeostatic system may likely lead to a direct or indirect dysregulation of the other, although some of its molecular actions are mutually exclusive, the eventual deleterious outcome is the same. Continued research into metal or cholesterol influences on AD pathology must take careful consideration of the other factor, given that they are intrinsically linked. For example, the study of an influential effect metals has on ApoE should always take into account its lipidation status, which affects its conformation and activity. Conversely, the study of Aβ aggregation in lipid rafts should consider the role metals play in the generation of ROS products and subsequent cell toxicity. Further research is needed to investigate molecular mechanisms that link metals and cholesterol with various players involved in AD pathogenesis. This knowledge is critical for future design and implementation of effective therapeutic strategies to treat AD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funding from the National Health and Medical Research Council of Australia (to Ashley I. Bush and James A. Duce), Australian Research Council (to Ashley I. Bush), Cooperative Research Centre for Mental Health (to Ashley I. Bush), Alzheimer’s Research UK (to James A. Duce), the Addi and Cassi Fund (to Ashley I. Bush and Ya Hui Hung), Perpetual Trustees [Harry Secomb Foundation and Payne L and Heather Medical Research Charitable Foundation (to Ashley I. Bush)], The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant.
References
Acevedo, K. M., Hung, Y. H., Dalziel, A. H., Li, Q. X., Laughton, K., Wikhe, K., et al. (2011). Copper promotes the trafficking of the amyloid precursor protein. J. Biol. Chem. 286, 8252–8262. doi: 10.1074/jbc.M110.128512
Ali, F. E., Separovic, F., Barrow, C. J., Cherny, R. A., Fraser, F., Bush, A. I., et al. (2005). Methionine regulates copper/hydrogen peroxide oxidation products of Abeta. J. Pept. Sci. 11, 353–360. doi: 10.1002/psc.626
Angeletti, B., Waldron, K. J., Freeman, K. B., Bawagan, H., Hussain, I., Miller, C. C., et al. (2005). BACE1 cytoplasmic domain interacts with the copper chaperone for superoxide dismutase-1 and binds copper. J. Biol. Chem. 280, 17930–17937. doi: 10.1074/jbc.M412034200
Area-Gomez, E., Del Carmen Lara Castillo, M., Tambini, M. D., Guardia-Laguarta, C., De Groof, A. J., Madra, M., et al. (2012). Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106–4123. doi: 10.1038/emboj.2012.202
Arispe, N., and Doh, M. (2002). Plasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease AbetaP (1-40) and (1-42) peptides. FASEB J. 16, 1526–1536. doi: 10.1096/fj.02-0829com
Armendariz, A. D., Gonzalez, M., Loguinov, A. V., and Vulpe, C. D. (2004). Gene expression profiling in chronic copper overload reveals upregulation of Prnp and App. Physiol. Genomics 20, 45–54. doi: 10.1152/physiolgenomics.00196.2003
Atwood, C. S., Moir, R. D., Huang, X., Scarpa, R. C., Bacarra, N. M., Romano, D. M., et al. (1998). Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 273, 12817–12826. doi: 10.1074/jbc.273.21.12817
Atwood, C. S., Perry, G., Zeng, H., Kato, Y., Jones, W. D., Ling, K. Q., et al. (2004). Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-beta. Biochemistry 43, 560–568. doi: 10.1021/bi0358824
Atwood, C. S., Scarpa, R. C., Huang, X., Moir, R. D., Jones, W. D., Fairlie, D. P., et al. (2000). Characterization of copper interactions with Alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42. J. Neurochem. 75, 1219–1233. doi: 10.1046/j.1471-4159.2000.0751219.x
Bales, K. R., Liu, F., Wu, S., Lin, S., Koger, D., Delong, C., et al. (2009). Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009
Bales, K. R., Verina, T., Cummins, D. J., Du, Y., Dodel, R. C., Saura, J., et al. (1999). Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 96, 15233–15238. doi: 10.1073/pnas.96.26.15233
Bales, K. R., Verina, T., Dodel, R. C., Du, Y., Altstiel, L., Bender, M., et al. (1997). Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat. Genet. 17, 263–264. doi: 10.1038/ng1197-263
Baltes, C., Princz-Kranz, F., Rudin, M., and Mueggler, T. (2011). Detecting amyloid-beta plaques in Alzheimer’s disease. Methods Mol. Biol. 711, 511–533. doi: 10.1007/978-1-61737-992-5_26
Barnham, K. J., Haeffner, F., Ciccotosto, G. D., Curtain, C. C., Tew, D., Mavros, C., et al. (2004). Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease beta-amyloid. FASEB J. 18, 1427–1429. doi: 10.1096/fj.04-1890fje
Barnham, K. J., Mckinstry, W. J., Multhaup, G., Galatis, D., Morton, C. J., Curtain, C. C., et al. (2003). Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J. Biol. Chem. 278, 17401–17407. doi: 10.1074/jbc.M300629200
Barrett, P. J., Song, Y., Van Horn, W. D., Hustedt, E. J., Schafer, J. M., Hadziselimovic, A., et al. (2012). The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171. doi: 10.1126/science.1219988
Bayer, T. A., Schafer, S., Simons, A., Kemmling, A., Kamer, T., Tepest, R., et al. (2003). Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 100, 14187–14192. doi: 10.1073/pnas.2332818100
Beel, A. J., Sakakura, M., Barrett, P. J., and Sanders, C. R. (2010). Direct binding of cholesterol to the amyloid precursor protein: an important interaction in lipid-Alzheimer’s disease relationships? Biochim. Biophys. Acta 1801, 975–982. doi: 10.1016/j.bbalip.2010.03.008
Beffert, U., Aumont, N., Dea, D., Lussier-Cacan, S., Davignon, J., and Poirier, J. (1998). Beta-amyloid peptides increase the binding and internalization of apolipoprotein E to hippocampal neurons. J. Neurochem. 70, 1458–1466. doi: 10.1046/j.1471-4159.1998.70041458.x
Beffert, U., Aumont, N., Dea, D., Lussier-Cacan, S., Davignon, J., and Poirier, J. (1999). Apolipoprotein E isoform-specific reduction of extracellular amyloid in neuronal cultures. Brain Res. Mol. Brain Res. 68, 181–185. doi: 10.1016/S0169-328X(99)00073-X
Bellingham, S. A., Ciccotosto, G. D., Needham, B. E., Fodero, L. R., White, A. R., Masters, C. L., et al. (2004a). Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J. Neurochem. 91, 423–428. doi: 10.1111/j.1471-4159.2004.02731.x
Bellingham, S. A., Lahiri, D. K., Maloney, B., La Fontaine, S., Multhaup, G., and Camakaris, J. (2004b). Copper depletion down-regulates expression of the Alzheimer’s disease amyloid-beta precursor protein gene. J. Biol. Chem. 279, 20378–20386. doi: 10.1074/jbc.M400805200
Bertram, L., Mcqueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 39, 17–23. doi: 10.1038/ng1934
Bertram, L., and Tanzi, R. E. (2005). The genetic epidemiology of neurodegenerative disease. J. Clin. Invest. 115, 1449–1457. doi: 10.1172/JCI24761
Biere, A. L., Ostaszewski, B., Zhao, H., Gillespie, S., Younkin, S. G., and Selkoe, D. J. (1995). Co-expression of beta-amyloid precursor protein (betaAPP) and apolipoprotein E in cell culture: analysis of betaAPP processing. Neurobiol. Dis. 2, 177–187. doi: 10.1006/nbdi.1995.0019.
Bjorkhem, I. (2013). Five decades with oxysterols. Biochimie 95, 448–454. doi: 10.1016/j.biochi.2012.02.029
Bjorkhem, I., Lutjohann, D., Diczfalusy, U., Stahle, L., Ahlborg, G., and Wahren, J. (1998). Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 39, 1594–1600.
Blacker, M., Noe, M. C., Carty, T. J., Goodyer, C. G., and Leblanc, A. C. (2002). Effect of tumor necrosis factor-alpha converting enzyme (TACE) and metalloprotease inhibitor on amyloid precursor protein metabolism in human neurons. J. Neurochem. 83, 1349–1357. doi: 10.1046/j.1471-4159.2002.01228.x
Bodovitz, S., and Klein, W. L. (1996). Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J. Biol. Chem. 271, 4436–4440. doi: 10.1074/jbc.271.8.4436
Borbon, I. A., and Erickson, R. P. (2011). Interactions of Npc1 and amyloid accumulation/deposition in the APP/PS1 mouse model of Alzheimer’s. J. Appl. Genet. 52, 213–218. doi: 10.1007/s13353-010-0021-1
Bouillot, C., Prochiantz, A., Rougon, G., and Allinquant, B. (1996). Axonal amyloid precursor protein expressed by neurons in vitro is present in a membrane fraction with caveolae-like properties. J. Biol. Chem. 271, 7640–7644. doi: 10.1074/jbc.271.13.7640
Brown, M. S., and Goldstein, J. L. (1999). A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. U.S.A. 96, 11041–11048. doi: 10.1073/pnas.96.20.11041
Bryleva, E. Y., Rogers, M. A., Chang, C. C., Buen, F., Harris, B. T., Rousselet, E., et al. (2010). ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. U.S.A. 107, 3081–3086. doi: 10.1073/pnas.0913828107
Buckley, C. A., Rouhani, F. N., Kaler, M., Adamik, B., Hawari, F. I., and Levine, S. J. (2005). Amino-terminal TACE prodomain attenuates TNFR2 cleavage independently of the cysteine switch. Am. J. Physiol. Lung Cell. Mol. Physiol. 288, L1132–L1138. doi: 10.1152/ajplung.00429.2004
Bulloj, A., Leal, M. C., Surace, E. I., Zhang, X., Xu, H., Ledesma, M. D., et al. (2008). Detergent resistant membrane-associated IDE in brain tissue and cultured cells: relevance to Abeta and insulin degradation. Mol. Neurodegener. 3, 22. doi: 10.1186/1750-1326-3-22
Burns, M., Gaynor, K., Olm, V., Mercken, M., Lafrancois, J., Wang, L., et al. (2003). Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J. Neurosci. 23, 5645–5649.
Bush, A. I., Multhaup, G., Moir, R. D., Williamson, T. G., Small, D. H., Rumble, B., et al. (1993). A novel zinc(II) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer’s disease. J. Biol. Chem. 268, 16109–16112.
Bush, A. I., Pettingell, W. H. Jr., De Paradis, M., Tanzi, R. E., and Wasco, W. (1994a). The amyloid beta-protein precursor and its mammalian homologues. Evidence for a zinc-modulated heparin-binding superfamily. J. Biol. Chem. 269, 26618–26621.
Bush, A. I., Pettingell, W. H. Jr., Paradis, M. D., and Tanzi, R. E. (1994b). Modulation of A beta adhesiveness and secretase site cleavage by zinc. J. Biol. Chem. 269, 12152–12158.
Bush, A. I., Pettingell, W. H., Multhaup, G., d Paradis, M., Vonsattel, J. P., Gusella, J. F., et al. (1994c). Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 265, 1464–1467. doi: 10.1126/science.8073293
Buxbaum, J. D., Liu, K. N., Luo, Y., Slack, J. L., Stocking, K. L., Peschon, J. J., et al. (1998). Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273, 27765–27767. doi: 10.1074/jbc.273.43.27765
Caccamo, A., Oddo, S., Sugarman, M. C., Akbari, Y., and Laferla, F. M. (2005). Age- and region-dependent alterations in Abeta-degrading enzymes: implications for Abeta-induced disorders. Neurobiol. Aging 26, 645–654. doi: 10.1016/j.neurobiolaging.2004.06.013
Cao, D., Fukuchi, K., Wan, H., Kim, H., and Li, L. (2006). Lack of LDL receptor aggravates learning deficits and amyloid deposits in Alzheimer transgenic mice. Neurobiol. Aging 27, 1632–1643. doi: 10.1016/j.neurobiolaging.2005.09.011
Carson, J. A., and Turner, A. J. (2002). Beta-amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases? J. Neurochem. 81, 1–8. doi: 10.1046/j.1471-4159.2002.00855.x
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., Demattos, R. B., Patterson, B. W., et al. (2011). Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 3, 89ra57. doi: 10.1126/scitranslmed.3002156
Cedazo-Minguez, A., Wiehager, B., Winblad, B., Huttinger, M., and Cowburn, R. F. (2001). Effects of apolipoprotein E (apoE) isoforms, beta-amyloid (Abeta) and apoE/Abeta complexes on protein kinase C-alpha (PKC-alpha) translocation and amyloid precursor protein (APP) processing in human SH-SY5Y neuroblastoma cells and fibroblasts. Neurochem. Int. 38, 615–625. doi: 10.1016/S0197-0186(00)00128-5
Chen, J. H., Lin, K. P., and Chen, Y. C. (2009). Risk factors for dementia. J. Formos. Med. Assoc. 108, 754–764. doi: 10.1016/S0929-6646(09)60402-2
Cho, H. H., Cahill, C. M., Vanderburg, C. R., Scherzer, C. R., Wang, B., Huang, X., et al. (2010). Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 285, 31217–31232. doi: 10.1074/jbc.M110.149161
Choo-Smith, L. P., Garzon-Rodriguez, W., Glabe, C. G., and Surewicz, W. K. (1997). Acceleration of amyloid fibril formation by specific binding of Abeta-(1-40) peptide to ganglioside-containing membrane vesicles. J. Biol. Chem. 272, 22987–22990. doi: 10.1074/jbc.272.37.22987
Ciccotosto, G. D., Tew, D., Curtain, C. C., Smith, D., Carrington, D., Masters, C. L., et al. (2004). Enhanced toxicity and cellular binding of a modified amyloid beta peptide with a methionine to valine substitution. J. Biol. Chem. 279, 42528–42534. doi: 10.1074/jbc.M406465200
Cirrito, J. R., Deane, R., Fagan, A. M., Spinner, M. L., Parsadanian, M., Finn, M. B., et al. (2005). P-glycoprotein deficiency at the blood–brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Invest. 115, 3285–3290. doi: 10.1172/JCI25247
Ciuculescu, E. D., Mekmouche, Y., and Faller, P. (2005). Metal-binding properties of the peptide APP170-188: a model of the ZnII-binding site of amyloid precursor protein (APP). Chemistry 11, 903–909. doi: 10.1002/chem.200400786
Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., et al. (2005). Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84. doi: 10.1038/nn1372
Cole, G. M., and Ard, M. D. (2000). Influence of lipoproteins on microglial degradation of Alzheimer’s amyloid beta-protein. Microsc. Res. Tech. 50, 316–324. doi: 10.1002/1097-0029(20000815)50:4<316::AID-JEMT11>3.0.CO;2-E
Cole, T. B., Wenzel, H. J., Kafer, K. E., Schwartzkroin, P. A., and Palmiter, R. D. (1999). Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. U.S.A. 96, 1716–1721. doi: 10.1073/pnas.96.4.1716
Collingwood, J. F., Mikhaylova, A., Davidson, M., Batich, C., Streit, W. J., Terry, J., et al. (2005). In situ characterization and mapping of iron compounds in Alzheimer’s disease tissue. J. Alzheimers Dis. 7, 267–272.
Connor, J. R., Snyder, B. S., Beard, J. L., Fine, R. E., and Mufson, E. J. (1992). Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J. Neurosci. Res. 31, 327–335. doi: 10.1002/jnr.490310214
Coon, K. D., Myers, A. J., Craig, D. W., Webster, J. A., Pearson, J. V., Lince, D. H., et al. (2007). A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J. Clin. Psychiatry 68, 613–618. doi: 10.4088/JCP.v68n0419
Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. Y., Karlo, J. C., Zinn, A. E., et al. (2012). ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506. doi: 10.1126/science.1217697
Crameri, A., Biondi, E., Kuehnle, K., Lutjohann, D., Thelen, K. M., Perga, S., et al. (2006). The role of seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and Abeta generation in vivo. EMBO J. 25, 432–443. doi: 10.1038/sj.emboj.7600938
Cross, J. B., Duca, J. S., Kaminski, J. J., and Madison, V. S. (2002). The active site of a zinc-dependent metalloproteinase influences the computed pK(a) of ligands coordinated to the catalytic zinc ion. J. Am. Chem. Soc. 124, 11004–11007. doi: 10.1021/ja0201810
Crouch, P. J., Tew, D. J., Du, T., Nguyen, D. N., Caragounis, A., Filiz, G., et al. (2009). Restored degradation of the Alzheimer’s amyloid-beta peptide by targeting amyloid formation. J. Neurochem. 108, 1198–1207. doi: 10.1111/j.1471-4159.2009.05870.x
Curtain, C. C., Ali, F., Volitakis, I., Cherny, R. A., Norton, R. S., Beyreuther, K., et al. (2001). Alzheimer’s disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 276, 20466–20473. doi: 10.1074/jbc.M100175200
Dahlgren, K. N., Manelli, A. M., Stine, W. B. Jr., Baker, L. K., Krafft, G. A., et al. (2002). Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053. doi: 10.1074/jbc.M201750200
Dahms, S. O., Konnig, I., Roeser, D., Guhrs, K. H., Mayer, M. C., Kaden, D., et al. (2012). Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain. J. Mol. Biol. 416, 438–452. doi: 10.1016/j.jmb.2011.12.057
Danielsson, J., Pierattelli, R., Banci, L., and Graslund, A. (2007). High-resolution NMR studies of the zinc-binding site of the Alzheimer’s amyloid beta-peptide. FEBS J. 274, 46–59. doi: 10.1111/j.1742-4658.2006.05563.x
Davis, C. D., Milne, D. B., and Nielsen, F. H. (2000). Changes in dietary zinc and copper affect zinc-status indicators of postmenopausal women, notably, extracellular superoxide dismutase and amyloid precursor proteins. Am. J. Clin. Nutr. 71, 781–788.
Davis, W. Jr. (2008). The cholesterol transport inhibitor U18666a regulates amyloid precursor protein metabolism and trafficking in N2aAPP “Swedish” cells. Curr. Alzheimer Res. 5, 448–456. doi: 10.2174/156720508785908900
Delacourte, A., and Defossez, A. (1986). Alzheimer’s disease: tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J. Neurol. Sci. 76, 173–186. doi: 10.1016/0022-510X(86)90167-X
Donovan, A., Lima, C. A., Pinkus, J. L., Pinkus, G. S., Zon, L. I., Robine, S., et al. (2005). The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 1, 191–200. doi: 10.1016/j.cmet.2005.01.003
Duce, J. A., and Bush, A. I. (2010). Biological metals and Alzheimer’s disease: implications for therapeutics and diagnostics. Prog. Neurobiol. 92, 1–18. doi: 10.1016/j.pneurobio.2010.04.003
Duce, J. A., Tsatsanis, A., Cater, M. A., James, S. A., Robb, E., Wikhe, K., et al. (2010). Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142, 857–867. doi: 10.1016/j.cell.2010.08.014
Eckert, A., Hauptmann, S., Scherping, I., Meinhardt, J., Rhein, V., Drose, S., et al. (2008). Oligomeric and fibrillar species of beta-amyloid (A beta 42) both impair mitochondrial function in P301L tau transgenic mice. J. Mol. Med. (Berl.) 86, 1255–1267. doi: 10.1007/s00109-008-0391-6
Eckert, G. P., Vardanian, L., Rebeck, G. W., and Burns, M. P. (2007). Regulation of central nervous system cholesterol homeostasis by the liver X receptor agonist TO-901317. Neurosci. Lett. 423, 47–52. doi: 10.1016/j.neulet.2007.05.063
Eckert, G. P., Wood, W. G., and Muller, W. E. (2010). Lipid membranes and beta-amyloid: a harmful connection. Curr. Protein Pept. Sci 11, 319–325. doi: 10.2174/138920310791330668
Edbauer, D., Winkler, E., Regula, J. T., Pesold, B., Steiner, H., and Haass, C. (2003). Reconstitution of gamma-secretase activity. Nat. Cell Biol. 5, 486–488. doi: 10.1038/ncb960
Ehehalt, R., Keller, P., Haass, C., Thiele, C., and Simons, K. (2003). Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160, 113–123. doi: 10.1083/jcb.200207113
Faller, P., and Hureau, C. (2009). Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-beta peptide. Dalton Trans. 1080–1094. doi: 10.1039/b813398k
Fan, J., Donkin, J., and Wellington, C. (2009). Greasing the wheels of Abeta clearance in Alzheimer’s disease: the role of lipids and apolipoprotein E. Biofactors 35, 239–248. doi: 10.1002/biof.37
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356. doi: 10.1002/biof.37
Fenton, H. J. H. (1894). Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 65, 899–911. doi: 10.1039/ct8946500899
Ferrera, P., Mercado-Gomez, O., Silva-Aguilar, M., Valverde, M., and Arias, C. (2008). Cholesterol potentiates beta-amyloid-induced toxicity in human neuroblastoma cells: involvement of oxidative stress. Neurochem. Res. 33, 1509–1517. doi: 10.1007/s11064-008-9623-y
Frears, E. R., Stephens, D. J., Walters, C. E., Davies, H., and Austen, B. M. (1999). The role of cholesterol in the biosynthesis of beta-amyloid. Neuroreport 10, 1699–1705. doi: 10.1007/s11064-008-9623-y
Friedlich, A. L., Lee, J. Y., Van Groen, T., Cherny, R. A., Volitakis, I., Cole, T. B., et al. (2004). Neuronal zinc exchange with the blood vessel wall promotes cerebral amyloid angiopathy in an animal model of Alzheimer’s disease. J. Neurosci. 24, 3453–3459. doi: 10.1523/JNEUROSCI.0297-04.2004
Gaggelli, E., Grzonka, Z., Kozlowski, H., Migliorini, C., Molteni, E., Valensin, D., et al. (2008). Structural features of the Cu(II) complex with the rat Abeta(1-28) fragment. Chem. Commun. (Camb.) 341–343. doi: 10.1523/JNEUROSCI.0297-04.2004
Ganz, T. (2005). Cellular iron: ferroportin is the only way out. Cell Metab. 1, 155–157. doi: 10.1016/j.cmet.2005.02.005
Garai, K., Sengupta, P., Sahoo, B., and Maiti, S. (2006). Selective destabilization of soluble amyloid beta oligomers by divalent metal ions. Biochem. Biophys. Res. Commun. 345, 210–215. doi: 10.1016/j.bbrc.2006.04.056
Ghribi, O., Golovko, M. Y., Larsen, B., Schrag, M., and Murphy, E. J. (2006). Deposition of iron and beta-amyloid plaques is associated with cortical cellular damage in rabbits fed with long-term cholesterol-enriched diets. J. Neurochem. 99, 438–449. doi: 10.1111/j.1471-4159.2006.04079.x
Glenner, G. G., and Wong, C. W. (1984a). Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135. doi: 10.1016/0006-291X(84)91209-9
Glenner, G. G., and Wong, C. W. (1984b). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/S0006-291X(84)80190-4
Gong, J. S., Morita, S. Y., Kobayashi, M., Handa, T., Fujita, S. C., Yanagisawa, K., et al. (2007). Novel action of apolipoprotein E (ApoE): ApoE isoform specifically inhibits lipid-particle-mediated cholesterol release from neurons. Mol. Neurodegener. 2, 9. doi: 10.1186/1750-1326-2-9
Goodman, L. (1953). Alzheimer’s disease; a clinico-pathologic analysis of twenty-three cases with a theory on pathogenesis. J. Nerv. Ment. Dis. 118, 97–130. doi: 10.1097/00005053-195308000-00001
Greenough, M. A., Volitakis, I., Li, Q. X., Laughton, K., Evin, G., Ho, M., et al. (2011). Presenilins promote the cellular uptake of copper and zinc and maintain copper chaperone of SOD1-dependent copper/zinc superoxide dismutase activity. J. Biol. Chem. 286, 9776–9786. doi: 10.1074/jbc.M110.163964
Haass, C., Schlossmacher, M. G., Hung, A. Y., Vigo-Pelfrey, C., Mellon, A., Ostaszewski, B. L., et al. (1992). Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 359, 322–325. doi: 10.1038/359322a0
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Haber, F., and Weiss, J. (1934). The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. Lond. A. Math. Phys. Sci. 147, 332–351. doi: 10.1098/rspa.1934.0221
Haeffner, F., Smith, D. G., Barnham, K. J., and Bush, A. I. (2005). Model studies of cholesterol and ascorbate oxidation by copper complexes: relevance to Alzheimer’s disease beta-amyloid metallochemistry. J. Inorg. Biochem. 99, 2403–2422. doi: 10.1016/j.jinorgbio.2005.09.011
Hama, E., Shirotani, K., Iwata, N., and Saido, T. C. (2004). Effects of neprilysin chimeric proteins targeted to subcellular compartments on amyloid beta peptide clearance in primary neurons. J. Biol. Chem. 279, 30259–30264. doi: 10.1074/jbc.M401891200
Hayashi, H., Igbavboa, U., Hamanaka, H., Kobayashi, M., Fujita, S. C., Wood, W. G., et al. (2002). Cholesterol is increased in the exofacial leaflet of synaptic plasma membranes of human apolipoprotein E4 knock-in mice. Neuroreport 13, 383–386. doi: 10.1097/00001756-200203250-00004
Hesse, L., Beher, D., Masters, C. L., and Multhaup, G. (1994). The beta A4 amyloid precursor protein binding to copper. FEBS Lett. 349, 109–116. doi: 10.1016/0014-5793(94)00658-X
Himes, R. A., Park, G. Y., Siluvai, G. S., Blackburn, N. J., and Karlin, K. D. (2008). Structural studies of copper(I) complexes of amyloid-beta peptide fragments: formation of two-coordinate bis(histidine) complexes. Angew. Chem. Int. Ed. Engl. 47, 9084–9087. doi: 10.1002/anie.200803908
Hirsch-Reinshagen, V., Maia, L. F., Burgess, B. L., Blain, J. F., Naus, K. E., Mcisaac, S. A., et al. (2005). The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J. Biol. Chem. 280, 43243–43256. doi: 10.1074/jbc.M508781200
Hoke, D. E., Tan, J. L., Ilaya, N. T., Culvenor, J. G., Smith, S. J., White, A. R., et al. (2005). In vitro gamma-secretase cleavage of the Alzheimer’s amyloid precursor protein correlates to a subset of presenilin complexes and is inhibited by zinc. FEBS J. 272, 5544–5557. doi: 10.1111/j.1742-4658.2005.04950.x
Holmes, C. (2002). Genotype and phenotype in Alzheimer’s disease. Br. J. Psychiatry 180, 131–134. doi: 10.1192/bjp.180.2.131
Holtzman, D. M., Fagan, A. M., Mackey, B., Tenkova, T., Sartorius, L., Paul, S. M., et al. (2000). Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann. Neurol. 47, 739–747. doi: 10.1002/1531-8249(200006)47:6<739::AID-ANA6>3.0.CO;2-8
Huang, L., Kirschke, C. P., and Gitschier, J. (2002). Functional characterization of a novel mammalian zinc transporter, ZnT6. J. Biol. Chem. 277, 26389–26395. doi: 10.1074/jbc.M200462200
Huang, X., Atwood, C. S., Moir, R. D., Hartshorn, M. A., Tanzi, R. E., and Bush, A. I. (2004). Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Abeta peptides. J. Biol. Inorg. Chem. 9, 954–960. doi: 10.1007/s00775-004-0602-8
Huang, X., Atwood, C. S., Moir, R. D., Hartshorn, M. A., Vonsattel, J. P., Tanzi, R. E., et al. (1997). Zinc-induced Alzheimer’s Abeta1-40 aggregation is mediated by conformational factors. J. Biol. Chem. 272, 26464–26470. doi: 10.1074/jbc.272.42.26464
Huang, X., Cuajungco, M. P., Atwood, C. S., Hartshorn, M. A., Tyndall, J. D., Hanson, G. R., et al. (1999). Cu(II) potentiation of Alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 274, 37111–37116. doi: 10.1074/jbc.274.52.37111
Hung, Y. H., Bush, A. I., and La Fontaine, S. (2013). Links between copper and cholesterol in Alzheimer’s disease. Front. Physiol. 4:111. doi: 10.3389/fphys.2013.00111
Hung, Y. H., Faux, N. G., Killilea, D. W., Yanjanin, N., Firnkes, S., Volitakis, I., et al. (2014). Altered transition metal homeostasis in Niemann–Pick disease, type C1. Metallomics 6, 542–553. doi: 10.1039/c3mt00308f
Hung, Y. H., Robb, E. L., Volitakis, I., Ho, M., Evin, G., Li, Q. X., et al. (2009). Paradoxical condensation of copper with elevated beta-amyloid in lipid rafts under cellular copper deficiency conditions: implications for Alzheimer disease. J. Biol. Chem. 284, 21899–21907. doi: 10.1074/jbc.M109.019521
Hur, J. Y., Welander, H., Behbahani, H., Aoki, M., Franberg, J., Winblad, B., et al. (2008). Active gamma-secretase is localized to detergent-resistant membranes in human brain. FEBS J. 275, 1174–1187. doi: 10.1111/j.1742-4658.2008.06278.x
Hureau, C., and Faller, P. (2009). Abeta-mediated ROS production by Cu ions: structural insights, mechanisms and relevance to Alzheimer’s disease. Biochimie 91, 1212–1217. doi: 10.1016/j.biochi.2009.03.013
Irizarry, M. C., Cheung, B. S., Rebeck, G. W., Paul, S. M., Bales, K. R., and Hyman, B. T. (2000). Apolipoprotein E affects the amount, form, and anatomical distribution of amyloid beta-peptide deposition in homozygous APP(V717F) transgenic mice. Acta Neuropathol. 100, 451–458. doi: 10.1007/s004010000263
Irizarry, M. C., Deng, A., Lleo, A., Berezovska, O., Von Arnim, C. A., Martin-Rehrmann, M., et al. (2004). Apolipoprotein E modulates gamma-secretase cleavage of the amyloid precursor protein. J. Neurochem. 90, 1132–1143. doi: 10.1111/j.1471-4159.2004.02581.x
Iuliano, L. (2011). Pathways of cholesterol oxidation via non-enzymatic mechanisms. Chem. Phys. Lipids 164, 457–468. doi: 10.1016/j.chemphyslip.2011.06.006
Iwatsubo, T. (2004). The gamma-secretase complex: machinery for intramembrane proteolysis. Curr. Opin. Neurobiol 14, 379–383. doi: 10.1016/j.conb.2004.05.010
Janssen, J. C., Beck, J. A., Campbell, T. A., Dickinson, A., Fox, N. C., Harvey, R. J., et al. (2003). Early onset familial Alzheimer’s disease: mutation frequency in 31 families. Neurology 60, 235–239. doi: 10.1212/01.WNL.0000042088.22694.E3
Jiao, Y., and Yang, P. (2007). Mechanism of copper(II) inhibiting Alzheimer’s amyloid beta-peptide from aggregation: a molecular dynamics investigation. J. Phys. Chem. B 111, 7646–7655. doi: 10.1021/jp0673359.
Jin, L. W., Shie, F. S., Maezawa, I., Vincent, I., and Bird, T. (2004). Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann–Pick type C defects is associated with endosomal abnormalities. Am. J. Pathol. 164, 975–985. doi: 10.1016/S0002-9440(10)63185-9
Kaden, D., Munter, L. M., Joshi, M., Treiber, C., Weise, C., Bethge, T., et al. (2008). Homophilic interactions of the amyloid precursor protein (APP) ectodomain are regulated by the loop region and affect beta-secretase cleavage of APP. J. Biol. Chem. 283, 7271–7279. doi: 10.1074/jbc.M708046200
Kakio, A., Nishimoto, S., Yanagisawa, K., Kozutsumi, Y., and Matsuzaki, K. (2002). Interactions of amyloid beta-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 41, 7385–7390. doi: 10.1021/bi0255874
Kalvodova, L., Kahya, N., Schwille, P., Ehehalt, R., Verkade, P., Drechsel, D., et al. (2005). Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 280, 36815–36823. doi: 10.1074/jbc.M504484200
Karr, J. W., and Szalai, V. A. (2008). Cu(II) binding to monomeric, oligomeric, and fibrillar forms of the Alzheimer’s disease amyloid-beta peptide. Biochemistry 47, 5006–5016. doi: 10.1021/bi702423h
Kayed, R., Head, E., Thompson, J. L., Mcintire, T. M., Milton, S. C., Cotman, C. W., et al. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. doi: 10.1126/science.1079469
Kelleher, S. L., and Lonnerdal, B. (2002). Zinc transporters in the rat mammary gland respond to marginal zinc and vitamin A intakes during lactation. J. Nutr. 132, 3280–3285.
Kienlen-Campard, P., Tasiaux, B., Van Hees, J., Li, M., Huysseune, S., Sato, T., et al. (2008). Amyloidogenic processing but not amyloid precursor protein (APP) intracellular C-terminal domain production requires a precisely oriented APP dimer assembled by transmembrane GXXXG motifs. J. Biol. Chem. 283, 7733–7744. doi: 10.1074/jbc.M707142200
Kim, J., Castellano, J. M., Jiang, H., Basak, J. M., Parsadanian, M., Pham, V., et al. (2009). Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron 64, 632–644. doi: 10.1016/j.neuron.2009.11.013
Kim, S., Jeon, T. J., Oberai, A., Yang, D., Schmidt, J. J., and Bowie, J. U. (2005). Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 14278–14283. doi: 10.1073/pnas.0501234102
Kim, S. I., Yi, J. S., and Ko, Y. G. (2006). Amyloid beta oligomerization is induced by brain lipid rafts. J. Cell. Biochem. 99, 878–889. doi: 10.1002/jcb.20978
Kodam, A., Maulik, M., Peake, K., Amritraj, A., Vetrivel, K. S., Thinakaran, G., et al. (2010). Altered levels and distribution of amyloid precursor protein and its processing enzymes in Niemann–Pick type C1-deficient mouse brains. Glia 58, 1267–1281. doi: 10.1002/glia.21001
Kojro, E., Gimpl, G., Lammich, S., Marz, W., and Fahrenholz, F. (2001). Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10. Proc. Natl. Acad. Sci. U.S.A. 98, 5815–5820. doi: 10.1073/pnas.081612998
Koldamova, R., Staufenbiel, M., and Lefterov, I. (2005). Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 280, 43224–43235. doi: 10.1074/jbc.M504513200
Kosicek, M., Malnar, M., Goate, A., and Hecimovic, S. (2010). Cholesterol accumulation in Niemann–Pick type C (NPC) model cells causes a shift in APP localization to lipid rafts. Biochem. Biophys. Res. Commun. 393, 404–409. doi: 10.1016/j.bbrc.2010.02.007
Kosik, K. S., Joachim, C. L., and Selkoe, D. J. (1986). Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 83, 4044–4048. doi: 10.1073/pnas.83.11.4044
Kuhn, P. H., Wang, H., Dislich, B., Colombo, A., Zeitschel, U., Ellwart, J. W., et al. (2010). ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032. doi: 10.1038/emboj.2010.167
Lammich, S., Kojro, E., Postina, R., Gilbert, S., Pfeiffer, R., Jasionowski, M., et al. (1999). Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3927. doi: 10.1073/pnas.96.7.3922
Ledesma, M. D., Abad-Rodriguez, J., Galvan, C., Biondi, E., Navarro, P., Delacourte, A., et al. (2003). Raft disorganization leads to reduced plasmin activity in Alzheimer’s disease brains. EMBO Rep. 4, 1190–1196. doi: 10.1038/sj.embor.7400021
Lee, J. Y., Cole, T. B., Palmiter, R. D., Suh, S. W., and Koh, J. Y. (2002). Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 7705–7710. doi: 10.1073/pnas.092034699
Lee, S. J., Liyanage, U., Bickel, P. E., Xia, W., Lansbury, P. T. Jr., and Kosik, K. S. (1998). A detergent-insoluble membrane compartment contains A beta in vivo. Nat. Med. 4, 730–734. doi: 10.1038/nm0698-730
Lee, V. M., Balin, B. J., Otvos, L. Jr., and Trojanowski, J. Q. (1991). A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251, 675–678. doi: 10.1126/science.1899488
Lei, P., Ayton, S., Finkelstein, D. I., Spoerri, L., Ciccotosto, G. D., Wright, D. K., et al. (2012). Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295. doi: 10.1038/nm.2613
Lesne, S., Kotilinek, L., and Ashe, K. H. (2008). Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience 151, 745–749. doi: 10.1016/j.neuroscience.2007.10.054
Lichtenthaler, S. F. (2011). alpha-secretase in Alzheimer’s disease: molecular identity, regulation and therapeutic potential. J. Neurochem. 116, 10–21. doi: 10.1111/j.1471-4159.2010.07081.x
Lin, M. S., Chen, L. Y., Wang, S. S., Chang, Y., and Chen, W. Y. (2008). Examining the levels of ganglioside and cholesterol in cell membrane on attenuation the cytotoxicity of beta-amyloid peptide. Colloids Surf. B Biointerfaces 65, 172–177. doi: 10.1016/j.colsurfb.2008.03.012
Lind, S. E., Mcdonagh, J. R., and Smith, C. J. (1993). Oxidative inactivation of plasmin and other serine proteases by copper and ascorbate. Blood 82, 1522–1531.
Ling, Y., Morgan, K., and Kalsheker, N. (2003). Amyloid precursor protein (APP) and the biology of proteolytic processing: relevance to Alzheimer’s disease. Int. J. Biochem. Cell Biol. 35, 1505–1535. doi: 10.1016/S1357-2725(03)00133-X
Linkous, D. H., Adlard, P. A., Wanschura, P. B., Conko, K. M., and Flinn, J. M. (2009). The effects of enhanced zinc on spatial memory and plaque formation in transgenic mice. J. Alzheimers. Dis. 18, 565–579. doi: 10.3233/JAD-2009-1162
Linkous, D. H., Flinn, J. M., Koh, J. Y., Lanzirotti, A., Bertsch, P. M., Jones, B. F., et al. (2008). Evidence that the ZNT3 protein controls the total amount of elemental zinc in synaptic vesicles. J. Histochem. Cytochem. 56, 3–6. doi: 10.1369/jhc.6A7035.2007
Liu, B., Moloney, A., Meehan, S., Morris, K., Thomas, S. E., Serpell, L. C., et al. (2011). Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J. Biol. Chem. 286, 4248–4256. doi: 10.1074/jbc.M110.158980
Lovell, M. A., Robertson, J. D., Teesdale, W. J., Campbell, J. L., and Markesbery, W. R. (1998). Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 158, 47–52. doi: 10.1016/S0022-510X(98)00092-6
Lovell, M. A., Smith, J. L., Xiong, S., and Markesbery, W. R. (2005). Alterations in zinc transporter protein-1 (ZnT-1) in the brain of subjects with mild cognitive impairment, early, and late-stage Alzheimer’s disease. Neurotox. Res. 7, 265–271. doi: 10.1007/BF03033884
Lund, E. G., Guileyardo, J. M., and Russell, D. W. (1999). cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. U.S.A. 96, 7238–7243. doi: 10.1073/pnas.96.13.7238
Lutjohann, D., and von Bergmann, K. (2003). 24S-hydroxycholesterol: a marker of brain cholesterol metabolism. Pharmacopsychiatry 36(Suppl. 2), S102–S106. doi: 10.1055/s-2003-43053
Mainous, A. G. III, Eschenbach, S. L., Wells, B. J., Everett, C. J., and Gill, J. M. (2005). Cholesterol, transferrin saturation, and the development of dementia and Alzheimer’s disease: results from an 18-year population-based cohort. Fam. Med. 37, 36–42.
Malnar, M., Kosicek, M., Lisica, A., Posavec, M., Krolo, A., Njavro, J., et al. (2012). Cholesterol-depletion corrects APP and BACE1 misstrafficking in NPC1-deficient cells. Biochim. Biophys. Acta 1822, 1270–1283. doi: 10.1016/j.bbadis.2012.04.002
Malnar, M., Kosicek, M., Mitterreiter, S., Omerbasic, D., Lichtenthaler, S. F., Goate, A., et al. (2010). Niemann–Pick type C cells show cholesterol dependent decrease of APP expression at the cell surface and its increased processing through the beta-secretase pathway. Biochim. Biophys. Acta 1802, 682–691. doi: 10.1016/j.bbadis.2010.05.006
Mantyh, P. W., Ghilardi, J. R., Rogers, S., Demaster, E., Allen, C. J., Stimson, E. R., et al. (1993). Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J. Neurochem. 61, 1171–1174. doi: 10.1111/j.1471-4159.1993.tb03639.x
Marquer, C., Devauges, V., Cossec, J. C., Liot, G., Lecart, S., Saudou, F., et al. (2011). Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J. 25, 1295–1305. doi: 10.1096/fj.10-168633
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., Mcdonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
Matsuzaki, K., Kato, K., and Yanagisawa, K. (2010). Abeta polymerization through interaction with membrane gangliosides. Biochim. Biophys. Acta 1801, 868–877. doi: 10.1016/j.bbalip.2010.01.008
Maulik, M., Westaway, D., Jhamandas, J. H., and Kar, S. (2013). Role of cholesterol in APP metabolism and its significance in Alzheimer’s disease pathogenesis. Mol. Neurobiol. 47, 37–63. doi: 10.1007/s12035-012-8337-y
Maynard, C. J., Cappai, R., Volitakis, I., Cherny, R. A., White, A. R., Beyreuther, K., et al. (2002). Overexpression of Alzheimer’s disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J. Biol. Chem. 277, 44670–44676. doi: 10.1074/jbc.M204379200
Michikawa, M., Fan, Q. W., Isobe, I., and Yanagisawa, K. (2000). Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J. Neurochem. 74, 1008–1016. doi: 10.1046/j.1471-4159.2000.0741008.x
Miller, L. M., Wang, Q., Telivala, T. P., Smith, R. J., Lanzirotti, A., and Miklossy, J. (2006). Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 155, 30–37. doi: 10.1016/j.jsb.2005.09.004
Miyashita, N., Straub, J. E., Thirumalai, D., and Sugita, Y. (2009). Transmembrane structures of amyloid precursor protein dimer predicted by replica-exchange molecular dynamics simulations. J. Am. Chem. Soc. 131, 3438–3439. doi: 10.1021/ja809227c
Miyata, M., and Smith, J. D. (1996). Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 14, 55–61. doi: 10.1038/ng0996-55
Moir, R. D., Atwood, C. S., Romano, D. M., Laurans, M. H., Huang, X., Bush, A. I., et al. (1999). Differential effects of apolipoprotein E isoforms on metal-induced aggregation of A beta using physiological concentrations. Biochemistry 38, 4595–4603. doi: 10.1021/bi982437d
Morris, M. C., Evans, D. A., Bienias, J. L., Tangney, C. C., Bennett, D. A., Aggarwal, N., et al. (2003). Dietary fats and the risk of incident Alzheimer disease. Arch. Neurol. 60, 194–200. doi: 10.1001/archneur.60.2.194
Morris, M. C., Evans, D. A., Bienias, J. L., Tangney, C. C., and Wilson, R. S. (2004). Dietary fat intake and 6-year cognitive change in an older biracial community population. Neurology 62, 1573–1579. doi: 10.1212/01.WNL.0000123250.82849.B6
Morris, M. C., Evans, D. A., Tangney, C. C., Bienias, J. L., Schneider, J. A., Wilson, R. S., et al. (2006). Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch. Neurol. 63, 1085–1088. doi: 10.1001/archneur.63.8.1085
Muller-Spahn, F., and Hock, C. (1999). Risk factors and differential diagnosis of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 249(Suppl. 3), 37–42. doi: 10.1007/PL00014172
Multhaup, G., Schlicksupp, A., Hesse, L., Beher, D., Ruppert, T., Masters, C. L., et al. (1996). The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to copper(I). Science 271, 1406–1409. doi: 10.1126/science.271.5254.1406
Munter, L. M., Voigt, P., Harmeier, A., Kaden, D., Gottschalk, K. E., Weise, C., et al. (2007). GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 26, 1702–1712. doi: 10.1038/sj.emboj.7601616
Murray, I. V., Sindoni, M. E., and Axelsen, P. H. (2005). Promotion of oxidative lipid membrane damage by amyloid beta proteins. Biochemistry 44, 12606–12613. doi: 10.1021/bi050926p
Needham, B. E., Ciccotosto, G. D., and Cappai, R. (2014). Combined deletions of amyloid precursor protein and amyloid precursor-like protein 2 reveal different effects on mouse brain metal homeostasis. Metallomics 6, 598–603. doi: 10.1039/c3mt00358b
Nelson, T. J., and Alkon, D. L. (2005). Oxidation of cholesterol by amyloid precursor protein and beta-amyloid peptide. J. Biol. Chem. 280, 7377–7387. doi: 10.1074/jbc.M409071200
Noda, Y., Asada, M., Kubota, M., Maesako, M., Watanabe, K., Uemura, M., et al. (2013). Copper enhances APP dimerization and promotes Abeta production. Neurosci. Lett. 547, 10–15. doi: 10.1016/j.neulet.2013.04.057
Okada, T., Ikeda, K., Wakabayashi, M., Ogawa, M., and Matsuzaki, K. (2008). Formation of toxic Abeta(1-40) fibrils on GM1 ganglioside-containing membranes mimicking lipid rafts: polymorphisms in Abeta(1-40) fibrils. J. Mol. Biol. 382, 1066–1074. doi: 10.1016/j.jmb.2008.07.072
Opazo, C., Huang, X., Cherny, R. A., Moir, R. D., Roher, A. E., White, A. R., et al. (2002). Metalloenzyme-like activity of Alzheimer’s disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2). J. Biol. Chem. 277, 40302–40308. doi: 10.1074/jbc.M206428200
Osenkowski, P., Ye, W., Wang, R., Wolfe, M. S., and Selkoe, D. J. (2008). Direct and potent regulation of gamma-secretase by its lipid microenvironment. J. Biol. Chem. 283, 22529–22540. doi: 10.1074/jbc.M801925200
Panchal, M., Loeper, J., Cossec, J. C., Perruchini, C., Lazar, A., Pompon, D., et al. (2010). Enrichment of cholesterol in microdissected Alzheimer’s disease senile plaques as assessed by mass spectrometry. J. Lipid Res. 51, 598–605. doi: 10.1194/jlr.M001859
Park, I. H., Jung, M. W., Mori, H., and Mook-Jung, I. (2001). Zinc enhances synthesis of presenilin 1 in mouse primary cortical culture. Biochem. Biophys. Res. Commun. 285, 680–688. doi: 10.1006/bbrc.2001.5243
Parkin, E. T., Turner, A. J., and Hooper, N. M. (1999). Amyloid precursor protein, although partially detergent-insoluble in mouse cerebral cortex, behaves as an atypical lipid raft protein. Biochem. J. 344(Pt 1), 23–30. doi: 10.1042/0264-6021:3440023
Perrone, L., Mothes, E., Vignes, M., Mockel, A., Figueroa, C., Miquel, M. C., et al. (2010). Copper transfer from Cu-Abeta to human serum albumin inhibits aggregation, radical production and reduces Abeta toxicity. Chembiochem 11, 110–118. doi: 10.1002/cbic.200900474
Phinney, A. L., Drisaldi, B., Schmidt, S. D., Lugowski, S., Coronado, V., Liang, Y., et al. (2003). In vivo reduction of amyloid-beta by a mutant copper transporter. Proc. Natl. Acad. Sci. U.S.A. 100, 14193–14198. doi: 10.1073/pnas.2332851100
Pierrot, N., Tyteca, D., D’Auria, L., Dewachter, I., Gailly, P., Hendrickx, A., et al. (2013). Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol. Med. 5, 608–625. doi: 10.1002/emmm.201202215
Poirier, J. (2003). Apolipoprotein E and cholesterol metabolism in the pathogenesis and treatment of Alzheimer’s disease. Trends Mol. Med. 9, 94–101. doi: 10.1016/S1471-4914(03)00007-8
Poirier, J., Baccichet, A., Dea, D., and Gauthier, S. (1993). Cholesterol synthesis and lipoprotein reuptake during synaptic remodelling in hippocampus in adult rats. Neuroscience 55, 81–90. doi: 10.1016/0306-4522(93)90456-P
Puglielli, L., Friedlich, A. L., Setchell, K. D., Nagano, S., Opazo, C., Cherny, R. A., et al. (2005). Alzheimer disease beta-amyloid activity mimics cholesterol oxidase. J. Clin. Invest. 115, 2556–2563. doi: 10.1172/JCI23610
Ramstedt, B., and Slotte, J. P. (2006). Sphingolipids and the formation of sterol-enriched ordered membrane domains. Biochim. Biophys. Acta 1758, 1945–1956. doi: 10.1016/j.bbamem.2006.05.020
Raux, G., Guyant-Marechal, L., Martin, C., Bou, J., Penet, C., Brice, A., et al. (2005). Molecular diagnosis of autosomal dominant early onset Alzheimer’s disease: an update. J. Med. Genet. 42, 793–795. doi: 10.1136/jmg.2005.033456
Reiman, E. M., Chen, K., Liu, X., Bandy, D., Yu, M., Lee, W., et al. (2009). Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 106, 6820–6825. doi: 10.1073/pnas.0900345106
Riddell, D. R., Zhou, H., Comery, T. A., Kouranova, E., Lo, C. F., Warwick, H. K., et al. (2007). The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol. Cell. Neurosci. 34, 621–628. doi: 10.1016/j.mcn.2007.01.011
Rogers, J. T., Randall, J. D., Cahill, C. M., Eder, P. S., Huang, X., Gunshin, H., et al. (2002). An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 277, 45518–45528. doi: 10.1074/jbc.M207435200
Roychaudhuri, R., Yang, M., Hoshi, M. M., and Teplow, D. B. (2009). Amyloid beta-protein assembly and Alzheimer disease. J. Biol. Chem. 284, 4749–4753. doi: 10.1074/jbc.R800036200
Rozga, M., Protas, A. M., Jablonowska, A., Dadlez, M., and Bal, W. (2009). The Cu(II) complex of Abeta40 peptide in ammonium acetate solutions. Evidence for ternary species formation. Chem. Commun. (Camb.) 1374–1376. doi: 10.1039/b819616h
Runz, H., Rietdorf, J., Tomic, I., De Bernard, M., Beyreuther, K., Pepperkok, R., et al. (2002). Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J. Neurosci. 22, 1679–1689.
Sato, K., Tanabe, C., Yonemura, Y., Watahiki, H., Zhao, Y., Yagishita, S., et al. (2012). Localization of mature neprilysin in lipid rafts. J. Neurosci. Res. 90, 870–877. doi: 10.1002/jnr.22796
Sato, T., Tang, T. C., Reubins, G., Fei, J. Z., Fujimoto, T., Kienlen-Campard, P., et al. (2009). A helix-to-coil transition at the epsilon-cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis. Proc. Natl. Acad. Sci. U.S.A. 106, 1421–1426. doi: 10.1073/pnas.0812261106
Scheuermann, S., Hambsch, B., Hesse, L., Stumm, J., Schmidt, C., Beher, D., et al. (2001). Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J. Biol. Chem. 276, 33923–33929. doi: 10.1074/jbc.M105410200
Schneider, A., Rajendran, L., Honsho, M., Gralle, M., Donnert, G., Wouters, F., et al. (2008). Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J. Neurosci. 28, 2874–2882. doi: 10.1523/JNEUROSCI.5345-07.2008
Schneider, A., Schulz-Schaeffer, W., Hartmann, T., Schulz, J. B., and Simons, M. (2006). Cholesterol depletion reduces aggregation of amyloid-beta peptide in hippocampal neurons. Neurobiol. Dis. 23, 573–577. doi: 10.1016/j.nbd.2006.04.015
Schubert, D., and Chevion, M. (1995). The role of iron in beta amyloid toxicity. Biochem. Biophys. Res. Commun. 216, 702–707. doi: 10.1006/bbrc.1995.2678
Shankar, G. M., and Walsh, D. M. (2009). Alzheimer’s disease: synaptic dysfunction and Abeta. Mol. Neurodegener. 4, 48. doi: 10.1186/1750-1326-4-48
Shearer, J., and Szalai, V. A. (2008). The amyloid-beta peptide of Alzheimer’s disease binds Cu(I) in a linear bis-his coordination environment: insight into a possible neuroprotective mechanism for the amyloid-beta peptide. J. Am. Chem. Soc. 130, 17826–17835. doi: 10.1021/ja805940m
Shinall, H., Song, E. S., and Hersh, L. B. (2005). Susceptibility of amyloid beta peptide degrading enzymes to oxidative damage: a potential Alzheimer’s disease spiral. Biochemistry 44, 15345–15350. doi: 10.1021/bi050650l
Shoji, M. (2002). Cerebrospinal fluid Abeta40 and Abeta42: natural course and clinical usefulness. Front. Biosci. 7:d997–d1006. doi: 10.2741/shoji
Simons, A., Ruppert, T., Schmidt, C., Schlicksupp, A., Pipkorn, R., Reed, J., et al. (2002). Evidence for a copper-binding superfamily of the amyloid precursor protein. Biochemistry 41, 9310–9320. doi: 10.1021/bi0258647
Simons, M., Keller, P., De Strooper, B., Beyreuther, K., Dotti, C. G., and Simons, K. (1998). Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 95, 6460–6464. doi: 10.1073/pnas.95.11.6460
Simons, M., Keller, P., Dichgans, J., and Schulz, J. B. (2001). Cholesterol and Alzheimer’s disease: is there a link? Neurology 57, 1089–1093. doi: 10.1212/WNL.57.6.1089
Sinha, S., Anderson, J. P., Barbour, R., Basi, G. S., Caccavello, R., Davis, D., et al. (1999). Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402, 537–540. doi: 10.1038/990114
Smith, D. G., Ciccotosto, G. D., Tew, D. J., Perez, K., Curtain, C. C., Boas, J. F., et al. (2010). Histidine 14 modulates membrane binding and neurotoxicity of the Alzheimer’s disease amyloid-beta peptide. J. Alzheimers Dis. 19, 1387–1400. doi: 10.3233/JAD-2010-1334
Smith, D. P., Smith, D. G., Curtain, C. C., Boas, J. F., Pilbrow, J. R., Ciccotosto, G. D., et al. (2006). Copper-mediated amyloid-beta toxicity is associated with an intermolecular histidine bridge. J. Biol. Chem. 281, 15145–15154. doi: 10.1074/jbc.M600417200
Smith, M. A., Perry, G., Richey, P. L., Sayre, L. M., Anderson, V. E., Beal, M. F., et al. (1996). Oxidative damage in Alzheimer’s. Nature 382, 120–121. doi: 10.1038/382120b0
Smith, M. A., Richey Harris, P. L., Sayre, L. M., Beckman, J. S., and Perry, G. (1997). Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J. Neurosci. 17, 2653–2657.
Song, Y., Hustedt, E. J., Brandon, S., and Sanders, C. R. (2013). Competition between homodimerization and cholesterol binding to the C99 domain of the amyloid precursor protein. Biochemistry 52, 5051–5064. doi: 10.1021/bi400735x
Sparks, D. L., Petanceska, S., Sabbagh, M., Connor, D., Soares, H., Adler, C., et al. (2005). Cholesterol, copper and Abeta in controls, MCI, AD and the AD cholesterol-lowering treatment trial (ADCLT). Curr. Alzheimer Res. 2, 527–539. doi: 10.2174/156720505774932296
Sparks, D. L., Scheff, S. W., Hunsaker, J. C. III, Liu, H., Landers, T., and Gross, D. R. (1994). Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 126, 88–94. doi: 10.1006/exnr.1994.1044
Sparks, D. L., and Schreurs, B. G. (2003). Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 100, 11065–11069. doi: 10.1073/pnas.1832769100
Spoerri, L., Vella, L. J., Pham, C. L., Barnham, K. J., and Cappai, R. (2012). The amyloid precursor protein copper binding domain histidine residues 149 and 151 mediate APP stability and metabolism. J. Biol. Chem. 287, 26840–26853. doi: 10.1074/jbc.M112.355743
Sponne, I., Fifre, A., Koziel, V., Oster, T., Olivier, J. L., and Pillot, T. (2004). Membrane cholesterol interferes with neuronal apoptosis induced by soluble oligomers but not fibrils of amyloid-beta peptide. FASEB J. 18, 836–838. doi: 10.1096/fj.03-0372fje
Stefani, M., and Liguri, G. (2009). Cholesterol in Alzheimer’s disease: unresolved questions. Curr. Alzheimer Res. 6, 15–29. doi: 10.2174/156720509787313899
Stoltenberg, M., Bruhn, M., Sondergaard, C., Doering, P., West, M. J., Larsen, A., et al. (2005). Immersion autometallographic tracing of zinc ions in Alzheimer beta-amyloid plaques. Histochem. Cell Biol. 123, 605–611. doi: 10.1007/s00418-005-0787-0
Stoltenberg, M., Bush, A. I., Bach, G., Smidt, K., Larsen, A., Rungby, J., et al. (2007). Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience 150, 357–369. doi: 10.1016/j.neuroscience.2007.09.025
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., et al. (1993). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 1977–1981. doi: 10.1073/pnas.90.5.1977
Suh, S. W., Jensen, K. B., Jensen, M. S., Silva, D. S., Kesslak, P. J., Danscher, G., et al. (2000). Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 852, 274–278. doi: 10.1016/S0006-8993(99)02096-X
Swaiman, K. F., and Machen, V. L. (1984). Iron uptake by mammalian cortical neurons. Ann. Neurol. 16, 66–70. doi: 10.1002/ana.410160113
Syme, C. D., Nadal, R. C., Rigby, S. E., and Viles, J. H. (2004). Copper binding to the amyloid-beta (Abeta) peptide associated with Alzheimer’s disease: folding, coordination geometry, pH dependence, stoichiometry, and affinity of Abeta-(1-28): insights from a range of complementary spectroscopic techniques. J. Biol. Chem. 279, 18169–18177. doi: 10.1074/jbc.M313572200
Syme, C. D., and Viles, J. H. (2006). Solution 1H NMR investigation of Zn2+ and Cd2+ binding to amyloid-beta peptide (Abeta) of Alzheimer’s disease. Biochim. Biophys. Acta 1764, 246–256. doi: 10.1016/j.bbapap.2005.09.012
Tabner, B. J., Turnbull, S., El-Agnaf, O. M., and Allsop, D. (2002). Formation of hydrogen peroxide and hydroxyl radicals from A(beta) and alpha-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radic. Biol. Med. 32, 1076–1083. doi: 10.1016/S0891-5849(02)00801-8
Tanzi, R. E., and Bertram, L. (2005). Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. doi: 10.1016/j.cell.2005.02.008
Tougu, V., Karafin, A., and Palumaa, P. (2008). Binding of zinc(II) and copper(II) to the full-length Alzheimer’s amyloid-beta peptide. J. Neurochem. 104, 1249–1259. doi: 10.1111/j.1471-4159.2007.05061.x
Tougu, V., Karafin, A., Zovo, K., Chung, R. S., Howells, C., West, A. K., et al. (2009). Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-beta (1-42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J. Neurochem. 110, 1784–1795. doi: 10.1111/j.1471-4159.2009.06269.x
Valensin, D., Mancini, F. M., Luczkowski, M., Janicka, A., Wisniewska, K., Gaggelli, E., et al. (2004). Identification of a novel high affinity copper binding site in the APP(145–155) fragment of amyloid precursor protein. Dalton Trans. 16–22. doi: 10.1039/b312411h
Vance, J. E. (2006). Lipid imbalance in the neurological disorder, Niemann–Pick C disease. FEBS Lett. 580, 5518–5524. doi: 10.1016/j.febslet.2006.06.008
Vanier, M. T., and Millat, G. (2003). Niemann–Pick disease type C. Clin. Genet. 64, 269–281. doi: 10.1034/j.1399-0004.2003.00147.x
Vanmierlo, T., Rutten, K., Dederen, J., Bloks, V. W., Van Vark-Van Der Zee, L. C., Kuipers, F., et al. (2011). Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol. Aging 32, 1262–1272. doi: 10.1016/j.neurobiolaging.2009.07.005
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Vekrellis, K., Ye, Z., Qiu, W. Q., Walsh, D., Hartley, D., Chesneau, V., et al. (2000). Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 20, 1657–1665.
Venti, A., Giordano, T., Eder, P., Bush, A. I., Lahiri, D. K., Greig, N. H., et al. (2004). The integrated role of desferrioxamine and phenserine targeted to an iron-responsive element in the APP-mRNA 5′-untranslated region. Ann. N. Y. Acad. Sci. 1035, 34–48. doi: 10.1196/annals.1332.003
Vetrivel, K. S., Cheng, H., Lin, W., Sakurai, T., Li, T., Nukina, N., et al. (2004). Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279, 44945–44954. doi: 10.1074/jbc.M407986200
Vigo-Pelfrey, C., Lee, D., Keim, P., Lieberburg, I., and Schenk, D. B. (1993). Characterization of beta-amyloid peptide from human cerebrospinal fluid. J. Neurochem. 61, 1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x
Wahrle, S., Das, P., Nyborg, A. C., Mclendon, C., Shoji, M., Kawarabayashi, T., et al. (2002). Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23. doi: 10.1006/nbdi.2001.0470
Wahrle, S. E., Jiang, H., Parsadanian, M., Hartman, R. E., Bales, K. R., Paul, S. M., et al. (2005). Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 280, 43236–43242. doi: 10.1074/jbc.M508780200
Wahrle, S. E., Jiang, H., Parsadanian, M., Kim, J., Li, A., Knoten, A., et al. (2008). Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest. 118, 671–682. doi: 10.1172/JCI33622
Wahrle, S. E., Jiang, H., Parsadanian, M., Legleiter, J., Han, X., Fryer, J. D., et al. (2004). ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 279, 40987–40993. doi: 10.1074/jbc.M407963200
Waldron, K. J., Rutherford, J. C., Ford, D., and Robinson, N. J. (2009). Metalloproteins and metal sensing. Nature 460, 823–830. doi: 10.1038/nature08300
Walkley, S. U., and Suzuki, K. (2004). Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta 1685, 48–62. doi: 10.1016/j.bbalip.2004.08.011
Walter, J., Schindzielorz, A., Hartung, B., and Haass, C. (2000). Phosphorylation of the beta-amyloid precursor protein at the cell surface by ectocasein kinases 1 and 2. J. Biol. Chem. 275, 23523–23529. doi: 10.1074/jbc.M002850200
Wang, D. S., Iwata, N., Hama, E., Saido, T. C., and Dickson, D. W. (2003). Oxidized neprilysin in aging and Alzheimer’s disease brains. Biochem. Biophys. Res. Commun. 310, 236–241. doi: 10.1016/j.bbrc.2003.09.003
Wang, S. S., Rymer, D. L., and Good, T. A. (2001). Reduction in cholesterol and sialic acid content protects cells from the toxic effects of beta-amyloid peptides. J. Biol. Chem. 276, 42027–42034. doi: 10.1074/jbc.M102834200
Weidemann, A., Konig, G., Bunke, D., Fischer, P., Salbaum, J. M., Masters, C. L., et al. (1989). Identification, biogenesis, and localization of precursors of Alzheimer’s disease A4 amyloid protein. Cell 57, 115–126. doi: 10.1016/0092-8674(89)90177-3
White, A. R., Multhaup, G., Maher, F., Bellingham, S., Camakaris, J., Zheng, H., et al. (1999a). The Alzheimer’s disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J. Neurosci. 19, 9170–9179.
White, A. R., Reyes, R., Mercer, J. F., Camakaris, J., Zheng, H., Bush, A. I., et al. (1999b). Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 842, 439–444. doi: 10.1016/S0006-8993(99)01861-2
Whitehouse, P. J. (1997). Genesis of Alzheimer’s disease. Neurology 48, S2–S7. doi: 10.1212/WNL.48.5_Suppl_7.2S
Wood, W. G., Schroeder, F., Avdulov, N. A., Chochina, S. V., and Igbavboa, U. (1999). Recent advances in brain cholesterol dynamics: transport, domains, and Alzheimer’s disease. Lipids 34, 225–234. doi: 10.1007/s11745-999-0357-9
Wu, W. H., Lei, P., Liu, Q., Hu, J., Gunn, A. P., Chen, M. S., et al. (2008). Sequestration of copper from beta-amyloid promotes selective lysis by cyclen-hybrid cleavage agents. J. Biol. Chem. 283, 31657–31664. doi: 10.1074/jbc.M804722200
Xu, X., and London, E. (2000). The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry 39, 843–849. doi: 10.1021/bi992543v
Yamauchi, K., Tozuka, M., Hidaka, H., Nakabayashi, T., Sugano, M., and Katsuyama, T. (2002). Isoform-specific effect of apolipoprotein E on endocytosis of beta-amyloid in cultures of neuroblastoma cells. Ann. Clin. Lab. Sci. 32, 65–74.
Yamauchi, K., Tozuka, M., Hidaka, H., Nakabayashi, T., Sugano, M., Kondo, Y., et al. (2000). Effect of apolipoprotein AII on the interaction of apolipoprotein E with beta-amyloid: some apo(E-AII) complexes inhibit the internalization of beta-amyloid in cultures of neuroblastoma cells. J. Neurosci. Res. 62, 608–614. doi: 10.1002/1097-4547(20001115)62:4<608::AID-JNR16>3.0.CO;2-4
Yan, R., Bienkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., Pauley, A. M., et al. (1999). Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 402, 533–537. doi: 10.1038/990107
Yang, D. S., Small, D. H., Seydel, U., Smith, J. D., Hallmayer, J., Gandy, S. E., et al. (1999). Apolipoprotein E promotes the binding and uptake of beta-amyloid into Chinese hamster ovary cells in an isoform-specific manner. Neuroscience 90, 1217–1226. doi: 10.1016/S0306-4522(98)00561-2
Yip, C. M., Elton, E. A., Darabie, A. A., Morrison, M. R., and Mclaurin, J. (2001). Cholesterol, a modulator of membrane-associated Abeta-fibrillogenesis and neurotoxicity. J. Mol. Biol. 311, 723-734. doi: 10.1006/jmbi.2001.4881
You, H., Tsutsui, S., Hameed, S., Kannanayakal, T. J., Chen, L., Xia, P., et al. (2012). Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 1737–1742. doi: 10.1073/pnas.1110789109
Zampagni, M., Evangelisti, E., Cascella, R., Liguri, G., Becatti, M., Pensalfini, A., et al. (2010). Lipid rafts are primary mediators of amyloid oxidative attack on plasma membrane. J. Mol. Med. (Berl.) 88, 597–608. doi: 10.1007/s00109-010-0603-8
Zelcer, N., Khanlou, N., Clare, R., Jiang, Q., Reed-Geaghan, E. G., Landreth, G. E., et al. (2007). Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 10601–10606. doi: 10.1073/pnas.0701096104
Zheng, H., and Koo, E. H. (2011). Biology and pathophysiology of the amyloid precursor protein. Mol. Neurodegener. 6, 27. doi: 10.1186/1750-1326-6-27
Zhou, Y., and Richardson, J. S. (1996). Cholesterol protects PC12 cells from beta-amyloid induced calcium disordering and cytotoxicity. Neuroreport 7, 2487–2490. doi: 10.1097/00001756-199611040-00017
Keywords: Alzheimer’s disease, amyloid precursor protein, Aβ, cholesterol, metals, iron, copper, zinc
Citation: Wong BX, Hung YH, Bush AI and Duce JA (2014) Metals and cholesterol: two sides of the same coin in Alzheimer’s disease pathology. Front. Aging Neurosci. 6:91. doi: 10.3389/fnagi.2014.00091
Received: 28 February 2014; Accepted: 28 April 2014;
Published online: 15 May 2014.
Edited by:
Paul Adlard, The Mental Health Research Institute, AustraliaReviewed by:
Carsten Culmsee, Philipps University of Marburg, GermanyJurgen Gotz, The University of Sydney, Australia
Copyright © 2014 Wong, Hung, Bush and Duce. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James A. Duce, School of Molecular and Cellular Biology, Faculty of Biological Sciences, University of Leeds, Leeds LS2 9JT, North Yorkshire, UK e-mail: j.a.duce@leeds.ac.uk