Our “energy-Ca2+ signaling deficits” hypothesis and its explanatory potential for key features of Alzheimer’s disease
 Ming Chen1,2*
Ming Chen1,2*  Huey T. Nguyen1
Huey T. Nguyen1- 1Aging Research Laboratory, Research and Development Service, Bay Pines Veterans Affairs Healthcare System, Bay Pines, FL, USA
- 2Department of Molecular Pharmacology and Physiology, University of South Florida, Tampa, FL, USA
Sporadic Alzheimer’s disease (sAD) has not been explained by any current theories, so new hypotheses are urgently needed. We proposed that “energy and Ca2+ signaling deficits” are perhaps the earliest modifiable defects in brain aging underlying memory decline and tau deposits (by means of inactivating Ca2+-dependent protease calpain). Consistent with this hypothesis, we now notice that at least eight other known calpain substrates have also been reported to accumulate in aging and AD. Thus, protein accumulation or aggregation is not a “pathogenic” event, but occurs naturally and selectively to a peculiar family of proteins, and is best explained by calpain inactivation. Why are only calpain substrates accumulated and how can they stay for decades in the brain without being attacked by many other non-specific proteases there? We believe that these long-lasting puzzles can be explained by calpain’s unique properties, especially its unusual specificity and exclusivity in substrate recognition, which can protect the substrates from other proteases’ attacks after calpain inactivation. Interestingly, our model, in essence, may also explain tau phosphorylation and the formation of amyloid plaques. Our studies suggest that α-secretase is an energy-/Ca2+-dual dependent protease and is also the primary determinant for Aβ levels. Therefore, β- and γ-secretases can only play secondary roles and, by biological laws, they are unlikely to be “positively identified”. This study thus raises serious questions for policymakers and researchers and these questions may help explain why sAD can remain an enigma today.
A Conceptual Crisis in Alzheimer’s Disease Study
The study of Alzheimer’s disease (AD) is in crisis today. Although progress has been made in understanding early-onset AD (a rare disease mostly caused by gene mutations), late-onset sporadic AD (LOAD or sAD), which affects the vast majority of AD victims and is the main threat to modern society, has remained an enigma after nearly 40 years of intensive studies (Holtzman et al., 2012). This problem has motivated policymakers in Congress and funding agencies to call for increased funding and innovative researches.
But we think it is more important to understand why the crisis has happened. To this end, we have undertaken an independent analysis of the current issues and realized that the official definition for sAD, a senile disorder, as a “discrete disease” by the National Institute on Aging (NIA) may be the root problem. This definition has ignored the fundamental differences between senile disorders and discrete diseases in origin, study paradigm and intervention approach. Our study emphasizes that sAD, a devastating disease in social impact, but is also a normality in its biological nature (like hearing loss and heart failure at advanced age). This should be a new conceptual basis to understand the disorder (Chen and Fernandez, 2000, 2001a; Chen et al., 2011a,b).
As the NIA definition has overlooked the unique features of sAD, it has consequently confined the studies to the prominent “pathological” lesions (e.g., plaques and tangles) and presumed “abnormal” pathways (e.g., gene mutations or Ca2+ overactivation). Such studies, though well-intentioned and highly productive, may never explain the basic features of sAD (e.g., why it is a result of demographic changes and why it increases exponentially with age). As such, these studies, though being praised enthusiastically by the AD study field and mass media, have never been accepted by the general medical community (e.g., NIH independent committee consensus statements 2002 and 2010; see NIA website).
This may be why, after so many years with over 130,000 research papers published and many of which are in prestigious journals, sAD can remain a conceptual enigma accompanied by repetitive failures in clinical trials, a crisis unseen in medical history. Thus, a high priority today is to synthesize current data into novel hypotheses that can explain sAD features better than the existing ones, thereby guiding future studies in a new direction.
Our Hypothesis for the Origins of sAD
In this context, we and a growing number of other investigators have started to think that sAD should be understood from a new perspective, i.e., aging (Chen, 1998; Swerdlow, 2007; Yankner et al., 2008; Castellani et al., 2009; Herrup, 2010; Sperling et al., 2011; Korczyn, 2012). From this ground, we have proposed a new hypothesis for the natural history of sAD (Figure 1). This hypothesis is unique in that it divides the widely called “AD process” into two distinctive stages: normal aging and cell-death stages. Emphasizing the normal aging stage is because no sAD case occurs without having passed through a long aging process. Also, unlike other models that focus on the prominent “pathological” lesions or cellular impairments in the latter stage (Holtzman et al., 2012), our hypothesis considers the former stage as the primary study focus because we believe that similar to other senile disorders, sAD is initiated from invisible and normal changes during natural aging, and only such changes are the reasonable drug targets for its intervention (Chen et al., 2011b).
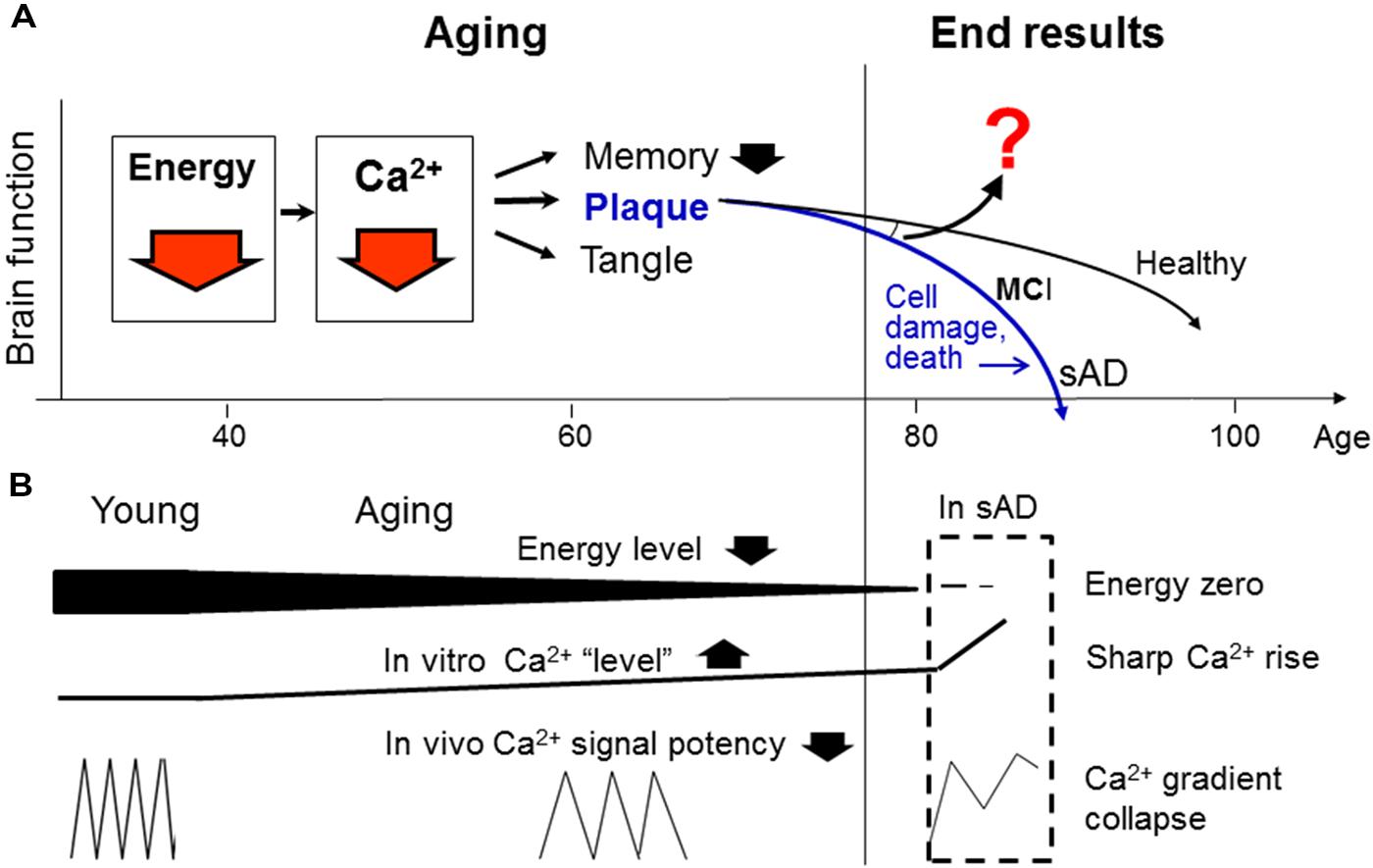
FIGURE 1. An integrative model for the “natural history” of sporadic Alzheimer’s disease (sAD). (A) Proposes that energy deficiency is the earliest modifiable defect in brain aging, which causes, among other things, a Ca2+ signaling deficit underlying inefficient memory and the formation of plaques and tangles (by means of inactivating Ca2+-dependent proteases and phosphatases; not shown, see text below). (B) Explains why energy deficit decreases Ca2+ signal potency (by reducing Ca2+ wave frequency), and why this change can manifest as slow rising Ca2+ “levels” during aging and sharp Ca2+ rises in cell death. As the aging process continues into advanced stage, it will diverge into various end results in the elderly population ranging from healthy brain to MCI or sAD. Thus, the reasons for the divergence should be a supreme question to answer (question mark; see text below). Despite the remaining questions, our model points to energy and Ca2+ deficits as two ideal points of entry for early sAD intervention (red arrows), whereas targeting the prominent lesions themselves (plaques and tangles) or numerous cell death-associated impairments will not have any therapeutic values. The current “amyloid hypothesis” (blue color) is also shown for comparison.
But, among the myriad changes in the aging brain, which one(s) should receive our primary attention? In this regard, we (Chen and Fernandez, 2001a) and others (Beal, 1998; Lannert and Hoyer, 1998; McDaniel et al., 2003; Höglinger et al., 2005; Moreira et al., 2010; Swerdlow, 2012) have suggested that energy/mitochondrial dysfunction is perhaps the earliest modifiable defect. Indeed, from the viewpoint of bioenergetics, life itself is merely a process of energy generation and consumption, and free energy is the ultimate driving force for physiological activities, especially neurotransmission and memory (Chen and Fernandez, 2000; Chen et al., 2011b; Figure 1). We also believe that energy deficit in most elderly is a naturally-occurring event, one that is not triggered by any “pathogenic/erroneous” factors, except for the passage of time.
By what mechanism can energy affect memory? We reasoned that because Ca2+ is a central regulator in neurotransmission and Ca2+ signaling is highly energy-dependent, energy deficit must cause memory inefficiency primarily by means of inactivating Ca2+ signaling, amongst its many consequences in the body (e. g., free radicals, bone loss, etc.) (Chen et al., 2011b). This will happen with concomitant formations of plaques and tangles (by means of inactivating Ca2+-dependent proteases such as calpain; tau is a known calpain substrate; plaques may be formed by a similar mechanism, see below; Chen and Fernandez, 2001b; Chen et al., 2011b; Figure 1). It thus appears that the proposed energy and Ca2+ deficits can uniformly explain the three diagnostic markers of sAD: memory decline, plaques and tangles.
The model also suggests that additional factors (question mark; Figure 1) are required to diverge the aging process into various end results in the elderly (see below). In contrast, the current “amyloid hypothesis” (blue color) does not explain two basic questions: (i) what has caused the plaque formation; and (ii) why many elderly remain healthy despite the presence of plaques and tangles.
Controversies in Ca2+ Changes in Aging
Interestingly, while energy depletion is a well-accepted concept, the proposed “Ca2+ deficit”, a logical consequence of energy deficit, has nevertheless become a primary point of controversy, because it confronts the current “Ca2+ overload/activation” hypothesis (Khachaturian, 1994), a doctrine that has been taken as granted by numerous studies (e.g., Manya et al., 2002; Sloane et al., 2003; Ferreira, 2012). As we have discussed extensively (Chen and Fernandez, 1999, 2001a,b; Chen et al., 2011b), however, the latter hypothesis is questionable because it: (i) derives from the flawed “disease” definition of sAD and thus rests on a presumptive “abnormal” pathway as its “cause”; (ii) defies the commonsense knowledge (e.g., how can the energy-dependent Ca2+ signaling be activated in the energy-depleted aging process?); and (iii) has not been supported by consistent clinical data after many trials, but is challenged by at least three studies reporting that calcium antagonists exhibit negative effects on cognition in the elderly (Heckbert et al., 1997; Maxwell et al., 1999; Wagner et al., 2012).
In contrast to this hypothesis, our model is in line with the clinical studies (Heckbert et al., 1997; Maxwell et al., 1999; Ritchie et al., 2007; Newhouse et al., 2012; Wagner et al., 2012) and also with several important reports showing that a number of Ca2+-dependent enzymes/factors are inactivated during aging (Baudry et al., 1986; Guttmann et al., 1997; Gao et al., 1998; Pahlavani and Vargas, 1999; Agbas et al., 2005; Moriguchi et al., 2006; Pascale et al., 2007; Rice et al., 2014; Sun et al., 2014). Most importantly, the model can logically explain several basic features of sAD (see above).
In support of this model, our experimental studies have recently demonstrated that the Ca2+ signaling efficacy and calpain activity are both decreased in old human fibroblasts, despite the elevated steady-state Ca2+ levels there. While much more studies are required for definitively resolving this issue, our findings suggest that higher static Ca2+ “levels” in aged cells result from energy depletion and lead to lower Ca2+ signaling potency. This intriguing phenomenon may happen because the frequency and amplitude of the Ca2+ waves in vivo may reduce by aging, but this change can manifest as a “Ca2+ overstay” or elevated steady-state “levels” in vitro (Figure 1B). For this reason, the rational approach to reduce the observed “higher Ca2+ levels” in the aging brain cells should make use of energy metabolism stimulators and Ca2+ agonists, not antagonists as currently believed (Nguyen et al., 2013).
Furthermore, we have also found that Ca2+/calpain is indeed sharply activated in the dying cells (Nguyen et al., 2013), suggesting that Ca2+/calpain undergoes a “bi-phasic” change in the “sAD process”: first inactivated during aging, then dramatically activated at the cell-death stage, as observed in the AD brain (Saito et al., 1993). The latter change, however, may not have any therapeutic values (Figures 1A,B).
Selective Accumulation of Calpain Substrates in Aging
Is this hypothesis reasonable? Despite the supportive data, it must be kept in mind that the dynamic Ca2+ changes in the living brain have not been directly determined by current technologies, so any hypotheses about them should be scrutinized with caution. One way to do this is by examining their corollaries. An important corollary of our model is that if the Ca2+/calpain deficit causes the deposition of tau, then it should also cause the concomitant depositions of many other known calpain substrates. Is this the case? This would be a key test for our model.
To this end we undertook a review of literature and found that, amazingly, at least eight other proteins have also been reported to accumulate or aggregate in aging and AD. They are: spectrin, crystalline (in cataracts), filamin, myosin, MAP2, calcineurin, tubulin, and neurofilament proteins (Johnson et al., 1991; Ashford et al., 1998; Biswas et al., 2004; Agbas et al., 2005; Puig et al., 2005; Feuillette et al., 2010; Yan et al., 2012), which are all well-known calpain substrates (Goll et al., 2003; a comprehensive review). These accumulated proteins, if considered together, would have profound implications for the origins of sAD and for our proposal, as it suggests that protein accumulation during aging is not a “pathogenic” event, but occurs naturally and preferentially to a peculiar protein family.
But discrepancies exist. For example, proteins other than calpain substrates have also been reported to accumulate in the AD brain (Griffin et al., 1989). However, it appears that only calpain substrates have been consistently reported by various laboratories to progressively accumulate from early aging and throughout the aging and AD process.
Also, because the accumulated proteins are the breakdown products of well-known calpain substrates, they have been widely called “calpain-cleaved products,” thereby suggesting “calpain overactivation” (Biswas et al., 2004; Feuillette et al., 2010; Yan et al., 2012). This view, however, needs to be revisited for two reasons. First, it implies “the more protease cleavage, the more substrate accumulation,” a scheme that sounds illogical. Second, it has overlooked the fact that calpain is essential for physiological activities such as cell division and growth. As such it should reach maximum activity in the young, but none of its products is accumulated there. So it is reasonable to believe that genuine calpain-cleaved products will not accumulate even at their peak levels. Therefore, the accumulated fragments in aging and AD brains should be the alternatively cleaved products by other proteases after calpain inactivation. Sequencing of these products and comparing them with genuine calpain-cleaved ones will confirm this view.
Yet, it is also widely believed today that the accumulated proteins may not result from insufficient proteolysis, but from abnormal aggregation or phosphorylation by abnormally activated protein kinases (e.g., tau; Chung, 2009; Bolshette et al., 2014). We have reservations for these views because they have not explained why abnormal events can happen in normal early aging during which the proteins start to aggregate, and especially why the aggregation occurs selectively to calpain substrates.
The issues are still open for debate together with many other alternative views [e.g., Ca2+/calpain is unchanged in normal aging (Saito et al., 1993; Ito et al., 1994); tau is a caspase substrate (Fasulo et al., 2000); or aggregation is due to protein misfolding or autophagy (Nixon, 2013; Bolshette et al., 2014)]. It must be noted, however, that comparing with these other competing models, our hypothesis may offer the simplest and coherent explanation for the accumulation of several calpain substrates by a uniform mechanism.
Why are Calpain Substrates Selectively Accumulated?
Now, why are calpain substrates selectively accumulated, how can they stay for decades in the brain without being attacked by many non-specific proteases there? And why does this not happen to other proteins? These long-standing puzzles need to be explained, but notably, they may not be easily explained by current models such as protein misfolding or autophagy (for their lack of selectivity). Thus we think that a clue should lie in the unique properties of calpain.
It is well-known that unlike most proteases that act non-specifically or randomly, calpain only makes limited cleavages on certain enzymes/proteins at designated sites and activates them for physiological functions. A well-known example is the cleavage of PKC by calpain to activate the PKC-related signal pathways (Kishimoto et al., 1989). Notably, such a cleavage in vivo must be highly specific, perhaps also exclusive, because if PKC is cleaved by any other proteases at the same site, disruption of signal transductions would result. We think that this could be a critical but as-yet-undocumented feature of the calpain-substrate interaction.
How can calpain ensure such a specificity and exclusivity in substrate recognition, given that it does not have a strong sequence recognition preference on its substrates (Friedrich and Bozóky, 2005)? For this puzzle, it has been speculated that calpain may recognize a unique and as-yet-unknown configuration in the secondary/tertiary structures of its substrates (Carafoli and Molinari, 1998; Cuerrier et al., 2005).
A “Ligand-Receptor” Model for Calpain-Substrate Recognition
Based on the current information, we further notice that the specificity and exclusivity of calpain-substrate interaction is quite similar to that of ligand-receptor (or antigen–antibody). From this new angle, we now propose that calpain substrates in vivo may assume an unusual, receptor-like configuration that is exclusively accessible by calpain. Upon activation, calpain may change its conformation in such a way that allows itself to fit into the substrate. Such a key-and-lock fitting would safeguard the specificity and exclusivity of their interactions free of interferences by any other proteases (Figure 2A; using PKC as an example).
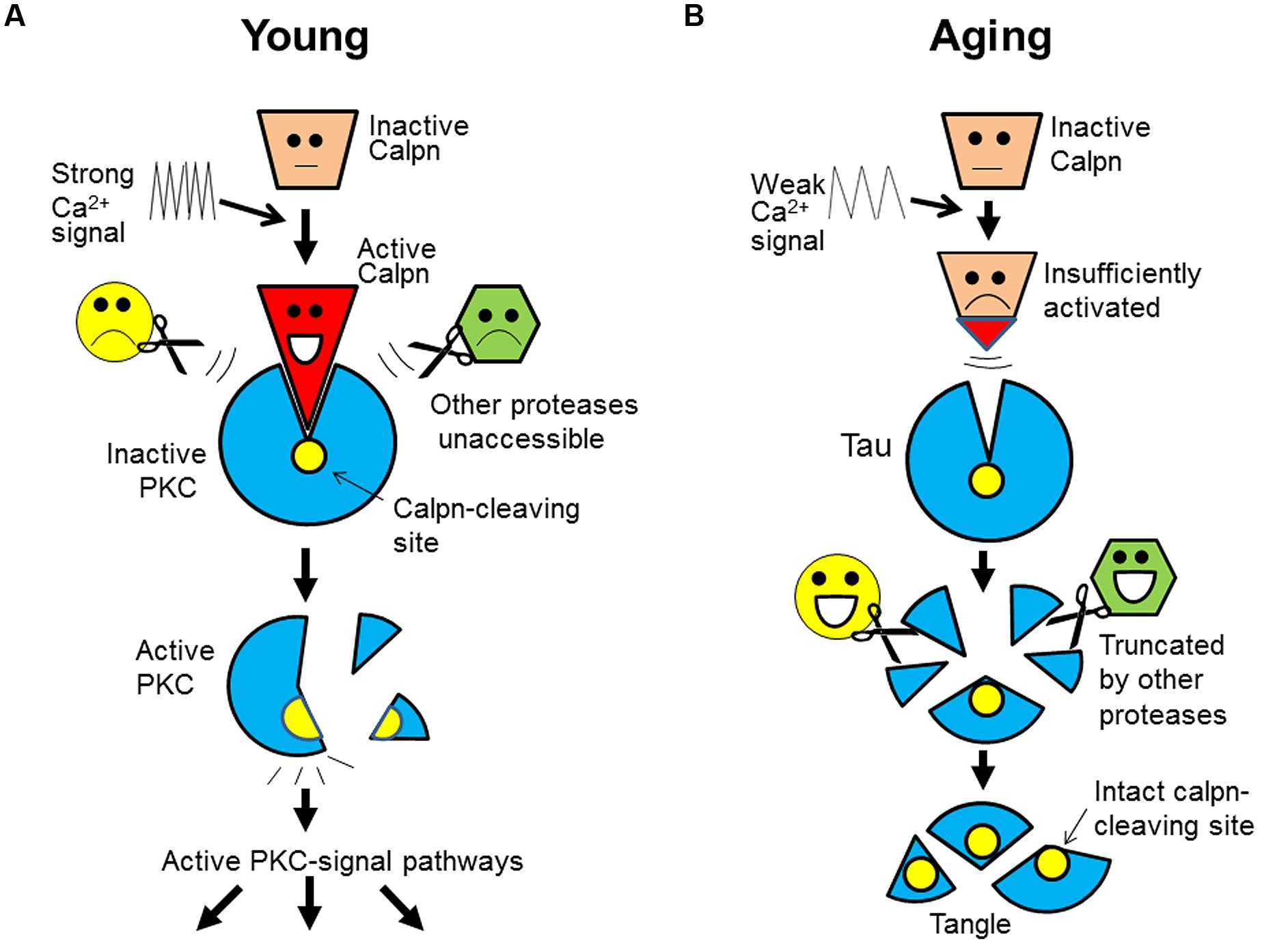
FIGURE 2. A “ligand-receptor” model for calpain-substrate interaction and selective accumulation of calpain substrates in brain aging. (A) In the young, calpain substrate (e.g., PKC) assumes a “receptor-like” configuration that is exclusively accessible by active calpain (as ligand). This unique relationship safeguards the specificity and exclusivity of their interaction, which allows a faithful cleavage/activation of PKC in the signal transduction free of interferences by any other proteases. (B) During aging, however, calpain is insufficiently activated as a result of energy and Ca2+ signaling deficits (slowed Ca2+ waves). So, calpain substrates (e.g., tau) will remain intact because other proteases will not cleave it at the calpain-leaving sites. Over time, tau will be truncated by other proteases at other sites, but its core fragments encompassing the calpain-cleaving sites will be spared and deposited (as tangles). Calpn, calpain.
Furthermore, of great interest is that this model may also explain the selective accumulation of calpain substrates. The receptor-like configuration of the substrates implies that after calpain inactivation, its substrates (such as tau) would not be attacked by any other proteases at the same sites. For this reason, the substrates would accumulate and then be truncated by other proteases at other sites, but their core fragments encompassing the calpain-cleaving sites (perhaps the domains recognized by calpain) would be spared and deposited over time, as undigested protein remnants (e.g., tangles; Figure 2B).
This model can explain: why only calpain substrates are protease-resistant; why the deposited tau is truncated at both ends (Guillozet-Bongaarts et al., 2005) and contains the predicted calpain-cleaving sites (Yang and Ksiezak-Reding, 1995); why the accumulated breakdown products from genuine calpain substrates are not genuine calpain-cleaved products, but are alternatively cleaved ones (see above); and more importantly, why protein accumulation is invariably accompanying memory decline during aging (both events are regulated by Ca2+), but targeting the deposited proteins themselves has not delayed the progression of dementia in clinical trials (protein deposits are secondary to the initial defects; Figure 1).
Can the proposed “ligand-receptor” model be experimentally tested? The model predicts that (i) distinct calpain substrates may share a unique secondary/tertiary configuration complementary to that of calpain in their crystal structures; (ii) deleting or mutating key amino acids in the unique configuration will abolish the substrates’ sensitivity to calpain; and (iii) such a configuration may not exist in other proteins. These corollaries may aid experimental validation of the model.
Why is Tau Phosphorylated?
Very interestingly yet, our “ligand-receptor” model in its essence may also explain tau phosphorylation from a new perspective. Recall that there have been two basic scenarios for tau phosphorylation: overactivation of protein kinases, or inactivation of protein phosphatases (Trojanowski and Lee, 1995). Although the former is a current favor, questions remain as to whether kinases can be “activated” in normal aging where most enzymes loss activities (see above and below).
So the latter scenario is more reasonable. But, which phosphatase among numerous in the brain should we focus on? This key question has long limited the progress of the study area with a standing dilemma: if any phosphatase is inactivated, the loss will be compensated by many other non-specific phosphatases, so how can tau stay phosphorylated for decades in the brain?
These and other considerations thus lead us to a unique protein phosphatase, calcineurin, a Ca2+-dependent enzyme, of which tau is a well-known substrate (Kayyali et al., 1997; Yu et al., 2006; Karch et al., 2013). As a regulated enzyme, its substrate recognition should also be specific and exclusive to safeguard the integrity of signal transduction (similar to calpain-substrate interaction; Figure 2). Thus, as a physiological substrate of both calpain and calcineurin, tau may also assume a receptor-like configuration that is exclusively accessible by calcineurin. This would prevent tau from dephosphorylation by any other phosphatases at the same sites after calcineurin inactivation.
Thus, as a result of Ca2+ signal down-regulation during aging, tau would deposit and stay phosphorylated in the brain (Chen and Fernandez, 2001b). The two natural events can reinforce each other to render tangles a prominent feature of sAD, and thus there is no need to assume an abnormal mechanism for it.
Meanwhile, if other phosphatases are believed to be responsible for tau overphosphorylation, then it needs to explain why their activity loss is not compensated by many non-specific phosphatases in the aging brain.
How are Amyloid Plaques Formed?
While our model may explain tangles and other protein deposits, it also raises a cardinal question: is amyloid-β precursor protein (APP) also a calpain substrate? This touches a sensitive issue because if it is, then calpain would mediate its normal processing, or as α-secretase as we suggested but controversial (Chen et al., 2000; Chen and Fernandez, 2004, 2005). But if it is not, then plaques would remain unexplained, because a key prediction of our model is that only the substrates of regulated proteases will deposit during natural aging (Figure 2).
So, the cardinal question becomes: is α-secretase a signal transduction-regulated protease? This point should be clear by now because numerous studies have shown that α-secretase is sensitively regulated by many signal transduction pathways, most notably glutamatergic, cholinergic, ERK/MAPK-, and PKC-related pathways (Buxbaum et al., 1990; Efthimiopoulos et al., 1996; Slack et al., 1997; Jolly-Tornetta et al., 1998).
But, while these elegant studies point to α-secretase as a regulated protease, they have not suggested a reasonable target for sAD intervention, since it is difficult to target all those pathways in practice. So a unifying model is needed, which should make fewest assumptions in order to parsimoniously and coherently explain most reports, thereby providing a simple drug target for intervention.
In this context, we proposed several years ago that α-secretase is a Ca2+-dependent protease, since Ca2+ appears to be a common denominator for those pathways (Chen, 1997). This view, though controversial, has since been strongly corroborated by an array of more recent reports, which have directly or indirectly linked α-secretase activity to Ca2+ (Mathews et al., 2002; Adlerz et al., 2007; Marcello et al., 2007; Dreses-Werringloer et al., 2008; Hoey et al., 2009; Vingtdeux et al., 2010; Suh et al., 2011; Zeiger et al., 2013; Kyratzi and Efthimiopoulos, 2014).
We thus believe that the concept of a Ca2+-dependent α-secretase is established and this offers a practical means for intervention even though its identity remains elusive. It is also noteworthy that because Ca2+ is the most sensitive and most exquisite regulator in the body, this can explain why APP α-processing is also sensitive to innumerable other elements such as cholesterol, cytokins, sirtuin, nardilysin, tetraspanin, and TIMP3 (Lichtenthaler, 2011; review).
Contrary to our view, however, an influential report has suggested that Aβ genesis, which changes in opposite direction to α-secretase, is a “Ca2+-dependent process” (Querfurth and Selkoe, 1994). But, it should be pointed out that physiological Ca2+-dependent processes [neurotransmission, cell growth, muscle contraction, etc. (Berridge et al., 1998)] are all reducing their activities during aging. Thus it is highly unlikely that Aβ genesis, which increases with aging, would be a physiological Ca2+-dependent process, even though it can be elevated by a Ca2+ stimulator at certain concentrations in the test tube.
Additional to its Ca2+-dependent feature, we and others have shown that α-secretase is also an energy-dependent protease, because its activity fluctuates intimately with cellular energy levels (Gasparini et al., 1997; Hoyer et al., 2005; Sawmiller et al., 2012). This view, at first glance, seems incompatible with the knowledge that proteolysis in vivo is generally a spontaneous event, and it is protein synthesis that is energy-dependent. So how can α-secretase be energy-driven? This question can be explained by the fact that Ca2+ signaling per se is highly energy-dependent (Walsh et al., 2009). This means that energy and Ca2+ signaling in vivo will undergo changes hand-in-hand. Moreover, APP α-processing, as part of protein secretions essential for cell maintenance and growth, will not occur spontaneously but must be controlled by energy supply and physiological demand. The energy-/Ca2+-dual dependent features of α-secretase provide a mechanism for such a control and, thus, they should be the “signature traits” of the enzyme to aid its identification.
Additional Evidence for α-Secretase Being Energy-/Ca2+-Dual Dependent
The proposed “signature traits” for α-secretase have touched a central yet most controversial issue in the sAD study: the mechanism of the plaque formation. So we further examined them by measuring α-secretase activity in response to five agents in a comprehensive way. The agents are ATP, nicotine, glutamate, epidermal growth factor (EGF), and phorbol 12,13-dibutylester (PDBu), which represent the five most important signal pathways regulating α-secretase: energy-, cholingeric, glutamatergic, ERK/MAPK-, and PKC-related pathways, respectively, (Buxbaum et al., 1990; Efthimiopoulos et al., 1996; Slack et al., 1997; Jolly-Tornetta et al., 1998; Sawmiller et al., 2012).
In cultured SH-SY5Y cells we found that these five agents all robustly enhanced the release of sAPPα (secreted APP by α-secretase) to various degrees (Figure 3). But these effects were abolished by BAPTA-AM, an intracellular Ca2+ chelator and, at the same time, the effects were also blocked by rotenone, a respiratory chain complex 1 inhibitor (Figure 3). This suggests that α-secretase is both energy- and Ca2+-dependent.
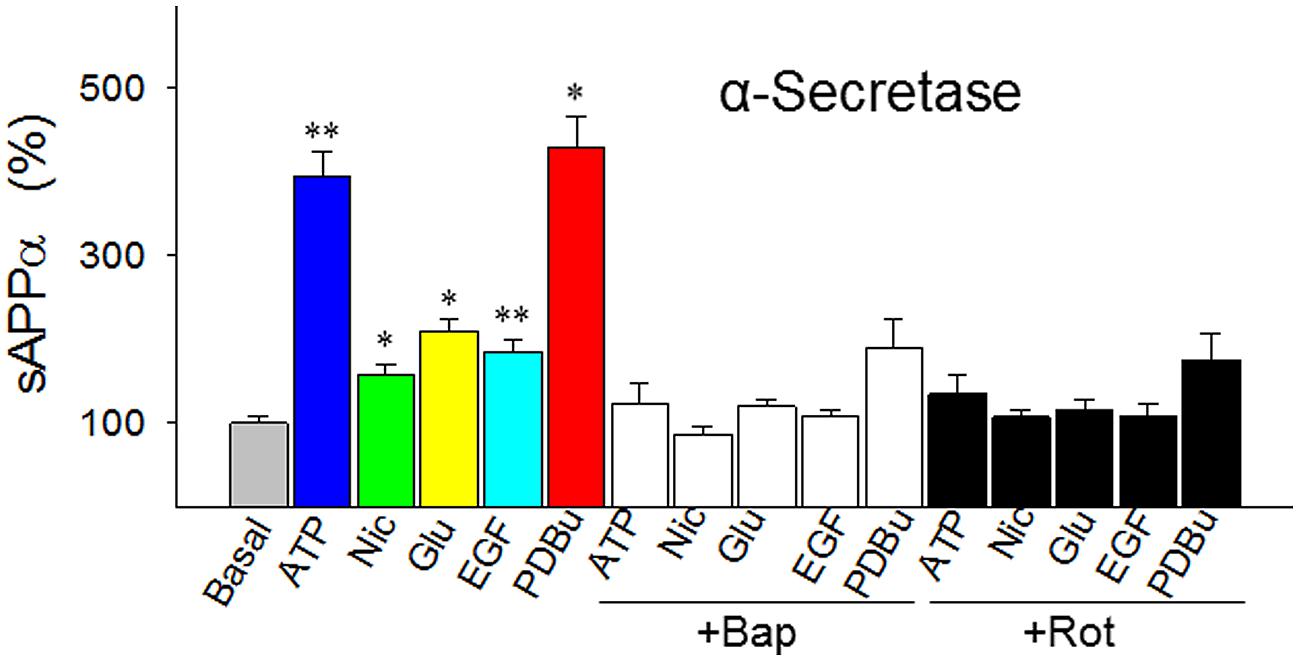
FIGURE 3. Responses of α-secretase activity (measured as sAPPα levels) to five agents that represent the five best known metabolic/signal pathways regulating α-secretase (energy-, cholinergic, glutamatergic, ERK/MAPK- and PKC-related pathways, respectively). The agents were tested in the absence or presence of intracellular Ca2+ chelator BAPTA-AM (Bap, 10 nM) or energy metabolism inhibitor rotenone (Rot, 1 μM) in cultured SH-SY5Y cells. In the experiments testing the inhibitors, the cells were preincubated with Bap or Rot for 10 min before the addition of the agents. All values are means + SEM from at least five independent experiments. *p < 0.01; **p < 0.001, versus basal level. Nic, nicotine; Glu, glutamate; EGF, epidermal growth factor; PDBu, phorbol 12, 13-dibutylester. Methods for Western blotting to determine the sAPPα levels were as previously described (Chen and Fernandez, 2004).
Furthermore, of the five agents we tested, the mechanism of PDBu needs to be further clarified, because the mode of actions of phorbol esters, the strongest stimulator for α-secretase when tested in vitro, has only been attributed to PKC activation today. Since PKC does not directly explain the proteolytic cleavage of APP, we speculated that phorbol esters may also activate α-secretase by mobilizing Ca2+, as PKC is a Ca2+-dependent enzyme (Kikkawa et al., 1982). To test this possibility, we carried out Ca2+ imaging and found that PDBu evoked Ca2+ transients in a concentration-dependent manner in cultured SH-SY5Y cells, an effect that was completely blocked by BAPTA-AM (Figure 4). Thus, among other actions, PDBu is also a potent Ca2+ activator.
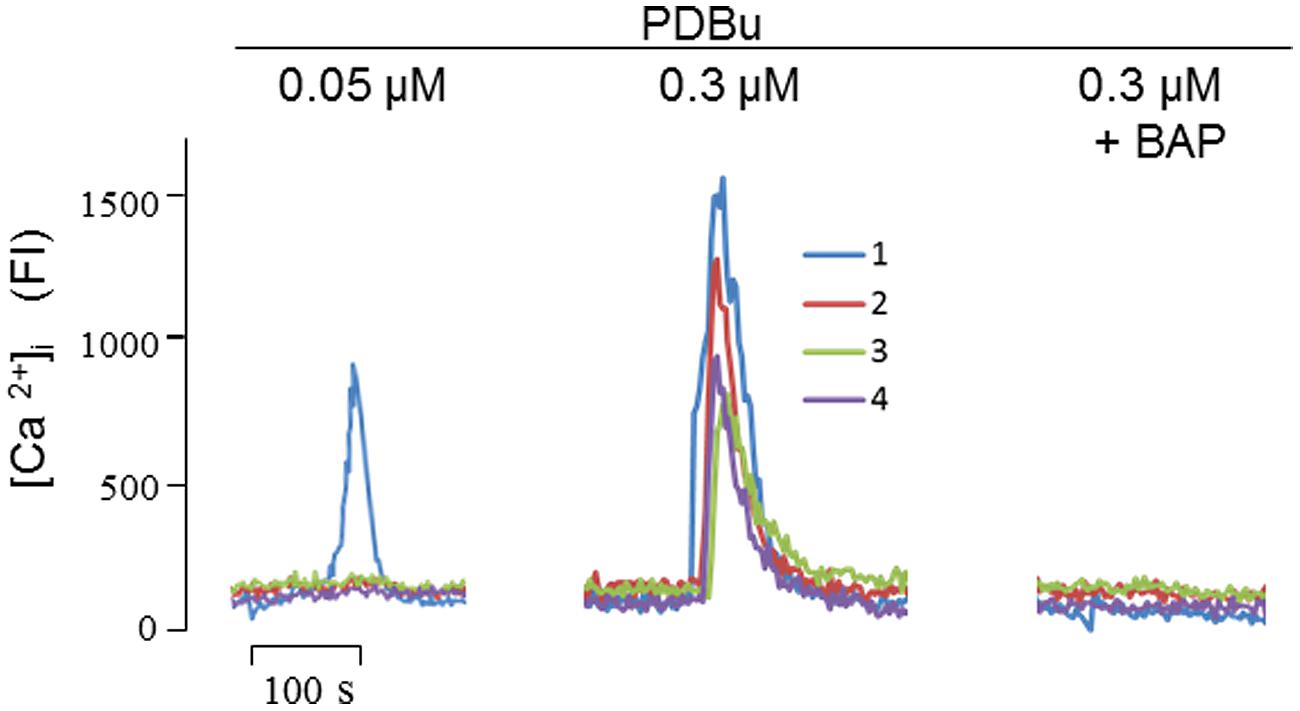
FIGURE 4. Phorbol 12,13-dibutylester (PDBu) mobilized intracellular Ca2+ signals in cultured SH-SY5Y cells. At the indicated concentrations, PDBu evoked fluorescent signals in sequential stimulations. In testing the inhibitory effect of Bap (10 nM), cells were preincubated with Bap for 10 min before the addition of PDBu. Four fluorescence traces were recorded corresponding to the four single cells focused by the laser scanning confocal microscope. FI, fluorescence intensity. Methods for intracellular Ca2+ imaging were described previously (Nguyen et al., 2013).
Energy-Ca2+ Deficits Can Also Explain Amyloid Plaques
The Ca2+-dependent feature of α-secretase has prompted us to suggest calpain as its candidate (Chen et al., 2000; Chen and Fernandez, 2004, 2005). However, question remains, because APP is a transmembrane protein, whereas calpain lacks a hydrophobic segment in its primary sequence, as we noted (Chen and Fernandez, 1998).
But, there are other proteases that are both Ca2+-dependent and transmembrane. In fact, furin or PC7, two members of the proprotein convertase family, have been reported to have both features and also are directly or indirectly involved in APP α-processing (Lopez-Perez et al., 1999; Hwang et al., 2006; Turpeinen et al., 2013). If such a protease (be it furin, PC7 or another Ca2+-dependent protease) is eventually proven to be α-secretase, then our model predicts that it should display biochemical properties similar to calpain, and that it must also be specific and exclusive in substrate recognition (otherwise Ca2+ signaling would be disrupted) and inactivated by aging. We thus believe that amyloid plaques are formed by a mechanism similar to the depositions of tau and other calpain substrates: Ca2+ signaling deficit (Figure 2).
The unsolved identity of α-secreatse thus should attract the attention of the research field with focused studies on it. Although it is no longer of key importance for sAD intervention (activating Ca2+ will activate it), we still believe that if this issue is resolved, then the three major diagnostic markers of sAD would be mechanistically understood in scientific terms. Unfortunately, however, most research attentions today are devoted instead to other competing models: metalloproteases (Buxbaum et al., 1998; Lammich et al., 1999) and β-/γ-secretases (Citron et al., 1997; Vassar et al., 1999). As discussed below, however, these other models, though popular, may face severe theoretical obstacles.
The Theoretical Obstacles for Metalloproteases as α-Secretase
(a) Metalloproteases require a metal as their catalytic center, not an activity regulator, so they belong to unregulated proteases (though they can be affected by the signal pathways), thus may not be the most reasonable candidates for α-secretase. (b) More importantly, if α-secretase is really an unregulated protease, then it would be difficult or impossible to modify its activity in the brain by common approaches (such as physical exercises). (c) None of the many other substrates of metalloproteases (Vingtdeux and Marambaud, 2012) is known to accumulate in aging (as our model predicted). (d) Metalloproteases initially appear to be α-secretase candidates by their “specific” cleavage on Aβ peptide in vitro (Buxbaum et al., 1998; Lammich et al., 1999), yet α-secretase is not sequence-specific but “distance-specific” (the distance of the cleavage site to the membrane; Maruyama et al., 1991; Sisodia, 1992). This key feature is imposed by the “double-anchorage” of both APP and α-secretase in the membrane (Chen, 1997), but difficult to mimic in vitro.
Can β- and γ-Secretases be Responsible for Aβ Genesis?
Over the years, however, the key importance of α-secretase – whatever its identity – has been severely overshadowed by a much more popular concept that Aβ is solely generated via an abnormal “amyloidogenic” pathway by “β- and γ-secretases.” So inhibiting them would cure AD without touching α-secretase (Selkoe, 2005). As the concept is so appealing, the β-/γ-secretases have been claimed to be “positively identified” and published in top-tier journals (Citron et al., 1997; Vassar et al., 1999), and thousands of studies have since followed.
We, however, stick to the biological laws first. It has come to our attention that such enthusiasms stem from the “disease” definition of sAD, so they may face hard questions. For example, if they are solely responsible for Aβ genesis, then β- and γ-secretases must be progressively activated by aging, that is, activated by free radicals, energy crisis and other age-related insults.
Can proteases be activated by such insults in vivo? This fundamental question now stands in front of us – regardless our subjective perceptions of sAD or philosophic faith – and demands a definitive answer. Further, if Aβ is generated by β-/γ-secretases, then how many other “β-/γ-secretases” would have to be assumed for tau and other deposited protein fragments? Are they all activated by aging?
Proteases and kinases (see above) are known to be activated by their increased quantity or functionality via up-regulated DNA replication, protein synthesis or signal transductions, etc. during physiological activities such as cell growth and memory formation. Now, if they are also “activated” by aging, an opposite process that diminishes those activities leading inevitably to a full stop in the end, then cogent mechanisms compatible with the established biological principles must be provided for why and how it can happen.
Perhaps, aging inactivates an intrinsic protease inhibitor, thereby activating the protease (Rao et al., 2008)? This scenario needs to explain in the first place why aging affects only one member of the pair. Yet, maybe proteases are activated by age-related abnormalities such as epigenetic changes, cell death, DNA damages, inflammations, immune defects or many other possible metabolic errors?
Again, the “disease” definition of sAD has allowed countless such “abnormalities,” replicas of pathogenic factors in discrete diseases, to be assumed and pursued. But it is a conceptually confused journey, because plaques and tangles, the common features of the normal aging brains, have to be logically explained by normal changes in aging, similar to cholesterol deposition.
α-Secretase is the Primary Determinant for Aβ Levels
If age-related cholesterol deposition simply results from an inefficient lipid degradation – a normal event in aging, rather than from any “abnormal” lipid “depository pathway” or “activated” lipid “deposit-ases” – then plaques and tangles would also be formed by a similar and simple mechanism: insufficient proteolysis, at least at their initial phase.
It is well-known by now that most intact APP is α-processed in the young, leaving only a tiny amount of it to generate Aβ. Therefore, unless intact APP is somehow increased, there would be no chance whatsoever for Aβ to increase (likewise, unless lipid catabolism slows down, no cholesterol will deposit). Thus, α-secretase activity, which controls the levels of intact APP, would be the primary determinant or rate-limiting step for Aβ levels (i.e., other proteases can only play secondary roles; similar to tangle formation; Figure 2B). Yet, Ca2+-regulated α-secretase may not allow any other unregulated proteases to act more sensitively to “compete” with it for alternative APP cleavages (i.e., they have a chance only after α-secretase inactivation).
This concept, in fact, has been well-established by numerous in vitro and in vivo studies showing that activating α-secretase, alone, reduces Aβ; and conversely, inhibiting the enzyme, alone, increases Aβ [Chen, 1997; Chen and Fernandez, 2001b; and references therein]. Why is this knowledge being ignored in the pursuing of β-/γ-secreatses? Perhaps because it points to Aβ overproduction as a natural event in aging, contrasting with the “disease” definition.
The following considerations further suggest that “identification of β-/γ-secretases” is a problematic concept in inception: (i) most proteases are non-specific, so most peptides they produced in our body are unlikely to have specific and singular – thus identifiable – “secretases”; (ii) Aβ is no exception, as it exists in vivo not as Aβ1-40/42 only, but a mixture of many peptides with their N-termini starting from -4 -3, -2, +3 up to +9, and C-termini from 34, 38, 44, and up to 46 (Masters et al., 1985; Haass and Selkoe, 1993; Kim et al., 2001; Zhao et al., 2004). Such heterogeneous peptides are unlikely to be generated by two single proteases; (iii) indeed, several “β-site cleaving proteases” have been identified today, proving the multiplicity of “β-secretase.” If so, then what are the values for identifying one or a few of them? And (iv) if γ-secretase is a presenilin, then not only would it be “overactivated” by aging (no one has provided a reason for this assumption), but also by each of its near-200 gene mutations – a wild “gain of function” model that contrasts with a “loss of function” (memory) disease [note that our “mutations cause inefficient Ca2+ channeling” model better explains the roles of the mutations (Chen and Fernandez, 2001b; Chen et al., 2011b), which has been supported by an important recent study (Sun et al., 2014)].
We are aware that current models are corroborated by mountains of evidence, but they have not explained sAD. So it is necessary for us to keep an open mind. The supreme judge in science is reason, not only “evidence” (also note that not all published data can be called “evidence,” unless they offer a reasonable explanation for the disease).
A Broad New Frontier for sAD Prevention
Our model (Figure 1) points to energy and Ca2+ deficits as the two primary drug targets for early sAD intervention. As such, numerous energy-promoting and Ca2+-activating agents/practices would be useful. These include but are not limited to: physical exercise, glucose catabolism stimulators, energy-rich substances, growth factors, hormones, neurotransmitter receptor agonists, neurotrophic factors, and metabolic enhancers (Nguyen et al., 2013; Nguyen and Chen, 2014).
This would be a broad and new research frontier (compared to current dominant anti-amyloid and anti-calcium strategies). Notably, similar approaches have been successfully used to delay or prevent other senile disorders such as atherosclerosis and osteoporosis, and some of them have been shown to exhibit neuroprotective effects in sAD studies (Pettegrew et al., 2000; McDaniel et al., 2003; Chan and Shea, 2007; Ritchie et al., 2007; Fisher, 2012; Newhouse et al., 2012). Thus, it is reasonable to anticipate that early use of such drugs (or their more effective derivatives or their combinations), especially when enhanced by targeting many risk factors in late life via improved social supports, will significantly delay or even prevent sAD.
At the same time, our study warns that current enthusiastic clinical trials on anti-amyloid and anti-mutation drugs in high risky individuals for EOAD and FTD are not the most effective use of the invaluable resources, because they will not benefit, but only distract, our study focus on sAD, the most severe social threatening disease.
Conclusion
Sporadic Alzheimer’s disease differs fundamentally from discrete diseases. This is the watershed where their study paradigms diverge but is also where commonsense and illusion collide.
In this work we further examined our “energy/Ca2+ deficits” model for its explanatory potential for several key sAD features in comparison with current models (e.g., Ca2+ overactivation, amyloid hypothesis, tau kinase activation, protein misfolding, and β-/γ-secretases). It appears that despite the controversies and paradoxes, our model can explain the sAD features better than other models.
Our model (Figure 1) also rests on several assumptions including a “ligand-receptor” relationship between calpain and substrates, “alternative breakdown” of calpain substrates, energy-/Ca2+-dual dependent α-secretase and its rate-limiting role in Aβ genesis. These assumptions may subject to modifications as new findings emerge in the future, but our starting point for reasoning – sAD initiates from natural aging – and the key intervention targets it suggests – energy and Ca2+ signaling deficits – may stand the test of time and reason.
Nevertheless, our model leaves a supreme question at large: if memory inefficiency, plaques and tangles all result from normal aging as we suggested, then what on earth can explain the devastating sAD only in some but not in other elderly (Figure 1; question mark)? This question should have been guiding the sAD research from the beginning, yet unfortunately, it has never been explicitly asked by NIA. As we discussed elsewhere (Chen and Fernandez, 2000; Chen et al., 2011b), this question can be hopelessly complicated to answer if sAD is as defined today, but it may be much simpler in concept if it is merely one of many senile disorders, as viewed by Alois Alzheimer and his colleagues by commonsense. NIA needs to return to commonsense in the perception of sAD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by the Merit Review program of the U.S. Department of Veterans Affairs and the Bay Pines VA Healthcare System. It is also supported by the CWS Foundation. The contents do not represent the views of the Department of Veterans Affairs or the United States Governments.
References
Adlerz, L., Holback, S., Multhaup, G., and Iverfeldt, K. (2007). IGF-1-induced processing of the amyloid precursor protein family is mediated by different signaling pathways. J. Biol. Chem. 282, 10203–10209. doi: 10.1074/jbc.M611183200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Agbas, A., Zaidi, A., and Michaelis, E. K. (2005). Decreased activity and increased aggregation of brain calcineurin during aging. Brain Res. 1059, 59–71. doi: 10.1016/j.brainres.2005.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ashford, J. W., Soultanian, N. S., Zhang, S. X., and Geddes, J. W. (1998). Neuropil threads are collinear with MAP2 immunostaining in neuronal dendrites of Alzheimer brain. J. Neuropathol. Exp. Neurol. 57, 972–978. doi: 10.1097/00005072-199810000-00009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baudry, M., DuBrin, R., Beasley, L., Leon, M., and Lynch, G. (1986). Low levels of calpain activity in Chiroptera brain: implications for mechanisms of aging. Neurobiol. Aging 7, 255–258. doi: 10.1016/0197-4580(86)90004-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beal, M. F. (1998). Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta 1366, 211–223. doi: 10.1016/S0005-2728(98)00114-5
Berridge, M. J., Bootman, M. D., and Lipp, P. (1998). Calcium – a life and death signal. Nature 395, 645–648. doi: 10.1038/27094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Biswas, S., Harris, F., Dennison, S., Singh, J., and Phoenix, D. A. (2004). Calpains: targets of cataract prevention? Trends Mol. Med. 10, 78–84. doi: 10.1016/j.molmed.2003.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bolshette, N. B., Thakur, K. K., Bidkar, A. P., Trandafir, C., Kumar, P., and Gogoi, R. (2014). Protein folding and misfolding in the neurodegenerative disorders: a review. Rev. Neurol. (Paris) 170, 151–161. doi: 10.1016/j.neurol.2013.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buxbaum, J. D., Gandy, S. E., Cicchetti, P., Ehrlich, M. E., Czernik, A. J., Fracasso, R. P.,et al. (1990). Processing of Alzheimer beta/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 87, 6003–6006. doi: 10.1073/pnas.87.15.6003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buxbaum, J. D., Liu, K. N., Luo, Y., Slack, J. L., Stocking, K. L., Peschon, J. J.,et al. (1998). Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 273, 27765–27767. doi: 10.1074/jbc.273.43.27765
Carafoli, E., and Molinari, M. (1998). Calpain: a protease in search of a function? Biochem. Biophys. Res. Commun. 247, 193–203. doi: 10.1006/bbrc.1998.8378
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Castellani, R. J., Zhu, X., Lee, H. G., Smith, M. A., and Perry, G. (2009). Molecular pathogenesis of Alzheimer’s disease: reductionist versus expansionist approaches. Int. J. Mol. Sci. 10, 1386–1406. doi: 10.3390/ijms10031386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chan, A., and Shea, T. B. (2007). Effects of dietary supplementation with N-acetyl cysteine, acetyl-L-carnitine and S-adenosyl methionine on cognitive performance and aggression in normal mice and mice expressing human ApoE4. Neuromolecular Med. 9, 264–269. doi: 10.1007/s12017-007-8005-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, M. (1997). Alzheimer’s α-secretase may be a calcium-dependent protease. FEBS Lett. 417, 163–167. doi: 10.1016/S0014-5793(97)01214-3
Chen, M. (1998). The Alzheimer’s plaques, tangles and memory deficits may have a common origin. Part I: a calcium deficit hypothesis. Front. Biosci. 3:a27–a31.
Chen, M., Durr, J., and Fernandez, H. L. (2000). Possible role of calpain in normal processing of Alzheimer’s beta-amyloid precursor protein in human platelets. Biochem. Biophys. Res. Commun. 273, 170–175. doi: 10.1006/bbrc.2000.2919
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, M., and Fernandez, H. L. (1998). The Alzheimer’s plaques, tangles and memory deficits may have a common origin. Part IV: can calpain act as α-secretase? Front. Biosci. 3:a66–a75.
Chen, M., and Fernandez, H. L. (1999). Ca2+ signaling down-regulation in aging and Alzheimer’s disease: Why is Ca2+ so difficult to measure? Cell Calcium 26, 149–153. doi: 10.1054/ceca.1999.0067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, M., and Fernandez, H. L. (2000). Revisiting Alzheimer’s disease from a new perspective: can “risk factors” play a key role? J. Alzheimers Dis. 2, 97–108.
Chen, M., and Fernandez, H. L. (2001a). Alzheimer’s disease revisited 25 years later: is it a “disease” or senile condition? Front. Biosci. 6:e30–e40.
Chen, M., and Fernandez, H. L. (2001b). Where do Alzheimer’s plaques and tangles come from? Aging-induced protein degradation inefficiency. Front. Biosci. 6:e1–e11. doi: 10.2741/Chen
Chen, M., and Fernandez, H. L. (2004). Stimulation of beta-amyloid precursor protein alpha-processing by phorbol ester involves calcium and calpain activation. Biochem. Biophys. Res. Commun. 316, 332–340. doi: 10.1016/j.bbrc.2004.02.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, M., and Fernandez, H. L. (2005). mu-Calpain is functionally required for alpha-processing of Alzheimer’s beta-amyloid precursor protein. Biochem. Biophys. Res. Commun. 330, 714–721. doi: 10.1016/j.bbrc.2005.03.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, M., Maleski, J. J., and Sawmiller, D. R. (2011a). Scientific truth or false hope? Understanding Alzheimer’s disease from an aging perspective. J. Alzheimers Dis. 24, 3–10.
Chen, M., Nguyen, H. T., and Sawmiller, D. R. (2011b). What to look for beyond “pathogenic” factors in senile dementia? A functional deficiency of Ca2+ signaling. J. Alzheimers Dis. 27, 3–10. doi: 10.3233/JAD-2011-111142
Chung, S. H. (2009). Aberrant phosphorylation in the pathogenesis of Alzheimer’s disease. BMB Rep. 42, 467–474. doi: 10.5483/BMBRep.2009.42.8.467
Citron, M., Westaway, D., Xia, W., Carlson, G., Diehl, T., Levesque, G.,et al. (1997). Mutant presenilins of Alzheimer’s disease increase production of 43-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 3, 67–72. doi: 10.1038/nm0197-67
Cuerrier, D., Moldoveanu, T., and Davies, P. L. (2005). Determination of peptide substrate specificity for mu-calpain by a peptide library-based approach: the importance of primed side interactions. J. Biol. Chem. 280, 40632–40641. doi: 10.1074/jbc.M506870200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dreses-Werringloer, U., Lambert, J. C., Vingtdeux, V., Zhao, H., Vais, H., Siebert, A.,et al. (2008). A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 133, 1149–1161. doi: 10.1016/j.cell.2008.05.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Efthimiopoulos, S., Vassilacopoulou, D., Ripellino, J. A., Tezapsidis, N., and Robakis, N. K. (1996). Cholinergic agonists stimulate secretion of soluble full-length amyloid precursor protein in neuroendocrine cells. Proc. Natl. Acad. Sci. U.S.A. 12, 8046–8050. doi: 10.1073/pnas.93.15.8046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fasulo, L., Ugolini, G., Visintin, M., Bradbury, A., Brancolini, C., Verzillo, V.,et al. (2000). The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J. Neurochem. 75, 624–633. doi: 10.1046/j.1471-4159.2000.0750624.x
Ferreira, A. (2012). Calpain dysregulation in Alzheimer’s disease. ISRN Biochem. 2012:728571. doi: 10.5402/2012/728571
Feuillette, S., Deramecourt, V., Laquerriere, A., Duyckaerts, C., Delisle, M. B., Maurage, C. A.,et al. (2010). Filamin-A and Myosin VI colocalize with fibrillary Tau protein in Alzheimer’s disease and FTDP-17 brains. Brain Res. 1345, 182–189. doi: 10.1016/j.brainres.2010.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fisher, A. (2012). Cholinergic modulation of amyloid precursor protein processing with emphasis on M1 muscarinic receptor: perspectives and challenges in treatment of Alzheimer’s disease. J. Neurochem. 120(Suppl. 1), 22–33. doi: 10.1111/j.1471-4159.2011.07507.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Friedrich, P., and Bozóky, Z. (2005). Digestive versus regulatory proteases: on calpain action in vivo. Biol. Chem. 386, 609–612. doi: 10.1515/BC.2005.071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, J., Yin, D., Yao, Y., Williams, T. D., and Squier, T. C. (1998). Progressive decline in the ability of calmodulin isolated from aged brain to activate the plasma membrane Ca-ATPase. Biochemistry 37, 9536–9548. doi: 10.1021/bi9803877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gasparini, L., Racchi, M., Benussi, L., Curti, D., Binetti, G., Bianchetti, A.,et al. (1997). Effect of energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neurosci. Lett. 231, 113–117. doi: 10.1016/S0304-3940(97)00536-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goll, D. E., Thompson, V. F., Li, H., Wei, W., and Cong, J. (2003). The calpain system. Physiol. Rev. 83, 731–801.
Griffin, W. S. T., Stanley, L. C., Ling, C., White, C. L. III, and Araoz, C. (1989). Brain interleukin-1 and S100 immunoreactivity elevated in Down syndrome and Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 86, 7611–7615. doi: 10.1073/pnas.86.19.7611
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guillozet-Bongaarts, A. L., Garcia-Sierra, F., Reynolds, M. R., Horowitz, P. M., Fu, Y., Wang, T.,et al. (2005). Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol. Aging 26, 1015–1022. doi: 10.1016/j.neurobiolaging.2004.09.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guttmann, R. P., Elce, J. S., Bell, P. D., Isbell, J. C., and Johnson, G. V. (1997). Oxidation inhibits substrate proteolysis by calpain I but not autolysis. J. Biol. Chem. 272, 2005–2012. doi: 10.1074/jbc.272.3.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haass, C., and Selkoe, D. J. (1993). Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 75, 1039–1042. doi: 10.1016/0092-8674(93)90312-E
Heckbert, S. R., longstreth, W. T. Jr., Psaty, B. M., Murros, K. E., Smith, N. L., Newman, A. B.,et al. (1997). The association of antihypertensive agents with MRI white matter findings and with modified mini-mental state examination in older adults. J. Am. Geriatr. Soc. 45, 1423–1433.
Herrup, K. (2010). Reimagining Alzheimer’s disease – an age-based hypothesis. J. Neurosci. 30, 16755–16762. doi: 10.1523/JNEUROSCI.4521-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoey, S. E., Williams, R. J., and Perkinton, M. S. (2009). Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J. Neurosci. 29, 4442–4460. doi: 10.1523/JNEUROSCI.6017-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Höglinger, G. U., Lannuzel, A., Khondiker, M. E., Michel, P. P., Duyckaerts, C., Féger, J.,et al. (2005). The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J. Neurochem. 95, 930–939. doi: 10.1111/j.1471-4159.2005.03493.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holtzman, D. M., Mandelkow, E., and Selkoe, D. J. (2012). Alzheimer disease in 2020. Cold Spring Harb. Perspect. Med. 2:a011585. doi: 10.1101/cshperspect.a011585
Hoyer, A., Bardenheuer, H. J., Martin, E., and Plaschke, K. (2005). Amyloid precursor protein (APP) and its derivatives change after cellular energy depletion. An in vitro-study. J. Neural Transm. 112, 239–253. doi: 10.1007/s00702-004-0176-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hwang, E. M., Kim, S. K., Sohn, J. H., Lee, J. Y., Kim, Y., Kim, Y. S.,et al. (2006). Furin is an endogenous regulator of alpha-secretase associated APP processing. Biochem. Biophys. Res. Commun. 349, 654–659. doi: 10.1016/j.bbrc.2006.08.077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ito, E., Oka, K., Etcheberrigaray, R., Nelson, T. J., McPhie, D. L., Tofel-Grehl, B.,et al. (1994). Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 91, 534–538. doi: 10.1073/pnas.91.2.534
Johnson, G. V., Greenwood, J. A., Costello, A. C., and Troncoso, J. C. (1991). The regulatory role of calmodulin in the proteolysis of individual neurofilament proteins by calpain. Neurochem. Res. 16, 869–873. doi: 10.1007/BF00965535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jolly-Tornetta, C., Gao, Z. Y., Lee, V. M., and Wolf, B. A. (1998). Regulation of amyloid precursor protein secretion by glutamate receptors in human Ntera 2 neurons. J. Biol. Chem. 273, 14015–14021. doi: 10.1074/jbc.273.22.14015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karch, C. M., Jeng, A. T., and Goate, A. M (2013). Calcium phosphatase calcineurin influences tau metabolism. Neurobiol. Aging 34, 374–386. doi: 10.1016/j.neurobiolaging.2012.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kayyali, U. S., Zhang, W., Yee, A. G., Seidman, J. G., and Potter, H. (1997). Cytoskeletal changes in the brains of mice lacking calcineurin A alpha. J. Neurochem. 68, 1668–1678. doi: 10.1046/j.1471-4159.1997.68041668.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khachaturian, Z. S. (1994). Calcium hypothesis of Alzheimer’s disease and brain ageing. Ann. N. Y. Acad. Sci. 747, 1–11. doi: 10.1111/j.1749-6632.1994.tb44398.x
Kikkawa, U., Takai, Y., Minakuchi, R., Inohara, S., and Nishizuka, Y. (1982). Calcium-activated, phospholipid-dependent protein kinase from rat brain. Subcellular distribution, purification, and properties. J. Biol. Chem. 257, 13341–13348.
Kim, S. H., Leem, J. Y., Lah, J. J., Slunt, H. H., Levey, A. I., Thinakaran, G.,et al. (2001). Multiple effects of aspartate mutant presenilin 1 on the processing and trafficking of amyloid precursor protein. J. Biol. Chem. 276, 43343–43350. doi: 10.1074/jbc.M108245200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kishimoto, A., Mikawa, K., Hashimoto, K., Yasuda, I., Tanaka, S., Tominaga, M.,et al. (1989). Limited proteolysis of protein kinase C subspecies by calcium-dependent neutral protease (calpain). J. Biol. Chem. 264, 4088–4092.
Korczyn, A. D. (2012). Why have we failed to cure Alzheimer’s disease? J. Alzheimers Dis. 29, 275–282.
Kyratzi, E., and Efthimiopoulos, S. (2014). Calcium regulates the interaction of amyloid precursor protein with Homer3 protein. Neurobiol. Aging 35, 2053–2063. doi: 10.1016/j.neurobiolaging.2014.03.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lammich, S., Kojro, H., Postina, R., Gilbert, S., Pfeiffer, R., Jasionowski, M.,et al. (1999). Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3927. doi: 10.1073/pnas.96.7.3922
Lannert, H., and Hoyer, S. (1998). Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav. Neurosci. 112, 1199–1208. doi: 10.1037/0735-7044.112.5.1199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lichtenthaler, S. F. (2011). Alpha-secretase in Alzheimer’s disease: molecular identity, regulation and therapeutic potential. J. Neurochem. 116, 10–21. doi: 10.1111/j.1471-4159.2010.07081.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lopez-Perez, E., Seidah, N. G., and Checler, F. (1999). Proprotein convertase activity contributes to the processing of the Alzheimer’s beta-amyloid precursor protein in human cells: evidence for a role of the prohormone convertase PC7 in the constitutive alpha-secretase pathway. J. Neurochem. 73, 2056–2062.
Manya, H., Inomata, M., Fujimori, T., Dohmae, N., Sato, Y., Takio, K.,et al. (2002). Klotho protein deficiency leads to overactivation of mu-calpain. J. Biol. Chem. 277, 35503–35508. doi: 10.1074/jbc.M206033200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marcello, E., Gardoni, F., Mauceri, D., Romorini, S., Jeromin, A., Epis, R.,et al. (2007). Synapse-associated protein-97 mediates alpha-secretase ADAM10 trafficking and promotes its activity. J. Neurosci. 27, 1682–1691. doi: 10.1523/JNEUROSCI.3439-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maruyama, K., Kametani, F., Usami, M., Yamao-Harigaya, W., and Tanaka, K. (1991). “Secretase,” Alzheimer amyloid protein precursor secreting enzyme is not sequence-specific. Biochem. Biophys. Res. Commun. 179, 1670–1676. doi: 10.1016/0006-291X(91)91767-7
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
Mathews, P. M., Jiang, Y., Schmidt, S. D., Grbovic, O. M., Mercken, M., and Nixon, R. A. (2002). Calpain activity regulates the cell surface distribution of amyloid precursor protein. Inhibition of calpains enhances endosomal generation of beta-cleaved C-terminal APP fragments. J. Biol. Chem. 277, 36415–36424. doi: 10.1074/jbc.M205208200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maxwell, C. J., Hogan, D. B., and Ebly, E. M. (1999). Calcium-channel blockers and cognitive function in elderly people: results from the Canadian study of health and aging. CMAJ 161, 501–506.
McDaniel, M. A., Maier, S. F., and Einstein, G. O. (2003). “Brain-specific” nutrients: a memory cure? Nutrition 19, 957–975. doi: 10.1016/S0899-9007(03)00024-8
Moreira, P. I., Carvalho, C., Zhu, X., Smith, M. A., and Perry, G., (2010). Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 1802, 2–10. doi: 10.1016/j.bbadis.2009.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moriguchi, S., Han, F., Nakagawasai, O., Tadano, T., and Fukunaga, K. (2006). Decreased calcium/calmodulin-dependent protein kinase II and protein kinase C activities mediate impairment of hippocampal long-term potentiation in the olfactory bulbectomized mice. J. Neurochem. 97, 22–29. doi: 10.1111/j.1471-4159.2006.03710.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Newhouse, P., Kellar, K., Aisen, P., White, H., Wesnes, K., Coderre, E.,et al. (2012). Nicotine treatment of mild cognitive impairment: a 6-month double-blind pilot clinical trial. Neurology 78, 91–101. doi: 10.1212/WNL.0b013e31823efcbb
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nguyen, H. T., and Chen, M. (2014). High-energy compounds mobilize intracellular Ca2+ and activate protease calpain in cultured cells: is calpain an energy-dependent protease? Brain Res. Bull. 102, 9–14. doi: 10.1016/j.brainresbull.2014.01.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nguyen, H. T., Sawmiller, D. R., Markov, O., and Chen, M. (2013). Elevated [Ca2+]i levels occur with decreased calpain activity in aged human fibroblasts and their reversal by high-energy compounds: new paradigm for Alzheimer’s disease prevention. J. Alzheimers Dis. 37, 835–848.
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. doi: 10.1038/nm.3232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pahlavani, M. A., and Vargas, D. M. (1999). Age-related decline in activation of calcium/calmodulin-dependent phosphatase calcineurin and kinase CaMK-IV in rat T cells. Mech. Ageing Dev. 112, 59–74. doi: 10.1016/S0047-6374(99)00077-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pascale, A., Amadio, M., Govoni, S., and Battaini, F. (2007). The aging brain, a key target for the future: the protein kinase C involvement. Pharmacol. Res. 55, 560–569. doi: 10.1016/j.phrs.2007.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pettegrew, J. W., Levine, J., and McClure, R. J. (2000). Acetyl-L-carnitine physical-chemical, metabolic, and therapeutic properties: relevance for its mode of action in Alzheimer’s disease and geriatric depression. Mol. Psychiatry 5, 616–632. doi: 10.1038/sj.mp.4000805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Puig, B., Ferrer, I., Ludueña, R. F., and Avila, J. (2005). BetaII-tubulin and phospho-tau aggregates in Alzheimer’s disease and Pick’s disease. J. Alzheimers Dis. 7, 213–220.
Querfurth, H. W., and Selkoe, D. J. (1994). Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry 33, 4550–4561. doi: 10.1021/bi00181a016
Rao, M. V., Mohan, P. S., Peterhoff, C. M., Yang, D. S., Schmidt, S. D., Stavrides, P. H.,et al. (2008). Marked calpastatin (CAST) depletion in Alzheimer’s disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. J. Neurosci. 28, 12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rice, R. A., Berchtold, N. C., Cotman, C. W., and Green, K. N. (2014). Age-related downregulation of the CaV3.1 T-type calcium channel as a mediator of amyloid beta production. Neurobiol. Aging 35, 1002–1011. doi: 10.1016/j.neurobiolaging.2013.10.090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ritchie, K., Carrière, I., de Mendonca, A., Portet, F., Dartigues, J. F., Rouaud, O.,et al. (2007). The neuroprotective effects of caffeine: a prospective population study (the Three City Study). Neurology 69, 536–545. doi: 10.1212/01.wnl.0000266670.35219.0c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saito, K., Elce, J. S., Hamos, J. E., and Nixon, R. A. (1993). Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 90, 2628–2632. doi: 10.1073/pnas.90.7.2628
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sawmiller, D. R., Nguyen, H. T., Markov, O., and Chen, M. (2012). High-energy compounds promote physiological processing of Alzheimer’s amyloid-β precursor protein and boost cell survival in culture. J. Neurochem. 123, 525–531. doi: 10.1111/j.1471-4159.2012.07923.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Selkoe, D. J. (2005). Defining molecular targets to prevent Alzheimer’s disease. Arch. Neurol. 62, 192–195. doi: 10.1001/archneur.62.2.192
Sisodia, S. S. (1992). Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl. Acad. Sci. U.S.A. 89, 6075–6079. doi: 10.1073/pnas.89.13.6075
Slack, B. E., Breu, J., Muchnicki, L., and Wurtman, R. J. (1997). Rapid stimulation of amyloid precursor protein release by epidermal growth factor: role of protein kinase C. Biochem. J. 327, 245–249.
Sloane, J. A., Hinman, J. D., Lubonia, M., Hollander, W., and Abraham, C. R. (2003). Age-dependent myelin degeneration and proteolysis of oligodendrocyte proteins is associated with the activation of calpain-1 in the rhesus monkey. J. Neurochem. 84, 157–168. doi: 10.1046/j.1471-4159.2003.01541.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sperling, R. A., Jack, C. R. Jr., and Aisen, P. S. (2011). Testing the right target and right drug at the right stage. Sci. Transl. Med. 3:111. doi: 10.1126/scitranslmed.3002609
Suh, J., Lyckman, A., Wang, L., Eckman, E. A., and Guénette, S. Y. (2011). FE65 proteins regulate NMDA receptor activation-induced amyloid precursor protein processing. J. Neurochem. 119, 377–388. doi: 10.1111/j.1471-4159.2011.07419.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, S., Zhang, H., Liu, J., Popugaeva, E., Xu, N. J., Feske, S.,et al. (2014). Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82, 79–93. doi: 10.1016/j.neuron.2014.02.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Swerdlow, R. H. (2007). Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging? Neurobiol. Aging 28, 1465–1480. doi: 10.1016/j.neurobiolaging.2006.06.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Swerdlow, R. H. (2012). Mitochondria and cell bioenergetics: increasingly recognizedcomponents and a possible etiologic cause of Alzheimer’s disease. Antioxid. Redox Signal. 16, 1434–1455. doi: 10.1089/ars.2011.4149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trojanowski, J. Q., and Lee, V. M. (1995). Phosphorylation of paired helical filament tau in Alzheimer’s disease neurofibrillary lesions: focusing on phosphatases. FASEB J. 9, 1570–1576.
Turpeinen, H., Ortutay, Z., and Pesu, M. (2013). Genetics of the first seven proprotein convertase enzymes in health and disease. Curr. Genomics 14, 453–467. doi: 10.2174/1389202911314050010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P.,et al. (1999). Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Vingtdeux, V., Giliberto, L., Zhao, H., Chandakkar, P., Wu, Q., Simon, J. E.,et al. (2010). AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 285, 9100–9113. doi: 10.1074/jbc.M109.060061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vingtdeux, V., and Marambaud, P. (2012). Identification and biology of α-secretase. J. Neurochem. 120(Suppl. 1), 34–45. doi: 10.1111/j.1471-4159.2011.07477.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagner, G., Icks, A., Abholz, H. H., Schröder-Bernhardi, D., Rathmann, W., and Kostev, K. (2012). Antihypertensive treatment and risk of dementia: a retrospective database study. Int. J. Clin. Pharmacol. Ther. 50, 195–201. doi: 10.5414/CP201284
Walsh, C., Barrow, S., Voronina, S., Chvanov, M., Petersen, O. H., and Tepikin, A. (2009). Modulation of calcium signalling by mitochondria. Biochim. Biophys. Acta 1787, 1374–1382. doi: 10.1016/j.bbabio.2009.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, X. X., Jeromin, A., and Jeromin, A. (2012). Spectrin breakdown products (SBDPs) as potential biomarkers for neurodegenerative diseases. Curr. Transl. Geriatr. Exp. Gerontol. Rep. 1, 85–93. doi: 10.1007/s13670-012-0009-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, L. S., and Ksiezak-Reding, H. (1995). Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. Eur. J. Biochem. 233, 9–17. doi: 10.1111/j.1432-1033.1995.009_1.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yankner, B. A., Lu, T., and Loerch, P. (2008). The aging brain. Annu. Rev. Pathol. 3, 41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044
Yu, D. Y., Luo, J., Bu, F., Song, G. J., Zhang, L. Q., and Wei, Q. (2006). Inhibition of calcineurin by infusion of CsA causes hyperphosphorylation of tau and is accompanied by abnormal behavior in mice. Biol. Chem. 387, 977–983. doi: 10.1515/BC.2006.121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zeiger, W., Vetrivel, K. S., Buggia-Prévot, V., Nguyen, P. D., Wagner, S. L.,et al. (2013). Ca2+ influx through store-operated Ca2+ channels reduces Alzheimer disease β-amyloid peptide secretion. J. Biol. Chem. 288, 26955–26966. doi: 10.1074/jbc.M113.473355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, G., Mao, G., Tan, J., Dong, Y., Cui, M. Z., Kim, S. H.,et al. (2004). Identification of a new presenilin-dependent zeta-cleavage site within the transmembrane domain of amyloid precursor protein. J. Biol. Chem. 279, 50647–50650. doi: 10.1074/jbc.C400473200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Alzheimer’s disease, energy, calcium, amyloid, tau
Citation: Chen M and Nguyen HT (2014) Our “energy-Ca2+ signaling deficits” hypothesis and its explanatory potential for key features of Alzheimer’s disease. Front. Aging Neurosci. 6:329. doi: 10.3389/fnagi.2014.00329
Received: 24 July 2014; Accepted: 10 November 2014;
Published online: 03 December 2014.
Edited by:
Rodrigo Orlando Kuljiš, Zdrav Mozak Limitada, ChileReviewed by:
Changiz Geula, Northwestern University, USAAnna Maria Colangelo, University of Milano-Bicocca, Italy
Copyright © 2014 Chen and Nguyen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Chen, Aging Research Laboratory, Research and Development Service, Bay Pines Veterans Affairs Medical Center, 10000 Bay Pines Boulevard R & D/151, Bay Pines, FL 33744, USA e-mail: ming.chen@va.gov