Clearance of amyloid β-protein and its role in the spreading of Alzheimer's disease pathology
 Dietmar R. Thal
Dietmar R. Thal- Laboratory of Neuropathology – Institute of Pathology, Center for Biomedical Research, University of Ulm, Ulm, Germany
Amyloid β-protein (Aβ) containing amyloid plaques and abnormal phosphorylated τ-protein containing neurofibrillary tangles (NFTs) are hallmark lesions of Alzheimer's disease. Both Aβ plaques and NFTs show hierarchical patterns in which the areas of the brain are subsequently affected by Aβ plaques and NFTs, respectively (Braak and Braak, 1991; Thal et al., 2002). Aβ plaques start to develop in the neocortex (phase 1) and spread from there into allocortical regions (phase 2), diencephalon, basal forebrain and striatum (phase 3), midbrain and medulla oblongata (phase 4), and finally into the pons and the cerebellum (phase 5) (Thal et al., 2002). The first NFTs in the brain hemispheres are found in the transentorhinal cortex (stage I), then in the entorhinal cortex (stage II), the hippocampus (stage III), the temporal cortex (stage IV), further neocortical areas except the primary fields (stage V), and, finally, also in primary cortical areas, such as the primary visual cortex (stage VI) (Braak and Braak, 1991). Axonal connections between subsequently affected brain regions suggest that AD pathology spreads along neuronal pathways (Thal et al., 2002; Braak and Del Tredici, 2011).
Insufficient clearance of Aβ has been considered to play an essential role in the pathogenesis of AD. Clearance mechanisms that contribute to Aβ elimination from brain are cellular enzymatic proteolysis in glial cells, neurons or in the extracellular space (Qiu et al., 1998; Yamaguchi et al., 1998; Iwata et al., 2000; Thal et al., 2000; Farris et al., 2003), transport through the blood-brain barrier (Shibata et al., 2000; Ito et al., 2007), and perivascular drainage (Weller et al., 2008) (Figure 1A).
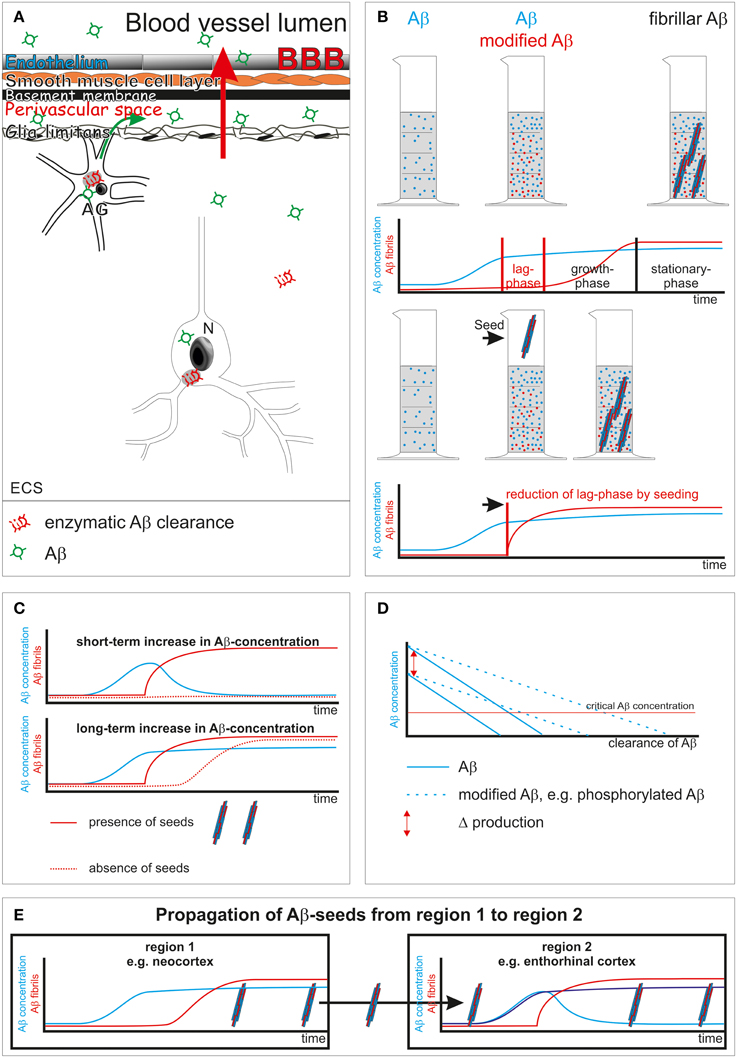
Figure 1. Schematic representation of Aβ clearance and propagation. (A) Aβ clearance mechanisms: enzymatic clearance within neurons (N) and glial cells [here shown in the example is an astrocyte (AG)] or in the extracellular space (ECS) (Qiu et al., 1998; Yamaguchi et al., 1998; Iwata et al., 2000; Thal et al., 2000; Farris et al., 2003); transport through the blood-brain barrier (BBB) into the blood (red arrow) (Shibata et al., 2000; Ito et al., 2007); drainage into the perivascular space (green arrow) (Weller et al., 2008). (B) Aβ aggregation into fibrils shows a lag-phase that is reduced by the presence of seeds (Thal et al., 2015). (C) Initiation of Aβ fibril formation in the presence of seeds starts immediately after a critical concentration is passed even for a short period of time. In the absence of seeds a longer increase of Aβ concentration may be necessary to pass the lag-phase for initiating Aβ aggregation. (D) Influence of clearance rate, Aβ-production and its composition [presence of modified forms of Aβ that are cleared less effectively than normal Aβ (Russo et al., 2002; Kumar et al., 2012)] on a potentially critical concentration of Aβ for disease progression. N-terminal truncated, pyroglutamate-modified Aβ is less soluble than non-modified full-length Aβ (Schlenzig et al., 2009). In other words, this modification of Aβ modified its critical concentration for aggregation. (E) Propagation of AD pathology, e.g., Aβ plaques, takes place when seeds of Aβ are transported to a second primarily non-involved brain region. As soon as the concentration of Aβ passes a critical level Aβ aggregation and fibril formation takes place, leading to the deposition of Aβ plaques in the secondarily affected brain region.
Here, I will discuss the potential impact of impaired Aβ clearance on propagation mechanisms for Aβ and τ.
Biophysical and Biochemical Prerequisites for Aβ and τ Protein Aggregation
Biophysically, protein aggregation takes place once a critical concentration of proteins has been passed. Fibril formation sets in after a concentration-dependent lag-phase, i.e., the time interval between passing a critical concentration and forming fibrils (Figure 1B) (Chirita et al., 2005). Within the lag-phase, assembly of proteins into non-fibrillar intermediates, i.e., oligomers of all sizes, precedes fibril formation (Thal et al., 2015). Proteins in general differ in their capability to aggregate, and their assembly can be further modulated by chaperones (Gething and Sambrook, 1992). Posttranslational modifications of proteins, such as N-terminal truncation and pyroglutamate formation, phosphorylation, or glycation increase the tendency of Aβ or τ to form aggregates (Necula and Kuret, 2004; Schlenzig et al., 2009; Kumar et al., 2011). Seeds of preaggregated fibrils can reduce the lag-phase dramatically and trigger immediate aggregation (Figure 1B).
In other words, a prerequisite for Aβ and/or τ protein aggregation is either a sufficient concentration of Aβ or τ over a period of time long enough to pass the respective lag-phase or the presence of preaggregated seeds to start aggregation as soon as the critical protein concentration has been reached.
Aβ Clearance Modulates Concentration and Onset of Fibril Formation of Aβ
Sufficient Aβ clearance is required to prevent an increase of the Aβ concentration. In the absence of preaggregated Aβ seeds a short-term increase of Aβ concentration not exceeding the lag-phase may not start the aggregation process. Accordingly, a “long-term” elevation of Aβ concentration is required to initiate the process of Aβ aggregation under normal conditions (Figures 1B,C). As soon as preaggregated Aβ seeds are present aggregation of Aβ will be initiated once the critical Aβ concentration has been passed as demonstrated in animal models for Aβ deposition (Meyer-Luehmann et al., 2006) (Figure 1C).
Impairment of Aβ Clearance by Modified Forms and/or Aβ Intermediates
Posttranslationally modified forms of Aβ are cleared less efficiently from the brain as shown for N-terminal truncated and pyroglutamate-modified Aβ and phosphorylated Aβ (Russo et al., 2002; Kumar et al., 2012) (Figure 1D). Oligomeric Aβ intermediates are stable and quite resistant to degeneration (Viola and Klein, 2015). Moreover, Aβ oligomers alter proteasomal clearance (Cecarini et al., 2012). Accordingly, it is tempting to speculate that even low amounts of Aβ intermediates and fibrils as well as posttranslational modifications of Aβ foster disease progression not only by acting as seeds but also by their property of impairing physiological Aβ clearance and, thereby, increasing Aβ concentration.
Spreading of Aβ Pathology
Spreading of Aβ pathology means that Aβ aggregation and deposition already took place at least in the neocortex and a second region becomes involved in this process. Preaggregated neocortical Aβ may be transported into secondarily affected brain regions by glial cells or neurons or by diffusion (Guo and Lee, 2014; Thal et al., 2015). In the event that seeds prevail in a given brain region Aβ aggregation will be initiated as soon as a critical concentration has been passed (Figure 1E). Given the prevalence of cortical Aβ plaques (i.e., aggregated Aβ) in most elderly individuals (Braak et al., 2011) it is tempting to speculate that progression of Aβ pathology into further brain regions can be triggered by insufficient local clearance and subsequently increased Aβ levels and by seeding its aggregation. If so, continuously lowering Aβ concentration to avoid even short-term increases might be effective in preventing propagation of Aβ pathology, similar to the inactivation of Aβ seeds as shown for continuous Aβ antibody treatment in Aβ-producing mice (Paganetti et al., 2013).
Aβ and τ
Animal experiments in τ-transgenic mice have demonstrated that injecting Aβ or crossbreeding these animals with Aβ-producing amyloid precursor protein transgenic mice accelerates and increases τ-pathology (Gotz et al., 2001; Lewis et al., 2001). Moreover, anti-Aβ antibody treatment reduced τ-pathology in an Aβ and τ-pathology producing mouse model (Oddo et al., 2004). As such, it is tempting to speculate that there is cross-seeding of τ-pathology by Aβ aggregates in vivo similar as in vitro (Lasagna-Reeves et al., 2010). Arguments against relevant cross-seeding of Aβ and τ in the AD brain may be (a) that τ aggregates are intracellular aggregates while Aβ plaques are extracellular protein aggregates, and (b) Aβ plaques develop first in the neocortex whereas NFTs are found in this part of the brain only in advanced stages of AD. However, Aβ also occurs intracellularly (Gouras et al., 2000) and τ aggregates are transported through the extracellular space (Kfoury et al., 2012), indicating that an interaction of τ and Aβ may be possible either intra- or extracellularly. Moreover, as discussed for Aβ, a sufficient protein concentration is essential for the initiation of protein aggregation. Accordingly, cross-seeding of τ by Aβ cannot take place if there is not enough aggregation-prone τ protein even in the presence of huge amounts of Aβ seeds. Such a constellation may apply for neocortical brain regions in early stages of AD. Therefore, it seems to be likely that cross-seeding of Aβ and τ contributes to the development of AD and may be modulated by changing the clearance of Aβ. Cross-seeding of Aβ and τ does not exclude the independent aggregation of τ, as τ-aggregates have been shown to trigger the initiation of τ-pathology (Clavaguera et al., 2009).
Conclusion
The hypothesis that insufficient Aβ clearance contributes to the development of AD does not contradict a major role of preaggregated Aβ and/or τ seeds in the propagation of the disease. Moreover, improving Aβ clearance, e.g., by enhancing its enzymatic degradation or vaccination strategies, may be capable of slowing down disease propagation given the relevance of Aβ as a substrate for the protein aggregation process in AD.
Conflict of Interest Statement
The author discloses the following potential conflicts of interest: DRT received consultancies from Covance Laboratories (UK) and GE-Healthcare (UK), a speaker honorarium from GE-Healthcare (UK) and collaborated with Novartis Pharma Basel (Switzerland).
Acknowledgments
The author received support for his scientific projects by DFG-grant TH624/6-1 and Alzheimer Forschung Initiative grants #10810, 13803.
References
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Braak, H., and Del Tredici, K. (2011). Alzheimer's pathogenesis: is there neuron-to-neuron propagation? Acta Neuropathol. 121, 589–595. doi: 10.1007/s00401-011-0825-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathological process in Alzheimer's disease: age categories 1 year to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/NEN.0b013e318232a379
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cecarini, V., Bonfili, L., Cuccioloni, M., Mozzicafreddo, M., Rossi, G., Buizza, L., et al. (2012). Crosstalk between the ubiquitin-proteasome system and autophagy in a human cellular model of Alzheimer's disease. Biochim. Biophys. Acta 1822, 1741–1751. doi: 10.1016/j.bbadis.2012.07.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chirita, C. N., Congdon, E. E., Yin, H., and Kuret, J. (2005). Triggers of full-length tau aggregation: a role for partially folded intermediates. Biochemistry 44, 5862–5872. doi: 10.1021/bi0500123
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. doi: 10.1038/ncb1901
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4162–4167. doi: 10.1073/pnas.0230450100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gething, M. J., and Sambrook, J. (1992). Protein folding in the cell. Nature 355, 33–45. doi: 10.1038/355033a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gotz, J., Chen, F., Van Dorpe, J., and Nitsch, R. M. (2001). Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293, 1491–1495. doi: 10.1126/science.1062097
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gouras, G. K., Tsai, J., Naslund, J., Vincent, B., Edgar, M., Checler, F., et al. (2000). Intraneuronal Abeta42 accumulation in human brain. Am. J. Pathol. 156, 15–20. doi: 10.1016/S0002-9440(10)64700-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, J. L., and Lee, V. M. (2014). Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 20, 130–138. doi: 10.1038/nm.3457
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ito, S., Ohtsuki, S., Kamiie, J., Nezu, Y., and Terasaki, T. (2007). Cerebral clearance of human amyloid-beta peptide (1-40) across the blood-brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J. Neurochem. 103, 2482–2490. doi: 10.1111/j.1471-4159.2007.04938.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iwata, N., Tsubuki, S., Takaki, Y., Watanabe, K., Sekiguchi, M., Hosoki, E., et al. (2000). Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. 6, 143–150. doi: 10.1038/77399
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kfoury, N., Holmes, B. B., Jiang, H., Holtzman, D. M., and Diamond, M. I. (2012). Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 287, 19440–19451. doi: 10.1074/jbc.M112.346072
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kumar, S., Rezaei-Ghaleh, N., Terwel, D., Thal, D. R., Richard, M., Hoch, M., et al. (2011). Extracellular phosphorylation of the amyloid beta-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer's disease. EMBO J. 30, 2255–2265. doi: 10.1038/emboj.2011.138
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kumar, S., Singh, S., Hinze, D., Josten, M., Sahl, H. G., Siepmann, M., et al. (2012). Phosphorylation of amyloid-beta peptide at serine 8 attenuates its clearance via insulin-degrading and angiotensin-converting enzymes. J. Biol. Chem. 287, 8641–8651. doi: 10.1074/jbc.M111.279133
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Guerrero-Muoz, M. J., Jackson, G. R., and Kayed, R. (2010). Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041. doi: 10.1021/bi1016233
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lewis, J., Dickson, D. W., Lin, W. L., Chisholm, L., Corral, A., Jones, G., et al. (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491. doi: 10.1126/science.1058189
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meyer-Luehmann, M., Coomaraswamy, J., Bolmont, T., Kaeser, S., Schaefer, C., Kilger, E., et al. (2006). Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 313, 1781–1784. doi: 10.1126/science.1131864
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Necula, M., and Kuret, J. (2004). Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro. J. Biol. Chem. 279, 49694–49703. doi: 10.1074/jbc.M405527200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oddo, S., Billings, L., Kesslak, J. P., Cribbs, D. H., and Laferla, F. M. (2004). Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43, 321–332. doi: 10.1016/j.neuron.2004.07.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paganetti, P., Reichwald, J., Bleckmann, D., Abramowski, D., Ammaturo, D., Barske, C., et al. (2013). Transgenic expression of beta1 antibody in brain neurons impairs age-dependent amyloid deposition in APP23 mice. Neurobiol. Aging 34, 2866–2878. doi: 10.1016/j.neurobiolaging.2013.06.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qiu, W. Q., Walsh, D. M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M. B., et al. (1998). Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J. Biol. Chem. 273, 32730–32738. doi: 10.1074/jbc.273.49.32730
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Russo, C., Violani, E., Salis, S., Venezia, V., Dolcini, V., Damonte, G., et al. (2002). Pyroglutamate-modified amyloid beta-peptides—AbetaN3(pE)—strongly affect cultured neuron and astrocyte survival. J. Neurochem. 82, 1480–1489. doi: 10.1046/j.1471-4159.2002.01107.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schlenzig, D., Manhart, S., Cinar, Y., Kleinschmidt, M., Hause, G., Willbold, D., et al. (2009). Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry 48, 7072–7078. doi: 10.1021/bi900818a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499. doi: 10.1172/JCI10498
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thal, D. R., Rüb, U., Orantes, M., and Braak, H. (2002). Phases of Abeta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/WNL.58.12.1791
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thal, D. R., Schultz, C., Dehghani, F., Yamaguchi, H., Braak, H., and Braak, E. (2000). Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathol. 100, 608–617. doi: 10.1007/s004010000242
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thal, D. R., Walter, J., Saido, T. C., and Fändrich, M. (2015). Neuropathology and biochemistry of Abeta and its aggregates in Alzheimer's disease. Acta Neuropathol. 129, 167–182. doi: 10.1007/s00401-014-1375-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Viola, K. L., and Klein, W. L. (2015). Amyloid beta oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 129, 183–206. doi: 10.1007/s00401-015-1386-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weller, R. O., Subash, M., Preston, S. D., Mazanti, I., and Carare, R. O. (2008). Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol. 18, 253–266. doi: 10.1111/j.1750-3639.2008.00133.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamaguchi, H., Sugihara, S., Ogawa, A., Saido, T. C., and Ihara, Y. (1998). Diffuse plaques associated with astroglial amyloid beta protein, possibly showing a disappearing stage of senile plaques. Acta Neuropathol. 95, 217–222. doi: 10.1007/s004010050790
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: amyloid β-protein, tau-protein, perivascular drainage, propagation of protein aggregates, cellular clearance
Citation: Thal DR (2015) Clearance of amyloid β-protein and its role in the spreading of Alzheimer's disease pathology Front. Aging Neurosci. 7:25. doi: 10.3389/fnagi.2015.00025
Received: 26 January 2015; Paper pending published: 17 February 2015;
Accepted: 22 February 2015; Published: 09 March 2015.
Edited by:
Roxana Octavia Carare, University of Southampton, UKReviewed by:
Lary C. Walker, Emory University, USACopyright © 2015 Thal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dietmar R. Thal, dietmar.thal@uni-ulm.de