Catecholamine-based treatment in AD patients: expectations and delusions
 Alessandro Stefani1,2*
Alessandro Stefani1,2*  Enrica Olivola1
Enrica Olivola1
 Claudio Liguori3
Claudio Liguori3
 Atticus H. Hainsworth4
Atticus H. Hainsworth4
 Valentina Saviozzi1
Valentina Saviozzi1
 Giacoma Angileri1
Giacoma Angileri1
 Vincenza D’Angelo1
Vincenza D’Angelo1
 Salvatore Galati5
Salvatore Galati5  Mariangela Pierantozzi1
Mariangela Pierantozzi1
- 1Department of System Medicine, Università di Roma Tor Vergata, Rome, Italy
- 2IRCCS Fondazione Santa Lucia, Rome, Italy
- 3Policlinico Tor Vergata, Neurofisiopatologia, Rome, Italy
- 4St George’s University of London, London, UK
- 5Neurocenter Svizzera Italiana, Lugano, Switzerland
In Alzheimer disease, the gap between excellence of diagnostics and efficacy of therapy is wide. Despite sophisticated imaging and biochemical markers, the efficacy of available therapeutic options is limited. Here we examine the possibility that assessment of endogenous catecholamine levels in cerebrospinal fluid (CSF) may fuel new therapeutic strategies. In reviewing the available literature, we consider the effects of levodopa, monoamine oxidase inhibitors, and noradrenaline (NE) modulators, showing disparate results. We present a preliminary assessment of CSF concentrations of dopamine (DA) and NE, determined by HPLC, in a small dementia cohort of either Alzheimer’s disease (AD) or frontotemporal dementia patients, compared to control subjects. Our data reveal detectable levels of DA, NE in CSF, though we found no significant alterations in the dementia population as a whole. AD patients exhibit a small impairment of the DA axis and a larger increase of NE concentration, likely to represent a compensatory mechanism. While waiting for preventive strategies, a pragmatic approach to AD may re-evaluate catecholamine modulation, possibly stratified to dementia subtypes, as part of the therapeutic armamentarium.
Introduction
In Alzheimer’s disease (AD), efficacious therapies are elusive. Currently approved molecules include acetylcholinesterase inhibitors (AChE-I, such as donepezil, rivastigmine, and galantamine) and the uncompetitive N-methyl-d-aspartate (NMDA) glutamate antagonist memantine. These all have demonstrated only weak effects on cognitive functions. Nevertheless, in the absence of evidence-based novel approaches, these drugs remain central to dementia care. To note, recent reports confirm their safe profile, even in Parkinson’s disease (PD) dementia complex (Emre et al., 2014).
In addition, some innovations have been proposed, including newly designed hybrids or alternative routes of administration (Bhavna et al., 2014; Thiratmatrakul et al., 2014). While some authors advocate a more extensive utilization of memantine, definitive data on the glutamatergic transmission involvement in AD are incomplete. Moreover, clinical results have indicated only temporarily limited or disappointing effects of memantine. In an ample series of mild to moderate AD, there were no significant differences in the groups receiving memantine alone or memantine plus alpha tocopherol (Dysken et al., 2014). Thus, the dictum remains that, unfortunately: “the more the disease progresses, the more the ineffectiveness of anti-dementia drugs emerges” (Esposito et al., 2013).
The modern diagnostic paradigm has fueled huge investments in strategies to immunize against the amyloid burden. But, as recently stated, even “BACE inhibitors might reveal as a watershed despite premises and hopes” (Yan and Vassar, 2014); and inhibitors of γ-secretase, including tarenflurbil or semagacestat, “were discontinued due to their lack of cognitive improvement or unacceptable side effects” (Mancuso et al., 2011).
That said, we are witnessing an impressive increase of our diagnostic capability. FDG- and amyloid-PET imaging (Lista et al., 2014) have improved the “early – and differential – diagnosis of dementia” (Perani et al., 2014). Cerebrospinal fluid (CSF) biomarkers that reflect the core pathology of AD (De Souza et al., 2014) appear to facilitate an etiological diagnosis even in the prodromal stages of the disorder.
On one hand, an early diagnosis might induce behavior change (e.g., abolition of risk factors, changes in life style). The identification of individuals at risk of developing dementia among people with subjective cognitive complaints or mild cognitive impairment would certainly influences social habits. On the other hand, such a diagnosis will create expectancies that may be difficult to fulfill.
Does the capability “to tackle initial steps of the deranged pathway decades before the clinical explosion” translate into better prognosis? Hampel et al. (2014) claims: “there is a mounting consensus that such disease-modifying compounds and/or interventions are more likely to be effectively administered as early as possible in the cascade of pathogenic processes preceding and underlying the clinical expression of AD.” Well, which molecules became available and were marketed in the last decade?
Several concomitant biochemical alterations appear to be more realistic targets. These include the malfunctioning insulin signaling (Dar et al., 2014; Wirz et al., 2014), the dysfunction of mitochondria-associated membranes, cerebrovascular changes with altered permeability (Diomedi and Stefani, 2014), and disturbed energy metabolism. More radically, some leaders in the functional neurosurgery world are proposing deep brain stimulation of anterior cingulate cortex (or the nucleus basalis of Meynert) (Hardenacke et al., 2013; Laxton and Lozano, 2013). This is not the appropriate context to discuss these initiatives (so far, limited to a few patients).
The central hypothesis governing this manuscript is that we might profitably investigate catecholaminergic transmission in AD patients.
It is already established that deficit in endogenous catechol pathways affects cortical plasticity. It was shown, in routine studies with paired pulse protocols, that levodopa and rotigotine might partially revert electrophysiological disturbances attributable to cholinergic deficit. Martorana and Koch recently suggested “the dopaminergic system may well be involved in the occurrence of cognitive decline, often being predictive of rapidly progressive forms of AD” (Koch et al., 2012; Martorana et al., 2013).
These recent statements have returned our attention to catecholamine-mediated functions in aging brain disease. In addition, we are aware of the putative key role of noradrenaline (NE) in shaping the so-called “cognitive reserve” (Robertson, 2013) and the well-known correlation of nigrostriatal impairment with dementia in movement disorders (McKeith et al., 2007; Tatsch and Poepper, 2013). These reflections inspired the current manuscript.
Catecholamine Therapy in AD
MAO Inhibitors
The therapeutic potential of monoamine oxidase inhibitors (MAOI) in AD has been suggested in light of their neuroprotective properties and augmenting effect on monoaminergic transmission (Klegeris and McGeer, 2000). In the early 90s, Agnoli et al. (1992) performed one of the first pilot studies, followed by several groups (Tolbert and Fuller, 1996; Freedman et al., 1998; Tariot et al., 1998) but results were quite inconsistent (Thomas, 2000).
A promising double-blind, randomized, multicenter trial, for example, provided some encouraging results (Sano et al., 1997, although the baseline score on MMSE was biased).
A 2003 Cochrane review on selegiline and AD (Birks and Flicker, 2003) admitted that, “despite its initial promise, and its role in the treatment of PD sufferers, selegiline for AD has been proved disappointing.” The authors concluded “no evidence of a significant adverse event profile” but also “no evidence of a clinically meaningful benefit for AD” (Birks and Flicker, 2003).
Recent experience is suggesting new potential avenues. We are now aware of the limitations hampering old trials, such as the methodological problems in complex disease like dementia, and the difficulty in interpreting results in the absence of reliable biomarkers (some of which are now available). In other words, it is now possible to pursue trials with: (a) small cohorts (enrolling specific disease subtype, avoiding the bias of non-specific inclusion of mixed dementia type), (b) correlation between biomarkers and cognitive scores – not dominated by mere analogic scoring (such as MMSE scale).
In addition, rasagiline, whose proteiform neuroprotective abilities are undisputed, acquired the potential status of a disease-modifying agent in PD (Hanagasi et al., 2011; Kupershmidt et al., 2012). Recent years are witnessing the possibility that these agents induce protective effects on cognitive performance not merely in PD, but also in normal brain aging.
Not surprisingly, the pipeline of several small companies is recently developing hybrids, combining propargylamine-derived molecules with AChE-I (Zheng et al., 2012; Lu et al., 2013).
This strategy is summarized by Bolea et al. (2013): the paradigm by the Food and Drug Administration-approved drugs based on the “one drug, one target” (donepezil, galantamine, and rivastigmine) might be rephrased into the “one drug, multiple targets” (hence, “a variety of hybrid compounds acting on very diverse targets”) (Schneider et al., 1993; Bolea et al., 2013).
Levodopa and Dopamine Agonists
A direct involvement of dopamine in AD was inferred by several pre-clinical, experimental contributions. Himeno presented striking data, showing that the DA agonist apomorphine, in a transgenic murine AD model, not only accelerated amyloid degradation and protected hippocampal neurons from oxidative stress, but also “restored the memory dysfunctions and improved the major pathological hallmarks” (Himeno et al., 2011). In another seminal paper (Kadowaki Horita et al., 2013), bilateral lesions of the dopaminergic input to the prefrontal cortex (PFC) were produced in rats using 6-hydroxydopamine (6-OHDA). Cognitive performance was improved by istradefylline treatment, by increasing dopamine levels in the PFC in both normal and PFC-lesioned rats.
Several different groups have suggested dopamine as a key player in the clinical course of AD (Itoh et al., 1996; Kemppainen et al., 2003; Kumar and Patel, 2007; Mura et al., 2010). Kemppainen et al. examined the DA receptor-binding potential [through PET with the D2/D3 antagonist (11)c FLB 457]; it was found a reduced binding in the right hippocampus, with a positive correlation with verbal memory performance and picture naming (at the Boston Test), supporting studies “to evaluate the efficiency of dopaminergic medication on patients with early AD.”
Mura et al. (2010) stressed the possibility that pathological Abeta oligomers may exert a dysfunctional impact on endogenous catecholaminergic transmission, even before the obvious neurodegenerative structural alterations.
Martorana’s group has highlighted a potential DA deficit (D1-mediated, at least in part) underlying cognitive impairment. More intriguingly, levodopa partially restored cortical transmission (evaluated by TMS tools, Martorana et al., 2008, 2009, 2010, 2011). Levodopa and/or dopamine agonists, such as rotigotine, whose endogenous binding is not limited to D2-like preferring sites, can promote modest but significant cognitive amelioration (Martorana et al., 2013). These findings are not surprising per se. A large corpus of pharmacological experiments in 6-OHDA-lesioned rodents demonstrated the efficacy of apomorphine in reverting perturbed behavioral tasks (such as water-maze), thus championing the D2–D1 mixed agonist profile. In our experience with mild PD subjects, levodopa proved effective (or at least without cognitive counterbalance), whilst selective D2 agonists might impair short-term verbal memory, attentional-executive functions, and verbal fluency (Brusa et al., 2003).
Noradrenaline Modulators
Dysfunction of locus coeruleus (LC) is a hallmark of AD (Zarow et al., 2003; Bekar et al., 2012). It was postulated long ago that damage to LC noradrenergic neurons (deposition of neurofibrillary tangles) might contribute to disease progression (Zweig et al., 1989).
Noticeably, NE may participate in the pathogenesis of the amyloid cascade, as suggested in transgenic APP23 mice, when the classical α(1)-adrenoceptor antagonist prazosin reduced the generation of amyloid β in N2a cells, and promoted a cascade of molecular events culminating in benefits to memory function (Katsouri et al., 2013).
Yet, experimental setting has provided conflicting results. For example, 1-month treatment with l-DOPS, NE precursor, improved learning in the Morris water-maze test compared with vehicle-treated mice. l-DOPS increased CNS NA levels, and average latency times in the water-maze test were inversely correlated to NA levels (Kalinin et al., 2012).
In patients, prazosin improved behavioral symptoms in AD patients with agitation/aggression (Wang et al., 2009). Yu et al. (2011), in a population-based study of individuals with incident AD, demonstrated that beta-blockers are also associated with delay of functional decline. In contrast, Gliebus and Lippa (2007) showed that “a trend for worse delayed memory retrieval occurred in patients who were on CNS-active beta-blockers.” Further, beta-adrenergic agonists were anecdotally shown as “memory rescuing” (Gibbs et al., 2010). These inconsistent findings may be attributed to two major biases; (a) inconsistent inclusion criteria and/or (b) concomitant loss of LC functions and ongoing compensatory mechanisms (with variable degree depending on AD stage).
In favor of a strong compensatory mechanism is previous evidence, showing that the increase in NE, following alpha-2 adrenoreceptor blockade in both aging and AD, is detected in absence of metabolite increase (Raskind et al., 1999). This is consistent with partial loss of CNS noradrenergic neurons, with compensatory activation of surviving noradrenergic projections.
A previous elegant work (performed in AD and DLB patients) corroborated this assumption, showing compensation through an increase in tyrosine hydroxylase (TH) mRNA expression in the remaining LC neurons, sprouting of dendrites into the peri-LC dendritic zone, and sprouting of axonal projections to the hippocampus as determined by alpha-2-receptor binding (Szot et al., 2006).
In contrast, some reports (e.g., Fitzgerald, 2010) suggested that, independent of potential loss of LC cells, brain NE levels may be elevated in some persons with AD, representing an etiological factor in some cases of AD, and not merely an epiphenomenon of the disease.
A recent epigenetic study (Mustapic et al., 2013) favors the prevalence of compensatory mechanisms, given that the activity and levels of dopamine beta-hydroxylase was reduced since the early stages AD (advancing the hypothesis for an indication to NA reuptake inhibitors).
Preliminary Data
Since 2012, we have started to evaluate CSF catecholamine concentrations in dementia patients. Routine lumbar puncture (LP, Stefani et al., 2005, 2006, 2009, 2012; Chiaravalloti et al., 2014; Liguori et al., 2014) was performed in patients during brief admission to our clinic. We also assessed neurocognitive impairment, exclusion of concomitant iatrogenic or metabolic risk factors, and carried out neuroimaging (MRI and/or FDG-PET). Dementia history was <4 years, in line with mild to moderate stages (Table 1 for major epidemiological features).
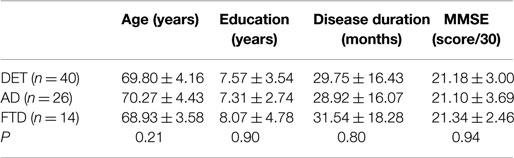
Table 1. Epidemiological main features of the studied populations (as a whole and split into AD-like vs. FTD-like).
In this relatively small cohort of patients, we analyzed the CSF concentration of DA and NE [plus some major metabolites of DA and 5-hydroxytriptamine (5-HT) namely homovanillic acid (HVA) and 5-hydroxyindolacetic acid (5-HIAA), previously indicated as possible biomarkers correlate with behavioral disturbances].
An age-matched control cohort was composed of patients with other neurological diseases (mostly radiculopathies) and manifesting no cognitive decline (mean MMSE 29.7, data not shown).
For the meantime, the first analysis revealed non-significant alterations in catecholamine CSF levels; nevertheless, demented patients showed the tendency toward a decreased DA/NE ratio with respect to controls.
In light of patients’ clinical profile ascertained along 2-year follow-up, and after FGD-PET completion and the acquisition of the complete CSF profile (including the assessment of Abeta 1–42, total tau, and phospho-tau proteins), the dementia group was subdivided into: AD-type (n = 26) and frontotemporal dementia (FTD) (n = 14; see Table 2). Table 2 reflects this more selective analysis.
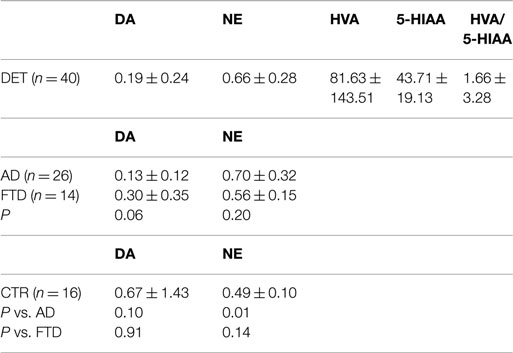
Table 2. Main CSF results (concentrations are expressed in nanomoles per liter).
The tendency for a lower CSF DA concentration was strictly confined to the AD-like subtype. More importantly, the AD group showed a significant increase of NE concentration; conversely, the HVA/5-HIAA ratios, investigated in previous papers as a potential biomarker for behavioral FTD subtype, in our hands revealed no difference among studied groups.
Discussion and Conclusion
1. This manuscript has tried to shed new lights on catecholamine-based treatment in AD. Asides from the opportunity to modulate dopamine-sensitive signs in patients attributable to LBD (not addressed here), recent data support a renewed interest around the usefulness of dopaminergic agents in handling cortical dementia, particularly AD-type.
This brief literature review imposes an extreme caution. Arguments to build up a therapeutic indication are still poor. After all, levodopa-centered therapy as add-on treatment in a cocktail regimen for AD patients has been tested only in observational clinical studies (or centered on electrophysiological indexes) and in very limited trials.
Our CSF data indicate that AD patients have increased NE levels, relative to aged, neurological controls without dementia. These findings suggest compensatory changes, but do not offer a simple therapeutic answer. In fact, they could be attributed, per se, to non-specific processes, representing a mere correlate of senescence (in light of a well-known intrinsic vulnerability of VTA/SN/LC regions in any aging brain).
In addition, a precise pathogenesis role of catecholamine along the course of AD is still questionable. Strong is the evidence, for instance, on a perturbed catecholamine vesicular system shared by different movement disorders, but not playing relevant role in AD (Goldstein et al., 2013).
2. In the short term, as sample size increases, we will assess the relation of CSF pattern to specific clinical phenotypes; in this content, longitudinal assessment may facilitate the understanding of the impact of DA- or NE-based therapy on specific behavioral traits.
Our students recall the clinical presentation of Alois Alzheimer’s patient, D. Frau Auguste, dominated by emotional distress and infidelity/excessive jealousy, before or in combination with cognitive symptoms (Geda et al., 2013). Still nowadays, despite the fact that we gather a clinical diagnosis of “probable AD” decades in advance, psychic alterations are prominent and critical, mostly in early-onset AD.
Although disease-modifying therapies are the “Holy Grail” to pursue symptomatic therapies, addressing neuropsychiatric aspects are also important for the quality of life of patients and caregivers (Geerts et al., 2013). Amongst the unmet needs, are the relevance of delusions (not simply attributable to psychotic derangement, Ismail et al., 2011.) and the severity of apathy. Borroni et al. (2010) in reminding that >50% AD patients manifest psychiatric disturbances such as psychosis, depression, agitation, disinhibition, aggression, hyperactivity, and socially intrusive behaviors, consider likely candidates the involvement of DA- or serotonin-related pathways and associated genetic variabilities. Yet, these assumptions do not translate into operative pharmacological strategies. As a matter of fact, the current attitude shared by general practitioners (adding SSRI or SNRI or trazodone) is just empirical (Mizukami et al., 2009; Sepehry et al., 2012).
Particularly elusive is the treatment of apathy. As reviewed by Vilalta-Franch et al. (2013), apathy syndrome in Alzheimer’s disease (ASAD) has a prevalence and incidence/year respectively 21 and 10.6%; it has been reported that ASAD persisted in 61.2% of patients after 1 year, causing an increased functional disability, but no relationship with cognitive impairment or increased caregiver burden was detected. Apathy treatment, despite occasional trial with modafinil or methylphenidate, remains a challenge (Herrmann et al., 2008; Padala et al., 2010; Frakey et al., 2012).
3. A comprehensive review (Schneider et al., 2014), examining late-stage AD drug development from 1984 to 2013, reminds that “the predominant drug targets have been the cholinergic system and the amyloid cascade.” Yet, adjourned “regulatory guidance and oversight have evolved to allow for enrichment of early-stage AD trial samples using biomarkers and phase-specific outcomes.” Hence, although “validated drug targets for AD remain to be developed (and only drugs that affect an aspect of cholinergic function have shown consistent, but modest, clinical effects in late-phase trials), there is opportunity for substantial improvements in drug discovery and clinical development methods.”
It is uncertain whether this hope of catching the disease in its presymptomatic phase (Wirz et al., 2014) is realistic. Of course, we should agree with opinion leader as Hampel when declaring that treatments “need to be applied before various molecular mechanisms converge into an irreversible pathway leading to morphological/structural damages” (Hampel et al., 2014). Yet, most modern health systems cannot tolerate decades of diagnostic protocols whose therapeutic effectiveness is unvalidated. The cost of one new drug ranges from an optimistic $2.0 billion to $5.7 billion (95% CI $1.5–2.9 billion) (Scott et al., 2014).
In other words, we doubt that “advances in the study of pre-clinical AD have driven the recognition that efficacy of at least some AD therapies may depend on initiation of treatment before clinical manifestation of disease, leading to a new era of AD prevention research” (Langbaum et al., 2013).
Truly enough, upcoming years will deal with a much more pragmatic vision of AD therapy, which explore treatable impairment such as vascular co-morbidity (Diomedi and Stefani, 2014) or insulin dysfunction (Wirz et al., 2014).
In conclusion, and despite conflicting findings, there is the opportunity to foresee extended and blind trials dedicated to clarify to what extent MAOI, dopamine agonists, levodopa, or NE modulators may act synergically with AChE-I since the first AD stages and/or in selected behavioral phenotypes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agnoli, A., Fabbrini, G., Fioravanti, M., and Martucci, N. (1992). CBF and cognitive evaluation of Alzheimer type patients before and after IMAO-B treatment: a pilot study. Eur. Neuropsychopharmacol. 2, 31–35. doi: 10.1016/0924-977X(92)90033-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bekar, L. K., Wei, H. S., and Nedergaard, M. (2012). The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. J. Cereb. Blood Flow Metab. 32, 2135–2145. doi:10.1038/jcbfm.2012.115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhavna, Md, S., Ali, M., Ali, R., Bhatnagar, A., Baboota, S., et al. (2014). Donepezil nanosuspension intended for nose to brain targeting: in vitro and in vivo safety evaluation. Int. J. Biol. Macromol. 67, 418–425. doi:10.1016/j.ijbiomac.2014.03.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Birks, J., and Flicker, L. (2003). Selegiline for Alzheimer’s disease. Cochrane Database Syst. Rev. 1:CD000442. doi:10.1002/14651858.CD000442
Bolea, I., Gella, A., and Unzeta, M. (2013). Propargylamine-derived multitarget-directed ligands: fighting Alzheimer’s disease with monoamine oxidase inhibitors. J. Neural Transm. 120, 893–902. doi:10.1007/s00702-012-0948-y
Borroni, B., Costanzi, C., and Padovani, A. (2010). Genetic susceptibility to behavioral and psychological symptoms in Alzheimer disease. Curr. Alzheimer Res. 7, 158–164. doi:10.2174/156720510790691173
Brusa, L., Bassi, A., Stefani, A., Pierantozzi, M., Peppe, A., Caramia, M. D., et al. (2003). Pramipexole in comparison to l-dopa: a neuropsychological study. J. Neural Transm. 110, 373–380. doi:10.1007/s00702-002-0811-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiaravalloti, A., Stefani, A., Fiorentini, A., Lacanfora, A., Stanzione, P., and Schillaci, O. (2014). Do CSF levels of t-Tau, p-Tau and β1–42 amyloid correlate with dopaminergic system impairment in patients with a clinical diagnosis of Parkinson disease? A 123I-FP-CIT study in the early stages of the disease. Eur. J. Nucl. Med. Mol. Imaging 41, 2137–2143. doi:10.1007/s00259-014-2841-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dar, T. A., Sheikh, I. A., Ganie, S. A., Ali, R., Singh, L. R., Gan, S. H., et al. (2014). Molecular linkages between diabetes and Alzheimer’s disease: current scenario and future prospects. CNS Neurol. Disord. Drug Targets 13, 290–298. doi:10.2174/18715273113126660135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Souza, L. C., Sarazin, M., Teixeira-Júnior, A. L., Caramelli, P., Santos, A. E., and Dubois, B. (2014). Biological markers of Alzheimer’s disease. Arq. Neuropsiquiatr. 72, 227–231. doi:10.1590/0004-282X20130233
Diomedi, M., and Stefani, A. (2014). Hemodynamic changes in Alzheimer’s disease: a leading role behind the curtain? Commentary on “CSF Aβ1-42 combined with neuroimaging biomarkers in the early detection, diagnosis and prediction of Alzheimer’s disease” by Lista et al. Alzheimers Dement. doi:10.1016/j.jalz.2014.10.004
Dysken, M. W., Sano, M., and Asthana, S. (2014). Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA 311, 33–44. doi:10.1001/jama.2013.282834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Emre, M., Poewe, W., De Deyn, P. P., Barone, P., Kulisevsky, J., Pourcher, E., et al. (2014). Long-term safety of rivastigmine in Parkinson disease dementia: an open-label, randomized study. Clin. Neuropharmacol. 37, 9–16. doi:10.1097/WNF.0000000000000010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Esposito, Z., Belli, L., Toniolo, S., Sancesario, G., Bianconi, C., and Martorana, A. (2013). Amyloid β, glutamate, excitotoxicity in Alzheimer’s disease: are we on the right track? The more the disease progresses, the more the uneffectiveness of antidementia drugs emerges. CNS Neurosci. Ther. 19, 549–555. doi:10.1111/cns.12095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fitzgerald, P. J. (2010). Is elevated norepinephrine an etiological factor in some cases of Alzheimer’s disease? Curr. Alzheimer Res. 7, 506–516. doi:10.2174/156720510792231775
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frakey, L. L., Salloway, S., Buelow, M., and Malloy, P. J. (2012). A randomized, double-blind, placebo-controlled trial of modafinil for the treatment of apathy in individuals with mild-to-moderate Alzheimer’s disease. J. Clin. Psychiatry 73, 796–801. doi:10.4088/JCP.10m06708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Freedman, M., Rewilak, D., Xerri, T., Cohen, S., Gordon, A. S., Shandling, M., et al. (1998). L-deprenyl in Alzheimer’s disease: cognitive and behavioral effects. Neurology 50, 660–668. doi:10.1212/WNL.50.3.660
Geda, Y. E., Schneider, L. S., Gitlin, L. N., Miller, D. S., Smith, G. S., Bell, J., et al. (2013). Neuropsychiatric symptoms in Alzheimer’s disease: past progress and anticipation of the future. Alzheimers Dement. 9, 602–608. doi:10.1016/j.jalz.2012.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geerts, H., Roberts, P., Spiros, A., and Carr, R. (2013). A strategy for developing new treatment paradigms for neuropsychiatric and neurocognitive symptoms in Alzheimer’s disease. Front. Pharmacol. 4:47. doi:10.3389/fphar.2013.00047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibbs, M. E., Maksel, D., Gibbs, Z., Hou, X., Summers, R. J., and Small, D. H. (2010). Memory loss caused by beta-amyloid protein is rescued by a beta(3)-adrenoceptor agonist. Neurobiol. Aging 31, 614–624. doi:10.1016/j.neurobiolaging.2008.05.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gliebus, G., and Lippa, C. F. (2007). The influence of beta-blockers on delayed memory function in people with cognitive impairment. Am. J. Alzheimers Dis. Other Demen. 22, 57–61. doi:10.1177/1533317506295889
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goldstein, D. S., Sullivan, P., Holmes, C., Miller, G. W., Alter, S., Strong, R., et al. (2013). Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J. Neurochem. 126, 591–603. doi:10.1111/jnc.12345
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hampel, H., Lista, S., Teipel, S. J., Garaci, F., Nisticò, R., Blennow, K., et al. (2014). Perspective on future role of biological markers in clinical therapy trials of Alzheimer’s disease: a long-range point of view beyond 2020. Biochem. Pharmacol. 88, 426–449. doi:10.1016/j.bcp.2013.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanagasi, H. A., Gurvit, H., Unsalan, P., Horozoglu, H., Tuncer, N., Feyzioglu, A., et al. (2011). The effects of rasagiline on cognitive deficits in Parkinson’s disease patients without dementia: a randomized, double-blind, placebo-controlled, multicenter study. Mov. Disord. 26, 1851–1858. doi:10.1002/mds.23738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hardenacke, K., Kuhn, J., Lenartz, D., Maarouf, M., Mai, J. K., Bartsch, C., et al. (2013). Stimulate or degenerate: deep brain stimulation of the nucleus basalis Meynert in Alzheimer dementia. World Neurosurg. 80, e35–e43. doi:10.1016/j.wneu.2012.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herrmann, N., Rothenburg, L. S., Black, S. E., Ryan, M., Liu, B. A., Busto, U. E., et al. (2008). Methylphenidate for the treatment of apathy in Alzheimer disease: prediction of response using dextroamphetamine challenge. J. Clin. Psychopharmacol. 28, 296–301. doi:10.1097/JCP.0b013e318172b479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Himeno, E., Ohyagi, Y., Ma, L., Nakamura, N., Miyoshi, K., Sakae, N., et al. (2011). Apomorphine treatment in Alzheimer mice promoting amyloid-b degradation. Ann. Neurol. 69, 248–256. doi:10.1002/ana.22319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ismail, Z., Nguyen, M. Q., Fischer, C. E., Schweizer, T. A., Mulsant, B. H., and Mamo, D. (2011). Neurobiology of delusions in Alzheimer’s disease. Curr. Psychiatry Rep. 13, 211–218. doi:10.1007/s11920-011-0195-1
Itoh, A., Nitta, A., Nadai, M., Nishimura, K., Hirose, M., Hasegawa, T., et al. (1996). Dysfunction of cholinergic and dopaminergic neuronal systems in beta-amyloid protein-infused rats. J. Neurochem. 66, 1113–1117. doi:10.1046/j.1471-4159.1996.66031113.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kadowaki Horita, T., Kobayashi, M., Mori, A., Jenner, P., and Kanda, T. (2013). Effects of the adenosine A2A antagonist istradefylline on cognitive performance in rats with a 6-OHDA lesion in prefrontal cortex. Psychopharmacology (Berl.) 230, 345–352. doi:10.1007/s00213-013-3158-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kalinin, S., Polak, P. E., Lin, S. X., Sakharkar, A. J., Pandey, S. C., and Feinstein, D. L. (2012). The noradrenaline precursor L-DOPS reduces pathology in a mouse model of Alzheimer’s disease. Neurobiol. Aging 33, 1651–1663. doi:10.1016/j.neurobiolaging.2011.04.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Katsouri, L., Vizcaychipi, M. P., McArthur, S., Harrison, I., Suárez-Calvet, M., Lleo, A., et al. (2013). Prazosin, an α(1)-adrenoceptor antagonist, prevents memory deterioration in the APP23 transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 1105–1115. doi:10.1016/j.neurobiolaging.2012.09.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kemppainen, N., Laine, M., Laakso, M. P., Kaasinen, V., Någren, K., Vahlberg, T., et al. (2003). Hippocampal dopamine D2 receptors correlate with memory functions in Alzheimer’s disease. Eur. J. Neurosci. 18, 149–154. doi:10.1046/j.1460-9568.2003.02716.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klegeris, A., and McGeer, P. L. (2000). R-(-)-Deprenyl inhibits monocytic THP-1 cell neurotoxicity independently of monoamine oxidase inhibition. Exp. Neurol. 166, 458–464. doi:10.1006/exnr.2000.7517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koch, G., Di Lorenzo, F., Bonnì, S., Ponzo, V., Caltagirone, C., and Martorana, A. (2012). Impaired LTP- but not LTD-like cortical plasticity in Alzheimer’s disease patients. J. Alzheimers Dis. 31, 593–599. doi:10.3233/JAD-2012-120532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumar, U., and Patel, S. C. (2007). Immunohistochemical localization of dopamine receptor subtypes (D1ReD5R) in Alzheimer’s disease brain. Brain Res. 1131, 187–196. doi:10.1016/j.brainres.2006.10.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kupershmidt, L., Amit, T., Bar-Am, O., Weinreb, O., and Youdim, M. B. (2012). Multi-target, neuroprotective and neurorestorative M30 improves cognitive impairment and reduces Alzheimer’s-like neuropathology and age-related alterations in mice. Mol. Neurobiol. 46, 217–220. doi:10.1007/s12035-012-8304-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Langbaum, J. B., Fleisher, A. S., Chen, K., Ayutyanont, N., Lopera, F., Quiroz, Y. T., et al. (2013). Ushering in the study and treatment of preclinical Alzheimer disease. Nat. Rev. Neurol. 9, 371–381. doi:10.1038/nrneurol.2013.107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laxton, A. W., and Lozano, A. M. (2013). Deep brain stimulation for the treatment of Alzheimer disease and dementias. World Neurosurg. 80, e1–e8. doi:10.1016/j.wneu.2012.06.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liguori, C., Stefani, A., Sancesario, G., Sancesario, G. M., Marciani, M. G., and Pierantozzi, M. (2014). CSF lactate levels, τ proteins, cognitive decline: a dynamic relationship in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatr. doi:10.1136/jnnp-2014-308577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lista, S., Garaci, F. G., Ewers, M., Teipel, S., Zetterberg, H., Blennow, K., et al. (2014). CSF Aβ1-42 combined with neuroimaging biomarkers in the early detection, diagnosis and prediction of Alzheimer’s disease. Alzheimers Dement. 10, 381–392. doi:10.1016/j.jalz.2013.04.506
Lu, C., Zhou, Q., Yan, J., Du, Z., Huang, L., and Li, X. (2013). A novel series of tacrine-selegiline hybrids with cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 62, 745–753. doi:10.1016/j.ejmech.2013.01.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mancuso, C., Siciliano, R., Barone, E., Butterfield, D. A., and Preziosi, P. (2011). Pharmacologists and Alzheimer disease therapy: to boldly go where no scientist has gone before. Expert Opin. Investig. Drugs 20, 1243–1261. doi:10.1517/13543784.2011.601740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martorana, A., Stefani, A., Palmieri, M. G., Esposito, Z., Bernardi, G., Sancesario, G., et al. (2008). L-dopa modulates motor cortex excitability in Alzheimer’s disease patients. J. Neural Transm. 115, 1313–1319. doi:10.1007/s00702-008-0082-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martorana, A., Mori, F., Esposito, Z., Kusayanagi, H., Monteleone, F., Codecà, C., et al. (2009). Dopamine modulates cholinergic cortical excitability in Alzheimer’s disease patients. Neuropsychopharmacology 34, 2323–2328. doi:10.1038/npp.2009.60
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martorana, A., Esposito, Z., and Koch, G. (2010). Beyond the cholinergic hypothesis: do current drugs work in Alzheimer’s disease? CNS Neurosci. Ther. 16, 235–245. doi:10.1111/j.1755-5949.2010.00175.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martorana, A., Semprini, R., and Koch, G. (2011). “Clinical profile of Alzheimer’s disease non-responder patient,” in Advanced Understanding of Neuro- degenerative Diseases, ed. C. Raymond Chuen-Chung (Rijeka: InTech), 156–168. [ISBN 978-953-307-529-7].
Martorana, A., Di Lorenzo, F., Esposito, Z., Lo Giudice, T., Bernardi, G., Caltagirone, C., et al. (2013). Dopamine D2-agonist rotigotine effects on cortical excitability and central cholinergic transmission in Alzheimer’s disease patients. Neuropharmacology 64, 108–113. doi:10.1016/j.neuropharm.2012.07.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McKeith, I., O’Brien, J., Walker, Z., Tatsch, K., Booij, J., Darcourt, J., et al. (2007). Sensitivity and specificity of dopamine transporter imaging with 123I.FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicenter study. Lancet Neurol. 6, 305–313. doi:10.1016/S1474-4422(07)70057-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mizukami, K., Hatanaka, K., Tanaka, Y., Sato, S., and Asada, T. (2009). Therapeutic effects of the selective serotonin noradrenaline reuptake inhibitor milnacipran on depressive symptoms in patients with Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 33, 349–352. doi:10.1016/j.pnpbp.2008.12.019
Mura, E., Lanni, C., Preda, S., Pistoia, F., Sarà, M., Racchi, M., et al. (2010). Beta-amyloid: a disease target or a synaptic regulator affecting age-related neurotransmitter changes? Curr. Pharm. Des. 16, 672–683. doi:10.2174/138161210790883723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mustapic, M., Presecki, P., Pivac, N., Mimica, N., Hof, P. R., Simic, G., et al. (2013). Genotype-independent decrease in plasma dopamine beta-hydroxylase activity in Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 1, 94–99. doi:10.1016/j.pnpbp.2013.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Padala, P. R., Burke, W. J., Shostrom, V. K., Bhatia, S. C., Wengel, S. P., Potter, J. F., et al. (2010). Methylphenidate for apathy and functional status in dementia of the Alzheimer type. Am. J. Geriatr. Psychiatry 18, 371–374. doi:10.1097/JGP.0b013e3181cabcf6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perani, D., Schillaci, O., Padovani, A., Nobili, F. M., Iaccarino, L., Della Rosa, P. A., et al. (2014). A survey of FDG- and amyloid-PET imaging in dementia and GRADE analysis. Biomed Res. Int. 2014, 785039. doi:10.1155/2014/785039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raskind, M. A., Peskind, E. R., Holmes, C., and Goldstein, D. S. (1999). Patterns of cerebrospinal fluid catechols support increased central noradrenergic responsiveness in aging and Alzheimer’s disease. Biol. Psychiatry 46, 756–765. doi:10.1016/S0006-3223(99)00008-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robertson, I. H. (2013). A noradrenergic theory of cognitive reserve: implications for Alzheimer’s disease. Neurobiol. Aging 34, 298–308. doi:10.1016/j.neurobiolaging.2012.05.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sano, M., Ernesto, C., Thomas, R. G., Klauber, M. R., Schafer, K., Grundman, M., et al. (1997). A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s disease cooperative study. N. Engl. J. Med. 336, 1216–1222. doi:10.1056/NEJM199704243361704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneider, L. S., Mangialasche, F., Andreasen, N., Feldman, H., Giacobini, E., Jones, R., et al. (2014). Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. Intern. Med. 275, 251–283. doi:10.1111/joim.12191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneider, L. S., Olin, J. T., and Pawluczyk, S. (1993). A double-blind crossover pilot study of l-deprenyl (selegiline) combined with cholinesterase inhibitor in Alzheimer’s disease. Am. J. Psychiatry 150, 321–323. doi:10.1176/ajp.150.2.321
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scott, T. J., O’Connor, A. C., Link, A. N., and Beaulieu, T. J. (2014). Economic analysis of opportunities to accelerate Alzheimer’s disease research and development. Ann. N. Y. Acad. Sci. 1313, 17–34. doi:10.1111/nyas.12417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sepehry, A. A., Lee, P. E., Hsiung, G. Y., Beattie, B. L., and Jacova, C. (2012). Effect of selective serotonin reuptake inhibitors in Alzheimer’s disease with comorbid depression: a meta-analysis of depression and cognitive outcomes. Drugs Aging 29, 793–806. doi:10.1007/s40266-012-0012-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stefani, A., Bernardini, S., Panella, M., Pierantozzi, M., Nuccetelli, M., Koch, G., et al. (2005). AD with subcortical white matter lesions and vascular dementia: CSF markers for differential diagnosis. J. Neurol. Sci. 237, 83–88. doi:10.1016/j.jns.2005.05.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stefani, A., Martorana, A., Bernardini, S., Panella, M., Mercati, F., Orlacchio, A., et al. (2006). CSF markers in Alzheimer disease patients are not related to the different degree of cognitive impairment. J. Neurol. Sci. 251, 124–128. doi:10.1016/j.jns.2006.09.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stefani, A., Sancesario, G., Pierantozzi, M., Leone, G., Galati, S., Hainsworth, A. H., et al. (2009). CSF biomarkers, impairment of cerebral hemodynamics and degree of cognitive decline in Alzheimer’s and mixed dementia. J. Neurol. Sci. 283, 109–115. doi:10.1016/j.jns.2009.02.343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stefani, A., Brusa, L., Olivola, E., Pierantozzi, M., and Martorana, A. (2012). CSF and clinical hallmarks of subcortical dementias: focus on DLB and PDD. J. Neural Transm. 119, 861–875. doi:10.1007/s00702-012-0820-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szot, P., White, S. S., Greenup, J. L., Leverenza, J. B., Peskind, E. R., and Raskind, M. A. (2006). Compensatory changes in the noradrenergic nervous system in the locus coeruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J. Neurosci. 26, 467–478. doi:10.1523/JNEUROSCI.4265-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tariot, P. N., Goldstein, B., Podgorski, C. A., Cox, C., and Frambes, N. (1998). Short-term administration of selegiline for mild-to-moderate dementia of the Alzheimer’s type. Am. J. Geriatr. Psychiatry 6, 145–154. doi:10.1097/00019442-199821000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tatsch, K., and Poepper, I. (2013). Nigrostriatal dopamine terminal imaging with dopamine transporter SPCET: an update. J. Nucl. Med. 54, 1331–1338. doi:10.2967/jnumed.112.105379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thiratmatrakul, S., Yenjai, C., Waiwut, P., Vajragupta, O., Reubroycharoen, P., Tohda, M., et al. (2014). Synthesis, biological evaluation and molecular modeling study of novel tacrine-carbazole hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 75, 21–30. doi:10.1016/j.ejmech.2014.01.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomas, T. (2000). Monoamine oxidase-B inhibitors in the treatment of Alzheimer’s disease. Neurobiol. Aging 21, 343–348. doi:10.1016/S0197-4580(00)00100-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tolbert, S. R., and Fuller, M. A. (1996). Selegiline in treatment of behavioral and cognitive symptoms of Alzheimer disease. Ann. Pharmacother. 30, 1122–1129.
Vilalta-Franch, J., Calvó-Perxas, L., Garre-Olmo, J., Turró-Garriga, O., and López-Pousa, S. J. (2013). Apathy syndrome in Alzheimer’s disease epidemiology: prevalence, incidence, persistence, and risk and mortality factors. J. Alzheimers Dis. 33, 535–543. doi:10.3233/JAD-2012-120913
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, L. Y., Shofer, J. B., Rohde, K., Hart, K. L., Hoff, D. J., McFall, Y. H., et al. (2009). Prazosin for the treatment of behavioral symptoms in patients with Alzheimer disease with agitation and aggression. Am. J. Geriatr. Psychiatry 17, 744–751. doi:10.1097/JGP.0b013e3181ab8c61
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wirz, K. T., Keitel, S., Swaab, D. F., Verhaagen, J., and Bossers, K. (2014). Early molecular changes in Alzheimer disease: can we catch the disease in its presymptomatic phase? J. Alzheimers Dis. 38, 719–740. doi:10.3233/JAD-130920
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, R., and Vassar, R. (2014). Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 13, 319–329. doi:10.1016/S1474-4422(13)70276-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, J. T., Wang, N. D., Ma, T., Jiang, H., Guan, J., and Tan, L. (2011). Roles of β-adrenergic receptors in Alzheimer’s disease: implications for novel therapeutics. Brain Res. Bull. 84, 111–117. doi:10.1016/j.brainresbull.2010.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zarow, C., Lyness, S. A., Mortimer, J. A., and Chui, H. C. (2003). Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 60, 337–341. doi:10.1001/archneur.60.3.337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, H., Amit, T., Bar-Am, O., Fridkin, M., Youdim, M. B., and Mandel, S. A. (2012). From anti-Parkinson’s drug rasagiline to novel multitarget iron chelators with acetylcholinesterase and monoamine oxidase inhibitory and neuroprotective properties for Alzheimer’s disease. J. Alzheimers Dis. 30, 1–16. doi:10.3233/JAD-2012-120013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Alzheimer’s disease, CSF, catecholamine, dopamine, monoaminooxidase inhibitors
Citation: Stefani A, Olivola E, Liguori C, Hainsworth AH, Saviozzi V, Angileri G, D’Angelo V, Galati S and Pierantozzi M (2015) Catecholamine-based treatment in AD patients: expectations and delusions. Front. Aging Neurosci. 7:67. doi: 10.3389/fnagi.2015.00067
Received: 23 February 2015; Accepted: 17 April 2015;
Published: 04 May 2015
Edited by:
P. Hemachandra Reddy, Texas Tech University, USAReviewed by:
Koteswara Rao Valasani, The University of Kansas, USAErnesto Fedele, University of Genoa, Italy
Copyright: © 2015 Stefani, Olivola, Liguori, Hainsworth, Saviozzi, Angileri, D’Angelo, Galati and Pierantozzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Stefani, Università di Roma Tor Vergata, Viale Oxford, 81, Rome 00133, Italy, stefani@uniroma2.it