Huntington’s disease and striatal signaling
- 1 UMRS 952, INSERM, UMR 7224, CNRS Université Pierre et Marie Curie – Paris-6, Paris, France
- 2 Pôle des Maladies du Système Nerveux, Département de Neurologie, Hopital Pitié-Salpétrière, Assistance publique-Hôpitaux de Paris, Paris, France
- 3 Department of Neurology, First Faculty of Medicine, Charles University in Prague and General University Hospital, Prague, Czech Republic
Huntington’s Disease (HD) is the most frequent neurodegenerative disease caused by an expansion of polyglutamines (CAG). The main clinical manifestations of HD are chorea, cognitive impairment, and psychiatric disorders. The transmission of HD is autosomal dominant with a complete penetrance. HD has a single genetic cause, a well-defined neuropathology, and informative pre-manifest genetic testing of the disease is available. Striatal atrophy begins as early as 15 years before disease onset and continues throughout the period of manifest illness. Therefore, patients could theoretically benefit from therapy at early stages of the disease. One important characteristic of HD is the striatal vulnerability to neurodegeneration, despite similar expression of the protein in other brain areas. Aggregation of the mutated Huntingtin (HTT), impaired axonal transport, excitotoxicity, transcriptional dysregulation as well as mitochondrial dysfunction, and energy deficits, are all part of the cellular events that underlie neuronal dysfunction and striatal death. Among these non-exclusive mechanisms, an alteration of striatal signaling is thought to orchestrate the downstream events involved in the cascade of striatal dysfunction.
Clinical Aspects
Huntington’s disease (HD) is a fatal disorder with a general prevalence of about 10 per 100,000 births, with some regions of the world having a higher prevalence of up to 700 per 100,000 (Harper, 1992; Paradisi et al., 2008). The typical age of onset is usually between 35 and 50 years but is very variable ranging from 1 to 85 years or more. The disease begins before 30 years of age in about 15% of patients and is then referred to as juvenile HD. The duration of the disease from onset to death is about 15–20 years. Clinical features of HD can be divided into three groups: movement disorders, cognitive impairment, and psychiatric manifestations. Chorea is the most characteristic movement disorder of classical HD and is characterized by brief, involuntary, abnormal movements, which appear unpredictably in all the parts of the body (Quinn and Schrag, 1998). As the disease progresses, the choreic movements generally tend to diminish and be replaced by akineto-rigid parkinsonism that can be associated with dystonic postures (Penney et al., 1990; Kremer et al., 1992). At an advanced stage of the disease, a high proportion of HD patients have falls (Busse et al., 2009), and a loss of independent ambulation, which may precipitate admission to nursing homes (Wheelock et al., 2003). Other movement disorders can be occasionally observed during the course of the disease, including tics (Kerbeshian et al., 1991; Jankovic et al., 1995) and myoclonia (Vogel et al., 1991; Carella et al., 1993; Thompson et al., 1994). Cognitive impairment plays a major role in the functional decline and loss of autonomy of the patients. It can precede motor symptoms or occur during the course of the disease, and usually leads, in turn, to dementia. Cognitive alteration has a sub-cortical profile with predominant impairment of executive/attention functions (Caine et al., 1978; Bamford et al., 1995; Ho et al., 2003; Peinemann et al., 2005). Instrumental functions (language, praxia, and gnosis) and memory are generally better preserved in HD than in other types of dementia (Caine et al., 1977; Hodges et al., 1990; Pillon et al., 1993, 1994). Neurobehavioral symptoms are very frequent. They can be the initial manifestations of the disease or occur at any time during the course of the disease (Shiwach and Norbury, 1994; Kirkwood et al., 2002). Irritability, agitation, apathy, anxiety, social withdrawal, impulsiveness, alcohol abuse, obsessive–compulsive disorder (Cummings and Cunningham, 1992; Patzold and Brune, 2002), hostility, and sexual disorders are common (Pflanz et al., 1991; Fedoroff et al., 1994; Paulsen et al., 2001). Mood disorders are very frequent along HD, including depression (Caine and Shoulson, 1983; Folstein et al., 1983; Di Maio et al., 1993; Cummings, 1995; Jensen et al., 1998) and manic episodes (Mendez, 2000). Various types of psychotic disorders can also be observed (Cummings, 1995; Rosenblatt and Leroi, 2000). HD patients have a risk of suicide that is 10 times higher than in the general population. Sleep disorders and weight loss are also frequently encountered in HD patients (Morton et al., 2005; Arnulf et al., 2008; Aziz et al., 2008; Videnovic et al., 2009).
Contrary to patients with typical HD, those with the juvenile form (or the Westphal variant) do not display chorea. These forms are characterized by a combination of progressive akineto-rigid parkinsonism, dystonia, ataxia, dementia, and psychiatric disorders (Siesling et al., 1997; Gonzalez-Alegre and Afifi, 2006). Seizures can also occur. Patients with childhood-onset HD can develop non-specific encephalopathy resulting in seizures, myoclonus, and rapid cognitive deterioration (Siesling et al., 1997; Gambardella et al., 2001; Gonzalez-Alegre and Afifi, 2006).
Genetics
The mutation responsible for HD is located at the 5′ terminal part of the HTT gene on chromosome 4p16.3 (The Huntington Collaborative Research Group, 1993). The mutation consists of an unstable expansion of the CAG repeat sequence, located in exon 1, at the NH2-terminal part of the protein. The mutated protein causes neuronal dysfunction and death, particularly in the striatum and cortex, although it is ubiquitously expressed. The penetrance of the mutation is almost complete. The HTT gene is normal when it contains less than 27 CAG repeats. Between 27 and 35 CAG repeats do not cause HD but may expand in successive generations. Intermediate alleles (between 36 and 39 repeats) repetitions are usually associated with late onset disease and may express a variable penetrance as the patient may die before disease onset. Individuals with 39 CAG repeats or greater will develop symptoms of HD (Kenney et al., 2007; Reynolds, 2008; Semaka et al., 2008). About 10% of HD patients have no family history of HD (Goldberg et al., 1993; Davis et al., 1994), with some of these patients receiving the mutant allele from an asymptomatic father with an intermediate allele. Such alleles do not cause HD but show instability on replication and tend to expand in successive generation with greater instability in spermatogenesis than in oogenesis (Zuhlke et al., 1993; Ranen et al., 1995). This instability of increased number of CAG repeats over successive generations explains the phenomenon of genetic anticipation, which is defined by the tendency of an earlier disease onset in successive generations (Goldberg et al., 1993; Myers et al., 1993; Alford et al., 1996). The age of onset cannot be predicted from the CAG repeat length in clinical practice. However, the number of repeats inversely correlates with the age of onset (Andrew et al., 1993; Duyao et al., 1993; Snell et al., 1993; Wexler et al., 2004; Andresen et al., 2007).
Neuropathology
Brain weight may be reduced by as much as 25–30% in advanced HD cases. Gross pathology of HD is limited to the brain, with atrophy predominating in the caudate–putamen and to a lesser extent, the cerebral cortex. The neuropathological signature of HD is the prominent striatal neuron loss and the presence of intranuclear inclusion bodies, which mainly consist of the accumulation of abnormal expansion of polyglutamines [Exp-Huntingtin (HTT)]. A grading system for the striatal neuropathology was established using macroscopic and microscopic criteria (Vonsattel’s grade; Vonsattel et al., 1985). It defines five grades ranging from 0 to 4 with increasing severity. The grade correlates closely with the extent of clinical disability. The most vulnerable neuronal population is the medium spiny neurons (MSNs) of the striatum. According to the Vonsattel’s grade, the striato-pallidal MSNs, which express enkephalin and dopaminergic D2 receptors, degenerate first (grade 2). Then striato-nigral MSNs which express substance P and dopaminergic D1 receptors degenerate (grade 3). The degeneration of MSNs occurs according to a dorso-ventral and medio-lateral gradient and is associated with a reduced expression of substance P, leu-enkephalin, calcineurin, calbindin, histamine H2-receptors, dopamine receptors, cannabinoid receptors, and Adenosine A2 receptors (Goto et al., 1989; Martinez-Mir et al., 1991; Richfield et al., 1991; Richfield and Herkenham, 1994). The striatal interneurons, aspiny striatal cholinergic, and somatostatine containing neurons, are relatively spared (Lange et al., 1976; Dawbarn et al., 1985; Ferrante et al., 1985, 1987, 1991). Another characteristic neuropathological change is a modification of the dendritic arborization of spiny neurons, with an axonal retraction before cell death (Graveland et al., 1985; Kiechle et al., 2002).
Molecular Mechanisms of the Disease
Whether neuronal degeneration in HD is due to the loss of normal HTT properties or a gain of toxic functions, or both, is not fully elucidated. In addition, age-related “normal” alterations in cellular functioning may accelerate HD pathogenesis (Diguet et al., 2009). Importantly, HTT is required for normal embryonic development as the loss of the protein leads to lethality of mouse embryos around day 8.5 (Duyao et al., 1995; Zeitlin et al., 1995) and the selective knockdown of the protein in neurons and testis produces apoptosis in these tissues (Dragatsis et al., 2000). The wild-type HTT is a ubiquitous protein, expressed in most cells of the organism and within virtually all cellular compartments (DiFiglia et al., 1997; Gutekunst et al., 1999; Kegel et al., 2002, 2005; Hoffner et al., 2005; Caviston et al., 2007; Rockabrand et al., 2007; Strehlow et al., 2007). Cell-based assays have focused on striatal specific cell-autonomous effects of Exp-HTT on HD neuropathology, since striatal but not cortical neurons in culture, show spontaneous degeneration in presence of Exp-HTT (Saudou et al., 1998; Arrasate et al., 2004; Garcia et al., 2004). Evidence in favor of cell-autonomous effects also include that a lentiviral mediated delivery of Exp-HTT to rat striatum results in a progressive pathology characterized by the appearance of ubiquitinated HTT aggregates, loss of dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) staining, and cell death (De Almeida et al., 2002). Nevertheless, in a genetic model with striatal specific expression of Exp-HTT, cell-autonomous nuclear accumulation of Exp-HTT aggregates in striatal neurons were observed, but no significant locomotor deficits nor striatal neuropathology, were found (Gu et al., 2007). This work suggested a “two-hit” hypothesis in which both cell-autonomous toxicity and pathological cell–cell interactions are critical to HD pathogenesis.
Huntingtin has multiple interacting partners, some of them exhibiting enhanced binding with Exp-HTT, while a handful prefer associating with the wild-type HTT (Harjes and Wanker, 2003; Li and Li, 2004). Among the binding partners of HTT are a dozen transcription factors which appear to affect the transcriptional profile in HD brain tissue or cells (Luthi-Carter et al., 2000, 2002; Sipione et al., 2002; Sugars and Rubinsztein, 2003; Zhai et al., 2005; Kuhn et al., 2007). HTT is also considered as a scaffold protein, orchestrating the intracellular trafficking, signaling pathways, and transcriptional activity that are required for neuronal homeostasis; these functions being strongly hindered by the Exp-HTT expression (see below). Exp-HTT is cleaved by various intracellular proteases, including caspases, and this proteolytic processing plays a key role in the pathophysiology of HD since the cleaved N-terminal fragment is much more toxic than the full-length mutant protein. These cleaved versions of Exp-HTT can also undergo aggregate formation (Scherzinger et al., 1997; Huang et al., 1998; Perutz et al., 2002), which is thought to interfere with normal cellular functioning. Aggregates of insoluble proteins are found in the brain of HD patients and in various HD models. They are enriched in striatal neurons and first appear in the neuropil of striatal MSNs (Li et al., 2000, 2001; Lee et al., 2004). By sequestering the wild-type HTT (Martindale et al., 1998) or associated proteins involved in transcription (Kazantsev et al., 1999; Steffan et al., 2000; Nucifora et al., 2001) or transport (Gunawardena et al., 2003; Trushina et al., 2004), these aggregates are thought to alter the fate of neuronal cells. Aggregate-mediated toxicity could be attributed to defects in RNA synthesis, cell survival, microtubule-dependent trafficking, or the ubiquitin–proteasome system (DiFiglia et al., 1997; Harjes and Wanker, 2003; Li and Li, 2004; Bennett et al., 2007). Nevertheless, the toxicity of aggregates still remains an issue of debate. Several studies have indicated that Exp-HTT aggregates are connected with neurodegeneration or cytotoxicity (Davies et al., 1997; DiFiglia et al., 1997; Ordway et al., 1997), whereas other studies have suggested that aggregate formation is either not intimately correlated with cytotoxicity (Saudou et al., 1998; Arrasate et al., 2004; Slow et al., 2005; Sawada et al., 2007; Mitra et al., 2009) or plays a protective role in cells (Arrasate et al., 2004; Mitra et al., 2009). Some authors argue that smaller oligomeric aggregates may be more toxic than larger ones (Gong et al., 2008). Furthermore, Exp-HTT can misfold into distinct β-sheet aggregates, called amyloids, which can be either toxic or non-toxic depending on their conformation (Nekooki-Machida et al., 2009). Aggregate toxicity can also depend on the cellular compartment in which they are localized, for example they could be more toxic in the neurites than in the nucleus (Li et al., 2000, 2001; Lee et al., 2004). Importantly, aggregates cannot be cleared by the ubiquitin–proteasome system (Bence et al., 2001; Jana et al., 2001; Verhoef et al., 2002; Venkatraman et al., 2004) and alter the normal efficacy of the clearance-machinery (Bennett et al., 2007; Hunter et al., 2007). The lower basal activity of the ubiquitin–proteasome system in neurons as compared to glial cells may account for the preferential accumulation of aggregates in neurons (Tydlacka et al., 2008). It is noteworthy that inhibiting the aggregation of Exp-HTT can alleviate the symptoms in various models of HD (Carmichael et al., 2000; Jana et al., 2000; Vacher et al., 2005; Ravikumar et al., 2006; Chopra et al., 2007; Herbst and Wanker, 2007; Perrin et al., 2007; Sarkar et al., 2007; Seo et al., 2007).
Changes in Axonal Transport and Synaptic Dysfunction
Vesicular transport is altered in HD and is linked to a subsequent synaptic dysfunction (Gunawardena et al., 2003; Szebenyi et al., 2003; Gauthier et al., 2004; Trushina et al., 2004; Caviston and Holzbaur, 2009; Sinadinos et al., 2009). These defaults of trafficking are mainly due to an impaired interaction between Exp-HTT and motor proteins (Szebenyi et al., 2003; Lee et al., 2004), but can also be a consequence of neuritic aggregates that act as physical roadblocks (Gunawardena et al., 2003; Szebenyi et al., 2003; Trushina et al., 2004). According to the non-autonomous theory, the “neurotrophin disorder hypothesis” is the most widely accepted (Zuccato and Cattaneo, 2009). The release of brain derived neurotrophic factor (BDNF) from cortical afferences, which provides an important neurotrophic support to striatal neurons, is impaired in HD. Transcriptional dysregulation of BDNF in cortical neurons was first described (Zuccato et al., 2001; see Transcriptional Dysregulation chapter below). Then, altered axonal transport deficit of BDNF from cortical neurons, was shown to participate in the default of BDNF release in the striatum (Gauthier et al., 2004). Wild-type HTT interacts with the molecular motor complex that transports organelles along the microtubules in axons (Gauthier et al., 2004; Caviston and Holzbaur, 2009). This interaction is altered in HD, due to Exp-HTT expression, which decreases axonal transport and the release of BDNF from cortical neurons to their terminals within the striatum (Figure 1). This is thought to participate in the striatal vulnerability in HD. One approach to compensate for the defective BDNF transport is to mimic the phosphorylation of Exp-HTT on Ser421, in order to restore the interaction between Exp-HTT and dynactin, and their association with microtubules (Colin et al., 2008). Interestingly, increasing tubulin acetylation using a specific histone deacetylase (HDAC6) inhibitor restores the recruitment of motor proteins, including kinesin-1 and dynein to microtubules, and increases BDNF axonal transport in cortical neurons (Dompierre et al., 2007).
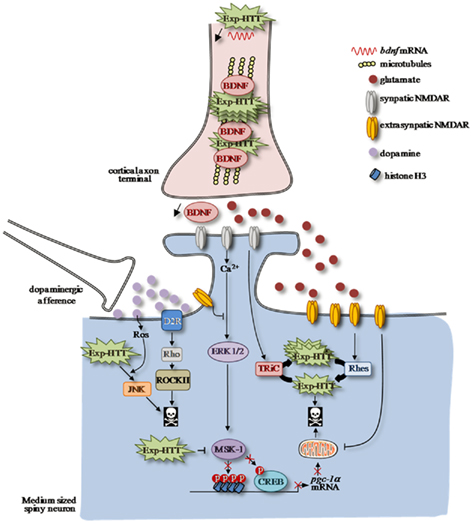
Figure 1. Altered striatal signaling pathways in HD. Afferent corticostriatal and nigro-striatal projections modulate striatal signaling, which is impaired in Huntington’s Disease (HD). Neurotrophic factor BDNF release from cortical afferences is decreased in cortico-striatal synapses as a consequence of Exp-HTT-mediated down-regulation of bdnf transcription and axonal transport in cortical neurons. A shift from synaptic to extrasynaptic NMDAR-dependent signaling participates to striatal neurons to death. In a physiological condition, calcium influx through synaptic NMDAR promotes activation of the MAPkinase/ERK signaling pathway along with its nuclear target, the MSK-1 protein, which phosphorylates the transcription factor CREB. MSK-1 activation promotes chromatin remodeling, which is crucial for CREB-mediated pgc-1α transcription, a key gene involved in mitochondria biogenesis. Synaptic NMDARs also promote formation of non-toxic Exp-HTT inclusion via TRIC. In HD, localization and activity of extrasynaptic NMDAR are enhanced by Exp-HTT, which promotes neuronal cell injury and death. The toxic effect of extrasynaptic NMDAR is partly due to (i) the upregulation of Rhes expression that disaggregates the non-toxic Exp-HTT inclusions (ii) to an impairment of mitochondrial functions and (iii) to a decrease of the ERK/MSK-1/CREB signaling module to the pgc-1α promoter. Dopamine release from nigro-striatal inputs promotes oxidative stress via the production of reactive oxygen species (ROS), which potentiates activation of the pro-apoptotic JNK pathway induced by Exp-HTT. In addition, Dopamine potentiates Exp-HTT-mediated striatal neurons death through activation of the Rho/ROCK signaling pathway downstream D2R.
Huntingtin is also involved in the trafficking and secretion of vesicles from the Golgi apparatus (Strehlow et al., 2007). In particular, it promotes, acting in concert with transglutaminase 2 (TGase 2) and HJSJ1b, the budding of vesicles containing BDNF from the Golgi to the cytoplasm (Borrell-Pages et al., 2006a; Del Toro et al., 2006). Modulation of TGase 2 and HJSJ1b expression by cystamine or cysteamine increases the release of BDNF in mice and monkey models of HD (Borrell-Pages et al., 2006b). The trafficking of neural proteins from the Golgi is also regulated by HTT-interacting protein 14 (HIP14), which normally interacts with HTT to regulate the trafficking of neuronal proteins and their synaptic release through palmitoylation of cysteine string protein (CSP; Yanai et al., 2006; Ohyama et al., 2007). Finally, Exp-HTT increases c-Jun-Kinase3 (JNK3) activity, which phosphorylates kinesin on Ser176 and reduces its binding to microtubules (Morfini et al., 2009). Thus, inhibition of the neuron-specific JNK3 can also protect from defects in fast axonal transport induced by Exp-HTT.
Excitotoxicity and Striatal Signaling
In HD, increased glutamate levels within the striatum are thought to arise from reduction of glial glutamate uptake (Liévens et al., 2001; Behrens et al., 2002). This is because of selective down-regulation of the glutamate transporter GLT1, which is mainly expressed in astrocytes. In Drosophila glia, Exp-HTT antagonize epidermal growth factor receptor (EGFR) Ras-extracellular signal-regulated kinase (ERK) signaling pathway, resulting in down-regulation of the glutamate transporter levels (Liévens et al., 2006). In addition, the expression of glutamine synthetase, an enzyme that converts glutamate to glutamine in glia, was also found to be altered (Liévens et al., 2001; Behrens et al., 2002). These findings demonstrated that the HD mutation results in a progressively deranged glutamate handling in the brain, beginning before the onset of symptoms in mice. They also provided evidence for a contribution of excitotoxicity to the pathophysiology of HD.
NMDAR-Mediated Excitotoxicity in HD
Ineffective management of Ca2+ homeostasis by striatal neurons ultimately leads to cell death via numerous signaling pathways. This excitotoxicity mostly implicates the N-Methyl-D-Aspartate type glutamate receptors (NMDAR) due to their high calcium permeability and slow activation/deactivation kinetics (Rothman and Olney, 1995; Dingledine et al., 1999; Cull-Candy et al., 2001). The earliest evidence in support of a role for NMDAR-mediated excitotoxicity in HD came from rodent studies where the administration of NMDAR agonists mimicked some clinical and pathological features of the disease (Beal et al., 1986; Sanberg et al., 1989; Ferrante et al., 1993). Studies have since switched from these chemical HD models to genetic mouse models where the expression of Exp-HTT protein has been linked to NMDAR-mediated excitotoxicity (Levine et al., 1999; Cepeda et al., 2001; Zeron et al., 2004). However, comparisons between HD models are complicated as the mRNA and protein expression levels of the NMDAR subunits vary between models and the disease state (Fan et al., 2007). Furthermore, age-related dependence to excitotoxicity has been recently highlighted in the transgenic YAC128 HD mice model, which display enhanced sensitivity to excitotoxicity in the early phase of the disease, prior to development of cognitive dysfunction and motor abnormalities, and resistance to excitotoxic stress as the disease progresses (Graham et al., 2009).
Targeting NMDAR Subunits
Pharmacological analyses have indicated that striatal NMDARs are most commonly formed from heterotetramers containing two obligatory NMDAR type 1 subunits (GluN1, previously known as NR1) plus a combination of two NMDAR type 2 subunits, GluN2A and GluN2B, as heterodimers or as heterotrimers (GluN1 plus GluN2A and GluN2B). Some literature points to NMDARs containing principally GluN2A as being pro-survival whereas GluN2B-containing NMDARs act as the excitotoxic mediators (Zeron et al., 2002; Liu et al., 2007). As the expression of GluN2B is significantly higher than GluN2A in the striatum than in other brain regions (Landwehrmeyer et al., 1995; Kuppenbender et al., 2000), a GluN2B-mediated excitotoxicity is consistent with the striatal vulnerability for neurodegeneration in HD. Interestingly, recent work has demonstrated a greater contribution by GluN2A containing NMDAR in the D1 expressing MSNs than D2 expressing MSNs which degenerate first in HD (Jocoy et al., 2011). In the YAC transgenic FVB/N mouse model of HD, NMDA-induced cell death is prevented by a specific GluN2B antagonist ifenprodil (Zeron et al., 2002). Likewise, double mutant mice expressing Exp-HTT and overexpressing GluN2B displayed an exacerbated striatal degeneration (Heng et al., 2009). The logical conclusion from these studies is that selective antagonists of GluN2B such as ifenprodil, RO-25,6981, and CP101,606 should offer some neuroprotection. However, the use of three different GluN2B antagonists had no beneficial effects in the R6/2 mouse model of HD (Tallaksen-Greene et al., 2010). In addition, both ifenprodil and RO-25,6981 increase cell death induced by oxidative stress (Papadia et al., 2008) and blockade of spontaneous GluN2B activity exacerbates staurosporine-induced cell death (Martel et al., 2009). Therefore, as the outcome depends on the considered model and the particular excitotoxic insult the argument is left open as to whether GluN2B merits specific targeting in HD excitotoxicity.
Glutamate Receptor Localization and Excitotoxicity
Instead of a definite GluN2B subunit-specific implication for excitotoxic signaling, work by Hardingham and Bading fueled the hypothesis that activation of extrasynaptic NMDAR (those located at the cell body, dendritic shaft or on the neck of spines) promotes cell death whereas stimulation of synaptic NMDAR promotes cell survival (Hardingham and Bading, 2010). These hypotheses may not be in complete opposition as extrasynaptic NMDAR contain more GluN2B than synaptic ones (Tovar and Westbrook, 1999; Steigerwald et al., 2000; Jocoy et al., 2011). Stimulation of calcium entry via synaptic NMDAR is well tolerated and coupled to activation of the MAPkinase/ERK signaling pathway along with its target the transcription factor cyclic AMP-response element binding (CREB), which regulates genes such as bdnf (Hardingham et al., 2001, 2002). On the other hand these pro-survival mediators are dominantly opposed by the activation of extrasynaptic NMDAR which leads to the loss of mitochondrial membrane potential, inhibition of ERK signaling, CREB shut off (Vanhoutte and Bading, 2003), and an induction of a distinct genomic program including the gene clca1a, which encodes a calcium-activated chloride channel sufficient to kill neurons (Zhang et al., 2007). In HD, the striatal specific signaling partners of synaptic versus extrasynaptic NMDAR have begun to be analyzed. Ras homolog enriched in striatum (Rhes) is localized in the striatum, where it specifically bind to Exp-HTT and elicits both a decrease in ubiquitination and an increase in sumoylation of Exp-HTT, which leads to its disaggregation and favors the formation of neurotoxic soluble microaggregates (Subramaniam et al., 2009). Okamoto et al. (2009 found that Rhes expression is reduced when extrasynaptic receptors are blocked, whereas antagonism of synaptic NMDAR leads to a decrease in Exp-HTT aggregates and cell death. This study distinguished between the two receptor pools by the use of the uncompetitive NMDAR antagonist memantine, which selectively antagonizes extrasynaptic NMDAR at low concentrations (Rammes et al., 2008). The formation of non-toxic Exp-HTT inclusions is dependent on synaptic NMDAR activation and the induction of the T-complex-1 (TCP-1) subunit of TCP-1 ring complex (TRiC), which associates with heat shock protein 70 (Hsp 70) to favor the formation of these inclusions. By contrast, activation of extrasynaptic NMDAR in the presence of Exp-HTT down-regulates the protective PPAR alpha-co-activator-1α (PGC-1α) cascade (see below) via an inhibition of ERK activation and CREB phosphorylation (Okamoto et al., 2009). Inhibition of extrasynaptic NMDAR after in vivo treatment with a low dose of memantine over a period of 8 months increased TCP-1 protein levels, inclusion formation, and improved the motor function of YAC128 HD mice. By using the same approach, Milnerwood et al. (2010) showed that a blockade of extrasynaptic NMDAR restores basal levels of CREB activation and significantly overcomes motor learning deficits of YAC128 mice. Even at pre-symptomatic stages, YAC128 mice express more NMDAR subunits at extrasynaptic sites than YAC18 control mice, and consequently have a heightened response to glutamate spillover dependent on an extrasynaptic GluN2B-containing NMDAR (Milnerwood et al., 2010).
In addition to lateral NMDAR receptor localization, altered NMDA receptor trafficking may also participate to neuronal excitotoxicity in the striatum. Accelerated NMDA receptor trafficking and increased expression at the cell surface were found in striatal neurons from the YAC72 HD mouse model (Fan et al., 2007). This phenomenon can be related to an altered interaction between Exp-HTT and postsynaptic density 95 (PSD-95), a scaffold protein necessary for NMDAR stability, which has been previously described (Roche et al., 2001; Sun et al., 2001). Recently, association of PSD-95 with GluN2B in striatal tissue has been shown to be enhanced by Exp-HTT (Fan et al., 2009). Treatment of cultured MSNs with a TAT coupled peptide that blocks binding of GluN2B with PSD-95, reduces NMDAR surface expression in both YAC transgenic and WT MSN and rescues cells from NMDAR excitotoxicity.
The HIP1, which normally interacts with HTT, is involved in the intracellular trafficking of glutamate receptors of the AMPA subtypes. Exp-HTT, which interacts less efficiently with HIP1 than its normal counterpart, participates to increased membranal expression of AMPA receptors and hence to excitotoxic neuronal death in HD (Metzler et al., 2007). Altered calcium homeostasis in HD could be due to an abnormal interaction between Exp-HTT and the type 1 inositol 1,4,5-trisphosphate receptor (InsP3R1), which regulates the cytoplasmic calcium clearance by the endoplasmic reticulum (Tang et al., 2004, 2009). Disrupting the interaction between InsP3R1 and Exp-HTT normalizes calcium signaling, protects from glutamate-induced apoptosis in striatal neurons in vitro, and reduces neuronal pathology and motor deficits in a mouse model of HD in vivo.
Striatal Vulnerability: The Dopaminergic Hypothesis
In addition to the glutamatergic stimulation, some evidence indicate that Dopamine (DA) stimulation may play a key role in excitotoxicity in HD (Reynolds et al., 1998; Charvin et al., 2005, 2008; Cyr et al., 2006; Stack et al., 2007; Tang et al., 2007; Benchoua et al., 2008); Knock-out mice for the DA transporter (DAT) show spontaneous striatal death accompanied by behavioral alterations that resemble HD specifically during aging (Cyr et al., 2003). In an elegant study, a double mutant mouse strain with both enhanced dopamine transmission and endogenous expression of a mutant HTT gene was generated. This strain was generated by crossing the DAT knock-out mouse with a knock-in mouse model of HD containing 92 CAG repeats (Cyr et al., 2006). These double mutant mice exhibited increased behavioral and neuropathological hallmarks of HD, including neuropil aggregates in MSN projection neurons. DA released from nigro-striatal inputs, is present in high concentrations within the striatum and enhances sensitivity to glutamatergic inputs. It may also produce oxidative stress via the production of reactive oxygen species (ROS), a cellular process that increases with aging (Jakel and Maragos, 2000). In particular, cultures of striatal neurons from R6/2 HD mice are sensitized to DA-induced oxidative stress, leading to neuronal autophagy (Petersen et al., 2001). ROS produced by low doses of DA potentiate activation of the pro-apoptotic JNK pathway induced by Exp-HTT (Garcia et al., 2004; Charvin et al., 2005), the pharmacological inhibition of which is neuroprotective in the R6/2 transgenic mouse model of HD (Apostol et al., 2008). DA may also render striatal neurons more vulnerable to Exp-HTT via dopamine receptor-mediated mechanisms (Charvin et al., 2005, 2008). Depending on the cell line model of HD, expressing either the full length or truncated versions of Exp-HTT, D1, or D2 receptors stimulation seems to be different. Full-length Exp-HTT is required for alteration of calcium signaling (Zhang et al., 2008). In striatal neurons from YAC128 transgenic or Q111 knock-in mice, both expressing full-length Exp-HTT, D1 receptor stimulation potentiates calcium influx via NMDA receptors, and hence excitotoxic processes, including mitochondrial depolarization, and caspase activation (Cepeda et al., 2001; Zeron et al., 2002, 2004; Starling et al., 2005; Tang et al., 2007). More recently, a calcium-dependent activation of calpain was shown to be involved in striatal death after convergent activation of NMDA and D1 receptors in HD cell models (Paoletti et al., 2008). Increases in calcium influx lead to the cleavage of Cdk5 co-activator p35 into p25, which enables an aberrant toxic activation of Cdk5 (Paoletti et al., 2008). By contrast, when associated with p35 as a co-activator, Cdk5 is known to be protective via phosphorylation of Exp-HTT and the blockade of caspase-induced cleavage, resulting in attenuated aggregate formation and toxicity (Luo et al., 2005; Anne et al., 2007). Consistently, Cdk5/p35 suppresses the formation of aggregates induced by a short fragment of Exp-HTT (exon 1), via a new, unexpected role on microtubule stability and hence inclusion formation (Kaminosono et al., 2008). Together, these data highlight the complexity of Cdk5 activity and the importance of targeting selectively p25 to block Exp-HTT-induced inclusion formation and neuronal death.
A central role of DARPP-32 (Dopaminergic and cAMP-regulated phosphoprotein) has also been proposed (Metzler et al., 2010). DARPP-32 phosphorylation at Thr34 is induced by a D1 agonist (SKF 81297) and inhibits in turn the activity of PP1, a phosphatase that dephosphorylates the Ser421 residue of the HTT protein. Of interest, inhibition of PP1 can offer protection from NMDA-induced excitotoxicity in YAC128 mice, through increased phosphorylation of Ser421-HTT. The loss of DARPP-32 expression described in HD results in an increase of PP1 activity followed by a decrease of Ser421-Exp-HTT phosphorylation (Metzler et al., 2010). As its cleaved version, Exp-HTT is not sensitive to D1 agonists, probably because it is not sensitive to calcium overload and calcium-dependent proteolytic processes produced by D1 receptor stimulation. By contrast, D2 receptors stimulation potentiates Exp-HTT-induced aggregate formation, deficiency of mitochondrial complex II protein activity, and neuronal death (Charvin et al., 2005; Benchoua et al., 2008). In vivo, in a rat model of HD based on lentiviral-mediated expression of Exp-HTT exon 1 in the striatum (De Almeida et al., 2002), an early and chronic treatment with the D2 antagonist, haloperidol decanoate, protects striatal neurons from Exp-HTT-induced dysfunction, and aggregates formation (Charvin et al., 2008). Striatal signaling mediated by D2 receptor stimulation on Exp-HTT toxicity has been recently elucidated. Inhibition of the Rho/ROCK pathway using selective inhibitors or knockdown of ROCK-II expression reversed D2 agonist-mediated aggregate formation, neuritic retraction and neuronal death induced by Exp-HTT (Deyts et al., 2009).
Mitochondrial Dysfunction and Energy Deficits
Defects in energy metabolism in brain and muscles, has long been proposed to be involved in HD, from clinical (Djousse et al., 2002; Hamilton et al., 2004) biochemical (Arenas et al., 1998; Turner et al., 2007) and neuroimaging studies (Jenkins et al., 1998). In HD patients there is strong evidence for reduced glucose consumption in the brain, more specifically in the basal ganglia (Grafton et al., 1992; Kuwert et al., 1993) as well as increased lactate concentrations in the basal ganglia and occipital cortex (Jenkins et al., 1993), and lactate-to-pyruvate levels in the CSF (Jenkins et al., 1998). Various mechanisms that underlie the energy deficit in the HD brain have been proposed (Mochel et al., 2007; Mochel and Haller, 2011). They include impaired oxidative phosphorylation, oxidative stress, impaired mitochondrial calcium handling, abnormal mitochondria trafficking, decreased glycolysis, and transcriptional deregulation of PGC-1α. A deficiency of respiratory chain complex II, i.e., succinate dehydrogenase (SDH), in HD has been proposed since the observation that accidental ingestion of 3-nitropropionic acid (3-NP), an irreversible inhibitor of SDH, reproduces the clinical and neuropathological characteristics of HD in humans (Brouillet et al., 1999). In rodents, systemic 3-NP administration reproduces selective striatal degeneration, despite an altered SDH activity in multiple brain regions (Brouillet et al., 1998). Conversely, restoration of the complex II activity level is neuroprotective in a cellular model of HD (Benchoua et al., 2006). Activation of the pro-apoptotic JNK pathway is observed selectively in striatal neurons of 3-NP-administered rats in vivo, and overexpression of a dominant negative, non-phosphorylable version of c-Jun in vitro inhibits striatal degeneration induced by 3-NP (Garcia et al., 2002). DA signaling regulates SDH enzymatic activity (Benchoua et al., 2008) and hence may account for the vulnerability of striatal neurons in HD. Furthermore, as detailed in the next section Exp-HTT-induced transcriptional dysregulation is now thought to contribute to altered bioenergetics.
Transcriptional Dysregulation
Transcriptional dysregulation is an early event in the neuropathological process. Altered levels of dopaminergic receptor and neuropeptide mRNAs observed in patient’s brain tissues (Augood et al., 1996, 1997) are also observed in pre-symptomatic HD’s transgenic mice, suggesting that changes in transcription underlie neurodegeneration rather than reflecting non-specific degradation of all RNAs in affected neurons (Cha et al., 1998, 1999). Subsequently, multiple genes encoding neurotransmitter receptors, enzymes, and proteins involved in neuron structure, stress responses, and axonal transport were found to be dysregulated (Luthi-Carter et al., 2000, 2002; Sugars and Rubinsztein, 2003; Cha, 2007; Runne et al., 2007), with overlaps of altered transcripts between various mouse models of HD and brain of HD patients (Kuhn et al., 2007). Interestingly, more than 80% of classically admitted striatal-enriched genes (genes with higher relative expression in the striatum compared with other brain regions) are decreased in a mouse model of HD as well as in human HD postmortem brain (Desplats et al., 2006). A down-regulation of novel striatal-enriched genes involved in vesicle transport and trafficking, tryptophan metabolism, and neuroinflammation were identified more recently in both HD mouse striatum and caudate from HD patients (Mazarei et al., 2010). Of interest, most of HD-induced dysregulation of the striatal transcriptome can be largely attributed to the intrinsic effects of mutant HTT, in the absence of expression in cortical neurons (Thomas et al., 2011).
Transcriptional dysregulation can be found in large genomic regions in a coordinated fashion and this dysregulation is associated with disease progression. Attempts were made to use transcriptional dysregulation as a biomarker in HD. Genome-wide expression profiling of the blood from HD’s patients revealed significant differences in symptomatic patients (Borovecki et al., 2005), but not moderate-stage patients (Runne et al., 2007). Thus, these biomarkers need to be further validated before their widespread use in clinical trials.
Molecular Mechanisms of Transcriptional Dysregulation in HD
Within the nucleus Exp-HTT, under its soluble or aggregated form, interacts with and inhibits the activity of proteins involved in the normal transcriptional machinery. These include TATA binding protein (TBP), transcription factor II F (TFIIF), and tyrosine-aminotransferase II (TATII) 130 (Shimohata et al., 2000; Suhr et al., 2001; Dunah et al., 2002; Li et al., 2002). Exp-HTT also sequesters transcription factors involved in cell viability, including CREB protein (CBP), p53, specificity protein 1 (Sp1), nuclear factor-kappa B (NF-κB), nuclear receptor co-repressor (NCoR), and CA150 (Boutell et al., 1999; Li et al., 2000; Steffan et al., 2000; Nucifora et al., 2001; Dunah et al., 2002; Bae et al., 2005; Arango et al., 2006). Expression levels of BDNF, and its receptor TrkB, are decreased in the striatum of HD patient’s, suggesting a deficit in cortical neurotrophic support of the striatum (Ferrer et al., 2000; Zuccato et al., 2001; Lynch et al., 2007; Strand et al., 2007). In animal models of HD, cortical BDNF expression is reduced (Zuccato et al., 2001). Moreover, downregulating BDNF in striatum in mice worsens the HD phenotype, whereas elevating BDNF expression in the forebrain alleviates the HD phenotype (Canals et al., 2004; Strand et al., 2007; Gharami et al., 2008; Xie et al., 2010). The molecular mechanisms by which Exp-HTT drives the down-regulation of BDNF expression in cortical neurons have been unraveled. Wild-type HTT sequesters R element-1 silencing transcription factor (REST), a transcriptional repressor of neuronal survival factors including BDNF, within the cytoplasm. The HTT mutation leads to REST release within the nucleus, where it exerts a potent inhibitory role on the transcription of BDNF and other neuronal genes (Zuccato et al., 2001, 2003, 2007). Furthermore, REST mRNA levels are increased in R6/2 mouse model of HD and NG108 neuronal-like model of HD. At the transcriptional level, Sp1 binds to the Sp factor binding sites contained in the promoter of REST and contributes to Exp-HTT-mediated REST upregulation (Ravache et al., 2010). BDNF expression is also a component of the neuroprotective transcriptional response mediated by NF-κB in neurons (Lipsky et al., 2001). In addition, the loss of BDNF expression and low levels of NF-κB activity in neurons could lead to impairments of cognitive functions (Kaltschmidt et al., 2005; Meffert and Baltimore, 2005), a common feature of neurodegenerative disorders such as HD.
A Link between Transcriptional Dysregulation and Energy Deficits in HD
A link between Exp-HTT-induced transcriptional dysregulation and energy deficits has been recently described. Exp-HTT binds to the tumor suppression gene p53 more avidly than wild-type HTT and has been reported to increase p53 protein levels, nuclear localization and transcriptional activity in neuronal cultures and transgenic mice (Bae et al., 2005). Augmented p53 activity mediates mitochondrial membrane depolarization and decreases complex IV activity, and p53 inhibition or genetic deletion ameliorates these changes in a cell culture model. PGC-1α is a transcriptional co-activator that regulates mitochondrial biogenesis and oxidative phosphorylation (Cui et al., 2006; Weydt et al., 2006). A role of PGC-1α in HD pathogenesis was suspected from the observation of selective striatal lesions in PGC-1α knock-mice (Lin et al., 2004). A direct link between CREB phosphorylation and transcriptional regulation at the PGC-1α promoter has been observed in neuronal cells (Cui et al., 2006). Impairment of the CREB/PGC-1α signaling cascade by suppression of excitatory synaptic activity or by stimulation of extracellular NMDA receptors increases the vulnerability of HD neuronal cells (Okamoto et al., 2009). Mitogen and stress-activated protein kinase-1 (MSK-1), a nuclear protein kinase activated downstream of ERK, was shown to control the expression levels of PGC-1α via increased binding of phosphorylated-CREB and Histone H3 at the promoter region of PGC-1α. Overexpression of MSK-1 was protective against Exp-HTT-induced striatal dysfunctions in vitro (Roze et al., 2008) and in vivo (Martin et al., 2011; Figure 1).
Acting on Chromatin Remodeling to Improve Transcriptional Dysregulation in HD
Chromatin remodeling that underlies DNA decompaction was first described in dividing cells, but is also one of the prime events of transcription in post-mitotic mature neurons (Taniura et al., 2007). This “above the genome” molecular mechanism, also called an epigenetic mechanism, gates DNA access, and hence transcription. It is critically controlled by post-translational modifications of histones (H2A and H2B, H3 and H4), a group of highly basic proteins tightly linked to DNA. In particular, the methylation and acetylation state of histones is closely linked to the regions of transcriptional activity by regulating transcription factor access to promoter regions in the DNA. Histone acetylation at a promoter generally increases transcription and the enzymes that catalyze these reactions are histone acetyltransferases (HATs). By contrast, HDACs catalyze deacetylation. Exp-HTT interacts with CBP and blocks its intrinsic HAT activity (Steffan et al., 2001). Administration of HDACs inhibitors, including SAHA, sodium butyrate, and phenylbutyrate, has demonstrated their therapeutic role in several HD models (Steffan et al., 2000; Ferrante et al., 2003; Hockly et al., 2003; Gardian et al., 2005), as they improved behavioral performance and neuronal survival. Interestingly, administration of a new benzamide-type HDAC inhibitor with lower potential toxicity than previous HDAC inhibitors, HDACi 4b, also restores the transcription of critical striatal genes and improves motor and neuropathological phenotype of R6/2 HD mice (Thomas et al., 2008). All these HDAC inhibitors act broadly across various classes of HDACs (Lesort et al., 1999). Inhibitors targeting a specific class of HDACs may result in a better benefit to side effect ratio (Pallos et al., 2008). Finally, it must be emphasized that the levels of acetylated histones are not decreased globally in HD mice models, but rather selectively in the promoters of genes that are specifically down-regulated in HD (Sadri-Vakili et al., 2007).
Methylation of histones plays the opposite, inhibitory role on transcription. One of the proteins involved in methyltransferase activity at histone H3 (K9) is ERG-associated protein with SET domain (ESET). ESET expression is increased in HD patients and R6/2 HD mice (Ryu et al., 2006). Sp1 acts as a transcriptional activator of the ESET promoter at guanosine–cytosine (GC)-rich DNA binding sites (Yang et al., 2003). Inhibiting Sp1 binding to these sites using mitramycin (a clinically approved antitumor antibiotic) suppressed basal ESET promoter activity in a dose-dependent manner. The combined pharmacological treatment of mithramycin and cystamine, down-regulates ESET gene expression and hypertrimethylation of histone H3. This treatment significantly ameliorates the behavioral and neuropathological phenotype of R6/2 HD mice and improves their survival. Owing to its HEAT repeat α-solenoid structure, HTT acts as a facilitator of the epigenetic silencer polycomb repressive complex 2 (PRC2; Seong et al., 2010). The polyglutamine region augments PRC2 stimulation and hence H3 trimethylation on specific promoters, including Hoxb9. In general, the DNA/RNA binding agents anthracyclines are thought to provide a significant therapeutic potential by correcting the pathological nucleosome changes and realigning transcription. Two such agents, chromomycin and mithramycin, were found to improve altered nucleosomal homeostasis, and by virtue of normalizing the shift in the balance between methylation and acetylation in HD mice, could alter a subset of down-regulated genes accompanied by a significant improvement of the behavioral and neuropathological phenotype observed in HD mice (Stack et al., 2007).
The transcriptional co-repressor transglutaminase 2 (TG2), which is up-regulated in HD, interacts physically with Histone H3 and could contribute to gene silencing via hyperpolyamination of histone tails (McConoughey et al., 2010). The inhibition of TG2 increases PGC-1α expression, but also that of 40% of genes that are dysregulated (Karpuj et al., 1999; Lesort et al., 1999) in HD striatal neurons. Histone H2A ubiquitinylation is increased in R6/2 HD mice and the association of ubiquitinylated H2A with the promoters of down-regulated genes is increased in an in vitro model of HD (Kim et al., 2008). This transcriptional repression is rescued by restoration of the ubiquitinylated H2A level. In addition, histone H2B ubiquitinylation is decreased in R6/2 HD mice and association of ubiquitinylated H2B with promoters positively correlates with transcriptional level in R6/2 mice. This transcriptional modulation by H2 ubiquitinylation is thought to occur through a subsequent interference with methylation of histone H3 (Kim et al., 2008).
Histone H3 phosphorylation is also critical to induce the nucleosomal response and gene transcription at some promoters. MSK-1 is critically involved in Histone H3 phosphorylation in the striatum (Brami-Cherrier et al., 2005, 2009). It is deficient in the striatum of R6/2 mice and postmortem caudate of HD patients (Roze et al., 2008). Restoring MSK-1 expression and subsequent striatal H3 phosphorylation in an in vitro model system of HD protects against neuronal alteration induced by the Exp-HTT including neuritic retraction, aggregate formation, and neuronal death (Roze et al., 2008). In vivo, in a rat model of HD based on striatal lentiviral expression of Exp-HTT, overexpression of MSK-1 induced hyperphosphorylation of H3 and CREB, along with an overexpression of PGC-1α (Martin et al., 2011). Similarly to PGC-1α, MSK-1 protects from Exp-HTT-induced striatal dysfunctions, including DARPP-32 down-regulation and neuronal death. Furthermore MSK-1 knock-out mice are more susceptible to 3-NP-induced striatal lesion, and aging MSK-1 knock-out mice show spontaneous striatal degeneration (Martin et al., 2011).
Conclusion
The regulation of neuronal death has been intensely investigated and, due to its paramount implications for neurodegenerative disease, has sparked one of the most prolific research fields in the past decades. In particular, basic research has provided novel insights into the molecular machinery of neuronal signaling, and how it mediates neuronal dysfunction and death in progressive neurodegenerative diseases. HD involves a complex pathological cascade with multiple deleterious mechanisms, affecting predominantly the striatal neurons. This striatal vulnerability to HD-induced neuronal dysfunction reflects both the particular characteristics of striatal neurons (cell-autonomous alterations) and their location within the functional neuronal networks (non-cell-autonomous alterations). The various pathological events are not proceeding in succession but instead take place in parallel with continuous reciprocal interactions. This parallel and interactive nature should be taken into account for both the general understanding of the HD-related neurodegeneration and any therapeutic approach.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Centre National pour la Recherche Scientifique, Université Pierre and Marie Curie, Ministère de la Recherche Scientifique. Jocelyne Caboche was funded by Fondation pour la Recherche sur le Cerveau and Hereditary Disease Foundation. Emma Cahill is supported by the Ecole des Neurosciences de Paris, Elodie Martin was a recipient from Ministère de la Recherche Scientifique, and Fondation Huntington France. Cecilia Bonnet is supported by the Czech Ministry of Education, Grantove Agentury UK and IGA.
References
Alford, R. L., Ashizawa, T., Jankovic, J., Caskey, C. T., and Richards, C. S. (1996). Molecular detection of new mutations, resolution of ambiguous results and complex genetic counseling issues in Huntington disease. Am. J. Med. Genet. 66, 281–286.
Andresen, J. M., Gayan, J., Cherny, S. S., Brocklebank, D., Alkorta-Aranburu, G., Addis, E. A., Cardon, L. R., Housman, D. E., and Wexler, N. S. (2007). Replication of twelve association studies for Huntington’s disease residual age of onset in large Venezuelan kindreds. J. Med. Genet. 44, 44–50.
Andrew, S. E., Goldberg, Y. P., Kremer, B., Telenius, H., Theilmann, J., Adam, S., Starr, E., Squitieri, F., Lin, B., Kalchman, M. A., Graham, R. K., and Hayden, M. R. (1993). The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 4, 398–403.
Anne, S. L., Saudou, F., and Humbert, S. (2007). Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J. Neurosci. 27, 7318–7328.
Apostol, B. L., Simmons, D. A., Zuccato, C., Illes, K., Pallos, J., Casale, M., Conforti, P., Ramos, C., Roarke, M., Kathuria, S., Cattaneo, E., Marsh, J. L., and Thompson, L. M. (2008). CEP-1347 reduces mutant huntingtin-associated neurotoxicity and restores BDNF levels in R6/2 mice. Mol. Cell. Neurosci. 39, 8–20.
Arango, M., Holbert, S., Zala, D., Brouillet, E., Pearson, J., Regulier, E., Thakur, A. K., Aebischer, P., Wetzel, R., Deglon, N., and Neri, C. (2006). CA150 expression delays striatal cell death in overexpression and knock-in conditions for mutant huntingtin neurotoxicity. J. Neurosci. 26, 4649–4659.
Arenas, J., Campos, Y., Ribacoba, R., Martin, M. A., Rubio, J. C., Ablanedo, P., and Cabello, A. (1998). Complex I defect in muscle from patients with Huntington’s disease. Ann. Neurol. 43, 397–400.
Arnulf, I., Nielsen, J., Lohmann, E., Schiefer, J., Wild, E., Jennum, P., Konofal, E., Walker, M., Oudiette, D., Tabrizi, S., and Durr, A. (2008). Rapid eye movement sleep disturbances in Huntington disease. Arch. Neurol. 65, 482–488.
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810.
Augood, S. J., Faull, R. L., and Emson, P. C. (1997). Dopamine D1 and D2 receptor gene expression in the striatum in Huntington’s disease. Ann. Neurol. 42, 215–221.
Augood, S. J., Faull, R. L., Love, D. R., and Emson, P. C. (1996). Reduction in enkephalin and substance P messenger RNA in the striatum of early grade Huntington’s disease: a detailed cellular in situ hybridization study. Neuroscience 72, 1023–1036.
Aziz, N. A., Van Der Burg, J. M., Landwehrmeyer, G. B., Brundin, P., Stijnen, T., and Roos, R. A. (2008). Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 71, 1506–1513.
Bae, B. I., Xu, H., Igarashi, S., Fujimuro, M., Agrawal, N., Taya, Y., Hayward, S. D., Moran, T. H., Montell, C., Ross, C. A., Snyder, S. H., and Sawa, A. (2005). p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 47, 29–41.
Bamford, K. A., Caine, E. D., Kido, D. K., Cox, C., and Shoulson, I. (1995). A prospective evaluation of cognitive decline in early Huntington’s disease: functional and radiographic correlates. Neurology 45, 1867–1873.
Beal, M. F., Kowall, N. W., Ellison, D. W., Mazurek, M. F., Swartz, K. J., and Martin, J. B. (1986). Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature 321, 168–171.
Behrens, P. F., Franz, P., Woodman, B., Lindenberg, K. S., and Landwehrmeyer, G. B. (2002). Impaired glutamate transport and glutamate–glutamine cycling: downstream effects of the Huntington mutation. Brain 125, 1908–1922.
Bence, N. F., Sampat, R. M., and Kopito, R. R. (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555.
Benchoua, A., Trioulier, Y., Diguet, E., Malgorn, C., Gaillard, M. C., Dufour, N., Elalouf, J. M., Krajewski, S., Hantraye, P., Deglon, N., and Brouillet, E. (2008). Dopamine determines the vulnerability of striatal neurons to the N-terminal fragment of mutant huntingtin through the regulation of mitochondrial complex II. Hum. Mol. Genet. 17, 1446–1456.
Benchoua, A., Trioulier, Y., Zala, D., Gaillard, M. C., Lefort, N., Dufour, N., Saudou, F., Elalouf, J. M., Hirsch, E., Hantraye, P., Deglon, N., and Brouillet, E. (2006). Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol. Biol. Cell 17, 1652–1663.
Bennett, E. J., Shaler, T. A., Woodman, B., Ryu, K. Y., Zaitseva, T. S., Becker, C. H., Bates, G. P., Schulman, H., and Kopito, R. R. (2007). Global changes to the ubiquitin system in Huntington’s disease. Nature 448, 704–708.
Borovecki, F., Lovrecic, L., Zhou, J., Jeong, H., Then, F., Rosas, H. D., Hersch, S. M., Hogarth, P., Bouzou, B., Jensen, R. V., and Krainc, D. (2005). Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 102, 11023–11028.
Borrell-Pages, M., Canals, J. M., Cordelieres, F. P., Parker, J. A., Pineda, J. R., Grange, G., Bryson, E. A., Guillermier, M., Hirsch, E., Hantraye, P., Cheetham, M. E., Neri, C., Alberch, J., Brouillet, E., Saudou, F., and Humbert, S. (2006a). Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J. Clin. Invest. 116, 1410–1424.
Borrell-Pages, M., Zala, D., Humbert, S., and Saudou, F. (2006b). Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell. Mol. Life Sci. 63, 2642–2660.
Boutell, J. M., Thomas, P., Neal, J. W., Weston, V. J., Duce, J., Harper, P. S., and Jones, A. L. (1999). Aberrant interactions of transcriptional repressor proteins with the Huntington’s disease gene product, huntingtin. Hum. Mol. Genet. 8, 1647–1655.
Brami-Cherrier, K., Roze, E., Girault, J. A., Betuing, S., and Caboche, J. (2009). Role of the ERK/MSK1 signalling pathway in chromatin remodelling and brain responses to drugs of abuse. J. Neurochem. 108, 1323–1335.
Brami-Cherrier, K., Valjent, E., Herve, D., Darragh, J., Corvol, J. C., Pages, C., Arthur, S. J., Girault, J. A., and Caboche, J. (2005). Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J. Neurosci. 25, 11444–11454.
Brouillet, E., Conde, F., Beal, M. F., and Hantraye, P. (1999). Replicating Huntington’s disease phenotype in experimental animals. Prog. Neurobiol. 59, 427–468.
Brouillet, E., Guyot, M. C., Mittoux, V., Altairac, S., Conde, F., Palfi, S., and Hantraye, P. (1998). Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J. Neurochem. 70, 794–805.
Busse, M. E., Wiles, C. M., and Rosser, A. E. (2009). Mobility and falls in people with Huntington’s disease. J. Neurol. Neurosurg. Psychiatr. 80, 88–90.
Caine, E. D., Ebert, M. H., and Weingartner, H. (1977). An outline for the analysis of dementia. The memory disorder of Huntingtons disease. Neurology 27, 1087–1092.
Caine, E. D., Hunt, R. D., Weingartner, H., and Ebert, M. H. (1978). Huntington’s dementia. Clinical and neuropsychological features. Arch. Gen. Psychiatry 35, 377–384.
Caine, E. D., and Shoulson, I. (1983). Psychiatric syndromes in Huntington’s disease. Am. J. Psychiatry 140, 728–733.
Canals, J. M., Pineda, J. R., Torres-Peraza, J. F., Bosch, M., Martin-Ibanez, R., Munoz, M. T., Mengod, G., Ernfors, P., and Alberch, J. (2004). Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J. Neurosci. 24, 7727–7739.
Carella, F., Scaioli, V., Ciano, C., Binelli, S., Oliva, D., and Girotti, F. (1993). Adult onset myoclonic Huntington’s disease. Mov. Disord. 8, 201–205.
Carmichael, J., Chatellier, J., Woolfson, A., Milstein, C., Fersht, A. R., and Rubinsztein, D. C. (2000). Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 97, 9701–9705.
Caviston, J. P., and Holzbaur, E. L. (2009). Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 19, 147–155.
Caviston, J. P., Ross, J. L., Antony, S. M., Tokito, M., and Holzbaur, E. L. (2007). Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. U.S.A. 104, 10045–10050.
Cepeda, C., Ariano, M. A., Calvert, C. R., Flores-Hernandez, J., Chandler, S. H., Leavitt, B. R., Hayden, M. R., and Levine, M. S. (2001). NMDA receptor function in mouse models of Huntington disease. J. Neurosci. Res. 66, 525–539.
Cha, J. H. (2007). Transcriptional signatures in Huntington’s disease. Prog. Neurobiol. 83, 228–248.
Cha, J. H., Frey, A. S., Alsdorf, S. A., Kerner, J. A., Kosinski, C. M., Mangiarini, L., Penney, J. B. Jr., Davies, S. W., Bates, G. P., and Young, A. B. (1999). Altered neurotransmitter receptor expression in transgenic mouse models of Huntington’s disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 981–989.
Cha, J. H., Kosinski, C. M., Kerner, J. A., Alsdorf, S. A., Mangiarini, L., Davies, S. W., Penney, J. B., Bates, G. P., and Young, A. B. (1998). Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc. Natl. Acad. Sci. U.S.A. 95, 6480–6485.
Charvin, D., Roze, E., Perrin, V., Deyts, C., Betuing, S., Pages, C., Regulier, E., Luthi-Carter, R., Brouillet, E., Deglon, N., and Caboche, J. (2008). Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo. Neurobiol. Dis. 29, 22–29.
Charvin, D., Vanhoutte, P., Pages, C., Borrelli, E., and Caboche, J. (2005). Unraveling a role for dopamine in Huntington’s disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc. Natl. Acad. Sci. U.S.A. 102, 12218–12223.
Chopra, V., Fox, J. H., Lieberman, G., Dorsey, K., Matson, W., Waldmeier, P., Housman, D. E., Kazantsev, A., Young, A. B., and Hersch, S. (2007). A small-molecule therapeutic lead for Huntington’s disease: preclinical pharmacology and efficacy of C2-8 in the R6/2 transgenic mouse. Proc. Natl. Acad. Sci. U.S.A. 104, 16685–16689.
Colin, E., Zala, D., Liot, G., Rangone, H., Borrel-Pagès, M., Li, X. J., Saudou, F., and Humbert, S. (2008). Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 27, 2124–2134.
Cui, L., Jeong, H., Borovecki, F., Parkhurst, C. N., Tanese, N., and Krainc, D. (2006). Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69.
Cull-Candy, S., Brickley, S., and Farrant, M. (2001). NMDA receptor subunits: diversity, development and disease. Curr. Opin. Neurobiol. 11, 327–335.
Cummings, J. L. (1995). Behavioral and psychiatric symptoms associated with Huntington’s disease. Adv. Neurol. 65, 179–186.
Cummings, J. L., and Cunningham, K. (1992). Obsessive-compulsive disorder in Huntington’s disease. Biol. Psychiatry 31, 263–270.
Cyr, M., Beaulieu, J. M., Laakso, A., Sotnikova, T. D., Yao, W. D., Bohn, L. M., Gainetdinov, R. R., and Caron, M. G. (2003). Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc. Natl. Acad. Sci. U.S.A. 100, 11035–11040.
Cyr, M., Sotnikova, T. D., Gainetdinov, R. R., and Caron, M. G. (2006). Dopamine enhances motor and neuropathological consequences of polyglutamine expanded huntingtin. FASEB J. 20, 2541–2543.
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A., Scherzinger, E., Wanker, E. E., Mangiarini, L., and Bates, G. P. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548.
Davis, M. B., Bateman, D., Quinn, N. P., Marsden, C. D., and Harding, A. E. (1994). Mutation analysis in patients with possible but apparently sporadic Huntington’s disease. Lancet 344, 714–717.
Dawbarn, D., De Quidt, M. E., and Emson, P. C. (1985). Survival of basal ganglia neuropeptide Y-somatostatin neurones in Huntington’s disease. Brain Res. 340, 251–260.
De Almeida, L. P., Ross, C. A., Zala, D., Aebischer, P., and Deglon, N. (2002). Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J. Neurosci. 22, 3473–3483.
Del Toro, D., Canals, J. M., Gines, S., Kojima, M., Egea, G., and Alberch, J. (2006). Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J. Neurosci. 26, 12748–12757.
Desplats, P. A., Kass, K. E., Gilmartin, T., Stanwood, G. D., Woodward, E. L., Head, S. R., Sutcliffe, J. G., and Thomas, E. A. (2006). Selective deficits in the expression of striatal-enriched mRNAs in Huntington’s disease. J. Neurochem. 96, 743–757.
Deyts, C., Galan-Rodriguez, B., Martin, E., Bouveyron, N., Roze, E., Charvin, D., Caboche, J., and Betuing, S. (2009). Dopamine D2 receptor stimulation potentiates PolyQ-Huntingtin-induced mouse striatal neuron dysfunctions via Rho/ROCK-II activation. PLoS ONE 4, e8287. doi: 10.1371/journal.pone.0008287
Di Maio, L., Squitieri, F., Napolitano, G., Campanella, G., Trofatter, J. A., and Conneally, P. M. (1993). Onset symptoms in 510 patients with Huntington’s disease. J. Med. Genet. 30, 289–292.
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., and Aronin, N. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993.
Diguet, E., Petit, F., Escartin, C., Cambon, K., Bizat, N., Dufour, N., Hantraye, P., Deglon, N., and Brouillet, E. (2009). Normal aging modulates the neurotoxicity of mutant huntingtin. PLoS ONE 4, e4637. doi: 10.1371/journal.pone.0004637
Dingledine, R., Borges, K., Bowie, D., and Traynelis, S. F. (1999). The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61.
Djousse, L., Knowlton, B., Cupples, L. A., Marder, K., Shoulson, I., and Myers, R. H. (2002). Weight loss in early stage of Huntington’s disease. Neurology 59, 1325–1330.
Dompierre, J. P., Godin, J. D., Charrin, B. C., Cordelieres, F. P., King, S. J., Humbert, S., and Saudou, F. (2007). Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 27, 3571–3583.
Dragatsis, I., Dietrich, P., and Zeitlin, S. (2000). Expression of the Huntingtin-associated protein 1 gene in the developing and adult mouse. Neurosci. Lett. 282, 37–40.
Dunah, A. W., Jeong, H., Griffin, A., Kim, Y. M., Standaert, D. G., Hersch, S. M., Mouradian, M. M., Young, A. B., Tanese, N., and Krainc, D. (2002). Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science 296, 2238–2243.
Duyao, M., Ambrose, C., Myers, R., Novelletto, A., Persichetti, F., Frontali, M., Folstein, S., Ross, C., Franz, M., Abbott, M., Gray, J., Conneally, P., Young, A., Penney, J., Hollingsworth, Z., Shoulson, I., Lazzarini, A., Falek, A., Koroshetz, W., Sax, D., Bird, E., Vonsattel, J., Bonilla, E., Alvir, J., Bickham Conde, J., Cha, J.-H., Dure, L., Gomez, F., Ramos, M., Sanchez-Ramos, J., Snodgrass, S., de Young, M., Wexler, N., Moscowitz, C., Penchaszadeh, G., MacFarlane, H., Anderson, M., Jenkins, B., Srinidhi, J., Barnes, G., Gusella, J., and MacDonald, M. (1993). Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Genet. 4, 387–392.
Duyao, M. P., Auerbach, A. B., Ryan, A., Persichetti, F., Barnes, G. T., Mcneil, S. M., Ge, P., Vonsattel, J. P., Gusella, J. F., Joyner, A. L., and MacDonald, M. E. (1995). Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 269, 407–410.
Fan, J., Cowan, C. M., Zhang, L. Y., Hayden, M. R., and Raymond, L. A. (2009). Interaction of postsynaptic density protein-95 with NMDA receptors influences excitotoxicity in the yeast artificial chromosome mouse model of Huntington’s disease. J. Neurosci. 29, 10928–10938.
Fan, M. M., Fernandes, H. B., Zhang, L. Y., Hayden, M. R., and Raymond, L. A. (2007). Altered NMDA receptor trafficking in a yeast artificial chromosome transgenic mouse model of Huntington’s disease. J. Neurosci. 27, 3768–3779.
Fedoroff, J. P., Peyser, C., Franz, M. L., and Folstein, S. E. (1994). Sexual disorders in Huntington’s disease. J. Neuropsychiatry Clin. Neurosci. 6, 147–153.
Ferrante, R. J., Kowall, N. W., Beal, M. F., Martin, J. B., Bird, E. D., and Richardson, E. P. Jr. (1987). Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 46, 12–27.
Ferrante, R. J., Kowall, N. W., Beal, M. F., Richardson, E. P. Jr., Bird, E. D., and Martin, J. B. (1985). Selective sparing of a class of striatal neurons in Huntington’s disease. Science 230, 561–563.
Ferrante, R. J., Kowall, N. W., Cipolloni, P. B., Storey, E., and Beal, M. F. (1993). Excitotoxin lesions in primates as a model for Huntington’s disease: histopathologic and neurochemical characterization. Exp. Neurol. 119, 46–71.
Ferrante, R. J., Kowall, N. W., and Richardson, E. P. Jr. (1991). Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J. Neurosci. 11, 3877–3887.
Ferrante, R. J., Kubilus, J. K., Lee, J., Ryu, H., Beesen, A., Zucker, B., Smith, K., Kowall, N. W., Ratan, R. R., Luthi-Carter, R., and Hersch, S. M. (2003). Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 23, 9418–9427.
Ferrer, I., Goutan, E., Marin, C., Rey, M. J., and Ribalta, T. (2000). Brain-derived neurotrophic factor in Huntington disease. Brain Res. 866, 257–261.
Folstein, S. E., Franz, M. L., Jensen, B. A., Chase, G. A., and Folstein, M. F. (1983). Conduct disorder and affective disorder among the offspring of patients with Huntington’s disease. Psychol. Med. 13, 45–52.
Gambardella, A., Muglia, M., Labate, A., Magariello, A., Gabriele, A. L., Mazzei, R., Pirritano, D., Conforti, F. L., Patitucci, A., Valentino, P., Zappia, M., and Quattrone, A. (2001). Juvenile Huntington’s disease presenting as progressive myoclonic epilepsy. Neurology 57, 708–711.
Garcia, M., Charvin, D., and Caboche, J. (2004). Expanded huntingtin activates the c-Jun terminal kinase/c-Jun pathway prior to aggregate formation in striatal neurons in culture. Neuroscience 127, 859–870.
Garcia, M., Vanhoutte, P., Pages, C., Besson, M. J., Brouillet, E., and Caboche, J. (2002). The mitochondrial toxin 3-nitropropionic acid induces striatal neurodegeneration via a c-Jun N-terminal kinase/c-Jun module. J. Neurosci. 22, 2174–2184.
Gardian, G., Browne, S. E., Choi, D. K., Klivenyi, P., Gregorio, J., Kubilus, J. K., Ryu, H., Langley, B., Ratan, R. R., Ferrante, R. J., and Beal, M. F. (2005). Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 280, 556–563.
Gauthier, L. R., Charrin, B. C., Borrell-Pages, M., Dompierre, J. P., Rangone, H., Cordelieres, F. P., De Mey, J., Macdonald, M. E., Lessmann, V., Humbert, S., and Saudou, F. (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118, 127–138.
Gharami, K., Xie, Y., An, J. J., Tonegawa, S., and Xu, B. (2008). Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington’s disease phenotypes in mice. J. Neurochem. 105, 369–379.
Goldberg, Y. P., Rommens, J. M., Andrew, S. E., Hutchinson, G. B., Lin, B., Theilmann, J., Graham, R., Glaves, M. L., Starr, E., McDonald, H., Nasir, J., Schappert, K., Kalchman, M. A., Clark, L. A., and Hayden, M. R. (1993). Identification of an Alu retrotransposition event in close proximity to a strong candidate gene for Huntington’s disease. Nature 362, 370–373.
Gong, B., Lim, M. C., Wanderer, J., Wyttenbach, A., and Morton, A. J. (2008). Time-lapse analysis of aggregate formation in an inducible PC12 cell model of Huntington’s disease reveals time-dependent aggregate formation that transiently delays cell death. Brain Res. Bull. 75, 146–157.
Gonzalez-Alegre, P., and Afifi, A. K. (2006). Clinical characteristics of childhood-onset (juvenile) Huntington disease: report of 12 patients and review of the literature. J. Child Neurol. 21, 223–229.
Goto, S., Hirano, A., and Rojas-Corona, R. R. (1989). Immunohistochemical visualization of afferent nerve terminals in human globus pallidus and its alteration in neostriatal neurodegenerative disorders. Acta Neuropathol. 78, 543–550.
Grafton, S. T., Mazziotta, J. C., Pahl, J. J., St George-Hyslop, P., Haines, J. L., Gusella, J., Hoffman, J. M., Baxter, L. R., and Phelps, M. E. (1992). Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington’s disease. Arch. Neurol. 49, 1161–1167.
Graham, R. K., Pouladi, M. A., Joshi, P., Lu, G., Deng, Y., Wu, N.-P., Figueroa, B. E., Metzler, M., André, V. M., Slow, E. J., Raymond, L., Friedlander, R., Levine, M. S., Leavitt, B. R., and Hayden, M. R. (2009). Differential susceptibility to excitotoxic stress in YAC128 mouse models of HD between initiation and progression of disease. J. Neurosci. 29, 2193–2204.
Graveland, G. A., Williams, R. S., and DiFiglia, M. (1985). Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 227, 770–773.
Gu, X., Andre, V. M., Cepeda, C., Li, S. H., Li, X. J., Levine, M. S., and Yang, X. W. (2007). Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington’s disease. Mol. Neurodegener. 2, 8.
Gunawardena, S., Her, L. S., Brusch, R. G., Laymon, R. A., Niesman, I. R., Gordesky-Gold, B., Sintasath, L., Bonini, N. M., and Goldstein, L. S. (2003). Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40, 25–40.
Gutekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S., Jones, R., Rye, D., Ferrante, R. J., Hersch, S. M., and Li, X. J. (1999). Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J. Neurosci. 19, 2522–2534.
Hamilton, J. M., Wolfson, T., Peavy, G. M., Jacobson, M. W., and Corey-Bloom, J. (2004). Rate and correlates of weight change in Huntington’s disease. J. Neurol. Neurosurg. Psychiatr. 75, 209–212.
Hardingham, G. E., Arnold, F. J., and Bading, H. (2001). Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat. Neurosci. 4, 261–267.
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696.
Hardingham, G. E., Fukunaga, Y., and Bading, H. (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5, 405–414.
Harjes, P., and Wanker, E. E. (2003). The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem. Sci. 28, 425–433.
Heng, M. Y., Detloff, P. J., Wang, P. L., Tsien, J. Z., and Albin, R. L. (2009). In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J. Neurosci. 29, 3200–3205.
Herbst, M., and Wanker, E. E. (2007). Small molecule inducers of heat-shock response reduce polyQ-mediated huntingtin aggregation. A possible therapeutic strategy. Neurodegener. Dis. 4, 254–260.
Ho, A. K., Sahakian, B. J., Brown, R. G., Barker, R. A., Hodges, J. R., Ane, M. N., Snowden, J., Thompson, J., Esmonde, T., Gentry, R., Moore, J. W., and Bodner, T. (2003). Profile of cognitive progression in early Huntington’s disease. Neurology 61, 1702–1706.
Hockly, E., Richon, V. M., Woodman, B., Smith, D. L., Zhou, X., Rosa, E., Sathasivam, K., Ghazi-Noori, S., Mahal, A., Lowden, P. A., Steffan, J. S., Marsh, J. L., Thompson, L. M., Lewis, C. M., Marks, P. A., and Bates, G. P. (2003). Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 100, 2041–2046.
Hodges, J. R., Salmon, D. P., and Butters, N. (1990). Differential impairment of semantic and episodic memory in Alzheimer’s and Huntington’s diseases: a controlled prospective study. J. Neurol. Neurosurg. Psychiatr. 53, 1089–1095.
Hoffner, G., Island, M. L., and Djian, P. (2005). Purification of neuronal inclusions of patients with Huntington’s disease reveals a broad range of N-terminal fragments of expanded huntingtin and insoluble polymers. J. Neurochem. 95, 125–136.
Huang, C. C., Faber, P. W., Persichetti, F., Mittal, V., Vonsattel, J. P., Macdonald, M. E., and Gusella, J. F. (1998). Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell Mol. Genet. 24, 217–233.
Hunter, J. M., Lesort, M., and Johnson, G. V. (2007). Ubiquitin-proteasome system alterations in a striatal cell model of Huntington’s disease. J. Neurosci. Res. 85, 1774–1788.
Jakel, R. J., and Maragos, W. F. (2000). Neuronal cell death in Huntington’s disease: a potential role for dopamine. Trends Neurosci. 23, 239–245.
Jana, N. R., Tanaka, M., Wang, G., and Nukina, N. (2000). Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 9, 2009–2018.
Jana, N. R., Zemskov, E. A., Wang, G., and Nukina, N. (2001). Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 10, 1049–1059.
Jankovic, J., Beach, J., and Ashizawa, T. (1995). Emotional and functional impact of DNA testing on patients with symptoms of Huntington’s disease. J. Med. Genet. 32, 516–518.
Jenkins, B. G., Koroshetz, W. J., Beal, M. F., and Rosen, B. R. (1993). Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology 43, 2689–2695.
Jenkins, B. G., Rosas, H. D., Chen, Y. C., Makabe, T., Myers, R., Macdonald, M., Rosen, B. R., Beal, M. F., and Koroshetz, W. J. (1998). 1H NMR spectroscopy studies of Huntington’s disease: correlations with CAG repeat numbers. Neurology 50, 1357–1365.
Jensen, P., Fenger, K., Bolwig, T. G., and Sorensen, S. A. (1998). Crime in Huntington’s disease: a study of registered offences among patients, relatives, and controls. J. Neurol. Neurosurg. Psychiatr. 65, 467–471.
Jocoy, E. L., Andre, V. M., Cummings, D. M., Rao, S. P., Wu, N., Ramsey, A. J., Caron, M. G., Cepeda, C., and Levine, M. S. (2011). Dissecting the contribution of individual receptor subunits to the enhancement of N-methyl-d-aspartate currents by dopamine D1 receptor activation in striatum. Front. Syst. Neurosci. 5:28. doi: 10.3389/fnsys.2011.00004
Kaltschmidt, B., Widera, D., and Kaltschmidt, C. (2005). Signaling via NF-kappaB in the nervous system. Biochim. Biophys. Acta 1745, 287–299.
Kaminosono, S., Saito, T., Oyama, F., Ohshima, T., Asada, A., Nagai, Y., Nukina, N., and Hisanaga, S. (2008). Suppression of mutant Huntingtin aggregate formation by Cdk5/p35 through the effect on microtubule stability. J. Neurosci. 28, 8747–8755.
Karpuj, M. V., Garren, H., Slunt, H., Price, D. L., Gusella, J., Becher, M. W., and Steinman, L. (1999). Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington’s disease brain nuclei. Proc. Natl. Acad. Sci. U.S.A. 96, 7388–7393.
Kazantsev, A., Preisinger, E., Dranovsky, A., Goldgaber, D., and Housman, D. (1999). Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 96, 11404–11409.
Kegel, K. B., Meloni, A. R., Yi, Y., Kim, Y. J., Doyle, E., Cuiffo, B. G., Sapp, E., Wang, Y., Qin, Z. H., Chen, J. D., Nevins, J. R., Aronin, N., and DiFiglia, M. (2002). Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J. Biol. Chem. 277, 7466–7476.
Kegel, K. B., Sapp, E., Yoder, J., Cuiffo, B., Sobin, L., Kim, Y. J., Qin, Z. H., Hayden, M. R., Aronin, N., Scott, D. L., Isenberg, G., Goldmann, W. H., and DiFiglia, M. (2005). Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol. Chem. 280, 36464–36473.
Kenney, C., Powell, S., and Jankovic, J. (2007). Autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov. Disord. 22, 127–130.
Kerbeshian, J., Burd, L., Leech, C., and Rorabaugh, A. (1991). Huntington disease and childhood-onset Tourette syndrome. Am. J. Med. Genet. 39, 1–3.
Kiechle, T., Dedeoglu, A., Kubilus, J., Kowall, N. W., Beal, M. F., Friedlander, R. M., Hersch, S. M., and Ferrante, R. J. (2002). Cytochrome C and caspase-9 expression in Huntington’s disease. Neuromolecular Med. 1, 183–195.
Kim, M. O., Chawla, P., Overland, R. P., Xia, E., Sadri-Vakili, G., and Cha, J. H. (2008). Altered histone monoubiquitylation mediated by mutant huntingtin induces transcriptional dysregulation. J. Neurosci. 28, 3947–3957.
Kirkwood, S. C., Siemers, E., Viken, R., Hodes, M. E., Conneally, P. M., Christian, J. C., and Foroud, T. (2002). Longitudinal personality changes among presymptomatic Huntington disease gene carriers. Neuropsychiatry Neuropsychol. Behav. Neurol. 15, 192–197.
Kremer, B., Weber, B., and Hayden, M. R. (1992). New insights into the clinical features, pathogenesis and molecular genetics of Huntington disease. Brain Pathol. 2, 321–335.
Kuhn, A., Goldstein, D. R., Hodges, A., Strand, A. D., Sengstag, T., Kooperberg, C., Becanovic, K., Pouladi, M. A., Sathasivam, K., Cha, J. H., Hannan, A. J., Hayden, M. R., Leavitt, B. R., Dunnett, S. B., Ferrante, R. J., Albin, R., Shelbourne, P., Delorenzi, M., Augood, S. J., Faull, R. L., Olson, J. M., Bates, G. P., Jones, L., and Luthi-Carter, R. (2007). Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 16, 1845–1861.
Kuppenbender, K. D., Standaert, D. G., Feuerstein, T. J., Penney, J. B. Jr., Young, A. B., and Landwehrmeyer, G. B. (2000). Expression of NMDA receptor subunit mRNAs in neurochemically identified projection and interneurons in the human striatum. J. Comp. Neurol. 419, 407–421.
Kuwert, T., Lange, H. W., Boecker, H., Titz, H., Herzog, H., Aulich, A., Wang, B. C., Nayak, U., and Feinendegen, L. E. (1993). Striatal glucose consumption in chorea-free subjects at risk of Huntington’s disease. J. Neurol. 241, 31–36.
Landwehrmeyer, G. B., Standaert, D. G., Testa, C. M., Penney, J. B. Jr., and Young, A. B. (1995). NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J. Neurosci. 15, 5297–5307.
Lange, H., Thorner, G., Hopf, A., and Schroder, K. F. (1976). Morphometric studies of the neuropathological changes in choreatic diseases. J. Neurol. Sci. 28, 401–425.
Lee, W. C., Yoshihara, M., and Littleton, J. T. (2004). Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 3224–3229.
Lesort, M., Chun, W., Johnson, G. V., and Ferrante, R. J. (1999). Tissue transglutaminase is increased in Huntington’s disease brain. J. Neurochem. 73, 2018–2027.
Levine, M. S., Klapstein, G. J., Koppel, A., Gruen, E., Cepeda, C., Vargas, M. E., Jokel, E. S., Carpenter, E. M., Zanjani, H., Hurst, R. S., Efstratiadis, A., Zeitlin, S., and Chesselet, M. F. (1999). Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J. Neurosci. Res. 58, 515–532.
Li, H., Li, S. H., Johnston, H., Shelbourne, P. F., and Li, X. J. (2000). Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat. Genet. 25, 385–389.
Li, H., Li, S. H., Yu, Z. X., Shelbourne, P., and Li, X. J. (2001). Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J. Neurosci. 21, 8473–8481.
Li, S. H., Cheng, A. L., Zhou, H., Lam, S., Rao, M., Li, H., and Li, X. J. (2002). Interaction of Huntington disease protein with transcriptional activator Sp1. Mol. Cell. Biol. 22, 1277–1287.
Li, S. H., and Li, X. J. (2004). Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 20, 146–154.
Liévens, J. C., Rival, T., Iche, M., Chneiweiss, H., and Birman, S. (2006). Expanded polyglutamine peptides disrupt EGF receptor signaling and glutamate transporter expression in Drosophila. Hum. Mol. Gen. 14, 713–724.
Liévens, J. C., Woodman, B., Mahal, A., Spasic-Boscovic, O., Samuel, D., Kerkerian-Le Goff, L., and Bates, G. P. (2001). Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol. Dis. 8, 807–821.
Lin, J., Wu, P. H., Tarr, P. T., Lindenberg, K. S., St-Pierre, J., Zhang, C. Y., Mootha, V. K., Jager, S., Vianna, C. R., Reznick, R. M., Cui, L., Manieri, M., Donovan, M. X., Wu, Z., Cooper, M. P., Fan, M. C., Rohas, L. M., Zavacki, A. M., Cinti, S., Shulman, G. I., Lowell, B. B., Krainc, D., and Spiegelman, B. M. (2004). Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119, 121–135.
Lipsky, R. H., Xu, K., Zhu, D., Kelly, C., Terhakopian, A., Novelli, A., and Marini, A. M. (2001). Nuclear factor kappaB is a critical determinant in N-methyl-D-aspartate receptor-mediated neuroprotection. J. Neurochem. 78, 254–264.
Liu, Y., Wong, T. P., Aarts, M., Rooyakkers, A., Liu, L., Lai, T. W., Wu, D. C., Lu, J., Tymianski, M., Craig, A. M., and Wang, Y. T. (2007). NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 27, 2846–2857.
Luo, S., Vacher, C., Davies, J. E., and Rubinsztein, D. C. (2005). Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J. Cell Biol. 169, 647–656.
Luthi-Carter, R., Strand, A., Peters, N. L., Solano, S. M., Hollingsworth, Z. R., Menon, A. S., Frey, A. S., Spektor, B. S., Penney, E. B., Schilling, G., Ross, C. A., Borchelt, D. R., Tapscott, S. J., Young, A. B., Cha, J. H., and Olson, J. M. (2000). Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 9, 1259–1271.
Luthi-Carter, R., Strand, A. D., Hanson, S. A., Kooperberg, C., Schilling, G., La Spada, A. R., Merry, D. E., Young, A. B., Ross, C. A., Borchelt, D. R., and Olson, J. M. (2002). Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington’s disease mouse models reveal context-independent effects. Hum. Mol. Genet. 11, 1927–1937.
Lynch, G., Kramar, E. A., Rex, C. S., Jia, Y., Chappas, D., Gall, C. M., and Simmons, D. A. (2007). Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington’s disease. J. Neurosci. 27, 4424–4434.
Martel, M. A., Wyllie, D. J., and Hardingham, G. E. (2009). In developing hippocampal neurons, NR2B-containing N-methyl-D-aspartate receptors (NMDARs) can mediate signaling to neuronal survival and synaptic potentiation, as well as neuronal death. Neuroscience 158, 334–343.
Martin, E., Betuing, S., Pages, C., Cambon, K., Auregan, G., Deglon, N., Roze, E., and Caboche, J. (2011). Mitogen- and stress-activated protein kinase 1-induced neuroprotection in Huntington’s disease: role on chromatin remodeling at the PGC-1-alpha promoter. Hum. Mol. Genet. 20, 2422–2434.
Martindale, D., Hackam, A., Wieczorek, A., Ellerby, L., Wellington, C., Mccutcheon, K., Singaraja, R., Kazemi-Esfarjani, P., Devon, R., Kim, S. U., Bredesen, D. E., Tufaro, F., and Hayden, M. R. (1998). Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 18, 150–154.
Martinez-Mir, M. I., Probst, A., and Palacios, J. M. (1991). Adenosine A2 receptors: selective localization in the human basal ganglia and alterations with disease. Neuroscience 42, 697–706.
Mazarei, G., Neal, S. J., Becanovic, K., Luthi-Carter, R., Simpson, E. M., and Leavitt, B. R. (2010). Expression analysis of novel striatal-enriched genes in Huntington disease. Hum. Mol. Genet. 19, 609–622.
McConoughey, S. J., Basso, M., Niatsetskaya, Z. V., Sleiman, S. F., Smirnova, N. A., Langley, B. C., Mahishi, L., Cooper, A. J., Antonyak, M. A., Cerione, R. A., Li, B., Starkov, A., Chaturvedi, R. K., Beal, M. F., Coppola, G., Geschwind, D. H., Ryu, H., Xia, L., Iismaa, S. E., Pallos, J., Pasternack, R., Hils, M., Fan, J., Raymond, L. A., Marsh, J. L., Thompson, L. M., and Ratan, R. R. (2010). Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol. Med. 2, 349–370.
Meffert, M. K., and Baltimore, D. (2005). Physiological functions for brain NF-kappaB. Trends Neurosci. 28, 37–43.
Metzler, M., Gan, L., Mazarei, G., Graham, R. K., Liu, L., Bissada, N., Lu, G., Leavitt, B. R., and Hayden, M. R. (2010). Phosphorylation of huntingtin at Ser421 in YAC128 neurons is associated with protection of YAC128 neurons from NMDA-mediated excitotoxicity and is modulated by PP1 and PP2A. J. Neurosci. 30, 14318–14329.
Metzler, M., Gan, L., Wong, T. P., Liu, L., Helm, J., Liu, L., Georgiou, J., Wang, Y., Bissada, N., Cheng, K., Roder, J. C., Wang, Y. T., and Hayden, M. R. (2007). NMDA receptor function and NMDA receptor-dependent phosphorylation of huntingtin is altered by the endocytic protein HIP1. J. Neurosci. 27, 2298–2308.
Milnerwood, A. J., Gladding, C. M., Pouladi, M. A., Kaufman, A. M., Hines, R. M., Boyd, J. D., Ko, R. W., Vasuta, O. C., Graham, R. K., Hayden, M. R., Murphy, T. H., and Raymond, L. A. (2010). Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 65, 178–190.
Mitra, S., Tsvetkov, A. S., and Finkbeiner, S. (2009). Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in huntington disease. J. Biol. Chem. 284, 4398–4403.
Mochel, F., Charles, P., Seguin, F., Barritault, J., Coussieu, C., Perin, L., Le Bouc, Y., Gervais, C., Carcelain, G., Vassault, A., Feingold, J., Rabier, D., and Durr, A. (2007). Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS ONE 2, e647. doi: 10.1371/journal.pone.0000647
Mochel, F., and Haller, R. G. (2011). Energy deficit in Huntington disease: why it matters. J. Clin. Invest. 121, 493–499.
Morfini, G. A., You, Y. M., Pollema, S. L., Kaminska, A., Liu, K., Yoshioka, K., Bjorkblom, B., Coffey, E. T., Bagnato, C., Han, D., Huang, C. F., Banker, G., Pigino, G., and Brady, S. T. (2009). Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 12, 864–871.
Morton, A. J., Wood, N. I., Hastings, M. H., Hurelbrink, C., Barker, R. A., and Maywood, E. S. (2005). Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci. 25, 157–163.
Myers, R. H., Macdonald, M. E., Koroshetz, W. J., Duyao, M. P., Ambrose, C. M., Taylor, S. A., Barnes, G., Srinidhi, J., Lin, C. S., Whaley, W. L., Lazzarini, A. M., Schwarz, M., Wolff, G., Bird, E. D., Vonsattel, J.-P. G., and Gusella, J. F. (1993). De novo expansion of a (CAG)n repeat in sporadic Huntington’s disease. Nat. Genet. 5, 168–173.
Nekooki-Machida, Y., Kurosawa, M., Nukina, N., Ito, K., Oda, T., and Tanaka, M. (2009). Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9679–9684.
Nucifora, F. C. Jr., Sasaki, M., Peters, M. F., Huang, H., Cooper, J. K., Yamada, M., Takahashi, H., Tsuji, S., Troncoso, J., Dawson, V. L., Dawson, T. M., and Ross, C. A. (2001). Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291, 2423–2428.
Ohyama, T., Verstreken, P., Ly, C. V., Rosenmund, T., Rajan, A., Tien, A. C., Haueter, C., Schulze, K. L., and Bellen, H. J. (2007). Huntingtin-interacting protein 14, a palmitoyl transferase required for exocytosis and targeting of CSP to synaptic vesicles. J. Cell Biol. 179, 1481–1496.
Okamoto, S., Pouladi, M. A., Talantova, M., Yao, D., Xia, P., Ehrnhoefer, D. E., Zaidi, R., Clemente, A., Kaul, M., Graham, R. K., Zhang, D., Vincent Chen, H. S., Tong, G., Hayden, M. R., and Lipton, S. A. (2009). Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 15, 1407–1413.
Ordway, J. M., Tallaksen-Greene, S., Gutekunst, C. A., Bernstein, E. M., Cearley, J. A., Wiener, H. W., Dure, L. S. T., Lindsey, R., Hersch, S. M., Jope, R. S., Albin, R. L., and Detloff, P. J. (1997). Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 91, 753–763.
Pallos, J., Bodai, L., Lukacsovich, T., Purcell, J. M., Steffan, J. S., Thompson, L. M., and Marsh, J. L. (2008). Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 17, 3767–3775.
Paoletti, P., Vila, I., Rife, M., Lizcano, J. M., Alberch, J., and Gines, S. (2008). Dopaminergic and glutamatergic signaling crosstalk in Huntington’s disease neurodegeneration: the role of p25/cyclin-dependent kinase 5. J. Neurosci. 28, 10090–10101.
Papadia, S., Soriano, F. X., Leveille, F., Martel, M. A., Dakin, K. A., Hansen, H. H., Kaindl, A., Sifringer, M., Fowler, J., Stefovska, V., Mckenzie, G., Craigon, M., Corriveau, R., Ghazal, P., Horsburgh, K., Yankner, B. A., Wyllie, D. J., Ikonomidou, C., and Hardingham, G. E. (2008). Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 11, 476–487.
Paradisi, I., Hernandez, A., and Arias, S. (2008). Huntington disease mutation in Venezuela: age of onset, haplotype analyses and geographic aggregation. J. Hum. Genet. 53, 127–135.
Patzold, T., and Brune, M. (2002). Obsessive compulsive disorder in Huntington disease: a case of isolated obsessions successfully treated with sertraline. Neuropsychiatry Neuropsychol. Behav. Neurol. 15, 216–219.
Paulsen, J. S., Ready, R. E., Hamilton, J. M., Mega, M. S., and Cummings, J. L. (2001). Neuropsychiatric aspects of Huntington’s disease. J. Neurol. Neurosurg. Psychiatr. 71, 310–314.
Peinemann, A., Schuller, S., Pohl, C., Jahn, T., Weindl, A., and Kassubek, J. (2005). Executive dysfunction in early stages of Huntington’s disease is associated with striatal and insular atrophy: a neuropsychological and voxel-based morphometric study. J. Neurol. Sci. 239, 11–19.
Penney, J. B. Jr., Young, A. B., Shoulson, I., Starosta-Rubenstein, S., Snodgrass, S. R., Sanchez-Ramos, J., Ramos-Arroyo, M., Gomez, F., Penchaszadeh, G., Alvir, J., Esteves, J., DeQuiroz, I., Marsol, N., Moreno, H., Conneally, P. M., Bonilla, E., and Wexler, N. S. (1990). Huntington’s disease in Venezuela: 7 years of follow-up on symptomatic and asymptomatic individuals. Mov. Disord. 5, 93–99.
Perrin, V., Regulier, E., Abbas-Terki, T., Hassig, R., Brouillet, E., Aebischer, P., Luthi-Carter, R., and Deglon, N. (2007). Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington’s disease. Mol. Ther. 15, 903–911.
Perutz, M. F., Pope, B. J., Owen, D., Wanker, E. E., and Scherzinger, E. (2002). Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid beta-peptide of amyloid plaques. Proc. Natl. Acad. Sci. U.S.A. 99, 5596–5600.
Petersen, A. A., Larsen, K. E., Behr, G. G., Romero, N., Przedborski, S., Brundin, P., and Sulzer, D. (2001). Brain-derived neurotrophic factor inhibits apoptosis and dopamine-induced free radical production in striatal neurons but does not prevent cell death. Brain Res. Bull. 56, 331–335.
Pflanz, S., Besson, J. A., Ebmeier, K. P., and Simpson, S. (1991). The clinical manifestation of mental disorder in Huntington’s disease: a retrospective case record study of disease progression. Acta Psychiatr. Scand. 83, 53–60.
Pillon, B., Deweer, B., Agid, Y., and Dubois, B. (1993). Explicit memory in Alzheimer’s, Huntington’s, and Parkinson’s diseases. Arch. Neurol. 50, 374–379.
Pillon, B., Deweer, B., Michon, A., Malapani, C., Agid, Y., and Dubois, B. (1994). Are explicit memory disorders of progressive supranuclear palsy related to damage to striatofrontal circuits? Comparison with Alzheimer’s, Parkinson’s, and Huntington’s diseases. Neurology 44, 1264–1270.
Rammes, G., Zieglgansberger, W., and Parsons, C. G. (2008). The fraction of activated N-methyl-D-aspartate receptors during synaptic transmission remains constant in the presence of the glutamate release inhibitor riluzole. J. Neural Transm. 115, 1119–1126.
Ranen, N. G., Stine, O. C., Abbott, M. H., Sherr, M., Codori, A. M., Franz, M. L., Chao, N. I., Chung, A. S., Pleasant, N., Callahan, C., Kasch, L. M., Ghaffari, M., Chase, G. A., Kazazian, H. H., Brandt, J., Folstein, S. E., and Ross, C. A. (1995). Anticipation and instability of IT-15 (CAG)n repeats in parent-offspring pairs with Huntington disease. Am. J. Hum. Genet. 57, 593–602.
Ravache, M., Weber, C., Merienne, K., and Trottier, Y. (2010). Transcriptional activation of REST by Sp1 in Huntington’s disease models. PLoS ONE 5, e14311. doi: 10.1371/journal.pone.0014311
Ravikumar, B., Berger, Z., Vacher, C., O’Kane, C. J., and Rubinsztein, D. C. (2006). Rapamycin pre-treatment protects against apoptosis. Hum. Mol. Genet. 15, 1209–1216.
Reynolds, D. S., Carter, R. J., and Morton, A. J. (1998). Dopamine modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of Huntington’s disease. J. Neurosci. 18, 10116–10127.
Reynolds, N. (2008). Re: autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov. Disord. 23, 1795–1796; author reply 1793.
Richfield, E. K., and Herkenham, M. (1994). Selective vulnerability in Huntington’s disease: preferential loss of cannabinoid receptors in lateral globus pallidus. Ann. Neurol. 36, 577–584.
Richfield, E. K., O’Brien, C. F., Eskin, T., and Shoulson, I. (1991). Heterogeneous dopamine receptor changes in early and late Huntington’s disease. Neurosci. Lett. 132, 121–126.
Roche, K. W., Standley, S., Mccallum, J., Dune Ly, C., Ehlers, M. D., and Wenthold, R. J. (2001). Molecular determinants of NMDA receptor internalization. Nat. Neurosci. 4, 794–802.
Rockabrand, E., Slepko, N., Pantalone, A., Nukala, V. N., Kazantsev, A., Marsh, J. L., Sullivan, P. G., Steffan, J. S., Sensi, S. L., and Thompson, L. M. (2007). The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 16, 61–77.
Rosenblatt, A., and Leroi, I. (2000). Neuropsychiatry of Huntington’s disease and other basal ganglia disorders. Psychosomatics 41, 24–30.
Rothman, S. M., and Olney, J. W. (1995). Excitotoxicity and the NMDA receptor – still lethal after eight years. Trends Neurosci. 18, 57–58.
Roze, E., Betuing, S., Deyts, C., Marcon, E., Brami-Cherrier, K., Pages, C., Humbert, S., Merienne, K., and Caboche, J. (2008). Mitogen- and stress-activated protein kinase-1 deficiency is involved in expanded-huntingtin-induced transcriptional dysregulation and striatal death. FASEB J. 22, 1083–1093.
Runne, H., Kuhn, A., Wild, E. J., Pratyaksha, W., Kristiansen, M., Isaacs, J. D., Regulier, E., Delorenzi, M., Tabrizi, S. J., and Luthi-Carter, R. (2007). Analysis of potential transcriptomic biomarkers for Huntington’s disease in peripheral blood. Proc. Natl. Acad. Sci. U.S.A. 104, 14424–14429.
Ryu, H., Lee, J., Hagerty, S. W., Soh, B. Y., Mcalpin, S. E., Cormier, K. A., Smith, K. M., and Ferrante, R. J. (2006). ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 103, 19176–19181.
Sadri-Vakili, G., Bouzou, B., Benn, C. L., Kim, M. O., Chawla, P., Overland, R. P., Glajch, K. E., Xia, E., Qiu, Z., Hersch, S. M., Clark, T. W., Yohrling, G. J., and Cha, J. H. (2007). Histones associated with downregulated genes are hypo-acetylated in Huntington’s disease models. Hum. Mol. Genet. 16, 1293–1306.
Sanberg, P. R., Calderon, S. F., Giordano, M., Tew, J. M., and Norman, A. B. (1989). The quinolinic acid model of Huntington’s disease: locomotor abnormalities. Exp. Neurol. 105, 45–53.
Sarkar, S., Perlstein, E. O., Imarisio, S., Pineau, S., Cordenier, A., Maglathlin, R. L., Webster, J. A., Lewis, T. A., O’Kane, C. J., Schreiber, S. L., and Rubinsztein, D. C. (2007). Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 3, 331–338.
Saudou, F., Finkbeiner, S., Devys, D., and Greenberg, M. E. (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66.
Sawada, H., Ishiguro, H., Nishii, K., Yamada, K., Tsuchida, K., Takahashi, H., Goto, J., Kanazawa, I., and Nagatsu, T. (2007). Characterization of neuron-specific huntingtin aggregates in human huntingtin knock-in mice. Neurosci. Res. 57, 559–573.
Scherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach, B., Hasenbank, R., Bates, G. P., Davies, S. W., Lehrach, H., and Wanker, E. E. (1997). Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558.
Semaka, A., Warby, S., Leavitt, B. R., and Hayden, M. R. (2008). Re: autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov. Disord. 23, 1794–1795; author reply 1793.
Seo, H., Sonntag, K. C., Kim, W., Cattaneo, E., and Isacson, O. (2007). Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PLoS ONE 2, e238. doi: 10.1371/journal.pone.0000238
Seong, I. S., Woda, J. M., Song, J. J., Lloret, A., Abeyrathne, P. D., Woo, C. J., Gregory, G., Lee, J. M., Wheeler, V. C., Walz, T., Kingston, R. E., Gusella, J. F., Conlon, R. A., and Macdonald, M. E. (2010). Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 19, 573–583.
Shimohata, T., Nakajima, T., Yamada, M., Uchida, C., Onodera, O., Naruse, S., Kimura, T., Koide, R., Nozaki, K., Sano, Y., Ishiguro, H., Sakoe, K., Ooshima, T., Sato, A., Ikeuchi, T., Oyake, M., Sato, T., Aoyagi, Y., Hozumi, I., Nagatsu, T., Takiyama, Y., Nishizawa, M., Goto, J., Kanazawa, I., Davidson, I., Tanese, N., Takahashi, H., and Tsuji, S. (2000). Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat. Genet. 26, 29–36.
Shiwach, R. S., and Norbury, C. G. (1994). A controlled psychiatric study of individuals at risk for Huntington’s disease. Br. J. Psychiatry 165, 500–505.
Siesling, S., Vegter-Van Der Vlis, M., and Roos, R. A. (1997). Juvenile Huntington disease in the Netherlands. Pediatr. Neurol. 17, 37–43.
Sinadinos, C., Burbidge-King, T., Soh, D., Thompson, L. M., Marsh, J. L., Wyttenbach, A., and Mudher, A. K. (2009). Live axonal transport disruption by mutant huntingtin fragments in Drosophila motor neuron axons. Neurobiol. Dis. 34, 389–395.
Sipione, S., Rigamonti, D., Valenza, M., Zuccato, C., Conti, L., Pritchard, J., Kooperberg, C., Olson, J. M., and Cattaneo, E. (2002). Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum. Mol. Genet. 11, 1953–1965.
Slow, E. J., Graham, R. K., Osmand, A. P., Devon, R. S., Lu, G., Deng, Y., Pearson, J., Vaid, K., Bissada, N., Wetzel, R., Leavitt, B. R., and Hayden, M. R. (2005). Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc. Natl. Acad. Sci. U.S.A. 102, 11402–11407.
Snell, R. G., Macmillan, J. C., Cheadle, J. P., Fenton, I., Lazarou, L. P., Davies, P., Macdonald, M. E., Gusella, J. F., Harper, P. S., and Shaw, D. J. (1993). Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Genet. 4, 393–397.
Stack, E. C., Dedeoglu, A., Smith, K. M., Cormier, K., Kubilus, J. K., Bogdanov, M., Matson, W. R., Yang, L., Jenkins, B. G., Luthi-Carter, R., Kowall, N. W., Hersch, S. M., Beal, M. F., and Ferrante, R. J. (2007). Neuroprotective effects of synaptic modulation in Huntington’s disease R6/2 mice. J. Neurosci. 27, 12908–12915.
Starling, A. J., Andre, V. M., Cepeda, C., De Lima, M., Chandler, S. H., and Levine, M. S. (2005). Alterations in N-methyl-D-aspartate receptor sensitivity and magnesium blockade occur early in development in the R6/2 mouse model of Huntington’s disease. J. Neurosci. Res. 82, 377–386.
Steffan, J. S., Bodai, L., Pallos, J., Poelman, M., Mccampbell, A., Apostol, B. L., Kazantsev, A., Schmidt, E., Zhu, Y. Z., Greenwald, M., Kurokawa, R., Housman, D. E., Jackson, G. R., Marsh, J. L., and Thompson, L. M. (2001). Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413, 739–743.
Steffan, J. S., Kazantsev, A., Spasic-Boskovic, O., Greenwald, M., Zhu, Y. Z., Gohler, H., Wanker, E. E., Bates, G. P., Housman, D. E., and Thompson, L. M. (2000). The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. U.S.A. 97, 6763–6768.
Steigerwald, F., Schulz, T. W., Schenker, L. T., Kennedy, M. B., Seeburg, P. H., and Kohr, G. (2000). C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J. Neurosci. 20, 4573–4581.
Strand, A. D., Baquet, Z. C., Aragaki, A. K., Holmans, P., Yang, L., Cleren, C., Beal, M. F., Jones, L., Kooperberg, C., Olson, J. M., and Jones, K. R. (2007). Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J. Neurosci. 27, 11758–11768.
Strehlow, A. N., Li, J. Z., and Myers, R. M. (2007). Wild-type huntingtin participates in protein trafficking between the Golgi and the extracellular space. Hum. Mol. Genet. 16, 391–409.
Subramaniam, S., Sixt, K. M., Barrow, R., and Snyder, S. H. (2009). Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 324, 1327–1330.
Sugars, K. L., and Rubinsztein, D. C. (2003). Transcriptional abnormalities in Huntington disease. Trends Genet. 19, 233–238.
Suhr, S. T., Senut, M. C., Whitelegge, J. P., Faull, K. F., Cuizon, D. B., and Gage, F. H. (2001). Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J. Cell Biol. 153, 283–294.
Sun, Y., Savanenin, A., Reddy, P. H., and Liu, Y. F. (2001). Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J. Biol. Chem. 276, 24713–24718.
Szebenyi, G., Morfini, G. A., Babcock, A., Gould, M., Selkoe, K., Stenoien, D. L., Young, M., Faber, P. W., Macdonald, M. E., Mcphaul, M. J., and Brady, S. T. (2003). Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40, 41–52.
Tallaksen-Greene, S. J., Janiszewska, A., Benton, K., Ruprecht, L., and Albin, R. L. (2010). Lack of efficacy of NMDA receptor-NR2B selective antagonists in the R6/2 model of Huntington disease. Exp. Neurol. 225, 402–407.
Tang, T. S., Chen, X., Liu, J., and Bezprozvanny, I. (2007). Dopaminergic signaling and striatal neurodegeneration in Huntington’s disease. J. Neurosci. 27, 7899–7910.
Tang, T. S., Guo, C., Wang, H., Chen, X., and Bezprozvanny, I. (2009). Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington’s disease mouse model. J. Neurosci. 29, 1257–1266.
Tang, T. S., Tu, H., Orban, P. C., Chan, E. Y., Hayden, M. R., and Bezprozvanny, I. (2004). HAP1 facilitates effects of mutant huntingtin on inositol 1,4,5-trisphosphate-induced Ca release in primary culture of striatal medium spiny neurons. Eur. J. Neurosci. 20, 1779–1787.
Taniura, H., Sng, J. C., and Yoneda, Y. (2007). Histone modifications in the brain. Neurochem. Int. 51, 85–91.
The Huntington Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983.
Thomas, E. A., Coppola, G., Desplats, P. A., Tang, B., Soragni, E., Burnett, R., Gao, F., Fitzgerald, K. M., Borok, J. F., Herman, D., Geschwind, D. H., and Gottesfeld, J. M. (2008). The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 105, 15564–15569.
Thomas, E. A., Coppola, G., Tang, B., Kuhn, A., Kim, S., Geschwind, D. H., Brown, T. B., Luthi-Carter, R., and Ehrlich, M. E. (2011). In vivo cell-autonomous transcriptional abnormalities revealed in mice expressing mutant huntingtin in striatal but not cortical neurons. Hum. Mol. Genet. 20, 1049–1060.
Thompson, P. D., Bhatia, K. P., Brown, P., Davis, M. B., Pires, M., Quinn, N. P., Luthert, P., Honovar, M., O’Brien, M. D., Marsden, C. D., and Harding, A. E. (1994). Cortical myoclonus in Huntington’s disease. Mov. Disord. 9, 633–641.
Tovar, K. R., and Westbrook, G. L. (1999). The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J. Neurosci. 19, 4180–4188.
Trushina, E., Dyer, R. B., Badger, J. D. II, Ure, D., Eide, L., Tran, D. D., Vrieze, B. T., Legendre-Guillemin, V., Mcpherson, P. S., Mandavilli, B. S., Van Houten, B., Zeitlin, S., Mcniven, M., Aebersold, R., Hayden, M., Parisi, J. E., Seeberg, E., Dragatsis, I., Doyle, K., Bender, A., Chacko, C., and Mcmurray, C. T. (2004). Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 24, 8195–8209.
Turner, C., Cooper, J. M., and Schapira, A. H. (2007). Clinical correlates of mitochondrial function in Huntington’s disease muscle. Mov. Disord. 22, 1715–1721.
Tydlacka, S., Wang, C. E., Wang, X., Li, S., and Li, X. J. (2008). Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J. Neurosci. 28, 13285–13295.
Vacher, C., Garcia-Oroz, L., and Rubinsztein, D. C. (2005). Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 14, 3425–3433.
Vanhoutte, P., and Bading, H. (2003). Opposing roles of synaptic and extrasynaptic NMDA receptors in neuronal calcium signalling and BDNF gene regulation. Curr. Opin. Neurobiol. 13, 366–371.
Venkatraman, P., Wetzel, R., Tanaka, M., Nukina, N., and Goldberg, A. L. (2004). Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 14, 95–104.
Verhoef, L. G., Lindsten, K., Masucci, M. G., and Dantuma, N. P. (2002). Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum. Mol. Genet. 11, 2689–2700.
Videnovic, A., Leurgans, S., Fan, W., Jaglin, J., and Shannon, K. M. (2009). Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat. Disord. 15, 471–474.
Vogel, C. M., Drury, I., Terry, L. C., and Young, A. B. (1991). Myoclonus in adult Huntington’s disease. Ann. Neurol. 29, 213–215.
Vonsattel, J. P., Myers, R. H., Stevens, T. J., Ferrante, R. J., Bird, E. D., and Richardson, E. P. Jr. (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577.
Wexler, N. S., Lorimer, J., Porter, J., Gomez, F., Moskowitz, C., Shackell, E., Marder, K., Penchaszadeh, G., Roberts, S. A., Gayan, J., Brocklebank, D., Cherny, S. S., Cardon, L. R., Gray, J., Dlouhy, S. R., Wiktorski, S., Hodes, M. E., Conneally, P. M., Penney, J. B., Gusella, J., Cha, J. H., Irizarry, M., Rosas, D., Hersch, S., Hollingsworth, Z., Macdonald, M., Young, A. B., Andresen, J. M., Housman, D. E., De Young, M. M., Bonilla, E., Stillings, T., Negrette, A., Snodgrass, S. R., Martinez-Jaurrieta, M. D., Ramos-Arroyo, M. A., Bickham, J., Ramos, J. S., Marshall, F., Shoulson, I., Rey, G. J., Feigin, A., Arnheim, N., Acevedo-Cruz, A., Acosta, L., Alvir, J., Fischbeck, K., Thompson, L. M., Young, A., Dure, L., O’Brien, C. J., Paulsen, J., Brickman, A., Krch, D., Peery, S., Hogarth, P., Higgins, D. S. Jr., and Landwehrmeyer, B. (2004). Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. U.S.A. 101, 3498–3503.
Weydt, P., Pineda, V. V., Torrence, A. E., Libby, R. T., Satterfield, T. F., Lazarowski, E. R., Gilbert, M. L., Morton, G. J., Bammler, T. K., Strand, A. D., Cui, L., Beyer, R. P., Easley, C. N., Smith, A. C., Krainc, D., Luquet, S., Sweet, I. R., Schwartz, M. W., and La Spada, A. R. (2006). Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 4, 349–362.
Wheelock, V. L., Tempkin, T., Marder, K., Nance, M., Myers, R. H., Zhao, H., Kayson, E., Orme, C., and Shoulson, I. (2003). Predictors of nursing home placement in Huntington disease. Neurology 60, 998–1001.
Xie, Y., Hayden, M. R., and Xu, B. (2010). BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J. Neurosci. 30, 14708–14718.
Yanai, A., Huang, K., Kang, R., Singaraja, R. R., Arstikaitis, P., Gan, L., Orban, P. C., Mullard, A., Cowan, C. M., Raymond, L. A., Drisdel, R. C., Green, W. N., Ravikumar, B., Rubinsztein, D. C., El-Husseini, A., and Hayden, M. R. (2006). Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat. Neurosci. 9, 824–831.
Yang, L., Mei, Q., Zielinska-Kwiatkowska, A., Matsui, Y., Blackburn, M. L., Benedetti, D., Krumm, A. A., Taborsky, G. J. Jr., and Chansky, H. A. (2003). An ERG (ets-related gene)-associated histone methyltransferase interacts with histone deacetylases 1/2 and transcription co-repressors mSin3A/B. Biochem. J. 369, 651–657.
Zeitlin, S., Liu, J. P., Chapman, D. L., Papaioannou, V. E., and Efstratiadis, A. (1995). Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 11, 155–163.
Zeron, M. M., Fernandes, H. B., Krebs, C., Shehadeh, J., Wellington, C. L., Leavitt, B. R., Baimbridge, K. G., Hayden, M. R., and Raymond, L. A. (2004). Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington’s disease. Mol. Cell. Neurosci. 25, 469–479.
Zeron, M. M., Hansson, O., Chen, N., Wellington, C. L., Leavitt, B. R., Brundin, P., Hayden, M. R., and Raymond, L. A. (2002). Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron 33, 849–860.
Zhai, W., Jeong, H., Cui, L., Krainc, D., and Tjian, R. (2005). In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell 123, 1241–1253.
Zhang, H., Li, Q., Graham, R. K., Slow, E., Hayden, M. R., and Bezprozvanny, I. (2008). Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington’s disease. Neurobiol. Dis. 31, 80–88.
Zhang, S. J., Steijaert, M. N., Lau, D., Schutz, G., Delucinge-Vivier, C., Descombes, P., and Bading, H. (2007). Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron 53, 549–562.
Zuccato, C., Belyaev, N., Conforti, P., Ooi, L., Tartari, M., Papadimou, E., Macdonald, M., Fossale, E., Zeitlin, S., Buckley, N., and Cattaneo, E. (2007). Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci. 27, 6972–6983.
Zuccato, C., and Cattaneo, E. (2009). Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 5, 311–322.
Zuccato, C., Ciammola, A., Rigamonti, D., Leavitt, B. R., Goffredo, D., Conti, L., Macdonald, M. E., Friedlander, R. M., Silani, V., Hayden, M. R., Timmusk, T., Sipione, S., and Cattaneo, E. (2001). Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 293, 493–498.
Zuccato, C., Tartari, M., Crotti, A., Goffredo, D., Valenza, M., Conti, L., Cataudella, T., Leavitt, B. R., Hayden, M. R., Timmusk, T., Rigamonti, D., and Cattaneo, E. (2003). Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 35, 76–83.
Keywords: polyglutamine, Huntingtin, excitotoxicity, mitochondrial dysfunctions, transcriptional deregulation
Citation: Roze E, Cahill E, Martin E, Bonnet C, Vanhoutte P, Betuing S and Caboche J (2011) Huntington’s disease and striatal signaling. Front. Neuroanat. 5:55. doi: 10.3389/fnana.2011.00055
Received: 12 May 2011; Accepted: 04 August 2011;
Published online: 23 August 2011.
Edited by:
Emmanuel Valjent, Université Montpellier 1&2, FranceReviewed by:
Michelle E. Ehrlich, Mount Sinai School of Medicine, USASandrine Humbert, Institut Curie–INSERM U1005–CNRS UMR3306, France
Copyright: © 2011 Roze, Cahill, Martin, Bonnet, Vanhoutte, Betuing and Caboche. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jocelyne Caboche, Laboratoire de Physiopathologie des Maladies du Système Nerveux Central, Université Pierre et Marie Curie, 9 Quai Saint Bernard, 75005 Paris, France. e-mail: jocelyne.caboche@snv. jussieu.fr