Developmental disorders of the midbrain and hindbrain
- Department of Radiology and Biomolecular Imaging, Neuroradiology Section, University of California at San Francisco, San Francisco, CA, USA
Malformations of the midbrain (MB) and hindbrain (HB) have become topics of considerable interest in the neurology and neuroscience literature in recent years. The combined advances of imaging and molecular biology have improved analyses of structures in these areas of the central nervous system, while advances in genetics have made it clear that malformations of these structures are often associated with dysfunction or malformation of other organ systems. This review focuses upon the importance of communication between clinical researchers and basic scientists in the advancement of knowledge of this group of disorders. Disorders of anteroposterior (AP) patterning, cerebellar hypoplasias, disorders associated with defects of the pial limiting membrane (cobblestone cortex), disorders of the Reelin pathway, and disorders of the primary cilium/basal body organelle (molar tooth malformations) are the main focus of the review.
Introduction
For many years, anomalies of the cerebellum and brain stem were poorly reported in the scientific literature. The cerebellum was believed to have a minor role in brain function, while the brain stem was difficult to remove intact at autopsy and difficult to section. Radiologic analysis of these structures by pneumography, angiography, and X-ray computed tomography was poor. Recently, however, advances in developmental genetics, neurobiology, molecular biology, and neuroimaging have led to better understanding of developmental disorders of the embryonic midbrain (MB) and hindbrain (HB), which grow into the adult brainstem and cerebellum (Barkovich et al., 2007, 2009). Although malformations of the brainstem and cerebellum may be the only recognized abnormality in individuals with mental retardation or autism (Soto-Ares et al., 2003; Courchesne et al., 2005), they are more commonly identified in patients with malformations of the cerebrum. Among the most common of these are lissencephalies (Ross et al., 2001; Lecourtois et al., 2010), so-called “cobblestone malformations” of the cortex (formerly known as lissencephaly type II) resulting from defects in the pial limiting membrane (van Reeuwijk et al., 2006; Clement et al., 2008; Hewitt, 2009), anomalies of the cerebral commissures (Barkovich et al., 2007), and disorders of primary cilia function that include additional ocular, renal, hepatic, and limb bud anomalies (Lancaster et al., 2011; Sang et al., 2011).
The number and complexity of recognized malformations of the brainstem and cerebellum has been steadily increasing. These disorders were recently extensively reviewed and classified (Barkovich et al., 2009). This review will highlight a few MB-HB malformations and emphasize how knowledge of basic research in embryology, genetics, and cellular and molecular biology of the developing brain can be of importance in recognizing, understanding, and classifying these anomalies in humans.
Malformations Secondary to Early Patterning Defects
Malformations of the MB or HB secondary to defects in anteroposterior (AP) or dorsoventral (DV) patterning were nearly impossible to identify in neurology patients until magnetic resonance imaging became a commonly used tool in clinical diagnosis. The ability to acquire high resolution, high contrast, distortion-free images in sagittal and coronal planes allowed accurate gross assessment of MB-HB structures for the first time. However, the structures within the MB-HB are small and move with cardiac pulsations; therefore, physicians were slow to recognize subtle distortions in their structure and recognize their importance in developmental disorders. As a consequence, physicians have only recently begun to look for subtle variations in them. Only in the past 10 years have these malformations been fairly consistently identified and associated with normal developmental processes and their derangements (Jen et al., 2004; Bednarek et al., 2005; Moog et al., 2005; Sicotte et al., 2006; Barkovich et al., 2007; Barth et al., 2007; Jissendi-Tchofo et al., 2009).
Disorders of A-P Patterning
The easiest malformations to identify are those due to disturbed AP patterning, particularly at the MB-HB junction, which is defined in mice (and presumably in humans) by a balance of Otx2 and Gbx2 signaling that defines the position of the isthmus organizer (IsO) (Wassef and Joyner, 1997; Millet et al., 1999; Chizhikov and Millen, 2003). The IsO ultimately defines the posterior limit of the MB and the anterior limit of the cerebellum (Chizhikov and Millen, 2003) (Figure 1A). The combination of a shortened MB and elongated pons associated with an enlarged anterior vermis in humans (Figure 1B), therefore, presumably results from rostral displacement of the IsO, with loss of MB and gain of R1 [from which the cerebellum forms, with the vermis deriving from the most rostral portion (Broccoli et al., 1999; Chizhikov and Millen, 2003)]; this malformation is presumed to result from GBX2 predominance over OTX2 and consequent rostral malpositioning of the IsO (Chizhikov and Millen, 2003; Barkovich et al., 2009). This finding has been described in Opitz G/BBB syndrome (OS), an X-linked form of which is caused by loss of function mutations of the MID1 gene (Quaderi et al., 1997). MID1 plays a role in the ubiquitin-specific regulation of the microtubule associated catalytic subunit of protein phosphatase 1Ac (Aranda-Orgillés et al., 2008). Its role in the pathogenesis of the disease is not clear. Patients show variable clinical signs and symptoms affecting multiple organ systems. Imaging shows hypoplasia of the anterior cerebellar vermis (Pinson et al., 2004; Fontanella et al., 2008). Mid1-null mice show motor coordination defects and procedural learning impairments. Of note, in addition to cerebellar vermian hypoplasia, these mice show shortening of the posterior dorsal MB, rostralization of the MB-HB, and down-regulation of Fgf17, a key transcription factor in the region (Lancioni et al., 2010). This is another area in which applying learning from human disease is helping to understand development in the MB-HB region.
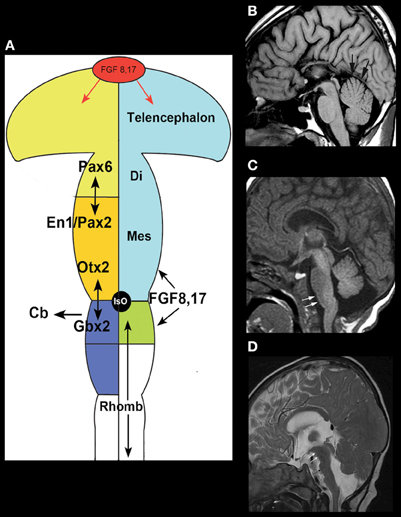
Figure 1. Disorders of AP patterning in the brain stem.(A) Anteroposterior patterning and the Isthmus Organizer. Regionalization of the brain starts with the formation of patterning centers that secrete signaling molecules such as the fibroblast growth factors (FGFs). Fgf8 and Fgr17 are important signaling molecules at both the anterior forebrain and the MB-HB junction. In the forebrain, it helps to direct formation of the prefrontal cortex and other rostral structures by inducing cells to secrete the transcription factor Pax6. At the MB-HB junction, the patterning center known as the isthmus organizer (IsO) is localized and induced to secrete Fgf8 and Fgf17 by the interaction of transcription factor Gbx2 from the rhombencephalon and Otx2 from the caudal mesencephalon. The secretion of Fgf8 and Fgf17 then induces further changes crucial to formation of the MB-HB junction and the formation of the cerebellum. The junction of the diencephalon (di) and mesencephalon (mes) is directed by the interaction of Pax6 from the diencephalon and En1/Pax2 from the rostral mesencephalon. Repression of Otx2 expression by FGF8 induces Gbx2 formation to establish the location of the MB-HB junction and can affect cerebellar formation, as the cerebellum forms from the most rostral portion of the HB. Similarly, alterations of Pax6 or En1/Pax2 will alter the location of the diencephalic-mesencephalic junction. (Adapted from Barkovich and Raybaud, 2012). (B) Sagittal T1 weighted image shows a short MB, long pons, and large superior vermis (black arrows), suggesting an abnormality of anteroposterior patterning with rostral misplacement of the Isthmus Organizer. In addition, the patient has agenesis of the corpus callosum. (C) Sagittal T1 weighted image shows a slightly small pons and a short, thick medulla (white arrows) with an abnormal pontomedullary transition. This is postulated to result from mixed gains and losses of rhombomere expression in the developing rhombencephalon or potentially a segmental shift of rhombomeres. (D) Sagittal T2 weighted image shows a very elongated MB (white arrows) with small, short pons (black arrow), and small cerebellar vermis, suggesting caudal displacement of the Isthmus Organizer due to abnormal anterioposterior patterning from overexpression of Otx2.
Shortening and thickening of the medulla (Figure 1C) (Barkovich et al., 2009) is postulated to result from mixed gains and losses of the eight rhombomeric segments within the pons and medulla or a segmental shift of rhombomeres; rhombomeres may be absent or they may be misexpressed, taking on characteristics of other rhombomeres. Such anomalies give the brain stem an abnormal shape (Figures 1C,D). Similar abnormalities result from murine embryo exposure to retinoic acid, which causes a dose-dependent anterior to posterior transformation of cell fate in which the HB is expanded at the expense of the MB and forebrain (Lumsden, 2004). Lesser changes in gradients of retinoic acid or other regionalizing molecules could result in transformations of the middle rhombomeres from pontine to medullary fate. Such changes are difficult to assess with current imaging techniques but may become possible as higher resolution/high field strength MR scanners are developed, along with better tractography programs.
Disorders with Cerebellar Hypoplasia
Just as rostral displacement of the MB-HB junction is expected to increase the size of rhombomere 1, caudal displacement of the IsO [presumably due to increased OTX2 (Broccoli et al., 1999)] would be expected to elongate the MB, and shorten the pons (particularly the R1 segment); the expected result would be cerebellar hypoplasia, particularly affecting the vermis (which is formed from the most rostral aspect of R1) (Chizhikov and Millen, 2003). Indeed, in the few cases observed clinically with an elongated MB and small pons, the cerebellum has always been small (Figure 1D). Cerebellar hypoplasias are common findings in autopsy studies and in clinical neuroimaging and have many causes (Barkovich et al., 2009). Although cerebellar hypoplasia may be an isolated finding, it is usually associated with other anomalies, which may be either supratentorial or infratentorial. A few examples follow.
The Dandy–Walker malformation and related disorders (cerebellar hypoplasia, mega cisterna magna, and Blake pouch cysts) are composed of a grouping of abnormalities of the cerebellum, its surrounding mesenchyme, and sometimes cerebral structures; this variable combination of features has generated considerable confusion and controversy (Raybaud, 1982; Raimondi et al., 1984; Barkovich et al., 1989; Tortori-Donati et al., 1996). The Dandy–Walker malformation, as initially defined, consists of an enlarged posterior fossa with a high position of the tentorium cerebelli, counterclockwise rotation and hypoplasia of the cerebellar vermis, and a dilated, cystic-appearing fourth ventricle that fills nearly the entire posterior fossa, presumably due to cyst-like expansion of the fourth ventricle (Figure 2A) (Hart et al., 1972; Raimondi et al., 1984). The cerebellar hemispheres are usually small and corpus callosal anomalies are found in as many as 20% of affected individuals (Barkovich et al., 1989). Significant variation in cerebellum, brain stem, surrounding CSF spaces, and associated supratentioral anomalies of all degrees may be found, however, in the malformation complex. Indeed, considerable variation can be seen in families with the same genetic mutation; the phenotype ranges from mild vermian hypoplasia to mega cisterna magna to varying severities of true Dandy–Walker malformation (Grinberg et al., 2004; Aldinger et al., 2009; Blank et al., 2011). It is noteworthy that FOXC1, which has been shown to cause this malformation complex in families, is expressed only in the mesenchyme overlying the cerebellum and not in the cerebellum itself (Aldinger et al., 2009). Similar ranges of posterior fossa anomalies have been described with deletion of 3q24 (loss of ZIC1–ZIC4) (Grinberg and Millen, 2005), duplication of 9p (Melaragno et al., 1992; Cazorla Calleja et al., 2003; Chen et al., 2005), deletion of 13q2 (McCormack et al., 2003; Ballarati et al., 2007), and deletion of 2q36.1 [which contains the PAX3 gene, strongly expressed in the developing cerebellum (Jalali et al., 2008)], as well as in neurocutaneous melanosis [a dysplasia of the leptomeninges that is most severe in the basal meninges around the brain stem and cerebellum (Narayanan et al., 1987; Barkovich et al., 1994; Acosta Jr., et al., 2005)]. Of note, MB-HB hypoplasia is only seen in neurocutaneous melanosis when melanosis is present in the meninges surrounding the brain stem and cerebellum, supporting the hypothesis that the developing leptomeninges have significant effects upon MB-HB development (Aldinger et al., 2009). Based upon all of these observations, it may be suggested that (1) the surrounding mesenchyme affects growth of the developing cerebellum during embryogenesis, and (2) mutations resulting in dysgenesis of both the cerebellum and its overlying mesenchyme are likely to be necessary for the entire Dandy–Walker malformation complex to form, with less severe dysgenesis resulting in malformations such as isolated cerebellar hypoplasia or isolated dysgenesis of the surrounding mesenchyme. In light of this information, it was suggested that the Dandy–Walker malformation be considered in the group of mesenchymal-neuroepithelial signaling defects (Barkovich et al., 2009). From this perspective, it follows that retrocerebellar arachnoid cysts and enlargement of the cisterna magna (called mega cisterna magna, an enlarged posterior fossa secondary to an enlarged cisterna magna, but a normal cerebellar vermis and fourth ventricle, Figure 2B) are a part of the Dandy–Walker spectrum from an embryogenesis perspective. In addition, the so-called persistent Blake pouch cyst, where the ependymal wall of the fourth ventricle extends through the foramen of Magendie and upwardly rotates a normal cerebellar vermis (Figure 2C), is sometimes considered a part of the spectrum. However, in the absence of mass effect (causing hydrocephalus) or associated cerebral/cerebellar dysgenesis, none of these findings seem to be of clinical significance; neurological and cognitive development seem to be related, instead, to the level of control of hydrocephalus, to the extent of associated supratentorial anomalies (Golden et al., 1987; Maria et al., 1987; Bindal et al., 1990–1991), and to the degree of cerebellar dysgenesis, manifested as lobulation of the vermis; normal lobulation is associated with good intellectual outcome whereas some have found that abnormal vermian lobulation is associated with poor intellectual outcome (Boddaert et al., 2003; Klein et al., 2003). The molecular biologic pathways involved in these disorders have not yet been identified.
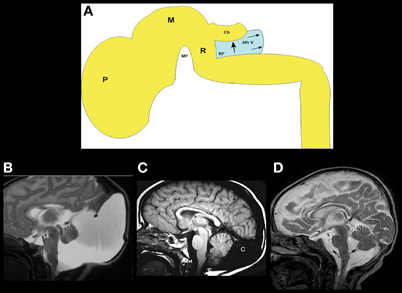
Figure 2. The concept and range of cerebellum/posterior fossa disorders in the “Dandy–Walker spectrum.”(A) Illustration of developing fourth ventricle/cerebellum and the impact of impaired egress of CSF. During normal development, the wall fourth ventricle normally thins, and the foramen of Magendie forms, in the midline. If the leptomeninges are abnormal, the cerebellum may be small and the outflow foramina of the fourth ventricle may not form. The fourth ventricle expands posteriorly (small black arrows) and superiorly, pushing the small cerebellar vermis (Cb) counterclockwise (large black arrow); the posterior fossa then enlarges. This combination of findings creates the classic Dandy–Walker malformation. Small black PF signifies pontine flexure, small black MF signifies mesencephalic flesure, large black P signifies prosencephalon, large black M signifies mesencephalon, large black R signifies rhombencephalon. (B) Classic Dandy–Walker malformation. Sagittal T2 weighted image shows the classic appearance with a small vermis (black arrow), rotated counterclockwise with abnormal foliation. The surrounding CSF spaces are markedly enlarged with abnormally enlarged posterior fossa and elevation of the tentorium cerebelli and the torcular Herophili. (C) Mega cisterna magna is a condition in which a collection of CSF in an enlarged cisterna magna (C) expands the posterior fossa but the midbrain and hindbrain are normal. It is seen in patients with gene mutations that, in siblings, cause the classic Dandy–Walker malformation. (D) Blake pouch cyst is a condition in which the ependymal wall of the fourth ventricle stretches out through the foramen of Magendie and causes enlargement of the foramen with mild rotation of the vermis (black arrow). It is considered by some to be an incidental finding and by others to be a mild form of the Dandy–Walker malformation.
MB-HB Anomalies Associated with Cerebral Anomalies
MB-HB anomalies are often found in association with supratentorial brain anomalies. Often, these are the result of similar processes occurring in both the forebrain and HB. For example, the so-called “cobblestone malformations” or “dystroglycanopathies” are disorders that are caused by impaired linkage of the endfeet of radial glial cells with the pial limiting membrane (Figure 3A), in many cases because of decreased O-glycosylation of a-dystroglycan, which impairs its binding to Laminin-2 in the basal lamina of that membrane (Saito et al., 2006; Godfrey et al., 2007; Clement et al., 2008; Li et al., 2008; Hewitt, 2009; Chan et al., 2010; Chiang et al., 2011). [These cerebral disorders were formerly called “lissencephaly type II” or “cobblestone lissencephaly.” That nomenclature resulted in considerable confusion with lissencephalies caused by undermigration of neurons destined for the cerebral cortex. As a result, the term cobblestone malformation is now preferred (Barkovich et al., 2012)]. Other causes of abnormal linkage include altered laminin deposition (Ackroyd et al., 2011), mutation of G protein-coupled receptor 56 (Gpr56) or its receptor collagen type III (Luo et al., 2011), and mutation of the ubiquitous basement membrane protein collagen type IV alpha 1 (Labelle-Dumais et al., 2011). The result of these abnormal linkages is that some migrating neurons leave the radial glial fiber before reaching their intended cortical layer and other neurons overmigrate through “gaps” form in the pial limiting membrane into the subarachnoid space (Figure 3A) (Martin, 2005). The resulting cerebral cortex is composed of radially oriented clumps of disoriented neurons, the subarachnoid space is filled with ectopic neurons that have overmigrated, and multiple nodules of undermigrated heterotopic neurons lie in the subcortical white matter (Friede, 1989; Norman et al., 1995; Haltia et al., 1997). Similar phenomena of gaps in basal lamina with over- and undermigrated neurons may be seen in the retina and the cerebellum (Friede, 1989; Norman et al., 1995; Haltia et al., 1997). Abnormal linkages using the same molecular structures are found in skeletal muscle, with the result being that many affected patients also have congenital muscular dystrophy (Moore et al., 2002; Martin, 2005; Kanagawa and Toda, 2006). In the cerebellum, the leptomeninges are of considerable importance for the normal migration of granule cells (Zarbalis et al., 2007; Koirala et al., 2009). Mice with dystroglycanopathies show widespread discontinuities in the pial basement membrane with disruption of the glial scaffolding and migration of granule cells into the subarachnoid space (Moore et al., 2002). In Gpr56 deficient mice, glial processes extend through and granule cells migrate through the aforementioned gaps in the glia limitans into the subarachnoid space (Koirala et al., 2009). In human studies, the effects upon the MB-HB vary considerably (Aida et al., 1994; Gelot et al., 1995; van der Knaap et al., 1997; Barkovich, 1998; Clement et al., 2008), with some patients having normal cerebella and others having mild dysgenesis resulting in mild vermian hypoplasia with some alteration of foliation; of note, the severity of cortical dysgenesis is sometimes similar in the cerebrum and cerebellum but the involvement may be discrepant, suggesting that some gene products have different roles in the forebrain and HB. Moderately severe cerebellar dysgenesis consists of a significantly dysmorphic cortex containing cyst-like structures that contain mesenchymal tissue (Figures 3B–F) and are connected to the surface via spaces containing penetrating blood vessels (Takada and Nakamura, 1990). This suggests a process similar to that in the cerebrum where cerebral tissues migrates outward and leptomeningeal tissues inward through the defects in the pial limiting membrane (Figure 3D). Most patients with cerebellar dysgenesis have a small pons with a ventral midline cleft (Figures 3B,C) (van der Knaap et al., 1997; Barkovich, 1998). In the most severe cases (Figures 3E,F), the cerebellum is extremely small with very dysmorphic cortex and disproportionately small vermis and a small brain stem with “kink” in the mid-pons that is best seen in the sagittal plane (Figure 3E); the reason for the small cerebellum might be an absence of dispersion of Purkinje cells (PCs) due to interruption of granule cell migration and consequent absence of Reelin secretion (see next section on cerebellar disorders due to abnormalities of the Reelin pathways). Nearly all affected patients also have abnormalities of the mesencephalic tectum, which is thickened without identifiable collicula due to overmigration of cells (Figures 3B,E), and dysmyelination; the reasons for these phenomena are not yet known.
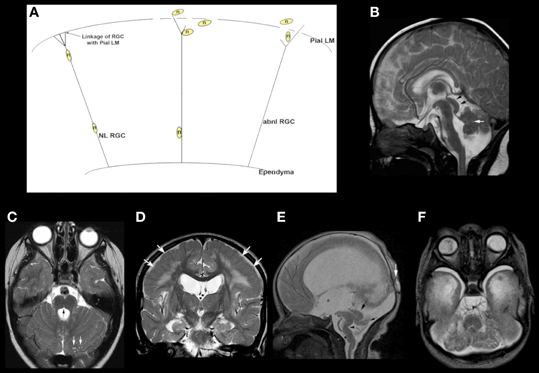
Figure 3. Array of findings in the midbrain and hindbrain of cobblestone malformations (formerly called Lissencephaly type II). Diagram (A) shows neuron (n) guided by normal radial glial cell (NL RGC) on the left, coursing from ependyma to an intact pial limiting membrane (Pial LM), where it attaches via a bridge made by beta dystroglycan, alpha dystroglycan, or GPR56, which attach to laminin-2 or collagen IV in the Pial LM. In the center and on the right, gaps are seen in the Pial LM; the RGCs do not attach properly due to defects of alpha dystroglycan or GPR56 in the leading process of the RGC, to laminin, or collagen IV, respectively, in the Pial LM. Neurons either detach prematurely or overmigrate through the gaps into the subarachnoid space. A relatively mild cerebellar anomaly is shown in the muscle-eye-brain phenotype shown in (B) and (C). Although the vermis is small and dysmorphic, the hemispheres have nearly normal foliation. A few small cysts are present (white arrows in B and C). The pons contains a midline cleft (black arrow in C). The midbrain tectum is large and smooth due to transpial migration of cells. A coronal image through the cerebrum (D) shows moderate ventricular enlargement, abnormal hyperintensity of subcortical, and deep white matter, and abnormal sulcation over the convexities; note that the cortex in this region (white arrows) is abnormally thick and seems to be formed of radially oriented bands of neurons. A much more severe Walker–Warburg phenotype is shown in (E) and (F). The sagittal image (E) shows a thin brain stem with a large kink in the mid pons (black arrowhead), resembling a persistent pontine flexure. The MB tectum is very large and rounded (large black arrow). Only a small vermis (small black arrow) is present. Massive hydrocephalus can be seen, as can a small occipital cephalocele (white arrow). The axial image (F) shows an extremely small, dysmorphic cerebellum with no vermis and many cysts within the irregular cortex. Both ocular globes are anomalous.
Another malformation complex involving both supra- and infratentorial structures is caused by mutations of the Reelin pathway. Reelin is a large glycoprotein that is secreted into the extracellular matrix by Cajal–Retzius cells in the marginal zone of the developing cerebral cortex, where its actions are thought to allow later migrating glutamatergic neurons to pass neurons in deeper cortical layers and to aid in detachment of the neurons from the radial glia at the proper cortical layer (D'Arcangelo et al., 1995; Ogawa et al., 1996; Trommsdorff et al., 1999; Hack et al., 2007; Sentürk et al., 2011). In the hippocampus, Reelin functions in the alignment of pyramidal neurons (Nakajima et al., 1997; Tissir and Goffinet, 2003), and in the cerebellum, Reelin is secreted by the external granular layer and cerebellar nuclear neurons during early development to aid in dispersion of PCs (Miyata et al., 1996; Trommsdorff et al., 1999; Hack et al., 2007; Larouche et al., 2008). In all regions, Reelin action is mediated by binding to specific receptors on target cells, ApoER2, VLDLR, (Miyata et al., 1997; D'Arcangelo et al., 1999; Hiesberger et al., 1999; Trommsdorff et al., 1999; Fink et al., 2006) and ephrin Bs, the transmembrane ligands for Eph receptors (Sentürk et al., 2011). Interaction with Reelin induces VLDLR and ApoER2 to bind the adaptor protein DAB1, which leads to activation of Src family tyrosine kinases (SFKs) and other kinases that phosphorylate DAB1 at its tyrosine residues (Howell et al., 1997; Sheldon et al., 1997; Rice et al., 1998; Bock and Herz, 2003). In the cerebellum, Reelin interacts with receptors on PCs, which respond by dispersing from their clusters in the central cerebellum and migrating to the cerebellar cortex (Trommsdorff et al., 1999; Hack et al., 2007; Sentürk et al., 2011). If the Reelin cascade within the PCs is disrupted, as by mutations of RELN, its receptors, or DAB1, neither the cerebrum nor the cerebellum form properly, although the precise histological details and mechanisms of the resulting malformations are debated (Bock and Herz, 2003; Larouche et al., 2008; Frotscher, 2010; Boyle et al., 2011; Honda et al., 2011; Sentürk et al., 2011).
Mutations of RELN in humans result in congenital lymphedema and hypotonia, impaired cognition, myopia, nystagmus, and generalized epilepsy. Imaging shows a very severe malformation, including thickened cortex, simplified sulcation, hippocampal dysmorphism, and profound cerebellar hypoplasia (Figures 4A,B) (Hong et al., 2000). Mutations of VLDLR produce a significantly less severe disorder, known as the disequilibrium syndrome (Boycott et al., 2005). In this disorder, children present with delayed motor development and cerebellar ataxia. MRI shows simplified cerebral sulcation (although sulcation is less simplified and cortex less thickened than with RELN mutations) and profound cerebellar hypoplasia (Figure 4C) (Glass et al., 2005). These patterns of cerebral and cerebellar dysgenesis are consistent with the observed findings in animal models with Vldlr knock-outs [Larouche et al., 2008 #3095];(Trommsdorff et al., 1999; Larouche et al., 2008; Honda et al., 2011; Sentürk et al., 2011). Vldlr mediates a “stop” signal for migrating cerebral cortical neurons; its absence allows overmigration with too many neurons in the molecular layer and mildly abnormal sulcation. Apoer2 is essential for migration of late generated cortical neurons past the earlier generated ones; its absence results in a thicker cortex with less sulcation, more resembling that seen with Reln mutations (Benhayon et al., 2003; Hack et al., 2007). In the cerebellum of Vldlr null animals, a large portion of PCs are not dispersed and remain as heterotopic clusters deep within the hemispheres, whereas Apoer2 null animals have ectopic PCs largely restricted to the anterior vermis, resulting in a much less severe cerebellar dysgenesis (Larouche et al., 2008). No human mutations of ApoER2 have been described in the literature. However, the author has seen MRIs of patients with RELN-like cerebral cortical dysgenesis but mildly abnormal cerebella (Figure 4D); testing is underway to determine whether these are caused by mutations of APOER2, DAB1, or some other, as yet unknown, component of the Reelin pathway.
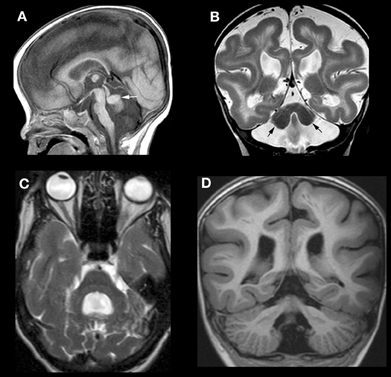
Figure 4. Array of findings in cerebrum and cerebellum of disorders of the Reelin pathway. The most severe situation, with severe RELN depletion results in a very small vermis (white arrow in A), small, smooth cerebellar hemispheres (black arrows in B), and a thick, pachygyric cerebral cortex (B). VLDLR mutation results in a less severe cerebral dysgenesis (cortex is thinner and more sulci are present) but the cerebellum is quite severely affected, both hypoplastic and smooth (C). Severe cerebral but less severe cerebellar involvement (note that the cerebellum is larger and cortex is less smooth) (D) can be seen, but the precise mechanism/mutation that results in this appearance is not known.
Disorders of Cerebellar Dysgenesis
Another well-defined syndrome of cerebellar dysgenesis is the Joubert Syndrome and its Related Disorders (JSRD, often called Molar Tooth Malformations). A familial syndrome of “agenesis of the cerebellar vermis” was first described by Joubert et al. in 1969; affected patients had episodic hyperpnea in infancy, abnormal eye movements, ataxia, and cognitive impairment (Joubert et al., 1969). The disorder was further elucidated in several papers by Boltshauser and his colleagues (Boltshauser and Isler, 1977; Boltshauser et al., 1981; Steinlin et al., 1997), who described a variable degree of vermian hypoplasia (rather than agenesis) and multiple other features, with variable outcomes, in affected patients. The advent of MRI revealed a characteristic “molar tooth” appearance of the MB (Figure 5) (Maria et al., 1997; Quisling et al., 1999). This appearance was soon found in many disorders with widely varying phenotypic features of other organs including the eyes, kidneys, liver, and extremities (Egger et al., 1982; Houdou et al., 1986; Chance et al., 1999; Satran et al., 1999; Haug et al., 2000; Gleeson et al., 2004), suggesting that these heretofore seemingly distinct disorders were in some way related. These disorders were very curious, as no common thread could be found among the processes involved, even within the same organs. Within the nervous system, it was difficult to find the common underlying cause connecting the disorders of retina development, vermian hypoplasia, aberrant white matter pathways (neither the corticospinal tracts nor the superior cerebellar peduncles decussate properly), occasional polymicrogyria and hypothalamic harmartomas (Figure 5) (Haug et al., 2000; Zaki et al., 2008; Giordano et al., 2009; Harting et al., 2011). It was also noteworthy that the characteristic molar tooth sign had many different appearances with the molar “roots” (composed of the superior cerebellar peduncles) sometimes thick, sometimes thin, sometimes straight and sometimes curved; clearly a lot of different processes were going on. Answers began to emerge when it was discovered that all of the genes implicated in these disorders are associated with the function of the primary cilium/basal body organelle, a structure that is present in many cell types, including renal tubule epithelial cells, retinal photoreceptors, chondrocytes, fibroblasts, and neurons (Arts et al., 2007; Chizhikov et al., 2007; Delous et al., 2007; Frank et al., 2008; Gorden et al., 2008; Spassky et al., 2008; Doherty, 2009). Ciliary membranes contain receptors and ion channel proteins mediating cell signaling, including roles for SHH, WNT, and PDGFa signaling pathways that control diverse processes (e.g., cell differentiation, migration, axonal pathfinding, and planar cell polarity). SHH binding to its transmembrane receptor PTCH abolishes the inhibitory effect of PTCH on SMO, resulting in localization of SMO to the primary cilium, and transduction of signals to the nucleus through the GLI transcription factors. The result is de-repression and activation of SHH target genes (Satir and Christensen, 2007). The SHH pathway is important for dorsal-ventral patterning of the neural tube and, later, for proliferation of cerebellar granule cells (Wechsler-Reya and Scott, 1999; Huangfu et al., 2003). However, reduction of granule cell proliferation cannot in itself explain the vermian hypoplasia seen in the MTMs (Chizhikov et al., 2007; Spassky et al., 2008), as the effect of SHH is diffuse, involving both vermis and hemispheres, but the vermis is very disproportionately involved in MTMs. A more likely explanation is decreased Wnt reporter activity, accompanied by reduced proliferation, at the site of hemispheric fusion, as was reported in the developing cerebellum of Ahi1-mutant mice (Lancaster et al., 2011); this phenotype was partially rescued by treatment with lithium, a Wnt pathway agonist (Lancaster et al., 2011). The wide phenotypic variation among families harboring mutations in genes encoding ciliary proteins also suggests that genetic modifiers are important in determining specific features within the ciliopathy spectrum (Davis et al., 2011; Zaki et al., 2011). This interesting group of disorders has much more to teach us about both development and disorders thereof.
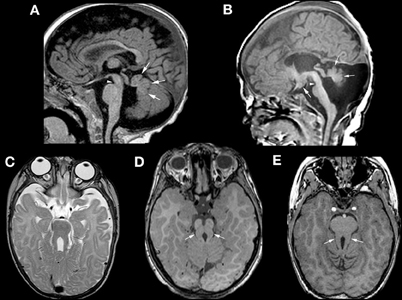
Figure 5. Neuroimaging findings in molar tooth malformations. The characteristic imaging findings of a small vermis (small white arrows, A,B) and narrow isthmus (small white arrowhead, A,B) are identified on sagittal images. A tuber cinereum hamartoma is seen in (B). The variable appearances of the “molar tooth,” resulting from the large, horizontal superior cerebellar peduncles, are shown (white arrows in C, D, and E).
Many other disorders of the MB-HB have been described in association with supratentorial anomies, including cerebellar hypoplasia associated with severe variants of cerebral lissencephaly secondary to alpha-A1 tubulin (TUBA1A) and other tubulin mutations (Poirier et al., 2007; Kumar et al., 2010), cerebellar hypoplasia associated with postmigrational microcephaly secondary to mutations of calcium modulated-dependent serine protein kinase (CASK), which is associated with X-linked mental retardation (Najm et al., 2008) and cerebellar hypoplasia associated with midline brain stem clefts and agenesis of the corpus callosum, presumably resulting from mutations preventing midline axonal crossing (Barkovich et al., 2009). Application of discoveries from developmental neuroscience will aid our understanding of these disorders, and what is learned from studying these disorders will help us to better understand brain development.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ackroyd, M. R., Whitmore, C., Prior, S., Kaluarachchi, M., Nikolic, M., Mayer, U., Muntoni, F., and Brown, S. C. (2011). Fukutin-related protein alters the deposition of laminin in the eye and brain. J. Neurosci. 31, 12927–12935.
Acosta, F. L. Jr., Binder, D. K., Barkovich, A. J., Frieden, I. J., and Gupta, N. (2005). Neurocutaneous melanosis presenting with hydrocephalus. Case report and review of the literature. J. Neurosurg. 102(Suppl. 1), 96–100.
Aida, N., Yagishita, A., Takada, K., and Katsumata, Y. (1994). Cerebellar MR in Fukuyama congenital muscular dystrophy: polymicrogyria with cystic lesions. AJNR. Am. J. Neuroradiol. 15, 1755–1759.
Aldinger, K. A., Lehmann, O. J., Hudgins, L., Chizhikov, V. V., Bassuk, A. G., Ades, L. C., Krantz, I. D., Dobyns, W. B., and Millen, K. J. (2009). FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat. Genet. 41, 1037–1042.
Aranda-Orgillés, B., Trockenbacher, A., Winter, J., Aigner, J., Köhler, A., Jastrzebska, E., Stahl, J., Müller, E.-C., Otto, A., Wanker, E., Schneider, R., and Schweiger, S. (2008). The Opitz syndrome gene product MID1 assembles a microtubule-associated ribonucleoprotein complex. Hum. Genet. 123, 163–176.
Arts, H. H., Doherty, D., van Beersum, S. E. c., Parisi, M. A., Letteboer, S. J. F., Gorden, N. T., Peters, T. A., Marker, T., Voesenek, K., Kartono, A., Ozyurek, H., Farin, F. M., Kroes, H. Y., Wolfrum, U., Brunner, H. G., Cremers, F. P. M., Glass, I. A., Knoers, N. V., and Roepman, R. (2007). Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 39, 882–888.
Ballarati, L., Rossi, E., Bonati, M. T., Gimelli, S., Maraschio, P., Finelli, P., Giglio, S., Lapi, E., Bedeschi, M. F., Guerneri, S., Arrigo, G., Patricelli, M. G., Mattina, T., Guzzardi, O., Pecile, V., Police, A., Scarano, G., Larizza, L., Zuffardi, O., and Giardino, D. (2007). 13q Deletion and central nervous system anomalies: further insights from karyotype-phenotype analyses of 14 patients. J. Med. Genet. 44:e60.
Barkovich, A. J. (1998). Neuroimaging manifestations and classification of congenital muscular dystrophies. AJNR. Am. J. Neuroradiol. 19, 1389–1396.
Barkovich, A., Frieden, I., and Williams, M. (1994). MR of neurocutaneous melanosis. AJNR. Am. J. Neuroradiol. 15, 859–867.
Barkovich, A. J., Guerrini, R., Kuzniecky, R. I., Jackson, G. D., and Dobyns, W. B. (2012). A developmental and genetic classification for malformations of cortical development: Update 2012. Brain (in press).
Barkovich, A. J., and Raybaud, C. A. (2012). Pediatric Neuroimaging, 5th edn. (Philadelphia: Lippincott Williams & Wilkins), 469.
Barkovich, A. J., Kjos, B. O., Norman, D., and Edwards, M. S. B. (1989). Revised classification of posterior fossa cysts and cyst-like malformations based on results of multiplanar MR imaging. AJNR. Am. J. Neuroradiol. 10, 977–988.
Barkovich, A. J., Millen, K. J., and Dobyns, W. B. (2007). A developmental classification of malformations of the brainstem. Ann. Neurol. 62, 625–639.
Barkovich, A. J., Millen, K. J., and Dobyns, W. B. (2009). A developmental and genetic classification for midbrain-hindbrain malformations. Brain 132, 3199–3230.
Barth, P. G., Majoie, C. B., Caan, M. W. A., Weterman, M. A. J., Kyllerman, M., Smit, L. M. E., Kaplan, R. A., Haas, R. H., Baas, F., Cobben, J.-M., and Poll-The, B. T. (2007). Pontine tegmental cap dysplasia: a novel brain malformation with a defect in axonal guidance. Brain 130, 2258–2266.
Bednarek, N., Scavarda, D., Mesmin, F., Sabouraud, P., Motte, J., and Morville, P. (2005). Midbrain disconnection: an aetiology of severe central neonatal hypotonia. Eur. J. Paediatr. Neurol. 9, 419–422.
Benhayon, D., Magdaleno, S., and Curran, T. (2003). Binding of purified Reelin to ApoER2 and VLDLR mediates tyrosine phosphorylation of Disabled-1. Brain Res. Mol. Brain Res. 112, 33–45.
Bindal, A. K., Storrs, B. B., and Mclone, D. G. (1990–1991). Management of the Dandy-Walker syndrome. Pediatr. Neurosci. 16, 163–169.
Blank, M. C., Grinberg, I., Aryee, E., Laliberte, C., Chizhikov, V. V., Henkelman, R. M., and Millen, K. J. (2011). Multiple developmental programs are altered by loss of Zic1 and Zic4 to cause Dandy-Walker malformation cerebellar pathogenesis. Development 138, 1207–1216.
Bock, H. H., and Herz, J. (2003). Reelin activates SRC family tyrosine kinases in neurons. Curr. Biol. 13, 18–26.
Boddaert, N., Klein, O., Ferguson, N., Sonigo, P., Parisot, D., Hertz-Pannier, L., Baraton, J., Emond, S., Simon, I., Chigot, V., Schmit, P., Pierre-Kahn, A., and Brunelle, F. (2003). Intellectual prognosis of the Dandy-Walker malformation in children: the importance of vermian lobulation. Neuroradiology 45, 320–324.
Boltshauser, E., Herdon, M., Dumermuth, G., and Isler, W. (1981). Joubert syndrome: clinical and polygraphic observations in a further case. Neuropediatrics 12, 181–191.
Boltshauser, E., and Isler, W. (1977). Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia associated with dysplasia of the cerebellar vermis. Neuropadiatrie 8, 57–66.
Boycott, K. M., Flavelle, S., Bureau, A., Glass, H. C., Fujiwara, T. M., Wirrell, E., Davey, K., Chudley, A. E., Scott, J. N., Mcleod, D. R., Parboosingh, J. S., and Boycott, K. M. (2005). Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am. J. Hum. Genet. 77, 477–483.
Boyle, M. P., Bernard, A., Thompson, C. L., Ng, L., Boe, A., Mortrud, M., Hawrylycz, M. J., Jones, A. R., Hevner, R. F., and Lein, E. S. (2011). Cell-type-specific consequences of reelin deficiency in the mouse neocortex, hippocampus, and amygdala. J. Comp. Neurol. 519, 2061–2089.
Broccoli, V., Boncinelli, E., and Wurst, W. (1999). The caudal limit of Otx2 expression positions the isthmic organizer. Nature 401, 164–168.
Cazorla Calleja, M. R., Verdu, A., and Felix, V. (2003). Dandy-Walker malformation in an infant with tetrasomy 9p. Brain Dev. 25, 220–223.
Chan, Y. M., Keramaris-Vrantsis, E., Lidov, H. G., Norton, J. H., Zinchenko, N., Gruber, H. E., Thresher, R., Blake, D. J., Ashar, J., Rosenfeld, J., and Lu, Q. L. (2010). Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum. Mol. Genet. 19, 3995–4006.
Chance, P. F., Cavalier, L., Satran, D., Pellegrino, J. E., Koenig, M., and Dobyns, W. B. (1999). Clinical nosologic and genetic aspects of Joubert and related syndromes. J. Child Neurol. 14, 660–666.
Chen, C. P., Chen, C. P., and Shih, J. C. (2005). Association of partial trisomy 9p and the Dandy-Walker malformation. Am. J. Med. Genet. A 132A, 111–112.
Chiang, N.-Y., Hsiao, C.-C., Huang, Y.-S., Chen, H.-Y., Hsieh, I.-J., Chang, G.-W., and Lin, H.-H. (2011). Disease-associated GPR56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. J. Biol. Chem. 286, 14215–14225.
Chizhikov, V. V., Davenport, J., Zhang, Q., Shih, E. K., Cabello, O. A., Fuchs, J. L., Yoder, B. K., and Millen, K. J. (2007). Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J. Neurosci. 27, 9780–9789.
Chizhikov, V., and Millen, K. J. (2003). Development and malformations of the cerebellum in mice. Mol. Genet. Metab. 80, 54–65.
Clement, E., Mercuri, E., Godfrey, C., Smith, J., Robb, S., Kinali, M., Straub, V., Bushby, K., Manzur, A., Talim, B., Cowan, F., Quinlivan, R., Klein, A., Longman, C., Mcwilliam, R., Topaloglu, H., Mein, R., Abbs, S., North, K., Barkovich, A. J., Rutherford, M., and Muntoni, F. (2008). Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann. Neurol. 64, 573–582.
Courchesne, E., Redcay, E., Morgan, J., and Kennedy, D. (2005). Autism at the beginning: microstructural and growth abnormalities underlying the cognitive and behavioral phenotype of autism. Dev. Psychopathol. 17, 577–597.
D'Arcangelo, G., Homayouni, R., Keshvara, L., Rice, D. S., Sheldon, M., and Curran, T. (1999). Reelin is a ligand for lipoprotein receptors. Neuron 24, 471–479.
D'Arcangelo, G., Miao, G. G., Chen, S. C., Soares, H. D., Morgan, J. I., and Curran, T. (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723.
Davis, E. E., Zhang, Q., Liu, Q., Diplas, B. H., Davey, L. M., Hartley, J., Stoetzel, C., Szymanska, K., Ramaswami, G., Logan, C. V., Muzny, D. M., Young, A. C., Wheeler, D. A., Cruz, P., Morgan, M., Lewis, L. R., Cherukuri, P., Maskeri, B., Hansen, N. F., Mullikin, J. C., Blakesley, R. W., Bouffard, G. G., Gyapay, G., Rieger, S., Tonshoff, B., Kern, I., Soliman, N. A., Neuhaus, T. J., Swoboda, K. J., Kayserili, H., Gallagher, T. E., Lewis, R. A., Bergmann, C., Otto, E. A., Saunier, S., Scambler, P. J., Beales, P. L., Gleeson, J. G., Maher, E. R., Attie-Bitach, T., Dollfus, H., Johnson, C. A., Green, E. D., Gibbs, R. A., Hildebrandt, F., Pierce, E. A., and Katsanis, N. (2011). TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189–196.
Delous, M., Baala, L., Salomon, R., Laclef, C., Vierkotten, J., Tory, K., Golzio, C., Lacoste, T., Besse, L., Ozilou, C., Moutkine, I., Hellman, N. E., Anselme, I., Silbermann, F., Vesque, C., Gerhardt, C., Rattenberry, E., Wolf, M. T. F., Gubler, M. C., Martinovic, J., Encha-Razavi, F., Boddaert, N., Gonzales, M., Macher, M. A., Nivet, H., Champion, G., Bertheleme, J. P., Niaudet, P., Mcdonald, F., Hildebrandt, F., Johnson, C. A., Vekemans, M., Antignac, C., Ruther, U., Schneider-Maunoury, S., Attie-Bitach, T., and Saunier, S. (2007). The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 39, 875–881.
Doherty, D. (2009). Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin. Pediatr. Neurol. 16, 143–154.
Egger, J., Bellman, M. H., Ross, E. M., and Baraitser, M. (1982). Joubert-Boltshauser syndrome with polydactyly in siblings. J. Neurol. Neurosurg. Psychiatr. 45, 737–739.
Fink, A. J., Englund, C., Daza, R. A., Pham, D., Lau, C., Nivison, M., Kowalczyk, T., and Hevner, R. F. (2006). Development of the deep cerebellar nuclei: transcription factors and cell migration from the rhombic lip. J. Neurosci. 26, 3066–3076.
Fontanella, B., Russolillo, G., and Meroni, G. (2008). MID1 mutations in patients with X-linked Opitz G/BBB syndrome. Hum. Mutat. 29, 584–594.
Frank, V., Hollander, A. I. D., Brüchle, N. O., Zonneveld, M. N., Nürnberg, G., Becker, C., Bois, G. D., Kendziorra, H., Roosing, S., Senderek, J., Nürnberg, P., Cremers, F. P. M., Zerres, K., and Bergmann, C. (2008). Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum. Mutat. 29, 45–52.
Frotscher, M. (2010). Role for Reelin in stabilizing cortical architecture. Trends Neurosci. 33, 407–414.
Gelot, A., Billette de Villemeur, T., Bordarier, C., Ruchoux, M. M., Moraine, C., and Ponsot, G. (1995). Developmental aspects of type II lissencephaly. Comparative study of dysplastic lesions in fetal and post-natal brains. Acta Neuropathol. 89, 72–84.
Giordano, L., Vignoli, A., Pinelli, L., Brancati, F., Accorsi, P., Faravelli, F., Gasparotti, R., Granata, T., Giaccone, G., Inverardi, F., Frassoni, C., Dallapiccola, B., Valente, E., and Spreafico, R. (2009). Joubert syndrome with bilateral polymicrogyria: clinical and neuropathological findings in two brothers. Am. J. Med. Genet. 149A, 1511–1515.
Glass, H. C., Boycott, K. M., Adams, C., Barlow, K., Scott, J. N., Chudley, A. E., Fujiwara, T. M., Morgan, K. E. Wirrell, E., and Mcleod, D. R. (2005). Autosomal recessive cerebellar hypoplasia in the Hutterite population. Dev. Med. Child Neurol. 47, 691–695.
Gleeson, J. G., Keeler, L. C., Parisi, M. A., Marsh, S. E., Chance, P. F., Glass, I. A., Graham Jr, J. M., Maria, B. L., Barkovich, A. J., and Dobyns, W. B. (2004). Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am. J. Med. Genet. A 125A, 125–134.
Godfrey, C., Clement, E., Mein, R., Brockington, M., Smith, J., Talim, B., Straub, V., Robb, S., Quinlivan, R., Feng, L., Jimenez-Mallebrera, C., Mercuri, E., Manzur, A. Y., Kinali, M., Torelli, S., Brown, S. C., Sewry, C. A., Bushby, K., Topaloglu, H., North, K., Abbs, S., and Muntoni, F. (2007). Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 130, 2725–2735.
Golden, J. A., Rorke, L. B., and Bruce, D. A. (1987). Dandy-Walker syndrome and associated anomalies. Pediatr. Neurosci. 13, 38–44.
Gorden, N. T., Arts, H. H., Parisi, M. A., Coene, K. L., Letteboer, S. J., Van Beersum, S. E., Mans, D. A., Hikida, A., Eckert, M., Knutzen, D., Alswaid, A. F., Ozyurek, H., Dibooglu, S., Otto, E. A., Liu, Y., Davis, E. E., Hutter, C. M., Bammler, T. K., Farin, F. M., Dorschner, M., Topcu, M., Zackai, E. H., Rosenthal, P., Owens, K. N., Katsanis, N., Vincent, J. B., Hildebrandt, F., Rubel, E. W., Raible, D. W., Knoers, N. V., Chance, P. F., Roepman, R., Moens, C. B., Glass, I. A., and Doherty, D. (2008). CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet. 83, 559–571.
Grinberg, I., and Millen, K. J. (2005). The ZIC gene family in development and disease. Clin. Genet. 67, 290–296.
Grinberg, I., Northrup, H., Ardinger, H., Prasad, C., Dobyns, W. B., and Millen, K. J. (2004). Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in Dandy-Walker malformation. Nat. Genet. 36, 1053–1055.
Hack, I., Hellwig, S., Junghans, D., Brunne, B., Bock, H. H., Zhao, S., and Frotscher, M. (2007). Divergent roles of ApoER2 and Vldlr in the migration of cortical neurons. Development 134, 3883–3891.
Haltia, M., Leivo, I., Somer, H., Pihko, H., Paetau, A., Kivelä, T., Tarkkanen, A., Tomé, F., Engvall, E., and Santavuori, P. (1997). Muscle-eye-brain disease: a neuropathological study. Ann. Neurol. 41, 173–180.
Hart, M. N., Malamud, N., and Ellis, W. G. (1972). The Dandy-Walker syndrome. A clinicopathological study based on 28 cases. Neurology 22, 771–780.
Harting, I., Kotzaeridou, U., Poretti, A., Seitz, A., Pietz, J., Bendszus, M., and Boltshauser, E. (2011). Interpeduncular heterotopia in joubert syndrome: a previously undescribed MR finding. AJNR. Am. J. Neuroradiol. 32, 1286–1289.
Haug, K., Khan, S., Fuchs, S., and Konig, R. (2000). OFD II, OFD VI, and Joubert syndrome manifestations in 2 sibs. Am. J. Med. Genet. 91, 135–137.
Hewitt, J. E. (2009). Abnormal glycosylation of dystroglycan in human genetic disease. Biochem. Biophys. Acta 1792, 853–861.
Hiesberger, T., Trommsdorff, M., Howell, B. W., Goffinet, A., Mumby, M. C., Cooper, J. A., and Herz, J. (1999). Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24, 481–489.
Honda, T., Kobayashi, K., Mikoshiba, K., and Nakajima, K. (2011). Regulation of cortical neuron migration by the Reelin signaling pathway. Neurochem. Res. 36, 1270–1279.
Hong, S. E., Shugart, Y. Y., Huang, D. T., Al Shahwan, S., Grant, P. E., Hourihane, J. O. B., Martin, N. D. T., and Walsh, C. A. (2000). Autosomal recessive lissencephaly with cerebellar hypoplasia (LCH) is associated with human reelin gene mutations. Nat. Genet. 26, 93–96.
Houdou, S., Ohno, K., Takashima, S., and Takeshita, K. (1986). Joubert syndrome associated with unilater ptosis and Leber congenital amaurosis. Pediatr. Neurol. 2, 102–105.
Howell, B. W., Gertler, F. B., and Cooper, J. A. (1997). Mouse disabled (mDab1): a Src binding protein implicated in neuronal development. EMBO J. 16, 121–132.
Huangfu, D., Liu, A., Rakeman, A. S., Murcia, N. S., Niswander, L., and Anderson, K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87.
Jalali, A., Aldinger, K., Chary, A., Mclone, D., Bowman, R., Le, L., Jardine, P., Newbury-Ecob, R., Mallick, A., Jafari, N., Russell, E. J., Curran, J., Nguyen, P., Ouahchi, K., Lee, C., Dobyns, W. B., Millen, K. J., Pina-Neto, J. M., Kessler, J. A., and Bassuk, A. G. (2008). Linkage to chromosome 2q36.1 in autosomal dominant Dandy-Walker malformation with occipital cephalocele and evidence for genetic heterogeneity. Hum. Genet. 123, 237–245.
Jen, J. C., Chan, W.-M., Bosley, T. M., Wan, J., Carr, J. R., Rub, U., Shattuck, D., Salamon, G., Kudo, L. C., Ou, J., Lin, D. D. M., Salih, M. A. M., Kansu, T., Al Dhalaan, H., Al Zayed, Z., Macdonald, D. B., Stigsby, B., Plaitakis, A., Dretakis, E. K., Gottlob, I., Pieh, C., Traboulsi, E. I., Wang, Q., Wang, L., Andrews, C., Yamada, K., Demer, J. L., Karim, S., Alger, J. R., Geschwind, D. H., Deller, T., Sicotte, N. L., Nelson, S. F., Baloh, R. W., and Engle, E. C. (2004). Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science 304, 1509–1513.
Jissendi-Tchofo, P., Kara, S., and Barkovich, A. J. (2009). Midbrain-hindbrain involvement in lissencephalies. Neurology 72, 410–418.
Joubert, M., Eisenring, J. J., Robb, J. P., and Andermann, F. (1969). Familial agenesis of the cerebellar vermis: a syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 19, 813–825.
Kanagawa, M., and Toda, T. (2006). The genetic and molecular basis of muscular dystrophy: roles of cell-matrix linkage in the pathogenesis. J. Hum. Genet. 51, 915–926.
Klein, O., Pierre-Kahn, A., Boddaert, N., Parisot, D., and Brunelle, F. (2003). Dandy-Walker malformation: prenatal diagnosis and prognosis. Childs Nerv. Syst. 19, 484–489.
Koirala, S., Jin, Z., Piao, X., and Corfas, G. (2009). GPR56-regulated granule cell adhesion is essential for rostral cerebellar development. J. Neurosci. 29, 7439–7449.
Kumar, R. A., Pilz, D. T., Babatz, T. D., Cushion, T. D., Harvey, K., Topf, M., Yates, L., Robb, S., Uyanik, G., Mancini, G. M. S., Rees, M. I., Harvey, R. J., and Dobyns, W. B. (2010). TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum. Mol. Genet. 19, 2817–2827.
Labelle-Dumais, C., Dilworth, D. J., Harrington, E. P., De Leau, M., Lyons, D., Kabaeva, Z., Manzini, M. C., Dobyns, W. B., Walsh, C. A., Michele, D. E., and Gould, D. B. (2011). COL4A1 mutations cause ocular dysgenesis, neuronal localization defects, and myopathy in mice and Walker-Warburg syndrome in humans. PLoS Genet. 7:e1002062. doi: 10.1371/journal.pgen.1002062
Lancaster, M. A., Gopal, D. J., Kim, J., Saleem, S. N., Silhavy, J. L., Louie, C. M., Thacker, B. E., Williams, Y., Zaki, M. S., and Gleeson, J. G. (2011). Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat. Med. 17, 726–731.
Lancioni, A., Pizzo, M., Fontanella, B., Ferrentino, R., Napolitano, L. M. R., De Leonibus, E., and Meroni, G. (2010). Lack of Mid1, the mouse ortholog of the Opitz syndrome gene, causes abnormal development of the anterior cerebellar vermis. J. Neurosci. 30, 2880–2887.
Larouche, M., Beffert, U., Herz, J., and Hawkes, R. (2008). The reelin receptors Apoer2 and Vldlr coordinate the patterning of purkinje cell topography in the developing mouse cerebellum. PLoS One 3:e1653. doi: 10.1371/journal.pone.0001653
Lecourtois, M., Poirier, K., Friocourt, G., Jaglin, X., Goldenberg, A., Saugier-Veber, P., Chelly, J., and Laquerrière, A. (2010). Human lissencephaly with cerebellar hypoplasia due to mutations in TUBA1A: expansion of the foetal neuropathological phenotype. Acta Neuropathol. 119, 779–789.
Li, S., Jin, Z., Koirala, S., Bu, L., Xu, L., Hynes, R. O., Walsh, C. A., Corfas, G., and Piao, X. (2008). GPR56 regulates pial basement membrane integrity and cortical lamination. J. Neurosci. 28, 5817–5826.
Lumsden, A. (2004). Segmentation and compartition in the early avian hindbrain. Mech. Dev. 121, 1081–1088.
Luo, R., Jeong, S.-J., Jin, Z., Strokes, N., Li, S., and Piao, X. (2011). G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination. Proc. Nat. Acad. Sci. U.S.A. 108, 12925–12930.
Maria, B., Hoang, K., Tusa, R., Mancuso, A., Hamed, L., Quisling, R., Hove, M., Fennell, E., Booth-Jones, M., Ringdahl, D., Yachnis, A., Creel, G., and Frerking, B. (1997). “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J. Child Neurol. 12, 423–430.
Maria, B. L., Zinreich, S. J., Carson, B. C., Rosenbaum, A. E., and Freeman, J. M. (1987). Dandy-Walker syndrome revisited. Pediatr. Neurosci. 13, 45–51.
Martin, P. T. (2005). The dystroglycanopathies: the new disorders of O-linked glycosylation. Semin. Pediatr. Neurol. 12, 152–158.
McCormack, W. M. Jr., Shen, J. J., Curry, S. M., Berend, S. A., Kashork, C., Pinar, H., Potocki, L., and Bejjani, B. A. (2003). Partial deletions of the long arm of chromosome 13 associated with holoprosencephaly and the Dandy-Walker malformation. Am. J. Med. Genet. A 118A, 384–389.
Melaragno, M. I., Brunoni, D., Patricio, F. R., Corbani, M., Mustacchi, Z., Dos Santos Rde, C., and Lederman, H. M. (1992). A patient with tetrasomy 9p, Dandy-Walker cyst and Hirschsprung disease. Ann. Genet. 35, 79–84.
Millet, S., Campbell, K., Epstein, D. J., Losos, K., Harris, E., and Joyner, A. L. (1999). A role for Gbx2 in repression of Otx2 and positioning the mid/hindbrain organizer. Nature 401, 161–164.
Miyata, T., Nakajima, K., Aruga, J., Takahashi, S., Ikenaka, K., Mikoshiba, K., and Ogawa, M. (1996). Distribution of a reeler gene-related antigen in the developing cerebellum: an immunohistochemical study with an allogeneic antibody CR-50 on normal and reeler mice. J. Comp. Neurol. 372, 215–228.
Miyata, T., Nakajima, K., Mikoshiba, K., and Ogawa, M. (1997). Regulation of Purkinje cell alignment by reelin as revealed with CR-50 antibody. J. Neurosci. 17, 3599–3609.
Moog, U., Jones, M. C., Bird, L. M., and Dobyns, W. B. (2005). Oculocerebrocutaneous syndrome: the brain malformation defines a core phenotype. J. Med. Genet. 42, 913–921.
Moore, S. A., Saito, F., Chen, J., Michele, D. E., Henry, M. D., Messing, A., Cohn, R. D., Ross-Barta, S. E., Westra, S., Williamson, R. A., Hoshi, T., and Campbell, K. P. (2002). Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 418, 422–425.
Najm, J., Horn, D., Wimplinger, I., Golden, J. A., Chizhikov, V. V., Sudi, J., Christian, S. I., Ullmann, R., Kuechler, A., Haas, C. A., Flubacher, A., Charnas, L. R., Uyanik, G., Frank, U., Klopocki, E., Dobyns, W. B., and Kutsche, K. (2008). Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 40, 1065–1067.
Nakajima, K., Mikoshiba, K., Miyata, T., Kudo, C., and Ogawa, M. (1997). Disruption of hippocampal development in vivo by CR-50 mAb against Reelin. Proc. Natl. Acad. Sci. U.S.A. 94, 8196–8201.
Narayanan, H. S., Gandhi, D. H., and Girimaji, S. R. (1987). Neurocutaneous melanosis associated with Dandy-Walker syndrome. Clin. Neurol. Neurosurg. 89, 197–200.
Norman, M. G., Mcgillivray, B. C., Kalousek, D. K., Hill, A., and Poskitt, K. J. (1995). Congenital Malformations of the Brain: Pathologic, Embryologic, Clinical, Radiologic and Genetic Aspects. Oxford: Oxford University Press.
Ogawa, M., Miyata, T., Nakajima, K., Yagyu, K., Seike, M., Ikenaka, K., Yamamoto, H., and Mikoshiba, K. (1996). The reeler gene associated antigen on Cajal-Retzius neurons is a crucial molecule for laminal organization of cortical neurons. Neuron 14, 899–912.
Pinson, L., Augé, J., Audollent, S., Mattéi, G., Etchevers, H., Gigarel, N., Razavi, F., Lacombe, D., Odent, S., Le Merrer, M., Amiel, J., Munnich, A., Meroni, G., Lyonnet, S., Vekemans, M., and Attié-Bitach, T. (2004). Embryonic expression of the human MID1 gene and its mutations in Opitz syndrome. J. Med. Genet. 41, 381–386.
Poirier, K., Keays, D. A., Francis, F., Saillour, Y., Bahi, N., Manouvrier, S., Fallet-Bianco, C., Pasquier, L., Toutain, A., Phan, F., Tuy, D., Bienvenu, T., Joriot, S., Odent, S., Ville, D., Desguerre, I., Goldenberg, A., Moutard, M.-L., Fryns, J.-P., van Esch, H., Harvey, R. J., Siebold, C., Flint, J., Beldjord, C., and Chelly, J. (2007). Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum. Mutat. 28, 1055–1064.
Quaderi, N. A., Schweiger, S., Gaudenz, K., Franco, B., Rugarli, E. I., Berger, W., Feldman, G. J., Volta, M., Andolfi, G., Gilgenkrantz, S., Marion, R. W., Hennekam, R. C., Opitz, J. M., Muenke, M., Ropers, H. H., and Ballabio, A. (1997). Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat. Genet. 17, 285–291.
Quisling, R., Barkovich, A., and Maria, B. (1999). Magnetic resonance imaging features and classification of central nervous system malformations in Joubert syndrome. J. Child Neurol. 14, 628–635.
Raybaud, C. (1982). Cystic malformations of the posterior fossa – abnormalities associated with development of the roof of the fourth ventricle and adjacent meningeal structures. J. Neuroradiol. 9, 103–133.
Rice, D. S., Sheldon, M., D'Arcangelo, G., Nakajima, K., Goldowitz, D., and Curran, T. (1998). Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development 125, 3719–3729.
Ross, M. E., Swanson, K., and Dobyns, W. B. (2001). Lissencephaly with cerebellar hypoplasia (LCH): a heterogeneous group of cortical malformations. Neuropediatrics 32, 256–263.
Saito, Y., Yamamoto, T., Mizuguchi, M., Kobayashi, M., Saito, K., Ohno, K., and Osawa, M. (2006). Altered glycosylation of alpha-dystroglycan in neurons of Fukuyama congenital muscular dystrophy brains. Brain Res. 1075, 223–228.
Sang, L., Miller, J. J., Corbit, K. C., Giles, R. H., Brauer, M. J., Otto, E. A., Baye, L. M., Wen, X., Scales, S. J., Kwong, M., Huntzicker, E. G., Sfakianos, M. K., Sandoval, W., Bazan, J. F., Kulkarni, P., Garcia-Gonzalo, F. R., Seol, A. D., O'Toole, J. F., Held, S., Reutter, H. M., Lane, W. S., Rafiq, M. A., Noor, A., Ansar, M., Devi, A. R. R., Sheffield, V. C., Slusarski, D. C., Vincent, J. B., Doherty, D. A., Hildebrandt, F., Reiter, J. F., and Jackson, P. K. (2011). Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 145, 513–528.
Satir, P., and Christensen, S. T. (2007). Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 69, 377–400.
Satran, D., Pierpont, M. E. M., and Dobyns, W. B. (1999). Cerebello-oculo-renal syndromes including Arima, Senior-Löken and COACH syndromes: more than just variants of Joubert syndrome. Am. J. Med. Genet. 86, 459–469.
Sentürk, A., Pfennig, S., Weiss, A., Burk, K., and Acker-Palmer, A. (2011). Ephrin Bs are essential components of the Reelin pathway to regulate neuronal migration. Nature 472, 356–360.
Sheldon, M., Rice, D. S., D'Arcangelo, G., Yoneshima, H., Nakajima, K., Mikoshiba, K., Howell, B. W., Cooper, J. A., Goldowitz, D., and Curran, T. (1997). Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 389, 730–733.
Sicotte, N. L., Salamon, G., Shattuck, D. W., Hageman, N., Rub, U., Salamon, N., Drain, A. E., Demer, J. L., Engle, E. C., Alger, J. R., Baloh, R. W., Deller, T., and Jen, J. C. (2006). Diffusion tensor MRI shows abnormal brainstem crossing fibers associated with ROBO3 mutations. Neurology 67, 519–521.
Soto-Ares, G., Yjoyes, B., Lemaitre, M., Vallee, L., and Pruvo, J. (2003). MRI in children with mental retardation. Pediatr. Radiol. 33, 334–345.
Spassky, N., Han, Y. G., Aguilar, A., Strehl, L., Besse, L., Laclef, C., Romaguera Ros, M., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (2008). Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev. Biol. 317, 246–259.
Steinlin, M., Schmid, M., Landau, K., and Boltshauser, E. (1997). Follow-up in children with Joubert syndrome. Neuropediatrics 28, 204–211.
Takada, K., and Nakamura, H. (1990). Cerebellar micropolygyria in Fukuyama congenital muscular dystrophy: observations in fetal and pediatric cases. Brain Dev. 12, 774–778.
Tissir, F., and Goffinet, A. M. (2003). Reelin and brain development. Nat. Rev. Neurosci. 4, 496–505.
Tortori-Donati, P., Fondelli, M., Rossi, A., and Carini, S. (1996). Cystic malformations of the posterior cranial fossa originating from a deficit of the posterior membranous area. mega cisterna magna and persisting Blake's pouch: two separate entities. Childs Nerv. Syst. 12, 303–308.
Trommsdorff, M., Gotthardt, M., Hiesberger, T., Shelton, J., Stockinger, W., Nimpf, J., Hammer, R., Richardson, J., and Herz, J. (1999). Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97, 689–701.
van der Knaap, M. S., Smit, L. M. E., Barth, P. G., Catsman-Berrevoets, C. E., Brouwer, O. F., Begeer, J. H., De Coo, I. F. M.,and (1997). MRI in classification of congenital muscular dystrophies with brain abnormalities. Ann. Neurol. 42, 50–59.
van Reeuwijk, J., Maugenre, S., van den Elzen, C., Verrips, A., Bertini, E., Muntoni, F., Merlini, L., Scheffer, H., Brunner, H. G., Guicheney, P., and van Bokhoven, H. (2006). The expanding phenotype of POMT1 mutations: from Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Hum. Mutat. 27, 453–459.
Wassef, M., and Joyner, A. (1997). Early mesencephalon/metencephalon patterning and development of the cerebellum. Perspect. Dev. Neurobiol. 5, 3–16.
Wechsler-Reya, R. J., and Scott, M. P. (1999). Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22, 103–114.
Zaki, M. S., Abdel-Aleem, A., Abdel-Salam, G. M. H., Marsh, S. E., Silhavy, J. L., Barkovich, A. J., Ross, M. E., Saleem, S. N., Dobyns, W. B., and Gleeson, J. G. (2008). The molar tooth sign: a new Joubert syndrome and related cerebellar disorders classification system tested in Egyptian families. Neurology 70, 556–565.
Zaki, M. S., Sattar, S., Massoudi, R. A., and Gleeson, J. G. (2011). Co-occurrence of distinct ciliopathy diseases in single families suggests genetic modifiers. Am. J. Med. Genet. A. 155A, 3042–3049.
Keywords: midbrain, hindbrain, cerebellum, malformations
Citation: Barkovich AJ (2012) Developmental disorders of the midbrain and hindbrain. Front. Neuroanat. 6:7. doi: 10.3389/fnana.2012.00007
Received: 14 December 2011; Paper pending published: 07 January 2012;
Accepted: 20 February 2012; Published online: 06 March 2012.
Edited by:
Salvador Martinez, University Miguel Hernandez, SpainReviewed by:
Nobuaki Tamamaki, Kumamoto University, JapanKazunori Nakajima, Keio University School of Medicine, Japan
Copyright: © 2012 Barkovich. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: A. James Barkovich, Department of Radiology and Biomolecular Imaging, Neuroradiology Section, Rm L371, University of California at San Francisco, 505 Parnassus Avenue, San Francisco, CA 94143-0628, USA. e-mail: james.barkovich@ucsf.edu