AA/12-lipoxygenase signaling contributes to inhibitory learning in Hermissenda Type B photoreceptors
- 1 Program in Neuroscience, Indiana University, Bloomington, IN, USA
- 2 Department of Psychological and Brain Sciences, Indiana University, Bloomington, IN, USA
Conditioned inhibition (CI) is a major category of associative learning that occurs when an organism learns that one stimulus predicts the absence of another. In addition to being important in its own right, CI is interesting because its occurrence implies that the organism has formed an association between stimuli that are non-coincident. In contrast to other categories of associative learning that are dependent upon temporal contiguity (pairings) of stimuli, the neurobiology of CI is virtually unexplored. We have previously described a simple form of CI learning in Hermissenda, whereby animals’ phototactic behavior is increased by repeated exposures to explicitly unpaired (EU) presentations of light and rotation. EU conditioning also produces characteristic reductions in the excitability and light response, and increases several somatic K+ currents in Type B photoreceptors. Type B photoreceptors are a major site of plasticity for classical conditioning in Hermissenda. Because arachidonic acid (AA) and/or its metabolites open diverse K+ channels in many cell types, we examined the potential contribution of AA to CI. Our results indicate that AA contributes to one of the major effects of EU-conditioning on Type B photoreceptors: decreases in light-evoked spike activity. We find that AA increases the transient (IA) somatic K+ current in Type B photoreceptors, further mimicking CI training. In addition, our results indicate that metabolism of AA by a 12-lipoxygenase enzyme is critical for these effects of AA, and further that 12-lipoxygenase metabolites are apparently generated during CI training.
Introduction
An important, but poorly understood, issue in the cellular analysis of learning and memory is how the stimulus relationship of “non-coincidence” is encoded and represented within the brain. Organisms not only learn that stimuli co-occur, as in standard forms of excitatory Pavlovian/classical conditioning (Domjan, 2010). But they also readily learn that certain stimuli do not co-occur, as in Pavlovian/classical conditioned inhibition learning. If the unconditioned stimulus (US) is an aversive one, such as electric shock, then the result of pairings (called excitatory conditioning) of a conditioned stimulus (CS) and US may be the conditioning of fear to that cue (Estes and Skinner, 1941). Alternatively, if a different CS reliably predicts the absence of electric shock, the learning that results may be “safety signal” learning, or the conditioning of “relief”, a central emotional state that is functionally opposite to that of fear. Indeed, a common characterization of inhibitory Pavlovian/classical conditioning regards the organism as learning that one stimulus (the CS) predicts the absence of the other (the US) (Rescorla, 1969; Domjan, 2010), just as the organism learns that a CS predicts the presence of a US during excitatory conditioning.
A common theme in cellular and molecular accounts of both classical and instrumental conditioning processes, where learning is critically dependent upon temporal contiguity of CS and US or response and consequence (viz., excitatory conditioning), is the involvement of molecular coincidence detection mechanisms (Bourne and Nicoll, 1995; Konnerth et al., 1996; Tsien, 2000). Some prominent examples include: (1) the NMDA subtypes of glutamatergic receptors that require both membrane depolarization and glutamate for activation of the receptor-coupled ion channel, (2) calcium-sensitive forms of adenylate cyclase that require coincident stimulation by G-protein alpha-S subunit (GαS) and Ca2+/calmodulin (Anholt, 1994) for full activity, and (3) certain protein kinase C (PKC) isoforms that require coincident Ca2+ and diacylglycerol (DAG) binding before PKC can phosphorylate its target substrates. The preceding list is only a partial one; numerous other molecules have also been implicated as coincidence detectors.
Studies of excitatory classical conditioning of the nudibranch mollusk Hermissenda crassicornis have provided extensive information about the behavioral, circuit-level, cellular, and molecular changes that occur during associative learning (Crow, 2004; Blackwell and Farley, 2008). Several molecular coincidence detectors (e.g., PKC) have been implicated in these changes (Farley and Auerbach, 1986; Farley and Schuman, 1991). Hermissenda have also been shown to exhibit inhibitory classical conditioning (Britton and Farley, 1999). Thus, Hermissenda provides the opportunity for detailed studies of inhibitory conditioning and mechanisms of non-coincidence detection, as well as explicit comparisons with mechanisms involved in excitatory conditioning.
Previous research on excitatory conditioning in Hermissenda has shown that these animals readily learn that light predicts the presence of turbulence (vestibular stimulation via high-speed circular or orbital rotation) when light, the CS, is repeatedly paired with high-speed rotation, the US (Crow and Alkon, 1978; Farley and Alkon, 1980). The behavioral consequences of this learning are decreases in several components of phototactic behavior (Farley and Alkon, 1982; Crow and Offenbach, 1983), as well as the elicitation of a “clinging” response to light, which is evoked only by rotation/turbulence prior to pairings (Lederhendler et al., 1986). Primary neural substrates of this form of excitatory conditioning involve persistent changes in the neuronal excitability and photoresponses of the ocular Type A and B photoreceptors (Crow and Alkon, 1980; Alkon et al., 1982, 1985; Farley, 1987a,b, 1988; Farley and Alkon, 1982), with the Type B photoreceptors receiving the most attention to date. Excitatory conditioning (light-rotation pairings) produces a number of changes in the neurophysiological properties of Type B photoreceptors, including increases in depolarizing light-induced generator potentials, spike frequencies, and resting input resistances (Crow and Alkon, 1980; Farley and Alkon, 1982; Farley, 1987a, 1988).
Just as pairings of light and rotation result in persistent increases in the excitability and light-responses of Type B photoreceptors, owing in part to decreases in somatic K+ currents (Alkon et al., 1982; Farley, 1988), explicitly-unpaired presentations (EU) of the same two stimuli (an inhibitory conditioning paradigm) result in persistent decreases in excitability and light-responses (light-induced generator potentials and spike frequencies) of Type B photoreceptors (Britton and Farley, 1999).
The question of how inhibitory conditioning, or more generally “non-coincidence” learning, is represented in the brain can be subdivided into: (1) what mechanisms account for expression of altered excitability and synaptic transmission due to inhibitory conditioning, (2) what mechanisms and signal transduction cascades are involved in the induction of these changes?
The present report is concerned with both issues. Several lines of research have focused our attention on arachidonic acid (AA) signaling pathways as critical mediators of expression of some of the alterations in the neurophysiological responses of Type B photoreceptors that are produced by EU light/rotation presentations in Hermissenda. First, in both invertebrate and vertebrate neurons, AA signaling has often been linked to increases in voltage- and/or calcium-dependent K+ currents. In Aplysia mechanosensory neurons, AA and 12(S)-HPETE metabolites activate the FRMF-amide stimulated S-K+ channel, and consequently reduce synaptic transmission (Piomelli et al., 1987b; Buttner et al., 1989). AA has also been found to increase the activity of large-conductance, Ca2+-dependent K+ channels in cardiac (Kim and Clapham, 1989; Ordway et al., 1991; Wallert et al., 1991), gastric (Ordway et al., 1989b), and pulmonary (Ordway et al., 1989a) smooth muscle cells. In mammalian neurons, AA enhances M-current (Kv7.2/3) K+ channels in hippocampal pyramidal (Schweitzer et al., 1990, 1993), sympathetic ganglion (Villarroel, 1994), as well as embryonic mesencephalic and hypothalamic neurons (Kim et al., 1995). Second, because AA can be metabolized into multiple bioactive metabolites, AA-signaling has the potential to produce diverse, combinatorial signaling effects. In Aplysia L14 neurons, for example, the 12(S)-HPETE metabolite of AA can produce both a rapid depolarization as well as a slower hyperpolarization (Piomelli et al., 1987a, 1989), mimicking the effects of histamine. These opposing effects on excitability appear to be mediated by distinct ion channels. In Hermissenda, AA has been reported to act synergistically with diacylglycerol (DAG) and Ca2+ to activate PKC (Lester et al., 1991), suggesting its involvement in excitatory conditioning (Talk et al., 1997; Muzzio et al., 2001). While the exact mechanism for the effect of AA on PKC is uncertain, it has been assumed that AA binds PKC directly (Lester et al., 1991).
Given the potential pleiotropic and opposing signaling effects of AA, its activation of diverse K+ channels in many cell types, and EU-produced increases in voltage- and Ca2+-dependent K+ channels in Hermissenda (Farley et al., 1999), AA signaling is an attractive candidate to mediate some of the inhibitory conditioning induced changes in Hermissenda Type B photoreceptors. We have examined that possibility here by assessing the effects of AA and various metabolites on photoresponses, spiking, and other measures of membrane excitability of Type B photoreceptors, to see if any of these are altered similar to the effects of behavioral EU conditioning. We first examined the effects of AA on Type B photoreceptors from untrained animals. We observed that AA decreased light-evoked spiking in these cells, similar to that produced by EU-conditioning. These effects of AA were attributable to lipoxygenase-metabolites; 12(S)-HPETE appears to be a critical one. We further determined that AA-signaling increased the amplitude of the transient, voltage-dependent A-current in Type B photoreceptors, similar to the effects of EU-conditioning (Farley et al., 1999). The A-current is a potent regulator of spiking in B photoreceptors, and its increased activation would be expected to decrease spike frequency. Subsequently, we also assessed the effects of AA on the light-evoked spiking of B photoreceptors from EU-conditioned animals. When AA was tested shortly after the conclusion of behavioral EU-training, we observed an apparent occlusion effect: AA failed to decrease spiking similar to its effect on B cells from untrained animals. When tested ∼24 h following conclusion of EU-training, we observed no occlusion effect. But pharmacological inhibition of the 12-lipoxygenase signaling pathway was found to reverse the EU-conditioning produced decreases in light-evoked spiking. These observations suggest that AA-signaling is one critical mediator of EU-conditioning effects in Hermissenda.
Materials and Methods
Animals
Adult Hermissenda were purchased from Sea Life Supply (Sand City, CA, USA). All Hermissenda were individually housed in perforated 50 ml sample tubes and kept in 50 gallon refrigerated (14°C) aquaria containing artificial seawater (ASW; Aqua Craft Bio-Sea Marinemix). All Hermissenda were subjected to a 12-h light/dark cycle and fed scallop pieces twice per week.
Behavioral Training
The procedure and equipment used for behavioral training have been previously described (Farley, 1987a; Britton and Farley, 1999). Animals were selected at random and moved from their numbered housing tubes to numbered training tubes containing ASW (14°C). The training tubes were then attached to a turntable that was mounted in a refrigerator (∼14°C). Vestibular stimulation was provided by high-speed rotation (100 RPM, ∼2.2 × g) on the turntable and ocular stimulation was provided by a light (∼300 μW/cm2) mounted above the turntable. Presentation of the stimuli was computer controlled. A single trial consisted of explicitly unpaired (EU) presentations of light and rotation: 30 s of light and 30 s of rotation separated by 4 min (a 9 min inter-trial interval; ITI). Animals received 3–6 days of training consisting of 30 trials per day. Animals were always trained during the middle 8 h of light in their 12-h light/dark cycle. At the end of each day’s training, the animals were returned to their home tubes and aquaria.
Electrophysiology
In preparation for electrophysiology, the circumesophageal nervous system was removed from an animal and pinned inside a 500-μl well comprised of petroleum jelly strips on a microscope slide. Electrode impalement was facilitated by incubating the nervous system in a non-specific protease solution (1 mg/ml, Sigma #P5380) for ∼9 min at room temperature. To remove the protease, the nervous system was thoroughly washed with cold (∼4°C) ASW. The well containing the nervous system was then filled with 500 μl of room temperature ASW (430 mM Na+, 10 mM K+, 10 mM Ca2+, 50 mM Mg2+, 10 mM Tris HCl, and 570 mM Cl−, pH = 7.6–7.8). For voltage-clamp experiments, all photoreceptor synaptic interactions and spike generation were eliminated via axotomy (Alkon and Fuortes, 1972).
The procedure used for sharp-electrode intracellular recording has been described (Farley, 1987a,b). Glass microelectrodes (A-M Systems #602000) were pulled to a tip resistance of ∼ 40 MΩ when filled with 1.5 M potassium acetate. After impaling a Type B photoreceptor and checking the initial light response to a brief flash of light, the cell was dark-adapted for 10 min. During the final 1–2 min of dark-adaptation, the resting input resistance (Rin) of the B photoreceptor was measured by using 500 ms current step injections (−0.5 nA to +0.1 nA in 0.1 nA increments). At the end of the 10-min dark-adaptation period, light stimulation began: 30 s presentations of light (∼300 μW/cm2) were delivered at 2 min inter-stimulus intervals (ISIs) until a total of 15 consecutive light steps had been presented. When drugs were applied acutely, they were added during the 4th light step. As described below, when inhibitors of AA signaling pathways were used, they were added 20–30 min prior to the start of electrophysiological recording. With the present light stimulation protocol, Type B photoreceptors reached a steady-state level of light-adaptation after 3–4 light steps, exhibiting constant peak- and steady-state generator potentials (SSGP), as well as spike frequencies (changes <5% over the course of the next 11–12 light steps). Following the 15 light steps, the cell was then dark-adapted again for 10 min, and Rin was re-measured. Because the final Rin check occurred at least 40 min after impalement, the recording electrode would occasionally block, leading to erroneous Rin measurements. Rin data from these cells were discarded. As mentioned above, drugs (except inhibitors) were applied during the fourth presentation of light and their effects assessed over the remaining light presentations. Because many inhibitors require time to diffuse and penetrate cell membranes within the ganglia and their concentration to reach equilibrium, nervous systems were incubated in the inhibitors for 20–30 min prior to recording.
The procedure used for two-electrode voltage-clamp has been described (Jin et al., 2009). The glass microelectrodes (A-M Systems #602000) were pulled to ∼15 MΩ when filled with 3 M KCl. After impaling a Type B photoreceptor with two electrodes, the nervous system was dark-adapted. The voltage-clamp protocol was a −60 mV holding potential with 400 ms voltage command steps from −60 mV to +5 mV in +5 mV steps. The command steps were separated by 2 s and a full voltage-clamp trial was administered every 5 min. Drugs were applied prior to the third voltage-clamp trial and their effect assessed over the remaining voltage trials.
Chemicals
All drugs, salts, solvents, etc. were reagent grade and all working solutions were prepared from stocks daily and stored at 4°C until used. Arachidonic acid (Sigma #A8798), 12(S)-HPETE (Cayman Chemical #44570), 5(S)-HPETE (Cayman Chemical #44230), and MK-886 (Sigma #M2692) were stored at 0.1 M in 100% ethanol at −70°C. Baicalein (Sigma #465119) was stored in powder form at 4°C. NDGA (nordihydroguaiaretic acid; Cayman Chemical #70300) was stored in powder form at -20°C. Daily working solutions of these drugs were prepared by diluting the required amount of stock to 10−2 M with dH2O. The final concentration of ethanol in all experiments never exceeded 0.1%. All other chemicals and solvents were stored and prepared according to the supplier’s recommendation.
Data Analysis
In this paper, we report data for several key measures of Type B photoreceptor light responses and excitability, including: light-evoked spike frequency, peak light response, SSGP, resting membrane potential (Vm), and Rin. The left panel of Figure 1E shows a typical response to a 30-s light step. Hermissenda Type B photoreceptors respond to light with a depolarizing generator potential with superimposed spike activity. Within ∼1 s following light onset, the generator potential reaches a maximal value. The difference between the resting membrane potential and this maximal value is defined as the peak light response. After a few seconds, the generator potential stabilizes (the SSGP) and does not change significantly over the remaining 20+ seconds of the light step. The increased spiking elicited by light is called the light-evoked spike frequency. Due to the rapidly varying changes in generator potential during the first few seconds, we only collect spike frequency data during the last 10 s (the stable portion) of the light response. Rin is described in Section “Electrophysiology.”
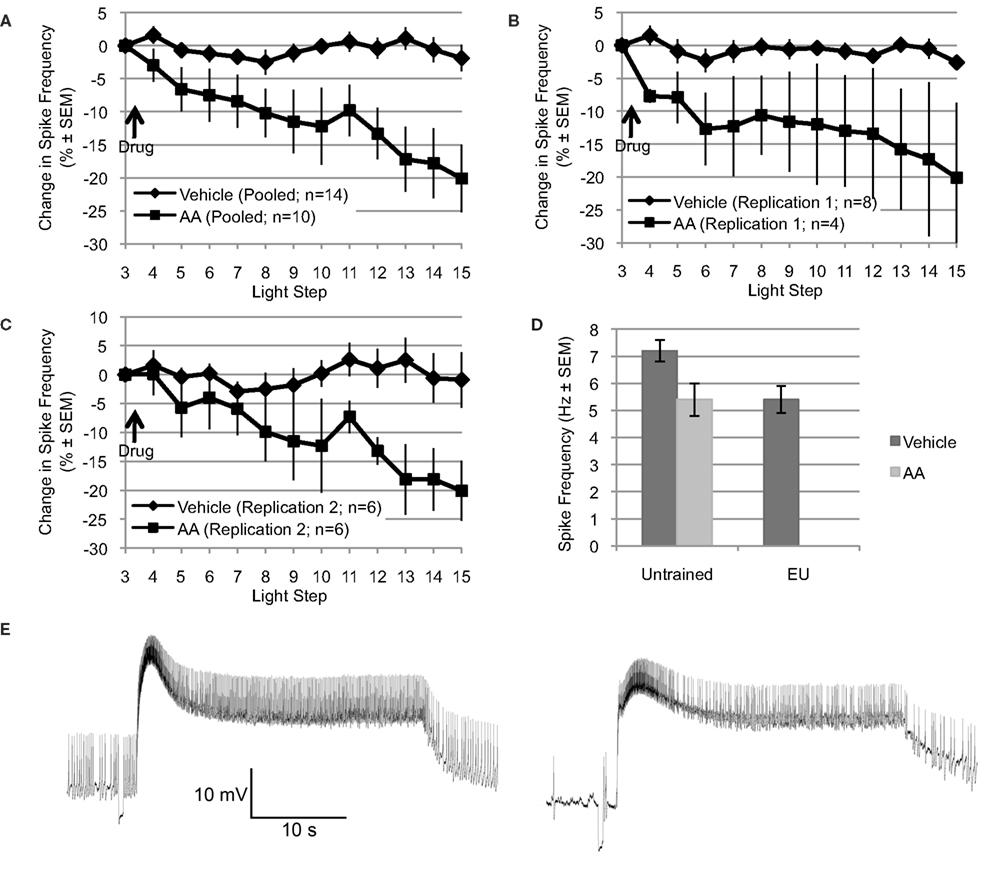
Figure 1. Arachidonic acid mimics the reduction in spike frequency observed following EU training. (A through C) Applied after the 3rd light step, AA (10−4 M) reduced light-evoked spiking over the remaining 12 light steps (∼22–24 min). In contrast, the spike frequency of Type B photoreceptors exposed to vehicle (dH2O + 0.1% ethanol) was essentially unchanged over the same period. Changes in spike frequency are expressed as percent change relative to that observed during the third light step, just prior to AA or vehicle addition. (A) Shows pooled data from two independent replications (shown in B and C). (D) The reduction in spike frequency produced by AA mimicked the reduction produced by EU training. During the 15th light step, Type B photoreceptors from untrained Hermissenda spiked at 7.2 Hz; however, when treated with AA, Type B photoreceptors from untrained Hermissenda spiked at 5.4 Hz. In comparison, after EU training, Type B photoreceptors spiked at 5.4 Hz. In A through D, as well as in all subsequent figures for which summary data are presented, each data point represents a mean ± SEM. (E) Representative light response from a Type B photoreceptor treated with AA: 3rd light step (left) and 15th light step (right). Note the reduction in spike frequency, which was 41% for this cell (during the last 10 s of the light response).
Unless stated, summary statistics are arithmetic mean ± SEM. The statistical significance of all comparisons reported in this paper was assessed using one-way or repeated measures analysis of variance (ANOVA) in conjunction with Tukey’s HSD test. For all comparisons, two-tailed tests were used and significance was only assumed when p < 0.05.
Results
AA Reduces Type B Photoreceptor Spike Frequency
Explicitly unpaired training decreases several key measures of Type B photoreceptor light responses and excitability, including: light-evoked spike frequency, peak light response, and SSGP (Britton and Farley, 1999). If AA signaling mediates any of these EU training related changes, then application of AA to nervous systems from untrained Hermissenda should mimic some or all of these effects of EU training. To test this, we applied AA (10−4 M) to nervous systems from untrained Hermissenda and found that AA reduced light-evoked spike frequency in Type B photoreceptors an average of 20.1 ± 5.1% (n = 10), while vehicle (dH2O in 0.1% ethanol) alone slightly decreased spiking by 1.9 ± 2.0% (n = 14). The difference in response of B cells to AA vs. vehicle was significant [F(1,22) = 11.09, p < 0.01] (Figure 1A).
The preceding data represent the pooled results from two separate and independent replications using different shipments of animals, different lots of AA, etc, separated by a period of 9–12 months. The principal results for the two separate replications were extremely similar (Figures 1B,C). For the first replication, AA reduced light-evoked spike frequency by 20.1 ± 11.4% (n = 4), while vehicle slightly decreased spiking by 2.6 ± 1.1 (n = 8) [F(1,10) = 5.47, p < 0.05]. For the second replication, conducted 9–12 months later (when the results of Figures 3–6 were collected), AA decreased light-evoked spiking 20.1 ± 4.8% (n = 6), while vehicle slightly decreased spiking by 0.9 ± 4.8% (n = 6) [F(1,10) = 5.01, p < 0.05]. Because there was no significant difference between the two replications, for either AA or controls, we combined the results of the two replications for added statistical power (Figure 1A). This pooled vehicle and AA data is used in all remaining comparisons and figures. We also note that the results of these two replications were very similar to our preliminary AA results collected years earlier by different experimenters using a different solvent (DMSO), chemicals/supplies, animals, data collection protocol, and equipment etc (Farley et al., 2003). In that study, AA-exposed (10−4 M) B cells (n = 7) showed a decrease of 17% after ∼8–10 min. The DMSO (0.1%) solvent-alone control cells (n = 4) showed no change in spike frequency. As in the present replications, neither AA nor solvent control had any effect on SSGP, Vm, or Rin. Thus, the effects of AA appear to be quite reliable. Figure 1E shows a representative trace of the effect of AA on a single Type B photoreceptor: before AA (3rd light step; left panel) and after AA (15th light step; right panel).
Because the reduction in spike frequency produced by AA qualitatively mimicked the reduced spike frequencies produced by EU training in previous studies (Britton and Farley, 1999; Farley et al., 1999), we also directly compared the effects of AA on cells from untrained animals with the effects of behavioral EU conditioning in the present study (Figure 1D). As described in Section “Behavioral training” and in additional detail below, a group of animals was exposed to multi-day EU training. AA stimulation of untrained cells and EU conditioning produced quantitatively similar reductions in spike frequency (Figure 1D). During the 15th light step, Type B photoreceptors from untrained Hermissenda treated with vehicle spiked at 7.2 ± 0.4 Hz (n = 14). When treated with AA, Type B photoreceptors from untrained Hermissenda spiked at 5.4 ± 0.6 Hz (n = 10) during the 15th light step. In comparison, 24-h after EU training (described in Section “Behavioral training”), Type B photoreceptors treated with vehicle spiked at 5.4 ± 0.5 Hz (n = 9) during the 15th light step. In Figure 1D, the mean spike frequencies for the vehicle- and AA-untrained conditions were taken from the pooled AA and vehicle data discussed in the previous paragraph. The mean spike frequency for the vehicle-EU condition was taken from the data presented in Figure 6 and discussed in Section “Involvement of AA in inhibitory conditioning.” As discussed in the previous paragraph, collection of the vehicle-EU data and the second replication of the vehicle- and AA-untrained data overlapped. For added statistical power, we compared the vehicle-EU data with the pooled vehicle- and AA-untrained data. There was a significant difference between these three conditions [F(2,32) = 4.47, p < 0.05]. The difference between the spike frequency of Type B photoreceptors from untrained Hermissenda that did not receive AA and the spike frequencies of the other two conditions was statistically different (p < 0.05 respectively; Tukey’s HSD). But the difference between cells from untrained Hermissenda that received AA, and cells from Hermissenda that received EU training only (no AA) was not different (p = 0.88; Tukey’s HSD). In summary, application of AA to Type B photoreceptors from untrained Hermissenda mimicked the effect of EU training on light-evoked spiking. While AA produced a moderate but clear and highly reliable decrease in spike frequency, AA had no significant effect on SSGP [an increase of 0.8 ± 0.7 mV; F(1,19) = 0.21, p = 0.65], or Vm [a depolarization of 1.1 ± 1.9 mV; F(1,19) = 0.05, p = 0.83].
We also examined the effects of AA on the characteristics of spontaneously occurring spikes. In this analysis, only a subset of cells (7 of 10) exposed to AA was used because the others failed to show spontaneous spikes before and/or after AA. The AA-exposed Type B cells included in this analysis showed no significant differences in spontaneous spike frequency (in the dark) during the time periods just prior to the 4th and 15th LS, when compared to vehicle-exposed cells. In addition, the reduction in spike frequency from the 4th to the 15th LS was comparable for AA- vs. vehicle-exposed cells: a 39.4 ± 23.2% reduction for AA (n = 7) compared to a 18.6 ± 22.5% reduction for vehicle (n = 5); p = 0.55, t-test. These same Type B cells showed no appreciable changes in spike amplitude (an increase of 0.6 ± 0.3 mV; p = 0.32), nor changes in the amplitude of the spike after-hyperpolarization (AHP) (in increase of 0.5 ± 0.7 mV; p = 0.52, t-test), for spikes measured during the time periods just prior to the 4th and 15th LS. However, there was a trend for spike duration to be shorter following AA addition, with five of seven cells showing a reduction of 20% or more. For all seven cells, spike duration was 9.63 ± 0.6 ms prior to the 4th LS; after AA (just before the 15th LS) spike duration was 8.28 ± 0.8. Although suggestive, and consistent with AA having enhanced a component of voltage-dependent K+ current (see below) involved in spike re-polarization, this difference was non-significant (p = 0.22, t-test).
Effects of Lipoxygenase and Cyclooxygenase Inhibitors
Arachidonic acid may affect ion channels or enzymes via direct binding. AA may also be metabolized by lipoxygenase or cyclooxygenase enzymes to produce active signaling molecules, like hydroperoxy-eicosatetraenoic acids (HPETEs) or leukotrienes. Many of these metabolites modulate various ion channels or bind to diverse intra- or extracellular receptors. To determine if metabolites of AA contribute to the effect of AA on Type B photoreceptor spike frequency, we applied AA in the presence of NDGA, a non-specific lipoxygenase inhibitor, and indomethacin, a non-specific cyclooxygenase inhibitor [significant main effect: F(2,18) = 4.015, p < 0.05] (Figure 2A). NDGA (10−4 M; n = 5) blocked the effect of AA (p < 0.05, Tukey’s HSD) without causing any conspicuous effect of its own on light-evoked spike frequency [7.3 ± 0.2 Hz vs. 7.3 ± 0.3 Hz for vehicle only; F(1,15) = 0.19, p = 0.67], SSGP [16.3 ± 0.9 mV vs. 20.7 ± 2.2 mV for vehicle only; F(1,13) = 1.39, p = 0.26], or Vm [−48.8 ± 1.7 mV vs. −49.3 ± 3.7 mV for vehicle only; F(1,13) = 0.01, p = 0.93]. This suggests that one or more lipoxygenase metabolites rather than AA itself mediated the reduction in Type B photoreceptor spike frequency. In contrast to NDGA, indomethacin (10−4 M; n = 4) had pronounced effects on the Type B photoreceptor light-response. Although the AA-produced reduction in light-evoked spike frequency was attenuated somewhat in the presence of indomethacin (a decrease of 10.7 ± 2.6% compared to the 20.1% decrease produced by AA alone [p = 0.194; Tukey’s HSD]), prior to AA application, Type B photoreceptors exposed to indomethacin exhibited SSGPs that were considerably larger (28 ± 2.9 mV vs. 18.2 ± 1.8 mV for vehicle only; F(1,14) = 10.64, p < 0.01) than those of Type B photoreceptors not exposed to indomethacin (Figure 2B). While indomethacin appeared to attenuate the reduction in spike frequency produced by AA, that attenuation may have been due to the increased light-response caused by indomethacin. Based on this data, we cannot exclude a contribution from cyclooxygenase metabolites, but the NDGA results and the results of the next section suggests that a lipoxygenase metabolite contributes the majority of AA’s effect on light-evoked spike frequency in Type B photoreceptors.
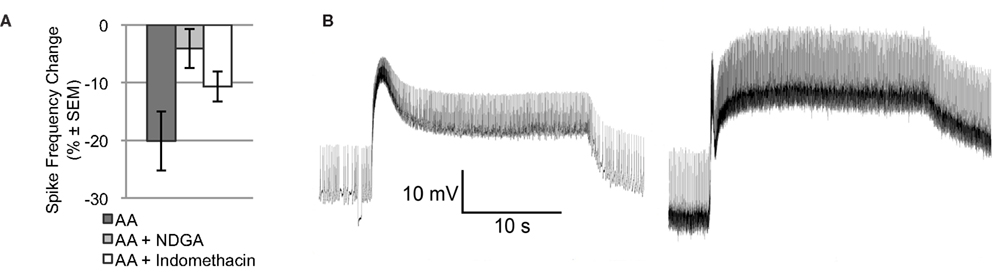
Figure 2. NDGA blocks the reduction in light-evoked spike frequency produced by AA. (A) The light-evoked spike frequency reduction produced by AA appears to be blocked by NDGA and indomethacin. (B) Representative light responses from a control Type B photoreceptor exposed to vehicle (left) and another cell exposed to indomethacin (right), both in the absence of AA. Note the enhanced light-response of the indomethacin treated cell. This enhanced light-response may account for the apparent attenuation of AA’s effect on light-evoked spike frequency.
Effects of AA are Mediated by a 12-Lipoxygenase Metabolite
Because the NDGA results (Figure 2A) suggest that AA’s effects are mediated by a lipoxygenase metabolite, we turned our attention to the lipoxygenase family of enzymes. Because presynaptic inhibition of synaptic transmission and opening of a K+ channel in Aplysia appear to be mediated by a metabolite of a 12-lipoxygenase enzyme (Piomelli et al., 1987b; Buttner et al., 1989), we next tested the ability of baicalein, a selective 12-lipoxygenase inhibitor, to block the effects of AA. We also tested the ability of 12(S)-HPETE, the metabolite produced by 12-lipoxygenase activity on AA, to mimic the effects of AA (Figure 3A). Clear differences between the treatment conditions in Figure 3A were apparent, consistent with AA’s effects being mediated by a 12-lipoxygenase metabolite: significant main effect of treatment F(3,32) = 4.85, p < 0.01. When Type B photoreceptors from untrained Hermissenda were incubated in baicalein (10−4 M; n = 7), baicalein blocked the effect of AA on Type B photoreceptor spiking (p < 0.05, Tukey’s HSD). When 12(S)-HPETE (10−4 M) was applied to nervous systems from untrained Hermissenda, 12(S)-HPETE reduced light-evoked spike frequency 24.4 ± 9.2% (n = 5; p < 0.05, Tukey’s HSD), mimicking the effect of AA on spiking (a reduction of 20.1%). Note: the comparisons in Figures 3A–C make use of pooled data for the vehicle and AA conditions. As discussed in Section “AA Reduces Type B Photoreceptor Spike Frequency,” we collected vehicle and AA data twice during this study. Data for the first replication were collected early in the study while data for the second replication were collected in conjunction with the other data shown in Figures 3A–C. As discussed in Section “AA Reduces Type B Photoreceptor Spike Frequency,” there was no difference between the two vehicle replications and there was no difference between the two AA replications. As a result, we pooled the data for the vehicle and AA conditions to increase the statistical power of all comparisons.
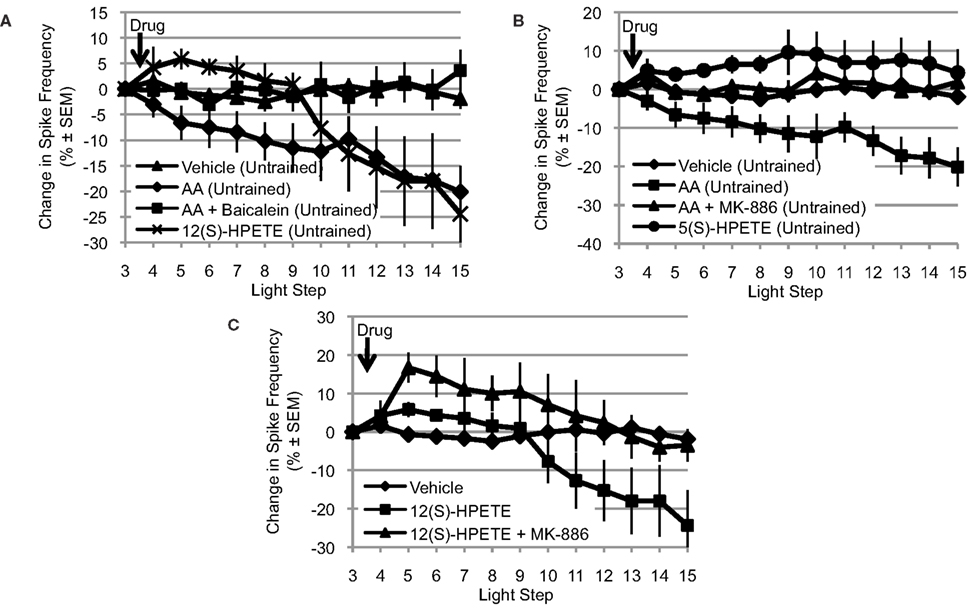
Figure 3. The effects of AA are mediated by a 12-lipoxygenase metabolite. (A) Baicalein (10−4 M, n = 7), a selective 12-lipoxygenase inhibitor, blocked the reduction in light-evoked spike frequency produced by AA. 12(S)-HPETE (10−4 M, n = 5) mimicked the effects of AA on light-evoked spike frequency. (B) MK-886 (10−4 M, n = 7), a reportedly selective inhibitor of 5-lipoxygenase activity, also blocked the effect of AA. However, 5(S)-HPETE (10−4 M, n = 6) failed to mimic the effect of AA. (C) MK-886 also blocked the effect of 12(S)-HPETE, suggesting that its actions are not restricted to inhibition of 5-lipoxygenase activity. (A–C) Use the pooled data depicted in Figure 1A; see Section “AA Reduces Type B Photoreceptor Spike Frequency” for a detailed description.
Interestingly, while the effects of AA and 12(S)-HPETE on spiking were nearly identical at the 15th light step, their time-courses were different. AA produced a slow, continuous decrease in spiking. In contrast, the effect of 12(S)-HPETE on spiking was delayed by many minutes. It is tempting to speculate that the different time-courses may reflect differential penetration of AA vs. 12(S)-HPETE into B cells. The pKa value of AA is ∼7, while the PKa value of 12(S)-HPETE is greater than 7 due to the peroxide group. Thus, the effect of 12(S)-HPETE may take longer to develop because 12(S)-HPETE would be expected to take more time to cross the Type B photoreceptor membrane (i.e., become protonated). Without additional research, we cannot say if the delayed response of 12(S)-HPETE represents a physiologically significant difference or is a consequence of pharmacokinetics.
Many enzymes may metabolize AA and more than one of these enzymes may be expressed in a given cell. Furthermore, if multiple enzymes are expressed, their effects could be cooperative, synergistic, or antagonistic. Because 5-lipoxygenase enzymes also metabolize AA and are another significant member of the lipoxygenase family of enzymes, we examined the potential contribution of this enzyme to AA’s effect on Type B photoreceptor spike frequency. Thus, we tested the ability of MK-886 (10−4 M), a reportedly selective inhibitor of 5-lipoxygenase activity, to block the effects of AA. We also tested the ability of 5(S)-HPETE (10−4 M), the metabolite produced by 5-lipoxygenase activity, to mimic the effect of AA (Figure 3B). Clear differences between the treatment conditions in Figure 3B were apparent: significant main effect of treatment, F(3,33) = 8.09, p < 0.01. When nervous systems from untrained Hermissenda were incubated in MK-886, AA failed to reduce the light-evoked spike frequency in Type B photoreceptors (a small increase of 1.9 ± 2.9% was observed, n = 7). The response to AA of MK-886 treated cells was significantly different from that of cells exposed to AA alone (p < 0.05, Tukey’s HSD), but not different from that of cells exposed to vehicle alone (p = 0.98, Tukey’s HSD). In contrast, 5(S)-HPETE failed to reduce light-evoked spike frequency (an increase of 4.3 ± 6.1%, n = 6) and thus was significantly different from AA alone (p < 0.01, Tukey’s HSD) but not vehicle (p = 0.26, Tukey’s HSD). There are several possible explanations for this inconsistency between the effects of the 5-lipoxygenase inhibitor (MK-886) and metabolite [5(S)-HPETE]. One possibility is that MK-886 acts directly on ion channels affected by AA or its metabolites. For example, MK-886 reportedly competes with AA or 12(S)-HPETE for a K+ channel binding site (Smirnov et al., 1998). If this were happening in Hermissenda Type B photoreceptors, it would suggest that MK-886’s block of AA’s effect might be because MK-886 interferes with 12(S)-HPETE’s activation of a K+ channel (e.g., IA channels, see below) rather than because of its inhibition of 5-lipoxygenase activity. To test this, we applied MK-886 prior to 12(S)-HPETE (n = 3) (Figure 3C), and observed significant differences in the response to 12(S)-HPETE depending upon whether cells had been treated with MK-886 or not: significant main effect of treatment: F(2,19) = 5.02, p < 0.05. We found that MK-886 blocked (p < 0.05, Tukey’s HSD) the effect of 12(S)-HPETE on light-evoked spike frequency. Thus, the inconsistency implied by MK-886’s block of AA’s effect and 5(S)-HPETE’s failure to mimic AA’s effect seems to arise from an effect of MK-886 that is independent of its inhibition of 5-lipoxygenase. MK-886 may block the activation of a K+ channel by 12(S)-HPETE, and/or interact with other components of the 12-lipoxygenase signaling pathway.
In summary, the consistent results obtained for baicalein (a 12-lipoxygenase inhibitor) and 12(S)-HPETE imply that a 12-lipoxygenase metabolite is critical for mediating AA’s reduction of Type B photoreceptors light-evoked spike frequency. The inconsistent results obtained for MK-886 (a 5-lipoxygenase inhibitor) and 5(S)-HPETE prevent us from inferring that a 5-lipoxygenase metabolite contributes to the effect of AA. However, we cannot exclude a possible role for 5-lipoxygenase at present.
Effects of AA on K+ Currents
Voltage clamp measurements indicated that AA-exposure (10−4 M) increased both the transient (IA) and sustained components of somatic voltage-dependent K+ current in Type B photoreceptors (n = 4) (Figure 4). On average, IA was increased by ∼18–44%, depending upon the test potential. Tests of IA values over the range of −30 to +5 mV, 12–15 min following AA-exposure, indicated that the currents were significantly increased [main effect of AA-exposure: F(1,2) = 17.93, p = 0.05]. The interaction between AA-exposure and test voltage was also significant, indicating that AA’s facilitation of IA increased with depolarization [F(7,14) = 12.54, p = 0.0005] (Figure 4B).
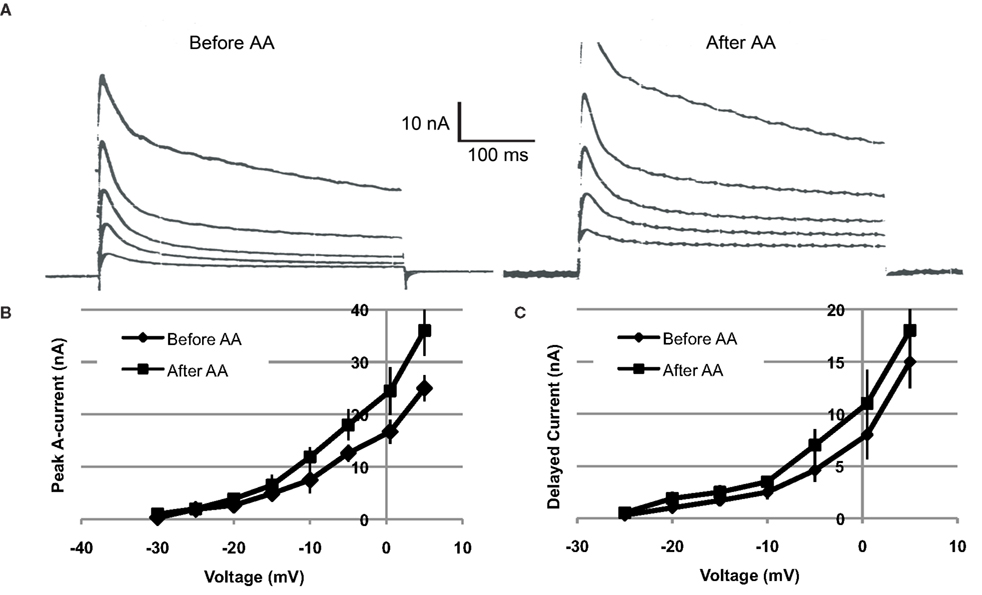
Figure 4. Arachidonic acid (10−4 M) increases K+ currents of Type B photoreceptors. (A) Left traces: Examples of currents elicited by depolarization of a voltage-clamped type B photoreceptor, just prior to AA exposure. Right traces: Currents from same cell recorded at same test potentials, 15 min following exposure to AA. Current traces depicted were elicited by depolarizations from −15 to +5 mV, in 5 mV increments. Both the transient and sustained components of K+ current were increased. (B,C) Summary current–voltage relationships for IA (B) and IDelayed (C), before and after (∼15 min) exposure of cells (n = 4) to AA. AA enhanced both IA and IDelayed, though the effect was greater and more reliable in the case of IA.
Further analysis indicated a trend for AA to have enhanced the sustained component of K+ current (IDelayed), measured at the end of the 400 ms clamp step (Figure 4C). On average, the effect of AA on IDelayed was weaker and less reliable than the facilitation of peak A current. Test of the main effect of AA indicated that it was non-significant [main effect of AA-exposure: F(1,2) = 14.80, p > 0.06]. However, the interaction between AA-exposure and test voltage was significant, indicating that AA facilitation of IDelayed was dependent upon the test potential [F(7,14) = 7.51, p = 0.001] (Figure 4C).
IDelayed is a composite current, consisting of residual IA, non-inactivating IK,v, and IK(Ca) (Alkon et al., 1984; Farley, 1988; Farley and Wu, 1989; Jin et al., 2009). On the basis of our present findings, we cannot be more precise as to which component(s) of IDelayed may have been affected by AA. It is possible that the small increases in IDelayed reflect the effects of AA on residual IA.
Involvement of AA in Inhibitory Conditioning
If AA or one of its metabolites [e.g., 12(S)-HPETE] mediates the reduction in light-evoked spike frequency produced by EU training, then we would expect a change in sensitivity to exogenous AA in Type B photoreceptors from Hermissenda that have been subjected to EU training. For example, EU training might occlude the reduction in spike frequency produced by exogenous AA. Alternatively, EU training may cause up- (or down-)regulation of enzymes involved in AA metabolism and therefore an increased (or decreased) response to exogenous AA, i.e., a further decrease in spike frequency. Because there are other possible EU training-related changes that may affect AA sensitivity, additional more complex changes in response to exogenous AA would not be entirely unexpected. To explore these issues, we examined the effects of AA on Type B photoreceptors at two different retention intervals, following the conclusion of standard EU training: immediately after (EU0), or 24 h after (EU24), the last training session.
For Type B photoreceptors from EU0 Hermissenda, we found that AA (10−4 M, n = 15) produced a small but reliable transient increase in light-evoked spike frequency. However, ∼25–30 min after AA application, the spike frequency of these cells had returned to baseline and there was no difference between them and Type B photoreceptors from EU0 Hermissenda exposed to vehicle (dH2O + 0.1% ethanol, n = 6) [F(1,16) = 0.743, p = 0.401] (Figure 5A). As was the case for Type B photoreceptors from untrained Hermissenda, AA did not produce a significant change in SSGP [an increase of 0.5 ± 1.3 mV; F(1,19) = 0.21, p = 0.65] or Vm [a depolarization of 1.7 ± 3.0 mV; F(1,19) = 0.12, p = 0.74]. During the 3rd light step, the spike frequency of Type B photoreceptors from EU0 Hermissenda was 6.4 ± 2.4 Hz. In contrast, the light-evoked spike frequency for Type B photoreceptors from untrained and EU24 Hermissenda were 7.0 ± 0.4 Hz and 4.9 ± 0.4 Hz, respectively (Figure 5B) [F(2,68) = 8.458, p < 0.01]. Due to the large variability between EU0 experiments, the spike frequency for B cells from EU0 animals was not significantly different from that of untrained Hermissenda (p = 0.22, Tukey’s HSD). That is, unlike EU24 Hermissenda, there was no clear reduction in light-evoked spiking for B cells immediately after training (EU0 Hermissenda).
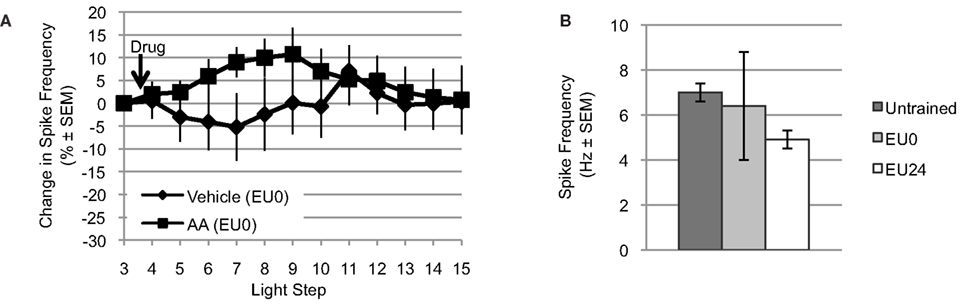
Figure 5. Exogenous AA fails to reduce light-evoked spike frequency in EU0 Hermissenda. (A) When tested immediately after the final training session, AA failed to reduce light-evoked spike frequency in Type B photoreceptors, suggesting an occlusion effect by EU conditioning. (B) The mean light-evoked spike frequency in B cells from EU0 Hermissenda fell between that of untrained and EU24 Hermissenda; however, it was not significantly different from either one.
For Type B photoreceptors from EU24 Hermissenda, we observed that light-evoked spike frequencies during the 3rd light step (prior to AA application) were significantly smaller than that of untrained preparations (4.9 ± 0.4 Hz, n = 9 vs. 7.0 ± 0.4 Hz, n = 15) (p < 0.01, Tukey’s HSD) (Figure 5B), which replicates our previous findings (Britton and Farley, 1999). We also tested the effects of AA and AA+ baicalein on Type B photoreceptors from EU24 Hermissenda (Figure 6A) [significant main effect: F(2,19) = 3.60, p < 0.05]. We found that AA (10−4 M) reduced the light-evoked spike frequency by 21.9 ± 10.5% (n = 9; p < 0.05, Tukey’s HSD) in B cells from EU24 animals, and further that the effect of AA on light-evoked spike frequency was blocked by prior incubation of nervous systems in baicalein (10−4 M; n = 6, p < 0.05, Tukey’s HSD). As was the case in the previous experiments in this report (Figure 1C), AA produced no significant effect on SSGP [an increase of 0.3 ± 1.2 mV; F(1,17) = 0.06, p = 0.82] or Vm [a depolarization of 2.9 ± 2.3 mV; F(1,17) = 0.88, p = 0.36].
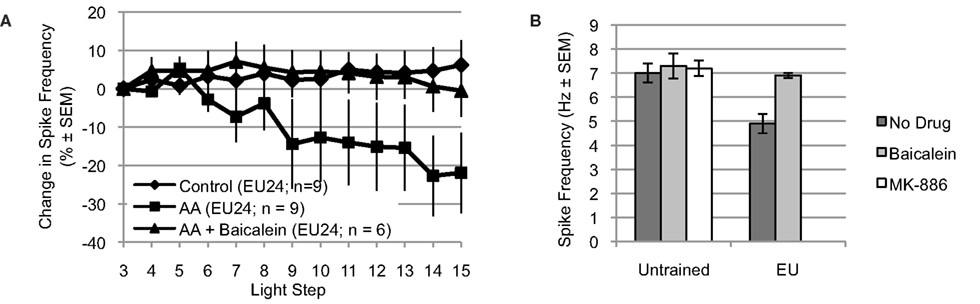
Figure 6. Arachidonic acid reduces spike frequency of B cells from EU trained animals, when tested 24 h post-conditioning (EU24 condition). The EU training related reduction in Type B photoreceptor light-evoked spike frequency is mediated by 12-lipoxygenase signaling. (A) AA reduced light-evoked spike frequency in Type B photoreceptors from EU24 Hermissenda. This reduction was blocked by incubation in baicalein. (B) Left half. For Type B photoreceptors from untrained Hermissenda, MK-886 and baicalein did not affect light-evoked spike frequency during the 3rd light step. Right half. For Type B photoreceptors from EU24 Hermissenda, light-evoked spike frequency during the 3rd light step was significantly reduced, relative to that observed in untrained preparations. Baicalein reversed the EU training-related reduction in light-evoked spike frequency.
Incubation of nervous systems from EU24 Hermissenda in baicalein also exposed a significant EU training-induced change in 12-lipoxygenase signaling (Figure 6B) [significant main effect: F(4,39) = 4.05, p < 0.01]. The reduced baseline (3rd light step) level of light-evoked spiking produced by EU training (4.9 ± 0.4 Hz, n = 9) was reversed by baicalein (6.9 ± 0.1 Hz, n = 6; p < 0.05, Tukey’s HSD), suggesting that a 12-lipoxygenase metabolite was responsible for the reduced spiking of Type B photoreceptors from EU24 Hermissenda. Neither baicalein (7.3 ± 0.5 Hz, n = 7; p = 1.00, Tukey’s HSD) nor MK-886 (7.2 ± 0.3 Hz, n = 7; p = 1.00, Tukey’s HSD) affected the baseline spike frequencies of Type B photoreceptors from untrained Hermissenda (7.0 ± 0.4 Hz, n = 15).
In summary, EU training apparently occluded the effects of AA when the latter was tested shortly after training (EU0 condition), suggesting that AA-metabolites are contributing to the training-produced differences in neurophysiological responses of Type B photoreceptors at this time, or training has inhibited 12-lipoxygenase activity. Twenty-four hours after EU training, however, AA reduced spike frequency by approximately the same amount as it did in untrained animals, and the 12-lipoxygenase inhibitor baicalein blocked this effect. One interpretation of this pattern is that AA signaling is not contributing differentially to Type B photoreceptor light-evoked spike frequencies of untrained vs. EU24 animals, and therefore is not responsible for the expression of maintained excitability reductions and spiking differences produced by EU training. However, the effects of baicalein-alone on light-evoked spike frequencies suggests otherwise. When applied to Type B photoreceptors from untrained animals, baicalein-alone had no effect on light-evoked spiking, suggesting that there is little constitutive 12-lipoxygenase activity in Type B photoreceptors from untrained animals. When applied to Type B photoreceptors from Hermissenda that had undergone EU training and were tested 24 h post-conditioning, however, baicalein-alone reversed the decrease in baseline light-evoked spiking produced by prior EU training. These results suggest that significant constitutive AA-metabolism and 12-lipoxygenase activity is occurring 24 h after EU training, such that the concentrations of 12-lipoxygenase metabolites [e.g., 12(S)-HPETE] are elevated, and contribute to the maintained decreases in light-evoked spiking. Thus, AA-signaling and 12-lipoxygenase metabolites may be contributing to the effects of EU-training on neurophysiological responses of Type B photoreceptors at both short- and long-term retention intervals, but in different ways.
Finally, comparison of the SSGP between the untrained, EU0, and EU24 conditions revealed another significant difference. The SSGP for B cells from untrained animals was 18.8 ± 1.3 mV (n = 10), while the SSGP for B cells from EU0 and EU24 animals were 13.4 ± 0.7 mV (n = 12) and 12.9 ± 0.7 mV (n = 9) respectively [significant main effect: F(2,30) = 11.49, p < 0.01]. The SSGP of B cells from untrained animals was significantly larger than that of B cells from the EU0 and EU24 conditions (p < 0.01 for both comparisons, Tukey’s HSD), but the difference between the EU0 and EU24 conditions was not significant (p = 0.92, Tukey’s HSD). The significantly smaller light response of B cells from EU24 animals (vs. Untrained) replicates our previous results (Britton and Farley, 1999). Interestingly, this same difference was apparent in cells from EU0 animals, despite the fact that these cells failed to show reduced spike frequencies. Interestingly, the large variability seen in light-evoked spiking in the EU0 condition was not observed in SSGPs. This pattern provides additional evidence for dissociation between the expression of EU-training effects on spike frequencies vs. photoresponse amplitude, as a function of retention interval, and may have its basis in the differential involvement of AA and lipoxygenase signaling pathways at these times.
Discussion
AA and a 12-Lipoxygenase Metabolite Reduced Spike Frequency in Type B Photoreceptors, Similar to the Effects of EU Conditioning
We observed that application of AA to Type B photoreceptors of untrained Hermissenda resulted in a ∼20% reduction in spiking during the SSGP photoresponse. This reduction was nearly identical to the magnitude of that produced by behavioral EU training (Britton and Farley, 1999). However, unlike behavioral EU training which also produces a 7–8 mV reduction in the SSGP photoresponse and modest reductions in Rin and Vm of B cells (Britton and Farley, 1999; Farley et al., 1999), AA failed to produce any detectable change in SSGP, or Vm. As discussed in greater detail ahead, this failure of AA to completely mimic the effects of EU conditioning is likely to be due to behavioral training’s stimulation of several signal transduction cascades in B cells, with AA being but one of these.
Arachidonic acid can affect ion channels and receptors through both direct as well as indirect means (Ordway et al., 1991; Meves, 1994). Among the more important indirect mechanisms are: (1) AA’s stimulation of protein kinases and phosphatases (Shearman et al., 1989; Golberg and Zidovetzki, 1998), (2) AA acting as a substrate for enzymatic modification by lipoxygenases, cyclooxygenases, cytochrome P450 enzymes etc (Samuelsson et al., 1987; Marks and Furstenberger, 1999). The latter class of mechanisms has been found to be especially critical for AA’s increase in K+ channel activity, and decreases in the strength of synaptic transmission (Northover, 1977; Piomelli et al., 1987b; Schwartz, 1991; Amin et al., 1999; Tegeder et al., 2001). Thus, we attempted to narrow the class of mechanisms by which AA affected spiking in Type B cells by testing AA’s effects in the presence of lipoxygenase and cyclooxygenase inhibitors.
Three sets of observations indicate that AA’s reduction of spiking in B cells is mediated by a lipoxygenase metabolite, probably 12(S)-HPETE. First, in addition to the block of AA’s effects by the non-specific lipoxygenase inhibitor NDGA, we also found that baicalein (a specific inhibitor of 12-lipoxygenase) blocked the effects of AA on spiking. Second, 12(S)-HPETE, the principal metabolite of 12-lipoxygenase, mimicked the effect of AA on B cell spiking. Third, while MK-886 (the 5-lipoxygenase inhibitor) appeared to block the effects of AA, 5(S)-HPETE failed to mimic AA and MK-886 blocked the effects of 12(S)-HPETE. None of the lipoxygenase inhibitors tested had any effects of their own on SSGP, Vm etc in B cells from untrained animals. Taken together, our results implicate the 12-lipoxygenase pathway as a critical mediator of AA’s effects. This conclusion obviously does not exclude the possible involvement of other lipoxygenases (e.g., the 8- and 15-lipoxygenases) and metabolites [15(S)-HPETE, 8(R)-HPETE, hepoxillins, other leukotrienes etc] that we have not yet examined. Lipoxygenase signaling pathways are notoriously complex, and many of the key enzymes exhibit only partial specificity insofar as their substrates and products are concerned (Marks and Furstenberger, 1999). There is ample opportunity and precedent for cross-talk between the different branches of lipoxygenase signaling, as occurs in platelet aggregation.
Indomethacin also partially attenuated (by ∼52%) the reduction of spiking by AA. This might suggest that some of AA’s effects were mediated by a cyclooxygenase (COX) metabolite. However, as noted earlier, indomethacin had such pronounced effects on SSGP, and other components of the photoresponse and excitability that we do not believe its partial abrogation of AA’s effects on spike frequency can be unambiguously attributed to inhibition of the generation of a COX metabolite. Indomethacin, as well as many other non-steroidal anti-inflammatory drugs (NSAIDs), has a variety of additional effects that are independent of its inhibition of COX activity (Northover, 1977; Singh et al., 1997; Amin et al., 1999; Tegeder et al., 2001). Some NSAIDs at high concentrations have also been reported to inhibit lipoxygenase activity, raising the possibility that indomethacin’s partial reduction of AA’s effect on spiking was due to inhibition of 12-lipoxygenase. While we cannot at present exclude a role for COX metabolites as playing a role in AA’s effects, we do not believe that the indomethacin results provide strong support for that possibility. Additional studies using COX metabolites such as prostaglandins and thromboxanes may help to clarify this issue.
Involvement of AA and Lipoxygenase Metabolites in EU-Conditioning
Two observations implicate AA- and lipoxygenase-signaling in EU conditioning. First, when tested shortly after the conclusion of EU conditioning, AA failed to reduce spiking in B cells from EU0 animals, an apparent occlusion effect. One anomalous result which we do not understand concerns the spiking rates of B cells from EU0 animals, measured prior to AA application, which was not significantly less than that of untrained control cells (∼6–7 Hz) (Figure 6B). If, during the course of EU-conditioning, 12-lipoxygenase activity had metabolized AA such that lipoxygenase metabolites were present and active shortly following training (thereby accounting for the occlusion effect), we would have expected the spike rates of B cells to be closer to that of EU24 animals (i.e., reduced relative to controls). Two possible explanations of this inconsistency are: (1) transient activation of an opposing signaling pathway by EU-conditioning stimulated AA metabolism, or another pathway, that increases excitability and spiking of B cells and masks the decreases produced by 12-lipoxygenase metabolites. One such candidate pathway is PKC. (2) A downregulation/desensitization/inhibition of AA-metabolic pathways because of prolonged stimulation during the lengthy EU-conditioning sessions (∼4.5–5 h), perhaps because of feedback inhibition from lipoxygenase metabolites, depletion of AA, and/or the redox state of 12-lipoxygenase (which strongly influences its activity) that might be influenced by conditioning. The conspicuous larger-than-normal variability in the spike rates for B cells from EU0 animals (Figure 5B) vs. those of either untrained or EU24 animals suggests a possible corresponding increase in the number of distinct signaling pathways that are active shortly after EU conditioning in EU0 animals.
A second result that supports the involvement of 12-lipoxygenase signaling during EU conditioning was the finding that baicalein reversed the EU-produced reductions in spiking of B cells from EU24 animals (measured prior to AA-application). This suggests that activity somewhere along the 12-lipoxgenase signaling pathway may be enhanced 24 h following training, such that constitutively produced 12(S)-HPETE is responsible for ongoing depression of spiking. According to this view, blocking 12-lipoxygenase activity might rapidly reduce the basal concentration of 12-lipoxygenase metabolites in B cells of previously conditioned EU24 animals, to levels not very different from untrained controls. However, if B cells (in the absence of baicalein) from EU24 animals have elevated 12(S)-HPETE levels, it is not obvious why they showed as strong a response to AA as untrained controls. One might instead expect an occlusion effect. However, if upregulation is occurring within the 12-lipoxygenase signaling pathway by EU conditioning, the result may not be entirely surprising. Upregulation of 12-lipoxygenase activity (e.g., increases in either the amount of 12-lipoxygenase expressed, or the Vmax or KM of the enzyme) might occur as a result of EU-conditioning, with the result that AA metabolism to 12-lipoxygenase metabolites is enhanced relative to untrained cells.
With the exception of the preceding results for baicalein on cells from EU24 animals, which are plausibly viewed as a test for the involvement of 12-lipoxygenase metabolites upon maintained expression of the reduction in spiking that is produced by EU conditioning, we have not yet tested for the involvement of AA/lipoxygenase metabolism in the induction of EU-conditioning produced alterations in B cells. Although the strategy of applying an inhibitor (e.g., baicalein) prior to and throughout EU-conditioning to see if it blocks the EU-reduction of spiking is straightforward in principle, technical difficulties have so far prevented us from attempting these experiments. These difficulties center on: (1) confining the actions of lipoxygenase inhibitors to Type B photoreceptors, when the inhibitor is applied to nervous systems of intact or semi-intact animals undergoing behavioral conditioning, or (2) the fact that current versions of in vitro EU-conditioning (using isolated nervous systems) are low-yield experiments, requiring at least 10 unpaired conditioning trials delivered over a lengthy interval (∼100 min) in order to observe EU-specific changes (J. Farley, unpublished observations). Although difficult to test at present, based on our current results, we would expect that specific 12-lipoxygenase inhibitors such as baicalein applied prior to and during EU conditioning might substantially prevent the reduction in spiking and increases in IA that are otherwise observed.
AA’s Effects on Voltage-Dependent K+ Channels in B Cells
In initial voltage-clamp experiments, AA was found to increase the rapid, transient component of voltage-dependent K+ current in B cells, IA. A small, but more variable and non-significant, enhancement of IDelayed was also observed. Comparisons of AA’s effects on K+ currents from untrained animals with the effects of behavioral EU conditioning are revealing. Behavioral EU training significantly increases both IA and IDelayed (Farley et al., 1999) and enhancement of IDelayed is largely due to enhancement of IK(Ca). The enhancement of IA by both AA and EU conditioning is particularly interesting, since this current is active in the subthreshold range of membrane potentials in B cells (Blackwell, 2006; Jin et al., 2009), and is a major determinant of spike frequency in many cells, including B cells.
This may provide a (partial) explanation for why AA, 12(S)-HPETE, and 12-lipoxygenase inhibitors all failed to affect the SSGP of B cells, while behavioral EU conditioning produces a clear and strong reduction. Our previous empirical results (Farley, 1988; Jin et al., 2009) indicate that combined changes in IA and IK(Ca) are required to substantially affect the SSGP in a manner that mimics the effects of paired (excitatory) behavioral training. Large changes in IA alone produce relatively small (∼2 mV) changes in SSGP. By analogy, perhaps EU-training produced increases in both IA and IDelayed (IK–Ca) are required to significantly reduce the SSGP response, while increases in IA alone may be sufficient to significantly reduce spiking. This dissociation between control of spiking and the SSGP is further supported by SSGP data for B cells from untrained, EU0, and EU24 animals. While the light-evoked spike frequency in B cells from EU0 animals was not significantly different from the untrained or EU24 conditions, the SSGP for B cells from EU0 animals was significantly different from that of untrained animals.
Thus, AA metabolism pathways may affect only one of the principal K+ currents affected by EU conditioning. Our present results obviously represent just an initial step in characterizing the effects of AA-signaling upon ionic conductance systems in Type B photoreceptors. Clearly, we also need to examine the effects of 12(S)-HPETE upon the K+ currents, and to also assess the effects of both AA and 12(S)-HPETE upon other ionic currents in B cells. We think it unlikely that AA-signaling is the only pathway to play a role in EU conditioning; the failure of AA and 12(S)-HPETE to affect the SSGP response underscores this point.
Potential Model for the Involvement of AA in Excitatory and Inhibitory Conditioning
While we present evidence in this paper that AA indirectly mediates the reduction in spike frequency produced by EU training, AA has also been reported to act synergistically with diacylglycerol (DAG) and Ca2+ to activate PKC in B cells (Lester et al., 1991), suggesting its involvement in excitatory conditioning (Talk et al., 1997; Muzzio et al., 2001).This potential dual role for AA signaling can be accommodated by several models. A simple one that we favor involves differential binding of AA with PKC vs. metabolism of AA by the 12-lipoxygenase enzyme. In this model, when AA is released in the presence of PKC activating-cofactors (DAG and Ca2+), AA facilitates activation of PKC and excitatory conditioning changes (reductions in K+ currents, enhanced light response, and spiking). In contrast, when AA is released in the absence of PKC activating-cofactors, metabolism of AA by a 12-lipoxygenase enzyme dominates, and some of the inhibitory conditioning changes occur (increases in IA and decreased light-evoked spiking). Furthermore, if AA is bound to PKC, this may preclude metabolism of AA by a 12-lipoxygenase enzyme. Alternatively, activation of PKC may block 12-lipoxygenase signaling. Although AA signaling may be common to both excitatory and inhibitory conditioning in Hermissenda, direct binding of AA to PKC may be an important process for excitatory conditioning, while 12-lipoxygenase metabolism of AA may be an important process for inhibitory conditioning.
Summary
The results of the present study are the first to demonstrate a signaling pathway (AA-lipoxygenase metabolism) that is implicated in conditioned inhibitory learning for any species. Analysis of safety-signal learning in mice has begun (Pollak et al., 2008) and it will be interesting to see if these two examples of inhibitory conditioning share common cellular and molecular mechanisms. At the moment, there is little apparent overlap. Our studies raise the provocative possibilities that AA-signaling may participate in both excitatory and inhibitory classical conditioning in Hermissenda, with the former occurring via direct binding and synergistic activation of PKC, and the latter occurring through AA-metabolism by 12-lipoxygenase. Our results suggest that similar to behavioral EU-conditioning, AA-signaling increases IA and thereby decreases light-evoked spiking in B cells. Because IA is decreased by pairings of light and rotation (Alkon et al., 1985; Farley, 1988) and contributes to the increased spiking of B cells from animals that have undergone excitatory conditioning, our results further suggest that bi-directional modulation of somatic K+ currents constitutes a flexible mechanism for encoding the inter-event relations of both coincidence as well as non-coincidence in Hermissenda Type B photoreceptors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by a grant from the National Institute of Neurological Disorders and Stroke (NIH NIDS NS48156). We thank the staff at Friday Harbor Laboratories (University of Washington, Friday Harbor, WA 98250) for providing excellent facilities, which facilitated the preparation of this manuscript. We thank the following people for their help with data collection and analysis: Jeff Johnson, Peter Puthenveetil, Kyle Smith, Doug Sparks, and Michael Teague.
References
Alkon, D. L., and Fuortes, M. G. (1972). Responses of photoreceptors in Hermissenda.J. Gen. Physiol. 60, 631–649.
Alkon, D. L., Farley, J., Sakakibara, M., and Hay, B. (1984). Voltage-dependent calcium and calcium-activated potassium currents of a molluscan photoreceptor. Biophys. J. 46, 605–614.
Alkon, D. L., Lederhendler, I., and Shoukimas, J. J. (1982). Primary changes of membrane currents during retention of associative learning.Science 215, 693–695.
Alkon, D. L., Sakakibara, M., Forman, R., Harrigan, J., Lederhendler, I., and Farley, J. (1985). Reduction of two voltage-dependent K+ currents mediates retention of a learned association. Behav. Neural Biol. 44, 278–300.
Amin, A. R., Attur, M. G., Pillinger, M., and Abramson, S. B. (1999). The pleiotropic functions of aspirin: mechanisms of action. Cell. Mol. Life Sci. 56, 305–312.
Anholt, R. R. (1994). Signal integration in the nervous system: adenylate cyclases as molecular coincidence detectors. Trends Neurosci. 17, 37–41.
Blackwell, K. T. (2006). Subcellular, cellular, and circuit mechanisms underlying classical conditioning in Hermissenda crassicornis. Anat. Rec.t B New Anat. 289, 25–37.
Bourne, H. R., and Nicoll, R. (1995). Molecular machines integrate coincident synaptic signals.Neuron 10, 65–75.
Britton, G., and Farley, J. (1999). Behavioral and neural bases of noncoincidence learning in Hermissenda. J. Neurosci. 19, 9126–9132.
Buttner, N., Siegelbaum, S. A., and Volterra, A. (1989). Direct modulation of Aplysia S-K + channels by a 12-lipoxygenase metabolite of arachidonic acid. Nature 342, 553–555.
Crow, T. (2004). Pavlovian conditioning of Hermissenda: current cellular, molecular, and circuit perspectives. Learn. Mem. 11, 229–238.
Crow, T., and Alkon, D. L. (1980). Associative behavioral modification in Hermissenda: cellular correlates. Science 209, 412–414.
Crow, T., and Offenbach, N. (1983). Modification of the initiation of locomotion in Hermissenda: behavioral analysis. Brain Res. 271, 301–310.
Crow, T. J., and Alkon, D. L. (1978). Retention of an associative behavioral change in Hermissenda. Science 201, 1239–1241.
Domjan, M. (2010). The Principles of Learning and Behavior. (Belmont, CA: Wadsworth Cengage Learning).
Estes, W. K., and Skinner, B. F. (1941). Some quantitative properties of anxiety.J. Exp. Psychol. 29, 390–400.
Farley, J. (1987a). Contingency learning and causal detection in Hermissenda: I. Behavior. Behav. Neurosci. 101, 13–27.
Farley, J. (1987b). Contingency learning and causal detection in Hermissenda: II. Cellular mechanisms. Behav. Neurosci. 101, 28–56.
Farley, J. (1988). Associative training results in persistent reductions in a calcium-activated potassium current in Hermissenda type B photoreceptors. Behav. Neurosci. 102, 748–802.
Farley, J., and Alkon, D. L. (1980). Neural organization predicts stimulus specificity for a retained associative behavioral change. Science 210, 1373–1375.
Farley, J., and Alkon, D. L. (1982). Associative neural and behavioral change in Hermissenda: consequences of nervous system orientation for light and pairing specificity. J. Neurophysiol. 48, 785–807.
Farley, J., and Auerbach, S. (1986). Protein kinase C activation induces conductance changes in Hermissenda photoreceptors like those seen in associative learning. Nature 319, 220–223.
Farley, J., Huang, H., and Kim, J. (1999). Non-coincidence learning in Hermissenda: ionic bases of decreased excitability of type B photoreceptors. Abstr. - Soc. Neurosci. 25, 645.9.
Farley, J., and Schuman, E. (1991). Protein kinase C inhibitors prevent induction and continued expression of cell memory in Hermissenda type B photoreceptors. Proc. Natl. Acad. Sci. U.S.A. 88, 2016–2020.
Farley, J., Smith, K., and Smith, B. (2003). Arachidonic Acid decreases spiking and increases IA in Hermissenda Type B cells. Abstr. - Soc. Neurosci. 29, 719.2.
Farley, J., and Wu, R. (1989). Serotonin modulation of Hermissenda type B photoreceptor light responses and ionic currents: implications for mechanisms underlying associative learning. Brain Res. Bull. 22, 335–351.
Golberg, E. M., and Zidovetzki, R. (1998). Synergistic effects of diacylglycerols and fatty acids on membrane structure and protein kinase C activity. Biochemistry 37, 5623–5632.
Jin, I., Huang, H., Smith, B., and Farley, J. (2009). Protein tyrosine kinase involvement in learning-produced changes in Hermissenda type B photoreceptors. J. Neurophysiol. 102, 3573–3595.
Kim, D., and Clapham, D. E. (1989). Potassium channels in cardiac cells activated by arachidonic acid and phospholipids. Science 244, 1174–1176.
Kim, D., Sladek, C. D., Aguado-Velasco, C., and Mathiasen, J. R. (1995). Arachidonic acid activation of a new family of K + channels in cultured rat neuronal cells. J. Physiol. 484 (Pt 3), 643–660.
Konnerth, A., Tsien, R. Y., Mikoshiba, K., and Altman, J. (1996). Coincidence Detection in the Nervous System: Molecular and Cellular Mechanisms. Consequences for Learning and Memory. Strasbourg: Human Frontier Science Program.
Lederhendler, I. I., Gart, S., and Alkon, D. L. (1986). Classical conditioning of Hermissenda: origin of a new response. J. Neurosci. 6, 1325–1331.
Lester, D. S., Collin, C., Etcheberrigaray, R., and Alkon, D. L. (1991). Arachidonic acid and diacylglycerol act synergistically to activate protein kinase C in vitro and in vivo. Biochem. Biophys. Res. Commun. 179, 1522–1528.
Marks, F., and Furstenberger, G. (1999). Prostaglandins, Leukotrienes and Other Eicosanoids: From Biogenesis to Clinical Application. Weinheim, New York, Chichester, Brisbane, Singapore, Toronto: Wiley-VCH.
Muzzio, I. A., Gandhi, C. C., Manyam, U., Pesnell, A., and Matzel, L. D. (2001). Receptor-stimulated phospholipase A(2) liberates arachidonic acid and regulates neuronal excitability through protein kinase C. J. Neurophysiol. 85, 1639–1647.
Ordway, R. W., Clapp, L. H., Gurney, A. M., Singer, J. J., and Walsh, J. V., Jr. (1989a). Fatty acids directly activate large conductance, calcium-activated K + channels in pulmonary artery smooth muscle cells from rabbit. J. Gen. Physiol. 94, 1176–1179.
Ordway, R. W., Walsh, J. V. Jr., and Singer, J. J. (1989b). Arachidonic acid and other fatty acids directly activate potassium channels in smooth muscle cells. Science 244, 1176–1179.
Ordway, R. W., Singer, J. J., and Walsh, J. V., Jr. (1991). Direct regulation of ion channels by fatty acids. Trends Neurosci. 14, 96–100.
Piomelli, D., Shapiro, E., Feinmark, S. J., and Schwartz, J. H. (1987a). Metabolites of arachidonic acid in the nervous system of Aplysia: possible mediators of synaptic modulation. J. Neurosci. 7, 3675–3686.
Piomelli, D., Volterra, A., Dale, N., Siegelbaum, S. A., Kandel, E. R., Schwartz, J. H., and Belardetti, F. (1987b). Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory cells. Nature 328, 38–43.
Piomelli, D., Shapiro, E., Zipkin, R., Schwartz, J. H., and Feinmark, S. J. (1989). Formation and action of 8-hydroxy-11, 12-epoxy-5,9,14-icosatrienoic acid in Aplysia: a possible second messenger in neurons. Proc. Natl. Acad. Sci. U.S.A. 86, 1721–1725.
Pollak, D. D., Monje, F. J., Zuckerman, L., Denny, C. A., Drew, M. R., and Kandel, E. R. (2008). An animal model of a behavioral intervention for depression. Neuron 60, 149–161.
Samuelsson, B., Dahlen, S., Lindgren, J. A., and Rouzer, C. A. (1987). Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science 237, 1171–1176.
Schwartz, J. H. (1991). Arachidonic acid metabolism: potential for diverse signalling within the same neuron. Biochem. Soc. Trans. 19, 387–390.
Schweitzer, P., Madamba, S., and Siggins, G. R. (1990). Arachidonic acid metabolites as mediators of somatostatin-induced increase of neuronal M-current.Nature 346, 464–467.
Schweitzer, P., Madamba, S., Champagnat, J., and Siggins, G. R. (1993). Somatostatin inhibition of hippocampal CA1 pyramidal neurons: mediation by arachidonic acid and its metabolites. J. Neurosci. 13, 2033–2049.
Shearman, M. S., Naor, Z., Sekiguchi, K., Kishimoto, A., and Nishizuka, Y. (1989). Selective activation of the gamma-subspecies of protein kinase C from bovine cerebellum by arachidonic acid and its lipoxygenase metabolites. FEBS Lett. 243, 177–182.
Singh, I. N., Sorrentino, G., Sitar, D. S., and Kanfer, J. N. (1997). Indomethacin and nordihydroguaiaretic acid inhibition of amyloid beta protein (25-35) activation of phospholipases A2 and D of LA-N-2 cells. Neurosci. Lett. 222, 5–8.
Smirnov, S. V., Knock, G. A., and Aaronson, P. I. (1998). Effects of the 5-lipoxygenase activating protein inhibitor MK886 on voltage-gated and Ca2 + -activated K + currents in rat arterial myocytes. Br. J. Pharmacol. 124, 572–578.
Talk, A. C., Muzzio, I. A., and Matzel, L. D. (1997). Phospholipases and arachidonic acid contribute independently to sensory transduction and associative neuronal facilitation in Hermissenda type B photoreceptors. Brain Res. 751, 196–205.
Tegeder, I., Pfeilschifter, J., and Geisslinger, G. (2001). Cyclooxygenase independent actions of cyclooxygenase inhibitors. FASEB J. 15, 2057–2072.
Tsien, J. Z. (2000). Linking Hebb’s coincidence-detection to memory formation. Curr. Opin. Neurobiol. 10, 266–273.
Villarroel, A. (1994). On the role of arachidonic acid in M-current modulation by muscarine in bullfrog sympathetic neurons. J. Neurosci. 14(11 Pt 2), 7053–7066.
Keywords: Hermissenda crassicornis, conditioned inhibition learning, arachidonic acid, lipoxygenase, HPETE, K+ channels
Citation: Walker TL, Campodonico JJ, Cavallo JS and Farley J (2010) AA/12-lipoxygenase signaling contributes to inhibitory learning in Hermissenda Type B photoreceptors. Front. Behav. Neurosci. 4:50. doi: 10.3389/fnbeh.2010.00050
Received: 02 March 2010;
Paper pending published: 26 March 2010;
Accepted: 15 July 2010;
Published online: 03 August 2010
Edited by:
Paul S. Katz, Georgia State University, USAReviewed by:
William Frost, Rosalind Franklin University, USAWayne S. Sossin, McGill University, Canada
Copyright: © 2010 Walker, Campodonico, Cavallo and Farley. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Joseph Farley, Program in Neuroscience, Indiana University, 1101 East 10th Street, Bloomington, IN 47405, USA. e-mail: farleyj@indiana.edu