L-DOPA-induced dyskinesia and abnormal signaling in striatal medium spiny neurons: focus on dopamine D1 receptor-mediated transmission
- Department of Neuroscience, Karolinska Institutet, Stockholm, Sweden
Dyskinesia is a serious motor complication caused by prolonged administration of L-DOPA to patients affected by Parkinson’s disease. Accumulating evidence indicates that L-DOPA-induced dyskinesia (LID) is primarily caused by the development of sensitized dopamine D1 receptor (D1R) transmission in the medium spiny neurons (MSNs) of the striatum. This phenomenon, combined with chronic administration of L-DOPA, leads to persistent and intermittent hyper-activation of the cAMP signaling cascade. Activation of cAMP signaling results in increased activity of the cAMP-dependent protein kinase (PKA) and of the dopamine- and cAMP-dependent phosphoprotein of 32 kDa (DARPP-32), which regulate several downstream effector targets implicated in the control of the excitability of striatal MSNs. Dyskinesia is also accompanied by augmented activity of the extracellular signal-regulated kinases (ERK) and the mammalian target of rapamycin complex 1 (mTORC1), which are involved in the control of transcriptional and translational efficiency. Pharmacological or genetic interventions aimed at reducing abnormal signal transduction at the level of these various intracellular cascades have been shown to attenuate LID in different animal models. For instance, LID is reduced in mice deficient for DARPP-32, or following inhibition of PKA. Blockade of ERK obtained genetically or using specific inhibitors is also able to attenuate dyskinetic behavior in rodents and non-human primates. Finally, administration of rapamycin, a drug which blocks mTORC1, results in a strong reduction of LID. This review focuses on the abnormalities in signaling affecting the D1R-expressing MSNs and on their potential relevance for the design of novel anti-dyskinetic therapies.
The striatum, which includes the caudate–putamen and the nucleus accumbens, is the major component of the basal ganglia, a set of interconnected subcortical structures critically involved in motor control. The GABAergic medium spiny neurons (MSNs) are the principal neuronal type in the striatum and represent the main receiving station of the basal ganglia. Their large dendritic arborization is innervated by excitatory glutamatergic inputs from cortical, thalamic, and limbic areas and modulatory dopaminergic inputs from midbrain neurons located in the substantia nigra pars compacta (SNc) and the ventral tegmental area.
In the caudate–putamen, which corresponds to the dorsal part of the striatum, dopamine regulates the activity of MSNs by acting on dopamine D1 receptors (D1Rs) and dopamine D2 receptors (D2Rs). It has become clear that D1Rs and D2Rs have a very distinct pattern of expression in the two projection pathways which connect the striatum to the output nuclei of the basal ganglia (i.e., substantia nigra pars reticulata and internal segment of the globus pallidus). In particular, it has been shown that D1Rs are located in the MSNs which directly innervate the substantia nigra pars reticulata and the internal segment of the globus pallidus. In contrast, D2Rs are present in the MSNs which project to these nuclei indirectly, via a circuit including the external segment of the globus pallidus and the subthalamic nucleus (Gerfen et al., 1990; Gerfen, 1992; Gong et al., 2003; Valjent et al., 2009).
The above distinction is at the basis of a commonly accepted model of basal ganglia transmission, which proposes that the activation of the neurons of the “direct” striatonigral pathway facilitates motor activity via disinhibition of thalamo-cortical neurons, whereas activation of the neurons of the “indirect” striatopallidal pathway reduces motor activity by increasing inhibition on thalamo-cortical neurons (Albin et al., 1989; Alexander and Crutcher, 1990; DeLong, 1990). The same model also posits that dopamine promotes motor activity by increasing the activity of striatonigral MSNs and, concomitantly, by inhibiting striatopallidal MSNs (Albin et al., 1989; Gerfen et al., 1990). These contrasting actions of dopamine depend on the selective expression of D1Rs and D2Rs in striatonigral and striatopallidal MSNs, as well as on the coupling of these receptors to different G-proteins. Thus, activation of D1Rs leads to Gαolf-mediated stimulation of adenylyl cyclase and increased cAMP, whereas activation of D2Rs leads to Gαi/o-mediated inhibition of adenylyl cyclase (Stoof and Kebabian, 1981; Herve et al., 1993; Zhuang et al., 2000).
Striatal MSNs are targeted by a large number of drugs, including addictive substances and antipsychotic drugs. Moreover, MSNs are the main target of medications used for the treatment of Parkinson’s disease (PD), a neurodegenerative disorder characterized by the progressive loss of the dopaminergic neurons of the SNc (Hornykiewicz, 1963; Braak et al., 2003). PD is commonly treated with the dopamine precursor L-DOPA, which efficiently counteracts the motor symptoms of the disease, i.e., rigidity, tremor, and hypokinesia (Cotzias et al., 1967; Birkmayer and Hornykiewicz, 1998). However, prolonged administration of L-DOPA results in the appearance of choreic, dystonic, and ballistic movements, collectively referred to as L-DOPA-induced dyskinesia (LID). These involuntary movements represent a serious limitation to the current pharmacotherapy for PD, particularly during the advanced stages of the disease (Obeso et al., 2000; Fabbrini et al., 2007).
The need for a therapy able to efficiently counteract LID has led to considerable progress in the understanding of the molecular basis of this motor disorder. In particular, striatal MSNs have become the subject of intense investigations, which identified a number of molecular abnormalities implicated in dyskinetic behavior. These abnormalities are primarily linked to the loss of dopamine input to the basal ganglia, which strongly enhances the responsiveness of MSNs to dopaminergic drugs. Such a sensitization is especially evident in the case of L-DOPA, which, following interruption of the nigrostriatal dopaminergic input, acquires the ability to affect multiple signaling pathways. One of the main goals of current research on LID is to study abnormal signaling processes induced by L-DOPA in the dopamine-depleted striatum and to determine their involvement in the emergence of pathological motor behavior.
Accumulating evidence indicates that LID develops in response to activation of sensitized D1Rs located on the MSNs of the direct striatonigral pathway. The effects produced by L-DOPA in these neurons are not limited to hyper-activation of the canonical cAMP/dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) cascade, but include modifications of signaling pathways not typically related to dopaminergic transmission, such as the extracellular signal-regulated kinases (ERK) and the mammalian target of rapamycin (mTOR) cascades. The present review focuses on these signaling abnormalities and on their potential relevance for the treatment of LID.
Mechanisms of D1R Sensitization in PD: Alterations in Receptor Distribution, G-Protein Coupling, and Adenylyl Cyclase Expression
In simple terms, the development of sensitized D1R transmission in PD can be regarded as a compensatory response to the lack of dopamine in the striatum. However, a clear understanding of the mechanisms implicated in this phenomenon has been elusive. Studies using 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), two toxins used to generate experimental models of PD, indicate that the number and affinity of D1Rs is unchanged following dopamine depletion (Breese et al., 1987; Savasta et al., 1988; Marshall et al., 1989; Joyce, 1991; Aubert et al., 2005). Similar results were obtained in postmortem samples from parkinsonian patients (Pimoule et al., 1985; Shinotoh et al., 1993; Hurley et al., 2001). However, work performed in non-human primates indicates that the loss of dopaminergic input to the striatum results in increased recruitment of D1Rs at the plasma membrane of MSNs (Guigoni et al., 2007), a phenomenon of potential relevance for the induction of LID.
Studies performed in MPTP lesioned monkeys have shown that repeated administration of L-DOPA increases the binding of the D1R antagonist SCH23390 to striatal membranes (Aubert et al., 2005). This effect is not accompanied by changes in the expression of D1R mRNA, suggesting that it may depend on alterations in the number of functionally available receptors. In support of this possibility, it has been shown that, in rodent and non-human primate models, LID is associated with increased localization of D1Rs at the cell surface, which may be caused by impaired receptor internalization and trafficking (Guigoni et al., 2007; Berthet et al., 2009).
The pathological enhancement in the number of D1Rs at the plasma membrane is likely to contribute to the increase in D1R transmission associated to LID and represents a potential target for therapeutic interventions. In line with this idea, recent findings have shown that LID is reduced by promoting G-protein coupled receptor (GPCR) desensitization. This process starts with the phosphorylation of the receptor by GPCR kinases (GRKs), followed by binding to arrestins and receptor internalization (Shenoy and Lefkowitz, 2003). Experiments performed in 6-OHDA-lesioned rats and MPTP lesioned monkeys show that lentiviral-mediated overexpression of the GRK6 in the striatum attenuates LID (Ahmed et al., 2010). In contrast, dyskinesia is worsened when GRK6 expression is reduced by transfection with a GRK6 miRNA. Moreover, GRK6 overexpression does not interfere with the anti-akinetic action of L-DOPA and may therefore represent a potential strategy to control dyskinetic behavior (Ahmed et al., 2010).
In cortical neurons, activation of the transcription factor cAMP response element-binding protein (CREB) is known to increase the levels of brain-derived neurotrophic factor (BDNF), which is involved in activity-dependent synaptic plasticity (Shieh et al., 1998; Tao et al., 1998). Interestingly, repeated administration of L-DOPA promotes BDNF expression in the frontal cortex of 6-OHDA-lesioned rats, via activation of dopamine D1-type receptors (Guillin et al., 2001). This effect, in concomitance with 6-OHDA-induced overexpression of BDNF TrκB receptors, is thought to increase the levels of dopamine D3 receptors (D3Rs) in the striatum (Guillin et al., 2001). Enhanced D3R expression in the MSNs of the direct pathway is associated to LID (Bordet et al., 1997, 2000; Guillin et al., 2001; Bezard et al., 2003) and may play a role in this condition by further exacerbating sensitized D1R transmission. In fact, D3Rs have been shown to exert a synergistic effect on D1R-mediated transmission through direct intramembrane interaction (Fiorentini et al., 2008; Marcellino et al., 2008). In support of this possibility, co-treatment with L-DOPA and the D3R antagonist ST 198 restores normal levels of membrane-bound D1Rs in dyskinetic animals (Berthet et al., 2009) and counteracts dyskinesia in experimental models of PD (Bezard et al., 2003; Kumar et al., 2009; Visanji et al., 2009; but see also Mela et al., 2010).
The increased recruitment of D1Rs at the plasma membrane may also be responsible for the increase in coupling of striatal D1Rs to Gαolf protein, which has been described in the striata of dyskinetic monkeys (Aubert et al., 2005). In connection to this observation, studies performed in 6-OHDA-lesioned rats and in postmortem samples from parkinsonian patients have demonstrated that loss of striatal dopamine is accompanied by increased levels of Gαolf (Herve et al., 1993; Corvol et al., 2004; Rangel-Barajas et al., 2011). However, Gαolf overexpression subsides during chronic L-DOPA administration (Corvol et al., 2004; Rangel-Barajas et al., 2011) and striatal levels of Gαolf in dyskinetic mice are similar to those of control animals (Rangel-Barajas et al., 2011).
Striatal MSNs express high levels of adenylyl cyclase type 5 (Glatt and Snyder, 1993; Mons and Cooper, 1994), which is stimulated in response to D1R-mediated activation of Gαolf (Herve et al., 1993; Zhuang et al., 2000). Evidence obtained using 6-OHDA-lesioned rats shows that dopamine depletion increases the levels of adenylyl cyclase type 5 in the striatum (Rangel-Barajas et al., 2011). A similar increase is also observed in the substantia nigra pars reticulata, which is innervated by the MSNs of the direct pathway (cf. above; Rangel-Barajas et al., 2011). Interestingly, these effects are maintained during repeated administration of L-DOPA, but only in animals displaying severe dyskinesia (Rangel-Barajas et al., 2011).
In summary, the work described above indicates that LID is associated to increased recruitment of D1Rs at the cell surface and to overexpression of adenylyl cyclase type 5 in the striatal MSNs of the direct pathway (Figure 1). These modifications are likely to influence dopaminergic transmission in the striatum and may underlie the enhancement in the ability of L-DOPA to increase the levels of cAMP, thereby activating cAMP-dependent protein kinase (PKA). The importance of augmented PKA activity in dyskinesia is indicated by the observation that, in 6-OHDA-lesioned rats, striatal infusion of the PKA inhibitor Rp-cAMPS attenuates LID (Lebel et al., 2010). The following sections will focus on the effects produced by L-DOPA-induced activation of the cAMP/PKA cascade and on their potential relevance for the development and expression of LID.
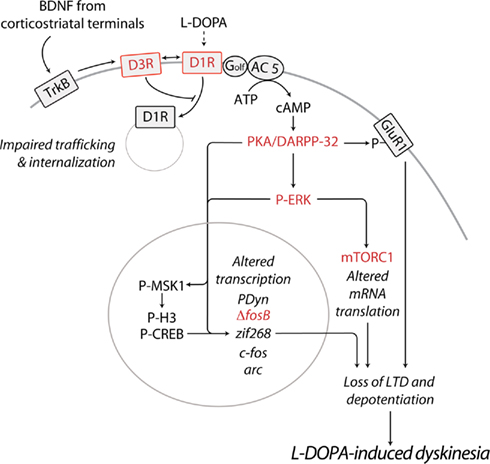
Figure 1. Schematic diagram illustrating some of the major abnormalities related to sensitized D1R-signaling and associated to LID. In PD, the loss of striatal dopamine leads to sensitization of D1Rs on the striatonigral MSNs of the direct pathway. Emerging evidence indicates that, if persistent, this phenomenon results in the appearance of dyskinesia. D1R sensitization may be caused by augmented D1R expression at the cell surface. Chronic administration of L-DOPA promotes the release of BDNF from corticostriatal neurons, leading to activation of TrκB receptors and increased expression of D3Rs, specifically in striatonigral MSNs. Direct interaction with D3Rs is likely to increase the levels of membrane-bound D1Rs, thereby exacerbating D1R sensitization and dyskinetic behavior. In line with this possibility, D3R antagonists have been found to counteract LID in experimental models of PD. Sensitized D1R transmission may also be caused by increased levels of adenylyl cyclase 5 (AC 5) in striatonigral MSNs. Increased responsiveness of the D1R/Gαolf/AC5 machinery to L-DOPA results in augmented synthesis of cAMP and hyper-activation of PKA and DARPP-32. Pharmacological inhibition of PKA, or genetic inactivation of DARPP-32 have been shown to reduce LID. Abnormal PKA/DARPP-32 signaling increases the phosphorylation of GluR1. This effect promotes the excitability of MSNs and may participate in the loss of corticostriatal LTD and depotentiation associated to LID. Sensitized D1R-mediated transmission leads also to activation of ERK, which controls transcriptional and translational processes. Both pharmacological and genetic suppression of ERK signaling counteracts the development and expression of LID. In the nucleus, PKA/DARPP-32 and ERK/MSK1 signaling leads to phosphorylation of CREB and histone H3, and increased expression of immediate early genes and prodynorphin. Reduced expression/activity of ΔfosB efficiently counteracts LID. Activation of ERK promotes mTORC1-dependent signaling, thereby accelerating mRNA translation. Blockade of mTORC1 with rapamycin has been found to attenuate the development of LID. Red color indicates receptors or signaling components whose targeting reduces LID. See text for abbreviations.
Downstream of cAMP and PKA: Role of DARPP-32 in Dyskinesia and Abnormal Corticostriatal Depotentiation
Dopamine D1 receptor-mediated transmission in striatal MSNs depends not only on PKA-dependent phosphorylation of downstream target proteins, but also on concomitant reduction of their dephosphorylation. This parallel mechanism is based on the ability of PKA to phosphorylate and activate DARPP-32, a potent inhibitor of protein phosphatase 1 (PP-1). Inhibition of PP-1 suppresses the dephosphorylation of several downstream targets of PKA, thereby amplifying behavioral responses produced by activation of cAMP signaling (Fienberg et al., 1998; Greengard, 2001; Borgkvist and Fisone, 2007).
The sensitization of D1Rs produced by dopamine depletion is reflected by the large increase in DARPP-32 phosphorylation observed in response to the administration of L-DOPA. In rodent models of PD L-DOPA-induced activation of the cAMP/PKA/DARPP-32 cascade has been associated to the emergence of dyskinesia (Picconi et al., 2003; Santini et al., 2007; Lebel et al., 2010). In MPTP lesioned non-human primates, increased phosphorylation of DARPP-32 has been shown to persist for up to 3 months of L-DOPA chronic administration, suggesting that DARPP-32 is involved not only in the development, but also in the maintenance and expression of LID (Santini et al., 2010a).
Genetic inactivation of DARPP-32 has proven to be an effective strategy to reduce experimental LID. Following 6-OHDA lesion and repeated administration of L-DOPA, DARPP-32 knock out mice display significantly less dyskinetic behavior in comparison to wild type littermates (Santini et al., 2007). Interestingly, LID is also reduced by cell-specific inactivation of DARPP-32 in the MSNs of the direct pathway. In contrast, selective inactivation of DARPP-32 in indirect MSNs does not affect the ability of L-DOPA to induce abnormal involuntary movements (Bateup et al., 2010). Taken together, these studies indicate not only the importance of PKA-induced activation of DARPP-32 in dyskinesia, but also the primary role played in this condition by the D1R-expressing MSNs of the direct pathway.
The abnormal activation of PKA and the concomitant hyper-phosphorylation of DARPP-32 observed in experimental models of LID lead to changes in the state of phosphorylation of target effector proteins, which may have profound repercussion on the excitability of striatal MSNs (Figure 1). High-frequency stimulation is known to induce long-term potentiation (LTP) at corticostriatal synapses (Calabresi et al., 1992b). Dopamine depletion abolishes LTP, which is rescued by systemic administration of L-DOPA (Centonze et al., 1999; Picconi et al., 2003, 2008). Once established, LTP can be reversed by low frequency stimulation (Picconi et al., 2003, 2008). This phenomenon, called depotentiation, is blocked by inhibition of PP-1 and, most importantly, is absent at the corticostriatal synapses of dyskinetic rats (Picconi et al., 2003, 2008). It has been proposed that depotentiation may prevent the generation of aberrant motor patterns, such as dyskinesia, by erasing non-essential information and normalizing striatal synaptic efficiency (Picconi et al., 2003, 2008; Calabresi et al., 2010). Thus, dyskinesia may be caused by L-DOPA through stimulation of sensitized D1Rs, hyper-activation of PKA, increased phosphorylation of DARPP-32, inhibition of PP-1, and abolishment of corticostriatal depotentiation (Picconi et al., 2003; Calabresi et al., 2010).
One possible mechanism by which inhibition of PP-1 by DARPP-32 may prevent depotentiation involves changes in the state of phosphorylation of the GluR1 subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor. Dyskinetic behavior correlates with the ability of L-DOPA to increase PKA-dependent phosphorylation of GluR1 at Ser845 (Santini et al., 2007). This effect is strictly dependent on concomitant phosphorylation of DARPP-32, since it is abolished in DARPP-32 knock out mice (Santini et al., 2007). Phosphorylation of GluR1 at Ser845 promotes glutamatergic transmission (Banke et al., 2000; Mangiavacchi and Wolf, 2004) and may participate in the block of depotentiation observed in dyskinetic rats (Picconi et al., 2003). Increased glutamatergic transmission may also be produced by augmented synaptic recruitment of AMPA receptor GluR2/3 subunits, which has been described in the striata of dyskinetic monkeys (Silverdale et al., 2010). The idea of the involvement in dyskinesia of enhanced AMPA receptor transmission is further supported by the observation that, in non-human primates, LID is increased by an AMPA receptor agonist and reduced by an AMPA receptor antagonist (Konitsiotis et al., 2000).
cGMP and Loss of Long-Term Depression in Dyskinesia
In the rat, depletion of striatal dopamine results in the loss of corticostriatal long-term depression (LTD). This effect is counteracted by D1R and D2R agonists, exogenous dopamine, or L-DOPA (Calabresi et al., 1992a; Picconi et al., 2008, 2011). Interestingly, recent evidence indicates that in dyskinesia the loss of LTD produced by 6-OHDA persists even after chronic L-DOPA administration (Picconi et al., 2011). This phenomenon, together with the loss of depotentiation (Picconi et al., 2003), is in line with the idea that LID is caused by impaired control of striatal excitatory transmission.
The lack of corticostriatal LTD associated with LID may be due to the decrease in cGMP signaling observed in dyskinetic animals (Giorgi et al., 2008). In the striatum, LTD depends on a group of interneurons, which express nitric oxide synthase (Kawaguchi et al., 1995; Calabresi et al., 1999). Nitric oxide activates a soluble form of guanylyl cyclase highly expressed in MSNs (Ding et al., 2004), leading to increased synthesis of cGMP and activation of cGMP-dependent protein kinase. Notably, cGMP signaling is implicated in LTD, which can be induced pharmacologically with zaprinast and UK-343664, two inhibitors of cGMP phosphodiesterase (the enzyme responsible for the conversion of cGMP to GMP; Calabresi et al., 1999; Picconi et al., 2011). Interestingly, local injection of these drugs in the striatum of dyskinetic rats has been found to rescue LTD and to reduce the dyskinetic response (Picconi et al., 2011). Further studies will be necessary to understand the molecular basis of the persistent loss of LTD associated to dyskinetic behavior.
Enhanced ERK Signaling in Dyskinesia
The changes in synaptic plasticity associated to LID, such as loss of depotentiation and LTD are likely to occur also in response to modifications in the activity of signaling pathways involved in the control of gene expression. ERK regulate transcriptional and translational processes (Thomas and Huganir, 2004; Costa-Mattioli et al., 2009) and have been implicated in the induction of striatal LTP (Xie et al., 2009). In neuronal cells, ERK signaling is promoted via Ca2+-dependent activation of the brain specific exchange factor Ras-guanyl nucleotide releasing factor 1 (Ras-GRF1, or CDC25Mm; Martegani et al., 1992; Shou et al., 1992). Ras-GRF1 induces the exchange of GDP for GTP on the small G-protein Ras (Farnsworth et al., 1995). A similar effect is produced by the calcium- and diacylglycerol-regulated guanine nucleotide exchange factor II (CalDAG-GEF II), which is highly enriched in striatal MSNs (Toki et al., 2001). Ras-GTP, in turn, activates the protein kinase Raf, leading to the phosphorylation of the mitogen-activated protein kinase/ERK kinase (MEK) and ERK (Figure 2).
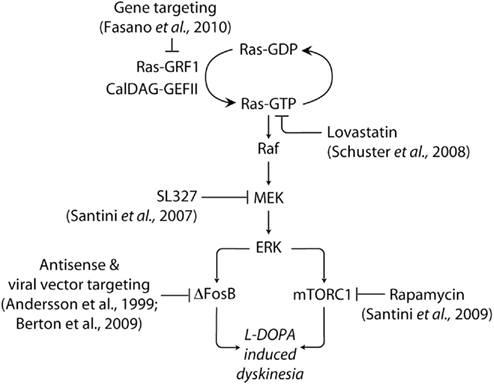
Figure 2. Targeting signaling upstream and downstream of ERK in LID. The abnormal activation of ERK produced by administration of L-DOPA in experimental models of PD is implicated in the emergence of dyskinetic behavior. ERK is activated by Ras-GRF1 and CalDAG-GEF II, which induce the exchange of GDP for GTP on the small G-protein Ras. Ras-GTP activates the protein kinase Raf, leading to the phosphorylation of MEK and ERK. Dyskinesia is attenuated in Ras-GRF1 knock out mice. A similar reduction is produced by inhibition of Ras, or MEK, achieved using lovastatin and SL327, respectively. ERK activation promotes the expression of the transcription factor ΔFosB, which is also implicated in LID. In addition, dyskinesia is associated to ERK-dependent activation of mTORC1, which is likely to accelerate local protein synthesis. Blockade of mTORC1 signaling with rapamycin has been found to reduce LID. See text for abbreviations.
It has been shown that substances that promote D1R transmission, such as cocaine and amphetamine, phosphorylate ERK via activation of the PKA/DARPP-32 signaling cascade. This effect is involved in the development of the locomotor sensitization produced by repeated administration of these drugs (Valjent et al., 2005). This finding suggests that changes in ERK phosphorylation may also be implicated in LID, which, in experimental models, develops in concomitance with a marked sensitization of the locomotor effect (i.e., turning behavior) produced by L-DOPA (Carey, 1991). Studies performed in 6-OHDA-lesioned rats show that depletion of striatal dopamine confers to a D1R agonist the ability to increase the phosphorylation of ERK (Gerfen et al., 2002). Similarly, L-DOPA, which is inactive when injected in naive or sham-lesioned animals, induces a large increase in ERK phosphorylation when administered to 6-OHDA-lesioned mice or MPTP intoxicated monkeys (Pavon et al., 2006; Santini et al., 2007, 2009a, 2010a).
The mechanism underlying this change in efficacy is likely to depend on sensitized D1R-mediated transmission and abnormal activation of cAMP signaling. Thus, blockade of PKA prevents the ability of L-DOPA to increase ERK phosphorylation in 6-OHDA-lesioned rats (Lebel et al., 2010). Moreover, in the dorsal striatum, the ability of L-DOPA to activate ERK appears to be attenuated by genetic inactivation of DARPP-32 (Santini et al., 2007). However, this last observation has been recently challenged (Gerfen et al., 2008) and further work will be necessary to assess the involvement of DARPP-32 in L-DOPA-induced ERK phosphorylation, particularly within the dorsolateral striatum.
The existence of a causal link between ERK and dyskinesia was first provided in a mouse model of LID. In particular, it was shown that blockade of ERK phosphorylation with SL327, an inhibitor of MEK, reduced the development of dyskinesia (Santini et al., 2007). Subsequent studies showed that lovastatin, which reduces Ras isoprenylation and activity (Mendola and Backer, 1990; Sebti et al., 1991), attenuates dyskinesia induced by L-DOPA in the 6-OHDA-lesioned rat (Schuster et al., 2008). In the same model, it was found that LID is associated to increased striatal expression of CalDAG-GEF II, which may represent a further mechanism accounting for increased ERK activation (Crittenden et al., 2009; cf. above; Figure 2).
The involvement in LID of the Ras–ERK pathway is further supported by recent evidence indicating that dyskinesia is attenuated in Ras-GRF1 knock out mice (Fasano et al., 2010; Figure 2). Moreover, in MPTP intoxicated monkeys, viral vector-mediated inhibition of Ras-GRF1 reverses pre-established LID (Fasano et al., 2010). This observation suggests that ERK signaling is involved not only in the development of LID but also in its expression/maintenance. It should be noted, however, that the phosphorylation of ERK produced by L-DOPA in the dopamine-depleted striatum appears to subside over prolonged drug administration. Thus, in dyskinetic MPTP lesioned monkeys, L-DOPA loses its ability to promote ERK phosphorylation following 3 months of chronic treatment (Santini et al., 2010a).
A similar phenomenon has been recently described in mice deficient for the transcription factor Pitx3 (Ding et al., 2011). These animals lack the dopaminergic innervation to the striatum and display akinesia reminiscent of parkinsonism (van den Munckhof et al., 2003; Hwang et al., 2005). In Ptix3 knock out mice, acute L-DOPA increases the number of phospho-ERK-positive MSNs. However, this effect disappears following 7 weeks of daily administration, in concomitance with the appearance of increased phospho-ERK immunoreactivity in cholinergic interneurons (Ding et al., 2011). The progressive reduction in the ability of L-DOPA to activate ERK in striatal MSNs was also observed in 6-OHDA-lesioned mice, further supporting the idea of a partial normalization in ERK signaling occurring over prolonged periods of drug administration (Ding et al., 2011). The increase in phospho-ERK observed in cholinergic neurons is difficult to appreciate using western immunoblotting, since these cells represent only about 2% of striatal neurons (Zhou et al., 2002). Nevertheless, in the striatum, cholinergic interneurons give rise to a particularly dense arborization and to a large number of synaptic contacts (Bolam et al., 1984; Phelps et al., 1985; Contant et al., 1996). Thus, augmented ERK signaling and increased activity in this specific neuronal population may have profound repercussions on striatal MSNs (Ding et al., 2011). In line with this idea, administration of the muscarinic acetylcholine receptor antagonist, dicyclomine, reduces dyskinetic behavior in Ptix3 knock out and 6-OHDA-lesioned mice (Ding et al., 2011).
Searching for Mechanisms of Dyskinesia Downstream of ERK
Taken together the studies described above indicate that LID can be counteracted by reducing the activity of the Ras–ERK cascade through pharmacological inhibition of Ras (Schuster et al., 2008) and MEK (Santini et al., 2007). However, this approach is likely to produce negative side-effects, due to the involvement of ERK in basic physiological processes, ranging from synaptic plasticity to cell survival (Orban et al., 1999; Thomas and Huganir, 2004; Kim and Choi, 2010). This limitation can be at least in part resolved by acting on Ras-GRF1, which is specifically expressed in neuronal cells and is not implicated in cell survival (Fasano et al., 2010). Alternatively, negative side-effects can be reduced by narrowing the specificity of intervention through acting downstream of ERK. This approach is particularly interesting, since ERK is involved in the control of a large number of target effector proteins, both in the nucleus and in the cytoplasm (Yoon and Seger, 2006; Figure 1).
The mitogen- and stress-activated protein kinase 1 (MSK1) is an important component of the ERK signaling cascade, specifically localized in the nucleus (Deak et al., 1998). LID is accompanied by a large increase in the phosphorylation of MSK1, which occurs selectively in the D1R-expressing neurons of the direct pathway (Santini et al., 2007, 2009a). In MSNs, activation of MSK1 leads to phosphorylation of CREB (Brami-Cherrier et al., 2005), which regulates the expression of genes involved in synaptic plasticity and drug addiction (Carlezon et al., 2005; Figure 1). Prolonged administration of L-DOPA has been found to increase CREB phosphorylation in a large proportion of striatal neurons. This effect is mimicked by a D1R agonist and is prevented by inhibition of PKA (Oh et al., 2003), which is known to regulate CREB. In cultured striatal neurons, PKA-dependent phosphorylation of CREB is blocked by inhibition of ERK (Zanassi et al., 2001). Moreover, the ability of cocaine to increase CREB phosphorylation in striatal MSNs is prevented by administration of SL327 (Brami-Cherrier et al., 2005). Therefore, it is likely that the increase in CREB phosphorylation induced in the striatum by repeated administration of L-DOPA is produced by concomitant activation of cAMP/PKA and ERK/MSK1 signaling. The potential implication of altered CREB phosphorylation in LID remains to be fully evaluated, particularly in view of the observation that striatal infusion of antisense oligonucleotide against CREB exacerbates the dyskinetic response to L-DOPA, rather than reducing it (Andersson et al., 2001).
Administration of L-DOPA to 6-OHDA-lesioned rodents results in a large increase in the state of phosphorylation of histone H3 at Ser10 (Santini et al., 2007, 2009a; Darmopil et al., 2009), which is known to depend on activation of MSK1 (Davie, 2003; Figure 1). This effect occurs in the MSNs of the direct pathway and correlates with the severity of dyskinetic movements (Santini et al., 2007, 2009a; Darmopil et al., 2009). Increased phosphorylation on Ser10 of histone H3 is thought to mediate transcriptional activation (Nowak and Corces, 2004) and may participate in changes in gene expression associated to LID.
ERK-Dependent Changes in Gene Expression Associated to Dyskinesia
Evidence obtained in rodent and non-human primate models indicates that LID is accompanied by increased levels of stable isoforms of ΔFosB (Andersson et al., 1999; Pavon et al., 2006; Berton et al., 2009; Darmopil et al., 2009; Fasano et al., 2010), a truncated splice variant of the immediate early gene fosB (Nestler et al., 2001; Figures 1 and 2). Importantly, increased ΔFosB has also been observed in postmortem striatal samples from parkinsonian patients treated with L-DOPA (Tekumalla et al., 2001). ΔFosB acts as a transcriptional activator by forming heterodimers with JunD, which bind to the activator protein-1 site of several late response genes. Enhanced expression of ΔFosB has been implicated in the long-term effects produced by substances of abuse, including dopaminergic drugs such as cocaine (Nestler et al., 2001).
The increase in ΔFosB associated to LID is restricted to the D1R-expressing MSNs of the direct pathway (Andersson et al., 1999; Pavon et al., 2006) and is mediated via activation of the D1R/cAMP cascade. Thus, genetic inactivation of D1Rs, or striatal infusion of Rp-cAMPS prevent the ability of L-DOPA to increase ΔFosB (Darmopil et al., 2009; Lebel et al., 2010). The accumulation of ΔFosB requires also PKA-catalyzed phosphorylation of DARPP-32 (Zachariou et al., 2006), which is strongly induced by administration of L-DOPA (Santini et al., 2007). ERK signaling is also involved in the regulation of ΔFosB, since inactivation of Ras-GRF1 (Fasano et al., 2010), or pharmacological inhibition of Ras (Schuster et al., 2008), abolish the increase in ΔFosB expression induced by L-DOPA.
Several lines of evidence indicate that the up-regulation of ΔFosB produced by chronic administration of L-DOPA participates in the development of dyskinesia. Studies performed in 6-OHDA-lesioned rats showed that LID is reduced by striatal injection of a fosB antisense oligonucleotide (Andersson et al., 1999; Figure 2). In the same model, it has been recently shown that viral vector-induced overexpression of ΔFosB enhances the ability of L-DOPA to induce dyskinetic behavior (Cao et al., 2010). Furthermore, experiments in MPTP lesioned macaques showed that overexpression of ΔJunD, a truncated variant of JunD lacking a transactivation domain and acting as a dominant negative inhibitor of ΔFosB, reduces dyskinesia (Berton et al., 2009; Figure 2).
Previous studies showed that repeated administration of a D1R agonist to 6-OHDA-lesioned rats increases the expression of mRNA coding for the opioid peptide prodynorphin in the MSNs of the direct pathway (Gerfen et al., 1990). A similar regulation was later found to correlate with dyskinesia and to depend on L-DOPA-induced expression of ΔFosB (Andersson et al., 1999; Figure 1). Although increased prodynorphin is regarded as a very robust marker of LID, a clear understanding of the role played by enhanced opioid transmission in dyskinesia is complicated by contrasting data obtained using opioid receptor antagonists (Samadi et al., 2006). Further studies will be necessary to fully characterize the significance of this and other effects dependent on ΔFosB for the development and expression of LID.
Another immediate early gene whose expression is increased in dyskinesia is that coding for the transcription factor Zif268 (or NGFI-A/Krox24/Egr1). Acute administration of L-DOPA to 6-OHDA-lesioned rats increases zif268 mRNA in both striatopallidal and striatonigral MSNs. Interestingly, repeated L-DOPA administration leading to dyskinesia normalizes the levels of zif268 mRNA in striatopallidal neurons, but not in striatonigral neurons (Carta et al., 2005). The lack of normalization of zif268 expression in the MSNs of the direct pathway may be due to the persistent activation of ERK observed in these cells in association with LID (Darmopil et al., 2009; Santini et al., 2009a). Indeed, ERK is required for cocaine-induced increase in zif268 expression (Valjent et al., 2006). Interestingly, Zif268 is involved in the sensitized motor response produced by repeated administration of cocaine (Valjent et al., 2006), suggesting that activation of the ERK/Zif268 cascade may represent a common mechanism implicated in abnormal motor behaviors (Figure 1).
In the hippocampus, Zif268 is required for the late phase of LTP (Jones et al., 2001). This regulation is thought to depend on the ability of Zif268 to induce Arc (or Arg3.1; Li et al., 2005), another immediate early gene involved in multiple forms of neuronal plasticity, including LTP and LTD (Bramham et al., 2008). Interestingly, increased expression of Arc has been found in the striata of dyskinetic rats, specifically at the level of the MSNs of the direct pathway (Sgambato-Faure et al., 2005). These observations raise the possibility that activation of the ERK/Zif268/Arc signaling pathway may participate in the plastic changes associated to LID. In particular, abnormal increase of Zif268 and Arc may promote or exacerbate LTP, leading to the loss of corticostriatal depotentiation associated to dyskinesia (Picconi et al., 2003; Figure 1).
ERK-Dependent Regulation of Mammalian Target of Rapamycin Complex 1
The involvement of ERK in LID may depend not only on the regulation of transcription factors (i.e., CREB, ΔFosB, and Zif268), but also on the modulation of local protein synthesis in the cytoplasm. In this regard, the mammalian target of rapamycin complex 1 (mTORC1), a key regulator of mRNA translation (Costa-Mattioli et al., 2009), is emerging as a particularly interesting subject of study (Santini et al., 2010b; Figure 1).
ERK has been proposed to activate mTORC1 through several mechanisms. For instance, ERK may promote the activation of the small GTPase, Ras homolog enriched in brain (Rheb), which stimulates mTORC1 (Long et al., 2005; Ma et al., 2005). In addition, ERK can phosphorylate and activate the regulatory associated protein of mTOR (or Raptor), an essential component of mTORC1, thereby promoting mTORC1-mediated signaling (Carriere et al., 2008).
Studies performed in the hippocampus showed that activated mTORC1 participates to the maintenance of LTP by promoting local protein synthesis immediately after synaptic stimulation (Tsokas et al., 2005). This effect, which requires activation of ERK (Tsokas et al., 2007), may be implicated in the maladaptive processes associated with LID, which affect the functioning of corticostriatal synapses (see above; Picconi et al., 2003, 2011; Calabresi et al., 2010). In line with this possibility, it has been found that administration of L-DOPA to 6-OHDA-lesioned mice promotes the phosphorylation of the p70 ribosomal S6 kinases (S6Ks) and the initiation factor 4E (eIF4E)-binding protein (4E-BP; Santini et al., 2009b), two major targets of mTORC1 (Thomas et al., 1979; Gingras et al., 2001; Ruvinsky and Meyuhas, 2006). These effects, which lead to activation of the eIF4E and to the phosphorylation of the ribosomal protein S6 (rpS6), are thought to accelerate protein synthesis (Richter and Sonenberg, 2005; Ruvinsky and Meyuhas, 2006; Roux et al., 2007).
The increase in mTORC1 signaling produced by L-DOPA occurs selectively in the MSNs of the direct pathway and requires concomitant activation of ERK signaling (Santini et al., 2009b). Blockade of mTORC1 does not interfere with the ability of L-DOPA to counteract the akinetic effect of the 6-OHDA lesion, but it diminishes the development of LID. Thus, combined administration of L-DOPA and rapamycin, a selective allosteric inhibitor of mTORC1 (Oshiro et al., 2004), leads to a substantial reduction in the emergence of abnormal involuntary movements in dopamine-depleted mice (Santini et al., 2009b; Figure 2). These results suggest that enhanced mRNA translation, leading to abnormal protein synthesis may participate in the development of LID. In this regard, it will be important to identify downstream effectors specifically affected by abnormal mTORC1 signaling (Klann, 2009; Santini et al., 2009b, 2010b).
Conclusion
The study of the molecular mechanisms at the basis of LID has unveiled a number of abnormalities in signal transduction associated to the development and manifestation of this condition (cf. Figures 1 and 2). Most importantly, it has been shown that drugs targeting specific intracellular components of the signaling machinery in striatal MSNs efficiently counteract the emergence of LID. These drugs include inhibitors of the Ras–ERK and mTORC1 cascades, such as SL327, lovastatin, and rapamycin (Figure 2), as well as inhibitors of the cGMP phosphodiesterase, such as zaprinast and UK-343664. Although some of these substances (e.g., lovastatin and rapamycin) have been tested in clinical settings, their use for the treatment of dyskinesia may present problems related to long-term side-effects at both central and peripheral level. This indicates the importance of testing additional substances interacting with signaling changes potentially implicated in LID and occurring downstream of the ERK and mTORC1 cascades.
This review focused on abnormal signaling at the level of the D1R-expressing MSNs of the direct striatonigral pathway. Whereas acting on these neurons seems to produce beneficial effects against dyskinetic behavior, other neuronal populations may represent equally feasible targets. For instance, pharmacological and genetic interventions aimed at modifying transmission in the D2R-expressing MSNs of the indirect pathway have been shown to produce anti-dyskinetic effects (Schwarzschild et al., 2006; Gold et al., 2007; Schuster et al., 2009). This is not surprising, since both direct and indirect MSNs act in large part by controlling the same set of structures (i.e., substantia nigra pars reticulata and internal segment of the globus pallidus), ultimately involved in the control exerted by the basal ganglia on motor function. Recent work indicates that other groups of striatal neurons are also involved in LID. In particular, it has been shown that dyskinesia may depend on changes in the activity of cholinergic interneurons, as well as of interneurons expressing nitric oxide synthase. One important question in the study of the mechanisms of dyskinesia will be to identify signaling abnormalities associated with LID and occurring at the level of these distinct neuronal populations, to understand their impact on basal ganglia transmission and to assess their contribution to dyskinetic behavior.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Gilberto Fisone was supported by Swedish Research Council grant 13482 and by the Swedish Brain Foundation. Alessandra Bonito-Oliva was supported by a fellowship from the Foundation Blanceflor Boncompagni-Ludovisi, née Bildt. Michael Feyder was supported by the National Institutes of Health – Karolinska Institutet Graduate Training Partnership Program.
References
Ahmed, M. R., Berthet, A., Bychkov, E., Porras, G., Li, Q., Bioulac, B. H., Carl, Y. T., Bloch, B., Kook, S., Aubert, I., Dovero, S., Doudnikoff, E., Gurevich, V. V., Gurevich, E. V., and Bezard, E. (2010). Lentiviral overexpression of GRK6 alleviates L-dopa-induced dyskinesia in experimental Parkinson’s disease. Sci. Transl. Med. 2, 28ra28.
Albin, R. L., Young, A. B., and Penney, J. B. (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375.
Alexander, G. E., and Crutcher, M. D. (1990). Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 13, 266–271.
Andersson, M., Hilbertson, A., and Cenci, M. A. (1999). Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson’s disease. Neurobiol. Dis. 6, 461–474.
Andersson, M., Konradi, C., and Cenci, M. A. (2001). cAMP response element-binding protein is required for dopamine-dependent gene expression in the intact but not the dopamine-denervated striatum. J. Neurosci. 21, 9930–9943.
Aubert, I., Guigoni, C., Hakansson, K., Li, Q., Dovero, S., Barthe, N., Bioulac, B. H., Gross, C. E., Fisone, G., Bloch, B., and Bezard, E. (2005). Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann. Neurol. 57, 17–26.
Banke, T. G., Bowie, D., Lee, H., Huganir, R. L., Schousboe, A., and Traynelis, S. F. (2000). Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 20, 89–102.
Bateup, H. S., Santini, E., Shen, W., Birnbaum, S., Valjent, E., Surmeier, D. J., Fisone, G., Nestler, E. J., and Greengard, P. (2010). Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc. Natl. Acad. Sci. U.S.A. 107, 14845–14850.
Berthet, A., Porras, G., Doudnikoff, E., Stark, H., Cador, M., Bezard, E., and Bloch, B. (2009). Pharmacological analysis demonstrates dramatic alteration of D1 dopamine receptor neuronal distribution in the rat analog of L-DOPA-induced dyskinesia. J. Neurosci. 29, 4829–4835.
Berton, O., Guigoni, C., Li, Q., Bioulac, B. H., Aubert, I., Gross, C. E., Dileone, R. J., Nestler, E. J., and Bezard, E. (2009). Striatal overexpression of DeltaJunD resets L-DOPA-induced dyskinesia in a primate model of Parkinson disease. Biol. Psychiatry 66, 554–561.
Bezard, E., Ferry, S., Mach, U., Stark, H., Leriche, L., Boraud, T., Gross, C., and Sokoloff, P. (2003). Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat. Med. 9, 762–767.
Birkmayer, W., and Hornykiewicz, O. (1998). The effect of l-3,4-dihydroxyphenylalanine (=DOPA) on akinesia in parkinsonism. Parkinsonism Relat. Disord. 4, 59–60.
Bolam, J. P., Wainer, B. H., and Smith, A. D. (1984). Characterization of cholinergic neurons in the rat neostriatum. A combination of choline acetyltransferase immunocytochemistry, Golgi-impregnation and electron microscopy. Neuroscience 12, 711–718.
Bordet, R., Ridray, S., Carboni, S., Diaz, J., Sokoloff, P., and Schwartz, J. C. (1997). Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc. Natl. Acad. Sci. U.S.A. 94, 3363–3367.
Bordet, R., Ridray, S., Schwartz, J. C., and Sokoloff, P. (2000). Involvement of the direct striatonigral pathway in levodopa-induced sensitization in 6-hydroxydopamine-lesioned rats. Eur. J. Neurosci. 12, 2117–2123.
Borgkvist, A., and Fisone, G. (2007). Psychoactive drugs and regulation of the cAMP/PKA/DARPP-32 cascade in striatal medium spiny neurons. Neurosci. Biobehav. Rev. 31, 79–88.
Braak, H., Del Tredici, K., Rub, U., De Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211.
Bramham, C. R., Worley, P. F., Moore, M. J., and Guzowski, J. F. (2008). The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J. Neurosci. 28, 11760–11767.
Brami-Cherrier, K., Valjent, E., Herve, D., Darragh, J., Corvol, J. C., Pages, C., Arthur, S. J., Girault, J. A., and Caboche, J. (2005). Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J. Neurosci. 25, 11444–11454.
Breese, G. R., Duncan, G. E., Napier, T. C., Bondy, S. C., Iorio, L. C., and Mueller, R. A. (1987). 6-Hydroxydopamine treatments enhance behavioral responses to intracerebral microinjection of D1- and D2-dopamine agonists into nucleus accumbens and striatum without changing dopamine antagonist binding. J. Pharmacol. Exp. Ther. 240, 167–176.
Calabresi, P., Di Filippo, M., Ghiglieri, V., Tambasco, N., and Picconi, B. (2010). Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol. 9, 1106–1117.
Calabresi, P., Gubellini, P., Centonze, D., Sancesario, G., Morello, M., Giorgi, M., Pisani, A., and Bernardi, G. (1999). A critical role of the nitric oxide/cGMP pathway in corticostriatal long-term depression. J. Neurosci. 19, 2489–2499.
Calabresi, P., Maj, R., Pisani, A., Mercuri, N. B., and Bernardi, G. (1992a). Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J. Neurosci. 12, 4224–4233.
Calabresi, P., Pisani, A., Mercuri, N. B., and Bernardi, G. (1992b). Long-term potentiation in the striatum unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur. J. Neurosci. 4, 929–935.
Cao, X., Yasuda, T., Uthayathas, S., Watts, R. L., Mouradian, M. M., Mochizuki, H., and Papa, S. M. (2010). Striatal overexpression of DeltaFosB reproduces chronic levodopa-induced involuntary movements. J. Neurosci. 30, 7335–7343.
Carey, R. J. (1991). Chronic L-dopa treatment in the unilateral 6-OHDA rat: evidence for behavioral sensitization and biochemical tolerance. Brain Res. 568, 205–214.
Carlezon, W. A. Jr., Duman, R. S., and Nestler, E. J. (2005). The many faces of CREB. Trends Neurosci. 28, 436–445.
Carriere, A., Cargnello, M., Julien, L. A., Gao, H., Bonneil, E., Thibault, P., and Roux, P. P. (2008). Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 18, 1269–1277.
Carta, A. R., Tronci, E., Pinna, A., and Morelli, M. (2005). Different responsiveness of striatonigral and striatopallidal neurons to L-DOPA after a subchronic intermittent L-DOPA treatment. Eur. J. Neurosci. 21, 1196–1204.
Centonze, D., Gubellini, P., Picconi, B., Calabresi, P., Giacomini, P., and Bernardi, G. (1999). Unilateral dopamine denervation blocks corticostriatal LTP. J. Neurophysiol. 82, 3575–3579.
Contant, C., Umbriaco, D., Garcia, S., Watkins, K. C., and Descarries, L. (1996). Ultrastructural characterization of the acetylcholine innervation in adult rat neostriatum. Neuroscience 71, 937–947.
Corvol, J. C., Muriel, M. P., Valjent, E., Feger, J., Hanoun, N., Girault, J. A., Hirsch, E. C., and Herve, D. (2004). Persistent increase in olfactory type G-protein alpha subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson disease. J. Neurosci. 24, 7007–7014.
Costa-Mattioli, M., Sossin, W. S., Klann, E., and Sonenberg, N. (2009). Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26.
Cotzias, G. C., Van Woert, M. H., and Schiffer, L. M. (1967). Aromatic amino acids and modification of parkinsonism. N. Engl. J. Med. 276, 374–379.
Crittenden, J. R., Cantuti-Castelvetri, I., Saka, E., Keller-Mcgandy, C. E., Hernandez, L. F., Kett, L. R., Young, A. B., Standaert, D. G., and Graybiel, A. M. (2009). Dysregulation of CalDAG-GEFI and CalDAG-GEFII predicts the severity of motor side-effects induced by anti-parkinsonian therapy. Proc. Natl. Acad. Sci. U.S.A. 106, 2892–2896.
Darmopil, S., Martin, A. B., De Diego, I. R., Ares, S., and Moratalla, R. (2009). Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 66, 603–613.
Davie, J. R. (2003). MSK1 and MSK2 mediate mitogen- and stress-induced phosphorylation of histone H3: a controversy resolved. Sci STKE 2003, PE33.
Deak, M., Clifton, A. D., Lucocq, L. M., and Alessi, D. R. (1998). Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 17, 4426–4441.
DeLong, M. R. (1990). Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 13, 281–285.
Ding, J. D., Burette, A., Nedvetsky, P. I., Schmidt, H. H., and Weinberg, R. J. (2004). Distribution of soluble guanylyl cyclase in the rat brain. J. Comp. Neurol. 472, 437–448.
Ding, Y., Won, L., Britt, J. P., Lim, S. A., Mcgehee, D. S., and Kang, U. J. (2011). Enhanced striatal cholinergic neuronal activity mediates L-DOPA-induced dyskinesia in parkinsonian mice. Proc. Natl. Acad. Sci. U.S.A. 108, 840–845.
Fabbrini, G., Brotchie, J. M., Grandas, F., Nomoto, M., and Goetz, C. G. (2007). Levodopa-induced dyskinesias. Mov. Disord. 22, 1379–1389; quiz 1523.
Farnsworth, C. L., Freshney, N. W., Rosen, L. B., Ghosh, A., Greenberg, M. E., and Feig, L. A. (1995). Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 376, 524–527.
Fasano, S., Bezard, E., D’Antoni, A., Francardo, V., Indrigo, M., Qin, L., Dovero, S., Cerovic, M., Cenci, M. A., and Brambilla, R. (2010). Inhibition of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) signaling in the striatum reverts motor symptoms associated with L-dopa-induced dyskinesia. Proc. Natl. Acad. Sci. U.S.A. 107, 21824–21829.
Fienberg, A. A., Hiroi, N., Mermelstein, P. G., Song, W., Snyder, G. L., Nishi, A., Cheramy, A., O’Callaghan, J. P., Miller, D. B., Cole, D. G., Corbett, R., Haile, C. N., Cooper, D. C., Onn, S. P., Grace, A. A., Ouimet, C. C., White, F. J., Hyman, S. E., Surmeier, D. J., Girault, J., Nestler, E. J., and Greengard, P. (1998). DARPP-32: regulator of the efficacy of dopaminergic neurotransmission. Science 281, 838–842.
Fiorentini, C., Busi, C., Gorruso, E., Gotti, C., Spano, P., and Missale, C. (2008). Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol. Pharmacol. 74, 59–69.
Gerfen, C. R. (1992). The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu. Rev. Neurosci. 15, 285–320.
Gerfen, C. R., Engber, T. M., Mahan, L. C., Susel, Z., Chase, T. N., Monsma, F. J. Jr., and Sibley, D. R. (1990). D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429–1432.
Gerfen, C. R., Miyachi, S., Paletzki, R., and Brown, P. (2002). D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J. Neurosci. 22, 5042–5054.
Gerfen, C. R., Paletzki, R., and Worley, P. (2008). Differences between dorsal and ventral striatum in Drd1a dopamine receptor coupling of dopamine- and cAMP-regulated phosphoprotein-32 to activation of extracellular signal-regulated kinase. J. Neurosci. 28, 7113–7120.
Gingras, A. C., Raught, B., Gygi, S. P., Niedzwiecka, A., Miron, M., Burley, S. K., Polakiewicz, R. D., Wyslouch-Cieszynska, A., Aebersold, R., and Sonenberg, N. (2001). Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 15, 2852–2864.
Giorgi, M., D’Angelo, V., Esposito, Z., Nuccetelli, V., Sorge, R., Martorana, A., Stefani, A., Bernardi, G., and Sancesario, G. (2008). Lowered cAMP and cGMP signalling in the brain during levodopa-induced dyskinesias in hemiparkinsonian rats: new aspects in the pathogenetic mechanisms. Eur. J. Neurosci. 28, 941–950.
Glatt, C. E., and Snyder, S. H. (1993). Cloning and expression of an adenylyl cyclase localized to the corpus striatum. Nature 361, 536–538.
Gold, S. J., Hoang, C. V., Potts, B. W., Porras, G., Pioli, E., Kim, K. W., Nadjar, A., Qin, C., Lahoste, G. J., Li, Q., Bioulac, B. H., Waugh, J. L., Gurevich, E., Neve, R. L., and Bezard, E. (2007). RGS9-2 negatively modulates L-3,4-dihydroxyphenylalanine-induced dyskinesia in experimental Parkinson’s disease. J. Neurosci. 27, 14338–14348.
Gong, S., Zheng, C., Doughty, M. L., Losos, K., Didkovsky, N., Schambra, U. B., Nowak, N. J., Joyner, A., Leblanc, G., Hatten, M. E., and Heintz, N. (2003). A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925.
Guigoni, C., Doudnikoff, E., Li, Q., Bloch, B., and Bezard, E. (2007). Altered D(1) dopamine receptor trafficking in parkinsonian and dyskinetic non-human primates. Neurobiol. Dis. 26, 452–463.
Guillin, O., Diaz, J., Carroll, P., Griffon, N., Schwartz, J. C., and Sokoloff, P. (2001). BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 411, 86–89.
Herve, D., Levi-Strauss, M., Marey-Semper, I., Verney, C., Tassin, J. P., Glowinski, J., and Girault, J. A. (1993). G(olf) and Gs in rat basal ganglia: possible involvement of G(olf) in the coupling of dopamine D1 receptor with adenylyl cyclase. J. Neurosci. 13, 2237–2248.
Hornykiewicz, O. (1963). Die topische lokalisation und das verhalten von noradrenalin und dopamin (3-Hydroxytyramin) in der substantia nigra des normalen und Parkinsonkranken menschen. Wien. Klin. Wochenschr. 56, 426–427.
Hurley, M. J., Mash, D. C., and Jenner, P. (2001). Dopamine D(1) receptor expression in human basal ganglia and changes in Parkinson’s disease. Brain Res. Mol. Brain Res. 87, 271–279.
Hwang, D. Y., Fleming, S. M., Ardayfio, P., Moran-Gates, T., Kim, H., Tarazi, F. I., Chesselet, M. F., and Kim, K. S. (2005). 3,4-dihydroxyphenylalanine reverses the motor deficits in Pitx3-deficient aphakia mice: behavioral characterization of a novel genetic model of Parkinson’s disease. J. Neurosci. 25, 2132–2137.
Jones, M. W., Errington, M. L., French, P. J., Fine, A., Bliss, T. V., Garel, S., Charnay, P., Bozon, B., Laroche, S., and Davis, S. (2001). A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 4, 289–296.
Joyce, J. N. (1991). Differential response of striatal dopamine and muscarinic cholinergic receptor subtypes to the loss of dopamine. I. Effects of intranigral or intracerebroventricular 6-hydroxydopamine lesions of the mesostriatal dopamine system. Exp. Neurol. 113, 261–276.
Kawaguchi, Y., Wilson, C. J., Augood, S. J., and Emson, P. C. (1995). Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 18, 527–535.
Kim, E. K., and Choi, E. J. (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405.
Konitsiotis, S., Blanchet, P. J., Verhagen, L., Lamers, E., and Chase, T. N. (2000). AMPA receptor blockade improves levodopa-induced dyskinesia in MPTP monkeys. Neurology 54, 1589–1595.
Kumar, R., Riddle, L., Griffin, S. A., Grundt, P., Newman, A. H., and Luedtke, R. R. (2009). Evaluation of the D3 dopamine receptor selective antagonist PG01037 on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology 56, 944–955.
Lebel, M., Chagniel, L., Bureau, G., and Cyr, M. (2010). Striatal inhibition of PKA prevents levodopa-induced behavioural and molecular changes in the hemiparkinsonian rat. Neurobiol. Dis. 38, 59–67.
Li, L., Carter, J., Gao, X., Whitehead, J., and Tourtellotte, W. G. (2005). The neuroplasticity-associated arc gene is a direct transcriptional target of early growth response (Egr) transcription factors. Mol. Cell. Biol. 25, 10286–10300.
Long, X., Lin, Y., Ortiz-Vega, S., Yonezawa, K., and Avruch, J. (2005). Rheb binds and regulates the mTOR kinase. Curr. Biol. 15, 702–713.
Ma, L., Chen, Z., Erdjument-Bromage, H., Tempst, P., and Pandolfi, P. P. (2005). Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121, 179–193.
Mangiavacchi, S., and Wolf, M. E. (2004). D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J. Neurochem. 88, 1261–1271.
Marcellino, D., Ferre, S., Casado, V., Cortes, A., Le Foll, B., Mazzola, C., Drago, F., Saur, O., Stark, H., Soriano, A., Barnes, C., Goldberg, S. R., Lluis, C., Fuxe, K., and Franco, R. (2008). Identification of dopamine D1-D3 receptor heteromers: indications for a role of synergistic D1-D3 receptor interactions in the striatum. J. Biol. Chem. 283, 26016–26025.
Marshall, J. F., Navarrete, R., and Joyce, J. N. (1989). Decreased striatal D1 binding density following mesotelencephalic 6-hydroxydopamine injections: an autoradiographic analysis. Brain Res. 493, 247–257.
Martegani, E., Vanoni, M., Zippel, R., Coccetti, P., Brambilla, R., Ferrari, C., Sturani, E., and Alberghina, L. (1992). Cloning by functional complementation of a mouse cDNA encoding a homologue of CDC25, a Saccharomyces cerevisiae RAS activator. EMBO J. 11, 2151–2157.
Mela, F., Millan, M. J., Brocco, M., and Morari, M. (2010). The selective D(3) receptor antagonist, S33084, improves parkinsonian-like motor dysfunction but does not affect L-DOPA-induced dyskinesia in 6-hydroxydopamine hemi-lesioned rats. Neuropharmacology 58, 528–536.
Mendola, C. E., and Backer, J. M. (1990). Lovastatin blocks N-ras oncogene-induced neuronal differentiation. Cell Growth Differ. 1, 499–502.
Mons, N., and Cooper, D. M. (1994). Selective expression of one Ca(2+)-inhibitable adenylyl cyclase in dopaminergically innervated rat brain regions. Brain Res. Mol. Brain Res. 22, 236–244.
Nestler, E. J., Barrot, M., and Self, D. W. (2001). DeltaFosB: a sustained molecular switch for addiction. Proc. Natl. Acad. Sci. U.S.A. 98, 11042–11046.
Nowak, S. J., and Corces, V. G. (2004). Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 20, 214–220.
Obeso, J. A., Olanow, C. W., and Nutt, J. G. (2000). Levodopa motor complications in Parkinson’s disease. Trends Neurosci. 23, S2–S7.
Oh, J. D., Chartisathian, K., Ahmed, S. M., and Chase, T. N. (2003). Cyclic AMP responsive element binding protein phosphorylation and persistent expression of levodopa-induced response alterations in unilateral nigrostriatal 6-OHDA lesioned rats. J. Neurosci. Res. 72, 768–780.
Orban, P. C., Chapman, P. F., and Brambilla, R. (1999). Is the Ras-MAPK signalling pathway necessary for long-term memory formation? Trends Neurosci. 22, 38–44.
Oshiro, N., Yoshino, K., Hidayat, S., Tokunaga, C., Hara, K., Eguchi, S., Avruch, J., and Yonezawa, K. (2004). Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells 9, 359–366.
Pavon, N., Martin, A. B., Mendialdua, A., and Moratalla, R. (2006). ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol. Psychiatry 59, 64–74.
Phelps, P. E., Houser, C. R., and Vaughn, J. E. (1985). Immunocytochemical localization of choline acetyltransferase within the rat neostriatum: a correlated light and electron microscopic study of cholinergic neurons and synapses. J. Comp. Neurol. 238, 286–307.
Picconi, B., Bagetta, V., Ghiglieri, V., Paille, V., Di Filippo, M., Pendolino, V., Tozzi, A., Giampa, C., Fusco, F. R., Sgobio, C., and Calabresi, P. (2011). Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain 134, 375–387.
Picconi, B., Centonze, D., Hakansson, K., Bernardi, G., Greengard, P., Fisone, G., Cenci, M. A., and Calabresi, P. (2003). Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 6, 501–506.
Picconi, B., Paille, V., Ghiglieri, V., Bagetta, V., Barone, I., Lindgren, H. S., Bernardi, G., Angela Cenci, M., and Calabresi, P. (2008). l-DOPA dosage is critically involved in dyskinesia via loss of synaptic depotentiation. Neurobiol. Dis. 29, 327–335.
Pimoule, C., Schoemaker, H., Reynolds, G. P., and Langer, S. Z. (1985). [3H]SCH 23390 labeled D1 dopamine receptors are unchanged in schizophrenia and Parkinson’s disease. Eur. J. Pharmacol. 114, 235–237.
Rangel-Barajas, C., Silva, I., Lopez-Santiago, L. M., Aceves, J., Erlij, D., and Floran, B. (2011). l-DOPA-induced dyskinesia in hemiparkinsonian rats is associated with up-regulation of adenylyl cyclase type V/VI and increased GABA release in the substantia nigra reticulata. Neurobiol. Dis. 41, 51–61.
Richter, J. D., and Sonenberg, N. (2005). Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480.
Roux, P. P., Shahbazian, D., Vu, H., Holz, M. K., Cohen, M. S., Taunton, J., Sonenberg, N., and Blenis, J. (2007). RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 282, 14056–14064.
Ruvinsky, I., and Meyuhas, O. (2006). Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem. Sci. 31, 342–348.
Samadi, P., Bedard, P. J., and Rouillard, C. (2006). Opioids and motor complications in Parkinson’s disease. Trends Pharmacol. Sci. 27, 512–517.
Santini, E., Alcacer, C., Cacciatore, S., Heiman, M., Herve, D., Greengard, P., Girault, J. A., Valjent, E., and Fisone, G. (2009a). L-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J. Neurochem. 108, 621–633.
Santini, E., Heiman, M., Greengard, P., Valjent, E., and Fisone, G. (2009b). Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2, ra36.
Santini, E., Sgambato-Faure, V., Li, Q., Savasta, M., Dovero, S., Fisone, G., and Bezard, E. (2010a). Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PLoS ONE 5, e12322. doi: 10.1371/journal.pone.0012322
Santini, E., Valjent, E., and Fisone, G. (2010b). mTORC1 signaling in Parkinson’s disease and L-DOPA-induced dyskinesia: a sensitized matter. Cell Cycle 9, 2713–2718.
Santini, E., Valjent, E., Usiello, A., Carta, M., Borgkvist, A., Girault, J. A., Herve, D., Greengard, P., and Fisone, G. (2007). Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J. Neurosci. 27, 6995–7005.
Savasta, M., Dubois, A., Benavides, J., and Scatton, B. (1988). Different plasticity changes in D1 and D2 receptors in rat striatal subregions following impairment of dopaminergic transmission. Neurosci. Lett. 85, 119–124.
Schuster, S., Doudnikoff, E., Rylander, D., Berthet, A., Aubert, I., Ittrich, C., Bloch, B., Cenci, M. A., Surmeier, D. J., Hengerer, B., and Bezard, E. (2009). Antagonizing L-type Ca2+ channel reduces development of abnormal involuntary movement in the rat model of L-3,4-dihydroxyphenylalanine-induced dyskinesia. Biol. Psychiatry 65, 518–526.
Schuster, S., Nadjar, A., Guo, J. T., Li, Q., Ittrich, C., Hengerer, B., and Bezard, E. (2008). The 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor lovastatin reduces severity of L-DOPA-induced abnormal involuntary movements in experimental Parkinson’s disease. J. Neurosci. 28, 4311–4316.
Schwarzschild, M. A., Agnati, L., Fuxe, K., Chen, J. F., and Morelli, M. (2006). Targeting adenosine A2A receptors in Parkinson’s disease. Trends Neurosci. 29, 647–654.
Sebti, S. M., Tkalcevic, G. T., and Jani, J. P. (1991). Lovastatin, a cholesterol biosynthesis inhibitor, inhibits the growth of human H-ras oncogene transformed cells in nude mice. Cancer Commun. 3, 141–147.
Sgambato-Faure, V., Buggia, V., Gilbert, F., Levesque, D., Benabid, A. L., and Berger, F. (2005). Coordinated and spatial upregulation of arc in striatonigral neurons correlates with L-dopa-induced behavioral sensitization in dyskinetic rats. J. Neuropathol. Exp. Neurol. 64, 936–947.
Shenoy, S. K., and Lefkowitz, R. J. (2003). Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem. J. 375, 503–515.
Shieh, P. B., Hu, S. C., Bobb, K., Timmusk, T., and Ghosh, A. (1998). Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 20, 727–740.
Shinotoh, H., Inoue, O., Hirayama, K., Aotsuka, A., Asahina, M., Suhara, T., Yamazaki, T., and Tateno, Y. (1993). Dopamine D1 receptors in Parkinson’s disease and striatonigral degeneration: a positron emission tomography study. J. Neurol. Neurosurg. Psychiatr. 56, 467–472.
Shou, C., Farnsworth, C. L., Neel, B. G., and Feig, L. A. (1992). Molecular cloning of cDNAs encoding a guanine-nucleotide-releasing factor for Ras p21. Nature 358, 351–354.
Silverdale, M. A., Kobylecki, C., Hallett, P. J., Li, Q., Dunah, A. W., Ravenscroft, P., Bezard, E., and Brotchie, J. M. (2010). Synaptic recruitment of AMPA glutamate receptor subunits in levodopa-induced dyskinesia in the MPTP-lesioned nonhuman primate. Synapse 64, 177–180.
Stoof, J. C., and Kebabian, J. W. (1981). Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature 294, 366–368.
Tao, X., Finkbeiner, S., Arnold, D. B., Shaywitz, A. J., and Greenberg, M. E. (1998). Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20, 709–726.
Tekumalla, P. K., Calon, F., Rahman, Z., Birdi, S., Rajput, A. H., Hornykiewicz, O., Di Paolo, T., Bedard, P. J., and Nestler, E. J. (2001). Elevated levels of DeltaFosB and RGS9 in striatum in Parkinson’s disease. Biol. Psychiatry 50, 813–816.
Thomas, G., Siegmann, M., and Gordon, J. (1979). Multiple phosphorylation of ribosomal protein S6 during transition of quiescent 3T3 cells into early G1, and cellular compartmentalization of the phosphate donor. Proc. Natl. Acad. Sci. U.S.A. 76, 3952–3956.
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183.
Toki, S., Kawasaki, H., Tashiro, N., Housman, D. E., and Graybiel, A. M. (2001). Guanine nucleotide exchange factors CalDAG-GEFI and CalDAG-GEFII are colocalized in striatal projection neurons. J. Comp. Neurol. 437, 398–407.
Tsokas, P., Grace, E. A., Chan, P., Ma, T., Sealfon, S. C., Iyengar, R., Landau, E. M., and Blitzer, R. D. (2005). Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J. Neurosci. 25, 5833–5843.
Tsokas, P., Ma, T., Iyengar, R., Landau, E. M., and Blitzer, R. D. (2007). Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J. Neurosci. 27, 5885–5894.
Valjent, E., Aubier, B., Corbille, A. G., Brami-Cherrier, K., Caboche, J., Topilko, P., Girault, J. A., and Herve, D. (2006). Plasticity-associated gene Krox24/Zif268 is required for long-lasting behavioral effects of cocaine. J. Neurosci. 26, 4956–4960.
Valjent, E., Bertran-Gonzalez, J., Herve, D., Fisone, G., and Girault, J. A. (2009). Looking BAC at striatal signaling: cell-specific analysis in new transgenic mice. Trends Neurosci. 32, 538–547.
Valjent, E., Pascoli, V., Svenningsson, P., Paul, S., Enslen, H., Corvol, J. C., Stipanovich, A., Caboche, J., Lombroso, P. J., Nairn, A. C., Greengard, P., Herve, D., and Girault, J. A. (2005). Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. U.S.A. 102, 491–496.
van den Munckhof, P., Luk, K. C., Ste-Marie, L., Montgomery, J., Blanchet, P. J., Sadikot, A. F., and Drouin, J. (2003). Pitx3 is required for motor activity and for survival of a subset of midbrain dopaminergic neurons. Development 130, 2535–2542.
Visanji, N. P., Fox, S. H., Johnston, T., Reyes, G., Millan, M. J., and Brotchie, J. M. (2009). Dopamine D3 receptor stimulation underlies the development of L-DOPA-induced dyskinesia in animal models of Parkinson’s disease. Neurobiol. Dis. 35, 184–192.
Xie, G. Q., Wang, S. J., Li, J., Cui, S. Z., Zhou, R., Chen, L., and Yuan, X. R. (2009). Ethanol attenuates the HFS-induced, ERK-mediated LTP in a dose-dependent manner in rat striatum. Alcohol. Clin. Exp. Res. 33, 121–128.
Yoon, S., and Seger, R. (2006). The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24, 21–44.
Zachariou, V., Sgambato-Faure, V., Sasaki, T., Svenningsson, P., Berton, O., Fienberg, A. A., Nairn, A. C., Greengard, P., and Nestler, E. J. (2006). Phosphorylation of DARPP-32 at Threonine-34 is required for cocaine action. Neuropsychopharmacology 31, 555–562.
Zanassi, P., Paolillo, M., Feliciello, A., Avvedimento, E. V., Gallo, V., and Schinelli, S. (2001). cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J. Biol. Chem. 276, 11487–11495.
Zhou, F. M., Wilson, C. J., and Dani, J. A. (2002). Cholinergic interneuron characteristics and nicotinic properties in the striatum. J. Neurobiol. 53, 590–605.
Keywords: cAMP, dopamine- and cAMP-regulated phosphoprotein 32 kDa, extracellular signal-regulated protein kinases, immediate early genes, mammalian target of rapamycin, Parkinson’s disease
Citation: Feyder M, Bonito-Oliva A and Fisone G (2011) L-DOPA-induced dyskinesia and abnormal signaling in striatal medium spiny neurons: focus on dopamine D1 receptor-mediated transmission. Front. Behav. Neurosci. 5:71. doi: 10.3389/fnbeh.2011.00071
Received: 23 August 2011; Accepted: 06 October 2011;
Published online: 24 October 2011.
Edited by:
Riccardo Brambilla, San Raffaele Scientific Institute and University, ItalyReviewed by:
Paolo Calabresi, Santa Maria della Misericordia Hospital, ItalyDenis Hervé, Institut du Fer-à-Moulin, France
Copyright: © 2011 Feyder, Bonito-Oliva and Fisone. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Gilberto Fisone, Department of Neuroscience, Karolinska Institutet, Retzius väg 8, 171 77 Stockholm, Sweden. e-mail: gilberto.fisone@ki.se